

Abstract
Uric acid (UA) is a purine metabolite that in most mammals is degraded by the hepatic enzyme uricase to allantoin. Epidemiological studies have shown that an elevated UA level predicts the development of cognition and memory deficits; however, there is no direct evidence of this relationship, and the underlying mechanism is largely undefined. Here, we show that a high-UA diet triggers the expression of proinflammatory cytokines, activates the Toll-like receptor 4 (TLR4)/nuclear factor (NF)-κB pathway, and increases gliosis in the hippocampus of Wistar rats. We, subsequently, identify a specific inhibitor of NF-κB, BAY11-7085, and show that stereotactic injections of the inhibitor markedly ameliorate UA-induced hippocampal inflammation and memory deficits in C57BL/6 mice. We also found that NF-κB is activated in the primary cultured hippocampal cells after UA administration. Additionally, C57BL/6 mice that lack TLR4 are substantially protected against UA-induced cognitive dysfunction, possibly due to a decrease in inflammatory gene expression in the hippocampus. Importantly, magnetic resonance imaging confirms that hyperuricemia in rats and humans is associated with gliosis in the hippocampus. Together, these results suggest that UA can cause hippocampal inflammation via the TLR4/NF-κB pathway, resulting in cognitive dysfunction. Our findings provide a potential therapeutic strategy for counteracting UA-induced neurodegeneration.
SIGNIFICANCE STATEMENT This work demonstrates that a high-uric acid (UA) diet triggers the expression of proinflammatory cytokines, activates the Toll-like receptor 4 (TLR4)/nuclear factor (NF)-κB pathway, and increases gliosis in the hippocampus of Wistar rats. Inhibition of the NF-κB signaling pathway markedly ameliorates UA-induced hippocampal inflammation and cognitive dysfunction in C57BL/6 mice. TLR4-knock-out mice are substantially protected against UA-induced cognitive dysfunction, possibly due to a decrease in inflammatory gene expression in the hippocampus. Moreover, magnetic resonance imaging confirms that hyperuricemia in rats and humans are associated with gliosis in the hippocampus. Together, this study suggests that there is an important link between UA-induced cognitive dysfunction and hippocampal inflammation in rodents and humans, which may have remarkable implications in the treatment of UA-induced neurodegeneration.
Keywords: cognitive dysfunction, hippocampal inflammation, hyperuricemia, TLR4/NF-κB signaling pathway, uric acid
Introduction
Uric acid (UA), an end product of purine metabolism, can be degraded by the hepatic enzyme uricase and converted to allantoin (So and Thorens, 2010). Epidemiological evidence strongly indicates that the prevalence of hyperuricemia is increasing worldwide (Conen et al., 2004; Zhu et al., 2011). Elevated UA levels commonly accompany hypertension, hyperlipidemia, obesity, renal disease, insulin resistance, and metabolic syndrome (So and Thorens, 2010). Along with the expansion of the aging population, the potential association between serum UA and a variety of neurodegenerative disorders has been of particular interest (Vannorsdall et al., 2008). Recent studies have consistently concluded that hyperuricemia may be an independent risk factor for white matter atrophy and memory deficits (Schretlen et al., 2007a; Afsar et al., 2011; Verhaaren et al., 2013). Laboratory studies have also indicated that older adults with high concentrations of serum UA exhibit impaired cognitive function and working memory (Schretlen et al., 2007b). Despite the understanding of the association of cognitive dysfunction with high UA, little is known regarding the pathological effect of UA on the brain and the underlying mechanism. Only a few studies have shown that an elevated UA level might affect cognitive functioning through cerebrovascular changes (Feig et al., 2008; Jin et al., 2012).
The hippocampus is the critical brain region for learning and memory, and hippocampal dysfunction plays an important role in altered cognitive ability (Hitti and Siegelbaum, 2014). Hippocampal inflammation is an early event in the development of neurodegenerative disorders, and can produce permanent functional changes during brain development (Giovanello et al., 2009). Long-term modifications of the hippocampus induced by inflammation have a profound impact on brain excitability that is associated with neurological dysfunctions (Boitard et al., 2014). UA is thought to be a direct and potent inflammasome activator, possessing proinflammatory activity (Chen et al., 2006; Martinon et al., 2006; Kono et al., 2010; Shi, 2010). Previous studies have reported that one of the inflammation markers, C-reactive protein, can be upregulated by UA in human vascular cells and endothelial cells (Kang et al., 2005). In addition, the Toll-like receptor 4 (TLR4)/nuclear factor (NF)-κB signaling pathway activation has been implicated in several diseases that are induced by UA. For instance, hyperuricemia causes pancreatic β-cell death and dysfunction through the NF-κB signaling (Jia et al., 2013). UA induces renal infiltration of inflammatory cells and tubule expression of inflammatory mediators via NF-κB activation (Zhou et al., 2012). Furthermore, NF-κB signaling pathway mediates hypothalamic inflammation, which is involved in metabolic syndrome (Zhang et al., 2008; Purkayastha et al., 2011).
TLRs are a family of pattern recognition receptors that play a critical role in the innate immune system by activating proinflammatory signaling in response to microbial pathogens (Medzhitov, 2001). The best-characterized TLR, TLR4, interacts with myeloid differentiation primary response gene 88 and initiates a downstream signaling cascade, causing the activation of NF-κB, which then activates the transcription of proinflammatory genes (Zuany-Amorim et al., 2002). Increasing evidence has indicated that TLRs are expressed mainly in neurons, astrocytes, and microglia in the brain, and mediate the responses to infection, stress, or injury (Rolls et al., 2007; Okun et al., 2011). TLR4, a widely studied TLR in the innate immune response to Gram-negative bacterial infection, can be activated by bacterial lipopolysaccharide (Hu et al., 2013). UA-induced activation of TLR4 can reduce hippocampal pyramidal neuron dendrite length and impair hippocampal-dependent spatial reference memory in an inflammation-dependent manner (Okun et al., 2012). TLR signaling pathways culminate in the activation of the transcription factor NF-κB, which controls the expression of an array of inflammatory cytokine genes (Kawai and Akira, 2007).
In the present study, we find that serum UA can pass through the blood–brain barrier (BBB) and act as a potent inflammatory stimulus, thereby resulting in TLR4/NF-κB pathway activation as well as the accumulation of gliosis in the hippocampus. Thus, we provide an important foundation for a new and potent therapeutic target for the treatment of UA-induced cognitive dysfunction.
Materials and Methods
Animals.
The TLR4 knock-out (TLR4−/−) mice used in this study were purchased from the The Jackson laboratory and were backcrossed six generations into the C57BL/6 strain. Wild-type (WT) littermates were used as controls.
Pathogen-free male Wistar rats (7–8 weeks old; weight, 250–300 g) and male C57BL/6 mice (7–8 weeks old; weight, 20–25 g) were obtained from the Beijing Animal Center (Beijing, People's Republic of China). The mice were housed five per cage in clear plastic cages with wire grid lids in a colony with a 12 h light/dark cycle (lights on from 7:00 A.M. to 7:00 P.M.) at a constant temperature. Access to food and water was unrestricted. The animals were acclimatized for 7 d before the experiment.
To establish the hyperuricemia model, rats were provided with a high-UA diet (HUAD) containing 2% oxonic acid (OA) and 2% UA for periods ranging from 1 d to 12 weeks, as previously described (Kim et al., 2000; Mazzali et al., 2001). All animal protocols in this study were performed in accordance with the guidelines established by the Association for Assessment and Accreditation of Laboratory Animal Care.
Antibodies and chemicals.
The primary antibodies used in immunofluorescence staining included the following: mouse monoclonal neuronal nuclei [neuronal-specific nuclear protein (NeuN); catalog #MAB377, Millipore]; rabbit monoclonal ionized calcium binding adaptor-1 (Iba-1; catalog #019-19741, Wako); goat polyclonal Iba-1 (catalog #ab5076, Abcam); mouse monoclonal glial fibrillary acidic protein (GFAP; catalog #C9205, Sigma-Aldrich); and rabbit monoclonal NF-κB (catalog #5970, Cell Signaling Technology). The following fluorescently labeled secondary antibodies were used: Cy-3 conjugate donkey anti-goat (catalog #A0502, Beyotime Biotechnology); Cy-3 conjugate goat anti-mouse (catalog #A0521, Beyotime Biotechnology); donkey anti-rabbit IgG heavy chain and light chain (H+L) Alexa Fluor 488 (catalog #ab150073, Abcam); and 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI; catalog #D9542, Sigma-Aldrich). A commercial kit for the extraction of total mRNA from the hippocampus was purchased from Axygen Scientific (catalog #AP-MN-MS-RNA-50). The reverse transcription kits and fluorescence quantification kits for quantitative real-time PCR (qRT-PCR) were purchased from Bio-Rad (iTaq Universal SYBR Green Supermix, catalog #1725121). The primers for qRT-PCR were designed and synthesized by Sangon Biotech. Reagents for serum biochemical analysis were purchased from Sichuan Maccura Biotechnology Co., Ltd. UA, OA, urea, and allantoin were purchased from Nanjing Chemlin Chemical Industry Co., Ltd., and BAY11-7085 (catalog #B4845) was purchased from TCI. All of the chemicals used in this study were of analytical grade.
Preparation of reagents.
The UA used in stereotactic injection administration in vivo or in cell culture was prepared as follows: UA was dissolved in saline, sonicated for 30 min, and filtered with a 0.22 μm membrane filter, followed by observation under a microscope to confirm the absence of UA crystals.
UA determination in hippocampal tissue.
Under general ketamine/xylazine anesthesia, hippocampal tissue was collected from rats with hyperuricemia and control rats after perfusion with 0.9% saline. Hippocampal tissues were then washed with 0.9% saline. The UA concentration in the hippocampal tissue was quantified by liquid chromatography tandem mass spectrometry (LC-MS/MS; Dai et al., 2007). The LC-MS/MS was composed of a Shimadzu high-performance liquid chromatography system consisting of two LC-20ADXR pumps (Shimadzu) and an AB Sciex 3200 QTRAP mass spectrometer. The mobile phases used included an acetonitrile and a water phase (10 mm ammonium acetate at pH 4.5), with a 5:95 volume ratio in the isocratic elution mode. The flow rate was 0.3 ml/min, and the injection volume was 5 μl. UA was detected by ESI-MS in negative ion mode with a collision energy of −20 V. The turbo heater temperature was set at 600°C. The nebulizing gas, turbo heater gas, and curtain gas flow rates were 45, 60, and 10 L/min, respectively. The ion spray voltage was −4 kV. The ions monitored for multiple reaction monitoring (MRM) were as follows: the parent ion of UA was mass-to-charge ratio (m/z) 166.9 and the monitored MRM ion was m/z 123.9. Using the LC-MS/MS method, the concentration of UA is linear from 10 to 8000 ng/ml.
Serum biochemistry analysis.
During the experimental period, the rats were fasted overnight in advance of sampling, and blood samples were obtained by cardiac puncture. Clinical chemistry analysis of the serum was performed on an Hitachi 7020 automatic biochemistry analyzer using appropriate kits with the following parameters: alanine aminotransferase (ALT), aspartate aminotransferase (AST), serum UA (SUA), serum glucose (GLU), blood urea nitrogen (BUN), serum creatinine (CREA).
Determination of superoxide dismutase activity.
Hippocampal tissues were homogenized by sonication in a sucrose buffer solution (10 mm Tris-HCl, pH 7.4, 0.25 m sucrose and 1 mm EDTA). The total superoxide dismutase (SOD) activity was determined in the supernatant obtained after centrifugation at 20,000 × g for 20 min at 4°C using SOD Assay Kit-WST (Dojindo Molecular Technologies Ltd.). The SOD concentration (in units per milliliter) that produced 50% inhibition of the WST (water-soluble tetrazolium salt) reaction was determined (IC50) using a standard SOD concentration (MP Biomedicals). Following this, the dilution rate of the rat hippocampus that established the IC50 was determined, and the unit concentration (in units per milliliter) of the extract was calculated. Each sample was analyzed in duplicate, and the results were expressed as enzyme activity per milligrams of protein.
RNA isolation and quantitative real-time PCR detection.
Total RNA was isolated from hippocampal tissue and primary cultured hippocampal cells with total RNA isolation kit (Axygen). RNA was quantified by spectrophotometry at 260 nm and subjected to cDNA synthesis using RevertAid First Strand cDNA Synthesis Kit for qPCR (Bio-Rad). Levels of mRNA for Nfkbia, Ikbkb, Ikbke, Il6, Il1b, Tnfa, Ccl2, Socs3, Tlr4, Hif-1α, and Gapdh (internal control) were measured using the CFX96 Real-Time System (Bio-Rad). The expression level of each gene was normalized to a housekeeping gene (Gapdh) and expressed as a percentage of chow-fed control or saline control. The primer sequences that were used for quantitative real-time PCR analyses are provided in Table 1.
Table 1.
List of all primer sequences used
| Species | Name | Sequence (forward) | Sequence (reverse) |
|---|---|---|---|
| Rats | Gapdh | AACGACCCCTTCATTGAC | TCCACGACATACTCAGCAC |
| Nfkbia | TGCCTGGCCAGTGTAGCAGTCTT | CAAAGTCACCAAGTGCTCCACGAT | |
| Tnfa | GCTCCCTCTCATCAGTTCCA | CTCCTCTGCTTGGTGGTTTG | |
| Il1b | TACAAGGAGAGACAAGCAACGACA | GATCCACACTCTCCAGCTGCA | |
| Il6 | CAGAGGATACCACCCACAACAGA | CAGTGCATCATCGCTGTTCATACA | |
| Ikbkb | AGGGTGACTAAGTCGAGAC | ACAGCCAGGATATGGTACG | |
| Ikbke | ACCACTAACTACCTGTGGCAT | ACTGCGAATAGCTTCACGATG | |
| Ccl2 | TGTTCACAGTTGCTGCCTGT | TGCTGCTGGTGATTCTCTTG | |
| Socs3 | GTCACCCACAGCAAGTTTCC | TCCAGTAGAATCCGCTCTCC | |
| Hif1a | CGATGACACGGAAACTGAAG | CAGAGGCAGGTAATGGAGACA | |
| Tlr4 | CCCTGCCACCATTTACAGTT | ATCAGAGTCCCAGCCAGATG | |
| Mice | Gapdh | GGCACAGTCAAGGCTGAGAATG | ATGGTGGTGAAGACGCCAGTA |
| Nfkbia | TCGCTCTTGTTGAAATGTGG | CTCTCGGGTAGCATCTGGAG | |
| Tnfa | CCACCATCAAGGACTCAAATG | GAGACAGAGGCAACCTGACC | |
| Il1b | CTCACAAGCAGAGCACAAGC | TCCAGCCCATACTTTAGGAAGA | |
| Il6 | CGGAGAGGAGACTTCACAGAG | CATTTCCACGATTTCCCAGA | |
| Ikbkb | GCCTTATGAACGAGGACGAG | CTGTCTGGGCTTCCACTCAC | |
| Ikbke | GCCATCCCAGGCAGTATCTA | TTCCAAGACCAGACCTCCAG | |
| Ccl2 | TCTCTCTTCCTCCACCACCAT | GCTCTCCAGCCTACTCATTGG | |
| Socs3 | GTCACCCACAGCAAGTTTCC | TCCAGTAGAATCCGCTCTCC | |
| Hif1a | CCCATTAGCAGGTGAAGGAA | CCAGAATCAAACCAAACCAA | |
| Tlr4 | GCACTGACACCTTCCTTTCC | GCCTTAGCCTCTTCTCCTTCA |
Immunofluorescence staining.
Indirect immunofluorescence staining was performed according to the procedures described previously. Briefly, cells or brain cryosections were incubated with the specific primary anti-p65 NF-κB antibody (1:400; Cell Signaling Technology), anti-NeuN antibody (1:100; Millipore), anti-Iba-1 antibody (1:500; Abcam), and anti-GFAP antibody (1:400; Sigma-Aldrich) followed by staining with donkey anti-rabbit IgG H&L (1:500; Alexa Fluor 488, Abcam), Cy3-label goat anti-mouse IgG H&L (1:500; Beyotime Biotechnology), and Cy3-label donkey anti-goat IgG H&L (1:500; Beyotime Biotechnology). Nuclear costaining was achieved by incubating sections in 1× PBS containing DAPI (2 g/ml) for 10 min. Slides were viewed with an Olympus BX53F microscope equipped with a digital camera (model DP80, Olympus). And NF-κB-positive cells were counted by ImageJ software.
Behavioral testing.
The Morris water maze paradigm was used to assess spatial learning by training rodents to locate a hidden platform (Vorhees and Williams, 2006). Briefly, the apparatus consists of a large, white, circular pool with a white platform that is submerged below the surface of the water. During training, the platform was hidden in one quadrant of the maze from the sidewall. The animal was gently placed into the water facing the wall at one of four randomly chosen quadrants that were separated by 90°. The time required (latency) to find the hidden platform with a 90 s limit was recorded by a blinded observer. Animals that failed to find the platform within 90 s were assisted to the platform. Animals were allowed to remain on the platform for 15 s on the first trial and 10 s on all subsequent trials. A probe trial of 90 s was given 24 h after the final learning trial. The percentage of time spent in the quadrant where the platform was previously located was recorded.
Stereotactic injection, cannulation, and administration.
Under general ketamine/xylazine anesthesia, C57BL/6 mice and TLR4−/− mice were positioned in a small-animal stereotaxic instrument, and the cranial surface was exposed (DiCarlo et al., 2001). A stainless steel guide cannula was stereotaxically implanted into the lateral cerebral ventricle of the mice (anteroposterior, −0.42 mm; mediolateral, ±1.5 mm; dorsoventral, −2.2 mm]. After cannulation, the mice were treated with ampicillin sodium (160,000 U/ml, i.m.; 0.1 ml/mice) for 4 d. After 4 d of antimicrobial therapy and a 3 d recovery period, the cannulated mice were treated daily with 2 μl of UA (600 ng/ml, 0.6 μl/min) for 4, 7, 14, or 21 d, respectively.
After 7 d of recovery, the cannulated animals were administered 2 μl of saline, UA (600 ng/ml), urea (600 ng/ml), or allantoin (600 ng/ml) daily for 21 d. To inhibit NF-κB activation in the hippocampus, 2 μl of BAY11-7085 (500 nm) was injected 30 min before the injection of UA. The infusion rates for saline, UA, urea, allantoin, and BAY11-7085 were 0.6 μl/min.
Primary culture of hippocampal neurons.
Primary cultured hippocampal neurons were prepared from the embryonic brains of rats at embryonic day 18 (E18) to E19 (Kim et al., 2006). Briefly, hippocampi were dissociated and stored on ice in DMEM/F12 (catalog #SH30023.01B, Hyclone) supplemented with 10% horse serum (catalog #26050088, Invitrogen). Tissues were transferred into 0.125% trypsin (catalog #2520056, Invitrogen) and incubated for 15–30 min in 37°C. Tissues were then gently pipetted 10–15 times, and uniform cellular dissociation was achieved. Cells were seeded into six-well culture plates coated with poly-l-lysine (catalog #P8954, Sigma-Aldrich). After 4–6 h, the medium was changed to adult neuronal growth medium consisting of neurobasal medium (catalog #A1371001, Invitrogen), B-27 supplement (catalog #12587010, Invitrogen), and 0.5 mm l-glutamine (catalog #25030149, Invitrogen). Neuronal cultures were maintained in an atmosphere of 10% CO2 at 37°C in a humidified incubator. Seven days after seeding, the primary culture of hippocampal neurons was exposed to UA (600 ng/ml) for 24 and 48 h.
Immunofluorescence for primary cultured cells.
Primary hippocampal neurons and glial cells were fixed with 4% paraformaldehyde for 5 min, followed by penetration with Triton X-100 (catalog #85111, Invitrogen). After blocking with 5% BSA (catalog #A70030, Sigma), the cells were incubated with primary antibodies to NeuN (1:100; Millipore), GFAP (1:400; Sigma-Aldrich), Iba-1 (1:500; Abcam), and p65/RelA (1:400; Cell Signaling Technology) at 4°C overnight, respectively. After washing, the cells were incubated with the appropriate secondary antibodies to donkey anti-rabbit IgG H&L (1:500; Alexa Fluor 488, Abcam), Cy3-label goat anti-mouse IgG H&L (1:500; Beyotime Biotechnology), or Cy3-label donkey anti-goat IgG H&L (1:500; Beyotime Biotechnology) at 37°C for 2 h. Nuclear staining was achieved by incubating sections in 1× PBS containing DAPI (2 g/ml) for 10 min. Images were captured with an Olympus BX53F microscope equipped with a digital camera (model DP80, Olympus).
Brain magnetic resonance imaging measurement.
Wistar rats were fed a standard laboratory chow or a HUAD for 12 weeks continuously. High-resolution magnetic resonance imaging (MRI) acquisitions were performed on a 7 T Avance 600 MHz/89 mm wide-bore vertical MR spectrometer (BioSpin, Bruker) using a 25-mm-inner diameter 1H birdcage coil. The 7 T MRI system was equipped with actively shielded gradient coils (maximum gradient strength, 100 G/cm) and a Paravision (version 5.1) console interface.
All rats underwent isoflurane anesthesia in an induction chamber. The rats were placed on a bite bar, and their heads were placed into a radiofrequency coil and secured to a cradle created specifically for the MRI system. The coil was then inserted vertically into a scanner heated to maintain thermoneutrality (32°C). The coil was equipped with an adjustable anesthetic flow and vacuum system to maintain sedation throughout the experiment. Total scan time was 1–1.5 h, during which anesthesia was titrated to ensure appropriate sedation. Following the imaging paradigm (Table 2), rats were removed from the coil and allowed to recover in their home cage (Pekny and Nilsson, 2005; Jeon et al., 2012).
Table 2.
High-resolution MRI protocol for quantitative assessment of hippocampal inflammation
| Sequence type | TR/TE (ms) | FOV (mm) | Acquisition/reconstruction resolutions | Acquisition time | |
|---|---|---|---|---|---|
| Triplot GE | FLASH | 100/6 | 80 × 80 | 12 s | |
| T2W | Multislice RARE | 2600/33, 24 slices (slice thickness, 1 mm), FA = 180° | 45 × 45 | 176 × 176 μm | 1 min 24 s |
| T1W | Multislice, multiecho | 500/8, 24 slices (slice thickness = 1 mm), FA = 180° | 45 × 45 | 234 × 234 μm | 2 min 24 s |
| T2Map | Multislice, multiecho | 2200/11–110, 24 slices (slice thickness = 1 mm), FA = 180° | 45 × 45 | 176 × 176 μm | 7 min 2 s |
| DTI | EPI | 4000/32, 30 diffusion directions, b = 670 s/mm2, slice thickness = 1 mm, FA = 90° | 45 × 45 | 351 × 351 μm | 9 min 20 s |
TR, Recycle delay; TE, echo time; FOV, field of view; GE, gradient echo; FA, flip angle; RARE, rapid acquisition with refocused echoes; T1W, T1 weighted; T2W, T2 weighted; DTI, diffusion tensor imaging; EPI, echo planar imaging.
Retrospective study of brain MRI in humans.
Magnetic resonance brain examinations that used hippocampus imaging protocols with coronal T2 fast spin echo (FES) fluid attenuated inversion recovery (FLAIR) sequences and that were performed at West China Hospital of Sichuan University were reviewed for two inclusion criteria: the availability of high quality coronal views of the hippocampus and the absence of clinical abnormalities that might confound interpretation. For the 51 subjects who met our inclusion criteria, their electronic medical records were viewed to determine serum UA concentration, gender, age, body weight, and final diagnosis. The exclusion criteria were as follows: absence of serum biochemical analysis for UA; age of <16 or >70 years; diagnosis of neurodegenerative disorder (e.g., multiple sclerosis); metabolic disease (hyperglycemia, hyperlipemia, fatty liver, hyperinsulinemia, diabetes, and obesity); serum biochemical parameter [including cholesterol, triglyceride, high-density lipoprotein, low-density lipoprotein (LDL), and glucose levels] abnormalities without definite metabolic disease; cerebral atrophy; and history of bariatric disease. Twenty-two subjects were excluded from the study [age, <16 years (n = 2); age, >70 years (n = 1); metabolic disease (n = 5); serum biochemical parameter abnormalities (n = 9); brain disease (n = 5)], yielding a total of 29 study participants. All subjects were classified into the following two groups: the hyperuricemia group (fasting serum UA concentration, >436 μm for men, and >378 μm for women); and the normal group. The basal characteristics of the study population are provided in Table 3. Regions of interest (ROIs) in the bilateral hippocampus were defined by a neuroradiologist, who was blinded to all clinical information. Mean ROI signal intensity, SD, and ROI area were measured using proprietary software on the PACS workstation (Centricity, GE Healthcare). Ratios were calculated by comparing the mean signal intensity in the hippocampus on each side with that in the ipsilateral hippocampus ROI. Our study was approved by the Medical Ethics Committee (Human) of West China Hospital and Sichuan University. All subjects had been informed and approved participation in this study, and “informed consent” was obtained from all subjects. The methods were performed in accordance with the approved guidelines.
Table 3.
Basal characteristics of the study population comparing control subjects and subjects with hyperuricemia
| Control subjects (n = 14) | Subjects with hyperuricemia (n = 15) | p Value | |
|---|---|---|---|
| Gender (n) | |||
| Female | 7 | 8 | |
| Male | 7 | 7 | |
| Age (years) | 46.21 ± 15.36 | 44.53 ± 18.02 | 0.3943 |
| Body weight (kg) | 57.23 ± 3.68 | 59.06 ± 2.57 | 0.2764 |
| UA (μmol/L) | 278.21 ± 48.60 | 439.07 ± 42.27 | <0.001 |
| TBIL (μmol/L) | 11.26 ± 1.08 | 12.98 ± 1.68 | 0.3445 |
| DBIL (μmol/L) | 3.87 ± 0.54 | 4.54 ± 0.76 | 0.4563 |
| IBIL (μmol/L) | 6.60 ± 0.65 | 8.56 ± 1.14 | 0.3429 |
| ALT (IU/L) | 24.34 ± 4.33 | 25.95 ± 4.18 | 0.9236 |
| AST (IU/L) | 22.54 ± 3.41 | 21.79 ± 2.53 | 0.3814 |
| TP (g/L) | 68.45 ± 1.08 | 69.26 ± 4.60 | 0.4713 |
| ALB (g/L) | 43.42 ± 0.75 | 46.76 ± 3.11 | 0.0578 |
| GLB (g/L) | 24.65 ± 0.94 | 23.17 ± 1.85 | 0.6732 |
| GLU (mmol/L) | 4.89 ± 0.14 | 4.78 ± 0.33 | 0.8571 |
| BUN (mmol/L) | 5.74 ± 0.35 | 4.93 ± 0.58 | 0.3429 |
| CREA (μmol/L) | 65.87 ± 3.67 | 83.18 ± 6.53 | 0.1056 |
| Cys-C (mg/L) | 0.89 ± 0.03 | 0.92 ± 0.06 | 0.5468 |
| TG (mmol/L) | 1.32 ± 0.24 | 1.35 ± 0.17 | 0.9543 |
| CHOL (mmol/L) | 4.16 ± 0.29 | 3.89 ± 0.34 | 0.6517 |
| HDL-CHOL (mmol/L) | 1.32 ± 0.13 | 1.39 ± 0.19 | 0.8345 |
| LDL-CHOL (mmol/L) | 2.13 ± 0.21 | 2.26 ± 0.24 | 0.6067 |
| ALP (IU/L) | 65.78 ± 4.58 | 76.13 ± 12.35 | 0.3542 |
| GGT (IU/L) | 21.50 ± 5.23 | 19.55 ± 3.28 | 0.4356 |
| CK (IU/L) | 91.45 ± 13.23 | 118.27 ± 15.69 | 0.5409 |
| LDH (IU/L) | 154.25 ± 10.93 | 169.91 ± 13.59 | 0.4321 |
| HBL (IU/L) | 124.67 ± 7.68 | 134.27 ± 12.34 | 0.5634 |
| Na (mmol/L) | 142.78 ± 5.79 | 142.35 ± 9.14 | 0.4843 |
| K (mmol/L) | 3.89 ± 0.09 | 3.92 ± 0.24 | 0.8254 |
| Cl (mmol/L) | 106.23 ± 0.85 | 105.48 ± 5.32 | 0.3489 |
| CO2 (mmol/L) | 24.67 ± 0.62 | 23.98 ± 1.56 | 0.1739 |
| β-HBA (mmol/L) | 0.15 ± 0.02 | 0.20 ± 0.03 | 0.5236 |
| Ca (mmol/L) | 2.19 ± 0.04 | 2.23 ± 0.05 | 0.1542 |
| Mg (mmol/L) | 0.91 ± 0.03 | 0.98 ± 0.06 | 0.0367 |
| PO4 (mmol/L) | 1.21 ± 0.04 | 1.22 ± 0.13 | 0.8659 |
The values are represented as the mean ± SEM. P < 0.001, compared with the control). TBIL, Total bilirubin; DBIL, Direct bilirubin; IBIL, Indirect bilirubin; TP, Total protein; ALB, Albumin; GLB, Globulin; Cys-C, Cystatin-C; TG, Triglyceride; CHOL, Cholesterol; HDL, High density lipoprotein; LDL, Low density lipoprotein; ALP, Alkaline phosphatase; GGT, Glutamyl transpeptadase; CK, Creatine kinase; LDH, Lactic dehydrogenase; HBL, Hemolysin BL; HBA, Hemoglobin A.
Statistical analysis.
Data are presented as the mean ± SEM. Differences between two means were assessed by unpaired, two-tailed Student's t test. Data involving more than two means were evaluated by one-way ANOVA followed by Tukey's post hoc tests (SigmaStat, SyStat; and GraphPad Prism, GraphPad Software). p values <0.05 were considered to be statistically significant. All values included in the figure legends represent the mean ± SEM (*p < 0.05; **p < 0.01; ***p < 0.001).
Results
Systemic hyperuricemia induces cognitive dysfunction in rodents
To investigate the effect of UA on learning and memory, a classic rat model of hyperuricemia was generated based upon a previously reported strategy (Kim et al., 2000; Mazzali et al., 2001). Rats were fed a HUAD that contained 2% OA and 2% UA continuously for 12 weeks. UA levels in rodents are lower than those in humans because of the presence of uricase, which is an hepatic enzyme that degrades UA into allantoin (Johnson et al., 2009). Therefore, OA, a uricase inhibitor, was added as a supplement to the diet. Because OA is a partial inhibitor with a short half-life and because rats that were treated with OA alone developed only mild hyperuricemia, we added UA as a supplement. As a result, there was a threefold increase in the serum UA concentration, from 75.8 μm in control rats to 258.3 μm in HUAD-fed rats (Fig. 1A, **p < 0.01), indicating that we successfully induced the hyperuricemia rat model. Likewise, the serum levels of ALT, AST, BUN, CREA, and GLU were measured using serum biochemistry analysis (Fig. 1B–F). The serum BUN and CREA levels in control rats were 7.2 mm and 29.2 μm, respectively, whereas they increased to 9.0 mm and 42.3 μm in the HUAD group, respectively (Fig. 1D,E, *p < 0.05). These results suggested the presence of kidney dysfunction induced by a HUAD. In addition, consistent with our previous study, the serum glucose level increased by 20% in HUAD-fed rats (Fig. 1F), suggesting that UA may induce insulin resistance and metabolic disorders (Masuo et al., 2003; Cheung et al., 2007).
Figure 1.

Systemic hyperuricemia induces cognitive dysfunction in rodents. A–F, The time course of serum UA (A), ALT (B), AST (C), BUN (D), CREA (E), and GLU (F) concentrations after the onset of HUAD feeding (n = 6 rats per group). G, The amount of UA in hippocampal tissue was determined in rats fed chow or a HUAD for up to 12 weeks (Student's t tests, **p < 0.01, n = 6 rats per group). H, I, Spatial learning of the hyperuricemia rats was assessed in a Morris water maze. Each trial is presented as the average of four individual tests, and four trials were performed with the escape latency to the platform being recorded (Student's t tests, *p < 0.05, n = 6 rats per group). J, Comparison of SOD activities in hippocampal tissue of hyperuricemia rats and control rats (n = 6 rats per group). All displayed values are the mean ± SEM. *p < 0.05, **p < 0.01 vs chow-fed controls.
To investigate whether UA could pass through the BBB and act on the brain directly, we assessed UA level in hippocampal tissue using LC-MS/MS. We found that the amount of UA in hippocampal tissue of HUAD-fed rats was 739.39 ng/g, whereas it was 309.55 ng/g in chow-fed controls (Fig. 1G, **p < 0.01). These results indicated that UA could pass through the BBB and be deposited at significant levels in hippocampal tissue. Additionally, the amount of UA in the hippocampus of HUAD-fed rats was significantly elevated.
The Morris water maze is one of the most widely used methods for studying the neural mechanisms of spatial learning and memory (Vorhees and Williams, 2006). Moreover, the hippocampus is the critical brain region for learning and memory, and hippocampus dysfunction plays an important role in altered cognitive ability (Hitti and Siegelbaum, 2014). To assess spatial learning and working memory after 12 weeks of HUAD administration, we used the Morris water maze paradigm, in which rats were trained to locate a hidden and submerged platform. Interestingly, the long-term HUAD-fed rats showed significantly reduced spatial learning and memory in an escape latency and probe trial (Fig. 1H,I, *p < 0.05), indicating cognitive dysfunction.
Oxidative stress is involved in the pathogenesis of cognitive impairment (Squadrito et al., 2000). We detected the activity of SOD, an important antioxidant enzyme, in the hippocampus of HUAD-fed rats by using a SOD Assay Kit (Dojindo Molecular Technologies Ltd.). The results showed that UA decreased SOD activity by 19% (Fig. 1J), suggesting that UA may induce oxidative stress.
Serum UA elevation leads to hippocampal chronic neuroinflammation and gliosis
Chronic hippocampal inflammation is associated with obesity-induced diabetes and is considered to be a risk factor for neurodegeneration (Jeon et al., 2012; Nguyen et al., 2014). We analyzed the transcription levels of inflammatory genes in the hippocampus of rats using qRT-PCR. The results showed that the expressions of the genes Nfkbia, Il1b, Il6, Ikbkb, and Tlr4 were significantly augmented after HUAD exposure for 12 weeks (Fig. 2G, *p < 0.05), indicating that systemic hyperuricemia induced inflammatory gene expression and TLR4/NF-κB signaling pathway activation in hippocampal tissue. Gliosis is a characteristic pathologic state in many brain disorders, wherein cytokines are the effectors (Thaler et al., 2012). Astrocyte activation and proliferation result in reactive gliosis, a reaction with specific structural and functional characteristics (Pekny and Nilsson, 2005). To investigate whether UA could cause astroglial responses in the hippocampus, the expression levels of microglia-specific markers GFAP and Iba-1 were analyzed by immunofluorescence. Within 12 weeks of HUAD administration, the fluorescence intensity of GFAP in the CA1, CA2, and CA3 regions of the hippocampus increased by approximately threefold and almost always remained at a high intensity (Fig. 2A–F; quantified on the right), suggesting an effect of UA in promoting astrocyte accumulation in this brain area. Furthermore, the astrocytes became increasingly enlarged, which is indicative of increased activity. We also found that the amount of the microglia-specific cytoplasmic marker Iba-1 clearly increased by more than twofold in the hippocampus after 12 weeks of HUAD exposure when compared with control rats. Concomitantly, microglia increased in size and became more active. Collectively, these results showed that hippocampal inflammation induced by HUAD was closely related to reactive gliosis in the hippocampus.
Figure 2.

Serum UA elevation results in chronic hippocampal neuroinflammation and gliosis. A–F, Hippocampal CA1 (A, B), CA2 (C, D), and CA3 (E, F) sections of the rats fed a standard chow or a HUAD were immunostained for GFAP (left) and Iba-1 (right). Immunofluorescence detection of the astrocytic marker GFAP protein and of the microglial marker Iba-1 in the rat hippocampus (4 μm) from rats fed either chow or a HUAD for 12 weeks. The image is displayed at 200× the original magnification and was used for the quantification of hippocampal astrocyte and microglia numbers. The mean numbers of hippocampal astrocytes and microglia in rats fed either chow or a HUAD were quantified in the regions displayed on the right (mean ± SEM; n = 6 rats per group). *p < 0.05; **p < 0.01; ***p < 0.001 vs chow-fed controls. G, Time course of the induction of mRNA expression of inflammatory mediators, including proinflammatory cytokines (Il6, Il1b, Tnfa, Socs3, and Ccl2) and TLR4/NF-κB signaling (Tlr4, Nfkbia, Ikbkb, and Ikbke) in the hippocampi of rats that were fed chow or a HUAD for up to 12 weeks (n = 6 rats per group). All mRNA species were quantified relative to the expression of the housekeeping gene Gapdh and are presented as fold changes relative to chow-fed controls. All displayed values are reported as the mean ± SEM. *p < 0.05 vs chow-fed control.
UA acts as an inflammatory stimulus and activates NF-κB in the hippocampus
Whether UA can directly induce the expression of proinflammatory cytokines in the hippocampus remains largely unknown. To address this issue, UA was stereotactically and continuously infused into the hippocampi of C57BL/6 mice. All mice were injected with 2 μl of UA (600 ng/ml) at a controlled rate of 0.6 μl/min. In addition, to exclude the possibility of chemical irritation of UA, an equal dose of urea and allantoin, metabolites of UA in rodents, was injected stereotactically. As expected, 21 d of stereotactic injection with UA produced a significant increase in the expression of proinflammatory cytokines in the hippocampus, whereas allantoin and urea did not induce such effects (Fig. 3E). These results suggest that UA could directly induce hippocampal inflammation rather than simply cause chemical irritation. Furthermore, we detected the expression of Nfkbia in the hippocampus and found that UA markedly increased the mRNA levels of proinflammatory cytokines and Nfkbia after 21 d of administration; however, urea and allantoin did not exhibit such effects (Fig. 3E). These results indicate that the pathogenic effect of UA, but not transient stimulation, may contribute to hippocampal inflammation.
Figure 3.
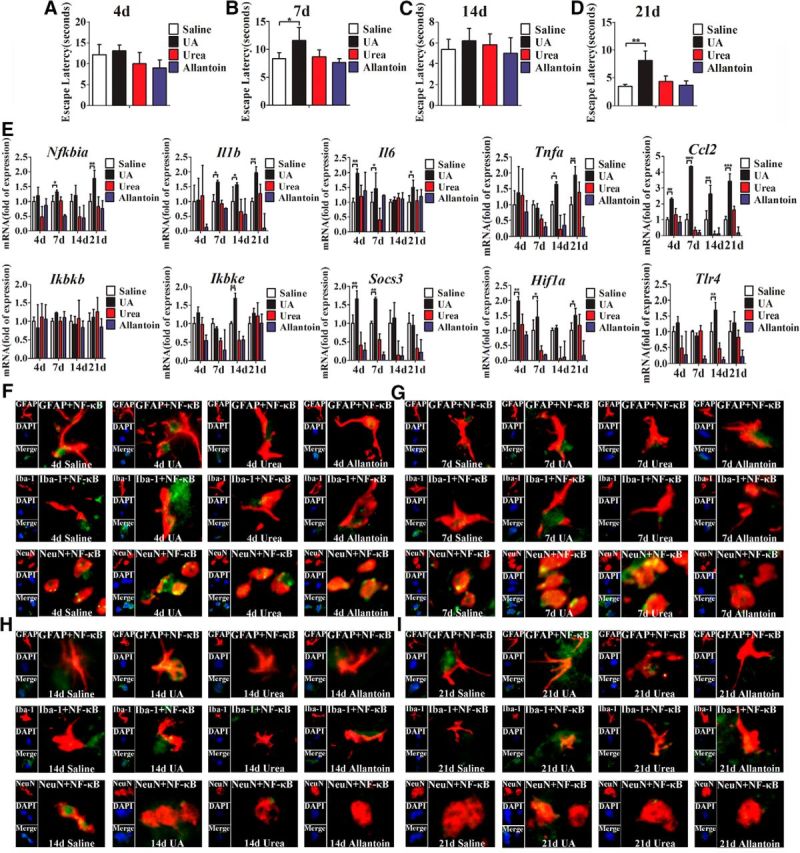
UA acts as an inflammatory stimulus and activates NF-κB in hippocampus. A–D, C57BL/6 mice were bilaterally injected with UA and implanted with a stainless steel guide cannula that targeted the hippocampus. Spatial learning of the mice was assessed in a Morris water maze after stereotactic injection of UA for 4 d (A), 7 d (B), 14 d (C), and 21 d (D), respectively. Each trial was presented as the average of four individual tests, and four trials were performed with the escape latency to the platform being recorded (Student's t tests, *p < 0.05; **p < 0.01, n = 6 rats per group). E, Time course of the induction of mRNA expression of inflammatory mediators, including proinflammatory cytokines (Il6, Il1b, Tnfa, Socs3, and Ccl2) and TLR4/NF-κB signaling (Tlr4, Nfkbia, Ikbkb, and Ikbke) in the hippocampi of mice that received stereotactic injection of saline, urea, allantoin, or 600 ng/ml UA for 4, 7, 14, and 21 d (n = 6 rats per group), respectively. All mRNA species were quantified relative to the expression of the housekeeping gene Gapdh and are presented as fold changes relative to saline controls. *p < 0.05; **p < 0.01; ***p < 0.001 vs normal saline control. F–I, Hippocampal tissues were immunostained for GFAP and RelA, Iba-1 and RelA, and NeuN and RelA (400×) after 4 d (F), 7 d (G), 14 d (H), or 21 d (I) of exposure to saline, urea, allantoin, or 600 ng/ml UA. RelA was used for the reporting of NF-κB. DAPI nuclear staining revealed all cells in the section. GFAP, DAPI, Merge (DAPI + NF-κB), GFAP + NF-κB; Iba-1, DAPI, Merge (DAPI + NF-κB), Iba-1 + NF-κB; and NeuN, DAPI, Merge (DAPI + NF-κB), NeuN + NF-κB. All displayed values are presented as the mean ± SEM.
As a critical mediator, NF-κB plays a central role in inducing inflammation and cognitive dysfunctions. We speculated that it may be involved in UA-induced hippocampal inflammation. To confirm this hypothesis, immunostaining for GFAP, Iba-1, NeuN, and the p65/RelA subunit of NF-κB was performed. As shown in Figure 3F–I, after 21 d of UA exposure (600 ng/ml, 2 μl), the expression of p65/RelA was activated in neurons, as characterized by p65/RelA translocation to the nucleus. Nevertheless, p65/RelA was barely activated in the glial cells. These results strongly support the idea that UA has the ability to activate NF-κB in the hippocampus.
NF-κB is central to the relationship between UA and cognitive dysfunction
To explore whether UA could induce cognitive dysfunction, the Morris water maze test was used to analyze the cognitive and learning ability of the mice that received an intrahippocampal injection of UA. As expected, the mice injected with UA exhibited a decrease in spatial learning and memory in an escape latency and probe trial (Fig. 3A–D); however, urea and allantoin did not affect cognitive function. To examine whether NF-κB plays an important role in UA-induced hippocampal inflammation, we injected BAY11-7085, a specific NF-κB inhibitor, into the hippocampus of mice 30 min before intrahippocampal injection of UA. Importantly, we found that BAY11-7085 significantly reversed the effect of UA on cognitive dysfunction (Fig. 4A,B). In addition, the upregulation of proinflammatory cytokines induced by UA was markedly inhibited (Fig. 4C). Furthermore, immunostaining for NeuN, GFAP, Iba-1, and the p65/RelA subunit of NF-κB in hippocampus was conducted to further test the effect of UA on hippocampal inflammation. We observed p65/RelA activation after exposure to 600 ng/ml UA for 21 d, which was characterized by p65/RelA translocation to the nucleus. Nevertheless, p65/RelA was barely activated following exposure to BAY11-7085 (Fig. 4D). Collectively, our findings indicate that hippocampal NF-κB activation may be an important signaling pathway that mediates UA-induced hippocampal inflammation.
Figure 4.
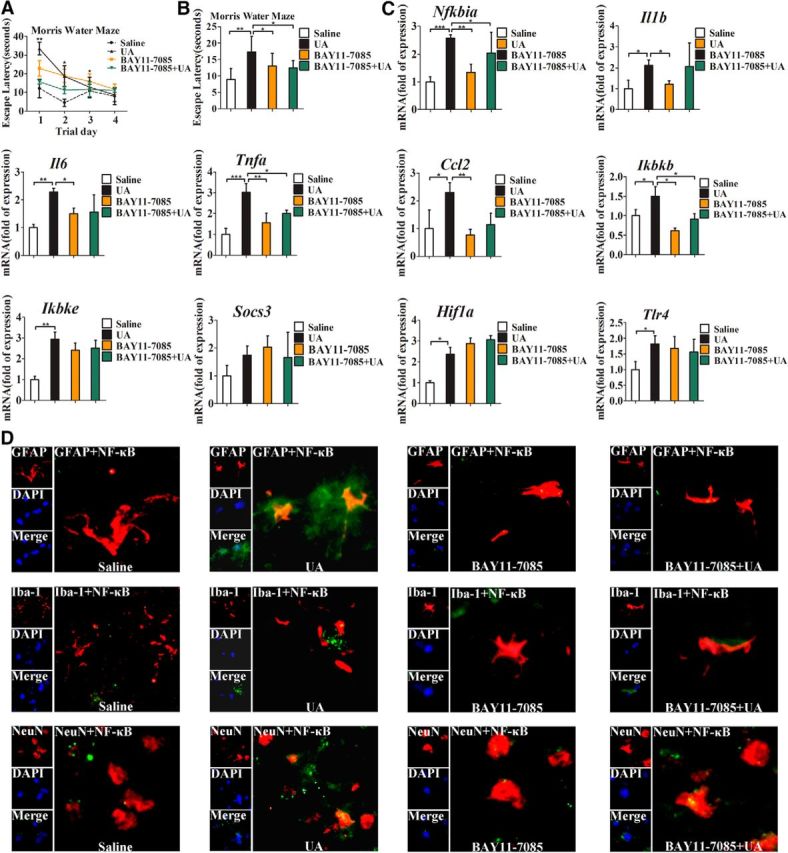
NF-κB is central to the relationship between UA and cognitive dysfunction. The tests were performed in C57BL/6 mice with stereotactic injection of normal saline or UA (600 ng/ml, 0.6 μl/min, 2 μl), with or without injection of BAY11-7085 (500 nm, 0.6 μl/min, 2 μl) 30 min before UA administration for 21 consecutive days (n = 6 mice per group). A, B, Spatial learning of the mice with stereotactic injection of normal saline or UA (600 ng/ml, 0.6 μl/min, 2 μl), with or without injection of BAY11-7085 (500 nm, 0.6 μl/min, 2 μl) 30 min before UA administration for 21 d, was assessed by a Morris water maze. Each trial was presented as the average of four individual tests, and four trials were performed with escape latency to the platform being recorded in a Morris water maze (Student's t tests, *p < 0.05; **p < 0.01, n = 6 rats per group). C, Expression levels of inflammatory mediators, including proinflammatory cytokines (Il6, Il1b, Tnfa, Socs3, and Ccl2) and TLR4/NF-κB signaling (Tlr4, Nfkbia, Ikbkb, and Ikbke), were determined by qRT-PCR in the hippocampi of the mice injected with saline, BAY11-7085 (500 nm, 2 μl), UA (600 ng/ml, 2 μl), or BAY11-7085 (500 nm, 2 μl) + UA (600 ng/ml, 2 μl) for 21 d (Student's t tests, *p < 0.05; **p < 0.01; ***p < 0.001, n = 6 rats per group). D, Hippocampal tissues was immunostained for GFAP and RelA, Iba-1 and RelA, and NeuN and RelA (400×) 21 d after being injected with saline, BAY11-7085 (500 nm, 2 μl), UA (600 ng/ml, 2 μl), or BAY11-7085 (500 nm, 2 μl) + UA (600 ng/ml, 2 μl). RelA was used for reporting on NF-κB. DAPI nuclear staining revealed all cells in the section. GFAP, DAPI, Merge (DAPI + NF-κB), GFAP + NF-κB; Iba-1, DAPI, Merge (DAPI + NF-κB), Iba-1 + NF-κB; and NeuN, DAPI, Merge (DAPI + NF-κB), and NeuN + NF-κB. All displayed values are the mean ± SEM.
UA induces NF-κB activation in primary cultured hippocampal cells in vitro
Based upon our aforementioned findings, hippocampal inflammation and reactive gliosis occurred at the late stage of the hyperuricemia rats. To further understand the molecular mechanisms of UA-induced hippocampal inflammation, the expression and localization of NeuN, GFAP, Iba-1, and p65/RelA were studied through immunostaining in primary cultured hippocampal cells. We found that p65/RelA translocation to the nucleus was markedly increased in the cells after exposure to UA for 48 h (Fig. 5B–D). To further study the role of UA in inducing inflammation, the mRNA levels of proinflammatory cytokines were detected. Consistently, the expression levels of Nfkbia, Il6, Il1b, Ikbkb, Ccl2, and Tlr4 were significantly increased in primary cultured hippocampal cells exposed to 600 ng/ml UA for 24 and 48 h, respectively (Fig. 5A).
Figure 5.

NF-κB is activated in the primary cultured hippocampal cells after UA administration. A, The mRNA levels of inflammatory mediators, including proinflammatory cytokines (Il6, Il1b, Tnfa, Socs3, and Ccl2) and TLR4/NF-κB signaling (Tlr4, Nfkbia, Ikbkb, and Ikbke), were determined in primary cultures of hippocampal cells after exposure to 600 ng/ml UA for 24 or 48 h. (Student's t tests, *p < 0.05; **p < 0.01 versus normal saline control). B–D, Hippocampal primary cultures were immunostained for NeuN and RelA (B), GFAP and RelA (C), and Iba-1 and RelA (D; 400×) after 24 or 48 h of exposure to 600 ng/ml UA. RelA was used for reporting NF-κB. DAPI nuclear staining revealed all cells in the section. NeuN, DAPI, Merge (DAPI + NF-κB), NeuN + NF-κB; GFAP, DAPI, Merge (DAPI + NF-κB), GFAP + NF-κB; and Iba-1, DAPI, Merge (DAPI + NF-κB), and Iba-1 + NF-κB. All displayed values are the mean ± SEM.
TLR4 loss ameliorates impaired cognitive dysfunction and hippocampal inflammation induced by HUAD
To assess the potential role of TLR4/NF-κB signaling in mediating hippocampal inflammation, we first quantified TLR4 mRNA expression in the hippocampus of rats that were fed a HUAD for 12 weeks. As mentioned above, TLR4 mRNA expression was significantly increased (Fig. 2G, *p < 0.05). As shown in Figure 6, A and B, the characteristics of these two types of mice were compared. The body weight increased steadily during the experiment, and no significant difference was observed between TLR4−/− and WT mice. However, the consumption of food slightly decreased along with the extension of HUAD feeding in the same type of mice. Moreover, the level of serum UA was elevated by >50% when compared with control mice fed a normal diet (Fig. 6C, **p < 0.01). To gain insight into the role of TLR4 in UA-induced cognitive dysfunction, the escape latencies of WT and TLR4−/− mice were compared in the Morris water maze paradigm. In comparison with WT mice, TLR4−/− mice showed enhanced spatial learning and memory in the escape latency and probe trial, suggesting that TLR4 could rescue cognitive dysfunction induced by UA (Fig. 6D,E). We further examined the expression levels of inflammatory genes in the hippocampus of mice fed with a HUAD for 12 weeks. In WT mice, the HUAD significantly induced the expression levels of Nfkbia, Tnfa, Il1b, Il6, Socs3, Ccl2, Ikbke, and Hif1a (hypoxia-inducible factor-1α); however, this effect was greatly attenuated in TLR4−/− mice (Fig. 6F). We then examined the nuclear translocation of NF-κB, a downstream signaling target of TLR4 activation. Consistently, HUAD-induced activation of NF-κB was prevented in hippocampal tissue of TLR4−/− mice (Fig. 6G–J). Together, these data suggest that TLR4/NF-κB signaling contributes, at least in part, to the HUAD-induced hippocampal inflammatory response.
Figure 6.
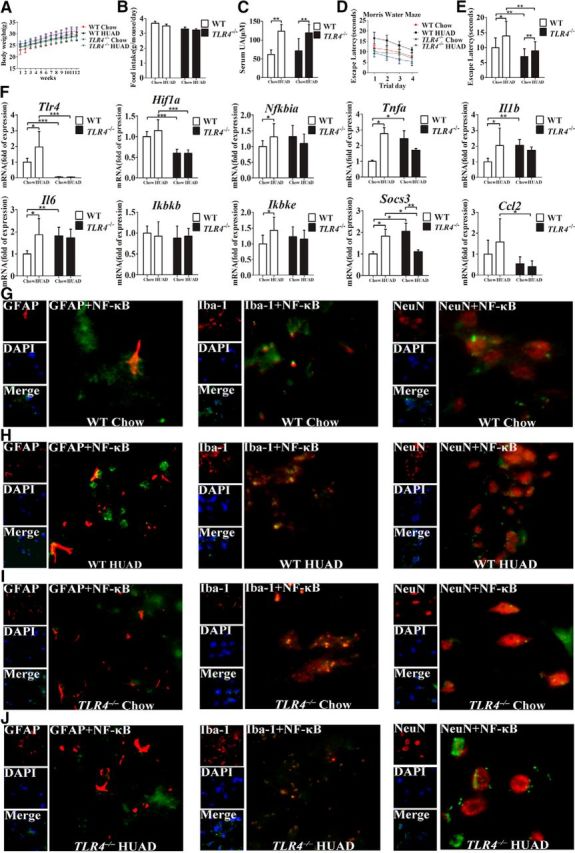
TLR4 loss ameliorates the impaired cognitive dysfunction and reduces hippocampal inflammation induced by a HUAD. A, B, The characteristics of TLR4−/ − and WT mice fed a HUAD for 12 weeks. A, Body weight. B, Food consumption. C, Time course of serum UA concentration after the onset of HUAD feeding (Student's t tests, **p < 0.01, n = 6 rats per group). D, E, Spatial learning of the mice after the onset of HUAD feeding, as demonstrated in the test of Morris water maze. Each trial presented was the average of four individual tests, and four trials were performed with the escape latency to the platform being recorded (Student's t tests, *p < 0.05; **p < 0.01, n = 6 rats per group). F, Expression levels of inflammatory mediators, including proinflammatory cytokines (Il6, Il1b, Tnfa, Socs3, and Ccl2) and TLR4/NF-κB signaling (Tlr4, Nfkbia, Ikbkb, and Ikbke), were determined by qRT-PCR in hippocampi of mice fed a HUAD for 12 weeks (Student's t tests, *p < 0.05; **p < 0.01; ***p < 0.001, n = 6 rats per group). G–J, Hippocampal tissues were immunostained for GFAP and RelA, Iba-1 and RelA, and NeuN and RelA (400×) after 12 weeks of being fed a HUAD. RelA was used for reporting on NF-κB. DAPI nuclear staining revealed all cells in the section. WT Chow (G), WT HUAD (H), TLR4−/− Chow (I), and TLR4−/− HUAD (J). GFAP, DAPI, Merge (DAPI + NF-κB), GFAP + NF-κB; Iba-1, DAPI, Merge (DAPI + NF-κB), Iba-1 + NF-κB; and NeuN, DAPI, Merge (DAPI + NF-κB), and NeuN + NF-κB. All displayed values are the mean ± SEM.
TLR4/NF-κB signaling mediates UA-induced hippocampal inflammation and cognitive dysfunction
To investigate the role of TLR4/NF-κB in mediating UA-induced inflammatory signaling in vivo, TLR4−/− mice and WT mice were then stereotactically injected with UA (600 ng/ml, 2 μl) into the hippocampus, using urea or allantoin as a control. UA potently stimulated mRNA expressions of the proinflammatory cytokines in the hippocampus of WT mice and, to a lesser extent, in TLR4−/− mice (Fig. 7D,E). Simultaneously, TLR4 deficiency prevented UA-induced NF-κB subunit p65/RelA translocation to the nucleus (Fig. 7F,G). In addition, comparison of cognitive dysfunction using the Morris water maze paradigm showed that the impairment of spatial learning and memory was inhibited to a much lower extent in TLR4−/− mice than in WT mice (Fig. 7A–C). These results indicate that TLR4/NF-κB signaling is required for mediating UA-induced cognitive dysfunction in the hippocampus.
Figure 7.

TLR4/NF-κB signaling mediates UA-induced hippocampal inflammatory signaling and cognitive dysfunction. A–C, Spatial learning of WT and TLR4−/− mice with UA, as demonstrated by a Morris water maze. Each trial is presented as the average of four individual tests, and four trials were performed with the escape latency to the platform being recorded. D, E, The mRNA levels of inflammatory mediators, including proinflammatory cytokines (Il6, Il1b, Tnfa, Socs3, and Ccl2) and TLR4/NF-κB signaling (Tlr4, Nfkbia, Ikbkb, and Ikbke), were measured in hippocampus after exposure to saline, urea, allantoin, or 600 ng/ml UA for 21 d. F, G, Hippocampal tissues were immunostained for GFAP and RelA, Iba-1 and RelA, and NeuN and RelA (400×) after stereotactic injection of saline, urea, allantoin, or 600 ng/ml UA. RelA was used for reporting NF-κB. DAPI nuclear staining revealed all cells in the section (Student's t tests, *p < 0.05; **p < 0.01; ***p < 0.001 vs WT saline control. #p < 0.05, ##p < 0.01, ###p < 0.001 vs WT normal control, n = 6 rats per group). GFAP, DAPI, Merge (DAPI + NF-κB), GFAP + NF-κB; Iba-1, DAPI, Merge (DAPI + NF-κB), Iba-1 + NF-κB; and NeuN, DAPI, Merge (DAPI + NF-κB), and NeuN + NF-κB. All displayed values are the mean ± SEM.
MRI-based quantitative assessment of hippocampal gliosis in rats and humans with hyperuricemia
Gliosis is a well characterized neural tissue response to injury from inflammation (Lee et al., 2013). In the aforementioned studies, we showed that a HUAD induced inflammation associated with gliosis in the hippocampus of rats. To histologically confirm that gliosis was induced by HUAD, we used high-field MRI to detect gliotic changes quantitatively. ROIs and example images from the two-dimensional sequence, T2 parametric map, and diffusion tensor imaging (DTI)-EPI sequence are shown (Fig. 8A,B). There was a difference in the T2 relaxation time in the hippocampal ROIs between hyperuricemia rats and chow-fed rats, indicating that UA could extend T2 relaxation times (Fig. 8C, *p < 0.05). Using DTI, values were compared for fractional anisotropy and tensor trace assessments. The tensor trace showed a pattern that was similar to T2 relaxation times, confirming that inflammation occurred (Fig. 8D); however, laterality (right vs left) showed a significant difference (*p < 0.05). Nevertheless, there was no significant difference in fractional anisotropy (Fig. 8E). These results indicated that gliosis occurred in the hippocampus of hyperuricemia rats.
Figure 8.

MRI-based quantitative assessment of hippocampal gliosis in rats and humans with hyperuricemia. A, B, Regions of interest (circles) and representative images of the normal rats (A) and rats with hyperuricemia (B). Aa–Bd, High-resolution, two-dimensional rapid acquisition (Aa, Ba); T2 map generated from a multiecho sequence (Ab, Bb); DTI for tensor trace measurement (Ac, Bc); and DTI for fractional anisotropy (Ad, Bd). C–E, Results of multiparametric quantitative assessment in rats with hyperuricemia and chow-fed rats, including T2 relaxation time (C), tensor trace (D), and fractional anisotropy (E; Student's t tests, *p < 0.05 vs control, n = 6 rats per group). F, G, Representative coronal T2 FLAIR images to diagnose FES through the hippocampus in a subject with normal serum UA levels (F) and a subject with hyperuricemia (G). Insets show the placement of ROIs (white circles) in the hippocampus. H, The concentration-dependent correlation between serum UA levels and hippocampus gliosis in humans (n = 29 subjects; r = 0.311). I, The concentration-dependent correlation between serum UA levels and hippocampus gliosis in rats (n = 12 rats; r = 0.871). All displayed values are the mean ± SEM.
A retrospective cohort study was performed to search for radiological evidence of hippocampus gliosis and to correlate our findings with hyperuricemia. We analyzed the data obtained from 15 hyperuricemia subjects with MRI examinations and from 14 normal subjects. The concentrations of serum UA in these hyperuricemia subjects ranged from 436 to 517 μm for men, and from 378 to 428 μm for women, whereas the average concentration was 278.2 μm in normal subjects. As mentioned above, we performed an initial inspection of the hippocampus for hyperintense signaling in T2 coronal sections. Hyperintense signaling in the hippocampus is a characteristic finding of gliosis in numerous inflammatory, ischemic, and degenerative neural disorders. To detect gliotic changes below the visual detection threshold and to eliminate the influence of changes in the background between different batches of imaging, ratios were created to compare the mean signal intensity between ROIs placed in the hippocampus and ROIs. ROIs are shown by white circles and arrows in the representative images from a normal subject and a subject with hyperuricemia (Fig. 8F,G). We also analyzed the concentration-dependent correlation between serum UA and hippocampus gliosis both in humans and in rats. T2 relaxation times were significantly different in the hippocampus, which was characterized by an increased density of both astrocytes and microglia (Fig. 8H,I). Collectively, our retrospective analysis suggested that hyperuricemia in humans and in rats was very likely associated with gliosis in the hippocampus.
Discussion
The results presented here provide compelling evidence that a high level of UA is associated with memory deficit due to enhanced proinflammatory cytokine gene expression and gliosis in the hippocampus (Fig. 9). We thus considered that chronic hippocampal inflammation could be a manifestation of long-term neuronal cell injury that triggers reactive gliosis involving microglial and astroglial cell populations, both of which are thought to limit the extent of inflammation and neuron loss in brain injury (Sofroniew, 2009). In addition, with sustained exposure to UA, the capacity of supportive glial cells to control damage is overwhelmed, thereby resulting in chronic inflammation and reactive gliosis in the hippocampus. Hippocampal NF-κB inhibition is able to protect mice against UA-induced neuroinflammation as well as memory deficit, which supports our hypothesis that a high level of UA can impair cognitive function through TLR4/NF-κB pathway. Finally, gliosis is closely associated with hyperuricemia in rodents and humans by MRI detection, suggesting a common mechanism of UA-induced hippocampal inflammation among different species.
Figure 9.

Proposed model: UA induces cognitive dysfunction through hippocampal inflammation.
Our findings are consistent with those of previous reports that a higher level of serum UA is associated cross-sectionally with poorer attention and working memory, psychomotor speed, and executive functioning (Schretlen et al., 2007a,b; Verhaaren et al., 2013). Moreover, our results are also supported by the findings that a higher level of serum UA correlates with greater white matter atrophy (Verhaaren et al., 2013) and cerebral ischemia, as measured by the volume of the hyperintense signal on T2-weighted brain MRI scans (Schretlen et al., 2007a,b; Vannorsdall et al., 2008). Nevertheless, a few studies suggested that UA may be related to better cognition or even to a decreased risk of dementia (Wu et al., 2013; Cao et al., 2015). Also, beneficial effects have been found for individuals with Parkinson's disease and its related cognitive decline (Wirdefeldt et al., 2011). Although serum UA possesses antioxidant properties, several harmful mechanisms have been proposed experimentally (Squadrito et al., 2000). UA may promote LDL oxidation in vitro (Schlotte et al., 1998), and may stimulate granulocyte adherence to the endothelium as well as peroxide and superoxide free radical liberation (Boogaerts et al., 1983). The net effect of these opposing effects on brain function remains unclear. We guess that the beneficial effect of uric acid may be present within its normal range, whereas the detrimental effect is more pronounced in conditions of hyperuricemia. Further research is needed to elucidate this issue.
Several mechanisms have been reported to explain the relationship between overnutrition and neurodegeneration involving the induction of neuroinflammation. This type of neurodegeneration is attributed to the neuronal TLR4/NF-κB pathway activation and the subsequent induction of neurotoxic inflammatory products (Dugan et al., 2009). In addition to its role as an inflammatory regulator, NF-κB signaling also controls cell survival, apoptosis, and synaptic plasticity (Mattson and Meffert, 2006). For example, NF-κB is activated in synapses in response to excitatory synaptic transmission and may play a pivotal role in processes such as learning and memory in the mature nervous system (Schmeisser et al., 2012). In the present study, the expression levels of the genes Ikbkb, Ikbke, and Nfkbia significantly increased after UA stimulation, suggesting that UA-induced inflammation may be mediated by the activation of the TLR4/NF-κB pathway. In fact, several researchers have shown that inhibitor of NF-κB kinase (IKKβ)/NF-κB disrupts hypothalamic neural stem cells in mediating a neurodegenerative mechanism in cases of dietary obesity and prediabetes (Li et al., 2012). Conversely, specific inhibition of IKKβ in hippocampal neurons significantly protects against cognitive impairment (Cai, 2013).
Some studies have reported that hypothalamic dysregulation induced by overnutrition involves a neuron-specific program through IKKβ/NF-κB and that inflammatory cytokines do not have strong effects in non-neuronal cells (Hummasti and Hotamisligil, 2010). Nevertheless, other studies have shown that under conditions of chronic inflammation induced by overnutrition and senescence, NF-κB is activated in microglia, which is regarded as the macrophage-like immune cells in the brain. Moreover, proinflammatory cytokines, such as TNF-α and IL-6, are released from microglia and target appetite-controlling neurons, which further activate NF-κB (Nguyen et al., 2014). Our in vitro study showed that in response to UA, NF-κB was activated mainly in neurons rather than in glial cells, which reflects a neuron-specific program involved in the TLR4/NF-κB pathway. However, crucially, the mechanism responsible for hippocampal inflammation induced by UA still needs to be studied, and a direct causal link between UA and TLR4/NF-κB paythway still needs to be demonstrated in primary hippocampal neurons.
There are several mechanisms that have been reported to explain overnutrition-induced inflammation in the brain, including the induction of endoplasmic reticulum stress and the activation of serine/threonine kinase (Velloso and Schwartz, 2011). In this study, we found that the loss of TLR4 protects mice from UA-induced cognitive dysfunction by downregulating hippocampal inflammatory gene expression. Indeed, it has been shown that UA released from injured cells constitutes a major endogenous danger signal that activates the NALP3 inflammasome, thereby resulting in increased IL-1β production (Denoble et al., 2011). Reducing the tissue UA level represents a novel therapeutic approach for controlling IL-1β production and chronic inflammatory lung pathology (Gasse et al., 2009). Moreover, a mixture of palmitate and oleic acid acts via TLR4 to induce NF-κB signaling in 293 cells (Shi et al., 2006). These results are consistent with our findings that UA may initiate inflammatory signaling through the activation of TLR4/NF-κB signaling in the hippocampus. Our results suggest that TLR4, a key molecular component of the innate immune system, may play an important role in sensing UA. Currently, very little is known about the roles of TLRs in cognition. Only one study reported that developmental TLR4 deficiency enhances spatial reference memory acquisition and memory retention, traits that are correlated with cAMP response element-binding protein upregulation in the hippocampus (Okun et al., 2012). Our findings suggest a relationship between the innate immune system and UA-induced cognitive dysfunction.
In recent years, multiparametric high-field MRI approaches have been used to assess and quantify gliosis in the hippocampus, based upon the techniques of T2 relaxation time or DTI (Briellmann et al., 2002). Hyperintensity is an indicator of gliosis. T2 relaxation time is significantly correlated with gliosis and is considered as a promising quantitative radiological marker of gliosis in the hippocampus, despite its minute size (Briellmann et al., 2002; Immonen et al., 2008). In the present study, T2 relaxation times were significantly different in the hippocampus of HUAD-induced hyperuricemia rats, which were characterized by an increased density of both astrocytes and microglia. We extended the finding of the elevated hippocampus T2 signal by associating it with high serum UA levels in humans. Nevertheless, through a retrospective cohort analysis of T2 MRI that was conducted on human subjects undergoing clinical examination, we found that the intensity of the T2 signal was significantly increased in the hippocampus of patients with hyperuricemia. This finding does not constitute definitive proof of increased gliosis because inflammatory edema, inflammation, hyperplasia, and tumors could have similar appearances. Nevertheless, subjects with pre-existing evidence of neurological abnormalities were excluded from the study, and these alternative explanations for increased T2 signal intensity are unlikely. Our results suggest that MRI-based techniques have translational value because they can be used to make comparable measurements of gliosis in human hippocampus. However, there are limitations in our study. In the retrospective study in humans, we compared only the hippocampal MRI signal intensity of the hyperuricemia group and the normal group. Due to time constraints, we did not track the prognosis of these subjects, which is likely to further illustrate the conclusions of this study.
In summary, although the suggestion that serum UA level is a risk factor for learning and memory is currently controversial, we show here that the consumption of a HUAD induces cognitive dysfunction through hippocampal inflammation and reactive gliosis in rodents. Experimental results indicate that hippocampal inflammation mediated by TLR4/NF-κB signaling may be implicated in the pathogenesis of cognitive dysfunction induced by UA. The understanding of the mechanisms by which a high UA level affects neuroplasticity and cognitive function provides a potential therapeutic approach to counteract hyperuricemia-related diseases.
Footnotes
This work was supported by National Science & Technology Major Project Grant 2012ZX09302-004, and National Natural Science Foundation of China Grants 81571301, 81271467, 81272459, 81402947, 81322035, and 30970938.
The authors declare no competing financials interests.
References
- Afsar B, Elsurer R, Covic A, Johnson RJ, Kanbay M. Relationship between uric acid and subtle cognitive dysfunction in chronic kidney disease. Am J Nephrol. 2011;34:49–54. doi: 10.1159/000329097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boitard C, Cavaroc A, Sauvant J, Aubert A, Castanon N, Layé S, Ferreira G. Impairment of hippocampal-dependent memory induced by juvenile high-fat diet intake is associated with enhanced hippocampal inflammation in rats. Brain Behav Immun. 2014;40:9–17. doi: 10.1016/j.bbi.2014.03.005. [DOI] [PubMed] [Google Scholar]
- Boogaerts MA, Hammerschmidt DE, Roelant C, Verwilghen RL, Jacob HS. Mechanisms of vascular damage in gout and oxalosis: crystal induced, granulocyte mediated, endothelial injury. Thromb Haemost. 1983;50:576–580. [PubMed] [Google Scholar]
- Briellmann RS, Kalnins RM, Berkovic SF, Jackson GD. Hippocampal pathology in refractory temporal lobe epilepsy—T2-weighted signal change reflects dentate gliosis. Neurology. 2002;58:265–271. doi: 10.1212/WNL.58.2.265. [DOI] [PubMed] [Google Scholar]
- Cai D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends Endocrinol Metab. 2013;24:40–47. doi: 10.1016/j.tem.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B, Wei QQ, Ou R, Yang J, Shang HF. Association of serum uric acid level with cognitive function among patients with multiple system atrophy. J Neurol Sci. 2015;359:363–366. doi: 10.1016/j.jns.2015.11.025. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, Akira S, Rock KL. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. 2006;116:2262–2271. doi: 10.1172/JCI28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ, Tzameli I, Pissios P, Rovira I, Gavrilova O, Ohtsubo T, Chen Z, Finkel T, Flier JS, Friedman JM. Xanthine oxidoreductase is a regulator of adipogenesis and PPAR gamma activity. Cell Metab. 2007;5:115–128. doi: 10.1016/j.cmet.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Conen D, Wietlisbach V, Bovet P, Shamlaye C, Riesen W, Paccaud F, Burnier M. Prevalence of hyperuricemia and relation of serum uric acid with cardiovascular risk factors in a developing country. BMC Public Health. 2004;4:9. doi: 10.1186/1471-2458-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai XH, Fang X, Zhang CM, Xu RF, Xu B. Determination of serum uric acid using high-performance liquid chromatography (HPLC)/isotope dilution mass spectrometry (ID-MS) as a candidate reference method. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;857:287–295. doi: 10.1016/j.jchromb.2007.07.035. [DOI] [PubMed] [Google Scholar]
- Denoble AE, Huffman KM, Stabler TV, Kelly SJ, Hershfield MS, McDaniel GE, Coleman RE, Kraus VB. Uric acid is a danger signal of increasing risk for osteoarthritis through inflammasome activation. Proc Natl Acad Sci U S A. 2011;108:2088–2093. doi: 10.1073/pnas.1012743108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo G, Wilcock D, Henderson D, Gordon M, Morgan D. Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol Aging. 2001;22:1007–1012. doi: 10.1016/S0197-4580(01)00292-5. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Ali SS, Shekhtman G, Roberts AJ, Lucero J, Quick KL, Behrens MM. IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS One. 2009;4:e5518. doi: 10.1371/journal.pone.0005518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med. 2008;359:1811–1821. doi: 10.1056/NEJMra0800885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasse P, Riteau N, Charron S, Girre S, Fick L, Pétrilli V, Tschopp J, Lagente V, Quesniaux VF, Ryffel B, Couillin I. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med. 2009;179:903–913. doi: 10.1164/rccm.200808-1274OC. [DOI] [PubMed] [Google Scholar]
- Giovanello KS, Schnyer D, Verfaellie M. Distinct hippocampal regions make unique contributions to relational memory. Hippocampus. 2009;19:111–117. doi: 10.1002/hipo.20491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitti FL, Siegelbaum SA. The hippocampal CA2 region is essential for social memory. Nature. 2014;508:88–92. doi: 10.1038/nature13028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Yu Y, Eugene Chin Y, Xia Q. The role of acetylation in TLR4-mediated innate immune responses. Immunol Cell Biol. 2013;91:611–614. doi: 10.1038/icb.2013.56. [DOI] [PubMed] [Google Scholar]
- Hummasti S, Hotamisligil GS. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ Res. 2010;107:579–591. doi: 10.1161/CIRCRESAHA.110.225698. [DOI] [PubMed] [Google Scholar]
- Immonen RJ, Kharatishvili I, Sierra A, Einula C, Pitkänen A, Gröhn OHJ. Manganese enhanced MRI detects mossy fiber sprouting rather than neurodegeneration, gliosis or seizure-activity in the epileptic rat hippocampus. Neuroimage. 2008;40:1718–1730. doi: 10.1016/j.neuroimage.2008.01.042. [DOI] [PubMed] [Google Scholar]
- Jeon BT, Jeong EA, Shin HJ, Lee Y, Lee DH, Kim HJ, Kang SS, Cho GJ, Choi WS, Roh GS. Resveratrol attenuates obesity-associated peripheral and central inflammation and improves memory deficit in mice fed a high-fat diet. Diabetes. 2012;61:1444–1454. doi: 10.2337/db11-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, Xing J, Ding Y, Shen Y, Shi X, Ren W, Wan M, Guo J, Zheng S, Liu Y, Liang X, Su D. Hyperuricemia causes pancreatic beta-cell death and dysfunction through NF-kappaB signaling pathway. PLoS One. 2013;8:e78284. doi: 10.1371/journal.pone.0078284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Yang F, Yang I, Yin Y, Luo JJ, Wang H, Yang XF. Uric acid, hyperuricemia and vascular diseases. Front Biosci (Landmark Ed) 2012;17:656–669. doi: 10.2741/3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RJ, Perez-Pozo SE, Sautin YY, Manitius J, Sanchez-Lozada LG, Feig DI, Shafiu M, Segal M, Glassock RJ, Shimada M, Roncal C, Nakagawa T. Hypothesis: could excessive fructose intake and uric acid cause type 2 diabetes? Endocr Rev. 2009;30:96–116. doi: 10.1210/er.2008-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DH, Park SK, Lee IK, Johnson RJ. Uric acid-induced C-reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells. J Am Soc Nephrol. 2005;16:3553–3562. doi: 10.1681/ASN.2005050572. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Kim MS, Pak YK, Jang PG, Namkoong C, Choi YS, Won JC, Kim KS, Kim SW, Kim HS, Park JY, Kim YB, Lee KU. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci. 2006;9:901–906. doi: 10.1038/nn1731. [DOI] [PubMed] [Google Scholar]
- Kim YG, Huang XR, Suga S, Mazzali M, Tang D, Metz C, Bucala R, Kivlighn S, Johnson RJ, Lan HY. Involvement of macrophage migration inhibitory factor (MIF) in experimental uric acid nephropathy. Mol Med. 2000;6:837–848. [PMC free article] [PubMed] [Google Scholar]
- Kono H, Chen CJ, Ontiveros F, Rock KL. Uric acid promotes an acute inflammatory response to sterile cell death in mice. J Clin Invest. 2010;120:1939–1949. doi: 10.1172/JCI40124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Thaler JP, Berkseth KE, Melhorn SJ, Schwartz MW, Schur EA. Longer T(2) relaxation time is a marker of hypothalamic gliosis in mice with diet-induced obesity. Am J Physiol Endocrinol Metab. 2013;304:E1245–E1250. doi: 10.1152/ajpendo.00020.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Tang Y, Cai D. IKKbeta/NF-kappaB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat Cell Biol. 2012;14:999–1012. doi: 10.1038/ncb2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- Masuo K, Kawaguchi H, Mikami H, Ogihara T, Tuck ML. Serum uric acid and plasma norepinephrine concentrations predict subsequent weight gain and blood pressure elevation. Hypertension. 2003;42:474–480. doi: 10.1161/01.HYP.0000091371.53502.D3. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Meffert MK. Roles for NF-kappa B in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–860. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- Mazzali M, Hughes J, Kim YG, Jefferson JA, Kang DH, Gordon KL, Lan HY, Kivlighn S, Johnson RJ. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension. 2001;38:1101–1106. doi: 10.1161/hy1101.092839. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- Nguyen JC, Killcross AS, Jenkins TA. Obesity and cognitive decline: role of inflammation and vascular changes. Front Neurosci. 2014;8:375. doi: 10.3389/fnins.2014.00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun E, Griffioen KJ, Mattson MP. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011;34:269–281. doi: 10.1016/j.tins.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun E, Barak B, Saada-Madar R, Rothman SM, Griffioen KJ, Roberts N, Castro K, Mughal MR, Pita MA, Stranahan AM, Arumugam TV, Mattson MP. Evidence for a developmental role for TLR4 in learning and memory. PLoS One. 2012;7:e47522. doi: 10.1371/journal.pone.0047522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Purkayastha S, Zhang G, Cai D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-beta and NF-kappaB. Nat Med. 2011;17:883–887. doi: 10.1038/nm.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, Schwartz M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. 2007;9:1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- Schlotte V, Sevanian A, Hochstein P, Weithmann KU. Effect of uric acid and chemical analogues on oxidation of human low density lipoprotein in vitro. Free Radic Biol Med. 1998;25:839–847. doi: 10.1016/S0891-5849(98)00160-9. [DOI] [PubMed] [Google Scholar]
- Schmeisser MJ, Baumann B, Johannsen S, Vindedal GF, Jensen V, Hvalby ØC, Sprengel R, Seither J, Maqbool A, Magnutzki A, Lattke M, Oswald F, Boeckers TM, Wirth T. IκB kinase/nuclear factor κB-dependent insulin-like growth factor 2 (Igf2) expression regulates synapse formation and spine maturation via Igf2 receptor signaling. J Neurosci. 2012;32:5688–5703. doi: 10.1523/JNEUROSCI.0111-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schretlen DJ, Inscore AB, Jinnah HA, Rao V, Gordon B, Pearlson GD. Serum uric acid and cognitive function in community-dwelling older adults. Neuropsychology. 2007a;21:136–140. doi: 10.1037/0894-4105.21.1.136. [DOI] [PubMed] [Google Scholar]
- Schretlen DJ, Inscore AB, Vannorsdall TD, Kraut M, Pearlson GD, Gordon B, Jinnah HA. Serum uric acid and brain ischemia in normal elderly adults. Neurology. 2007b;69:1418–1423. doi: 10.1212/01.wnl.0000277468.10236.f1. [DOI] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. Caught red-handed: uric acid is an agent of inflammation. J Clin Invest. 2010;120:1809–1811. doi: 10.1172/JCI43132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So A, Thorens B. Uric acid transport and disease. J Clin Invest. 2010;120:1791–1799. doi: 10.1172/JCI42344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squadrito GL, Cueto R, Splenser AE, Valavanidis A, Zhang H, Uppu RM, Pryor WA. Reaction of uric acid with peroxynitrite and implications for the mechanism of neuroprotection by uric acid. Arch Biochem Biophys. 2000;376:333–337. doi: 10.1006/abbi.2000.1721. [DOI] [PubMed] [Google Scholar]
- Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, Zhao X, Sarruf DA, Izgur V, Maravilla KR, Nguyen HT, Fischer JD, Matsen ME, Wisse BE, Morton GJ, Horvath TL, Baskin DG, Tschöp MH, Schwartz MW. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122:153–162. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannorsdall TD, Jinnah HA, Gordon B, Kraut M, Schretlen DJ. Cerebral ischemia mediates the effect of serum uric acid on cognitive function. Stroke. 2008;39:3418–3420. doi: 10.1161/STROKEAHA.108.521591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velloso LA, Schwartz MW. Altered hypothalamic function in diet-induced obesity. Int J Obes. 2011;35:1455–1465. doi: 10.1038/ijo.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaaren BF, Vernooij MW, Dehghan A, Vrooman HA, de Boer R, Hofman A, Witteman JC, Niessen WJ, Breteler MM, van der Lugt A, Ikram MA. The relation of uric acid to brain atrophy and cognition: the Rotterdam Scan Study. Neuroepidemiology. 2013;41:29–34. doi: 10.1159/000346606. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of Parkinson's disease: a review of the evidence. Eur J Epidemiol. 2011;26:S1–S58. doi: 10.1007/s10654-011-9581-6. [DOI] [PubMed] [Google Scholar]
- Wu Y, Zhang D, Pang Z, Jiang W, Wang S, Tan Q. Association of serum uric acid level with muscle strength and cognitive function among Chinese aged 50–74 years. Geriatr Gerontol Int. 2013;13:672–677. doi: 10.1111/j.1447-0594.2012.00962.x. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Fang L, Jiang L, Wen P, Cao H, He W, Dai C, Yang J. Uric acid induces renal inflammation via activating tubular NF-kappaB signaling pathway. PLoS One. 2012;7:e39738. doi: 10.1371/journal.pone.0039738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011;63:3136–3141. doi: 10.1002/art.30520. [DOI] [PubMed] [Google Scholar]
- Zuany-Amorim C, Hastewell J, Walker C. Toll-like receptors as potential therapeutic targets for multiple diseases. Nat Rev Drug Discov. 2002;1:797–807. doi: 10.1038/nrd914. [DOI] [PubMed] [Google Scholar]