

Abstract
α-Synuclein overexpression (ASOX) drives the formation of toxic aggregates in neurons vulnerable in Parkinson's disease (PD), including dopaminergic neurons of the substantia nigra (SN) and cholinergic neurons of the dorsal motor nucleus of the vagus (DMV). Just as these populations differ in when they exhibit α-synucleinopathies during PD pathogenesis, they could also differ in their physiological responses to ASOX. An ASOX-mediated hyperactivity of SN dopamine neurons, which was caused by oxidative dysfunction of Kv4.3 potassium channels, was recently identified in transgenic (A53T-SNCA) mice overexpressing mutated human α-synuclein. Noting that DMV neurons display extensive α-synucleinopathies earlier than SN dopamine neurons while exhibiting milder cell loss in PD, we aimed to define the electrophysiological properties of DMV neurons in A53T-SNCA mice. We found that DMV neurons maintain normal firing rates in response to ASOX. Moreover, Kv4.3 channels in DMV neurons exhibit no oxidative dysfunction in the A53T-SNCA mice, which could only be recapitulated in wild-type mice by glutathione dialysis. Two-photon imaging of redox-sensitive GFP corroborated the finding that mitochondrial oxidative stress was diminished in DMV neurons in the A53T-SNCA mice. This reduction in oxidative stress resulted from a transcriptional downregulation of voltage-activated (Cav) calcium channels in DMV neurons, which led to a reduction in activity-dependent calcium influx via Cav channels. Thus, ASOX induces a homeostatic remodeling with improved redox signaling in DMV neurons, which could explain the differential vulnerability of SN dopamine and DMV neurons in PD and could promote neuroprotective strategies that emulate endogenous homeostatic responses to ASOX (e.g., stressless pacemaking) in DMV neurons.
SIGNIFICANCE STATEMENT Overexpression of mutant α-synuclein causes Parkinson's disease, presumably by driving neurodegeneration in vulnerable neuronal target populations. However, the extent of α-synuclein pathology (e.g., Lewy bodies) is not directly related to the degree of neurodegeneration across various vulnerable neuronal populations. Here, we show that, in contrast to dopamine neurons in the substantia nigra, vagal motoneurons do not enhance their excitability and oxidative load in response to chronic mutant α-synuclein overexpression. Rather, by downregulating their voltage-activated calcium channels, vagal motoneurons acquire a stressless form of pacemaking that diminishes mitochondrial and cytosolic oxidative stress. Emulating this endogenous adaptive response to α-synuclein overexpression could lead to novel strategies to protect dopamine neurons and perhaps delay the onset of Parkinson's disease.
Keywords: acetylcholine, homeostasis, Lewy pathology, multiphoton microscopy, neurodegeneration, oxidative stress
Introduction
The formation of α-synuclein-laden intracellular inclusions in the brain, called Lewy pathologies (LPs), is a hallmark of Parkinson's disease (PD; Lewy, 1912; Braak et al., 2004). Braak and colleagues proposed that a subset of neuronal populations [e.g., vagal cholinergic motoneurons (DMVs), locus ceruleus (LC) adrenergic neurons, and substantia nigra (SN) dopamine neurons] acquires these LPs in a stereotypical temporal order ranging from premotor stages (DMV) to full clinical PD (Stage III), where loss of SN dopamine neurons is sufficient to cause the classical, levodopa-responsive motor symptoms (Braak et al., 2004; Seidel et al., 2015). Why certain neuronal populations are the targets of LPs and how this translates to different degrees of neurodegeneration is still not understood. Apart from sharing some potential morphological risk features (e.g., extraordinarily long nonmyelinated axonal arbors; Braak et al., 2004; Bolam and Pissadaki, 2012; Pissadaki and Bolam, 2013), these neuronal populations also share a metabolically costly electrophysiological phenotype, which is characterized by autonomous pacemaker firing, during which large amounts of activity-driven calcium enters the neuron via voltage activated (Cav) calcium channels (Surmeier et al., 2011). Given the low cytosolic calcium buffering in DMV neurons, calcium ions are readily taken up by the mitochondria, where they elevate oxidative stress, as monitored by optical redox sensors, such as mitochondrial targeted redox-sensitive GFP (mito-roGFP; Dooley et al., 2004; Guzman et al., 2010; Surmeier et al., 2011; Goldberg et al., 2012; Sanchez-Padilla et al., 2014). It is currently an open question whether this precarious electrical phenotype shared by vulnerable neurons has a physiological function [e.g., by allowing fast switching between tonic and burst states in vivo (Paladini and Roeper, 2014)]. Because there is ample evidence that LPs or α-synuclein overexpression (ASOX) are linked to elevated levels of oxidative stress (Hsu et al., 2000; Goedert, 2001; Sherer et al., 2001; Parihar et al., 2008), it might—by escalating the already high levels of activity-driven oxidative stress or possibly by potentiating Cav channels (Adamczyk and Strosznajder, 2006; Hettiarachchi et al., 2009)—have a particularly detrimental impact on these pacemaker neurons by combining innate (i.e., electrical phenotype) with acquired vulnerabilities due to toxic α-synuclein aggregates.
In accordance with this scheme, a recent study showed that mutant ASOX exacerbates the “stressful pacemaking” of SN dopamine neurons by speeding up their firing both in vivo and in vitro (Subramaniam et al., 2014). For SN dopamine neurons in middle-aged A53T-SNCA mice, this elevated discharge was caused by oxidative dysfunction of Kv4.3 channels, which normally dampens the pacemaker rate (Liss et al., 2001). In that study, the Kv4.3 protein was upregulated in the membrane of SN dopamine neurons in A53T-SNCA mice, indicating an incomplete homeostatic response aimed at reducing the α-synuclein-driven hyperactivity. However, the role of Cav channels was not investigated. In another study, transduction of cultured dopamine cells with preformed fibrils of α-synuclein led to elevated dendritic oxidative stress dependent on calcium flux via Cav channels (Dryanovski et al., 2013).
Motoneurons of the DMV are among the first cells to exhibit LPs (Lewy, 1912; Braak et al., 2004), suggesting that they are among particularly vulnerable neuronal populations. However, compared with the early onset and large extent of the LPs, the 30% cell loss (i.e., neurodegeneration) is relatively mild (Eadie, 1963; Seidel et al., 2010) and stands in stark contrast to the fast—and in some subregions almost complete—loss of SN dopamine neurons (Kordower et al., 2013). To test whether DMV neurons respond differently to a chronic challenge of mutant α-synuclein protein, we studied the electrophysiological properties of DMV neurons, the biophysics of Kv4.3 and Cav channels, and the redox state of DMV neurons in transgenic A53T-SNCA mouse models.
Materials and Methods
Slice preparation.
Experimental procedures adhered to and received prior written approval from the Hebrew University Institutional Animal Care and Use Committee and from the German Regierungspräsidium Darmstadt. Experiments were conducted on two mouse lines that overexpress the mutated human α-synuclein (A53T-SNCA) gene and their age-matched wild-type littermate controls. Measurements of the glutathione (GSH) rescue of Kv4.3-channel activity were conducted on 6–8-month-old A53T-SNCA mice (Fig. 1, A53T-SNCANbm) of both sexes on the C57BL/6J background, backcrossed for >6 generations from the original FVN background (Gispert et al., 2003; Subramaniam et al., 2014). Animals were deeply anesthetized with intraperitoneal injections of ketamine (200 mg/kg)–xylazine (23.32 mg/kg) and perfused transcardially with ice-cold modified artificial CSF (ACSF) oxygenated with 95% O2–5% CO2 and containing the following (in mm): 2.5 KCl, 125 NaCl, 25 NaHCO3, 1.25 Na2HPO4, 0.1 CaCl2, 6.2 MgSO4, 2.5 glucose, 2.96 kynurenic acid, and 50 sucrose. The cerebellum, pons, and medulla were rapidly removed, blocked in the coronal plane, and sectioned at a thickness of 240 μm in ice-cold modified ACSF. Slices were then submerged in ACSF, bubbled with 95% O2–5% CO2, and containing the following (in mm): 2.5 KCl, 125 NaCl, 25 NaHCO3, 1.25 Na2HPO4, 2 CaCl2, 2.1 MgSO4, 2.5 glucose, and 22.5 sucrose. Slices then were stored at 36°C for ≥90 min before recording. All the remaining experiments were conducted on 3–8-month-old or 11–15-month-old homozygous A53T-SNCA mice (Line M83; Fig. 1, A53T-SNCAVle; RRID:IMSR_JAX:004479) of both sexes on the C57BL/C3H background (Giasson et al., 2002). Animals in the older age group were used regardless of whether they exhibited motor abnormalities or not. All procedures were identical except that (1) the ice-cold modified ACSF in these experiments contained the following (in mm): 2.5 KCl, 26 NaHCO3, 1.25 Na2HPO4, 0.5 CaCl2, 10 MgSO4, 0.4 ascorbic acid, 10 glucose, and 210 sucrose; and (2) the ACSF in these experiments contained the following (in mm): 2.5 KCl, 126 NaCl, 26 NaHCO3, 1.25 Na2HPO4, 2 CaCl2, 2 MgSO4, and 10 glucose.
Figure 1.

α-Synuclein overexpression in cholinergic neurons of the DMV in A53T-SNCA mice. A, Human α-synuclein immunohistochemistry in coronal slices of the caudal brainstem in wild-typeNbm (left) and A53T-SNCANbm (right) mice. Nbm subscript denotes the modified Nussbaum model (Gispert et al., 2003; Subramaniam et al., 2014). XII n., Hypoglossal nucleus B. Human α-synuclein (green) and ChAT (red) immunohistochemistry in DMV in wild-typeVle (left) and A53T-SNCAVle (right) mice. Vle subscript denotes the Lee model (Giasson et al., 2002). C, Optical density measurements of the immunointensity of α-synuclein (green) expression within regions of interest determined from the ChAT (red) expression (dotted region is for illustration) reveals a rightward shift in the distribution of immunointensity values from A53T-SNCA relative to wild-type DMV neurons.
Slice visualization and electrophysiology.
The slices were transferred to the recording chamber mounted on an upright Zeiss Axioskop fixed-stage microscope and perfused with oxygenated ACSF at 32°C. A 60×, 0.9 numerical aperture water-immersion objective was used to examine the slice using standard infrared differential interference contrast video microscopy. Patch pipette resistance was typically 3–4.5 MΩ. For whole-cell current-clamp recordings and for voltage-clamp measurements of sodium conductances, the pipette contained the following (in mm): 135.5 KCH3SO3, 5 KCl, 2.5 NaCl, 5 Na-phosphocreatine, 10 HEPES, 0.2 EGTA, 0.21 Na2GTP, and 2 Mg1.5ATP, pH 7.3 with KOH (280–290 mOsm/kg). For whole-cell voltage-clamp recordings of calcium currents, the pipette contained the following (in mm): 111 CsCH3SO3, 12.5 CsCl, 1 MgCl2, 0.1 CaCl2, 10 HEPES, 1 EGTA, 0.21 Na2GTP, and 2 Mg1.5ATP, pH 7.3 with CsOH (280–290 mOsm/kg). For the whole-cell voltage-clamp recordings of the Kv4.3 currents, the pipette contained the following (in mm): 135 K-gluconate, 5 KCl, 2 MgCl2, 10 HEPES, 0.1 EGTA, 1 Li2GTP, 5 Na2ATP, pH 7.35 with KOH (270–300 mOsm/kg). Pipette solutions containing an additional 10 mm GSH were dialyzed into DMV neurons in the whole-cell configuration for testing redox modulation of Kv4.3 channels. Electrophysiological recordings were obtained with a Multiclamp 700B amplifier (Molecular Devices) or EPC-10 (HEKA). Signals were digitized at 20–100 kHz and logged onto a personal computer with the Signal 6 (Cambridge Electronic Design), pClamp9 (Molecular Devices; RRID:SCR_011323), or Patchmaster (HEKA; RRID:SCR_000034) software.
Unilateral injection of recombinant adeno-associated viral-mediated vector.
Mice were deeply anesthetized with isoflurane in a nonrebreathing system (2.5% induction, 1–1.5% maintenance) and placed in a stereotaxic frame (Kopf Instruments). Temperature was maintained at 35°C with a heating pad, artificial tears were applied to prevent corneal drying, and animals were hydrated with a bolus of injectable saline (10 ml/kg) mixed with analgesic (5 mg/kg carpofen). A total amount of 400 nl of an adeno-associated virus serotype 9 harboring a mitochondrial targeted redox-sensitive GFP expression construct (AAV9-CMV-MTS-roGFP; 2.3 × 1013 vg/ml; Guzman et al., 2010; Goldberg et al., 2012; Sanchez-Padilla et al., 2014) was injected into the DMV under aseptic conditions. After the injection, the needle was held in the brain for 5 more minutes before it was slowly retracted. All injections were performed using a microsyringe with a stainless steel needle (35 ga; WPI). The coordinates of the injection were as follows: anteroposterior, −7.5 mm; mediolateral, +0.33 mm; and dorsoventral, −4.45 mm, relative to bregma using a flat skull position (Paxinos and Franklin, 2004).
Wide-field calcium imaging.
Pipette solutions contained the following (in mm): 130 K-gluconate, 6 KCl, 8 NaCl, 10 HEPES, 2 Mg1.5ATP, 0.05 fluo-4, pH 7.3 with KOH (280–290 mOsm/kg). After rupture of the membrane patch, ≥5 min were allowed for equilibration of the indicator between the electrode and the somatic cytoplasm. Optical measurements were made using illumination at 470 nm (Mightex) and a cooled electron-multiplying CCD (Evolve 512 Delta, Photometrics). Pixel size was 0.4 μm and 2 × 2 binning was used to achieve high frame rates. Autofluorescence was corrected by subtracting the fluorescence level of a nearby region not containing any indicator-labeled process. Optical and electrophysiological data were obtained using custom-made Winfluor software (John Dempster, University of Strathclyde, UK).
Two-photon laser scanning microscopy.
The two-photon excitation source was a Chameleon Ultra 2 tunable laser system (680–1080 nm; Coherent). Optical signals were acquired using a 920 nm excitation beam to excite the roGFP fluorescence. The roGFP emission (490–560 nm) was detected by the Zeiss LSM 7 multiphoton system. We collected ≤60 frames of the roGFP signal (1.5–4 frames per second) in a single optical plane traversing the somata of the DMV neurons to determine the baseline fluorescence (F) of the cell. At the end of all experiments, 2 mm dithiothreitol (DTT) was applied for 10 min to reduce the mitochondria fully so as to measure the maximal fluorescence (FDTT), followed by 400 μm Aldrithiol (Ald) for 45 min to fully oxidize the mitochondria, so as to determine the minimal fluorescence (FAld). The relative oxidation was then calculated as (FDTT − F)/(FDTT − FAld) (Goldberg et al., 2012).
Immunohistochemistry.
Tissue preparation and the staining protocols were performed as described previously (Chiu et al., 2014). Briefly, animals were deeply anesthetized and perfused transcardially with 0.1 m phosphate buffer (PB) following ice-cold 4% PFA. Coronal sections of the DMV were cut at 30 μm on a cryostat microtome (Leica CM1950) in antifreeze buffer (1:1:3 volume ratio of ethyl glycerol, glycerol, and 0.1 m PB) and stored at −20°C before further analysis. The sections were preincubated in 5% normal horse serum and 0.3% Triton X-100 in 0.1 m PB for 40 min after washing steps, and incubated overnight at 4°C with the primary antibodies [goat anti-choline acetyltransferase (ChAT), 1:100 (Millipore; RRID:AB_262156); mouse anti-human α-synuclein, 1:1000 or 1:5000 (Thermo Fisher Scientific, RRID:AB_1954821); rabbit anti-rat Kv4.3 (1:1000; Alomone Labs, RRID:AB_2040178)]. On the second day, sections were incubated with fluorophore-conjugated species-specific secondary antibodies [donkey anti-mouse/anti-goat/anti-rat; 1:1000 (Abcam)] for 2 h at room temperature. Brain sections were rinsed in PBS and directly coverslipped by fluorescent mounting medium (Vectashield, Vector Laboratories). Multilabeling fluorescent immunostainings of juxtacellularly filled neurons were analyzed using a laser-scanning microscope (LSM 510 Meta, Zeiss, or Fluoview FV1000, Olympus) using either a 10×/0.3 numerical aperture or a 60×/1.4 numerical aperture differential interference contrast lens (10× zoom). For semiquantitative optical density analysis of the human α-synuclein transgene expression in DMV neurons, we used ImageJ software (http://rsbweb.nih.gov/ij/) to determine the mean immunosignal intensities of ChAT-positive regions of interest in the brainstem slices. We used three confocal slices through the DMV from each mouse with a total of three wild-type and three A53T-SNCA mice for the analysis. Each confocal slice was a collapsed z-stack of 10 adjacent 1-μm-thick optical slices chosen so that they arose primarily from within individual DMV neurons. Thus, while it is not possible to rule out the possibility that some of the fluorescence arises from other processes (e.g., glia), the majority of the signal is confined to ChAT-immunopositive neuronal somatodendritic domains in the DMV.
RNA extraction from mouse brain slices.
The DMV was microdissected out from 240-μm-thick brain slices. RNA extraction, cDNA synthesis, qPCR, and primer design were described previously (Cooper et al., 2015). The data were obtained using Bio-Rad CFX Manager 3.0 software. Also, for each primer pair, a dilution curve was created to calculate primer efficiency, which was then used to recalculate the expression results. Fixed values were normalized to the neuronal-specific TUBB3 as a neuronal marker.
Drugs and reagents.
For recording of autonomous discharge, we used a mixture of synaptic receptor blockers containing the following (in μm): 50 d-APV, 5 NBQX, 10 SR 95531, 1 CGP 55845, 10 mecamylamine, and 10 atropine. Voltage-activated sodium channels were blocked with 1 μm tetrodotoxin (TTX). Cav currents were antagonized with 1.8 mm cobalt (Goldberg et al., 2012). The acute effects of solution exchanges or drug applications were measured ≥5 min after wash on. TTX was obtained from Alomone Labs. Calcium indicators (fluo-4) were acquired from Invitrogen. The other drugs and reagents were obtained from Tocris Bioscience or Sigma-Aldrich.
Data and statistical analysis.
Electrophysiological data were analyzed and curve fitting was done using custom-made code on custom-made (Winfluor, Dr. John Dempster, University of Strathclyde; NUPver, Nicholas Schwarz, Northwestern University; oscilloscope, Dr. Charles Wilson, University of Texas at San Antonio) and commercial (Matlab, Mathworks; RRID:SCR_001622) software. The two-tailed Wilcoxon rank-sum test (RST) was used to test for changes of medians in two independent sample comparisons. The two-tailed Wilcoxon signed-rank test was used to test for changes of medians in matched-paired comparisons. ANCOVA was used to compare (1) the availability of voltage-activated sodium (Nav) channels in A53T-SNCA versus wild-type mice by measuring the dependence of maximal dV/dt of the action potential voltage trajectory on action potential number during a burst of 10 spikes; and (2) the percent change in Kv4.3 currents as a function of time since the beginning of intracellular dialysis with GSH. The null hypotheses were rejected if the P values were <0.05.
Results
Autonomous firing rates of DMV neurons are unchanged in A53T-SNCA mice
Immunohistochemical studies of coronal sections of the caudal brainstem revealed expression of mutant human α-synuclein in the DMV of transgenic A53T-SNCA mice, but not in their corresponding wild-type littermates (Fig. 1). Double staining for ChAT demonstrated that the mutant human protein is expressed in somata of DMV neurons (Fig. 1B), and in some case even more strongly in processes other than those of neuronal cell bodies (Fig. 1C, left). Nevertheless, quantitative optical density measurements reveal a 45-fold increase in the mean α-synuclein immunointensity in the somata of cholinergic motoneurons of the DMV from A53T-SNCA mice relative to wild-type mice (Fig. 1C, right), meaning that there is significant overexpression of α-synuclein in cholinergic DMV neurons only in the transgenic mouse. DMV neurons from either wild-type or A53T-SNCA mice recorded in the loose-patch or cell-attached modes (which do not disrupt the intracellular milieu and therefore preserve the autonomous firing patterns) in the presence of a mixture of glutamatergic, GABAergic, and cholinergic receptor antagonist demonstrated that the autonomous firing rate of DMV neurons was unaffected by ASOX (wild-type median: 1.21 spikes/s, n = 28; A53T-SNCA median: 1.28 spikes/s, n = 21; p > 0.99, RST; Fig. 2A). The dominant inward current activated in the subthreshold voltage range that drives autonomous pacemaking in DMV neurons is the persistent sodium current, which is significantly larger than the Cav1 calcium or the hyperpolarization/cyclic nucleotide gated cation (HCN) currents in these neurons (Goldberg et al., 2012). Direct measurement of the TTX-sensitive persistent sodium current in these neurons in A53T-SNCA mice (n = 7 neurons) versus wild-type mice (n = 7 neurons) demonstrated that it did not differ between the genotypes (p > 0.6, ANCOVA; Fig. 2B). This finding can therefore explain the lack of difference in the autonomous firing rates of DMV neurons between the two genotypes.
Figure 2.
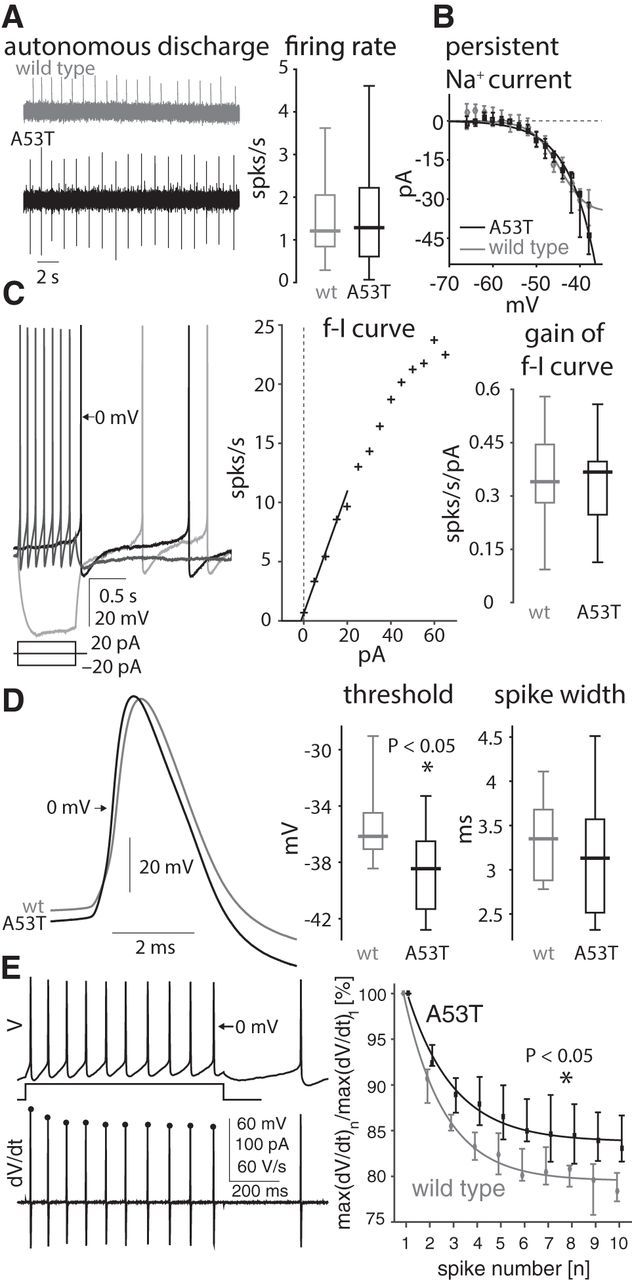
Firing properties of DMV neurons in A53T-SNCA mice. A, Left, Loose-patch recording of autonomous firing of DMV neurons in wild-type (wt; gray trace) and A53T-SNCA mice. Right, Distribution of autonomous DMV neurons firing rates in both genotypes (wild type: n = 28 neurons; A53T-SNCA: n = 21 neurons). B, Measurement of the persistent TTX-sensitive current in DMV neurons in response to long subthreshold voltage pulses reveals no difference between wild-type (gray, n = 7 neurons) and A53T-SNCA (black, n = 7 neurons) mice. C, Left, Whole-cell recording of the autonomous firing pattern (black trace) or in response to depolarizing (dark gray trace) and hyperpolarizing (light gray trace) pulses in a DMV neuron from an A53T-SNCA mouse. Middle, f–I curve of the cell depicted on the right, using the reciprocal of the first interspike interval. Straight line, Fit of the initial slope (gain) of the f–I curve. Right, Distribution of the gain of f–I curves from wild-type (n = 25) and A53T-SNCA (n = 21) neurons. D, Left, DMV neuron action potential waveform (during autonomous firing) in wild-type (gray trace, n = 10 neurons) and A53T-SNCA mice (black trace, n = 16 neurons). Right, Distributions of voltage threshold and width of action potentials in DMV neurons from wild-type (gray, n = 10 neurons) and A53T-SNCA mice (black, n = 15 neurons). E, Left, Voltage (V) and derivative of voltage (dV/dt,) trajectories in response to current injections that elicit 10 action potentials in an 800 ms interval. Distribution of the maximal dV/dt as a function of action potential number (n) normalized by the max dV/dt value of the first action potential reveals that Nav-channel availability is significantly larger for the A53T-SNCA mice (wild type: n = 19 neurons; A53T-SNCA: n = 19 neurons). In B and E, the medians of each measurement are presented with confidence intervals given by 50% × (1 ± 1/√k) percentiles where k is the sample size.
To determine whether ASOX might affect other aspects of the excitability of DMV neurons, we recorded their firing patterns in the whole-cell configuration. DMV neurons fired autonomously in the whole-cell configuration (Fig. 2C, left, black trace), as previously reported (Goldberg et al., 2012; Cooper et al., 2015). Long hyperpolarizing currents elicited the typical voltage sag, attributable to the HCN current, followed by the slow ramp up to voltage threshold, attributable to the A-type Kv4.3 potassium current (Fig. 2C, left, light gray trace). Long depolarizing currents sped up the cells and elicited a slow afterhyperpolarization (sAHP, Fig. 2C, left, dark gray trace; Sah and McLachlan, 1991; Hocherman et al., 1992; Goldberg et al., 2012). Measurement of the frequency–intensity (f–I) curve, using the reciprocal of the first interspike interval (Fig. 2C, middle), demonstrated that the firing rate initially rose linearly with current. However, the initial slope (gain) of the f–I curve did not differ between wild-type and A53T-SNCA neurons (wild-type median: 0.340 spikes/s/pA, n = 25; A53T-SNCA median: 0.367 spikes/s/pA, n = 21; p > 0.85, RST; Fig. 2C, right).
Availability of Nav channels is elevated in A53T-SNCA mice
Measurement of the trajectory of individual action potentials revealed that the voltage threshold of DMV neurons was slightly hyperpolarized in the A53T-SNCA mice (wild-type median: −36.2 mV, n = 10; A53T-SNCA median: −38.5 mV, n = 16; p < 0.04, RST), whereas the width of the action potential was unchanged (amplitude >70 mV; wild-type median: 3.35 ms, n = 10; A53T-SNCA median: 3.13 ms, n = 15; p > 0.35, RST; Fig. 2D). A hyperpolarized action potential voltage threshold is usually indicative of a higher degree of Nav-channel availability, which is also echoed in a steeper rate of rise of voltage (dV/dt) during the action potential upswing (Fig. 2D, left). A more sensitive biophysical measure of Nav-channel availability is the reduction in the maximal dV/dt during a train of action potentials (Hondeghem, 1978; Strichartz and Cohen, 1978; Fleidervish et al., 2008). Applying this technique, we found that the maximal dV/dt undergoes a smaller reduction during a train of action potentials in DMV neurons from A53T-SNCA mice compared with DMV neurons from wild-type littermate mice (wild-type n = 19 cells; A53T-SNCA n = 19 cells; p < 0.04, ANCOVA; Fig. 2E).
ASOX prevents oxidative dysfunction of Kv4.3 channels in DMV neurons
Because Nav-channel availability is elevated in reduced environments (Fukuda et al., 2005; Nakajima et al., 2010), our finding raises the intriguing possibility that the cytosol of DMV neurons in A53T-SNCA neurons is actually reduced relative to that of wild-type mice. If this is true, it could mean that Kv4.3 channels that give rise to a prominent A-type potassium current in DMV neurons (Yarom et al., 1985; Sah and McLachlan, 1992; Cooper et al., 2015) do not suffer the same oxidative impairment that reduces their amplitude in SN dopamine neurons in A53T-SNCA mice (Subramaniam et al., 2014). Immunohistochemical studies of Kv4.3 expression revealed strong expression in the DMV of both wild-type and A53T-SNCA mice (Fig. 3A). In contrast to SN dopamine neurons, where the Kv4.3-mediated A-type currents were impaired in the A53T-SNCA mice relative to the wild-type mice (Subramaniam et al., 2014), we found that Kv4.3 currents in DMV neurons from A53T-SNCA and wild-type mice showed no differences in amplitude (calculated by fitting a sigmoidal to the peak current as a function of voltage; wild-type median: 947 pA, n = 9 cells; A53T-SNCA median: 1171 pA, n = 9 cells; p > 0.86, RST; data not shown) or voltage-dependent gating (Fig. 3B).
Figure 3.

Glutathione (GSH) rescues Kv4.3 currents in DMV neurons in the wild-type but not the A53T-SNCA mice. A, Kv4.3 immunohistochemistry in DMV neurons in wild-type and A53T-SNCA mice. B, Measurement of transient outward (A-type) potassium currents in whole-cell voltage-clamp recordings reveals that activation and inactivation of Kv4.3 channels is unchanged in A53T-SNCA mice (n = 9 neurons) relative to wild-type controls (n = 9 neurons). C, Left, A-type current traces (in response to voltage steps to −40 mV as in the “activation” traces in B) during intracellular dialysis of 10 mm GSH reveal a rescue of the current in the wild-type (gray traces) but not the A53T-SNCA (black traces) mice. The traces are measured at 10 s intervals. Right, Graph showing the relative change of the median A-type potassium current during GSH dialysis on DMV neurons from control (gray circles, n = 6 neurons) and A53T-SNCA (black squares, n = 7 neurons) mice, respectively. The lines represent an exponential fit of median control data and a linear fit for the median A53T-SNCA data, respectively. Confidence intervals are calculated as in Figure 2.
In SN dopamine neurons, intracellular dialysis of the antioxidant GSH (10 mm) rescued the impaired Kv4.3-mediated A-type currents in the A53T-SNCA mice (Subramaniam et al., 2014). Surprisingly, we found that intracellular dialysis of GSH increased the amplitude of the A-type currents in wild-type DMV neurons by >60% (n = 6 cells, Fig. 3C, gray marks), which is indicative of a reversible oxidative dysfunction of Kv4.3 channels in these neurons under baseline conditions. In contrast, we found no evidence for oxidative dysfunction of Kv4.3 channels in DMV neurons from A53T-SNCA mice, because intracellular dialysis of 10 mm GSH did not alter the amplitude of A-type currents (n = 7 cells, Fig. 3C, black marks). The responses in the two genotypes to GSH dialysis differed significantly from each other (p < 10−9, ANCOVA).
Basal mitochondrial oxidative stress is reduced in DMV neurons in A53T-SNCA mice
The fact that GSH failed to rescue of Kv4.3 currents suggests that oxidative stress is reduced in DMV neurons from A53T-SNCA mice. To monitor oxidative levels directly in DMV neurons, we stereotaxically injected an adeno-associated virus serotype 9 harboring the mitochondrial-targeted, redox-sensitive GFP expression construct (AAV9-CMV-mito-roGFP) into the DMV of 7–11-month-old A53T-SNCA mice and their littermate controls (Fig. 4A). Two weeks following transfection, we used two-photon laser scanning microscopy (2PLSM) to image the level of mito-roGFP fluorescence in vagal motoneurons in coronal slices of DMVs. The relative oxidization level of each neuron was calibrated at the end of each experiment by comparing it to the levels of fluorescence attained with strong reducing and oxidizing agents (Goldberg et al., 2012). The distribution of values—estimated using viral delivery of mito-roGFP—of basal mitochondrial oxidative stress in DMV neurons of wild-type mice matched the values estimated from transgenic mice expressing mito-roGFP in DMV neurons under the CMV promoter (Fig. 4B, dotted boxplot; Goldberg et al., 2012). Importantly, our analysis revealed that basal mitochondrial oxidative stress in DMV neurons was significantly diminished in the A53T-SNCA mice (wild-type median: 40.4%, n = 7 cells from N = 4 mice; A53T-SNCA median: 8.6%, n = 10 cells from N = 4 mice; p < 0.02, RST; Fig. 4B).
Figure 4.

Basal mitochondrial oxidative stress is reduced in DMV neurons from A53T-SNCA mice. A, Confocal image of DMV neuron from a wild-type mouse expressing mito-roGFP 2 weeks after viral transfection with the AAV9-CMV-mito-roGFP. B, The relative oxidization was estimated by measuring the resting fluorescence of the mitochondria using 2PLSM (in the somata surrounding the nucleus) relative to post-treatment with strong reducing (2 mm dithiothreitol) and oxidizing (400 μm aldrithiol) agents. Basal mitochondrial oxidative stress is reduced in DMV neurons from A53T-SNCA mice (green boxplot, n = 10 neurons) relative to DMV neurons from wild-type mice (black boxplot, n = 7 neurons). The distribution of relative oxidization in DMV neurons from transgenic mice expressing mito-roGFP under the CMV promoter (dotted gray boxplot; Goldberg et al., 2012) is very similar to the distribution measured from the virally transfected wild-type mice in the present study.
Downregulated Cav currents reduced activity-dependent calcium levels in DMV neurons from A53T-SNCA mice
The degree of basal mitochondrial oxidative stress is dictated in DMV neurons (Goldberg et al., 2012) as well as in other neurons vulnerable in PD (Guzman et al., 2010; Surmeier et al., 2011; Sanchez-Padilla et al., 2014) by calcium influx via Cav channels during autonomous pacemaking. Therefore, finding a drastic reduction in basal mitochondrial oxidative stress in A53T-SNCA mice predicts reduced activity-dependent calcium levels and Cav currents in these mice relative to wild-type control mice. To compare activity-dependent intracellular calcium levels in DMV neurons from wild-type and A53T-SNCA mice, we used wide-field somatic calcium imaging of DMV neurons loaded with 50 μm of the fluorescent indicator, fluo-4 (Fig. 5A). Because the concentration of intracellular calcium depends on the neurons' firing rates (Goldberg et al., 2012), we measured the increase in ΔF/F0 while injecting a series of long current pulses to first silence and then activate the cells progressively faster (Fig. 5B; Cooper et al., 2015). As shown previously (Goldberg et al., 2012; Cooper et al., 2015), there is a robust linear relationship between ΔF/F0 and the neuron's firing rate (Fig. 5C). The slope of this linear relationship was significantly reduced by ∼25% in A53T-SNCA relative to wild-type mice [wild-type median: 0.104 (ΔF/F0)/(spikes/s), n = 19 cells from N = 3 mice; A53T-SNCA median: 0.078 (ΔF/F0)/(spikes/s), n = 19 cells from N = 3 mice; p < 0.05, RST; Fig. 5C], indicating that individual spikes contribute less to the basal calcium levels in DMV neurons in the A53T-SNCA relative to wild-type mice. This reduction in somatic calcium levels suggests that calcium currents via Cav channels, which are activated during spiking, are reduced in DMV neurons from A53T-SNCA mice relative to wild-type mice. To test this hypothesis, we measured cobalt-sensitive calcium currents with fast voltage ramps (240 mV/s), and found that the mean Cav currents (wild-type, n = 6 cells; A53T-SNCA, n = 12 cells) were indeed significantly smaller in 3–8-month-old (nominally presymptomatic) A53T-SNCA mice (the peak activation was reduced by more than half; Fig. 6A). In addition, the mean current waveform suggested that the voltage gating of the Cav current was shifted to more depolarized voltages (Fig. 6A). To quantify this effect at a cell-by-cell level, we estimated the half activation of the Cav current in each cell individually, and found that there was a significant rightward shift in the activation curve of Cav currents in the A53T-SNCA mice (wild-type median: −10.7 mV, n = 6 cells; A53T-SNCA median: −6.1 mV, n = 10 cells; p < 0.025, RST; Fig. 6B). Measurement of the average currents (data not shown) and half activation voltages in DMV neurons from 12–15-month-old mice revealed the same rightward shift (wild-type median: −13.3 mV, n = 4 cells from N = 3 mice; A53T-SNCA median: −4.7 mV, n = 4 cells from N = 3 mice; p < 0.03, RST; Fig. 6B).
Figure 5.

Spike-dependent calcium levels are reduced in DMV neurons in A53T-SNCA mice. A, A DMV neuron loaded with 50 μm fluo-4 for wide-field somatic calcium imaging. B, A sequence of 10-s-long current pulses from −20 pA to +60 pA, induces spiking at progressively higher rates in DMV neurons from wild-type (left) and A53T-SNCA (right) mice, which are accompanied by increases in somatic calcium levels, as measured by increased ΔF/F0. C, Left, The linear dependence of ΔF/F0 on firing rate in the DMV neuron depicted in B (average of final 5 s during each current step). Right, The distribution of the slope [(ΔF/F0)/(spikes/s)] of the linear relationship in A53T-SNCA (n = 19 neurons) and wild-type (n = 19 neurons) mice demonstrate that activity-dependent calcium levels are significantly reduced in A53T-SNCA relative to wild-type mice.
Figure 6.

Cav channels are downregulated in DMV neurons in A53T-SNCA mice. A, Mean cobalt-sensitive Cav currents evoked with a voltage ramp in 3-month-old A53T-SNCA mice (red, n = 12 neurons) is reduced and shifted to the right relative to littermate wild-type (wt) mice (black, n = 6 neurons). B, Left, Fitting activation curves to individual cobalt-sensitive current measurements yields their half-activation voltage (V1/2). Right, Distribution of half-activation voltages reveals a significant depolarization both in 3–8-month-old presymptomatic (wild type, n = 6 neurons; A53T-SNCA, n = 10 neurons) and 12–15-month-old symptomatic mice (wild type, n = 4 neurons; A53T-SNCA, n = 4 neurons). C, Cobalt-sensitive calcium action current evoked during an action potential voltage clamp is reduced leading to a significant reduction in the calcium charge fluxed during the clamp (inset; wild type, n = 6 neurons; A53T-SNCA, n = 16 neurons). D, Neuronal Cav channel α1 subunit mRNA expression normalized to TUBB3 in microdissected DMV from A53T-SNCA mice relative to wild-type controls (Cooper et al., 2015). The expression level is expressed in percentages of the median value of expression of Cav1.2 α1 subunit mRNA in wild-type mice (leftmost boxplot).
A rightward shift in the Cav activation curve implies that with each action potential there should be fewer channel openings and therefore a smaller flux of calcium ions in the A53T-SNCA mice. We used the action-potential clamp technique (Llinás et al., 1982; Bean, 2007; Goldberg et al., 2012) to estimate the calcium charge fluxed with each action potential (as the integral beneath the curve of the cobalt-sensitive calcium action current; Fig. 6C), and indeed found a reduction in this charge in the A53T-SNCA mice (wild-type median: 2.36 pC, n = 6 cells; A53T-SNCA median: 1.49 pC, n = 16 cells; p < 0.04, RST; Fig. 6C, inset), compatible with the reduction in the spike-triggered increases in calcium levels reported above (Fig. 5).
Cav1.2 and Cav2.3 α1 subunit transcripts are downregulated in A53T-SNCA mice
DMV neurons express a variety of Cav channels whose α1 subunits are mostly Cav1.2 or Cav2.3 (Cooper et al., 2015). Both these channels activate at slightly more hyperpolarized potentials than the rest of the high-voltage-activated Cav2 channels (Regan et al., 1991; Tottene et al., 1996; Magistretti et al., 2000; Lipscombe et al., 2004). We therefore hypothesized that the rightward shift, as well as the overall reduction in the current (Fig. 6A), could arise simply from a reduced expression levels of these subunits in the ASOX mice. Tissue level quantitative RT-PCR analysis in the DMV (normalized to neuronal markers) indeed revealed that transcripts of these two α1 subunits were significantly downregulated in the A53T-SNCA mice, while those of other Cav1 and Cav2 α1 subunits were not (Fig. 6D).
Discussion
α-Synuclein and selective vulnerability in PD
The causal role of α-synuclein in the pathogenesis of familial and sporadic PD has been firmly established (Goedert et al., 2013). Analysis of rare familial forms of PD identified either causal dominant mutations in the gene coding for α-synuclein (e.g., A53T-SNCA) or multiplications of the SNCA gene locus (Devine et al., 2011). In addition, genome-wide association studies in large cohorts of common sporadic forms of PD have demonstrated highly significant (p < 10−60) associations between increased risk for PD and certain single nucleotide polymorphisms (SNPs) within SNCA (Simón-Sánchez et al., 2009). These SNPs are associated with increased α-synuclein expression, which is in line with Mendelian forms of PD, where an increased α-synuclein load drives the disease process. There is also consensus that aggregation of α-synuclein to toxic fibrils—enhanced by load, mutations, and protein modifications—is a crucial step in PD pathogenesis (Farrer, 2006).
What is less understood is how these α-synuclein-related processes might manifest within the unique cellular context of each neuronal population vulnerable in PD. For example dopamine, its metabolites, and its transporters might exacerbate the toxic effect of α-synuclein (Tabrizi et al., 2000; Lotharius and Brundin, 2002; Wersinger and Sidhu, 2003; Sidhu et al., 2004; Sulzer, 2010; Oaks et al., 2013; Jain and Bhat, 2014; Jinsmaa et al., 2014; Tavassoly et al., 2014)—interactions unique to dopaminergic neurons. Neuromelanin may also interact with α-synuclein deleteriously (Dias et al., 2013; Xu and Chan, 2015), a process that may be relevant to LC neurons, as well. In summary, it is possible that cell-specific interactions predispose various neuronal types to divergent cell fates, which could explain why highly vulnerable (e.g., SN dopamine or LC) neurons massively degenerate in response to α-synucleinopathies while more resistant (e.g., DMV) neurons do so to a lesser extent.
α-Synuclein and oxidative dysfunction in vulnerable neurons
Mouse models, in which the load of mutant or wild-type α-synuclein expression can be experimentally regulated, can prove useful in addressing the question of cell-type-specific effects of α-synuclein. Nevertheless, few studies have looked at the physiological changes in defined neuronal populations in response to ASOX. Two recently published in vivo studies on SN dopamine neurons in ASOX mouse models offer conflicting results. Using a transgenic mouse model overexpressing human wild-type α-synuclein on a background of a C57BL6 α-synuclein knock-out mouse, Janezic and colleagues found ∼30% reduction of in vivo mean firing frequencies in old mice (18–22 months), where a significant loss of SN dopamine neurons was already present (Janezic et al., 2013). In contrast, Subramaniam and colleagues reported a twofold increase of in vivo mean firing frequencies in middle-aged (6–10 months) but not younger (3 months) adults (Subramaniam et al., 2014). The latter study also found that oxidative dysfunction of an A-type (Kv4.3) potassium channel was an important element of α-synuclein-induced hyperactivity. This finding is in agreement with other, more acute in vitro studies showing that α-synuclein enhances oxidative stress (Hsu et al., 2000; Sherer et al., 2001; Dryanovski et al., 2013).
In both SN dopamine neurons and vagal motoneurons, substantial calcium influx during autonomous pacemaking is sufficient to generate basal mitochondrial oxidative stress (Guzman et al., 2010; Goldberg et al., 2012). Multiple lines of evidence support the hypothesis that ASOX should elevate oxidative stress by elevating calcium influx. For example, ASOX may directly potentiate Cavs (Adamczyk and Strosznajder, 2006; Hettiarachchi et al., 2009) or may increase calcium influx by elevating autonomous firing rates, which will invariably increase activity-dependent calcium influx via Cav channels (Dryanovski et al., 2013; Subramaniam et al., 2014). Alternatively, neurons possess several homeostatic mechanisms controlling both synaptic and intrinsic excitability to maintain their physiological firing range even when challenged with stressors (Turrigiano, 2011; Dias et al., 2013), meaning that elevation of firing could be prevented and calcium currents could be kept at bay. Indeed, even SN dopamine neurons mount a partial homeostatic response to α-synuclein-induced hyperactivity by elevating expression and membrane delivery of Kv4.3 channels (Subramaniam et al., 2014).
Our findings suggest that DMV neurons, in contrast to SN dopamine neurons, are able to mount a successful homeostatic response to significant overexpression of α-synuclein (as validated by immunohistochemistry), and that these neurons are able to maintain their normal autonomously generated firing rates in vitro both in young (nominally presymptomatic) and in older adult mice. Interestingly, a recent study has reported a reduction in the autonomous firing rate of DMV neurons from aging triple synuclein knock-out (αβγ−/−) mice (Machhada et al., 2015). Our data demonstrate that the biophysical mechanism of the homeostatic response that maintains the firing rate in ASOX DMV neurons involved both Kv4.3 A-type channels, which are prominent determinants of DMV neuron firing rates (Cooper et al., 2015), and Cav channels, as explained presently.
Unlike its effect on SN dopamine neurons, GSH dialysis did not rescue Kv4.3-channel activity in DMV neurons in the A53T-SNCA mouse, indicating that the Kv4.3 activity was not impaired. Moreover, GSH dialysis demonstrated that Kv4.3 channels were impaired in wild-type DMV neurons, implying that not only did ASOX not exacerbate oxidative stress; rather it significantly alleviated it in the A53T-SNCA mice. Furthermore, direct visualization with 2PLSM of basal mitochondrial oxidative stress in DMV neurons with the virally delivered mito-roGFP reporter demonstrated that indeed oxidative stress was all but abolished in these neurons in the A53T-SNCA mice.
Oxidative stress and calcium homeostasis in vulnerable neurons
Given our previous study on the role of activity-dependent calcium influx via Cav channels in generating basal mitochondrial oxidative stress in DMV neurons (Goldberg et al., 2012), we looked for changes in calcium signaling in these cells. We identified significantly smaller mean Cav currents in A53T-SNCA compared with wild-type DMV neurons as early as 3 months after birth, and at least until 15 months. We also noted that the voltage-dependence of the mean Cav currents in A53T-SNCA DMV neurons, to which several Cav-channel subtypes—known to be coexpressed in DMV neurons (Cooper et al., 2015)—contribute. These reduced calcium currents also lead to measurable reductions in activity-dependent cytosolic calcium levels in A53T-SNCA DMV neurons. Comparative quantitative RT-PCR of the microdissected DMV nucleus demonstrated significantly lower mRNA levels of neuronal Cav1.2 and Cav2.3 α1 subunits in A53T-SNCA compared with wild-type mice. If translated into a matching reduction of functional Cav1.2 and Cav2.3 channel complexes, this result explains the observed reduction and apparent rightward shift of the global Cav currents (Regan et al., 1991; Tottene et al., 1996; Magistretti et al., 2000; Lipscombe et al., 2004).
In two recent studies (Goldberg et al., 2012; Cooper et al., 2015), we characterized the specific effect of various Cav-channel subunits on the autonomous firing of DMV neurons. We found that while Cav1.3 channels can speed up pacemaking, Cav2.2 and Cav1.2 channels slow down pacemaking presumably via their coupling to the small-conductance calcium-activated potassium current, and the current underlying the sAHP, respectively. Of these three subunits, only Cav1.2 is downregulated in A53T-SNCA DMV neurons, which might be expected to speed up their autonomous firing rate. However, the role of Cav1.2 channels was demonstrated by a paired comparison within individual DMV neurons of the firing rate before and after complete block of Cav1 currents (with 5 μm nifedipine; Cooper et al., 2015). Therefore, it is possible that in the current study when comparing A53T-SNCA versus wild-type DMV neurons, the expected small effect of the downregulation of Cav1.2 channels on the autonomous firing rate of the former population (which was estimated as an increase of the firing rate by 0.25 spikes/s), might have been lost within the normal firing rate variability of DMV neurons in both populations.
Antioxidative response in vagal motoneurons overexpressing α-synuclein
In essence, we discovered that DMV neurons initiated an antioxidative response upon chronic challenge with mutant α-synuclein without obvious impairment of their function—at least vis-à-vis in vitro pacemaking. This might explain why DMV neurons—although a locus of very early α-synucleinopathy in PD—do not degenerate at such an extent as the highly vulnerable SN dopamine neurons, at least based on the small number of quantitative human postmortem studies in the DMV (Eadie, 1963; Seidel et al., 2010). The unexpected and overshooting protective remodeling of redox signaling in DMV neurons might be executed on multiple levels ranging from changes in activity-mediated calcium influx, via enhanced calcium extrusion and buffering, to orchestrated transcriptional responses leading to an enhancement of endogenous antioxidative defense mechanisms [e.g., activation of the nuclear erythroid 2-related factor 2–antioxidant response element pathway, which restores GSH levels (Lim et al., 2014)]. The fact that the remodeling seems to be selective to DMV neurons may depend on their cholinergic specification, which in turn depends on the expression of brain region-specific transcription factors that influence Cav expression and that may be distinct from those present in SN dopamine neurons (Soreq, 2015). Deciphering the molecular details of this protective adaptation in DMV neurons might pave the way to novel treatments that could emulate these changes in SN dopamine neuron, to counter the tendency of ASOX to exacerbate their already high innate oxidative stress. Antagonizing Cav channels with brain-penetrant dihydropyridines, a treatment currently in a phase III clinical trial (“STEADY-PD III”, ClinicalTrials.gov identifier: NCT02168842), already follows the putative strategy of reducing oxidative stress and slowing the degeneration of SN dopamine neurons via manipulation of Cav channels. When challenged with ASOX, DMV neurons appear to employ the same Cav-current-reducing strategy by remodeling their biophysics toward stressless pacemaking.
Footnotes
This work was supported by grants from the Michael J. Fox Foundation (2013 Rapid Response Innovation Award to J.A.G.), the German-Israeli Foundation for Scientific Research and Development (no. I-1294-418.13/2015 to J.R. and J.A.G), and the Collaborative Research Center 815 “Redox Signaling” program to J.R. H.S. acknowledges the support of the Edmond and Lily Safra Center for Brain Sciences. The CMV-mito-roGFP adeno-associated virus was contributed by Dr. Jim Surmeier (Northwestern University). We thank Prof. Yoel Yaari and Dr. Alex Binshtok (the Hebrew University of Jerusalem) for the use of their equipment, and Anatoly Shapochnikov for excellent technical assistance.
The authors declare no competing financial interests.
References
- Adamczyk A, Strosznajder JB. Alpha-synuclein potentiates Ca2+ influx through voltage-dependent Ca2+ channels. Neuroreport. 2006;17:1883–1886. doi: 10.1097/WNR.0b013e3280115185. [DOI] [PubMed] [Google Scholar]
- Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8:451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- Bolam JP, Pissadaki EK. Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord. 2012;27:1478–1483. doi: 10.1002/mds.25135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- Chiu WH, Carlsson T, Depboylu C, Höglinger GU, Oertel WH, Ries V. Selegiline normalizes, while l-DOPA sustains the increased number of dopamine neurons in the olfactory bulb in a 6-OHDA mouse model of Parkinson's disease. Neuropharmacology. 2014;79:212–221. doi: 10.1016/j.neuropharm.2013.11.014. [DOI] [PubMed] [Google Scholar]
- Cooper G, Lasser-Katz E, Simchovitz A, Sharon R, Soreq H, Surmeier DJ, Goldberg JA. Functional segregation of voltage-activated calcium channels in motoneurons of the dorsal motor nucleus of the vagus. J Neurophysiol. 2015;114:1513–1520. doi: 10.1152/jn.00432.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MJ, Gwinn K, Singleton A, Hardy J. Parkinson's disease and α-synuclein expression. Mov Disord. 2011;26:2160–2168. doi: 10.1002/mds.23948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson's disease. J Parkinsons Dis. 2013;3:461–491. doi: 10.3233/JPD-130230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- Dryanovski DI, Guzman JN, Xie Z, Galteri DJ, Volpicelli-Daley LA, Lee VM, Miller RJ, Schumacker PT, Surmeier DJ. Calcium entry and α-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J Neurosci. 2013;33:10154–10164. doi: 10.1523/JNEUROSCI.5311-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eadie MJ. The pathology of certain medullary nuclei in Parkinsonism. Brain. 1963;86:781–792. doi: 10.1093/brain/86.4.781. [DOI] [PubMed] [Google Scholar]
- Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7:306–318. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]
- Fleidervish IA, Libman L, Katz E, Gutnick MJ. Endogenous polyamines regulate cortical neuronal excitability by blocking voltage-gated Na+ channels. Proc Natl Acad Sci U S A. 2008;105:18994–18999. doi: 10.1073/pnas.0803464105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K, Davies SS, Nakajima T, Ong BH, Kupershmidt S, Fessel J, Amarnath V, Anderson ME, Boyden PA, Viswanathan PC, Roberts LJ, 2nd, Balser JR. Oxidative mediated lipid peroxidation recapitulates proarrhythmic effects on cardiac sodium channels. Circ Res. 2005;97:1262–1269. doi: 10.1161/01.RES.0000195844.31466.e9. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/S0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Gispert S, Del Turco D, Garrett L, Chen A, Bernard DJ, Hamm-Clement J, Korf HW, Deller T, Braak H, Auburger G, Nussbaum RL. Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol Cell Neurosci. 2003;24:419–429. doi: 10.1016/S1044-7431(03)00198-2. [DOI] [PubMed] [Google Scholar]
- Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. 2013;9:13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- Goldberg JA, Guzman JN, Estep CM, Ilijic E, Kondapalli J, Sanchez-Padilla J, Surmeier DJ. Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson's disease. Nat Neurosci. 2012;15:1414–1421. doi: 10.1038/nn.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettiarachchi NT, Parker A, Dallas ML, Pennington K, Hung CC, Pearson HA, Boyle JP, Robinson P, Peers C. alpha-Synuclein modulation of Ca2+ signaling in human neuroblastoma (SH-SY5Y) cells. J Neurochem. 2009;111:1192–1201. doi: 10.1111/j.1471-4159.2009.06411.x. [DOI] [PubMed] [Google Scholar]
- Hocherman SD, Werman R, Yarom Y. An analysis of the long-lasting after-hyperpolarization of guinea-pig vagal motoneurones. J Physiol. 1992;456:325–349. doi: 10.1113/jphysiol.1992.sp019339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondeghem LM. Validity of Vmax as a measure of the sodium current in cardiac and nervous tissues. Biophys J. 1978;23:147–152. doi: 10.1016/S0006-3495(78)85439-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. alpha-Synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–410. doi: 10.1016/S0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain MK, Bhat R. Modulation of human α-synuclein aggregation by a combined effect of calcium and dopamine. Neurobiol Dis. 2014;63:115–128. doi: 10.1016/j.nbd.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Janezic S, Threlfell S, Dodson PD, Dowie MJ, Taylor TN, Potgieter D, Parkkinen L, Senior SL, Anwar S, Ryan B, Deltheil T, Kosillo P, Cioroch M, Wagner K, Ansorge O, Bannerman DM, Bolam JP, Magill PJ, Cragg SJ, Wade-Martins R. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci U S A. 2013;110:E4016–E4025. doi: 10.1073/pnas.1309143110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinsmaa Y, Sullivan P, Gross D, Cooney A, Sharabi Y, Goldstein DS. Divalent metal ions enhance DOPAL-induced oligomerization of alpha-synuclein. Neurosci Lett. 2014;569:27–32. doi: 10.1016/j.neulet.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, Bartus RT. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain. 2013;136:2419–2431. doi: 10.1093/brain/awt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewy FH. Paralysis agitans. I. Pathologische Anatomie (in German) In: Lewandowski B, editor. Handbuch der Neurologie. Berlin: Springer; 1912. pp. 920–933. [Google Scholar]
- Lim JL, Wilhelmus MM, de Vries HE, Drukarch B, Hoozemans JJ, van Horssen J. Antioxidative defense mechanisms controlled by Nrf2: state-of-the-art and clinical perspectives in neurodegenerative diseases. Arch Toxicol. 2014;88:1773–1786. doi: 10.1007/s00204-014-1338-z. [DOI] [PubMed] [Google Scholar]
- Lipscombe D, Helton TD, Xu W. L-type calcium channels: the low down. J Neurophysiol. 2004;92:2633–2641. doi: 10.1152/jn.00486.2004. [DOI] [PubMed] [Google Scholar]
- Liss B, Franz O, Sewing S, Bruns R, Neuhoff H, Roeper J. Tuning pacemaker frequency of individual dopaminergic neurons by Kv4.3L and KChip3.1 transcription. EMBO J. 2001;20:5715–5724. doi: 10.1093/emboj/20.20.5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Sugimori M, Simon SM. Transmission by presynaptic spike-like depolarization in the squid giant synapse. Proc Natl Acad Sci U S A. 1982;79:2415–2419. doi: 10.1073/pnas.79.7.2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotharius J, Brundin P. Impaired dopamine storage resulting from alpha-synuclein mutations may contribute to the pathogenesis of Parkinson's disease. Hum Mol Genet. 2002;11:2395–2407. doi: 10.1093/hmg/11.20.2395. [DOI] [PubMed] [Google Scholar]
- Machhada A, Ang R, Ackland GL, Ninkina N, Buchman VL, Lythgoe MF, Trapp S, Tinker A, Marina N, Gourine AV. Control of ventricular excitability by neurons of the dorsal motor nucleus of the vagus nerve. Heart Rhythm. 2015;12:2285–2293. doi: 10.1016/j.hrthm.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti J, Brevi S, de Curtis M. A blocker-resistant, fast-decaying, intermediate-threshold calcium current in palaeocortical pyramidal neurons. Eur J Neurosci. 2000;12:2376–2386. doi: 10.1046/j.1460-9568.2000.00125.x. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Davies SS, Matafonova E, Potet F, Amarnath V, Tallman KA, Serwa RA, Porter NA, Balser JR, Kupershmidt S, Roberts LJ., 3rd Selective γ-ketoaldehyde scavengers protect NaV1.5 from oxidant-induced inactivation. J Mol Cell Cardiol. 2010;48:352–359. doi: 10.1016/j.yjmcc.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oaks AW, Marsh-Armstrong N, Jones JM, Credle JJ, Sidhu A. Synucleins antagonize endoplasmic reticulum function to modulate dopamine transporter trafficking. PLoS One. 2013;8:e70872. doi: 10.1371/journal.pone.0070872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladini CA, Roeper J. Generating bursts (and pauses) in the dopamine midbrain neurons. Neuroscience. 2014;282:109–121. doi: 10.1016/j.neuroscience.2014.07.032. [DOI] [PubMed] [Google Scholar]
- Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci. 2008;65:1272–1284. doi: 10.1007/s00018-008-7589-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. 2nd ed. San Diego: Elsevier Academic; 2004. [Google Scholar]
- Pissadaki EK, Bolam JP. The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson's disease. Front Comput Neurosci. 2013;7:13. doi: 10.3389/fncom.2013.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan LJ, Sah DW, Bean BP. Ca2+ channels in rat central and peripheral neurons: high-threshold current resistant to dihydropyridine blockers and omega-conotoxin. Neuron. 1991;6:269–280. doi: 10.1016/0896-6273(91)90362-4. [DOI] [PubMed] [Google Scholar]
- Sah P, McLachlan EM. Ca(2+)-activated K+ currents underlying the afterhyperpolarization in guinea pig vagal neurons: a role for Ca(2+)-activated Ca2+ release. Neuron. 1991;7:257–264. doi: 10.1016/0896-6273(91)90264-Z. [DOI] [PubMed] [Google Scholar]
- Sah P, McLachlan EM. Potassium currents contributing to action potential repolarization and the afterhyperpolarization in rat vagal motoneurons. J Neurophysiol. 1992;68:1834–1841. doi: 10.1152/jn.1992.68.5.1834. [DOI] [PubMed] [Google Scholar]
- Sanchez-Padilla J, Guzman JN, Ilijic E, Kondapalli J, Galtieri DJ, Yang B, Schieber S, Oertel W, Wokosin D, Schumacker PT, Surmeier DJ. Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat Neurosci. 2014;17:832–840. doi: 10.1038/nn.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel K, Schöls L, Nuber S, Petrasch-Parwez E, Gierga K, Wszolek Z, Dickson D, Gai WP, Bornemann A, Riess O, Rami A, Den Dunnen WF, Deller T, Rüb U, Krüger R. First appraisal of brain pathology owing to A30P mutant alpha-synuclein. Ann Neurol. 2010;67:684–689. doi: 10.1002/ana.21966. [DOI] [PubMed] [Google Scholar]
- Seidel K, Mahlke J, Siswanto S, Krüger R, Heinsen H, Auburger G, Bouzrou M, Grinberg LT, Wicht H, Korf HW, den Dunnen W, Rüb U. The brainstem pathologies of Parkinson's disease and dementia with Lewy bodies. Brain Pathol. 2015;25:121–135. doi: 10.1111/bpa.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Greenamyre JT. Pathogenesis of Parkinson's disease. Curr Opin Investig Drugs. 2001;2:657–662. [PubMed] [Google Scholar]
- Sidhu A, Wersinger C, Vernier P. alpha-Synuclein regulation of the dopaminergic transporter: a possible role in the pathogenesis of Parkinson's disease. FEBS Lett. 2004;565:1–5. doi: 10.1016/j.febslet.2004.03.063. [DOI] [PubMed] [Google Scholar]
- Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Krüger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soreq H. Checks and balances on cholinergic signaling in brain and body function. Trends Neurosci. 2015;38:448–458. doi: 10.1016/j.tins.2015.05.007. [DOI] [PubMed] [Google Scholar]
- Strichartz G, Cohen I. Letter to the editor: Vmax as a measure of GNA in nerve and cardiac membranes. Biophys J. 1978;23:153–156. doi: 10.1016/S0006-3495(78)85440-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam M, Althof D, Gispert S, Schwenk J, Auburger G, Kulik A, Fakler B, Roeper J. Mutant α-synuclein enhances firing frequencies in dopamine substantia nigra neurons by oxidative impairment of A-type potassium channels. J Neurosci. 2014;34:13586–13599. doi: 10.1523/JNEUROSCI.5069-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D. Clues to how alpha-synuclein damages neurons in Parkinson's disease. Mov Disord 25 Suppl. 2010;1:S27–S31. doi: 10.1002/mds.22639. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Guzman JN, Sanchez-Padilla J, Goldberg JA. The origins of oxidant stress in Parkinson's disease and therapeutic strategies. Antioxid Redox Signal. 2011;14:1289–1301. doi: 10.1089/ars.2010.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Orth M, Wilkinson JM, Taanman JW, Warner TT, Cooper JM, Schapira AH. Expression of mutant α-synuclein causes increased susceptibility to dopamine toxicity. Hum Mol Genet. 2000;9:2683–2689. doi: 10.1093/hmg/9.18.2683. [DOI] [PubMed] [Google Scholar]
- Tavassoly O, Nokhrin S, Dmitriev OY, Lee JS. Cu(II) and dopamine bind to α-synuclein and cause large conformational changes. FEBS J. 2014;281:2738–2753. doi: 10.1111/febs.12817. [DOI] [PubMed] [Google Scholar]
- Tottene A, Moretti A, Pietrobon D. Functional diversity of P-type and R-type calcium channels in rat cerebellar neurons. J Neurosci. 1996;16:6353–6363. doi: 10.1523/JNEUROSCI.16-20-06353.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci. 2011;34:89–103. doi: 10.1146/annurev-neuro-060909-153238. [DOI] [PubMed] [Google Scholar]
- Wersinger C, Sidhu A. Attenuation of dopamine transporter activity by alpha-synuclein. Neurosci Lett. 2003;340:189–192. doi: 10.1016/S0304-3940(03)00097-1. [DOI] [PubMed] [Google Scholar]
- Xu S, Chan P. Interaction between neuromelanin and alpha-synuclein in Parkinson's disease. Biomolecules. 2015;5:1122–1142. doi: 10.3390/biom5021122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarom Y, Sugimori M, Llinás R. Ionic currents and firing patterns of mammalian vagal motoneurons in vitro. Neuroscience. 1985;16:719–737. doi: 10.1016/0306-4522(85)90090-9. [DOI] [PubMed] [Google Scholar]