

Abstract
The present study focuses on the function of cpg15, a neurotrophic factor, in ischemic neuronal recovery using transient global cerebral ischemic (TGI) mouse model and oxygen-glucose deprivation (OGD)-treated primary cultured cells. The results showed that expression of cpg15 proteins in astrocytes, predominantly the soluble form, was significantly increased in mouse hippocampus after TGI and in the cultured astrocytes after OGD. Addition of the medium from the cpg15-overexpressed astrocytic culture into the OGD-treated hippocampal neuronal cultures reduces the neuronal injury, whereas the recovery of neurite outgrowths of OGD-injured neurons was prevented when cpg15 in the OGD-treated astrocytes was knocked down, or the OGD-treated-astrocytic medium was immunoadsorbed by cpg15 antibody. Furthermore, lentivirus-delivered knockdown of cpg15 expression in mouse hippocampal astrocytes diminishes the dendritic branches and exacerbates injury of neurons in CA1 region after TGI. In addition, treatment with inhibitors of MEK1/2, PI3K, and TrkA decreases, whereas overexpression of p-CREB, but not dp-CREB, increases the expression of cpg15 in U118 or primary cultured astrocytes. Also, it is observed that the Flag-tagged soluble cpg15 from the astrocytes transfected with Flag-tagged cpg15-expressing plasmids adheres to the surface of neuronal bodies and the neurites. In conclusion, our results suggest that the soluble cpg15 from astrocytes induced by ischemia could ameliorate the recovery of the ischemic-injured hippocampal neurons via adhering to the surface of neurons. The upregulated expression of cpg15 in astrocytes may be activated via MAPK and PI3K signal pathways, and regulation of CREB phosphorylation.
SIGNIFICANCE STATEMENT Neuronal plasticity plays a crucial role in the amelioration of neurological recovery of ischemic injured brain, which remains a challenge for clinic treatment of cerebral ischemia. cpg15 as a synaptic plasticity-related factor may participate in promoting the recovery process; however, the underlying mechanisms are still largely unknown. The objective of this study is to reveal the function and mechanism of neuronal-specific cpg15 expressed in astrocytes after ischemia induction, in promoting the recovery of injured neurons. Our findings provided new mechanistic insight into the neurological recovery, which might help develop novel therapeutic options for cerebral ischemia via astrocytic-targeting interference of gene expression.
Keywords: astrocytes, cpg15, mouse cerebral ischemia, neurite outgrowth recovery, soluble
Introduction
Candidate plasticity-related gene 15 (cpg15, also termed neuritin) is a neurotropic factor gene that was first identified in the hippocampus as a gene induced by neural activity (Nedivi et al., 1993, 1996). The coding sequence of cpg15 gene contains a signal peptide at N terminus (amino acid residues 1–27) and a glycosylphosphoinositide (GPI) anchor sequence (amino acid residues 116–142) (Nedivi et al., 1996), which encodes a small, highly conserved protein. cpg15 protein is expressed as two forms: a soluble, secreted form and an extracellular membrane protein anchored to the cell surface via the (GPI) link in mammals (Nedivi et al., 1996; Naeve et al., 1997; Putz et al., 2005).
cpg15 protein plays a variety of important roles in the development of nervous system. During neural development, the expression of cpg15 is spatially and temporally correlated with synapse formation and activity-dependent plasticity (Nedivi et al., 1996; Corriveau et al., 1999; Lee and Nedivi, 2002), and neuronal migration (Antypa et al., 2011; Zito et al., 2014). Overexpression of cpg15 in neurons induces elaboration of dendritic and axonal arbors, as well as synaptic formation and maturation (Nedivi et al., 1998; Cantallops et al., 2000). The soluble form of cpg15 was found to be essential for the survival of undifferentiated cortical progenitors by preventing apoptosis during early development of nervous system (Putz et al., 2005).
The above functions of cpg15 in the neural plasticity during development of nervous system imply that cpg15 may also play roles in the neuronal injury rescue and recovery of neurological diseases, such as cerebral ischemia, evoked by stroke and cardiac arrest. After cerebral ischemia, neural plasticity plays a major role in the spontaneous recovery of injured neuronal network, including the dramatic reorganization and rewiring of surviving circuits, as well as local and long distance changes in axonal sprouting and dendritic arborization (Benowitz and Carmichael, 2010; Andres et al., 2011; George and Steinberg, 2015). Our previous study also showed that cpg15 was upregulated in ischemic hippocampus and might have promotion function in the reestablishment of neuronal network (Han et al., 2007). However, the exact function and mechanism of cpg15 in ischemic neuronal recovery remain unclear.
Astrocytes have been recognized to play multiple important roles in the recovery of ischemic neuronal network and neurogenesis via secreting neurotrophic factors after cerebral ischemia (Ransom et al., 2003; Swanson et al., 2004; Nedergaard and Dirnagl, 2005; Ouyang et al., 2007; Takano et al., 2009). As a neuronal-specific expressed neurotrophic factor, cpg15 hardly expresses in astrocytes at physiological status. However, here we noticed that the expression of cpg15 was upregulated dramatically in hippocampal astrocytes after mouse transient global cerebral ischemia. Our further investigation indicated that the ischemia-induced astrocytic cpg15, predominant in the soluble cpg15, could ameliorate the recovery of the ischemic-injured hippocampal neurons and may be activated via MAPK and PI3K signal pathways, and phosphorylation of CREB.
Materials and Methods
Experimental animals and treatments.
Adult male C57BL/6J mice 8–10 weeks of age were obtained from Shanghai Experimental Animal Center, Chinese Academy of Sciences, housed under conditions of constant temperature (25 ± 1°C) and humidity (55 ± 5%), a 12 h light-dark cycle, and were used to induce ischemia model. All animal experiments and surgical procedures were approved by the Institutional Animal Care and Fudan University Shanghai Medical College committee (IACUC Animal Project 20130227-026), used in strict accordance with the recommendations in the Guide for the care and use of laboratory animals of the National Institutes of Health. All surgery was performed under chloral hydrate anesthesia, and all efforts were made to minimize suffering.
Transient global cerebral ischemia.
Mice were subjected to transient global ischemia (TGI) by bilateral common carotid artery occlusion (BCCAO) for 30 min, followed by reperfusion. For sham surgery, mice were subjected to the same anesthesia and surgical procedures, except that the carotid arteries were not occluded. Body temperature was monitored and maintained at 37.5 ± 0.5°C with a rectal thermistor and heat lamp until the animal had fully recovered from anesthesia. Animals were killed by decapitation at different days after reperfusion.
For evaluation of histological damage, brain slices were stained with 0.1% cresyl violet according to the Nissl method. Viable neurons were defined as neurons in which a clear nucleus could be seen. With Nissl staining, ischemic damaged neurons exhibit features, including pyknosis and shrunken cell bodies. Hippocampal neuronal damage was evaluated qualitatively according to the method of Kawase et al. (1999): Grade 0, no damage to any hippocampal subregion; Grade 1, scattered ischemic neurons in CA1 subregion; Grade 2, moderate ischemic damage in CA1 subregion (less than half of pyramidal cells affected); Grade 3, severe damage to pyramidal cells in CA1 subregion (more than half of pyramidal cells affected); and Grade 4, extensive cell damage in all hippocampal subregions. Neuronal damage was evaluated by a researcher blinded to the studies.
Cell culture.
Mouse primary hippocampal neurons and astrocytes were isolated from postnatal 24 h and embryonic day17 (E17) mice embryos. The mixture of mouse primary hippocampal neurons and astrocytes was plated onto poly-L-lysine (Sigma)-coated dishes in Neurobasal with B27 (2%) and Gln (0.2%) at 37°C with 5% CO2. The mouse primary neurons were grown as the same protocol with additional adding of Ara-C (4 μm). The mouse primary astrocytes were grown in DMEM supplemented with 10% FCS (Invitrogen) on the poly-L-lysine-coated dishes at 37°C with 5% CO2. Mouse neuroblastoma N2a cells and human brain glioma cells, and U118 MG cells were grown in DMEM (Invitrogen), high glucose supplemented with 10% FCS (Invitrogen) at 37°C with 5% CO2.
Construct of lentivirus delivering knockdown of cpg15 expression.
Lentivirus vector containing the shRNA targeting cpg15 downstream of the CMV promoter was constructed by GeneChem using miR30-based shRNA system. In brief, the most effective (Putz et al., 2005) cpg15 shRNA sequence (5′-GGGCTTTTCAGACTGTTTG-3′) of three cDNA sequences spanning the cpg15 core domain and the scrambled shRNA construct (5′-TTCTCCGAACGTGTCACGT-3′) were synthesized, fused to a loop region, and then annealed to their antisense sequences and cloned separately into the pGV123-CMV-RNAi lentiviral vector, downstream of the CMV promoter. The vectors were then packed into lentivirus by GeneChem as described previously (Lois et al., 2002), and named LV-CMV-cpg15 shRNA and LV-CMV-Control shRNA; the former lentivirus delivers knockdown of cpg15 expression, and the latter acts as its control.
Construct of lentivirus delivering astrocyte-specific knockdown of cpg15 expression.
Astrocyte-specific cpg15-shRNA-delivering lentivirus was also constructed (GeneChem) for in vivo analysis of the function of astrocyte-expressed cpg15 in the recovery of ischemic-injured neurons. In brief, the CMV promoter of the above-mentioned cpg15 shRNA (or its control-shRNA)-containing lentiviral vectors was replaced with mouse GFAP promoter. The obtained lentiviruses were named LV-GFAP-cpg15 shRNA and LV-GFAP-Control shRNA, respectively; the former lentivirus delivers astrocyte-specific knockdown of cpg15 expression, and the latter acts as the control.
Lentiviral stereotaxic injection and cell infection.
In vivo injections of lentivirus were performed on C57 adult male mice 4 d before the BCCAO 30 min and reperfusion. In detail, 3 × 108 TU/ml, 0.5 μl of cpg15 shRNA lentivirus was stereotactically delivered into the hippocampus (coordinates from bregma: anteroposterior axis, −2 mm; lateromedial axis, ±1.8 mm; dorsoventral axis, −1.5 mm from the pial surface). An equal amount of scrambled sequence-containing lentivirus was also stereotactically delivered as the control group. After recovery from anesthesia, mice were returned to their cages and given ad libitum access to food and water until next operation. At 7, 14, and 21 d after BCCAO 30 min and reperfusion, mice were killed for further analysis.
For viral infection of primary cultured astrocytic and N2a cell cultures, primary cultured astrocytes isolated from postnatal 24 h C57 mice and N2a cells (from ATCC) were plated (at 80% confluence) in 24 well plates, and cpg15 shRNA lentivirus or scrambled control shRNA lentivirus (1 × 108 TU/ml, 5 μl) in the medium containing polybrene (5 μg/ml) was added per well on the second day. At 6–8 h later, the medium containing lentivirus was replaced by fresh culture medium. At 3–5 d after lentivirus infection, N2a cells were harvested for further analysis, astrocytes were treated by 4 h oxygen-glucose deprivation (OGD), and the culture medium was collected at reoxygenation 12 h and added into OGD-injured neurons. Experiments were performed for at least three times from independent cultures.
OGD.
The OGD treatment was applied on the primary cultured neurons, astrocytes, and N2a cells (in vitro) to mimic ischemia and reperfusion condition in vivo. After three washes with HBSS and one wash with DMEM without glucose and serum, astrocytic cultures were incubated with glucose/l-glutamine-free DMEM without serum in a hypoxic incubator chamber (Billups-Rothenberg) equilibrated with 95% N2/5% CO2 at 37°C for 4 h (for the mixed culture of mouse hippocampal neurons and astrocytes, or for independently cultured astrocytes), or for 8 h (for cultured N2a cells, or for independently cultured astrocytes). After OGD exposure, cells were maintained in glucose-containing DMEM under normoxic conditions for reoxygenation. Further analysis was performed at reoxygenation 0, 12, 24, 48, and 72 h after OGD.
Plasmids and DNA transfection.
For overexpression of cpg15 protein in cells, cDNA fragments encoding the full-length mouse cpg15 protein (the coding sequence that contains signal peptide, core domain, and GPI-anchored fraction) (NM_153529.2, Mus musculus neuritin 1-Nrn1, 188-616nt) were inserted into pAAV-internal ribosome entry site (IRES)-GFP vectors. For construction of pAAV vectors expressing secretory FLAG-tagged cpg15 (pAAV-Flag-cpg15-IRES-GFP), sequence encoding Flag peptide was inserted in frame between the upstream secretion signal sequence and the core domain sequence of cpg15 protein using a mutagenesis kit (Toyobo). For construction of vector expression of CREB protein in cells (pAAV-CREB-IRES-GFP), cDNA fragments encoding the full-length mouse CREB protein (NM_009952, 226-1251nt) were inserted into pAAV-IRES-GFP vectors. For construction of the CREB mutants in Ser133 phosphorylation site, coding sequence of serine133 in pAAV-CREB-IRES-GFP was replaced by that of glutamate to mimic the phosphorylated CREB, or replaced by that of alanine to mimic the dephosphorylated CREB using a mutagenesis kit (Toyobo). PCR products were completely sequenced, and all cDNA fragments and nucleotides were sequenced at the junction sites to confirm in-frame insertions.
Transfection of human brain glioma cells U118 MG and mouse primary cultured astrocytes was performed with Lipofectamine 2000 and LipofectamineLTX and Plus Reagents (Invitrogen) following the supplier's protocol. Briefly, cells were transfected with 0.5 μg of plasmid DNA per well of 24 well plate (60%–80% confluent cells), in the presence of 2 μl Lipofectamine 2000 reagent, or with 2.5 μg of plasmid DNA per well of 6 well plate, in the presence of 10 μl Lipofectamine 2000 reagent, or with 14 μg of plasmid DNA per well of 6 well plate, in the presence of 12 μl of LipofectamineLTX reagent. Cells were harvested 48 h after transfection for RT-PCR or Western blot analysis. Primary cultured astrocytes transfected with the null and CREB-overexpressing plasmid were separated from those untransfected by GFP FACS. The culture medium of primary astrocytes transfected with the null and Flag-cpg15-overexpressing plasmids was added into the primary neuronal culture to investigate whether the soluble cpg15 protein secreted from the astrocytes localizes on the surface of neurons.
Influence of the medium from cpg15-overexpressing astrocytes on OGD-induced neuronal injury.
Cultured U118 astrocytes were transfected with plasmids overexpressing cpg15 protein; 48 h after the transfection, the culture medium was collected and added into the cultured 4 h OGD-treated N2a cells at the time point of reoxygenation 0 h; N2a cells were then cultured under normoxic conditions for reoxygenation until the harvest time point. For the N2a cells that were harvested at reoxygenation 0 h, astrocytic medium was not added. Cell cytotoxicity and viability analysis were performed at reoxygenation 0, 12, 24, 48, and 72 h, respectively.
Influence of the medium from cpg15-silenced or cpg15 protein-immunoadsorbed astrocytic cultures on neurite outgrowth of OGD-injured neurons.
cpg15 small hairpin RNA (shRNA) was delivered by lentivirus for cpg15 silencing in hypoxia-induced primary astrocytes. Primary cultured astrocytes were infected with cpg15-silencing lentivirus (scramble shRNA letivirus as the control); 96 h later, astrocytes were treated with 4 h OGD; then at reoxygenation 12 h, the medium was harvested and added to OGD-injured primary hippocampal neurons at reoxygenation 12 h. Neurons were then cultured under normoxic conditions for reoxygenation. Further analysis of neurite outgrowth was performed at reoxygenation 24 and 48 h.
For cpg15 protein immunoadsorption, 1 μg of goat anti-cpg15 antibody (R&D Systems catalog #AF283, RRID: AB_2155109) preconjugated with 20 μl of rec-Protein G-Sepharose (Invitrogen), according to the supplier's instructions, was mixed with 500 μl of the medium from OGD-treated primary astrocytes at reoxygenation 12 h (equal amount of β-actin antibody as the control), after incubation for 4 h at 4°C, the rec-Protein G-Sepharose, along with the conjugated cpg15 antibodies and the immunoadsorbed cpg15 proteins, was removed from the medium by centrifugation (1000 × g, 5 min, 4°C), the medium was then added into OGD-injured primary neuronal culture at neuronal reoxygenation 12 h. At reoxygenation 24 and 48 h, the neurite outgrowth of neurons was analyzed by immunofluorescence triple staining followed by Image ProPlus software analysis. Experiments were performed at least three times from independent cultures.
Cell cytotoxicity and viability analysis.
It has been previously established that the release of LDH into the culture medium correlates linearly with the number of damaged cells after OGD. LDH release was assessed as an index of cell damaged and injury at different times after OGD using an LDH assay kit (Roche), according to the manufacturer's instructions. Cell viability analysis was performed by examining the number of living cells via Cell Counting Kit-8 (Dojindo) following the supplier's protocol.
Immunohistochemical staining and fluorescence immunolabeling of brain sections and cell.
Mice with deep anesthesia were intracardial perfused with saline followed by 4% PFA. Brains were removed, and coronal sections were cut with a freezing microtome (Leica) at a thickness of 20 μm. Coronal fresh frozen sections were postfixed with 4% PFA for 10 min at room temperature; after wash in PBS for three times, cryo-sections were treated with 0.3% H2O2 for 30 min, after three washes in PBS followed by incubation in blocking buffer containing 10% horse serum and 0.3% Triton X-100 in PBS for 30 min at 37°C, and then incubated for 2 h at 37°C and overnight at 4°C with goat anti-cpg15 (1:500, R&D Systems catalog #AF283, RRID: AB_2155109). After three washes in PBS, the sections were incubated with biotinylated secondary antibodies (1:200, Vector Laboratories) for 30 min at 37°C, followed by avidin-biotin-peroxidase (1:200, Vectastain Elite ABC kit, Vector Laboratories) for 45 min at 37°C. Immunoreactivity was visualized with 0.05% DAB (Sigma). The sections were then dehydrated, transparent and mounted on glass slides, and coverslipped with neutrophilia gum. The negative controls received the same treatments, except that primary antibodies were omitted and no unspecific staining was observed.
For brain fluorescent immunolabeling, sections were incubated in blocking buffer containing 10% horse serum and 0.3% Triton X-100 in PBS for 30 min at 37°C, then incubated for 2 h at 37°C and overnight at 4°C with goat anti-cpg15 (1:100, R&D Systems), mouse anti-NeuN (1:1000, Millipore catalog #MAB377 RRID:AB_2298772), and chicken anti-MAP2 (1:500, Millipore catalog #AB5543, RRID:AB_571049), or incubated 4 h at 4°C with rabbit anti-GFAP (1:500, Dako catalog #Z0334, RRID:AB_10013382). After three washes in PBS, the sections were incubated with corresponding fluorescence-conjugated second antibodies, such as AlexaFluor-488-conjugated donkey anti-goat IgG (1:500,Invitrogen), AlexaFluor-594-conjugated donkey anti-goat IgG (1:500, Invitrogen), AlexaFluor-647-conjugated donkey anti-mouse IgG (1:500, Invitrogen), AlexaFluor-546-conjugated goat anti-chicken IgG (1:500, Invitrogen), AlexaFluor-594-conjugated donkey anti-mouse IgG (1:500, Invitrogen), or AlexaFluor-647-conjugated donkey anti-rabbit IgG (1:500, Invitrogen) at 37°C for 1 h to reveal the positive signals. After washing, the sections were mounted on glass slides and coverslipped using Fluoromount medium (Sigma).
For cell immunofluorescence, cells grown on coverslips coated with poly-L-lysine were washed with PBS and fixed with 4% PFA for 30 min, followed by incubation for 5 min in 0.3% Triton X-100 (in PBS) to permeabilize the cells. The fixed cells were incubated in blocking buffer (10% horse serum) for 30 min at room temperature, followed by incubation with two or three kinds of the following antibodies: goat anti-cpg15 (1:100, R&D Systems), mouse anti-NeuN (1:1000, Millipore), and rabbit anti-GFAP (1:500, Dako), or rabbit anti-MAP2 (1:500, Millipore), rabbit anti-FLAG (1:200, Sigma-Aldrich catalog #F7425, RRID:AB_439687), and chicken anti-MAP2 (1:500, Millipore). After three washes with PBS, the sections were incubated for 1 h at room temperature with the corresponding fluorescence-conjugated secondary antibodies, such as AlexaFluor-488-conjugated donkey anti-goat IgG (1:500, Invitrogen), AlexaFluor-594-conjugated donkey anti-rabbit IgG (1:500, Invitrogen), AlexaFluor-647-conjugated donkey anti-mouse IgG (1:500, Invitrogen), AlexaFluor-488-conjugated donkey anti-rabbit IgG (1:500, Invitrogen), AlexaFluor-594-conjugated donkey anti-mouse IgG (1:500, Invitrogen), AlexaFluor-594-conjugated donkey anti-goat IgG (1:500, Invitrogen), or AlexaFluor-546-conjugated goat anti-chicken IgG (1:500, Invitrogen). Sections were then mounted onto glass slides and coverslipped using Fluoromount medium (Sigma). Fluorescence was detected by confocal laser-scanning microscopy (TCS SP8, Leica).
Western blot analysis and ELISA.
For protein semiquantitative analysis by Western blotting, brain tissues extracted from mouse hippocampus or cells were homogenized in ice-cold RIPA lysis buffer containing freshly added protease inhibitor mixture tablets (Roche). Samples (30–40 μg protein/lane) were separated by SDS-PAGE using 16.5% acrylamide gels and transferred to PVDF membranes (0.22 μm pore, Millipore) by wet electrophoresis for 2 h at 70 V. After washing in Tris-buffered saline for 5 min, the membranes were fixed by 0.25% glutaral in TBS buffer for 45 min and then washed extensively with TBS containing 0.1% Tween-20 (TBST) buffer. After being blocked by incubation with TBST containing 5% nonfat milk, the membranes were incubated for 2 h at room temperature followed by overnight at 4°C with the antibodies as follows: goat anti-cpg15 (1:500, R&D Systems), mouse monoclonal anti-β-actin (1:5000, Sigma-Aldrich catalog #A5441, RRID:AB_476744), rabbit anti-Akt (1:500, Cell Signaling Technology catalog #4691, RRID:AB_915783), rabbit anti-phospho-Akt (Ser473) (1:500, Cell Signaling Technology catalog #4060, RRID:AB_2315049), rabbit anti-p44/42 MAPK (Erk1/2) (137F5) (1:500, Cell Signaling Technology catalog #4695, RRID:AB_390779), rabbit anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (197G2) (1:500, Cell Signaling Technology catalog #4377, RRID:AB_331775), rabbit anti-CREB (1:500, Cell Signaling Technology catalog #9197, RRID:AB_331277), or rabbit anti-pCREB (1:500, Cell Signaling Technology catalog #9196, RRID:AB_331275). HRP-conjugated donkey anti-mouse IgG antibodies (1:2000, Kang Cheng), alkaline phosphatase-conjugated donkey anti-goat IgG antibody (1:100, Kang Cheng), or IRDye 800/700-conjugated antibody (1:8000, Odyssey) was used as a secondary antibody. Signals were developed by enhanced chemiluminescence substrate (Pierce) onto x-ray films (Kodak) for HRP and by BCIP/NBT substrates for alkaline phosphatase. The immunodensity values of each band were analyzed by Quantity One 1-D Analysis software (Bio-Rad).
ELISA kit (mouse Neuritin ELISA kit, Biovalue) was used to measure the amount of soluble cpg15 in the medium of OGD-treated primary astrocytic culture with or without lentivirus-delivered cpg15-silencing, at reoxygenation 0, 12, 24, 48, and 72 h, according to the manufacturer's instructions; the lentivirus-delivered scrambled shRNA was used as the nonsilencing control. The medium of nontreated astrocytes was used as another control for that of OGD-treated astrocytes. Experiments were performed in triplicates, and OD values were read at wavelength of 450 nm using a spectrophotometer (Bio-Rad). Experiments were performed for at least three times from independent cultures.
Immunoprecipitation.
For immunoprecipition, brain tissues extracted from mouse hippocampus were lysed in lysis buffer (RNase free, 50 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EDTA,10 mm NaF, 1% Triton X-100, 1% NP-40, 0.1% SDS, 1 U/μl RNasin, 1 mm PMSF, 0.01% DTT, protease inhibitor mixture), and then were incubated with goat anti-cpg15 antibody (1:100, R&D Systems) or goat IgG as negative control and rocking overnight at 4°C. The tissue lysate (10% lysate) was then incubated with Protein G-Sepharose beads (20 μl per IP reaction, Sigma), which were prewashed in washing buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EDTA, 10 mm NaF, 1 mm Na3VO4, 1 mm DTT, 1 U/ml ribonuclease inhibitor and protease inhibitors). After rocking for 1 h at 4°C, beads were centrifuged at 500 × g for 5 min at 4°C; then the pellets (beads) were washed five times in washing buffer, with a 5 min rocking interval between each washing. After washing, the IP beads were then analyzed by SDS-PAGE followed by silver staining or immunoblotting. Peptide sequencing and protein identification were performed by the Mass Spectrometry Core Facility of Institutes of Biomedical Sciences, Fudan University. Experiments were performed at least three times from independent cultures.
Reverse transcription (RT) and real-time PCR.
Total RNA was extracted from the cultured primary astrocytes or RIP samples using TRIzol reagent (Invitrogen) following the supplier's instructions. Reverse transcription was performed using M-MLV Reverse Transcriptase (Promega) with oligo dT primers. Real-time PCR was performed using the SYBR Green Real-time PCR system (Toyobo) on a MasterCycler Thermal Cyclers PCR (Eppendorf). The primers were as follows: GAPDH forward, 5′-GAA CCA CGA GAA ATA TGA CAA C-3′, reverse, 5′-ATG GCA TGG ACT GTG GTC A-3′; cpg15 forward, 5′-ATG GGA CTT AAG TTG AAC GG-3′, reverse, 5′-GTC TTG ATG TTC GTC TTG TCG T-3′; GAP20 forward, 5′-TGTGGAGGAAGTGGGAGAAG-3′, reverse, 5′-TACGGTGCCGCTTCTTTTCCA-3′; GAP22 forward, 5′-TGGTGGAGCAGTG TGTGGAC-3′, reverse, 5′-TGAGCAGGTTGTAGTTGGCC-3′; GAP29 forward, 5′-TTCTGCTGCTATTACAACTTC-3′, reverse, 5′-TTCACAGCTTTCACTTTT GC-3′; EP400 forward, 5′-AATGCCAACAGTGAACAACGA-3′, reverse, 5′-CACCACGAATTCCTCAGCTTT-3′. Each sample was analyzed in triplicate.
Statistical analysis.
For stereotaxic measurements of the densities of astrocytes expressing cpg15 proteins (both GFAP and cpg15-immunoreactive) in hippocampus, immunofluorescent labeled sections were observed by a fluorescence microscope and immunostained cells were counted in each section using Image Pro Plus 6.0 software. Every sixth (in total, 6 or 7 sections were analyzed) of the hippocampus-containing sections were selected, and the number of both GFAP and cpg15-immunoreactive cells in hippocampus for one mouse was represented with the average number for each section. Sections were numbered and analyzed blindly.
For neurite outgrowth analysis of primary neurons, images of 18 neurons were acquired from randomly selected fields of view. The length of the longest neurite for each cell was calculated using Image Pro Plus 6.0 software, as was a measure of total neurite outgrowth. A series of concentric circles was overlaid onto an image of each neuron, and the number of times neurites crossed each circle was calculated. This gave us a measure of both total neurite outgrowth (total number of crosspoints) and neurite branching structure (number of crosspoints related to distance from the cell body). All of these measurements were repeated in at least three independent cultures.
All results are reported as mean ± SEM, and analyses were performed using Origin software. Three experimental groups were compared by one-way ANOVA with Scheffe's post hoc pairwise analyses, and two experimental groups were compared with Student's unpaired, two-tailed t test.
Results
Global cerebral ischemic injury significantly increases cpg15 expression in astrocytes
It has been shown that cpg15 plays multiple roles during nervous system development, including promoting neurite outgrowth and synaptic maturation as well as regulating survival of cortical progenitors. Our previous study also showed that cpg15 expression is upregulated in ischemic hippocampus and cpg15 immunoabsorption from the culture medium increases the glutamate-induced injury of neurons (Han et al., 2007). Here, we used the mouse transient global cerebral ischemia (TGI) model to examine whether cpg15 is involved in the neuronal repair or recovery after brain ischemia. As shown in Fig. 1, Nissl staining showed the diffusely shrunken injured pyramidal neurons in hippocampal CA1 subregion (Fig. 1A, right) of mice after TGI, compared with the intact pyramidal neurons in the sham group (Fig. 1A, left). The severity of neuronal damage in hippocampal CA1 subregion aggravated from reperfusion 1 to 7 d, and then began to recover from reperfusion 7 d to 14 and 21 d (Fig. 1B). The expression of cpg15 protein in hippocampal CA1 subregion, especially the soluble form, was increased significantly at reperfusion 7 and 14 d after TGI, compared with the sham (Fig. 1C,D). Immunohistochemical analysis showed that cpg15 protein existed not only inside, but also outside, the pyramidal neurons in the injured hippocampal CA1 and DG subfields at reperfusion 7 and 14 d (Fig. 1E). Immunofluorescent double labeling showed that cpg15 protein was significantly increased in astrocytes, but not in neurons, during reperfusion 7 and 14 d after TGI (Fig. 1F,G). It indicated that TGI not only elicited the increase in expression level of cpg15 in whole hippocampal tissue, but also altered its cell-specific distribution pattern. Compared with the increased cpg15 level in neurons induced by slighter ischemic stimulation (BCCAO for 15 min) in our previous study (Han et al., 2007), the heavier ischemic injury (BCCAO for 30 min) in the present study induced high expression of cpg15 protein in astrocytes, which probably contributes to the reduction of neural injury or promotion of neural recovery from injury after ischemic damage.
Figure 1.
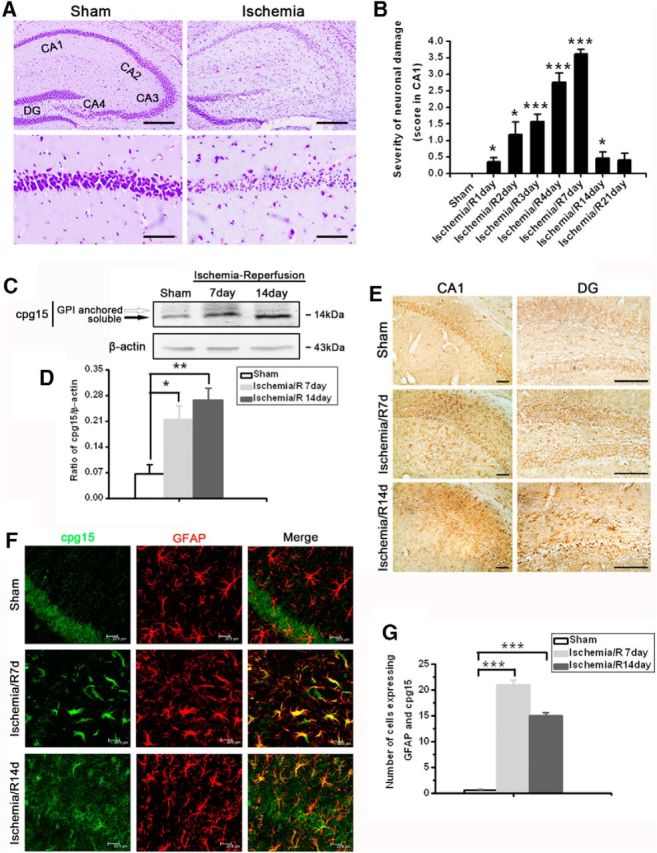
Global cerebral ischemic injury significantly increases cpg15 expression in astrocytes. A, Representative CV staining analysis of neuronal injury in mouse hippocampus after TGI. Bottom, Magnified from the Top images. Scale bars: Top, 50 μm; Bottom, 6 μm. B, Severity of hippocampal damage at reperfusion days 1–21 after TGI (Ischemia/R1–R21 d), assessed as described in Materials and Methods. Sham, n = 5; all other groups, n = 8. *p < 0.05, compared with the sham group. ***p < 0.001, compared with the sham group. DG, Dentate gyrus; CA1–4, cornus ammonis sectors 1–4. C, Representative Western blot images showing the expression of cpg15 protein in mouse hippocampus at reperfusion 7 and 14 d after TGI (two bands were detected, GPI-anchored and soluble cpg15). D, Relative amount of cpg15 protein in mouse hippocampus at reperfusion 7 and 14 d after TGI (statistical data from the Western blot images). β-Actin is used as the loading control. Sham, n = 3; all other groups, n = 6. *p < 0.05, compared with the sham. **p < 0.01, compared with the sham. E, Immunohistochemical analysis of cpg15 expression in mouse hippocampal CA1 and DG regions at reperfusion 7 and 14 d after TGI. Scale bars, 100 μm. F, Immunofluorescent double staining analysis of cpg15 (green) and astrocytic marker GFAP (red) showing that cpg15 was upregulated obviously in astrocytes in mouse hippocampal CA1 region at reperfusion 7 and 14 d after TGI. Scale bars, 20 μm. G, Statistical data from F showing the cell number expressing both GFAP and cpg15 proteins in mouse hippocampal CA1 region at reperfusion 7 and 14 d after TGI. Sham, n = 3; all other groups, n = 8. ***p < 0.001, compared with the sham. The hippocampus of sham-operated mice was used as the control.
The OGD elicits upregulation in the expression and secretion of cpg15 proteins in the cultured mouse primary hippocampal astrocytes
We further investigated the role of cpg15 protein expressed in astrocytes in the neuronal injury and recovery after ischemia using OGD-treated primary culture of hippocampal neurons and astrocytes. The results showed that, in the mixed culture of hippocampal neurons and astrocytes, cpg15 protein specifically expressed in neurons, but not in astrocytes in normal condition (Fig. 2A, Normal), whereas the cpg15 expression in astrocytes was significantly increased at reoxygenation 12 h after 4 h of OGD stimulation (Fig. 2A, OGD). In the separated cultured hippocampal astrocytes, the expression level of both cpg15 protein (Fig. 2B,C) and mRNA (Fig. 2D) in astrocytes increases significantly at reoxygenation 12, 24, 48, and 72 h after OGD treatment, and the OGD-induced significant increase of cpg15 protein in astrocytes was confirmed by immunofluorescence double labeling at reoxygenation 12, 24, and 48 h (Fig. 2E,F). These observations were consistent with those observed in an animal model (Fig. 1). There exist two protein forms of cpg15: soluble cpg15 and GPI-anchored cpg15. Both function in neuronal growth and survival (Nedivi et al., 1996, 1998; Putz et al., 2005). It is mainly the soluble cpg15 that increases significantly in mouse hippocampus after brain ischemia, as shown by Western blot analysis (Fig. 1C). To investigate whether soluble cpg15 also increases in OGD-stimulated astrocytes, ELISA analysis was performed. The results showed that the amount of soluble cpg15 in the culture medium of astrocytes increased significantly at reoxygenation 12, 24, 48, and 72 h after OGD treatment, with a peak level at 12, 24, and 48 h (Fig. 2G). The results indicate that soluble cpg15 can be expressed in and secreted outside the OGD-treated astrocytes and probably plays roles in reducing neuronal injury or promoting recovery of injured neurons.
Figure 2.
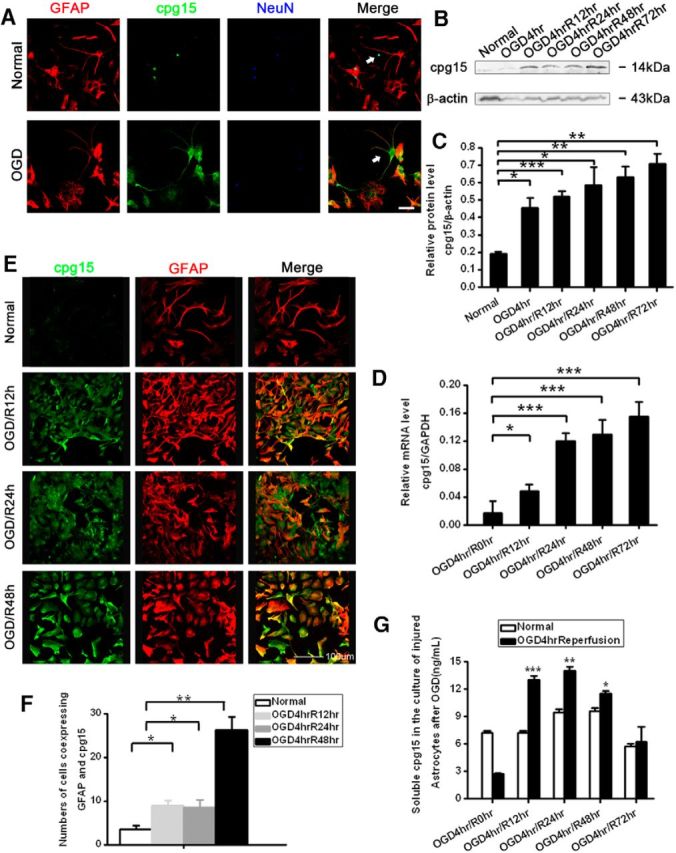
The OGD elicits upregulation in the expression and secretion of cpg15 proteins in the cultured mouse primary hippocampal astrocytes. A, Immunofluorescent triple staining of cpg15 (green), astrocytic marker GFAP (red), and neuronal marker NeuN (blue) in cocultured hippocampal neurons and astrocytes at 12 h after OGD. Scale bar, 50 μm. White arrows indicate the alteration of cpg15 distribution from neurons in normal status (top) to astrocytes after OGD (bottom). B, Representative Western blot analysis showing the expression of cpg15 protein in 4 h OGD-treated mouse hippocampal astrocytes at reoxygenation 0, 12, 24, 48, and 72 h. β-Actin as the loading control. C, Statistical data from B showing the ratio of cpg15 to β-actin. D, Expression levels of cpg15 mRNA measured by real-time PCR. GAPDH was used as the internal control. E, Immunofluorescent double staining of cpg15 (green) and astrocytic marker GFAP (red) in cultured hippocampal astrocytes at 12, 24, and 48 h after OGD. Scale bar, 100 μm. F, Statistical data from E showing the number of cells expressing both GFAP and cpg15. Cell numbers were counted by Image Pro-plus soft. G, Soluble cpg15 secreted into the culture medium, measured by ELISA. n = 6 for each group. *p < 0.05, compared with the normal group. **p < 0.01, compared with the normal group. ***p < 0.001, compared with the normal group.
Culture medium from astrocytes with overexpression of cpg15 reduced the OGD-induced neuronal injury
To determine whether soluble cpg15 secreted from OGD-stimulated astrocytes contributes to reducing neuronal OGD-induced injury, full-length cpg15 was overexpressed in U118 astrocytes by transfection of pAAV-cpg15-IRES-GFP plasmids. Then, the culture medium harvested from the U118 astrocytes was added into the OGD-injured N2a neurons. It was found that the medium from cpg15 overexpressed astrocytes has a significantly higher level of ability in decreasing the LDH release and increasing the cell viability of OGD-treated neurons at R12 h, R24 h, and R48 h, compared with that of the nonoverexpressed control (Fig. 3). The results indicate that cpg15 expressed in the astrocytes is likely secreted outside and reduces neuronal injury.
Figure 3.

Culture medium from astrocytes with overexpression of cpg15 reduces the OGD-induced neuronal injury. LDH release from (A) and cell viability of (B) 8 h-OGD-treated N2a cells cultured with the medium from U118 astrocytes with overexpression of cpg15 at reoxygenation 0, 12, 24, 48, and 72 h. No astrocytic medium was added into the R0hr harvested cells (OGD8hrR0hr). n = 12 for each group. *p < 0.05. **p < 0.01. ***p < 0.001. C, Western blot analysis confirming overexpression of cpg15 in U118 astrocytes transfected with pAAV-cpg15-IRES-GFP plasmid (pAAV-IRES-GFP as the control). β-Actin was used as the loading control.
Immunoadsorption of cpg15 from the culture medium of OGD-treated primary astrocytes suppresses dendritic outgrowth of OGD-injured primary hippocampal neurons and reestablishment of neural network
To investigate whether the OGD-induced expression of cpg15, especially the soluble cpg15 in the astrocytes, contributes to the recovery of OGD-injured neurons, the medium of OGD-treated astrocytes was immunoadsorbed with cpg15 antibody conjugated with Protein G-Sepharose for removal of the soluble cpg15, and then added into the medium of OGD-treated primary cultured hippocampal neurons (Fig. 4A). The immunoadsorption efficiency was confirmed by ELISA analysis. The results showed a significant decrease of soluble cpg15 concentration in the OGD-treated astrocytic medium (3.76 ± 0.88 ng/ml after cpg15 antibody immunoadsorption, compared with 13.30 ± 1.30 ng/ml after β-actin antibody absorption as the control, n = 3, p < 0.05). Immunofluorescent double labeling of the neural dentritic marker MAP-2 and neuronal nucleic marker NeuN showed that, after removal of the soluble cpg15 from the astrocytic medium, the dendritic outgrowth of the hippocampal neurons was suppressed significantly (compared with the control) at reoxygenation 48 h after OGD (Fig. 4B), as indicated by the statistical data of total neurite density, length of longest neurites, branch tips per neurite, and number of crosspoints at different distances from the cell bodies (Fig. 4C–F), whereas the survival of the neurons (total number of neurons) does not change significantly (Fig. 4G). The above results indicated that soluble cpg15 induced by OGD from astrocytes enhanced the neurite outgrowth of OGD-injured neurons and neuronal network reestablishment.
Figure 4.
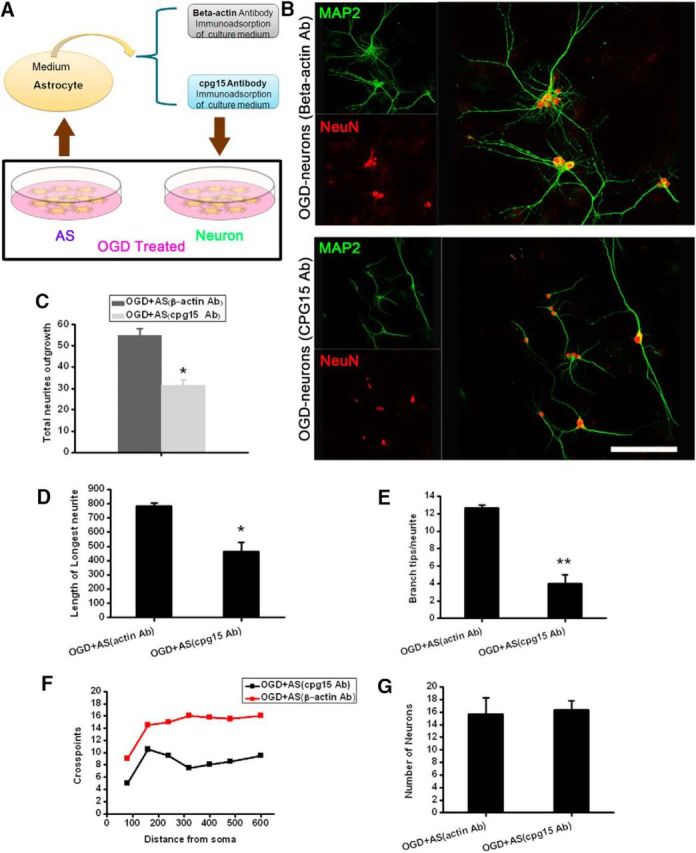
Immunoadsorption of cpg15 from the culture medium of OGD-treated hippocampal astrocytes suppresses dendritic outgrowth of OGD-injured primary hippocampal neurons and reestablishment of neural network. A, Schematic diagram showing the immunoadsorptional removal of soluble cpg15 from OGD-treated astrocyte culture medium. B, Representative immunofluorescence double staining of dendritic marker MAP2 (green) and neuronal marker NeuN (red) at reoxygenation 48 h in 4 h OGD-injured primary cultured neurons cultured with the cpg15-immunoadsorbed medium of OGD-treated astrocytes (cpg15 Ab). β-Actin immunoadsorbed medium as the control (β-actin Ab). C–G, Statistical data of total neurite density (C), length of longest neurites (D), branch tips per neurite (E), crosspoints (F), and number of total neurons (G) from B. Scale bars, 50 μm. n = 10 for each group. *p < 0.05. **p < 0.01.
Lentivirus-delivered knockdown of cpg15 expression in astrocytes suppressed the neurite outgrowth recovery of OGD-injured neurons and neuronal network reestablishment
To further investigate whether the OGD-induced expression of cpg15 in the astrocytes contributes to the recovery of OGD-injured neurons in the soluble form, we constructed the recombinant lentivirus encoding shRNA against cpg15 (LV-CMV-cpg15-shRNA) to knock down endogenous cpg15 expression in primary astrocytes. Lentivirus encoding scrambled shRNAs (LV-CMV-Control shRNA) was the negative control. As shown in Figure 5A, most of the primary cultured astrocytes were infected with the cpg15 shRNA-delivering lentivirus, with the GFP fluorescence as the reporter. The upregulation of the cpg15 expression was inhibited significantly in LV-CMV-cpg15 shRNA (abbreviated in Fig. 5A as “LV-cpg15 sh”)-infected astrocytes at reoxygenation 12 h after OGD treatment, compared with the control (LV-CMV-Ctrl-shRNA, abbreviated to “LV-Ctrl sh” in Fig. 5B,C), and the soluble cpg15 secreted from the LV-cpg15 shRNA-infected hippocampal astrocytes was also decreased significantly at reoxygenation 12, 24, 48, and 72 h after OGD treatment (Fig. 5D). Then the medium of OGD-treated astrocytes transfected with lentivirus was added into the OGD-treated hippocampal neurons to analyze the effect of astrocytic cpg15 knockdown. Immunofluorescent double labeling of the dentritic marker MAP-2 and neuronal marker NeuN showed that, after knockdown of the cpg15 expression in the astrocytes, the neurite extending of OGD-injured neurons, either in the neuronal cluster (Fig. 5E) or in the single neuron (Fig. 5F), was inhibited significantly, and the number of dendritic sprouts from the branches, whether at the dentritic stem (Fig. 5G, top) or at the branch ending (Fig. 5G, bottom), decreased significantly at reoxygenation 48 h after OGD, compared with the control (LV-Ctrl-shRNA). Statistical data of neurite outgrowth, such as average length of every primary neurite (Fig. 5H), branch tips per primary neurites (Fig. 5I), length of longest neurite (Fig. 5J), total neurite outgrowth crosspoints (Fig. 5K), and total neurite outgrowth (Fig. 5L), decreased significantly as well. In addition, LDH detection showed that astrocytic cpg15 knockdown significantly exacerbated the injury of primary cultured neurons by OGD (Fig. 5M) at reoxygenation 48 h. Together, these data indicate that the OGD treatment upregulates the cpg15 expression and secretion from the astrocytes and promotes the neurite recovery of OGD-injured neurons, such as neurite branching and extending.
Figure 5.
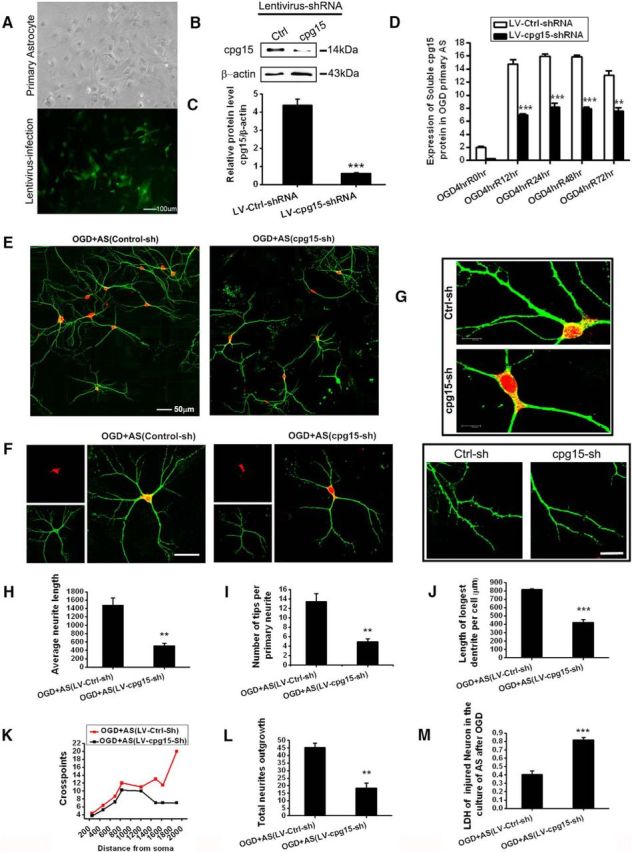
Knockdown cpg15 expression in primary cultured astrocytes via lentivirus-delivered shRNA depresses the neurite outgrowth recovery and neuronal network reestablishment of OGD-injured primary cultured neurons. A, Phase-contrast and fluorescent microscopy images of the primary cultured hippocampal astrocytes infected with cpg15 shRNA-delivering lentivirus (LV-CMV-cpg15-shRNA), showing the infection efficiency. Cells with GFP expression under fluorescent microscope were those infected by lentivirus. The phase-contrast microscope image of the same field shows the total cells. Scale bars, 100 μm. B, Representative Western blot image showing significantly inhibited expression of cpg15 protein by LV-CMV-cpg15-shRNA (abbreviated to LV-cpg15-sh) lentivirus infection in astrocytes at reoxygenation 12 h after 4 h OGD treatment. Scramble shRNA-delivering lentivirus (LV-CMV-Control-shRNA, abbreviated to LV-Ctrl-sh) was used as the negative control. C, Relative amount of cpg15 calculated from B. β-Actin as the loading control. n = 6 for each group. ***p < 0.001. D, Lentivirus-delivered cpg15 shRNA (LV-cpg15-shRNA) markedly reduces soluble cpg15 protein in the medium of astrocytes at reoxygenation 0, 12, 24, 48, and 72 h after 4 h OGD, as assayed by ELISA kit. The scrambled shRNA sequence as the control (LV-Ctrl-shRNA). n = 6 per group. **p < 0.01. ***p < 0.001. E–G, Immunofluorescent double staining of MAP2 (green), neuronal marker NeuN (red), at reoxygenation 48 h in OGD-treated primary hippocampal neurons added with the medium from OGD-treated primary astrocytes infected with LV-cpg15-shRNA, or LV-Ctrl shRNA (as the control). F, G, Representative images containing single neuron (F) or branch stem and tips (G), highlighting the difference of the neurite outgrowth between the cpg15 shRNA and the control groups. Scale bars: E, F, 50 μm; G, Top, 6 μm; G, Bottom, 20 μm. H–M, Statistical data of average neurite length (H), number of tips per primary neurite (I), length of longest neurite (J), total crosspoints (K), total neurite outgrowth (L), and LDH of OGD-injured neurons (M) in the cpg15 shRNA and the control groups. Data are expressed as at least three independent experiments. Each group, n = 6. ***p < 0.001.
Lentivirus-delivered knockdown of cpg15 expression in the astrocytes of mouse hippocampus decreases the dendritic branches and exacerbates injury of neurons in mouse hippocampal CA1 and DG regions after TGI
The above results of cultured cells showed that soluble cpg15 secreted from OGD-induced astrocytes promoted the neurite recovery of OGD-injured neurons. To verify this function of soluble cpg15 in vivo, cpg15-shRNA-delivering lentivirus and the control lentivirus (LV-CMV-cpg15 shRNA and LV-CMV-Control shRNA lentivirus) were inducted into the hippocampal CA1 subfield of wild-type mice via the stereotaxic injection 4 d before TGI treatment of the mice, and the brain tissues were analyzed at reperfusion 21 d. As shown in Results, injected lentivirus could be traced by GFP fluorescence in hippocampus and some adjacent regions of cerebral cortex (Fig. 6A,B), and the GFP protein-expressing lentivirus (green) distributed widely in mouse hippocampus; and in parallel, the cpg15 (red) expression in these regions decreased significantly in the cpg15-shRNA delivering lentivirus (cpg15-sh)-infected regions (Fig. 6C). Western blot analysis also showed significantly decreased expression of cpg15 in hippocampus injected with cpg15-shRNA-delivering lentivirus, compared with the scrambled-shRNA delivering lentivirus (Control-sh, Fig. 6D). In addition, the recovery of neuronal dendrites (as analyzed by MAP2 immunofluorescent staining) decreased significantly (Fig. 6E,F) in hippocampal CA1 and DG regions of cpg15-shRNA-delivering lentivirus infected mice at reperfusion 21 d after TGI. CV staining (Fig. 6G,H) also showed that lentivirus-delivered cpg15-shRNA (cpg15-sh) causes more severe damage in mouse hippocampus at reperfusion 21 d after TGI, compared with the control (Control-sh).
Figure 6.
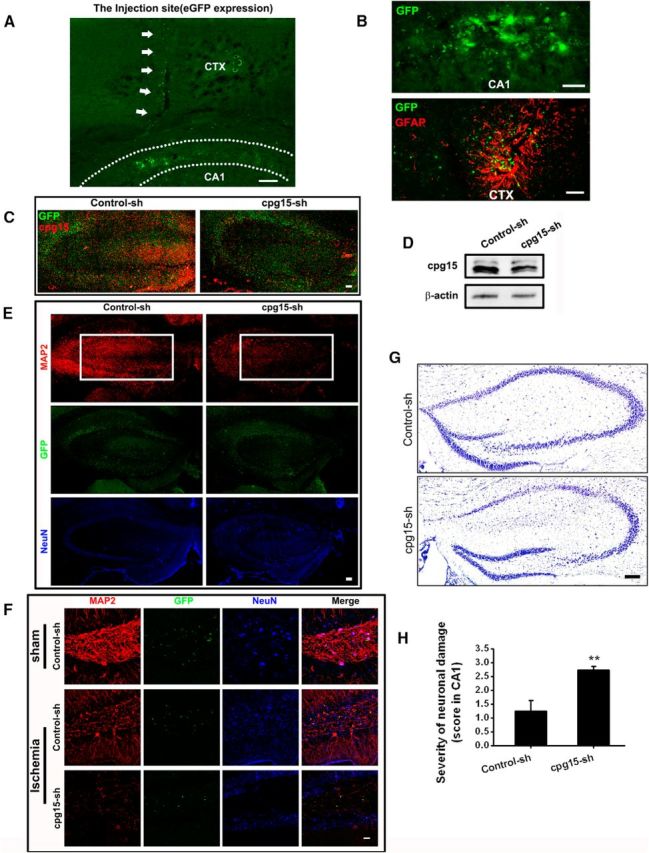
LV-CMV-cpg15 shRNA lentivirus-delivered knockdown of cpg15 expression in mouse hippocampal decreases the dendritic branches and exacerbates injury of neurons in hippocampal CA1 and DG regions at reperfusion 21 d after TGI. A, Representative fluorescent image of lentivirus-expressed GFP protein showing the injection trace (indicated by the white arrows) of lentivirus in the mouse hippocampal CA1 region. CTX, Cerebral cortex; CA1, cornus ammonis sector 1; DG, dentate gyrus. Scale bars, 50 μm. B, Lentivirus-mediated expression of GFP protein (green), indicating the distribution of injected lentivirus. GFAP (red) immunofluorescent staining was used for labeling astrocytes. C, Immunofluorescent staining of cpg15 (red) and the fluorescence of lentivirus-expressed GFP (green). D, Western blot analysis of cpg15 expression in the hippocampus of cpg15-shRNA-delivering lentivirus (LV-CMV-cpg15 shRNA, abbreviated to cpg15-sh)-injected mouse showing the efficiency of cpg15 silencing. Lentivirus mediating the scrambled shRNA (LV-CMV-Control shRNA, abbreviated to Control-sh) as the control. E, Immunofluorescent double staining of neuronal marker NeuN (blue) and dentritic marker MAP2 (red), and the fluorescence of lentivirus-expressed GFP (green) in mouse hippocampus at reperfusion 21 d after TGI. White boxes represent that the neuronal neurites in the hippocampus are significantly decreased by infection of cpg15 shRNA-delivering lentivirus. Scale bar, 50 μm. F, Immunofluorescent double staining of MAP2 (red) and NeuN (blue), and the fluorescence of lentivirus-expressed GFP (green) in sham and ischemic mouse hippocampal DG regions injected with cpg15 shRNA-delivering lentivirus. Scale bar, 25 μm. G, H, Lentivirus-mediated cpg15 knockdown (cpg15-sh) causes more severe damage in mouse hippocampus at reperfusion 21 d after TGI, as indicated by CV staining of mouse hippocampus (G) and the statistical hippocampal damage severity (H). Lentivirus delivering the scrambled shRNA as the control (Control-sh). Scale bar, 50 μm. n = 12 per group. **p < 0.01.
To further investigate the function of astrocyte-expressed cpg15 in the recovery of ischemic-injured hippocampal neurons, LV-GFAP-cpg15 shRNA lentivirus, which delivers astrocyte-specific knockdown of cpg15 expression, and its control (LV-GFAP-Control shRNA), was constructed and inducted into the hippocampal CA1 subfield of mice via the stereotaxic injection 4 d before TGI treatment, and the brain tissues were analyzed at reperfusion 14 and 21 d. As shown in Figure 7A, the expression of cpg15 was knocked down significantly by LV-GFAP-cpg15-sh lentivirus infection in cultured hippocampal astrocytes 12 h after 4 h OGD treatment (cpg15-sh, GFP-positive cells), compared with the control (Control-sh, GFP-positive cells). Furthermore, the lentivirus-delivered GFP protein was expressed specifically in the GFAP-immunopositive astrocytes of hippocampal CA1 and DG regions, for both LV-GFAP-cpg15 shRNA lentivirus (cpg15-sh) and LV-GFAP-Control shRNA (Control-sh) (Fig. 7B), and the cpg15 expression in the GFP-positive cells (as indicated by the arrows) was significantly reduced in the LV-GFAP-cpg15-sh lentivirus-injected hippocampus (Fig. 7C, cpg15-sh), compared with the control (Fig. 7C, Control-sh). Western blot analysis also showed significantly decreased cpg15 amount in the hippocampus injected with LV-GFAP-cpg15-sh lentivirus (cpg15-sh), compared with the LV-GFAP-Control-sh lentivirus (Control-sh) (Fig. 7D). In addition, the recovery of neuronal dendrites (as analyzed by MAP2 immunofluorescent staining) decreased significantly (Fig. 8A,B) in hippocampal CA1 regions of LV-GFAP-cpg15-sh lentivirus-infected mice at reperfusion 14 d after TGI, compared with the control. CV staining (Fig. 8C,D) also showed that LV-GFAP-cpg15-sh lentivirus causes more severe damage in mouse hippocampus at reperfusion 14 d after TGI, compared with the control.
Figure 7.
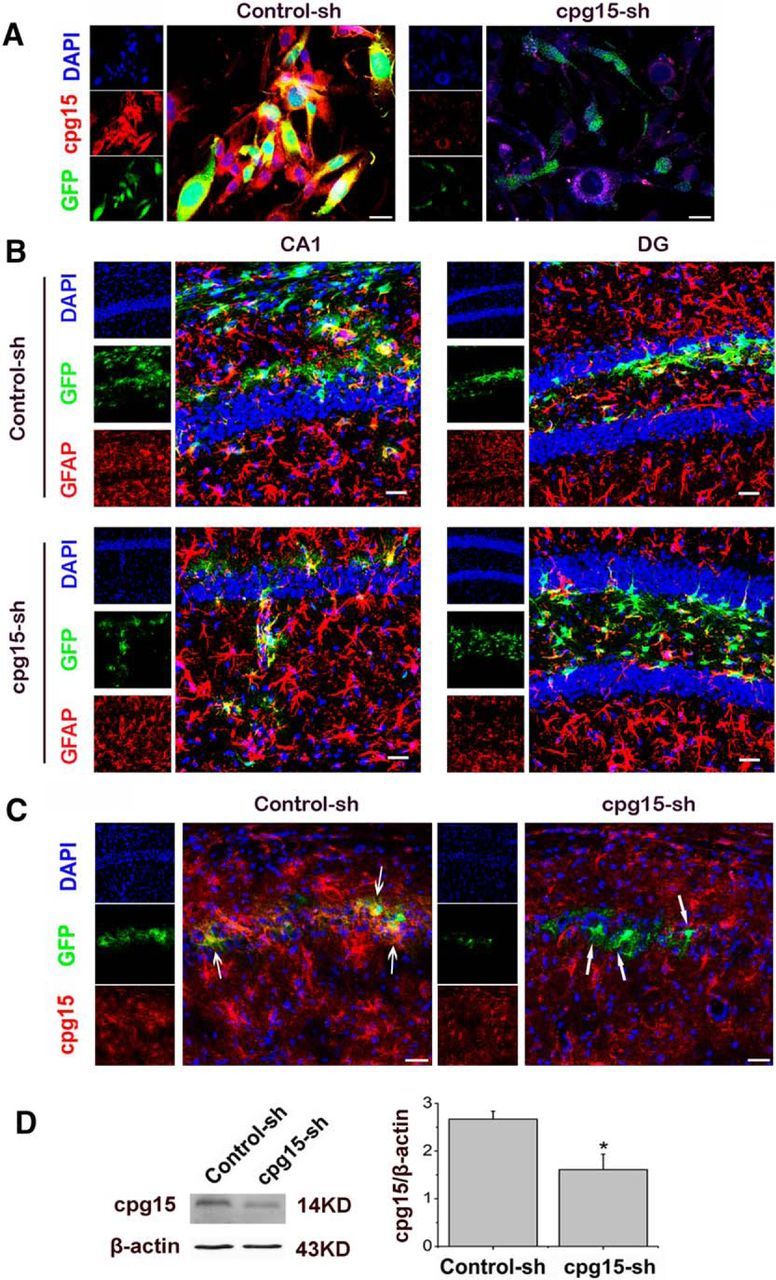
Astrocyte-specific knockdown of cpg15 expression in cultured cells and in mouse hippocampus by LV-GFAP-cpg15-shRNA lentivirus. A, Representative fluorescent images showing the cpg15 knockdown by LV-GFAP-cpg15-shRNA lentivirus (cpg15-sh) in cultured astrocytic cells 12 h after 4 h OGD treatment. Astrocytes infected with LV-GFAP-Control-shRNA lentivirus (Control-sh) as the control. Scale bar, 75 μm. B, Representative fluorescent images of lentivirus-expressed GFP (green) in the mouse hippocampal CA1 and DG regions at reperfusion 14 d after TGI, indicating that the LV-GFAP-cpg15-sh lentivirus (and its control, LV-GFAP-Control-sh lentivirus)-delivered GFP were expressed specifically in the astrocytes. Astrocytes were labeled by GFAP immunofluorescent staining (red). Scale bars, 25 μm. C, Lentivirus-mediated astrocyte-specific cpg15 knockdown in mouse hippocampal CA1 regions at reperfusion 14 d after TGI. Immunofluorescent staining of cpg15 (red) and the fluorescence of lentivirus-expressed GFP (green) were analyzed. The cpg15 expression was significantly reduced in the GFP-positive cells in the LV-GFAP-cpg15-sh lentivirus-injected hippocampus (cpg15-sh), but not in the LV-GFAP-control-sh lentivirus-injected hippocampus (Control-sh), as indicated by the arrows. Scale bars, 75 μm. D, Western blot analysis of cpg15 expression in the hippocampus of LV-GFAP-cpg15-sh lentivirus-injected mouse showing the efficiency of cpg15 knockdown. LV-GFAP-Control-sh lentivirus-injected hippocampus as the control. *p < 0.05.
Figure 8.
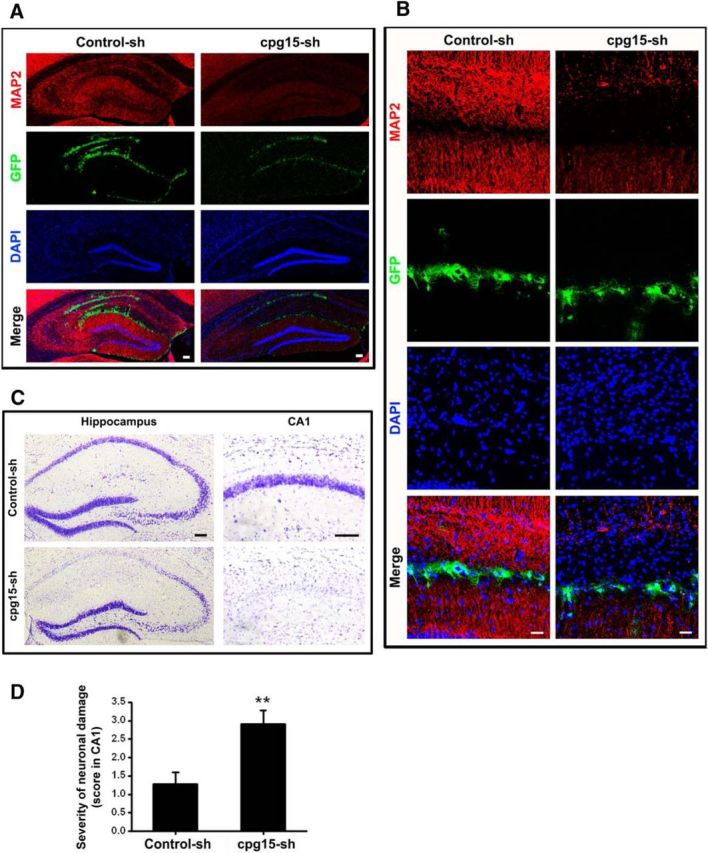
Lentivirus-delivered astrocyte-specific knockdown of cpg15 expression in mouse hippocampus decreases the dendritic branches and exacerbates injury of neurons in hippocampal CA1 region at reperfusion 14 d after TGI. A, B, Immunofluorescent staining of dentritic marker MAP2 (red), the fluorescence of lentivirus-expressed GFP (green) in mouse hippocampus at reperfusion 14 d after TGI. DAPI (blue) is used for nucleic staining. The results show that the neuronal neurites of CA1 region are significantly decreased by injection of LV-GFAP-cpg15-sh lentivirus (cpg15-sh). Scale bars: A, 100 μm; B, 25 μm. C, D, Lentivirus-mediated astrocyte-specific cpg15 knockdown (LV-GFAP-cpg15-sh, cpg15-sh) causes more severe damage in mouse hippocampus at reperfusion 14 d after TGI, as indicated by CV staining of mouse hippocampus (C) and the statistical hippocampal damage severity (D). LV-GFAP-Control-sh lentivirus-injected hippocampus (Control-sh) as the control. Scale bar, 100 μm. n = 10 per group. **p < 0.01.
These results are consistent with the above findings that OGD-induced upregulation of cpg15 expression in primary astrocytes promotes the neurite outgrowth and decreases the injury of OGD-treated primary hippocampal neurons. The results also indicate that the ischemia-induced upregulation of cpg15 expression in mouse hippocampal astrocytes plays a protective role in inhibiting the neuronal injury and promoting the recovery of injured neurons in mouse hippocampus after TGI.
OGD-induced upregulation of cpg15 expression in astrocytes was prevented by MAPK, PI3K, and TrkA signal pathway inhibitors
To investigate which factors cause the upregulation of cpg15 protein in astrocytes after ischemia, primary cultured astrocytes were subjected to 4 and 8 h of OGD to mimic the different extent of ischemic injury in vivo, and the soluble cpg15 in the culture medium was analyzed at reoxgeneration 12 and 24 h using ELISA. The results showed that the soluble cpg15 increased significantly in the medium of both 4 and 8 h OGD-treated astrocytes compared with the nontreated controls, and the elevation amplitude was dramatically higher for 8 h OGD treatment than 4 h treatment, at reoxygenation 12 h as well as 24 h (Fig. 9A). This result suggests that the aggravation of ischemic injury increases the cpg15 expression in astrocytes.
Figure 9.

Upregulation of cpg15 expression induced by OGD in astrocytes was prevented by MAPK, PI3K, and TrkA signal pathway inhibitors. A, Aggravated OGD injury increases the expression of soluble cpg15 in primary cultured astrocytes. Soluble cpg15 protein in the medium of primary cultured astrocytes at reoxygenation 12 and 24 h after 4 h OGD or 8 h OGD analyzed by ELISA. n = 8 for each group. **p < 0.01. ***p < 0.001. B, The expression levels of the phosphorylated (activated) proteins, p-Akt and p-Erk1/2, were upregulated in primary cultured astrocytes after 4 h OGD and reoxygenation. Left panels, Representative Western blot images. p-Akt (Ser473) and p-Erk1/2(Thr202/Tyr204) antibodies (Cell Signaling Technology,1:500) were used for p-Akt and p-Erk1/2 proteins, respectively. Akt and Erk1/2 antibodies (Cell Signaling Technology, 1:500) were used for the total Akt and Erk1/2 proteins, respectively. Analysis of GAPDH was used as the loading control. Right panels, Relative amount of p-Akt and p-Erk1/2 from the Western blot images. n = 3. *p < 0.05. ***p < 0.001. C, Relative expression levels of cpg15 mRNA in U118 astrocytes pretreated with increasing doses of the mitogen-activated kinase/ERK kinase (MEK-1/2) inhibitor (U0126), PI3K inhibitor (LY294002), TrkA inhibitor (K252a), or negative control U0124 (for U0126), analyzed by real-time RT-PCR. D, Relative expression levels of cpg15 mRNA in mouse primary astrocytes pretreated with 20 μm of the above inhibitors (or not treated as negative control), followed by 4 h OGD and 12 h reoxygenation. E, Representative Western blot analysis of cpg15 in the OGD-treated primary astrocytes pretreated with 20 μm of the above inhibitors (or not treated as negative control, OGD) at reoxygenation12 h after 4 h OGD. F, Relative amount of cpg15 protein from E. β-Actin is used as the loading control. n = 6 for each group. **p < 0.01. ***p < 0.001. G, The expression levels of p-Akt and p-Erk1/2 were suppressed by 20 μm of the inhibitors of PI3K (LY294002) and MEK1/2 (U0126), respectively, in primary cultured astrocytes at 12 h reoxygenation after 4 h OGD. Left panels, Representative Western blot images. Right panels, Relative amount of p-Akt and p-Erk1/2 from the Western blot images. n = 3. *p < 0.05. **p < 0.01.
Based on this result, we deduced that the injury-induced factors, such as neural growth factors or cytokines might be involved in the upregulation of cpg15 expression in astrocytes after ischemia, to further investigate whether the signal pathways downstream them (e.g., PI3K/Akt and MAPK pathways) are responsible for this regulation, we first investigated whether the Akt protein (in the PI3K/Akt pathway) and the Erk1/2 protein (in the MAPK pathway) were activated in astrocytes by OGD and reoxygenation. Cultured mouse hippocampal astrocytes were treated with 4 h OGD followed by different hours of reoxygenation, and the expression levels of the phosphorylated (activated) proteins p-Akt (Ser473-phosphorylated) and p-Erk1/2 (Thr202/Tyr204-phosphorylated) were analyzed by Western blot. The results showed that the levels of both p-Akt and p-Erk1/2 were significantly upregulated at R0hr and R12hr after OGD treatment (Fig. 9B), indicating that the PI3K and MAPK signal pathways were activated in the astrocytes by OGD and reoxygenation. To further investigate whether these signal pathways were involved in the regulation of cpg15 expression in the astrocytes, cultured U118 astrocytes were treated with different concentrations of U0126 (MEK-1/2 inhibitor), LY294002 (PI3K inhibitor), K252a (TrkA inhibitor), or U0124 (negative control for U0126), and the effect of these inhibitors on cpg15 expression was analyzed by real-time RT-PCR. The results showed that treatment of these inhibitors decreases the expression of cpg15 in a concentration-dependent model, although no significant difference exists between the control (U0124) and the non–inhibitor-treated (normal) group (Fig. 9C).
We further want to investigate the effect of these inhibitors on OGD-induced expression of cpg15 in primary cultured astrocytes. Astrocytes were pretreated with the optimized concentration (20 μm) of the inhibitors followed by 4 h OGD treatment, and the mRNA and protein levels of cpg15 were analyzed by real-time RT-PCR and Western blot at reoxygenation 12 h, the results showed that the expression of both mRNA (Fig. 9D) and protein (Fig. 9E,F) of cpg15 were suppressed significantly in primary cultured astrocytes pretreated with MEK-1/2, PI3K, and TrkA inhibitors (U0126, LY294002, and K252a), whereas no significant difference exists between the negative control (OGD+U0124) and the noninhibitor treated group (OGD). We also performed experiments to confirm whether these inhibitors actually work on the targeted kinases in the primary cultured hippocampal astrocytes treated by OGD and reoxygenation, by analyzing the phosphorylation (activation) level of the downstream proteins Akt and Erk1/2 using Western blot analysis. The results showed that the expression of p-Akt and p-Erk1/2 was indeed suppressed significantly by 20 μm of the inhibitors of PI3K (LY294002) and MEK1/2 (U0126), respectively, whereas the expression of total proteins of Akt and Erk1/2 has nonsignificant changes (Fig. 9G). In addition, the inhibitor K252a for the upstream TrkA also inhibits the Akt and Erk1/2 phosphorylation (data not shown) and the upregulation of cpg15 expression induced by OGD and reoxygenation (Fig. 9D–F). These results indicated that the upregulated expression of cpg15 in astrocytes induced by ischemia may be activated via PI3K and MAPK signal pathways, which could be activated by the injury-related factors, such as neural growth factors or cytokines.
CREB protein, especially the phosphorylated CREB, is involved in the OGD-induced upregulation of cpg15 expression in astrocytes
CREB is an important transcription factor and is reported to be involved in activity-dependent regulation of the cpg15 expression via binding the cpg15 promoter in neurons (Fujino et al., 2003). Here, we further want to investigate whether CREB also attributes to the OGD-induced upregulation of cpg15 expression in astrocytes. For this purpose, total CREB and phosphorylated CREB proteins, respectively, were analyzed by Western blot in OGD-stimulated primary cultured astrocytes at reoxygenation 0, 12, 24, 48, and 72 h. The results showed that both the total CREB (Fig. 10A) and the phosphorylated CREB (Fig. 10B) proteins were significantly increased in primary astrocytes from reoxygenation 12–72 h. We further overexpressed CREB proteins in primary cultured astrocytes by transfection of CREB-expressing vectors with GFP as the reporter. After FACS, the transfected cells containing GFP reporter were separated from those nontransfected ones and analyzed for cpg15 mRNA level with real-time RT-PCR and soluble cpg15 in the culture medium with ELISA. The results showed that overexpression of CREB proteins increases significantly the cpg15 mRNA in the astrocytes (Fig. 10C) as well as the soluble cpg15 in the medium (Fig. 10D), compared with the control (pAAV-GFP), indicating that CREB protein is involved in the promotion of cpg15 transcription in astrocytes. To further investigate whether the phosphorylated CREB is the key factor in promoting cpg15 transcription, the serine133 site of CREB coding sequence was mutagenized into glutamine to mimic the phosphorylated CREB, and mutagenized into alanine to mimic the dephosphorylated CREB, and the plasmids encoding these CREB mutants, along with the plasmid encoding the full-length CREB, were transfected into the U118 astrocytes for analyzing their functions. The results (Fig. 10E) showed that overexpression of the phosphomimetic CREB (pAAV-pCREB-GFP) increases dramatically the mRNA expression of cpg15 in astrocytes, with the elevation amplitude 5 times higher than that of total CREB overexpression (pAAV-CREB-GFP) and 10 times higher than that of the control (pAAV-GFP), whereas overexpression of dephosphomimetic CREB (pAAV-dpCREB-GFP) makes no difference from the control (pAAV-GFP). These results indicate that it is the phosphorylated CREB, not the dephosphorylated one, that promotes the transcription of cpg15 in astrocytes. The results also suggest that the phosphorylated CREB is attributed to the ischemia-induced upregulation of cpg15 expression in astrocytes via promoting its transcription.
Figure 10.

CREB protein, especially the phosphorylated form of CREB, is involved in the OGD-induced upregulation of cpg15 expression in astrocytes. Western blot images and their statistical data for expression of CREB (A) and phosphorylated CREB (p-CREB) (B) proteins in primary cultured astrocytes at reoxygenation 0, 12, 24, 48, and 72 h after OGD treatment. n = 6 for each group cells. **p < 0.01, compared with the non–OGD-treated group (Normal). ***p < 0.001, compared with the non–OGD-treated group (Normal). C, D, Overexpression of CREB protein (pAAV-CREB-GFP) significantly increases the expression of cpg15 mRNA in primary cultured astrocytes (C) and soluble cpg15 protein in the culture medium (D), analyzed by real-time RT-PCR (C) and ELISA (D), respectively. n = 6 for each group. ***p < 0.001, compared with empty plasmid transfected control (pAAV-GFP). E, Overexpression of p-CREB and CREB proteins significantly increases expression of cpg15 in U118 astrocytes. Constructed plasmid expressing CREB with Ser133Glu mutation is used to mimic p-CREB, and CREB with Ser133Ala mutation is used to mimic dp-CREB. Data are analyzed by real-time RT-PCR. n = 6 for each group. **p < 0.01. ***p < 0.001. ###p < 0.001.
Soluble Flag-tagged cpg15 expressed and secreted from primary cultured astrocytes adheres to the cell bodies and dendritic neurites of primary cultured hippocampal neurons
Two forms of cpg15 proteins are expressed in neural tissues: the soluble, secretory form and the membrane-bound form attached by a GPI link (Cantallops et al., 2000; Putz et al., 2005). The above results demonstrated that the soluble cpg15 secreted from OGD-stimulated astrocytes promotes the neurite outgrowth recovery and reduces the injury of OGD-treated neurons. To investigate how the soluble cpg15 secreted from the astrocytes functions on the neurons, we constructed the recombinant pAAV plasmid with the Flag tag inserted between the signal peptide and the coding sequence of cpg15 (Fig. 11A) for overexpression of the secretory Flag-tagged cpg15 in primary cultured astrocytes (Fig. 11B). The culture medium from the normal astrocytes with transfection was then added into the primary cultured hippocampal neurons, and the Flag-tagged cpg15 on neurons was analyzed by coimmunofluorescent labeling of Flag, MAP2 (the dentritic marker), and NeuN (the neuronal marker). The results showed that the soluble Flag-tagged cpg15 secreted from astrocytes aggregates and adheres to the cell bodies and dendritic neurites of primary cultured hippocampal neurons (Fig. 11D,E), whereas there was no Flag signal in the negative control (Fig. 11C). We also analyzed the location of Flag-tagged cpg15 using coimmunofluorescent labeling of Flag, MAP2, and GFAP (the astrocytic marker) in the primary cultured hippocampal neurons and astrocytes, and found that Flag-tagged cpg15 does not adhere to the astrocytes (data not shown), indicating that the adhering of Flag-cpg15 was neuron-specific. Combined with the above demonstration, this result indicates that the upregulated cpg15 protein in ischemia-stimulated astrocytes probably exerts its function to neurons via secreting as the soluble form and binding on the surface of neuronal cell bodies and outgrowth.
Figure 11.

Soluble Flag-cpg15 expressed and secreted from primary cultured astrocytes adheres to cell body and dendritic neurites of primary cultured hippocampal neurons. A, Schematic diagram of Flag-cpg15-IRES-hrGFP construct, which will express secretory Flag-tagged cpg15 protein. The Flag peptide coding sequence (yellow) was inserted between the secretion signal (ss) and the cpg15 coding sequence (cpg15-cd). This recombined fragment was then cloned upstream of an IRES and hrGFP sequence in pAAV plasmid. B, Western blot images verifying the overexpression of Flag-tagged cpg15 in primary cultured astrocytes transfected with empty, full-length cpg15, and Flag-tagged cpg15-expressing plasmid. C–E, Immunofluorescence triple staining of MAP2 (red), Flag-tagged protein (green), and neuronal marker NeuN (blue) in primary cultured neurons added with the medium from primary cultured astrocytes transfected with empty plasmid (pAAV) (C), or Flag-cpg15 plasmid (pAAV-Flag-cpg15) (D,E). White arrows indicate that soluble Flag-tagged cpg15 proteins expressed in the astrocytes gather along and adhere to the surface of primary cultured neuronal cell body and neurites.
BAIAP2 and LRFN2 proteins are coimmunoprecipitated with cpg15, and addition of cpg15 antibody into the culture medium regulates the expression of GAP20, GAP22, GAP29, and EP400 mRNA in cultured neurons
To explore the potential signal pathway through which cpg15 exerts its function on neurons, lysates of mouse brain tissues were immunoprecipitated by anti-cpg15 antibodies for detecting protein interaction with cpg15. The immunoprecipitates were analyzed by SDS-PAGE followed by silver staining. Two proteins, brain-specific angiogenesis inhibitor 1-associated protein 2-like protein 1 (BAIAP2, NP_080109.1) and leucine-rich repeat and fibronectin Type III domain-containing protein 2 (LRFN2, NP_081728.2), were found to be coimmunoprecipitated with the cpg15, as identified by mass spectrometry (Fig. 12A,B) and Western blot analysis (Fig. 12C). The results implied that BAIAP2 and LRFN2 might be involved in mediating the function of cpg15 on neurons. To further investigate which genes in neurons are regulated by cpg15, N2a neuronal cells were cultured in the medium added with cpg15 antibodies (1:100, R&D Systems, equal amount of β-actin antibody as the control), and the cells were harvested after further 12 h of culture for analyzing the regulated gene expression by whole-mouse genome microarray. The microarray data showed that the expressions of >1000 genes were significantly regulated. From them, we selected three GAPs (Rho GTPase-activating proteins, ARHGAP20, ARHGAP22, and ARHGAP29) and EP400 (E1A binding protein p400), the mRNA expressions of which were upregulated (ARHGAP20, ARHGAP22, ARHGAP29) or downregulated (EP400) significantly (as shown in Fig. 12D), for further confirmation by real-time RT-PCR analysis. The results showed that the regulation directions in RT-PCR data (Fig. 12E–H) were consistent with the microarray data for the four genes. The results implied that cpg15 protein probably interacts with BAIAP2 and LRFN2 proteins, which might mediate the intracellular signal and regulate the expression of downstream genes in neuronal cells, such as genes of GAP20, GAP22, GAP29, and EP400, and therefore participate in promoting neurite outgrowth and inhibiting the injury of neurons.
Figure 12.
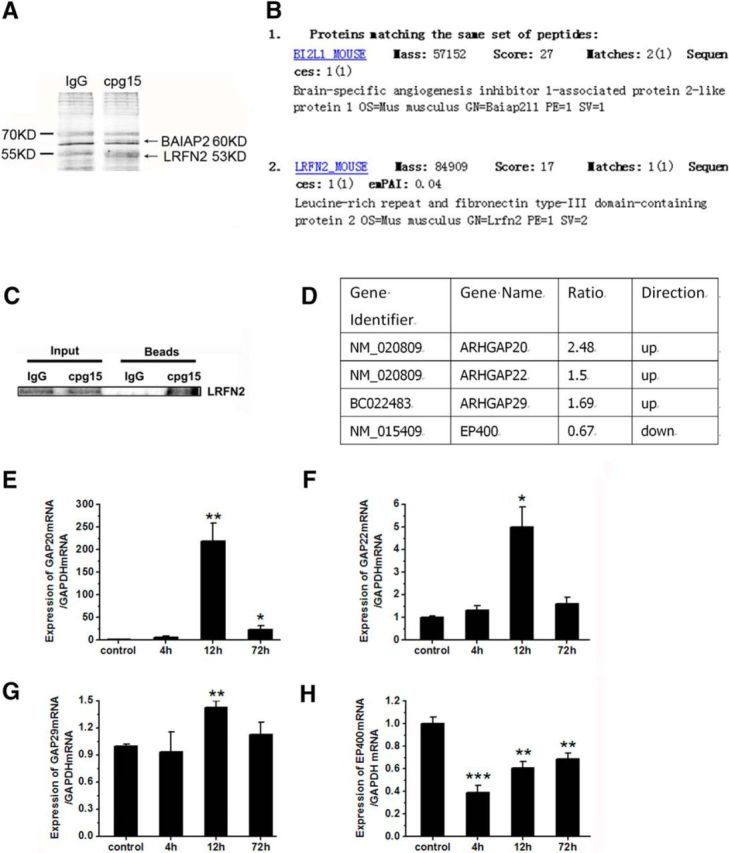
BAIAP2 and LRFN2 proteins are coimmunoprecipitated with cpg15 protein, and addition of cpg15 antibody in the culture medium regulates the expression of GAP20, GAP22, GAP29, and EP400 mRNA in neurons. A, Two protein bands (as indicated by the arrows) were coimmunoprecipitated with cpg15 protein in the lysate of mouse brain issue. SDS-PAGE followed with silver staining of immunoprecipitation complex by goat anti-cpg15 antibody was shown. Goat-IgG was used as the negative control. B, The identities of the two bands indicated by the arrows in A were BAIAP2 (also named BI2L1) and LRFN2, as obtained by mass spectrum (MS) analysis. C, Western blot images verifying LRFN2 protein coimmunoprecipitated with cpg15 protein in the lysate of mouse brain issue. D, Analysis from microarray data showing that the mRNA expression of GAP20, GAP22, GAP29 (ARHGAP20, ARHGAP22, ARHGAP29), and EP400 genes were regulated (up or down) significantly in N2a cells after addition of cpg15 antibody to the culture medium. Genes with significantly different expression patterns were generated using the two criteria: an adjusted p value from t test <0.05 and at least 1.5-fold difference between control and test samples. Three independent experiments were performed. E–H, Real-time RT-PCR analysis showing that the regulational direction of mRNA expression levels of GAP20 (E), GAP22 (F), GAP29 (G), and EP400 (H) genes were consistent with the microarray data. mRNA level in N2a cells at 4, 12, and 72 h after addition of cpg15 antibody into the culture medium were shown. n = 6 per group. *p < 0.05, compared with the control group. **p < 0.01, compared with the control group. ***p < 0.001, compared with the control group.
Discussion
Neurological recovery is an important process after cerebral ischemia; the underlying structural correlates of neuronal remodeling and regeneration process contain neuronal sprout, neurogenesis, and angiogenesis, which was similar to the development of nervous system (Hermann and Chopp, 2012). As an activity-regulated gene (Nedivi et al., 1993), cpg15 has been found to be involved in the promotion of a variety of events during the development of nervous system, such as neuronal growth, dendritic arbor growth of projection neurons (Nedivi et al., 1998), and shaping of dendritic arbors of target neurons during activity-dependent synaptic rearrangements (Corriveau et al., 1999). Considering the similarity of the neuronal process between the development of nervous system and the recovery after cerebral ischemia, we suppose that cpg15 may also play roles in the recovery of ischemic-injured brain.
Astrocytes have been recognized to have protective effects on ischemic injured neurons via secreting neurotrophins, such as BDNF (Zamanian et al., 2012; Miyamoto et al., 2015), GDNF (Yan et al., 2011), and CNTF (Kang et al., 2013), or extracellular matrix glycoproteins (Christopherson et al., 2005; Benner et al., 2013). In the present study, we observed that the expression of cpg15, a neuronal-specific expressed protein hardly observed in astrocytes in normal condition, was increased significantly in astrocytes, from reperfusion 1 d (data not shown) to 14 d after cerebral ischemia, and the cpg15 could be secreted outside the astrocytes as the soluble form. Considering the function of cpg15 protein in enhancing neuronal axonal sprouting, dendritic arbors, and synapses maturation during neural development (Nedivi et al., 1998; Corriveau et al., 1999; Lee and Nedivi, 2002; Javaherian and Cline, 2005), we want to know whether the astrocyte-expressed and secreted cpg15 protein can function as a neurotrophic factor in promoting the recovery or rescue of neurite outgrowth of the ischemic-impaired neurons. Our results in in vitro and in vivo experiments showed that the soluble cpg15 secreted from the ischemia-stimulated astrocytes dramatically reduces the ischemic injury of neurons and enhances the dendritic outgrowth recovery of injured neurons, probably via adhering to the surface of neuronal bodies and dendritic arbors. The function of astrocytic cpg15 in ischemic recovery is parallel to the plasticity mechanisms in the developing nervous system and those taking place in the adult brain after stroke (Murphy and Corbett, 2009). It is suggested that the reactive astrocyte-expressed and secreted cpg15 protein may act as an intercellular signal molecular in mediating the interaction between astrocytes and neurons after cerebral ischemia via accumulating around the injured neurons by binding to the neuronal membrane and promote the rewiring of ischemic neuronal circuits.
In addition to the soluble form, the membrane-bound GPI-anchored form of cpg15 expressed in the reactive astrocytes may also play important roles in the recovery of ischemic injured neurons. GPI-anchored proteins were known as an important element of lipid raft constituents (Guirland and Zheng, 2007; Miyamoto et al., 2015), which were found to play important roles in axonal growth and guidance. Thus, revealing the functional mechanism of lipid raft-interacting proteins might provide important insights into the intricate signaling interaction underlying neuronal wiring in the development of normal brain and the recovery of neural disease (Guirland and Zheng, 2007). There was evidence that an astrocytic membrane-spanning, cholesterol-binding protein, the σ-1 receptor, correlates with the lipid rafts and stimulates the neurological recovery after stroke by enhancing cellular transport of biomolecules required for brain repair (Ruscher et al., 2011). It is intriguing to consider that GPI-anchored cpg15 formed in the ischemic reactive astrocytes may function as neuronal axon guidance through enhancing axonal outgrowth and promoting wiring recovery of the ischemic injured neuronal connections. Actually, in the OGD-treated mixed culture of primary neurons and astrocytes, we have noticed that the neuronal axon extended along with the extending direction of the astrocytic filopodia, via triple immunofluorescent staining of cpg15, astrocytic and neuronal markers (data not shown). Our results imply that the GPI-anchored cpg15 in the ischemic reactive astrocytes might also function as a signal guidance for the axons of injured neurons and promote neuronal network reestablishment, just like the GPI-anchored cpg15 in the neurons (Cantallops et al., 2000; Javaherian and Cline, 2005).
Our results, that astrocytic cpg15 induced by ischemia promotes the neuronal recovery, imply that astrocyte-targeted overexpression of cpg15 might be a promising strategy for facilitating neuronal wiring establishment following cardiac arrest and resuscitation. There was also evidence showing that astrocyte-targeted overexpression of heat shock protein 72 (Hsp72) or superoxide dismutase 2 (SOD2) improved resistance of astrocytes to ischemic stress and reduced neuronal vulnerability to ischemia (Xu et al., 2010). In our in vivo analysis using lentivirus-delivered astrocyte-specific silencing of cpg15, we found that knockdown of cpg15 specifically in astrocytes in ischemic mouse hippocampus significantly suppressed the recovery of neurite outgrowth and aggravated the neuronal injury after cerebral ischemia. This finding confirmed the neuroprotective function of the upregulated cpg15 in astrocytes of the hippocampus after cerebral ischemia.
In investigating the pathways involved in the regulation of cpg15 expression in the ischemia-stimulated astrocytes, we identified that cpg15 was upregulated in astrocytes by MAPK, PI3K signal pathways and phosphorylated CREB proteins at the transcriptional level. As a signal pathway that could be activated in response to a variety of simulation, such as growth factors (Su et al., 2011) and ischemic injury (Boulos et al., 2007), the MAPK cascade is involved in many aspects of cellular physiology and the development of neurons and glia, including cell division, differentiation (Sweatt, 2001), and cell death (Fernandes et al., 2007). In addition, MEK, one of the key members in MAPK pathway, is essential for regulating gliogenesis (Li et al., 2012). Also, activation of the PI3K pathway was crucial in neuroprotection in the ischemic tolerance (Hashiguchi et al., 2004), and activation of TrkA pathway was important in the survival of CNS cells after brain injury (Lu et al., 2013). Our results, that astrocytic cpg15 was upregulated via MAPK, PI3K, and the upstream TrkA signal pathways, imply that the ischemia-induced astrocytic cpg15 may act as the downstream-regulated protein of these signal pathways and function on the gliogenesis, differentiation and survival of astrocytes, and even the glial scar formation (Chen et al., 2016).
CREB was a stimulus-induced transcription factor, activated by a variety of protein kinases, including MAPK, which could catalyze the phosphorylation of a particular residue, serine 133 (Ser133) of CREB, and Ser133 was required for the transcription activation (Shaywitz and Greenberg, 1999). As it has been found that CREB binds to the cpg15 promoter in neurons and partially regulated its activity-dependent expression (Fujino et al., 2003), we further investigated whether the CREB is also involved in the regulation of cpg15 expression in ischemic astrocytes. We found that the CREB protein, especially the Ser133-phosphorylated CREB, is involved in promoting the OGD-induced upregulation of cpg15 transcriptional expression in astrocytes, implying that the CREB is a transcription factor of cpg15 promoter not only in neurons, but also in astrocytes under ischemic stimulation as well.
Little has been known about the mechanisms of how cpg15 functions on neurons by far. Our results that cpg15 protein binds to the BAIAP2 and LRFN2 proteins of neurons, which are related to insulin receptor and postsynaptic membrane, respectively, imply that cpg15 may function on neurons through the insulin receptors or certain synapse-related molecules. The results that addition of cpg15 antibodies into the neuronal cell culture affects the expression of GAP20, GAP22, and GAP29 signal molecules and EP400 transcriptional factor imply that cpg15 may function on neurons via affecting these signal molecules and the transcription of downstream genes.
In conclusion, the present study indicates, for the first time, that the expression of cpg15, a neuronal-specific expressed protein, was increased significantly in astrocytes after cerebral ischemia. The cpg15 secreted as the soluble form from ischemia-stimulated astrocytes and promoted neuroprotection and recovery of neuronal neurite outgrowth probably via adhering to the surface of neurons. The upregulation of cpg15 expression in astrocytes may be activated via MAPK, PI3K, and the upstream TrkA signal pathways, and phosphorylation of CREB transcription factor. The astrocyte-secreted cpg15 protein probably interacts with BAIAP2 and LRFN2 proteins in neurons that might mediate the intracellular signal and regulate the expression of downstream genes in neuronal cells, such as genes of GAP20, GAP22, GAP29, and EP400, and therefore participate in promoting neurite outgrowth and inhibiting neuronal injury. As illustrated in Fig. 13, this study revealed a new mechanism for the recovery of ischemia-injured neurons via upregulating the expression of cpg15 and probably other neurotropic factors in astrocytes in which at normal status it does not express.
Figure 13.

A hypothetical model illustrating the function and mechanism of ischemia-induced astrocytic cpg15 on the neurological recovery after cerebral ischemia. The expression of cpg15, a neuronal-specific expressed protein, is increased significantly in astrocytes after cerebral ischemia. The cpg15 secreted as the soluble form from ischemia-stimulated astrocytes and promoted neuroprotection and recovery of neuronal neurite outgrowth probably via adhering to the surface of neurons. The upregulation of cpg15 expression in astrocytes may be activated via MAPK, PI3K, or TrkA signal pathways, and phosphorylation of CREB transcription factor. The astrocyte-secreted cpg15 protein probably interacts with BAIAP2 and LRFN2 proteins in neurons that might mediate the intracellular signal and regulate the expression of downstream genes in neuronal cells, such as genes of GAP20, GAP22, GAP29, and EP400, and therefore participate in promoting neurite outgrowth and inhibiting neuronal injury.
Footnotes
This work was supported by National Foundation of Natural Sciences of China Grants 31571037, 30971464, and 31370788, and the Shanghai Leading Academic Discipline Project B111. We thank Ke Qiao (Key Laboratory of Medical Molecular Virology, Ministry of Education and Public Health, Shanghai Medical School, Fudan University) for excellent technique in the confocal microscopy; and Prof. Bing-Qiao Zhao, Prof. Ping Zheng, and PhD students Ping Yang, Jiao-Jiao Song, Da Shao, and Jia-Lin Mo for kind help in the techniques of mouse brain ischemia and mouse stereotaxic injection.
The authors declare no competing financial interests.
References
- Andres RH, Horie N, Slikker W, Keren-Gill H, Zhan K, Sun G, Manley NC, Pereira MP, Sheikh LA, McMillan EL, Schaar BT, Svendsen CN, Bliss TM, Steinberg GK (2011) Human neural stem cells enhance structural plasticity and axonal transport in the ischaemic brain. Brain 134:1777–1789. 10.1093/brain/awr094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antypa M, Faux C, Eichele G, Parnavelas JG, Andrews WD (2011) Differential gene expression in migratory streams of cortical interneurons. Eur J Neurosci 34:1584–1594. 10.1111/j.1460-9568.2011.07896.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner EJ, Luciano D, Jo R, Abdi K, Paez-Gonzalez P, Sheng H, Warner DS, Liu C, Eroglu C, Kuo CT (2013) Protective astrogenesis from the SVZ niche after injury is controlled by Notch modulator Thbs4. Nature 497:369–373. 10.1038/nature12069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz LI, Carmichael ST (2010) Promoting axonal rewiring to improve outcome after stroke. Neurobiol Dis 37:259–266. 10.1016/j.nbd.2009.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulos S, Meloni BP, Arthur PG, Majda B, Bojarski C, Knuckey NW (2007) Evidence that intracellular cyclophilin A and cyclophilin A/CD147 receptor-mediated ERK1/2 signalling can protect neurons against in vitro oxidative and ischemic injury. Neurobiol Dis 25:54–64. 10.1016/j.nbd.2006.08.012 [DOI] [PubMed] [Google Scholar]
- Cantallops I, Haas K, Cline HT (2000) Postsynaptic CPG15 promotes synaptic maturation and presynaptic axon arbor elaboration in vivo. Nat Neurosci 3:1004–1011. 10.1038/79823 [DOI] [PubMed] [Google Scholar]
- Chen CH, Sung CS, Huang SY, Feng CW, Hung HC, Yang SN, Chen NF, Tai MH, Wen ZH, Chen WF (2016) The role of the PI3K/Akt/mTOR pathway in glial scar formation following spinal cord injury. Exp Neurol 278:27–41. 10.1016/j.expneurol.2016.01.023 [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA (2005) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120:421–433. 10.1016/j.cell.2004.12.020 [DOI] [PubMed] [Google Scholar]
- Corriveau RA, Shatz CJ, Nedivi E (1999) Dynamic regulation of cpg15 during activity-dependent synaptic development in the mammalian visual system. J Neurosci 19:7999–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes A, Falcão AS, Silva RF, Brito MA, Brites D (2007) MAPKs are key players in mediating cytokine release and cell death induced by unconjugated bilirubin in cultured rat cortical astrocytes. Eur J Neurosci 25:1058–1068. 10.1111/j.1460-9568.2007.05340.x [DOI] [PubMed] [Google Scholar]
- Fujino T, Lee WC, Nedivi E (2003) Regulation of cpg15 by signaling pathways that mediate synaptic plasticity. Mol Cell Neurosci 24:538–554. 10.1016/S1044-7431(03)00230-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George PM, Steinberg GK (2015) Novel stroke therapeutics: unraveling stroke pathophysiology and its impact on clinical treatments. Neuron 87:297–309. 10.1016/j.neuron.2015.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guirland C, Zheng JQ (2007) Membrane lipid rafts and their role in axon guidance. Adv Exp Med Biol 621:144–155. 10.1007/978-0-387-76715-4_11 [DOI] [PubMed] [Google Scholar]
- Han Y, Chen X, Shi F, Li S, Huang J, Xie M, Hu L, Hoidal JR, Xu P (2007) CPG15, a new factor upregulated after ischemic brain injury, contributes to neuronal network re-establishment after glutamate-induced injury. J Neurotrauma 24:722–731. 10.1089/neu.2006.0174 [DOI] [PubMed] [Google Scholar]
- Hashiguchi A, Yano S, Morioka M, Hamada J, Ushio Y, Takeuchi Y, Fukunaga K (2004) Up-regulation of endothelial nitric oxide synthase via phosphatidylinositol 3-kinase pathway contributes to ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab 24:271–279. 10.1097/01.WCB.0000110539.96047.FC [DOI] [PubMed] [Google Scholar]
- Hermann DM, Chopp M (2012) Promoting brain remodelling and plasticity for stroke recovery: therapeutic promise and potential pitfalls of clinical translation. Lancet Neurol 11:369–380. 10.1016/S1474-4422(12)70039-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javaherian A, Cline HT (2005) Coordinated motor neuron axon growth and neuromuscular synaptogenesis are promoted by CPG15 in vivo. Neuron 45:505–512. 10.1016/j.neuron.2004.12.051 [DOI] [PubMed] [Google Scholar]
- Kang K, Lee D, Hong S, Park SG, Song MR (2013) The E3 ligase Mind bomb-1 (Mib1) modulates Delta-Notch signaling to control neurogenesis and gliogenesis in the developing spinal cord. J Biol Chem 288:2580–2592. 10.1074/jbc.M112.398263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawase M, Murakami K, Fujimura M, Morita-Fujimura Y, Gasche Y, Kondo T, Scott RW, Chan PH (1999) Exacerbation of delayed cell injury after transient global ischemia in mutant mice with CuZn superoxide dismutase deficiency. Stroke 30:1962–1968. 10.1161/01.STR.30.9.1962 [DOI] [PubMed] [Google Scholar]
- Lee WC, Nedivi E (2002) Extended plasticity of visual cortex in dark-reared animals may result from prolonged expression of cpg15-like genes. J Neurosci 22:1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Newbern JM, Wu Y, Morgan-Smith M, Zhong J, Charron J, Snider WD (2012) MEK is a key regulator of gliogenesis in the developing brain. Neuron 75:1035–1050. 10.1016/j.neuron.2012.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D (2002) Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295:868–872. 10.1126/science.1067081 [DOI] [PubMed] [Google Scholar]
- Lu J, Frerich JM, Turtzo LC, Li S, Chiang J, Yang C, Wang X, Zhang C, Wu C, Sun Z, Niu G, Zhuang Z, Brady RO, Chen X (2013) Histone deacetylase inhibitors are neuroprotective and preserve NGF-mediated cell survival following traumatic brain injury. Proc Natl Acad Sci U S A 110:10747–10752. 10.1073/pnas.1308950110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto N, Maki T, Shindo A, Liang AC, Maeda M, Egawa N, Itoh K, Lo EK, Lok J, Ihara M, Arai K (2015) Astrocytes promote oligodendrogenesis after white matter damage via brain-derived neurotrophic factor. J Neurosci 35:14002–14008. 10.1523/JNEUROSCI.1592-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Corbett D (2009) Plasticity during stroke recovery: from synapse to behaviour. Nat Rev Neurosci 10:861–872. 10.1038/nrn2735 [DOI] [PubMed] [Google Scholar]
- Naeve GS, Ramakrishnan M, Kramer R, Hevroni D, Citri Y, Theill LE (1997) Neuritin: a gene induced by neural activity and neurotrophins that promotes neuritogenesis. Proc Natl Acad Sci U S A 94:2648–2653. 10.1073/pnas.94.6.2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Dirnagl U (2005) Role of glial cells in cerebral ischemia. Glia 50:281–286. 10.1002/glia.20205 [DOI] [PubMed] [Google Scholar]
- Nedivi E, Hevroni D, Naot D, Israeli D, Citri Y (1993) Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature 363:718–722. 10.1038/363718a0 [DOI] [PubMed] [Google Scholar]
- Nedivi E, Fieldust S, Theill LE, Hevron D (1996) A set of genes expressed in response to light in the adult cerebral cortex and regulated during development. Proc Natl Acad Sci U S A 93:2048–2053. 10.1073/pnas.93.5.2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedivi E, Wu GY, Cline HT (1998) Promotion of dendritic growth by CPG15, an activity-induced signaling molecule. Science 281:1863–1866. 10.1126/science.281.5384.1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG (2007) Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J Neurosci 27:4253–4260. 10.1523/JNEUROSCI.0211-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putz U, Harwell C, Nedivi E (2005) Soluble CPG15 expressed during early development rescues cortical progenitors from apoptosis. Nat Neurosci 8:322–331. 10.1038/nn1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom B, Behar T, Nedergaard M (2003) New roles for astrocytes (stars at last). Trends Neurosci 26:520–522. 10.1016/j.tins.2003.08.006 [DOI] [PubMed] [Google Scholar]
- Ruscher K, Shamloo M, Rickhag M, Ladunga I, Soriano L, Gisselsson L, Toresson H, Ruslim-Litrus L, Oksenberg D, Urfer R, Johansson BB, Nikolich K, Wieloch T (2011) The sigma-1 receptor enhances brain plasticity and functional recovery after experimental stroke. Brain 134:732–746. 10.1093/brain/awq367 [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68:821–861. 10.1146/annurev.biochem.68.1.821 [DOI] [PubMed] [Google Scholar]
- Su C, Underwood W, Rybalchenko N, Singh M (2011) ERK1/2 and ERK5 have distinct roles in the regulation of brain-derived neurotrophic factor expression. J Neurosci Res 89:1542–1550. 10.1002/jnr.22683 [DOI] [PubMed] [Google Scholar]
- Swanson RA, Ying W, Kauppinen TM (2004) Astrocyte influences on ischemic neuronal death. Curr Mol Med 4:193–205. 10.2174/1566524043479185 [DOI] [PubMed] [Google Scholar]
- Sweatt JD. (2001) The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem 76:1–10. 10.1046/j.1471-4159.2001.00054.x [DOI] [PubMed] [Google Scholar]
- Takano T, Oberheim N, Cotrina ML, Nedergaard M (2009) Astrocytes and ischemic injury. Stroke 40 [Suppl 3]:S8–S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Emery JF, Ouyang YB, Voloboueva LA, Giffard RG (2010) Astrocyte targeted overexpression of Hsp72 or SOD2 reduces neuronal vulnerability to forebrain ischemia. Glia 58:1042–1049. 10.1002/glia.20985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Dai H, Ding T, Dai A, Zhang F, Yu L, Chen G, Chen Z (2011) Effects of dexmedetomidine on the release of glial cell line-derived neurotrophic factor from rat astrocyte cells. Neurochem Int 58:549–557. 10.1016/j.neuint.2011.01.013 [DOI] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA (2012) Genomic analysis of reactive astrogliosis. J Neurosci 32:6391–6410. 10.1523/JNEUROSCI.6221-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito A, Cartelli D, Cappelletti G, Cariboni A, Andrews W, Parnavelas J, Poletti A, Galbiati M (2014) Neuritin 1 promotes neuronal migration. Brain Struct Funct 219:105–118. 10.1007/s00429-012-0487-1 [DOI] [PubMed] [Google Scholar]