

Abstract
Neuroinflammation is a major hallmark of amyotrophic lateral sclerosis (ALS), which is currently untreatable. Several anti-inflammatory compounds have been evaluated in patients and in animal models of ALS, but have been proven disappointing in part because effective targets have not yet been identified. Cyclophilin A, also known as peptidylprolyl cis-/trans-isomerase A (PPIA), as a foldase is beneficial intracellularly, but extracellularly has detrimental functions. We found that extracellular PPIA is a mediator of neuroinflammation in ALS. It is a major inducer of matrix metalloproteinase 9 and is selectively toxic for motor neurons. High levels of PPIA were found in the CSF of SOD1G93A mice and rats and sporadic ALS patients, suggesting that our findings may be relevant for familial and sporadic cases. A specific inhibitor of extracellular PPIA, MM218, given at symptom onset, rescued motor neurons and extended survival in the SOD1G93A mouse model of familial ALS by 11 d. The treatment resulted in the polarization of glia toward a prohealing phenotype associated with reduced NF-κB activation, proinflammatory markers, endoplasmic reticulum stress, and insoluble phosphorylated TDP-43. Our results indicates that extracellular PPIA is a promising druggable target for ALS and support further studies to develop a therapy to arrest or slow the progression of the disease in patients.
SIGNIFICANCE STATEMENT We provide evidence that extracellular cyclophilin A, also known as peptidylprolyl cis-/trans-isomerase A (PPIA), is a mediator of the neuroinflammatory reaction in amyotrophic lateral sclerosis (ALS) and is toxic for motor neurons. Supporting this, a specific extracellular PPIA inhibitor reduced neuroinflammation, rescued motor neurons, and extended survival in the SOD1G93A mouse model of familial ALS. Our findings suggest selective pharmacological inhibition of extracellular PPIA as a novel therapeutic strategy, not only for SOD1-linked ALS, but possibly also for sporadic ALS. This approach aims to address the neuroinflammatory reaction that is a major hallmark of ALS. However, given the complexity of the disease, a combination of therapeutic approaches may be necessary.
Keywords: amyotrophic lateral sclerosis, cyclophilin A, neuroinflammation
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating, incurable neurodegenerative disease that primarily affects motor neurons in the brain and spinal cord. Most cases are sporadic, with unknown etiology. Approximately 10% have a family history of a genetically dominant disorder in which mutations in Cu/Zn superoxide dismutase (SOD1) and C9ORF72 genes are the most common cause. Independently from the etiology, it is now established that ALS is a multifactorial disease involving different pathogenic mechanisms that require multiple non-neuronal cells for rapid disease progression and motor neuron death (Robberecht and Philips, 2013). In particular, astrocytes and microglia, in association with the neurodegenerative process, acquire a neuroinflammatory phenotype and elicit neuroinflammatory processes that contribute actively to motor neuron degeneration through a non-cell-autonomous mechanism (Boillée et al., 2006; Yamanaka et al., 2008).
Several compounds with anti-inflammatory properties have been evaluated in the mutant SOD1 mouse model of familial ALS. Many of those that had a positive effect on astrocytosis and microgliosis delayed the disease onset, but had only a mild or no effect on its progression (Drachman et al., 2002; Kriz et al., 2002; Van Den Bosch et al., 2002; Schütz et al., 2005; Kiaei et al., 2006; Neymotin et al., 2009). More promising results have been obtained in SOD1G93A mice by transgenic inhibition of microglial NF-kB (Frakes et al., 2014), the master regulator of the inflammatory response, and by knocking out matrix metalloproteinase 9 (MMP-9) (Kaplan et al., 2014), a NF-κB transcriptionally activated gene that contributes to the neuroinflammatory response in many neurological diseases (Yong et al., 2001).
Cyclophilin A, also known as peptidylprolyl cis-/trans-isomerase A (PPIA), is an enzyme acting as an acceleration factor in protein folding and assembly and is the main target of the immunosuppressive drug cyclosporine A (CsA) (Fischer et al., 1984; Handschumacher et al., 1984). It is expressed ubiquitously and abundantly, with the highest expression in the CNS (Göldner and Patrick, 1996). It is mainly cytoplasmic, but is also secreted extracellularly by different cell types, including neurons (Fauré et al., 2006). Its secretion in several cases is constitutive and increases under stress and/or pathological conditions (Hoffmann and Schiene-Fischer, 2014). Intracellular PPIA is beneficial: it protects cells from oxidative stress in various ways (Jäschke et al., 1998; Lee et al., 2001) and mitigates toxicity induced by mutant SOD1 protein aggregates (Lee et al., 1999). We reported recently that PPIA regulates key TAR DNA-binding protein 43 (TDP-43) functions, including the regulation of genes involved in the clearance of protein aggregates (Lauranzano et al., 2015). Moreover, knocking out PPIA exacerbated aggregation and accelerated disease progression in the SOD1G93A mouse model. However, extracellular PPIA has some detrimental functions, which are mediated by the extracellular matrix metalloproteinase inducer (EMMPRIN) receptor, also known as CD147/basigin, and depends on its peptidylprolyl cis-/trans-isomerase (PPIase) activity (Yurchenko et al., 2002; Malešević et al., 2013). It shows proinflammatory cytokine-like behavior, is a potent leukocyte chemoattractant and elicits inflammatory responses in vivo (Sherry et al., 1992; Xu et al., 1992). It induces the expression of matrix metalloproteinases (MMPs) and proinflammatory cytokines (Kim et al., 2005; Satoh et al., 2009; Seizer et al., 2010). Accordingly, PPIA has been linked to a number of human diseases (Nigro et al., 2013). Selective extracellular PPIA inhibitors have been introduced recently (Malešević et al., 2010; Malešević et al., 2013). These inhibitors are CsA derivatives, cell impermeable, and non-immunosuppressive and reduced the EMMPRIN-mediated effects of extracellular PPIA in a number of mouse models of chronic and acute inflammatory conditions by inhibiting its PPIase activity (Hoffmann and Schiene-Fischer, 2014).
We identified PPIA as an hallmark of disease in peripheral blood mononuclear cells and spinal cord of sporadic ALS patients and mutant SOD1 animal models (Massignan et al., 2007; Basso et al., 2009; Nardo et al., 2011). Here, we report that, under ALS conditions, high levels of extracellular PPIA exert a toxic effect specifically toward motor neurons through an EMMPRIN-dependent pathway. We have therefore developed a therapeutic strategy that, by inhibiting exclusively PPIA extracellularly, protects motor neurons and reduces the neuroinflammatory response.
Materials and Methods
Antibodies.
Antibodies for immunoblot (Western/dot blot) (IB), immunohistochemistry (IH), and immunocytochemistry (IC) were as follows: rabbit polyclonal anti-choline acetyltransferase (ChAT) antibody (1:1000 for IH; Immunological Science); mouse monoclonal anti-glial fibrillary acidic protein (GFAP) antibody (1:1000 for IH; Millipore, RRID:AB_94844); rat polyclonal anti-CD11b antibody (1:800 for IH; Millipore); rat monoclonal anti-CD68 (1:200 for IH; Serotec, RRID:AB_322219); mouse monoclonal anti-lamin A/C antibody (1:500 for IB; Millipore, RRID:AB_94752); mouse monoclonal anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (1:10000 for IB; Millipore, RRID:AB_10615768); rabbit polyclonal anti-mitochondrial import receptor subunit TOM20 (1:1000 for IB; Santa Cruz Biotechnology, RRID:AB_2207533); mouse monoclonal anti-cytochrome C (1:500 for IB; BD Biosciences, RRID:AB_396417); rabbit polyclonal anti-PPIA antibody (1:2500 for IB; 1:1000 for IH; Millipore, RRID:AB_2252847); mouse monoclonal anti-SMI32 antibody (1:2500 for IC; Covance, RRID:AB_509997); mouse monoclonal anti-NeuN antibody (1:250 for IC; Millipore, RRID:AB_2298772); goat polyclonal anti-mouse EMMPRIN antibody (1:1000 for IB; 1:500 for IC; Santa Cruz Biotechnology, RRID:AB_2066959); rabbit polyclonal anti-apoptosis-inducing factor (AIF) antibody (1:1000 IB; Cell Signaling Technology, RRID:AB_2224542); rabbit polyclonal anti-tumor necrosis factor α (TNFα) antibody (1:500 for IB; Abcam, RRID:AB_778525); mouse monoclonal anti-nitrotyrosine antibody (1:1000 for IB; Hycult Biotechnology, RRID:AB_533156); rabbit polyclonal anti-NF-κB p65 subunit antibody (1:1000 for IB; Cell Signaling Technology, RRID:AB_330561); rabbit polyclonal anti-phospo-NF-κB p65 (Ser536) antibody (1:1000 for IB; Cell Signaling Technology); rabbit polyclonal anti-78 kDa glucose-regulated protein (BiP) (1:500 for IB; Santa Cruz Biotechnology); mouse monoclonal anti-phospho Ser409/410 TDP-43 antibody (1:2000 for IB; Cosmo Bio, RRID:AB_1961900); goat anti-mouse or anti-rabbit peroxidase-conjugated secondary antibodies (1:5000 for IB; Santa Cruz Biotechnology); goat anti-rat, anti-mouse, or anti-rabbit biotinylated secondary antibodies (1:200 for IH; 1:500 for IC; Vector Laboratories); goat anti-rat biotinylated antibody followed by fluorescent signal coupling with streptavidine TSA amplification kit (cyanine 5, PerkinElmer); and goat Alexa Fluor 647 or 597 or 488 anti-mouse or anti-rabbit fluorophore-conjugated secondary antibodies (1:500 for IH and IC; Invitrogen).
Human samples.
The study with human samples was approved by the ethical committees of the Second University of Naples, Naples, Italy, and written informed consent was obtained from all participating subjects. CSF samples were from 28 sporadic ALS patients (13 males and 15 females) with definite ALS according to revised El Escorial criteria, aged 26–79 years (mean ± SD: 56 ± 12), with a duration of the disease of 11–48 months (mean ± SD: 20 ± 10) at the time of the lumbar puncture. Control CSF samples were from 28 patients (13 males and 15 females), aged 28–84 years (mean ± SD: 56 ± 14) with neurological conditions that do not result in neurodegeneration: multiple sclerosis (n = 16), chronic inflammatory demyelinating polyneuropathy (n = 4), hydrocephalus (n = 3), pseudotumor cerebri (n = 1), neoplasia (n = 1), vasculitis (n = 1), neurosyphilis (n = 1), and encephalitis (n = 1). CSF samples were collected, centrifuged at 450 × g for 10 min, and stored at −80°C.
Animal models.
Procedures involving animals and their care were conducted in conformity with the following laws, regulations, and policies governing the care and use of laboratory animals: Italian Governing Law (D.lgs 26/2014; Authorization 19/2008-A issued March 6, 2008 by Ministry of Health); Mario Negri Institutional Regulations and Policies providing internal authorization for persons conducting animal experiments (Quality Management System Certificate, UNI EN ISO 9001:2008, Reg. No. 6121); the National Institutes of Health's Guide for the Care and Use of Laboratory Animals (2011 edition), and European Union directives and guidelines (EEC Council Directive, 2010/63/UE). The Statement of Compliance (Assurance) with the Public Health Service (PHS) Policy on Human Care and Use of Laboratory Animals has been reviewed recently (9/9/2014) and will expire on September 30, 2019 (Animal Welfare Assurance #A5023-01). Animals were bred and maintained at the IRCCS–Istituto di Ricerche Farmacologiche Mario Negri, Milano, Italy, under standard conditions: temperature 21 ± 1°C, relative humidity 55 ± 10%, 12 h light schedule, and food and water ad libitum. Before every analysis, animals were deeply anesthetized with ketamine hydrochloride (IMALGENE, 100 mg/kg; Alcyon Italia) and medetomidine hydrochloride (DOMITOR, 1 mg/kg; Alcyon Italia) by intraperitoneal injection and killed by decapitation. Mice of the B6.Cg-Tg(SOD1-G93A)1Gur/J strain (RRID:IMSR_JAX:004435) obtained from The Jackson Laboratory, which express ∼20 copies of mutant human SOD1G93A, were used in the preclinical study. Female mice have disease onset at 111 ± 7 d of age and survive up to 163 ± 3 d of age. SOD1G93A mice and corresponding nontransgenic littermates on a homogeneous C57BL/6JOlaHsd genetic background were used for in vitro studies as described previously (Basso et al., 2013; Tortarolo et al., 2015). SOD1G93A transgenic mice were identified by PCR on DNA tail biopsies. PPIA−/− mice (strain 129S6/SvEvTac Ppiatm1Lubn/Ppiatm1Lbn; stock no. 005320) were obtained from The Jackson Laboratory and crossbred with SOD1G93A mice on a homogeneous 129S2/SvHsd genetic background (Marino et al., 2015). To generate the double-transgenic SOD1G93APPIA−/− mice, SOD1G93APPIA+/− male mice were crossbred with female nontransgenic PPIA+/− mice and F2 progeny were used for the study; SOD1G93APPIA+/+ mice have an onset at 101 ± 3 d of age and survive up to 130 ± 10 d, whereas SOD1G93APPIA−/− mice have an onset at 100 ± 3 d of age and survive up to 122 ± 8 d (Lauranzano et al., 2015). Genotyping for PPIA and SOD1G93A was done by standard PCR using primer sets designed by The Jackson Laboratory. Transgenic rats expressing ∼64 copies of mutant human SOD1G93A originally generated by Howland et al. (2002) were obtained from Taconic. Nontransgenic and SOD1G93A animals were bred and maintained on a Sprague Dawley rat strain. SOD1G93A rats were killed at 14–15 weeks of age (presymptomatic), 18–20 weeks of age (onset, when first symptoms of muscular dysfunction appeared), and 21–23 weeks of age (end stage). CSF was collected from the cisterna magna of mice and rats, centrifuged at 13.500 × g for 5 min at 4°C, and the supernatant was stored at −80°C until analysis.
Motor neurons.
Motor neurons were prepared as described previously (Sahawneh et al., 2010). Briefly, isolated ventral spinal cords from 15-d-old rat embryos were trypsinized and the tissue was disaggregated. The cell suspension was centrifuged on top of a 6% OptiPrep cushion (Sigma-Aldrich) and the motor neurons fraction was removed from the interface. Motor neurons were further purified by magnet-assisted cell separation (Miltenyi Biotec) using IgG 192 antibody against p75 low-affinity neurotrophin receptor (Millipore Bioscience Research Reagents). Motor neurons were plated on 96-well plates coated with poly-l-ornithine (Sigma-Aldrich) and laminin (Sigma-Aldrich) at a density of 500 cells/well. Motor neurons were maintained in Neurobasal medium supplemented with B27, heat-inactivated horse serum, glutamine, glutamate, 2-mercaptoethanol (all from Invitrogen), and trophic factors (1 ng/ml brain-derived neurotrophic factor, 0.1 ng/ml glial-derived neurotrophic factor, 10 ng/ml cardiotrophin-1) and incubated in a 5% CO2 humidified atmosphere at 37°C.
Cortical neurons.
Cortical neurons were prepared as described previously (Restelli et al., 2010). Briefly, cortices from 2-d-old animals were sliced into ∼1 mm pieces and incubated in cortical neuron dissociation medium (5.8 mm MgCl2, 0.5 mm CaCl2, 3.2 mm HEPES, 0.2 mm NaOH, 30 mm K2SO4, 0.5 μg/ml phenol red, pH 7.4; 292 mOsmol) containing 20 U/ml papain (Sigma-Aldrich) at 34°C for 30 min. Trypsin inhibitor (Sigma-Aldrich) was added to a final concentration of 0.5 mg/ml and the tissue was dissociated mechanically by passing through a flame-polished Pasteur pipette. Cells were plated at 150–250,000 cells/cm2 on poly-d-lysine-coated (25 μg/ml) plates and maintained in Neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen), penicillin/streptomycin, and glutamine 2 mm. To reduce the number of non-neuronal cells, aphidicolin (3.3 μg/ml; Sigma-Aldrich) was added to the medium 48 h after plating.
Primary astrocyte cultures.
Primary SOD1G93A and nontransgenic astrocytes were prepared as described previously (Basso et al., 2013). Briefly, cortices from 14-d-old transgenic mouse embryos were dissected and mechanically dissociated in HBSS containing 33 mm glucose. After centrifugation, the pellet was resuspended in culture medium prepared with DMEM/F12 containing 2 mm l-glutamine, 33 mm glucose, 5 μg/ml gentamycin, and 10% heat-inactivated horse serum and seeded (500,000 cells/ml) on 48-well plates (for coculture preparation) or 6-well plates (for conditioned medium analysis) coated with 1.5 μg/ml poly-l-ornithine and then treated with 10 μm AraC once they reached confluence. Collection of conditioned media was done essentially as described previously (Basso et al., 2013).
Primary microglial cultures.
Primary microglial cultures were derived from the cell preparation for primary astrocyte cultures. Once confluence was reached, mixed glial cells were washed with HBSS and shaked overnight at 200 rpm. Supernatant was collected and centrifuged to obtain microglia. The pellet was resuspended in culture medium prepared with 50% astrocyte conditioned medium, previously filtrated, and 50% of DMEM/F12 containing 2 mm l-glutamine, 33 mm glucose, 5 μg/ml gentamycin, and 10% heat-inactivated horse serum and seeded (500,000 cells/ml) onto 6-well plates coated with 1.5 μg/ml poly-l-ornithine. After 1 week in culture, fresh medium was conditioned for 24 h and collected. Conditioned media were centrifuged at 12,000 × g for 5 min at 4°C, as preclearing, and analyzed by dot blot.
Primary astrocyte–spinal neuron cocultures.
SOD1G93A and nontransgenic astrocyte–spinal neuron cocultures were prepared as described previously (Basso et al., 2013; Tortarolo et al., 2015). Briefly, spinal cords from 14-d-old embryos were dissected and dissociated mechanically in HBSS with 33 mm glucose. The cells were centrifuged onto a 4% BSA cushion and the pellet was resuspended in neuron culture medium: Neurobasal (Invitrogen), 2 mm l-glutamine, 33 mm glucose, 5 μg/ml gentamycin, 1 ng/ml brain-derived neurotrophic factor, 25 μg/ml insulin, 10 μg/ml putrescine, 30 nm sodium selenite, 2 μm progesterone, 100 μg/ml apo-transferrin, 10% heat-inactivated horse serum, and 10 μm AraC. Cells were seeded (1,000,000 cells/ml) on a preestablished astrocyte confluent layer. Cells were generally fixed with 4% paraformaldehyde (Merck) solution and after 3 washes in PBS 1 × were stored a 4°C. Conditioned medium was collected after 24 h or 6 d in culture.
Immunohistochemistry.
Mice were anesthetized and perfused transcardially with 50 ml of PBS followed by 100 ml of 4% paraformaldehyde (Sigma-Aldrich) solution in PBS. Spinal cords were rapidly removed, postfixed for 3 h, transferred to 20% sucrose in PBS overnight and then to 30% sucrose solution until they sank, frozen in N-pentane at −45°C and stored at −80°C. Before freezing, spinal cord was divided into cervical, thoracic, and lumbar segments and included in Tissue-tec OCT compound (Sakura). Coronal sections (30 μm) of lumbar spinal cord tract were subsequently sliced from L1 to L2 and analyzed. Immunohistochemistry for PPIA, ChAT, GFAP, and CD11b and CD68 was done as described previously (Tortarolo et al., 2003; Tortarolo et al., 2006). Sections were also stained with 0.5% cresyl violet to detect the Nissl substance of neuronal cells. Sections were examined under an Olympus BX61 light microscope. Images were collected with a camera using AnalySIS software (Soft Imaging Systems version 3.2). ChAT-immunopositive and Nissl-stained cells were counted in 20 slices, one every five sections (segments L1–L2), using TissueQuest analysis software (TissueGnostic) (Bigini et al., 2010). The number of motor neurons was calculated for each hemisection and the means used for statistical analysis. GFAP, CD11b, and CD68 signals were analyzed in six slices, one every 10 sections (segments L1–L2), using ImageJ software (Perego et al., 2011). CD11b- and CD68-stained sections were collected at 20 × by an Olympus BX-61 Virtual Stage microscope so to have complete stitching of the whole section, with a pixel size of 0.346 μm. Acquisition was done over 6-μm-thick stacks with a step size of 2 μm. The different focal planes were merged into a single stack by mean intensity projection to ensure consistent focus throughout the sample. CD11b and CD68 signals were analyzed for each hemisection over the whole gray or white matter by ImageJ software. Morphology of CD11b cells was analyzed based on circularity, solidity, and grid crossing (Zanier et al., 2015). Briefly, once segmented, the objects meeting the minimum size to be analyzed (35 μm2) were measured for circularity and solidity, both ranging from 0 (linear polygon) to 1 (perfect circular object). Mean single cell values for each parameter were used for statistics. To calculate grid crossings, segmented objects were superimposed on a grid image with horizontal and vertical lines distanced by 9 μm. The total number of object crossings on the grid was quantified and normalized for the total number of segmented objects; therefore, more crossings indicate more ramifications per cell.
Western/dot blot.
For WB, samples (30 μg) were separated in 12% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Millipore) as described previously (Basso et al., 2009). For dot blot, proteins (3 μg) were loaded directly onto nitrocellulose Trans-blot transfer membranes (0.2–0.45 μm; Bio-Rad) were done by depositing each sample on the membrane by vacuum filtration, as described previously (Massignan et al., 2007; Basso et al., 2009; Nardo et al., 2011). Dot blot was used when several samples were analyzed in parallel and exclusively after verification that the antibody used detected specific bands in WB. WB and dot blot membranes were blocked with 3% (w/v) BSA (Sigma-Aldrich) and 0.1% (v/v) Tween 20 in Tris-buffered saline, pH 7.5, and incubated with primary antibodies and then with peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology). Blots were developed with the Luminata Forte Western Chemiluminescent HRP Substrate (Millipore) on the ChemiDoc XRS system (Bio-Rad). Densitometry was done with Progenesis PG240 version 2006 software (Nonlinear Dynamics). The immunoreactivity of the different proteins was normalized to Ponceau Red staining (Fluka).
ELISA of PPIA.
The level of PPIA in conditioned culture medium and CSF was measured by ELISA kits for the mouse protein and the human protein (Wuhan USCN Business) following manufacturer's instructions.
AlphaLISA of MMP-9.
An AlphaLISA mouse MMP-9 Kit (PerkinElmer) was used to measure MMP-9 in lumbar spinal cord of mice and astrocyte–spinal neuron coculture conditioned medium. Lumbar spinal cord tissues were homogenized in lysis buffer (10 mm Tris-HCl, pH 7.6, 250 mm sucrose, 1 mm EDTA and 0.1 mm PMSF) and centrifuged at 845 × g for 20 min at 4°C; then, the supernatant was collected and analyzed according to the manufacturer's instructions. The signals were normalized to the total amount of protein as quantified by BCA protein assay (Pierce). Astrocyte–spinal neuron coculture medium was collected after 6 d of conditioning, centrifuged at 200 × g for 10 min to remove cell debris, and analyzed. AlphaLISA signals were measured using an Ensight Multimode Plate Reader (PerkinElmer).
Subcellular fractionation.
Mouse lumbar spinal cords were homogenized in buffer A (10 mm Tris-HCl, pH 7.4, 5 mm MgCl2, 25 mm KCl, 0.25 m sucrose, 0.5 mm DTT) containing a protease inhibitor mixture (Roche) and centrifuged at 800 × g for 10 min at 4°C. The supernatant was centrifuged twice at 800 × g for 10 min at 4°C (cytoplasmic fraction). The pellet was resuspended in 3 volumes of buffer A and centrifuged 3 times at 800 × g for 10 min at 4°C. The pellet was resuspended in one volume of buffer A and one volume of buffer B (10 mm Tris-HCl, pH 7.4, 5 mm MgCl2, 25 mm KCl, 2 m sucrose) containing a protease inhibitor mixture (Roche) and loaded on a layer of one volume of buffer B. Samples were ultracentrifuged at 100,000 × g for 45 min at 4°C. The pellet (nuclear fraction) was resuspended in 100 μl of buffer A, centrifuged at 800 × g for 10 min at 4°C, and resuspended in 40 μl of buffer A. GAPDH and lamin A/C were used, respectively, as cytoplasmic and nuclear markers. Cytochrome C and TOM20 were used as mitochondrial markers.
Cell treatments.
Motor neurons were treated at plating with 0.5 and 5 nm human recombinant PPIA (Sigma-Aldrich or R&D Systems) or purified from calf thymus (Sigma-Aldrich) dissolved in the culture medium and viability was assayed 24 h later. Cortical neurons were treated after 14 d in culture with 0.5 and 5 nm human recombinant PPIA (Sigma-Aldrich) dissolved in the culture medium and viability was assayed 24 and 72 h later. Nontransgenic astrocyte–spinal neuron cocultures were treated at neuron plating with 0.5 nm human recombinant PPIA (Sigma-Aldrich) or 0.5 nm MM218 (Malešević et al., 2010), dissolved in the culture medium or 0.5 nm CsA (Sigma-Aldrich), dissolved in ethanol, and then diluted with culture medium and cell viability was assayed 6 d later. SOD1G93A astrocyte–spinal neuron cocultures were treated at neuron plating with 0.5 nm human recombinant PPIA (Sigma-Aldrich) or 5 pm, 50 pm, 0.5 nm, 5 nm MM218, dissolved in the culture medium, and cell viability was assayed 6 d later.
Cell viability assays.
Rat motor neuron survival was determined by high-throughput image capture and analysis in 96-well plates using a Flash Cytometer (Trophos) as described previously (Sahawneh et al., 2010). Briefly, motor neurons plated in black 96-well plates (Greiner Bio-one) coated with poly-d-ornithine and laminin (Sigma-Aldrich) were treated with calcein-acetoxymethylester (calcein-AM; Invitrogen) in L15 medium (Invitrogen) for 1 h at 37°C. Fifteen microliters/well of 100 mg/ml hemoglobin (Sigma-Aldrich) in PBS 1 × was added to quench extracellular calcein and the plates were read using a Flash Cytometer.
Cortical neuron viability was assessed by measuring the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-dipheny ltetrazolium bromide (MTT) to formazan. Cells were incubated for 30 min at 37°C with 0.5 mg/ml MTT (Sigma-Aldrich). MTT was removed and cells were resuspended in DMSO and analyzed spectrophotometrically at 540 nm using automatic microplate reader (Infinite 200; Tecan).
Motor neuron survival in astrocyte–spinal neuron cocultures was determined by double immunocytochemistry for SMI32 and NeuN (Basso et al., 2013). Motor neuron survival was expressed as the ratio of the number of motor neurons (SMI32-positive cells) to the total neurons in the well (NeuN-positive cells). Wells were analyzed with an Olympus camera on a motorized microscope (Olympus). A reproducible grid of 9 × 9 frames (10 × enlargement) was created and 20 frames were acquired at 488 nm for NeuN and 648 nm for SMI32. NeuN-positive cells were automatically counted by ImageJ software and SMI32-positive cells were counted manually with Cell∧P software (Olympus), which identifies motor neurons as cells with extensive dendritic arborization and cell bodies ≥20 μm.
Preclinical study in the SOD1G93A mouse.
Starting at 98 d, SOD1G93A female mice received continuous intracerebroventricular infusions of vehicle (PBS) or MM218 inhibitor. A subcutaneously implanted osmotic minpump connected by polyvinylchloride tubing to a stainless steel cannula stereotaxically implanted into the lateral ventricle (Brain Infusion Kit 3; Alzet) was used. Mice received a subcutaneous dose (0.15 mg/kg body weight) of buprenorphine as analgesic immediately before and 12 h after the surgery. During the surgery, they were anesthesized by inhalational of isoflurane 3%. The minipump delivered vehicle or drug, 1 or 10 μm in the reservoir, for 28 d at a continuous rate of 0.11 μl/h. After 28 d, the minipump was replaced with another one to infuse the drug for a further 28 d. Pumps were weighed before implantation and at the end of the experiment to check complete delivery of their content. The effect of the MM218 inhibitor on the progression of the disease was assessed twice a week from 13 weeks of age on the basis of Rotarod performance (Pizzasegola et al., 2009). The Rotarod apparatus (Ugo Basile) was accelerated at a constant rate (4.2 rpm/min) from 7 to 28 rpm for a maximum of 5 min. The mice were given up to three attempts and the longest latency to fall was considered in statistical analysis. To observe the treatment effect at a late stage of the disease, paralysis was evaluated, assessed as the age at which the mice scored 0 in the Rotarod test, when the mice were not able to perform the test anymore. The mice were killed when they were unable to right themselves within 10 s after being placed on either side. The time was considered the end stage of the disease and was used to calculate survival. Life expectancy is considered the mean number of days of life remaining after beginning treatment.
Extraction and analysis of detergent-insoluble proteins.
Spinal cord tissues from treated and untreated SOD1G93A mice were processed as described previously (Basso et al., 2009). The fraction insoluble in 2% of Triton X-100 was resuspended in 50 mm Tris-HCl, pH 6.8, 1 mm DTT, and 2% SDS and analyzed by dot blot analysis. Immunoreactivity was normalized to protein loading (Ponceau red staining). The amount of Triton-resistant proteins isolated from the tissue was normalized to the soluble protein extracted. Proteins were quantified by the BCA protein assay (Pierce).
Results
PPIA is highly expressed by motor neurons and increases extracellularly in ALS
PPIA is highly concentrated in the brain and mainly localized in neurons (Göldner and Patrick, 1996). Figure 1A shows that PPIA is highly expressed especially by motor neurons, both in nontransgenic and SOD1G93A animals, as indicated by the intense PPIA immunoreactivity and clear-cut colocalization with the ChAT motor neuron marker in the mouse ventral horn spinal cord. PPIA was overexpressed in the spinal cord of SOD1G93A mice and rats compared with controls at a presymptomatic stage of the disease, specifically in lumbar spinal cord ventral horn (Nardo et al., 2011; Fig. 1A,B). As disease progresses, total PPIA protein level in ventral horns is apparently lower because of motor neuron death and colocalization with GFAP- and CD11b-positive cells is observed, especially at end-stage disease (Fig. 1C,D).
Figure 1.
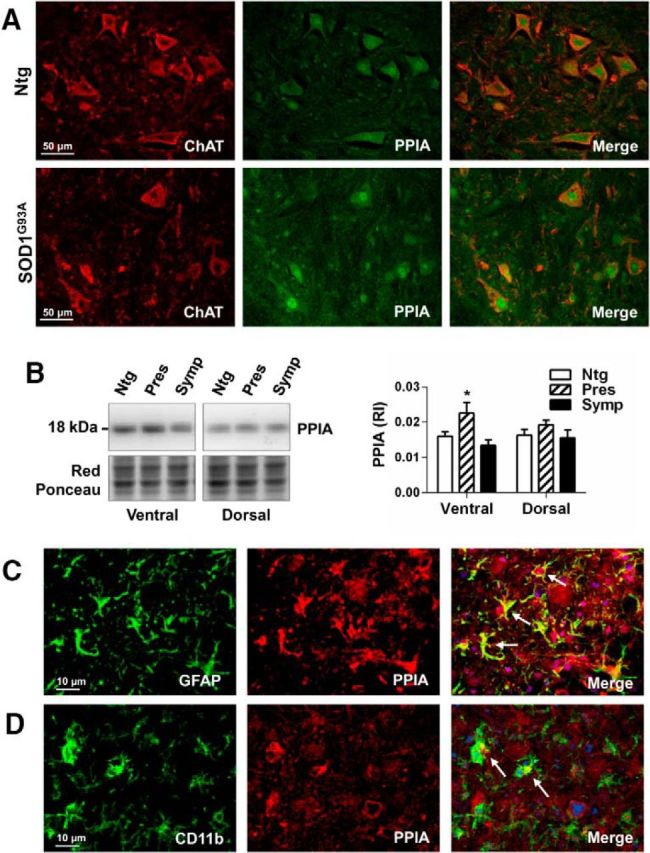
PPIA is upregulated in ALS. A, Representative confocal image of lumbar spinal cord ventral horns from nontransgenic (Ntg) and SOD1G93A mice (129Sv) at a presymptomatic stage of the disease (10 weeks) costained for ChAT (red staining) and PPIA (green staining). Scale bar, 50 μm. B, WB analysis for PPIA in lumbar spinal cord, ventral and dorsal horns, from Ntg and SOD1G93A mice (129Sv) at presymptomatic (Pres, 10 weeks of age) and symptomatic (Symp, 16 weeks of age) stages of the disease. Data, as relative immunoreactivities (RIs), are normalized to protein loading (Red Ponceau) and expressed as mean ± SEM (n = 4). *p < 0.05 versus Ntg and Symp by one-way ANOVA, Tukey's multiple-comparisons test. C, D, Colocalization of PPIA with astrocytes and microglia in lumbar spinal cord ventral horn from a SOD1G93A mouse at end stage (18 weeks of age). C, Representative confocal image of a section costained for GFAP (green staining), PPIA (red staining), and DAPI (blu staining). D, Representative confocal image of a section costained for CD11b (green staining), PPIA (red staining), and DAPI (blu staining). Scale bar, 10 μm. White arrows highlight colocalization between activated astrocytes or microglia and PPIA.
Oxidative stress and inflammatory conditions stimulate PPIA secretion (Sherry et al., 1992; Jin et al., 2000). PPIA levels were high in the conditioned medium of mouse glial cells, microglia (Fig. 2A), and astrocytes (Fig. 2B) and astrocyte–spinal neuron cocultures expressing SOD1G93A (Fig. 2C). High levels of PPIA were also found in the CSF of SOD1G93A mice and rats (Fig. 2D,E) compared with nontransgenic controls and of sporadic ALS patients compared with neurological controls (Fig. 2F). PPIA levels decrease in CSF at an advance stage of the disease, both in SOD1G93A mice and rats (Fig. 2D,E), and this correlates with the PPIA levels in the lumbar spinal cord ventral horn (Figs. 1B, 2G) and motor neuron death, confirming that PPIA is abundantly expressed by motor neurons and that these cells under ALS conditions contribute substantially to the high PPIA level extracellularly.
Figure 2.

High levels of extracellular PPIA under ALS conditions. A–C, PPIA levels in conditioned media of nontransgenic (Ntg) and SOD1G93A microglia (A), astrocytes (B), and astrocyte–spinal neuron cocultures (C) were analyzed by dot blot and immunoreactivity was normalized to total protein level in the cell lysates, assessed by BCA assay (A) or to protein loading (Red Ponceau; B, C). Data (mean ± SEM, n = 6) are shown as percentages of Ntg. *p < 0.05 by Student's t test. D, CSF samples from Ntg and SOD1G93A mice (B6.Cg) at the onset of symptoms (Onset, 16 weeks of age) and at a symptomatic stage (Symp, 20 weeks of age) of the disease were analyzed by ELISA. Each CSF sample is a pool from three mice. Data are shown as mean ± SEM (n = 6). *p < 0.05 versus Ntg and Symp by one-way ANOVA, Tukey's multiple-comparisons test. E, PPIA levels in CSF from Ntg and SOD1G93A rats were measured by WB at presymptomatic (Pres, 14–15 weeks of age; n = 8), onset (18–20 weeks of age; n = 6), and end (End, 21–23 weeks of age; n = 6) stages of the disease. Equal volumes of CSF samples (20 μl) were analyzed by WB. *p < 0.05 by one-way ANOVA, Fisher's least significant difference test. F, CSF samples from sporadic ALS patients (n = 28) and age- and sex-matched non-ALS neurological controls (n = 28) were analyzed by ELISA. *p < 0.05 by Student's t test. G, PPIA levels in lumbar spinal cord ventral horns from the same Ntg and SOD1G93A rats as in E were measured by dot blot. PPIA immunoreactivity was normalized to the actual amount of protein loaded (relative immunoreactivity, RI), detected after Red Ponceau staining. *p < 0.05 by one-way ANOVA, Fisher's least significant difference test.
Absence of PPIA preserves motor neurons in SOD1G93A mice
We crossbred SOD1G93A mice with PPIA−/− mice (Fig. 3A) and found that, in the absence of PPIA, motor neurons were protected, as assessed by counting the ChAT-positive neurons with a soma area > 250 μm2 in sections of ventral horn lumbar spinal cord (Fig. 3B). In particular, at the onset of symptoms, there was less motor neuron loss in SOD1G93APPIA−/− than in SOD1G93APPIA+/+ mice (22% versus 28%), with a greater difference at a later stage (28% vs 57%). In a PPIA−/− mouse model, neuroprotection has been linked to a decrease in PPIA-dependent apoptosis-inducing factor (AIF) translocation to the nucleus (Zhu et al., 2007). In the nuclear fraction, anti-AIF antibody detected two AIF isoforms, the active 57 kDa form and the 62 kDa mature protein, normally linked to the mitochondrial inner membrane, indicating that, in the nuclear fraction, there is a contamination of mitochondrial membrane components, confirmed by the presence of TOM20 (Fig. 3C,D). We indeed found a decrease in the nuclear translocation of the active 57 kDa-AIF form in the ventral horn lumbar spinal cord of SOD1G93APPIA−/− mice compared with SOD1G93APPIA+/+ mice. However, the effect was low and only at a symptomatic stage of the disease (Fig. 3C).
Figure 3.

The absence of PPIA preserves motor neurons in SOD1G93A mice. A, Representative images of Western blots for PPIA and actin from spinal cord of PPIA+/+, PPIA+/−, and PPIA−/− mice. B, Quantification of ChAT-stained motor neurons (MNs > 250 μm2) in lumbar spinal cord hemisections from SOD1G93APPIA+/+ and SOD1G93APPIA−/− (129Sv) mice at symptom onset (Onset, 14 weeks of age) and at a symptomatic (Symp, 16 weeks of age) stage and corresponding nontransgenic controls (PPIA+/+ and PPIA−/−). Data are expressed as mean ± SEM (n = 4); (ns) nonsignificant. *p < 0.05 by two-way ANOVA, Fisher's least significant difference test. Motor neuron loss as percentage of relative nontransgenic control is reported on the bar. C, Representative WB for AIF in the nuclear fraction of lumbar spinal cord tissue of SOD1G93APPIA+/+, SOD1G93APPIA−/− mice at symptom onset and symptomatic stages and corresponding nontransgenic controls (PPIA+/+ and PPIA−/−). The level of the active form of AIF (57 kDa) did not change at symptom onset (Onset, 14 weeks of age) and decreased in SOD1G93APPIA−/− mice compared with SOD1G93APPIA+/+ mice at a symptomatic (Symp, 16 weeks of age) stage. AIF immunoreactivity was normalized to protein loading (Red Ponceau) and to the mean number of ChAT-stained motor neurons (MNs > 250 μm2) in lumbar spinal cord hemisections. Data are shown as mean ± SEM (n = 3). *p < 0.05 by two-way ANOVA, Fisher's least significant difference test. D, Nuclear–cytoplasmic fractionation was evaluated using an array of markers: lamin A-C for the nucleus, GAPDH for the cytoplasm, TOM20 for the mitochondrial membrane component, and cytochrome C (CytC) for mitochondrial matrix. In the nuclear fraction, anti-AIF antibody detected two AIF isoforms, the active 57 kDa form and the 62 kDa mature protein, which are normally linked to the mitochondrial inner membrane, indicating that, in the nuclear fraction, there is a contamination of mitochondrial membrane components, which was confirmed by the presence of TOM20. E, AlphaLISA analysis of MMP-9 in lumbar spinal cord of SOD1G93APPIA−/− mice at presymptomatic (10 weeks of age) and at symptom onset (Onset, 14 weeks of age) stages compared with the corresponding nontransgenic (Ntg) controls showed lower expression of MMP-9 in the absence of PPIA. Data (mean ± SEM, n = 3), shown as percentages of the AlphaLISA signal counts in PPIA+/+, were normalized to protein concentration as quantified by BCA protein assay. *p < 0.05 by two-way ANOVA, Fisher's least significant difference test.
Extracellularly, PPIA interacting with the EMMPRIN receptor induces the expression of MMP-9 (Kim et al., 2005; Yuan et al., 2010), which can cause motor neuron death (Kaplan et al., 2014). We therefore measured the protein level of MMP-9 in lumbar spinal cord of SOD1G93APPIA−/− and SOD1G93APPIA+/+ mice. At a presymptomatic stage of the disease SOD1G93APPIA+/+ mice had a higher level of MMP-9 than nontransgenic controls, whereas SOD1G93APPIA−/− mice had a substantially lower level of MMP-9 than controls (Fig. 3E). At the onset of the disease, the level of MMP-9 was clearly high in ventral horn lumbar spinal cord of SOD1G93APPIA+/+ mice, whereas in the absence of PPIA, it was comparable to controls (Fig. 3E). This indicates that the absence of PPIA downregulates MMP-9 substantially and this could underlie the motor neuron protection in the SOD1G93APPIA−/− mice, as shown in SOD1G93A mice knocked out for MMP-9 (Kiaei et al., 2007; Kaplan et al., 2014). We conclude that the inhibition of specific extracellular functions of PPIA, for example, induction of MMP-9 expression through EMMPRIN receptor, may be a potential therapeutic strategy.
Motor neurons are vulnerable to extracellular PPIA toxicity
To test whether extracellular PPIA is toxic for motor neurons, we treated motor neuron cultures purified from nontransgenic and SOD1G93A rats with human recombinant PPIA. In both conditions, PPIA induced motor neuron death (Fig. 4A,B). Results were similar with different batches of recombinant protein and with the purified protein from calf thymus (data not shown). In contrast, human recombinant PPIA had no toxic effect on cortical (Fig. 4C) and cerebellar granule neurons (data not shown). We also tested the effect of PPIA in a more complex in vitro paradigm, astrocyte–spinal neuron cocultures expressing or not SOD1G93A, that is, spinal cord neurons, including large motor neurons and smaller rounded neurons, seeded on a preestablished astrocyte layer (Tortarolo et al., 2015). We found that recombinant PPIA at 0.5 nm, the concentration detected in the CSF of SOD1G93A mice at the onset of the disease, induced 30% motor neuron death in nontransgenic cocultures and a further loss compared with untreated cocultures in those expressing mutant SOD1 (Fig. 4D), whereas we detected no toxicity in NeuN-positive neuronal cells (data not shown), indicating that PPIA is specifically toxic for motor neurons. EMMPRIN is highly expressed by large motor neurons, as indicated by the intense EMMPRIN immunoreactivity and colocalization with SMI32 motor neuron marker (Fig. 4E). To look for possible mechanisms of the specific toxic effect toward motor neurons, we measured the protein level of EMMPRIN receptor in lumbar spinal cord of SOD1G93A mice and nontransgenic controls, in ventral horns, rich in motor neurons, and dorsal horns (Fig. 4F). The expression of EMMPRIN receptor was substantially higher in ventral horns compared with dorsal horns in nontransgenic mice, with a remarkable increase in mutant SOD1 mice at a symptomatic stage of the disease. Because, at this stage, there is significant motor neuron loss and gliosis, upregulation is also associated with glial cells, as described previously (Agrawal et al., 2011). Therefore, higher expression of EMMPRIN may underlie the toxic effect of extracellular PPIA toward motor neurons by a non-cell-autonomous mechanism.
Figure 4.
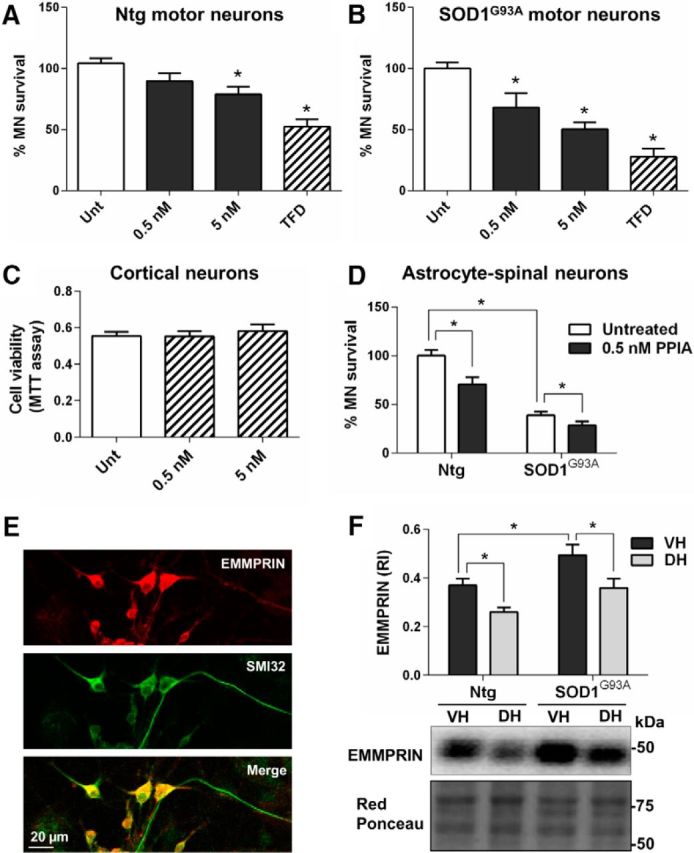
Exogenous PPIA is specifically toxic for motor neurons. A, B, Motor neuron survival was investigated in rat purified motor neurons (MNs) nontransgenic (Ntg; 16–19 wells per condition; A) and expressing SOD1G93A (4 per condition; B) treated for 24 h with 0.5 and 5 nm human recombinant PPIA. Data (mean ± SEM) are percentages of untreated (Unt). TFD, Trophic factor deprivation. *p < 0.05 compared with untreated (Unt) by one-way ANOVA, Dunnett's multiple-comparisons test. C, MTT assay performed in mouse cortical neurons treated for 72 h with 0.5 and 5 nm human recombinant PPIA revealed no change in cell viability compared with untreated (Unt). Results were similar after a 24 h of treatment. Data are expressed as mean ± SEM (n = 5 wells per condition). D, Motor neuron survival decreased both in nontransgenic (Ntg) and SOD1G93A expressing astrocyte–spinal neuron cocultures treated for 6 d with 0.5 nm human recombinant PPIA. Data (mean ± SEM, n = 6 wells per condition) are shown as the percentage of Ntg untreated. *p < 0.05 by one-way ANOVA, Tukey's multiple-comparisons test. E, Representative confocal image of primary spinal neuron culture costained for EMMPRIN (red staining) and SMI32 (green staining). Scale bar, 20 μm. F, Expression of EMMPRIN receptor was increased in ventral horn (VH) lumbar spinal cord of both nontransgenic (Ntg) and SOD1G93A mice (129Sv) at a symptomatic stage (16 weeks of age) compared with dorsal horn (DH) lumbar spinal cord. EMMPRIN immunoreactivity was normalized to protein loading (Red Ponceau). Data, shown as relative immunoreactivity (RI), are expressed as mean ± SEM (n = 5). *p < 0.05 by one-way ANOVA, Fisher's least significant difference test.
MM218 extracellular PPIA inhibitor protects motor neurons in an in vitro paradigm of ALS
To further assess whether extracellular PPIA could be a good therapeutic target for ALS, we tested the effect of a specific inhibitor, MM218 (Malešević et al., 2010), on astrocyte–spinal neuron cocultures expressing SOD1G93A, in which there is ∼50% spontaneous loss of motor neurons after 6 d in culture (Basso et al., 2013; Tortarolo et al., 2015). MM218 is a CsA derivative designed to be cell impermeable and therefore not as toxic for motor neuronal cultures as standard CsA (Van Den Bosch et al., 2004). We treated nontransgenic astrocyte–spinal neuron cocultures for 6 d to verify this. MM218 has an higher affinity for PPIA than CsA, with Ki values of 1.8 ± 0.6 and 8.4 ± 2.5 nm, respectively (Malešević et al., 2010). Nevertheless, CsA at 5 nm induced motor neuron death, whereas MM218 at the same concentration did not (Fig. 5A), confirming that general inhibition of PPIA, intracellularly and extracellularly, is not beneficial for motor neurons. We then treated SOD1G93A astrocyte–spinal neuron cocultures with the CsA derivative and determined motor neuron survival after 6 d in culture (Fig. 5B). Because the extracellular PPIA concentration in the culture medium was in the picomolar range (data not shown), we used MM218 at 5 pm to 0.5 nm. At 50 pm and 0.5 nm concentrations, MM218 fully rescued SOD1G93A motor neurons. Neurons were not affected, as detected by NeuN staining (data not shown).
Figure 5.
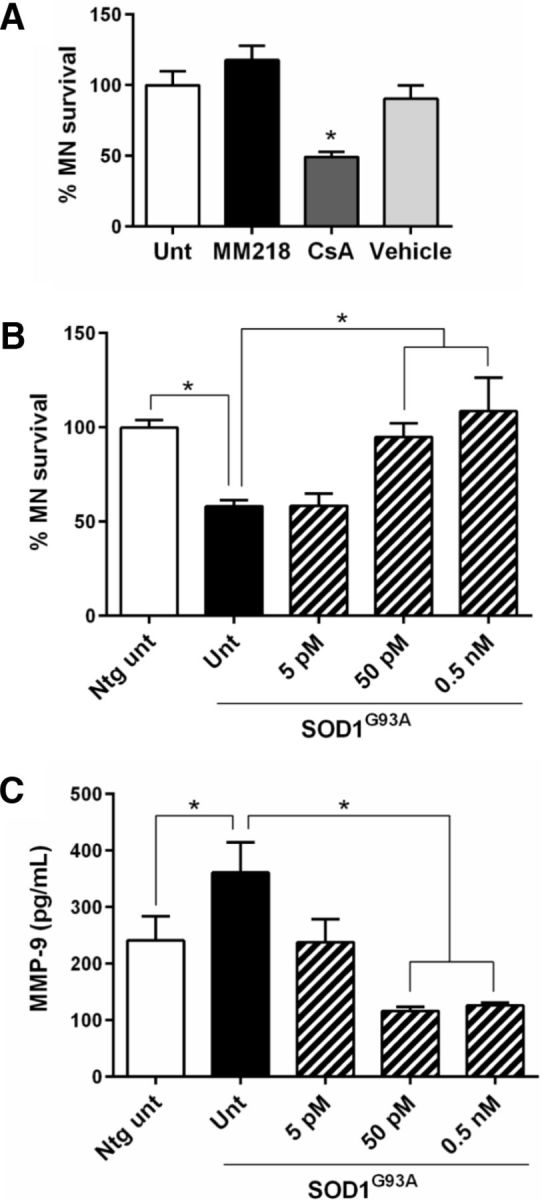
MM218 rescues motor neurons in SOD1G93A astrocyte–spinal neuron cocultures through inhibition of EMMPRIN activation. A, Nontransgenic astrocyte–spinal neuron cocultures treated for 6 d with 5 nm MM218 or CsA or ethanol (Vehicle). CsA, but not MM218, was toxic to motor neurons (MNs). Data (mean ± SEM, 6–8 wells per condition) are percentages of untreated (Unt). *p < 0.05 versus Unt by one-way ANOVA, Bonferroni's multiple-comparisons test. B, SOD1G93A astrocyte–spinal neuron cocultures treated for 6 d with increased concentrations of MM218. Data (mean ± SEM, n = 6–8 wells per condition) are percentages of nontransgenic untreated cocultures (Ntg Unt). *p < 0.05 by one-way ANOVA, Bonferroni's multiple-comparisons test. C, Levels of MMP-9 were measured by AlphaLISA in the medium of SOD1G93A astrocyte–spinal neuron cocultures treated for 6 d with increasing concentrations of MM218. Data (mean ± SEM, 6–8 wells per condition) are in picograms per milliliter. *p < 0.05 by one-way ANOVA, Fisher's least significant difference test. Experiments in A–C were repeated several times, with consistent results.
To test whether the protective effect of MM218, through specific inhibition of extracellular PPIA, was associated with less activation of EMMPRIN and a consequent decrease in MMP-9 induction, we measured MMP-9 protein levels in the coculture medium (Fig. 5C). There was a high level of MMP-9 in untreated cocultures expressing SOD1G93A compared with nontransgenic controls. The most effective MM218 concentrations, 50 pm and 0.5 nm, caused a significant decrease in MMP-9 in the conditioned medium compared with untreated transgenic cocultures. No effect on PPIA levels was observed (data not shown). We therefore concluded that MM218 had a protective effect in an in vitro paradigm of ALS through inhibition of EMMPRIN activation.
MM218 reduces neuroinflammation and prolongs survival in the SOD1G93A mouse model
To explore the therapeutic potential of MM218 in vivo, we ran a proof-of-concept preclinical trial in SOD1G93A mice. MM218 does not pass the blood–brain barrier (BBB), so the drug was administered intracerebroventricularly by continuous infusion using minipumps. We treated animals with MM218, 1 and 10 μm (in the minipump), which we estimated to correspond to low nanomolar concentrations in the CSF and a dose of 0.3–3.0 mg/kg /d, for 56 d starting from 98 d of age. We evaluated the effect of the extracellular PPIA inhibitor on the progression of the disease in SOD1G93A mice, considering survival and motor function after treatment (Fig. 6A–D). Both concentrations prolonged mean survival compared with the mice treated with vehicle (vehicle-treated 163 ± 10 d; 1 μm-treated 169 ± 9 d; 10 μm-treated 174 ± 7 d), but the effect was statistically significant only at 10 μm (Fig. 6A,B). When 50% of the vehicle-treated mice had been killed (7/13), 80% (10/12) of the MM218-treated mice were still alive, with the most long-lived surviving up to 183 d. There was a dose–response relationship, with increased survival with the higher drug concentration (Fig. 6B). At the highest dose, the treatment increased life expectancy by 17%. Unlike CsA, MM218 had no toxic effect after chronic intracerebroventricular infusion (Keep et al., 2001). Mice treated with 10 μm MM218 showed a tendency to better motor performance, as assessed by the Rotarod test (Fig. 6C), and, in the late stage of the disease, there was a significant delay (6 d) in paralysis (Fig. 6D). Treatment protected the motor neurons in the lumbar spinal cord of the mice, with significantly more spared motor neurons with a soma area >250 μm2 at disease end stage (Fig. 6E). The protective effect was also evident considering only the largest motor neurons (>400 μm2; data not shown). Investigating astrocytosis in ventral horn lumbar spinal cord of MM218- and vehicle-treated SOD1G93A mice by immunohistochemistry, we found a substantial decrease in the signal of GFAP in the treated animals (Fig. 7A). We then measured CD68 as a marker of microglia/macrophages activation and found that it increased significantly in the white and gray matter of the MM218-treated mice (Fig. 7B). CD68 is expressed on phagocytic cells that can adopt a prohealing phenotype (Girard et al., 2013). Accordingly, we detected a concomitant reduction in markers of neuroinflammation such as NF-κB activation, TNFα, nitrotyrosine, and MMP-9 in spinal cord tissues of treated animals (Fig. 8A–D). CD68 was expressed mainly by ramified CD11b+ cells in the gray and the white matter of the treated animals (Fig. 7C,D). Moreover, CD11b+ cells showed a decreased ameboid morphology (lower circularity and solidity) and more ramifications (higher grid crossing; Fig. 7E) in white and gray matter of the spinal cord of the treated mice, underlining a functional shift of microglia/macrophages upon PPIA inhibition (Fumagalli et al., 2015; Zanier et al., 2015). We also measured 78 kDa glucose-regulated protein (BiP), which is a marker of endoplasmic reticulum (ER) stress and found that it was lower in treated animals (Fig. 8E). Finally, we investigated whether the treatment interfered with PPIA intracellular functions, those linked to protein aggregation and TDP-43 function, such as regulation of histone deacetylase 6 expression (HDAC6), shown in previous works (Basso et al., 2009; Lauranzano et al., 2015). We found no changes in PPIA protein levels, soluble and detergent-insoluble aggregated SOD1G93A, and HDAC6 expression (data not shown). We observed a reduced level of insoluble phosphorylated TDP-43 in the spinal cord of the treated mice (Fig. 8F), indicating that MM218, by reducing neuroinflammation, may affect TDP-43 pathology.
Figure 6.
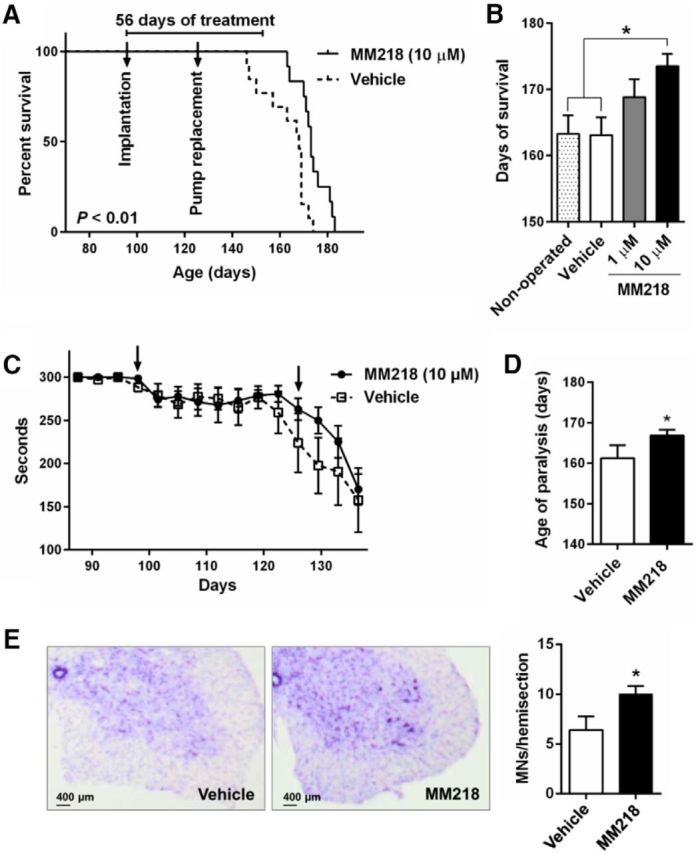
MM218 increases survival in SOD1G93A mice. A, Kaplan–Meier curve for survival of SOD1G93A mice (B6.Cg) treated with vehicle (n = 13) or 10 μm MM218 (n = 12) in the minipump (estimated concentrations in CSF are in the low nanomolar range). SOD1G93A mice received continuous intracerebroventricular infusion through osmotic minipump for 56 d starting from 98 d of age (implantation, first arrow), with a pump replacement after 28 d of treatment (second arrow). Log–rank Mantel–Cox test was done comparing vehicle-treated animals. B, Survival length of SOD1G93A mice treated with vehicle and MM218, 1 μm (n = 11) and 10 μm. MM218 increased life expectation in SOD1G93A mice with a dose–response effect (p < 0.01, post test for linear trend). There was no difference in survival between vehicle-treated and nontreated SOD1G93A mice (n = 12). Data are shown as mean ± SEM. *p < 0.05 by one-way ANOVA, Tukey's multiple-comparisons test. C, SOD1G93A mice treated with MM218 at 10 μm showed a tendency toward better motor function, as assessed by the Rotarod test. Data were analyzed up to 137 d, when all animals in each group were still alive. Data (mean ± SEM) are expressed in seconds and were evaluated by two-way ANOVA for repeated measures: p = 0.4, for treatment factor; p < 0.01, for age factor; p = 0.52, for interaction. D, Paralysis, assessed as the age at which the mice were no longer able to do the Rotarod test. The mean age was significantly different in the MM218-treated (167 ± 2) and vehicle-treated (161 ± 3) mice; *p < 0.05 by Student's t test. E, Quantification of Nissl-stained motor neurons (MNs > 250 μm2) in lumbar spinal cord hemisections from SOD1G93A mice treated with vehicle and MM218, 10 μm, at end-stage disease. Data are shown as mean ± SEM (n = 5). *p < 0.05 by Student's t test. Representative Nissl-stained lumbar spinal cord hemisections from vehicle (Veh)- and MM218-treated animals are shown. Scale bar, 400 μm.
Figure 7.
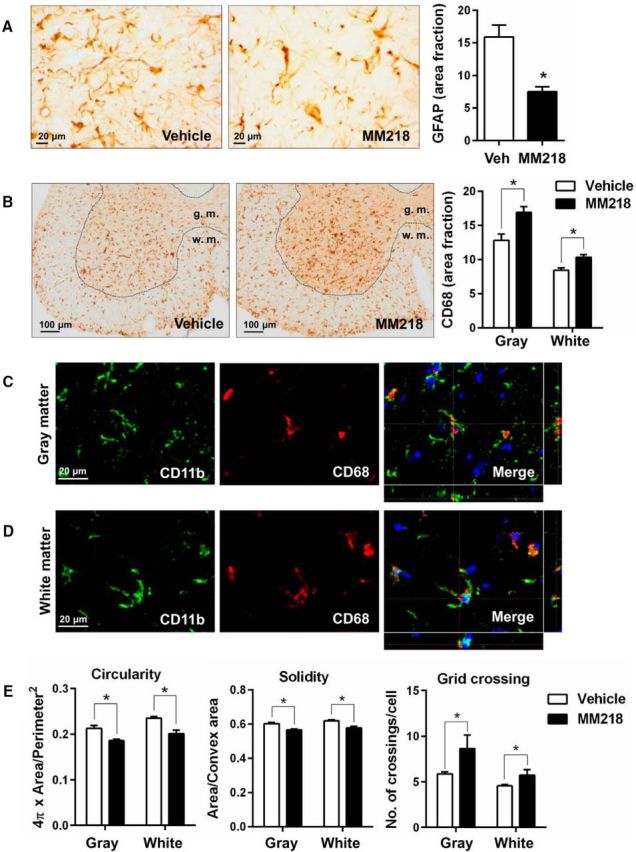
MM218 modulates glial activation. A, Activation of astrocytes was reduced in animals treated with 10 μm MM218, as assessed by GFAP immunostaining in lumbar spinal cord hemisections at end-stage disease. Representative GFAP-stained lumbar spinal cord hemisections from vehicle (Veh)- and MM218-treated animals are shown. Scale bar, 20 μm. B, Increased CD68 staining was detected in the white matter (w.m.) and gray matter (g.m.) of the lumbar spinal cord in the MM218-treated mice at end-stage disease. Representative CD68-stained lumbar spinal cord hemisections from vehicle- and MM218-treated animals are shown. Scale bar, 100 μm. C, D, CD68 staining colocalized exclusively with ramified CD11b+ cells in the gray (C) and the white (D) matter of the treated animals. Representative confocal image of lumbar spinal cord from vehicle- and MM218-treated animals at end-stage disease costained for CD11b (green staining) and CD68 (red staining). Scale bar, 20 μm. E, Quantitative analysis of shape descriptors (circularity, solidity, and grid crossing) indicates that CD11b+ cells were less ameboid and had more ramifications in lumbar spinal cord in MM218-treated mice at end-stage disease, both in the white and the gray matter. For A, B, and E, data are shown as mean ± SEM (n = 5). *p < 0.05 by Student's t test.
Figure 8.

MM218 reduces markers of neuroinflammation. A, F, Immnunoblot analysis of lysates from spinal cord of SOD1G93A mice treated with vehicle (Veh; n = 8) or 10 μm MM218 (n = 10). MM218 reduced NF-κB activation (A), proinflammatory cytokine TNFα (B), nitrotyrosine (C), MMP-9 (D), and ER stress chaperone BiP (E). Immunoreactivity, measured by dot blot (A, C, E) and Western blot (B) with the specific antibodies, was normalized to protein loading (Red Ponceau). NF-κB activation was quantified by normalization of the phospho (p)-NF-κB p65 signal to total NF-κB p65. Levels of MMP-9 were measured by AlphaLISA and were normalized to protein loading, as quantified by BCA assay, and to the mean number of Nissl-stained motor neurons (MNs > 400 μm2) in lumbar spinal cord hemisections. Data are shown as mean ± SEM. *p < 0.05, by Student's t test. F, Analysis of the Triton-insoluble fraction from spinal cord of SOD1G93A mice treated with vehicle (Veh; n = 8) or 10 μm MM218 (n = 10). Insoluble phosphorylated TDP-43 was measured by dot blot with the specific antibody. Immunoreactivity was normalized to protein loading (Red Ponceau). Data are shown as mean ± SEM. *p < 0.05, by Student's t test.
Discussion
The key findings of the present work are that extracellular PPIA is an unexpected mediator of the neuroinflammatory reaction in ALS and is toxic for motor neurons. Supporting this, a specific inhibitor of extracellular PPIA reduces neuroinflammation, protects motor neurons, and increases survival in SOD1G93A mice.
To decipher the role of PPIA in ALS pathogenesis, we crossbred the PPIA−/− mouse with the SOD1G93A mouse (Lauranzano et al., 2015). General depletion of PPIA exacerbated protein aggregation and affected key TDP-43 functions, hastening disease progression and shortening the lifespan. We concluded that PPIA has a beneficial role in ALS because of its intracellular functions in protein folding and complex assembly. Further studies, reported here, indicated that SOD1G93APPIA−/− mice have more spared motor neurons, confirming that motor neuron protection is not always predictive of an improved clinical response (Gould et al., 2006). This was consistent with reports of a possible role of PPIA in cell death pathways, by cooperating to AIF nuclear translocation (Zhu et al., 2007; Tanaka et al., 2011). However, we found a small decrease in AIF nuclear translocation in SOD1G93APPIA−/− mice only at a symptomatic stage of the disease. This suggested that motor neuron protection could not be explained solely by such a mechanism. One established function of extracellular PPIA mediated by the EMMPRIN receptor is to promote the induction and release of MMPs (Kim et al., 2005; Satoh et al., 2009; Seizer et al., 2010; Bahmed et al., 2012). MMPs can be neurotoxic through a number of mechanisms, including induction of ER stress (Kaplan et al., 2014). Most cells express low level of MMPs, but their expression can be induced by proinflammatory cytokines. High levels of MMPs have been found in tissues and biofluids of ALS patients (Lim et al., 1996; Beuche et al., 2000; Fang et al., 2009). Moreover, MMP-9 is a determinant of selective motor neuron degeneration (Kaplan et al., 2014). It is strongly expressed by vulnerable motor neurons in SOD1G93A mice and knocking out MMP-9 has led to motor neuron protection (Kiaei et al., 2007; Kaplan et al., 2014). We found that, in the absence of PPIA, the level of MMP-9 was substantially lower in SOD1G93A mice already at a presymptomatic stage. We therefore deduced that PPIA is a major inducer of MMP-9 and low levels of MMP-9 may contribute to motor neuron protection in SOD1G93APPIA−/− mice. Therefore, PPIA has distinctive and divergent roles in ALS depending on its interactors/substrates. For example, intracellularly, it is beneficial, acting as a foldase/chaperone for proteins such as TDP-43 and mutant SOD1 (Lauranzano et al., 2015), whereas extracellularly, it is detrimental by activating an EMMPRIN-dependent pathway.
The exact mechanism through which PPIA is secreted by cells has not yet been defined. Oxidative stress and inflammation stimulate PPIA secretion possibly by inducing posttranslational modifications and through a vesicular pathway (Suzuki et al., 2006; Soe et al., 2014). Increased levels of extracellular PPIA have been detected in many inflammatory conditions and its role in pathogenesis has been verified in a range of animal models (Hoffmann and Schiene-Fischer, 2014). We here report that high levels of extracellular PPIA are also associated with ALS, as detected in vitro in mouse glial cells and astrocyte–spinal neuron cocultures expressing SOD1G93A, and in vivo, in the CSF of SOD1G93A mice and rats and sporadic ALS patients. Therefore, our findings may be relevant for both the familial and the sporadic cases, which account for 90% of all patients. We found that motor neurons are more vulnerable than other neuronal types to PPIA toxicity. Extracellular functions of PPIA are mediated by EMMPRIN (Yurchenko et al., 2002; Malešević et al., 2013). EMMPRIN is highly expressed in CNS, especially in spinal cord gray matter (Fan et al., 1998). We found a strong staining for EMMPRIN in large motor neurons and a higher level of the protein in the ventral horn compared with the dorsal horn spinal cord, suggesting that EMMPRIN is highly expressed in motor neurons. PPIA is a major inducer of MMP-9, as shown in the SOD1G93APPIA−/− mouse studies. MMP-9 is expressed selectively by fast motor neurons and accelerates their degeneration by inducing ER stress (Kaplan et al., 2014). Therefore, increased secretion of PPIA by neurons and glial cells under pathological conditions could have a selective detrimental effect on vulnerable motor neurons in which the toxic EMMPRIN/MMP-9 pathway is upregulated.
Potent PPIA inhibitors, mainly CsA derivatives, have been developed for several clinical applications; however, all target both intracellular and extracellular PPIA. Our studies suggest that general PPIA inhibition is not recommended for ALS. We and others have shown that standard CsA is toxic for neurons and motor neurons in vitro (Maxwell et al., 2004; Van Den Bosch et al., 2004). Chronic intrathecal injection of CsA in mutant SOD1 mice improved survival, but had severe neurotoxic effects (Keep et al., 2001). Moreover, CsA administered systemically does not reach the CNS because of the poor BBB permeability and shows no benefit in ALS patients (Appel et al., 1988). We tested the cell-impermeable CsA derivative MM218 (Balsley et al., 2010; Malešević et al., 2010) in our in vitro paradigm of ALS, astrocyte–spinal neuron cocultures expressing SOD1G93A, in which the spontaneous loss of motor neurons is associated with increased extracellular PPIA and MMP-9 levels. In agreement with our hypothesis, MM218 protected motor neurons and concomitantly reduced MMP-9 induction, confirming the causal link between motor neuron death and activation of EMMPRIN. We therefore moved to a proof-of-concept preclinical trial in the SOD1G93A mouse model. The treatment started at approximately symptom onset affected disease progression with a dose–response relationship and the highest dose increased life expectancy by 17%. Motor performance, assessed by the Rotarod test, did not improve significantly, even if there was a clear tendency of amelioration. The test with mice implanted with a brain infusion cannula connected with an osmotic minipump may be challenging and not fully reliable. It is also possible that muscles in SOD1G93A mice are susceptible to extracellular PPIA toxicity. Under stress conditions, muscle cells secrete high levels of PPIA that promote inflammation in vivo (Satoh et al., 2009). It has been reported that an analog of MM218 reduced myocardial inflammation and fibrosis (Heinzmann et al., 2015). Given the tolerability of this drug, we would expect the clinical response to be greater with higher doses of MM218, possibly combining intracerebroventricular and systemic administration. The exclusive inhibition of extracellular PPIA by MM218 was sufficient to protect motor neurons in the SOD1G93A mice, as it was in vitro. PPIA activates through EMMPRIN a signaling pathway that is invariably NF-κB dependent (Kim et al., 2005; Satoh et al., 2009; Seizer et al., 2010; Bahmed et al., 2012). Here, we found that EMMPRIN is upregulated in ventral horn spinal cord of SOD1G93A mice. NF-κB is activated aberrantly in ALS patients and SOD1G93A mice and its absence or inhibition was beneficial in different mouse models of ALS (Swarup et al., 2011; Frakes et al., 2014; Patel et al., 2015). We found a reduced NF-κB activation in the spinal cord of the MM218-treated SOD1G93A mice. Therefore, we propose that extracellular PPIA, through EMMPRIN, contributes to the aberrant activation of NF-κB, so its pharmacological targeting by MM218 leads to a positive clinical response by inhibiting NF-κB signaling in motor neurons and neighboring glial cells through a non-cell-autonomous mechanism (Fig. 9A,B). This resulted in the polarization of glia toward a prohealing phenotype associated with decreased levels of neuroinflammatory factors such as TNFα and MMP-9. Extracellular PPIA, in addition to TNFα and MMP-9, induces expression of other proinflammatory cytokines/chemokines such as IL-8, MCP-1, and IL-1beta, depending on the cell type (Kim et al., 2005). TNFα was also reduced in SOD1G93A mice knocked out for MMP-9, which has been suggested to regulate the expression and release of soluble TNFα (Kiaei et al., 2007). However, our approach was more effective than the pharmacological inhibition of MMP-9 (Lorenzl et al., 2006; Kaplan et al., 2014), indicating that inhibiting the EMMPRIN-initiated pathway upstream is a better option, acting at the same time on a number of proinflammatory factors. Extracellular PPIA has a potent chemotactic activity toward leukocytes that is inhibited by MM218 (Yurchenko et al., 2002; Damsker et al., 2007; Malešević et al., 2010). Therefore, the protective effect of MM218 is unlikely to be linked to a recruitment of immunoregolatory cells to CNS that has been shown with other therapeutic strategies (Beers et al., 2011; Peviani et al., 2014; Kunis et al., 2015). We can hypothesize that the inhibition of extracellular PPIA limited the infiltration of certain toxic immune cells (Vaknin et al., 2011; Butovsky et al., 2012) and favored a protective activation of resident microglia. Consistent with this, MM218 treatment induced a morphological and phenotypical switch of microglia that may be associated with debris clearing and tissue remodeling (Vinet et al., 2012; Hong and Stevens, 2016).
Figure 9.
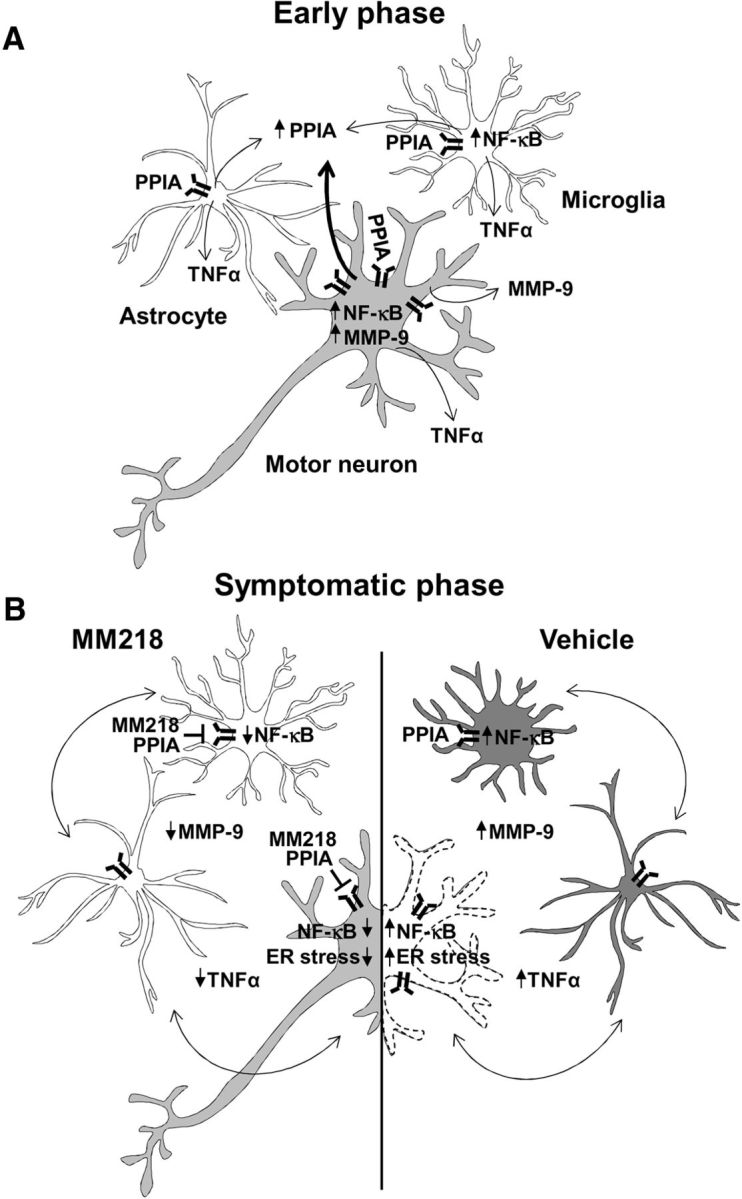
Model of the non-cell-autonomous mechanism by which extracellular PPIA mediates motor neuron death and MM218 is protective. A, At an early phase of the disease, PPIA is upregulated by motor neurons and neighboring cells as a protective response against pathogenic pathways induced by mutant SOD1 (protein misfolding, oxidative stress, etc.). As the disease progresses, motor neurons especially, but also glial cells, secrete PPIA, which activates an inflammatory response mediated by the EMMPRIN receptor, leading to NF-κB activation and induction of MMP-9 and proinflammatory cytokines (e.g., TNFα). B, The crosstalk between motor neurons and glial cells further exacerbates the proinflammatory response by increasing NF-κB activation, ER stress, and motor neuron death. Treatment with extracellular PPIA inhibitor MM218 at the onset of symptoms blocks the EMMPRIN-initiated pathway upstream, leading to polarization of glia toward a prohealing phenotype associated with reduced NF-κB activation, proinflammatory markers, and ER stress. This resulted in the protection of motor neurons and increased survival of SOD1G93A mice.
We also detected lower ER stress, like in the SOD1G93A mice knocked out for MMP-9 (Kaplan et al., 2014). The molecular mechanisms linking MMP-9 to ER stress have not been clarified. There is ample evidence of an interplay between ER stress and inflammation mediated by NF-κB activation (Hasnain et al., 2012) and pathological conditions in which inflammatory factors induce or exacerbate ER stress (Xue et al., 2005; Brozzi et al., 2015). Therefore, hypothetically blocking the EMMPRIN/NF-κB pathway by inhibiting extracellular PPIA may have a concomitant inhibitory effect on ER stress. Finally, the treatment did not affect PPIA levels and intracellular functions; it only reduced the level of detergent-insoluble phosphorylated TDP-43. This effect is probably linked to the reduced neuroinflammation. In fact, activation of the NF-κB pathway by inflammatory stimuli promoted TDP-43 pathology, which is associated with increased detergent-insoluble phosphorylated TDP-43 (Correia et al., 2015; Lauranzano et al., 2015).
Footnotes
This work was supported by Telethon Italy (Grant TCR08002 to V.B.), Italian Foundation for research on ALS (AriSLA) (Grant CypALS to V.B.), and the European Community's Health Seventh Framework Programme (FP7/2007–2013 under Grant 259867 to C.B). We thank Dr. Marco Marzo for contribution to the in vitro experiments, Dr. Giorgia Spano for help in collecting animal tissues, and Judith Baggott for editorial assistance.
The authors declare no competing financial interests.
References
- Agrawal SM, Silva C, Tourtellotte WW, Yong VW (2011) EMMPRIN: a novel regulator of leukocyte transmigration into the CNS in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neurosci 31:669–677. 10.1523/JNEUROSCI.3659-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel SH, Stewart SS, Appel V, Harati Y, Mietlowski W, Weiss W, Belendiuk GW (1988) A double-blind study of the effectiveness of cyclosporine in amyotrophic lateral sclerosis. Arch Neurol 45:381–386. 10.1001/archneur.1988.00520280027011 [DOI] [PubMed] [Google Scholar]
- Bahmed K, Henry C, Holliday M, Redzic J, Ciobanu M, Zhang F, Weekes C, Sclafani R, Degregori J, Eisenmesser E (2012) Extracellular cyclophilin-A stimulates ERK1/2 phosphorylation in a cell-dependent manner but broadly stimulates nuclear factor kappa B. Cancer Cell Int 12:19. 10.1186/1475-2867-12-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsley MA, Malešević M, Stemmy EJ, Gigley J, Jurjus RA, Herzog D, Bukrinsky MI, Fischer G, Constant SL (2010) A cell-impermeable cyclosporine A derivative reduces pathology in a mouse model of allergic lung inflammation. J Immunol 185:7663–7670. 10.4049/jimmunol.1001707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso M, Samengo G, Nardo G, Massignan T, D'Alessandro G, Tartari S, Cantoni L, Marino M, Cheroni C, De Biasi S, Giordana MT, Strong MJ, Estevez AG, Salmona M, Bendotti C, Bonetto V (2009) Characterization of detergent-insoluble proteins in ALS indicates a causal link between nitrative stress and aggregation in pathogenesis. PLoS One 4:e8130. 10.1371/journal.pone.0008130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso M, Pozzi S, Tortarolo M, Fiordaliso F, Bisighini C, Pasetto L, Spaltro G, Lidonnici D, Gensano F, Battaglia E, Bendotti C, Bonetto V (2013) Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J Biol Chem 288:15699–15711. 10.1074/jbc.M112.425066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Zhao W, Wang J, Huang A, Wen S, Liao B, Appel SH (2011) Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 134:1293–1314. 10.1093/brain/awr074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuche W, Yushchenko M, Mäder M, Maliszewska M, Felgenhauer K, Weber F (2000) Matrix metalloproteinase-9 is elevated in serum of patients with amyotrophic lateral sclerosis. Neuroreport 11:3419–3422. 10.1097/00001756-200011090-00003 [DOI] [PubMed] [Google Scholar]
- Bigini P, Steffensen KR, Ferrario A, Diomede L, Ferrara G, Barbera S, Salzano S, Fumagalli E, Ghezzi P, Mennini T, Gustafsson JA (2010) Neuropathologic and biochemical changes during disease progression in liver X receptor beta−/− mice, a model of adult neuron disease. J Neuropathol Exp Neurol 69:593–605. 10.1097/NEN.0b013e3181df20e1 [DOI] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW (2006) Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312:1389–1392. 10.1126/science.1123511 [DOI] [PubMed] [Google Scholar]
- Brozzi F, Nardelli TR, Lopes M, Millard I, Barthson J, Igoillo-Esteve M, Grieco FA, Villate O, Oliveira JM, Casimir M, Bugliani M, Engin F, Hotamisligil GS, Marchetti P, Eizirik DL (2015) Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia 58:2307–2316. 10.1007/s00125-015-3669-6 [DOI] [PubMed] [Google Scholar]
- Butovsky O, Siddiqui S, Gabriely G, Lanser AJ, Dake B, Murugaiyan G, Doykan CE, Wu PM, Gali RR, Iyer LK, Lawson R, Berry J, Krichevsky AM, Cudkowicz ME, Weiner HL (2012) Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest 122:3063–3087. 10.1172/JCI62636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia AS, Patel P, Dutta K, Julien JP (2015) Inflammation Induces TDP-43 Mislocalization and Aggregation. PLoS One 10:e0140248. 10.1371/journal.pone.0140248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsker JM, Bukrinsky MI, Constant SL (2007) Preferential chemotaxis of activated human CD4+ T cells by extracellular cyclophilin A. J Leukoc Biol 82:613–618. 10.1189/jlb.0506317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drachman DB, Frank K, Dykes-Hoberg M, Teismann P, Almer G, Przedborski S, Rothstein JD (2002) Cyclooxygenase 2 inhibition protects motor neurons and prolongs survival in a transgenic mouse model of ALS. Ann Neurol 52:771–778. 10.1002/ana.10374 [DOI] [PubMed] [Google Scholar]
- Fan QW, Yuasa S, Kuno N, Senda T, Kobayashi M, Muramatsu T, Kadomatsu K (1998) Expression of basigin, a member of the immunoglobulin superfamily, in the mouse central nervous system. Neurosci Res 30:53–63. 10.1016/S0168-0102(97)00119-3 [DOI] [PubMed] [Google Scholar]
- Fang L, Huber-Abel F, Teuchert M, Hendrich C, Dorst J, Schattauer D, Zettlmeissel H, Wlaschek M, Scharffetter-Kochanek K, Tumani H, Ludolph AC, Brettschneider J (2009) Linking neuron and skin: matrix metalloproteinases in amyotrophic lateral sclerosis (ALS). J Neurol Sci 285:62–66. 10.1016/j.jns.2009.05.025 [DOI] [PubMed] [Google Scholar]
- Fauré J, Lachenal G, Court M, Hirrlinger J, Chatellard-Causse C, Blot B, Grange J, Schoehn G, Goldberg Y, Boyer V, Kirchhoff F, Raposo G, Garin J, Sadoul R (2006) Exosomes are released by cultured cortical neurones. Mol Cell Neurosci 31:642–648. 10.1016/j.mcn.2005.12.003 [DOI] [PubMed] [Google Scholar]
- Fischer G, Bang H, Mech C (1984) Determination of enzymatic catalysis for the cis-trans-isomerization of peptide binding in proline-containing peptides. Biomed Biochim Acta 43:1101–1111. [PubMed] [Google Scholar]
- Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, Ladner KJ, Bevan AK, Foust KD, Godbout JP, Popovich PG, Guttridge DC, Kaspar BK (2014) Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron 81:1009–1023. 10.1016/j.neuron.2014.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli S, Perego C, Pischiutta F, Zanier ER, De Simoni MG (2015) The ischemic environment drives microglia and macrophage function. Front Neurol 6:81. 10.3389/fneur.2015.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard S, Brough D, Lopez-Castejon G, Giles J, Rothwell NJ, Allan SM (2013) Microglia and macrophages differentially modulate cell death after brain injury caused by oxygen-glucose deprivation in organotypic brain slices. Glia 61:813–824. 10.1002/glia.22478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göldner FM, Patrick JW (1996) Neuronal localization of the cyclophilin A protein in the adult rat brain. J Comp Neurol 372:283–293. [DOI] [PubMed] [Google Scholar]
- Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW (2006) Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci 26:8774–8786. 10.1523/JNEUROSCI.2315-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschumacher RE, Harding MW, Rice J, Drugge RJ, Speicher DW (1984) Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science 226:544–547. 10.1126/science.6238408 [DOI] [PubMed] [Google Scholar]
- Hasnain SZ, Lourie R, Das I, Chen AC, McGuckin MA (2012) The interplay between endoplasmic reticulum stress and inflammation. Immunol Cell Biol 90:260–270. 10.1038/icb.2011.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzmann D, Bangert A, Müller AM, von Ungern-Sternberg SN, Emschermann F, Schönberger T, Chatterjee M, Mack AF, Klingel K, Kandolf R, Malesevic M, Borst O, Gawaz M, Langer HF, Katus H, Fischer G, May AE, Kaya Z, Seizer P (2015) The novel extracellular cyclophilin A (CyPA) inhibitor MM284 reduces myocardial inflammation and remodeling in a mouse model of troponin I-induced myocarditis. PLoS One 10:e0124606. 10.1371/journal.pone.0124606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann H, Schiene-Fischer C (2014) Functional aspects of extracellular cyclophilins. Biol Chem 395:721–735. 10.1515/hsz-2014-0125 [DOI] [PubMed] [Google Scholar]
- Hong S, Stevens B (2016) Microglia: phagocytosing to clear, sculpt, and eliminate. Dev Cell 38:126–128. 10.1016/j.devcel.2016.07.006 [DOI] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD (2002) Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A 99:1604–1609. 10.1073/pnas.032539299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäschke A, Mi H, Tropschug M (1998) Human T cell cyclophilin18 binds to thiol-specific antioxidant protein Aop1 and stimulates its activity. J Mol Biol 277:763–769. 10.1006/jmbi.1998.1644 [DOI] [PubMed] [Google Scholar]
- Jin ZG, Melaragno MG, Liao DF, Yan C, Haendeler J, Suh YA, Lambeth JD, Berk BC (2000) Cyclophilin A is a secreted growth factor induced by oxidative stress. Circ Res 87:789–796. 10.1161/01.RES.87.9.789 [DOI] [PubMed] [Google Scholar]
- Kaplan A, Spiller KJ, Towne C, Kanning KC, Choe GT, Geber A, Akay T, Aebischer P, Henderson CE (2014) Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron 81:333–348. 10.1016/j.neuron.2013.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keep M, Elmér E, Fong KS, Csiszar K (2001) Intrathecal cyclosporin prolongs survival of late-stage ALS mice. Brain Res 894:327–331. 10.1016/S0006-8993(01)02012-1 [DOI] [PubMed] [Google Scholar]
- Kiaei M, Petri S, Kipiani K, Gardian G, Choi DK, Chen J, Calingasan NY, Schafer P, Muller GW, Stewart C, Hensley K, Beal MF (2006) Thalidomide and lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci 26:2467–2473. 10.1523/JNEUROSCI.5253-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiaei M, Kipiani K, Calingasan NY, Wille E, Chen J, Heissig B, Rafii S, Lorenzl S, Beal MF (2007) Matrix metalloproteinase-9 regulates TNF-alpha and FasL expression in neuronal, glial cells and its absence extends life in a transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol 205:74–81. 10.1016/j.expneurol.2007.01.036 [DOI] [PubMed] [Google Scholar]
- Kim H, Kim WJ, Jeon ST, Koh EM, Cha HS, Ahn KS, Lee WH (2005) Cyclophilin A may contribute to the inflammatory processes in rheumatoid arthritis through induction of matrix degrading enzymes and inflammatory cytokines from macrophages. Clin Immunol 116:217–224. 10.1016/j.clim.2005.05.004 [DOI] [PubMed] [Google Scholar]
- Kriz J, Nguyen MD, Julien JP (2002) Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 10:268–278. 10.1006/nbdi.2002.0487 [DOI] [PubMed] [Google Scholar]
- Kunis G, Baruch K, Miller O, Schwartz M (2015) Immunization with a myelin-derived antigen activates the brain's choroid plexus for recruitment of immunoregulatory cells to the CNS and attenuates disease progression in a mouse model of ALS. J Neurosci 35:6381–6393. 10.1523/JNEUROSCI.3644-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauranzano E, Pozzi S, Pasetto L, Stucchi R, Massignan T, Paolella K, Mombrini M, Nardo G, Lunetta C, Corbo M, Mora G, Bendotti C, Bonetto V (2015) Peptidylprolyl isomerase A governs TARDBP function and assembly in heterogeneous nuclear ribonucleoprotein complexes. Brain 138:974–991. 10.1093/brain/awv005 [DOI] [PubMed] [Google Scholar]
- Lee JP, Palfrey HC, Bindokas VP, Ghadge GD, Ma L, Miller RJ, Roos RP (1999) The role of immunophilins in mutant superoxide dismutase-1linked familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 96:3251–3256. 10.1073/pnas.96.6.3251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SP, Hwang YS, Kim YJ, Kwon KS, Kim HJ, Kim K, Chae HZ (2001) Cyclophilin a binds to peroxiredoxins and activates its peroxidase activity. J Biol Chem 276:29826–29832. 10.1074/jbc.M101822200 [DOI] [PubMed] [Google Scholar]
- Lim GP, Backstrom JR, Cullen MJ, Miller CA, Atkinson RD, Tökés ZA (1996) Matrix metalloproteinases in the neocortex and spinal cord of amyotrophic lateral sclerosis patients. J Neurochem 67:251–259. [DOI] [PubMed] [Google Scholar]
- Lorenzl S, Narr S, Angele B, Krell HW, Gregorio J, Kiaei M, Pfister HW, Beal MF (2006) The matrix metalloproteinases inhibitor Ro 28–2653 [correction of Ro 26–2853] extends survival in transgenic ALS mice. Exp Neurol 200:166–171. 10.1016/j.expneurol.2006.01.026 [DOI] [PubMed] [Google Scholar]
- Malešević M, Kühling J, Erdmann F, Balsley MA, Bukrinsky MI, Constant SL, Fischer G (2010) A cyclosporin derivative discriminates between extracellular and intracellular cyclophilins. Angew Chem Int Ed Engl 49:213–215. 10.1002/anie.200904529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malešević M, Gutknecht D, Prell E, Klein C, Schumann M, Nowak RA, Simon JC, Schiene-Fischer C, Saalbach A (2013) Anti-inflammatory effects of extracellular cyclosporins are exclusively mediated by CD147. J Med Chem 56:7302–7311. 10.1021/jm4007577 [DOI] [PubMed] [Google Scholar]
- Marino M, Papa S, Crippa V, Nardo G, Peviani M, Cheroni C, Trolese MC, Lauranzano E, Bonetto V, Poletti A, DeBiasi S, Ferraiuolo L, Shaw PJ, Bendotti C (2015) Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol Aging 36:492–504. 10.1016/j.neurobiolaging.2014.06.026 [DOI] [PubMed] [Google Scholar]
- Massignan T, Casoni F, Basso M, Stefanazzi P, Biasini E, Tortarolo M, Salmona M, Gianazza E, Bendotti C, Bonetto V (2007) Proteomic analysis of spinal cord of presymptomatic amyotrophic lateral sclerosis G93A SOD1 mouse. Biochem Biophys Res Commun 353:719–725. 10.1016/j.bbrc.2006.12.075 [DOI] [PubMed] [Google Scholar]
- Maxwell MM, Pasinelli P, Kazantsev AG, Brown RH Jr (2004) RNA interference-mediated silencing of mutant superoxide dismutase rescues cyclosporin A-induced death in cultured neuroblastoma cells. Proc Natl Acad Sci U S A 101:3178–3183. 10.1073/pnas.0308726100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardo G, Pozzi S, Pignataro M, Lauranzano E, Spano G, Garbelli S, Mantovani S, Marinou K, Papetti L, Monteforte M, Torri V, Paris L, Bazzoni G, Lunetta C, Corbo M, Mora G, Bendotti C, Bonetto V (2011) Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS One 6:e25545. 10.1371/journal.pone.0025545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neymotin A, Petri S, Calingasan NY, Wille E, Schafer P, Stewart C, Hensley K, Beal MF, Kiaei M (2009) Lenalidomide (Revlimid) administration at symptom onset is neuroprotective in a mouse model of amyotrophic lateral sclerosis. Exp Neurol 220:191–197. 10.1016/j.expneurol.2009.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro P, Pompilio G, Capogrossi MC (2013) Cyclophilin A: a key player for human disease. Cell Death Dis 4:e888. 10.1038/cddis.2013.410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel P, Julien JP, Kriz J (2015) Early-stage treatment with Withaferin A reduces levels of misfolded superoxide dismutase 1 and extends lifespan in a mouse model of amyotrophic lateral sclerosis. Neurotherapeutics 12:217–233. 10.1007/s13311-014-0311-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perego C, Fumagalli S, De Simoni MG (2011) Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J Neuroinflammation 8:174. 10.1186/1742-2094-8-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peviani M, Salvaneschi E, Bontempi L, Petese A, Manzo A, Rossi D, Salmona M, Collina S, Bigini P, Curti D (2014) Neuroprotective effects of the Sigma-1 receptor (S1R) agonist PRE-084, in a mouse model of motor neuron disease not linked to SOD1 mutation. Neurobiol Dis 62:218–232. 10.1016/j.nbd.2013.10.010 [DOI] [PubMed] [Google Scholar]
- Pizzasegola C, Caron I, Daleno C, Ronchi A, Minoia C, Carrì MT, Bendotti C (2009) Treatment with lithium carbonate does not improve disease progression in two different strains of SOD1 mutant mice. Amyotroph Lateral Scler 10:221–228. 10.1080/17482960902803440 [DOI] [PubMed] [Google Scholar]
- Restelli E, Fioriti L, Mantovani S, Airaghi S, Forloni G, Chiesa R (2010) Cell type-specific neuroprotective activity of untranslocated prion protein. PLoS One 5:e13725. 10.1371/journal.pone.0013725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht W, Philips T (2013) The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 14:248–264. 10.1038/nrn3430 [DOI] [PubMed] [Google Scholar]
- Sahawneh MA, Ricart KC, Roberts BR, Bomben VC, Basso M, Ye Y, Sahawneh J, Franco MC, Beckman JS, Estévez AG (2010) Cu, Zn-superoxide dismutase increases toxicity of mutant and zinc-deficient superoxide dismutase by enhancing protein stability. J Biol Chem 285:33885–33897. 10.1074/jbc.M110.118901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh K, Nigro P, Matoba T, O'Dell MR, Cui Z, Shi X, Mohan A, Yan C, Abe J, Illig KA, Berk BC (2009) Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat Med 15:649–656. 10.1038/nm.1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütz B, Reimann J, Dumitrescu-Ozimek L, Kappes-Horn K, Landreth GE, Schürmann B, Zimmer A, Heneka MT (2005) The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J Neurosci 25:7805–7812. 10.1523/JNEUROSCI.2038-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seizer P, Schönberger T, Schött M, Lang MR, Langer HF, Bigalke B, Krämer BF, Borst O, Daub K, Heidenreich O, Schmidt R, Lindemann S, Herouy Y, Gawaz M, May AE (2010) EMMPRIN and its ligand cyclophilin A regulate MT1-MMP, MMP-9 and M-CSF during foam cell formation. Atherosclerosis 209:51–57. 10.1016/j.atherosclerosis.2009.08.029 [DOI] [PubMed] [Google Scholar]
- Sherry B, Yarlett N, Strupp A, Cerami A (1992) Identification of cyclophilin as a proinflammatory secretory product of lipopolysaccharide-activated macrophages. Proc Natl Acad Sci U S A 89:3511–3515. 10.1073/pnas.89.8.3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soe NN, Sowden M, Baskaran P, Kim Y, Nigro P, Smolock EM, Berk BC (2014) Acetylation of cyclophilin A is required for its secretion and vascular cell activation. Cardiovasc Res 101:444–453. 10.1093/cvr/cvt268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Jin ZG, Meoli DF, Matoba T, Berk BC (2006) Cyclophilin A is secreted by a vesicular pathway in vascular smooth muscle cells. Circ Res 98:811–817. 10.1161/01.RES.0000216405.85080.a6 [DOI] [PubMed] [Google Scholar]
- Swarup V, Phaneuf D, Dupré N, Petri S, Strong M, Kriz J, Julien JP (2011) Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J Exp Med 208:2429–2447. 10.1084/jem.20111313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Shimazaki H, Kimura M, Izuta H, Tsuruma K, Shimazawa M, Hara H (2011) Apoptosis-inducing factor and cyclophilin A cotranslocate to the motor neuronal nuclei in amyotrophic lateral sclerosis model mice. CNS Neurosci Ther 17:294–304. 10.1111/j.1755-5949.2010.00180.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortarolo M, Veglianese P, Calvaresi N, Botturi A, Rossi C, Giorgini A, Migheli A, Bendotti C (2003) Persistent activation of p38 mitogen-activated protein kinase in a mouse model of familial amyotrophic lateral sclerosis correlates with disease progression. Mol Cell Neurosci 23:180–192. 10.1016/S1044-7431(03)00022-8 [DOI] [PubMed] [Google Scholar]
- Tortarolo M, Grignaschi G, Calvaresi N, Zennaro E, Spaltro G, Colovic M, Fracasso C, Guiso G, Elger B, Schneider H, Seilheimer B, Caccia S, Bendotti C (2006) Glutamate AMPA receptors change in motor neurons of SOD1G93A transgenic mice and their inhibition by a noncompetitive antagonist ameliorates the progression of amytrophic lateral sclerosis-like disease. J Neurosci Res 83:134–146. 10.1002/jnr.20715 [DOI] [PubMed] [Google Scholar]
- Tortarolo M, Vallarola A, Lidonnici D, Battaglia E, Gensano F, Spaltro G, Fiordaliso F, Corbelli A, Garetto S, Martini E, Pasetto L, Kallikourdis M, Bonetto V, Bendotti C (2015) Lack of TNF-alpha receptor type 2 protects motor neurons in a cellular model of amyotrophic lateral sclerosis and in mutant SOD1 mice but does not affect disease progression. J Neurochem 135:109–124. 10.1111/jnc.13154 [DOI] [PubMed] [Google Scholar]
- Vaknin I, Kunis G, Miller O, Butovsky O, Bukshpan S, Beers DR, Henkel JS, Yoles E, Appel SH, Schwartz M (2011) Excess circulating alternatively activated myeloid (M2) cells accelerate ALS progression while inhibiting experimental autoimmune encephalomyelitis. PLoS One 6:e26921. 10.1371/journal.pone.0026921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Bosch L, Tilkin P, Lemmens G, Robberecht W (2002) Minocycline delays disease onset and mortality in a transgenic model of ALS. Neuroreport 13:1067–1070. 10.1097/00001756-200206120-00018 [DOI] [PubMed] [Google Scholar]
- Van Den Bosch L, Storkebaum E, Vleminckx V, Moons L, Vanopdenbosch L, Scheveneels W, Carmeliet P, Robberecht W (2004) Effects of vascular endothelial growth factor (VEGF) on motor neuron degeneration. Neurobiol Dis 17:21–28. 10.1016/j.nbd.2004.06.004 [DOI] [PubMed] [Google Scholar]
- Vinet J, Weering HR, Heinrich A, Kälin RE, Wegner A, Brouwer N, Heppner FL, Rooijen Nv, Boddeke HW, Biber K (2012) Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J Neuroinflammation 9:27. 10.1186/1742-2094-9-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Leiva MC, Fischkoff SA, Handschumacher RE, Lyttle CR (1992) Leukocyte chemotactic activity of cyclophilin. J Biol Chem 267:11968–11971. [PubMed] [Google Scholar]
- Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, Mori K, Yagita H, Okumura K, Harding H, Nakano H (2005) Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J Biol Chem 280:33917–33925. 10.1074/jbc.M505818200 [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillée S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11:251–253. 10.1038/nn2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong VW, Power C, Forsyth P, Edwards DR (2001) Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci 2:502–511. 10.1038/35081571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Ge H, He B (2010) Pro-inflammatory activities induced by CyPA-EMMPRIN interaction in monocytes. Atherosclerosis 213:415–421. 10.1016/j.atherosclerosis.2010.09.033 [DOI] [PubMed] [Google Scholar]
- Yurchenko V, Zybarth G, O'Connor M, Dai WW, Franchin G, Hao T, Guo H, Hung HC, Toole B, Gallay P, Sherry B, Bukrinsky M (2002) Active site residues of cyclophilin A are crucial for its signaling activity via CD147. J Biol Chem 277:22959–22965. 10.1074/jbc.M201593200 [DOI] [PubMed] [Google Scholar]
- Zanier ER, Fumagalli S, Perego C, Pischiutta F, De Simoni MG (2015) Shape descriptors of the “never resting” microglia in three different acute brain injury models in mice. Intensive Care Med Exp 3:39. 10.1186/s40635-015-0039-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Wang X, Deinum J, Huang Z, Gao J, Modjtahedi N, Neagu MR, Nilsson M, Eriksson PS, Hagberg H, Luban J, Kroemer G, Blomgren K (2007) Cyclophilin A participates in the nuclear translocation of apoptosis-inducing factor in neurons after cerebral hypoxia-ischemia. J Exp Med 204:1741–1748. 10.1084/jem.20070193 [DOI] [PMC free article] [PubMed] [Google Scholar]