

Abstract
Cellular asymmetries play crucial roles in development and organ function. The planar cell polarity (PCP) signaling pathway is involved in the establishment of cellular asymmetry within the plane of a cell sheet. Inner ear sensory hair cells (HCs), which have several rows of staircase-like stereocilia and one kinocilium located at the vertex of the stereocilia protruding from the apical surface of each HC, exhibit a typical form of PCP. Although connections between cilia and PCP signaling in vertebrate development have been reported, their precise nature is not well understood. During inner ear development, several ciliary proteins are known to play a role in PCP formation. In the current study, we investigated a functional role for intestinal cell kinase (Ick), which regulates intraflagellar transport (IFT) at the tip of cilia, in the mouse inner ear. A lack of Ick in the developing inner ear resulted in PCP defects in the cochlea, including misorientation or misshaping of stereocilia and aberrant localization of the kinocilium and basal body in the apical and middle turns, leading to auditory dysfunction. We also observed abnormal ciliary localization of Ift88 in both HCs and supporting cells. Together, our results show that Ick ciliary kinase is essential for PCP formation in inner ear HCs, suggesting that ciliary transport regulation is important for PCP signaling.
SIGNIFICANCE STATEMENT The cochlea in the inner ear is the hearing organ. Planar cell polarity (PCP) in hair cells (HCs) in the cochlea is essential for mechanotransduction and refers to the asymmetric structure consisting of stereociliary bundles and the kinocilium on the apical surface of the cell body. We reported previously that a ciliary kinase, Ick, regulates intraflagellar transport (IFT). Here, we found that loss of Ick leads to abnormal localization of the IFT component in kinocilia, PCP defects in HCs, and hearing dysfunction. Our study defines the association of ciliary transport regulation with PCP formation in HCs and hearing function.
Keywords: cilia, hearing function, inner ear, intraflagellar transport, kinase, planar cell polarity
Introduction
Planar cell polarity (PCP) refers to coordinated polarization of cells within the two-dimensional plane of a cell sheet, entailing cellular asymmetries within the tissue plane. In vertebrates, PCP is required for diverse processes in development and organ function. In the inner ear, hair cells (HCs) display a distinctive form of PCP. The mammalian inner ear consists of the cochlea and the vestibule responsible for hearing and balance, respectively. The cochlea harbors the organ of Corti, which contains one row of inner HCs (IHCs), three rows of outer HCs (OHCs), and several types of supporting cells (SCs). PCP in HCs refers to the asymmetric structure formed by the stereociliary bundles and the kinocilium protruding from the apical surface of the cell body. The stereociliary bundles form a V-shaped array in OHCs and a semicircular shape in IHCs. The comprising stereocilia are aligned in a staircase pattern, with the highest stereocilia adjacent to a single kinocilium located at the lateral apex of the bundle. The staircase structure of stereociliary bundles is essential for mechanotransduction in HCs. Oscillations in air pressure generated by sound sources induce vibrations in the basilar membrane, followed by the deflection of stereociliary bundles. The stereociliary bundles deflect in a direction from the shortest stereocilia to the longest, leading to the opening of mechanotransduction channels (Schwander et al., 2010). The orientation of stereociliary bundles is thought to be directed by the kinocilium (Cotanche and Corwin, 1991; Tilney et al., 1992). At embryonic day 14.5 (E14.5) in mice, the kinocilium, assembled from the basal body, is located at the center of the HC and surrounded by stereocilia (Sobkowicz et al., 1995). As HCs begin to differentiate, the kinocilium moves to the periphery and the stereocilia nearest to the kinocilium begin to elongate (Tilney et al., 1992). By E18.5, the kinocilium reaches the lateral position along the mediolateral axis and stereociliary bundles are regularly organized (Kelly and Chen, 2007). Postnatally, kinocilia regress and disappear until postnatal day 21 (P21) (Lim and Anniko, 1985). SCs also possess a cilium on the apical surface of the cell body.
Ciliary proteins are known to be associated with PCP formation in the inner ear. Bardet–Biedl syndrome is a ciliopathy, which is a group of human disorders caused by defects in cilia function. Mutations in Bardet–Biedl syndrome 4 (Bbs4) or Mkks (also known as Bbs6) lead to misoriented or flattened sterociliary bundles in OHCs. In addition, Mkks−/− mutants display detachment of the kinocilium from sterociliary bundles (Ross et al., 2005). Mutant mice for Mks1 and Alsm1, causative genes for Meckel–Gruber syndrome and Alström syndrome in humans, respectively, exhibit misorientation of sterociliary bundles and abnormal localization of kinocilia (Cui et al., 2011; Jagger et al., 2011). Furthermore, mice defective in Ift88 or Kif3a, both of which are components of the intraflagellar transport (IFT) machinery, lack kinocilia and display misorientation or shape defects of stereociliary bundles (Jones et al., 2008; Sipe and Lu, 2011).
We and others have reported previously that intestinal cell kinase (Ick) and male-germ cell-associated kinase (Mak) are essential for the regulation of cilia formation in mammals (Omori et al., 2010; Chaya et al., 2014; Moon et al., 2014; Oud et al., 2016). Ick and Mak are serine-threonine kinases belonging to the MAP kinase superfamily (Miyata and Nishida, 1999; Togawa et al., 2000; Shinkai et al., 2002). Although Mak expression is cell-type specific, Ick is expressed in various tissues (Togawa et al., 2000). Two missense mutations in the human Ick gene, R272Q and G120C, were identified in patients with endocrine-cerebro-osteodysplasia (ECO) syndrome, a human recessive disorder associated with multiple anomalies involving the endocrine, cerebral, and skeletal systems (Lahiry et al., 2009; Oud et al., 2016). Auditory tests cannot be performed in ECO patients because the syndrome is neonatally lethal. In the current study, we investigated the role of Ick in the developing inner ear.
Materials and Methods
Animal care.
All procedures conformed to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Institutional Safety Committee on Recombinant DNA Experiments (approval ID 3380–6) and Animal Experimental Committees of Institute for Protein Research (approval ID 24–05-01), Osaka University. Mice were housed in a temperature-controlled room at 22°C with a 12 h light/dark cycle. Fresh water and rodent diet were available at all times. All animal experiments were performed on mice of either sex.
Generation of Ick flox (Ickflox) and Ick-null (Ick−/−) mice.
The generation of Ickflox mice and Ick−/− mice was as described previously (Chaya et al., 2014). Pax2-Cre mice (Ohyama and Groves, 2004) were crossed with Ickflox/+ mice to obtain Ickflox/+;Pax2-Cre+ mice. To generate inner ear Ick conditional knock-out (CKO) mice, we crossed Pax2-Cre;Ickflox/+ mice with Ickflox/flox mice.
In situ hybridization.
Whole brains of E15.5 and E18.5 embryos and inner ear tissues of P2, P9, and P28 mice were dissected and fixed in 4% paraformaldehyde (PFA) overnight at 4°C. Tissues were subsequently equilibrated in 30% sucrose overnight and cryosectioned at 25 μm thickness. Generation of the Ick riboprobe and in situ hybridization were performed as described previously (Chaya et al., 2014).
RT-PCR analysis.
Inner ear total RNAs from wild-type mice were isolated using TRIzol RNA extraction reagent (Invitrogen). RNA (2 μg) was reverse transcribed into cDNA with random hexamers using Superscript II (Invitrogen). The cDNAs were used as templates for PCRs by rTaq polymerase (Takara). The following sets of primers, which span an intron, were used: 5′-AAAGAATTCCCGACCCTGCAGTCCTCAGCAAAG-3′ (ICK, forward), 5′-AACCCTCTGCATGACAGAACCGAT-3′ (ICK, reverse) and 5′-CGTGCGTGACATCAAAGAGAA-3′ (β-actin, forward) and 5′-TGGATGCCACAGGATTCCAT-3′ (β-actin, reverse). The product sizes were 280 bp and 201 bp, respectively.
Immunohistochemistry.
Inner ears from E17.5 Ick KO mouse embryos, P1 Ick CKO mouse pups, and 6- to 7-week-old mice from both sexes were dissected and fixed in 4% PFA in PBS for 1–24 h at 4°C. Dissected inner ears were incubated with blocking solution (4% normal donkey serum and 0.02% Triton X-100 in PBS) for 1 h at room temperature. We incubated the inner ears in primary antibodies in blocking solution at 4°C overnight. The inner ears were then washed four times with PBS for 15 min each and incubated with a secondary antibody in the blocking solution for 30 min at room temperature. The inner ears were washed with PBS and mounted in gelvatol for examination. We used the following primary antibodies for immunostaining: anti-acetylated α-tublin (mouse, 6–11B-1, 1:1000; Sigma-Aldrich), anti-Pericentrin (rabbit, 923701, 1:400; BioLegend), anti-Ift88 (rabbit, 1:1000; a gift from Dr. G.J. Pazour, Pazour et al., 2002), anti-Vangl2 (rat, 1:200; a gift from Dr. J.P. Borg, Belotti et al., 2012), and anti-mPins (rabbit, 1:200, gift from Dr. M. Montcouquiol, Ezan et al., 2013) antibodies. Cy3-conjugated IgG (Jackson ImmunoResearch Laboratories, 1:400) and DyLight 649-conjugated IgG (Jackson ImmunoResearch Laboratories, 1:400) were used as secondary antibodies. Alexa Fluor 488-conjugated phalloidin (Invitrogen, 1:300) was used for staining the actin-rich stereocilia. The specimens were observed under a laser confocal microscope (LSM700; Carl Zeiss).
Scanning electron microscopy (SEM).
Inner ears from E17.5 Ick KO mouse embryos, P1 Ick CKO mouse pups, and 6- to 7-week-old mice from both sexes were dissected and fixed in 2% PFA and 2.5% glutaraldehyde in 0.1 m PB. Inner ears were dissected and postfixed with 1% OsO4 in H2O for 1 h on ice. Tissues were then dehydrated in an ethanol series and freeze-dried in t-butyl octanol or dried by isoamyl acetate in an HCP-2 critical-point dryer (Hitachi), followed by osmium tetroxide treatment, before observation with a scanning electron microscope (S4800; Hitachi).
Area measurements of IHCs in the apical turns of the cochlea.
The areas of IHCs labeled with palloidin in the apical turns of the cochlea were measured using ImageJ software. At least 70 cells were analyzed in each genotype.
Analysis of kinocilium position and stereociliary bundle orientation.
Kinocilium position and stereociliary bundle orientation were analyzed by SEM. Kinocilium positions were determined by drawing a line from the position of the base of the kinocilium to the center of the cell. We defined the angle of kinocilium position as the angle formed between this line and the line parallel to the mediolateral axis (α angle). In wild-type mice, this angle is close to 0° (see Fig. 5). The orientation of stereociliary bundles (β angle) was determined as described previously (Montcouquiol et al., 2003). When the stereociliary bundles are flat, the β angle was determined by the angle formed between the line parallel to the mediolateral axis and the line passing through the center of the line linking the ends of the stereociliary bundles and perpendicular to this line. Length of the kinocilium in IHCs and the cilium in SCs [Deiter's cells (DCs), outer pillar cells (OPCs), inner pillar cells (IPCs), and inner phalangeal cells (IPHs)] was analyzed by confocal microscopy. MetaMorph software (Molecular Devices) was used to measure cochlear length, kinocilium position, and stereociliary bundle orientation. ZEN software (Carl Zeiss) was used to measure the ciliary length of IHCs and SCs. Each row of HCs and SCs was divided into three regions according to the position along the length of the cochlea: basal, middle, and apical. At least 20 cells in each row in each region were analyzed. The kinocilium position (α angle) and the orientation of stereociliary bundles (β angle) were plotted (see Fig. 5B,C). The kinocilium position relative to the orientation of stereociliary bundles was analyzed by an absolute value of subdifference between α and β angles.
Figure 5.
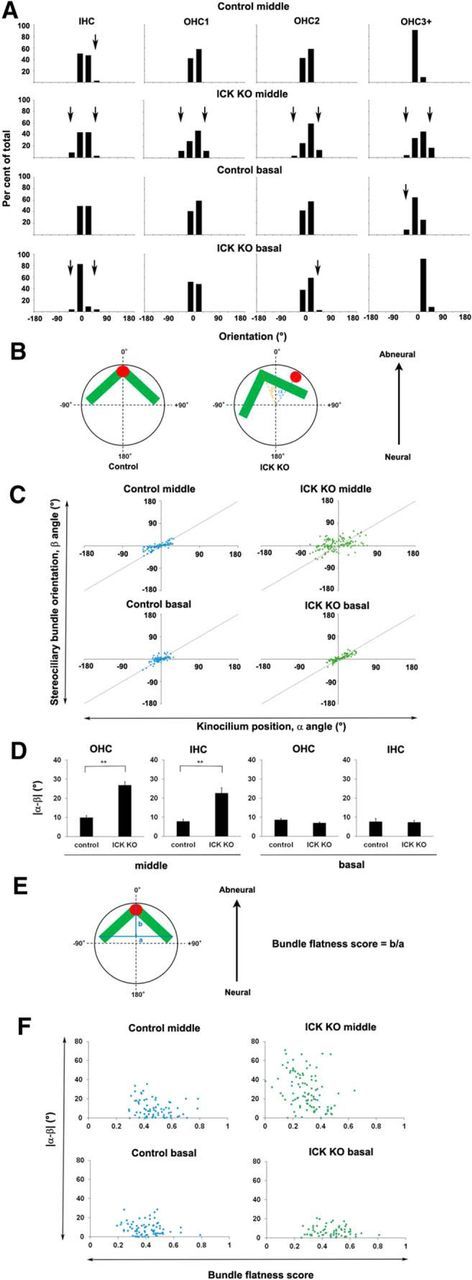
Ick is required for the normal kinocilium position and stereociliary bundle orientation. A, Distributions of stereociliary bundle orientations. Deviation of bundle orientations was more severe in OHC1 than in OHC2 and OHC3 in the middle turn of Ick−/− mice. Arrows show the percentages of the cells in which the absolute value of the angle of stereociliary bundle orientation is >30°. B, Schematic diagram of kinocilium position and stereociliary bundle orientation in HCs. α represents the angle of rotation from zero degrees to the kinocilium position; β represents the angle of rotation from zero degrees to the stereociliary bundle orientation, determined as the center of the stereociliary bundle. C, Scatter plot of the kinocilium position relative to the stereociliary bundle orientation. The kinocilium position was not correlated with the stereociliary bundle orientation in the middle turn of Ick−/− mice compared with that in control mice. However, in the basal turn of Ick−/− mice, the kinocilium position was correlated with the stereociliary bundle orientation similar to controls. D, Difference between α and β angles in IHCs and OHCs in the middle and the basal turns. In the middle turn, the difference between α and β angles was larger in Ick−/− mice than that in control mice. E, Schematic diagram of how to measure the bundle flatness score. F, Scatter plot of the bundle flatness score relative to the difference between α and β angles in OHCs. In the middle turn of Ick−/− cochlea, the bundle flatness score negatively correlated with the difference between α and β angles (correlation coefficient (r) = −0.35, p < 0.01). In contrast, in the middle and basal turns of control cochlea and in the basal turn of Ick−/− cochlea, no significant correlation between the bundle flatness score and the difference between α and β angles was detected. (r = −0.22, −0.16, and −0.11, p = 0.06, 0.17, and 0.38, respectively). Error bars indicate SE. **p < 0.01.
Auditory-brainstem response (ABR) and distortion product otoacoustic emission (DPOAE) measurements.
To assess hearing, mice were anesthetized with intraperitoneal administration of 2.5–10 mg/kg xylazine and 25–100 mg/kg of ketamine-HCl. Hearing was evaluated using ABR at 6–7 weeks of age. The ABR waveforms were recorded using BioSigRP software together with TDT system 3 Real-time Signal Processing System (Tucker Davis Technologies) under the condition using 30–3000 Hz band-pass filter settings and ABR waveforms from 500 click stimuli were averaged. Thresholds were defined as the sound intensity decreased by 10 dB steps. The intensity used was 0–100 dB SPL (and the threshold above this range was considered as 110 dB SPL). The number of animals tested was six control (Ickfloxflox; Pax2-Cre−, Ickflox/+; Pax2-Cre− and Ickflox/+; Pax2-Cre+) and five Ick CKO (Ickflox/flox; Pax2-Cre+) mice.
Measurements of DPOAE were tested bilaterally at the age of 6–7 weeks. The number of animals tested was five control and four Ick CKO mice. DPOAEs were measured by commercial instrumentation HearID Auditory Diagnostic System (Mimosa Acoustics) combined with CUBeDIS II version 2.40 software (Etymotic Research). DPOAE at the 2f1-f2 frequency was elicited using two tone stimuli, f1 and f2. A custom plastic ear tip attached to an ER-10C probe (Mimosa Acoustics) was inserted into the ear canal. DPOAE amplitudes were measured at f2 frequencies of 6, 12, 18, and 24 kHz and were plotted after substitution by noise floor amplitude.
H&E staining.
Inner ears were collected and fixed in 4% paraformaldehyde in PBS at 4°C overnight. After washing out the fixative with PBS, the tissues were decalcified in 0.12 m EDTA for 5 d at room temperature. The inner ears were embedded in paraffin, sectioned at 6 μm thickness, deparaffined, and stained with H&E.
Statistical analysis.
Statistical significance was evaluated using a Pearson's correlation coefficient test for Figure 5F or a Student's t test for other figures. Mean and SEs are shown.
Results
Ick is expressed in epithelial cells in the organ of Corti during development
To determine whether Ick is expressed in the inner ear, we performed in situ hybridization using developing and adult mouse inner ear sections (Fig. 1A–H). In the cochlea, Ick was highly expressed at E15.5 in epithelial cells, spiral ganglion, Reissner's membrane, stria vascularis, and spiral ligament (Fig. 1A). At E18.5, a steady signal in the same region was observed (Fig. 1B). The period from E14.5 to E18.5 is critical for stereociliary bundles and kinocilium polarization (Jones et al., 2008). At P2, Ick expression decreased in the epithelial cells in the organ of Corti (Fig. 1C,D). In the later stages of inner ear development, at P9 and P28, Ick expression decreased continuously (Fig. 1E–H). However, RT-PCR detected Ick expression at P28 (Fig. 1I). We then examined Ick expression in the developing vestibule (Fig. 1J–Q). In the vestibule, Ick signal was detected in epithelial cells in the crista and macula through developmental stages (Fig. 1J–M). Ick expression slightly decreased at P9 and became barely detectable at P28 (Fig. 1N–Q).
Figure 1.
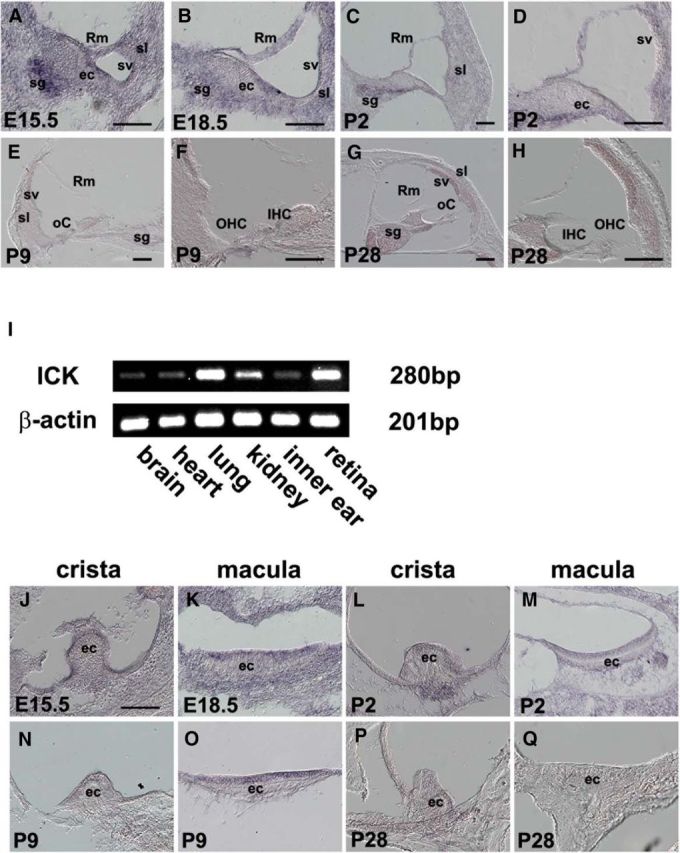
Ick expression in developing cochlear and vestibular cells. A–H, In situ hybridization analysis of mouse Ick in the developing and adult cochlear cross-sections. Ick was highly expressed at E15.5 (A) and E18.5 (B) in epithelial cells (ec), spiral ganglion (sg), Reissner's membrane (Rm), stria vascularis (sv), and spiral ligament (sl). After P2, Ick expression decreased in the organ of Corti (oC), including IHCs and OHCs, sg, Rm, sv, and sl (C–H). I, RT-PCR analysis of mouse Ick at P28. Ick expression is retained at P28. J–Q, In situ hybridization analysis of mouse Ick in developing and adult vestibule cross-sections. Ick was highly expressed in the crista at E15.5 (J) and in the macula at E18.5 (K). At P2, Ick signal was detected in the crista (L) and macula (M). At P9, Ick expression decreased in both the crista (N) and macula (O). At P28, Ick was barely detectable (P, Q). Scale bars, 100 μm.
Ick deletion causes a defect in cochlear extension
To determine a role for Ick in development of the organ of Corti, we first examined the length of the cochlea at late embryonic stages in control and Ick−/− mice. The cochlea is elongated from a shorter and thicker primordium through cellular rearrangement characteristic of convergent extension (Jones et al., 2008; Rida and Chen, 2009). According to the evidence to date, it appears that the PCP pathway regulates convergent extension in addition to the orientation of epithelial cells (Montcouquiol et al., 2003; Rida and Chen, 2009). At E17.5, the length of cochlear ducts in Ick−/− mice was significantly shorter than that in control littermates (Fig. 2A–C). In the apical and middle turns of the Ick−/− cochlea, OHCs were disorganized with the number of OHC rows increased to four or five rows (OHC3+) compared with the usual three rows of OHCs (OHC1–3s), consistent with a defect in convergent extension (Fig. 2D–G). In the apical turn, IHCs in Ick−/− mice were bigger than in control mice (control mice, 12.8 ± 0.35 μm2; Ick−/− mice, 14.3 ± 0.33 μm2, p < 0.01). These data suggest that Ick is required for normal convergent extension processes in inner ear development.
Figure 2.

Ick regulates cochlear extension. A, B, Stereo micrographs of whole cochlear ducts isolated from control (A) and Ick−/− mice (B) at E17.5. The length of cochlear ducts in Ick−/− mice (n = 3) was shorter than that in control mice (C). D–G, Confocal images of the surfaces of cochlear whole mounts in the apical and the middle turns isolated from control (D, F) and Ick−/− mice (E, G) at E17.5. In the Ick−/− cochlea, additional rows of OHCs were observed. Scale bars: A, B, 200 μm; D–G, 10 μm. Error bars indicate SE. *p < 0.05.
Ick deletion affects the kinocilium position and orientation of stereociliary bundles
To examine Ick function in inner ear development, we next performed SEM analysis on the middle and basal turns in the cochlea at E17.5 (Fig. 3A–L). In the middle and basal turns of the control cochlea, normal kinocilium position and orientation of stereociliary bundles were observed in both OHCs and IHCs (Fig. 3A,B, E–H). In contrast, compared with the V-shaped stereociliary bundles in the control cochlea, we observed flattened stereociliary bundles both in Ick−/− OHCs and IHCs (Fig. 3C,I, red arrowheads). In addition, the Ick−/− cochlea displayed aberrant localization of kinocilium and misorientation of stereociliary bundles in the middle turn (Fig. 3C,J, green arrows and blue arrowheads), suggesting that PCP is impaired in the Ick−/− cochlea. In contrast, in the basal turn of the Ick−/− cochlea, no significant PCP defect was observed in either OHCs or IHCs (Fig. 3D,K,L).
Figure 3.
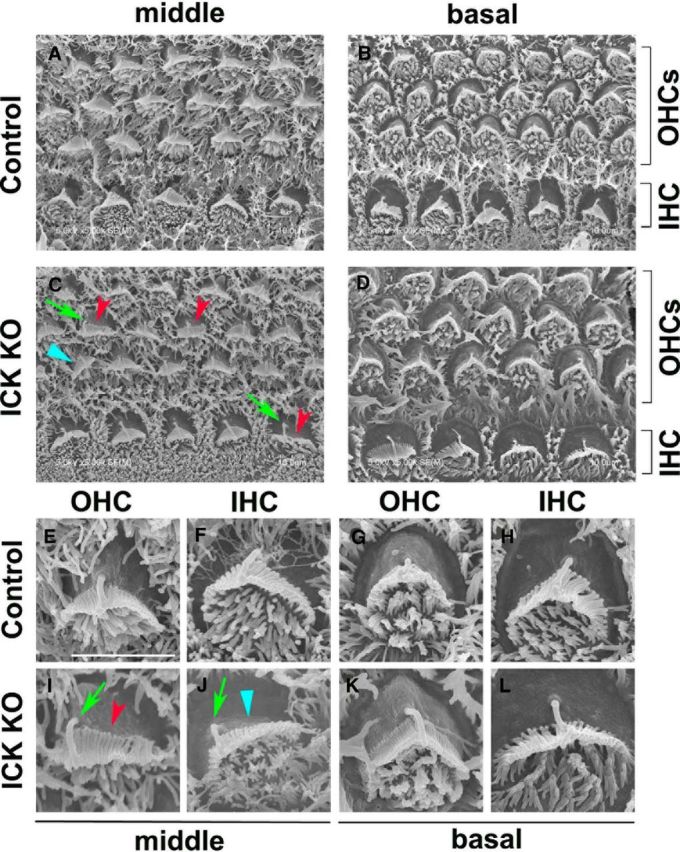
Ick is required for PCP establishment in cochlea development. A–L, SEM in the middle and the basal turns of the organ of Corti isolated from control (A, B, E–H) and Ick−/− (C, D, I–L) mice at E17.5. Ick−/− OHCs and IHCs displayed aberrant localization of the kinocilium (C, I, J, green arrows) and misoriented (C, J, blue arrowheads) or flattened stereociliary bundles (C, I̧ red arrowheads) in the middle turn. In the basal turns of Ick−/− OHCs and IHCs, these PCP defects were not observed (D, K, L). Scale bars: E–L, 3 μm.
To determine whether Ick is required for vestibular development, we observed the vestibule by SEM and immunostaining using antibodies against acetylated tubulins and phalloidin. As far as we examined, we observed no significant difference between control and Ick−/− vestibules (Fig. 4A–F). PCP formation in the vestibule was further assessed by immunostaining using an anti-pericentrin antibody and phalloidin. In the utricle of both control and Ick−/− mice, HCs were uniformly oriented with their polarized basal bodies labeled with pericentrin pointed toward the line of polarity reversal (Fig. 4G,H), suggesting that PCP is unaffected in the Ick−/− vestibule.
Figure 4.
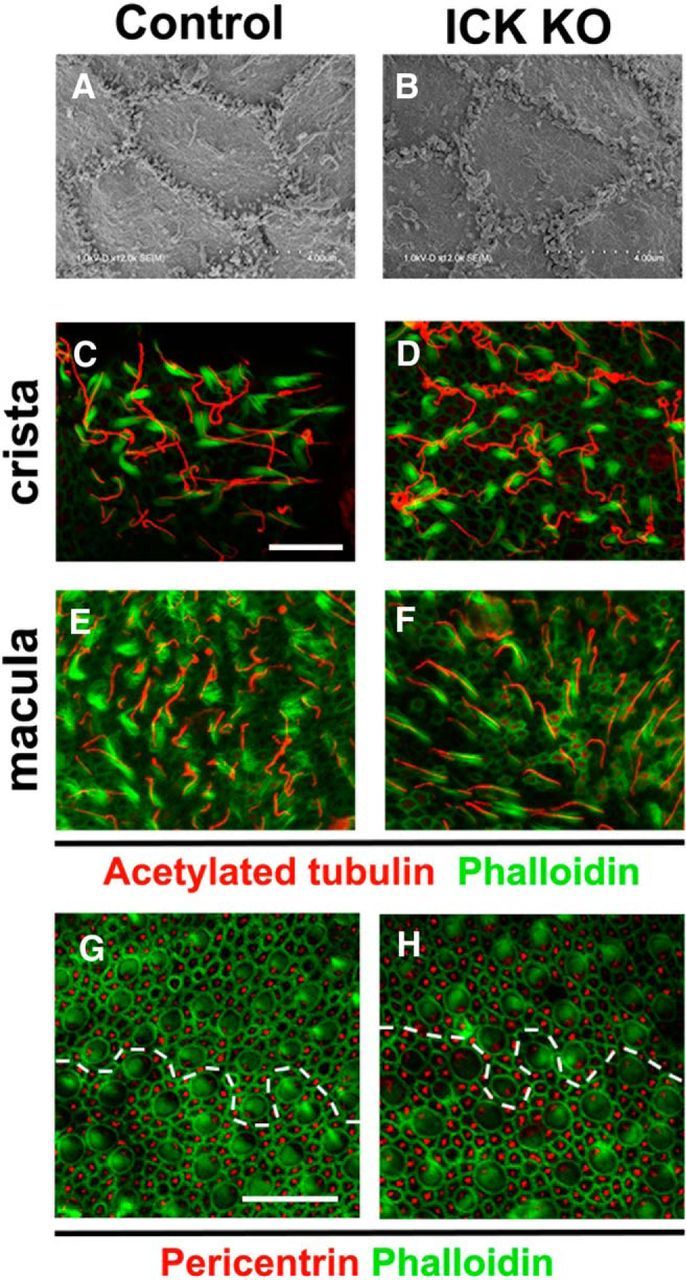
The Ick−/− vestibule is unaffected. A, B, SEMs from the apical surface of crista. C–F, Confocal images of the surfaces of vestibule whole mounts isolated from control (C, E) and Ick−/ − (D, F) mice at E17.5. In both crista (A–D) and macula (E, F), no significant difference between control and Ick−/− mice was observed. G, H, Confocal images of the surfaces of the whole-mount utricles isolated from control (G) and Ick−/ − (H) mice at E17.5. In both utricles, basal bodies in HCs pointed toward the line of polarity reversal (dotted lines). Scale bars: C–H, 20 μm.
To further investigate whether PCP signaling molecules are affected in the Ick−/− cochlea, we analyzed the deviation of stereociliary bundle orientation. The percentages of the cells in which an absolute value of the angle of stereociliary bundle orientation were >30° were 24% in OHC1, 16% in OHC2, and 21% in OHC3+, whereas in the control cochlea, those cells were barely observed, suggesting that the orientation defect was more severe in OHC1 than that in OHC2 and OHC3 (Fig. 5A). This phenotype is similar to the abnormal PCP phenotypes reported in the Ift88−/− cochlea, as well as in the cultured cochleae in which Gαi function is inhibited (Jones et al., 2008; Ezan et al., 2013), whereas it is opposite of the PCP phenotypes detected in homozygotes for the LP mutation (a spontaneous mutation in Vangl2) and Fz3−/−;Fz6−/− mice (Montcouquiol et al., 2003; Wang et al., 2006).
We then examined the correlation between orientation of stereociliary bundles and kinocilium position by quantifying the difference between the angle of kinocilium position (α angle) and the angle of stereociliary bundle orientation (β angle) in control and Ick−/− HCs (Fig. 5B). In control HCs, kinocilium position was consistent with the orientation of stereociliary bundles in both middle and basal turns. In contrast, the correlations between kinocilium position and stereociliary bundle orientation were inconsistent in the middle turn of Ick−/− HCs (Fig. 5C). The difference between α and β angles was significantly larger in both Ick−/− OHCs and IHCs than that in control HCs (Fig. 5D). These defects were not observed in the basal turn. These results suggest that Ick is required for coupling the kinocilium movement with the movement of stereociliary bundles.
Stereociliary bundle flatness was analyzed in OHCs, which have a distinct V-shape array in wild-type mice and thus are more suitable for quantification of the bundle flatness than IHCs. We measured the width (a) and the height (b) of a triangle with its base formed by the line connecting between the ends of the V-shaped array, where the bundle flatness score was calculated as b/a, which becomes 0 when the bundle is totally flat (Fig. 5E). We investigated the correlation of the bundle flatness with the difference between α and β angles in each cell. In the middle turn of cochlea, the bundle flatness score negatively correlated with the difference between α and β angles in Ick−/− mice but not in control mice, suggesting the correlation between PCP defects and bundle flatness. In contrast, no significant correlation was detected in the basal turn of the cochlea both in Ick−/− mice and in control mice (Fig. 5F).
Loss of Ick affects ciliary transport in both HCs and SCs
During cochlear PCP establishment, two PCP signaling pathways cooperate to control kinocilium migration and orientation of stereociliary bundles (Ezan et al., 2013). One is the core PCP signaling, which is required for neural-to-abneural gradient of cellular differentiation. The other is G-protein-dependent signaling, which is activated shortly after the PCP signaling pathway establishes the neural-to-abneural cues and instructs kinocilium migration. Furthermore, mice with cilium-associated gene mutations are known to exhibit PCP defects in the cochlea (Ross et al., 2005; Jones et al., 2008; Sipe and Lu, 2011).
To determine whether Ick genetically interacts with core PCP proteins, G-protein-dependent-proteins, or cilium-associated proteins, we performed immunostaining analysis of control and Ick−/− cochleae. We assessed whether the localizations of Vangl2, mammalian partner of inscuteable (mPins), and Ift88 are affected in Ick−/− HCs and SCs at E17.5. Ift88 is required for the assembly of primary cilia (Lehman et al., 2008). In the middle turn of the control cochlea, Ift88 was localized along the ciliary axoneme and at the base in HCs (Fig. 6A,B) and SCs (Fig. 6A,C). In contrast, in the middle turn of the Ick−/− cochlea, Ift88 localization along the axoneme decreased and Ift88 was enriched at the ciliary tip and base in both HCs (Fig. 6D,E) and SCs (Fig. 6D,F). The enrichment of Ift88 at the ciliary tip and the decrease of Ift88 along the ciliary axoneme were observed, not only in the middle turn, but also in the basal turn (Fig. 6G–L). A core PCP protein, Vangl2, and a G-protein-dependent-protein, mPins, were distributed normally both in control and Ick−/− middle turns of the cochlea (Fig. 6M–T). In the Ick−/− cochlea, cilia on the surface of SCs were abnormally elongated both in the middle and basal turns (Fig. 6D,J). These data suggest that Ick loss impairs ciliary transport in both HCs and SCs.
Figure 6.
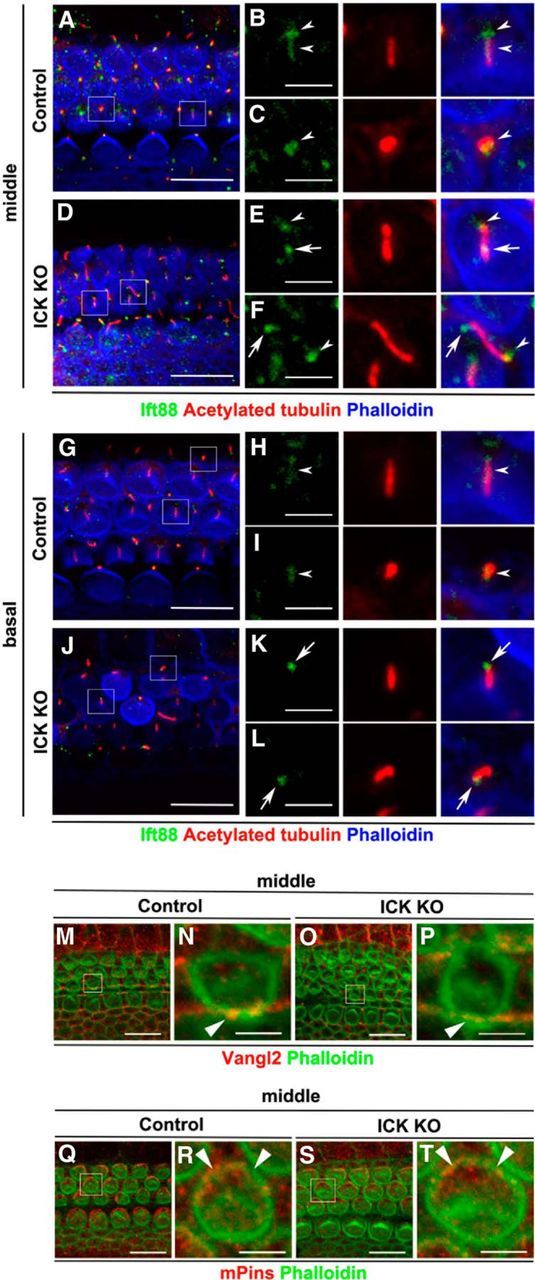
Loss of Ick impairs ciliary transport in both HCs and SCs. A–T, Confocal images of the surfaces of cochlear whole mounts isolated at E17.5 from control (A–C, G–I, M, N, Q, R) and Ick−/− mice (D–F, J–L, O, P, S, T). In the control middle turn of the cochlea, Ift88 was localized along the ciliary axoneme and at the base in HCs (A, B, arrowheads) and in OPCs (A, C, arrowhead). In the Ick−/− middle turn of the cochlea, Ift88 protein level localized along the axoneme decreased and Ift88 was enriched at the ciliary tip and at the base in HCs (D, E, arrow, ciliary tip; arrowhead, the base) and in OPCs (D, F, arrow, ciliary tip; arrowhead, the base). In the control basal turn of the cochlea, Ift88 was localized along the ciliary axoneme in HCs (G, H, arrowhead) and in DCs (G, I, arrowhead). In the Ick−/− basal turn of cochlea, the Ift88 protein level localized along the axoneme markedly decreased in HCs as well as in OPCs (J–L). Ift88 was enriched at the ciliary tip in HCs (J, K, arrow) and in DCs (J, L, arrow). Localization of a core PCP protein, Vangl2 (M–P, arrowheads), and a G-protein-dependent-protein, mPins (Q–T, arrowheads), was normal in both control and Ick−/− middle turns of the cochlea. Scale bars: A, D, G, J, M, O, Q, S, 10 μm; B, C, E, F, H, I, K, L, N, P, R, T, 2 μm.
Ick-deficient mice show elongated cilia in inner ear SCs
To further examine the possible role of Ick in the regulation of cilia length in inner ear development, we generated mice with a conditional deletion of Ick in the developing inner ear because the Ick KO mutation is lethal at P0. We crossed Ick-floxed mice (Chaya et al., 2014) with a transgenic mouse line expressing Cre recombinase under the control of the Pax2 locus in the otocyst at E9.5 (Soriano, 1999; Ohyama and Groves, 2004) to generate ICKflox/flox;Pax2-Cre+ (Ick CKO) mice. We performed immunostaining to analyze the Ick CKO cochlea at P1 because kinocilium disappears until P21. Ciliary length in SCs in Ick CKO cochlea was longer than that in the control cochlea (Fig. 7A–F). The mammalian organ of Corti contains several types of interdigitating SCs: three rows of DCs (DC1–3s), OPCs, IPCs, and IPHs (Fig. 7G). Along the total length of the cochlea, cilia were significantly elongated in DCs, OPCs, and IPCs (Fig. 7H–L). In contrast, ciliary length of IPHs in the Ick CKO cochlea were significantly longer than that in the control cochlea only in the apical turn (Fig. 7M) and kinocilia in IHCs were elongated in the Ick CKO cochlea only in the basal turn (Fig. 7N). Furthermore, in SEM analysis, aberrant localization of kinocilium and misorientation or flattened shape of stereociliary bundles in HCs were observed from the apical to the middle turn (Fig. 7O–R). In addition, the tips of cilia on the surface of SCs bulged in the Ick CKO cochlea, suggesting defects of the retrograde IFT (Fig. 7P–R). These data suggest that Ick has an essential role for ciliogenesis in cochlear development.
Figure 7.
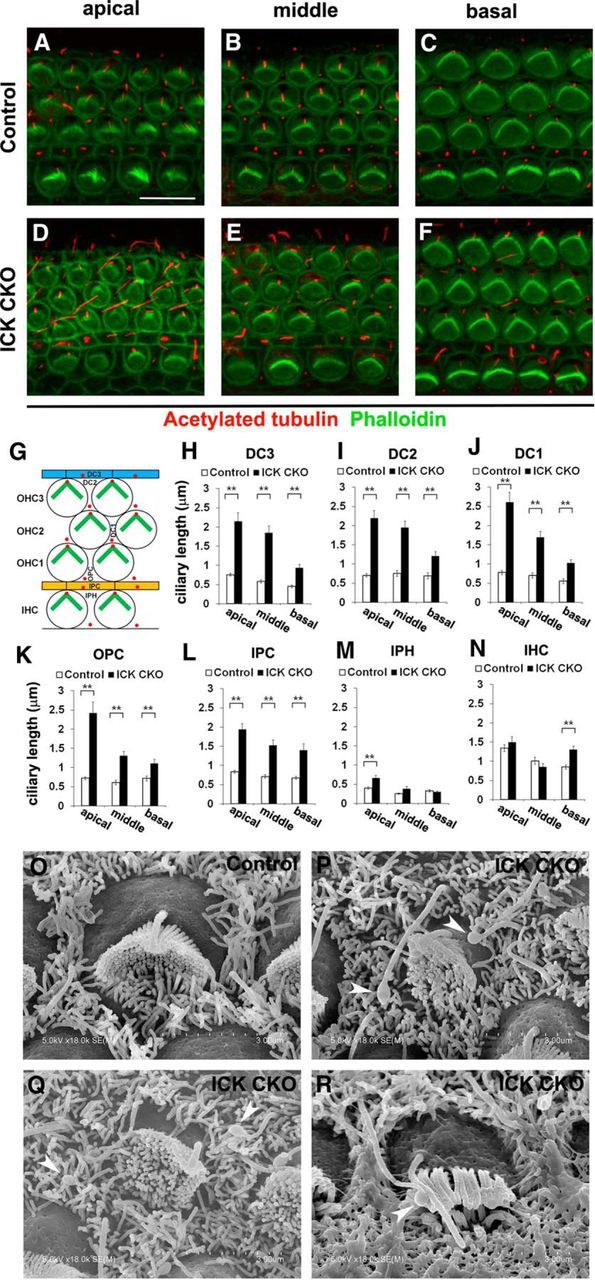
Disruption of Ick causes elongated cilia in SCs. A–F, Confocal images of the surfaces of cochlear whole mounts in the apical, middle, and basal turns isolated from control (A–C) and Ick CKO (D–F) mice at P1. In all regions of the cochlea, cilia in SCs appeared elongated. G, Schematic diagram of specialized cell types organizing the organ of Corti including DC1–3s, OPCs, IPCs, IPHs, OHC1–3s, and a single row of IHCs. H–N, Ciliary length of SCs (H–M) and IHCs (N). In DC1–3s, OPCs, and IPCs of Ick CKO mice, cilia were elongated in all regions of the cochlea (H–L). In IPHs of Ick CKO mice, cilia were significantly elongated in only the apical region (M). In IHCs of Ick CKO mice, kinocilia were elongated in the basal region (N). O–R, SEMs of HCs isolated from control (O) and Ick CKO (P–R) mice at P1. P, Q, Ick CKO apical OHCs. R, Ick CKO middle IHC. Aberrant localization of kinocilium and misorientation or flattening of stereociliary bundles were observed in the apical and the middle turns of Ick CKO mice. In addition, the tips of cilia on the surface of SCs bulged in Ick CKO mice (arrowheads). Scale bars: A–F, 10 μm. Error bars indicate SE. **p < 0.01.
Mature Ick CKO mice display auditory dysfunction and PCP defects in HCs
To examine a role for Ick in auditory function, we first tested ABRs of control and Ick CKO mice. We tested hearing in 6- to 7-week-old Ick CKO and control mice using ABR, with broadband click stimuli corresponding to the low frequencies at 2–4 kHz and tone-burst stimuli at 8, 16, 24, and 32 kHz. As shown by click stimuli and tone-burst stimuli at low frequencies of 8 kHz and 16 kHz, Ick CKO mice exhibited significantly higher thresholds compared with those of control littermates (Fig. 8A,B). In contrast, tone-burst stimuli at 24 and 32 kHz evoked no significant difference in the threshold between control and Ick CKO mice (Fig. 8B). We further tested DPOAEs to study OHC function. At stimulating frequencies of 6, 12, and 18 kHz, a significant decrease in the DPOAE level was detected in Ick CKO mice and no significant decrease was observed with stimulus at 24 kHz between Ick CKO and control littermates (Fig. 8C).
Figure 8.
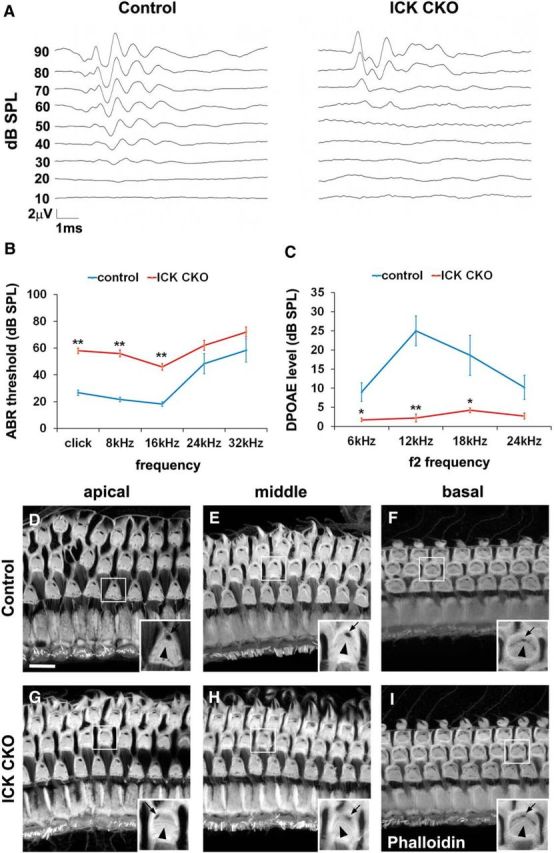
Loss of Ick causes auditory dysfunction and PCP defects in HCs. A, Representative ABR waveforms from an Ick CKO mouse and a control mouse at 6 weeks of age. B, ABR thresholds of 6- to 7-week-old Ick CKO mice (n = 5) (red) and controls (n = 6) (blue). ABR thresholds of Ick CKO mice were higher than those in controls at the frequencies <24 kHz. C, DPOAE levels of 6- to 7-week-old Ick CKO mice (n = 4) (red) and controls (n = 5) (blue). DPOAE levels of Ick CKO mice decreased compared with those of control mice at frequencies <24 kHz. D–I, Confocal images of the surfaces of cochlear whole mounts in the apical, middle and basal turns isolated from 6- to 7-week-old controls (D–F) and Ick CKO mice (G–I). Arrows indicate the position of the basal body and arrowheads show the orientation of stereociliary bundles. In the apical and middle turns of the Ick CKO cochlea, OHCs exhibited aberrant positions of the basal body, and misorientation or flattening of stereociliary bundles (G, H). Scale bars, 10 μm. Error bars indicate SE. *p < 0.05; **p < 0.01.
Next, we performed whole-mount phalloidin staining of control and Ick CKO cochleae to ascertain morphological changes in HCs. The 6- to 7-week-old control cochlea showed a normal position of the basal body (Fig. 8D–F, arrows), which is located at the vertex of the V-shaped stereociliary bundles, and a normal orientation of stereociliary bundles (Fig. 8D–F, arrowheads). In contrast, Ick CKO cochleae showed the aberrant positioning of the basal body (Fig. 8G,H, arrows), as well as the misorientation or flattened shape of stereociliary bundles (Fig. 8G,H, arrowheads), in the apical and middle turns. These PCP defects were not observed in the basal turn of the Ick CKO cochleae (Fig. 8I). We further performed H&E staining to explore other morphological changes in control and Ick CKO cochleae except HCs. No significant difference was observed in the spiral ganglion, stria vascularis, spiral ligament, or Reissner's membrane (Fig. 9A–F).
Figure 9.
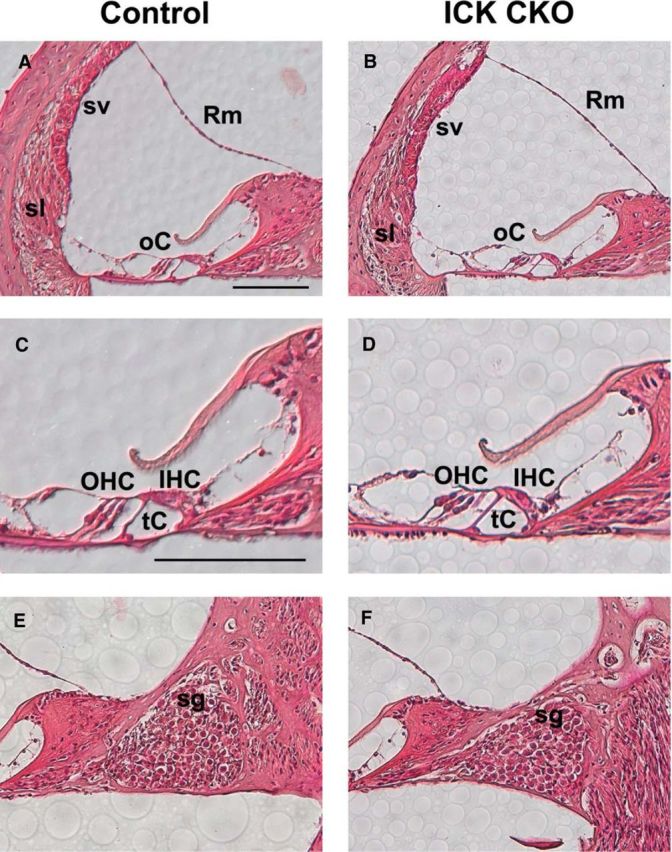
Ick CKO cochlea shows normal morphology. A–F, H&E analysis of the 6-week-old control (A, C, E) and Ick CKO middle turns of cochlea (B, D, F). C, D, Close-up images of the organ of Corti. No significant morphological difference between control and Ick CKO cochleae was observed in stria vascularis (sv), spiral ligament (sl), and Reissner's membrane (Rm), the tunnel of Corti (tC), or spiral ganglion (sg). Scale bars, 100 μm.
Discussion
PCP defects cause auditory dysfunction in Ick CKO mice
In the current study, we found that Ick CKO mice exhibit auditory dysfunction and PCP defects from the apical to middle turn in the cochlea. Do PCP defects cause auditory dysfunction in Ick CKO mice? The planar polarity of HCs depends on cellular differentiation at early stages and the reorientation process at later stages during development. The differentiation gradient spreads along both the basal-to-apical and neural-to-abneural axis of the cochlea (Dabdoub and Kelley, 2005). The later reorientation process aligns the stereociliary bundles correctly along the mediolateral axis of the organ of Corti. During inner ear development, there is a critical period during which planar polarity defects are amended to some extent (Copley et al., 2013). If PCP defects are not amended during this critical period, then the defects remain fixed and the consequence of the PCP defects on hearing function can be assessed. There have been several studies on the correlation between PCP defects and auditory dysfunction in Mkks, Alms1, and Vangl2 mutant mice (Ross et al., 2005; Jagger et al., 2011; Copley et al., 2013) (Table 1). Briefly, in all of these mutant mice, PCP defects including misorientation of stereociliary bundles were observed. Furthermore, in the Mkks and Alms1 mutant mice, flattened stereociliary bundles and kinocilia mislocalization were detected. Loss of HCs, a morphological change in addition to PCP defects, was observed in Alms1−/− mice. In all of these mutant mice, auditory dysfunction was detected, but it is difficult to conclude whether PCP defects caused the auditory dysfunction because the chochlea regions where PCP defects were observed did not correspond to the frequency spacing of the auditory dysfunction detected. Therefore, it is unclear whether auditory dysfunctions in these mutant mice are caused by PCP defects in the cochlea. In the current study, the Ick−/− mice also showed kinocilia mislocalization and the misorientation or flattening of stereociliary bundles as observed in Mkks, Alms1, and Vangl2 mutant mice. Interestingly, the Ick CKO cochlea displayed significant PCP defects consistent with those observed in the Ick−/− cochlea from the apical to middle turn and auditory dysfunction in Ick CKO mice was detected at frequencies <24 kHz. According to the mouse cochlear place-frequency map, the apical and middle regions of the cochlea tonotopically correspond to frequency spacing <24 kHz (Müller et al., 2005). In addition, in the 6-week-old Ick CKO cochlea, no significant morphological change except for the PCP defects in the apical and middle turns was observed. These observations suggest that auditory dysfunction in the Ick CKO mice is caused by PCP defects in the cochlea.
Table 1.
Summary of phenotypes in mouse models related to PCP
| Gene | Mutant | Stage | Region in cochlea | Cell type | PCP defects |
Loss of HCs | Range of auditory dysfunction | Reference | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Misorientation of bundles | Flattened bundles | Kinocilia mislocalization | ||||||||
| Mkks | Mkks−/− | Embryo | NA | OHCs | NA | + | + | NA | NA | Ross et al., 2005 |
| Adult | NA | OHCs | + | + | NA | NA | NA | |||
| Alsm1 | Alms1−/− | Pup | Whole | OHCs | + | NA | + | + | NA | Jagger et al., 2011 |
| Adult | Apical | OHCs | + | NA | NA | + | High | |||
| Vangl2 | Pax2-Cre+;Vangl2ΔTMs/LoxP | Embryo | Whole | OHCs/IHCs | + | NA | NA | - | NA | Copley et al., 2013 |
| Adult | Apical | OHCs/IHCs | + | NA | NA | - | Low-high | |||
| Ick | Pax2-Cre;Ickflox/flox | Pup | Apical/middle | OHCs/IHCs | + | + | + | - | NA | Present study |
| Adult | Apical/middle | OHCs/IHCs | + | + | + | - | Low-middle | |||
| Ick−/− | Embryo | Apical/middle | OHCs/IHCs | + | + | + | - | NA | ||
NA, Not available
Ick is essential for PCP establishment and cilia formation in the cochlea
We found that Ick is required for PCP establishment in HCs by showing that Ick controls the position of kinocilium and orientation of stereociliary bundles. We also confirmed a role for Ick in cilia formation in the organ of Corti by demonstrating that Ick controls ciliary transport in both HCs and SCs. Here, a matter of great concern is whether PCP defects are caused by ciliary dysfunction. Several studies have connected the primary cilium and PCP formation. The cilium restricts canonical Wnt signaling (Corbit et al., 2008) and acts as a switch from canonical Wnt signaling to noncanonical Wnt/PCP signaling (Simons et al., 2005). Vangl2 and Dvl are observed within or at the base of the cilia (Ross et al., 2005; Park et al., 2008). Furthermore, mice defective in ciliary proteins, including Mkks, Mks1, Alsm1, and Kif3a, display the mislocation of kinocilium and/or basal body relative to the stereociliary bundle vertex as was observed in Ick mutant mice (Ross et al., 2005; Cui et al., 2011; Jagger et al., 2011; Sipe and Lu, 2011). Although mice defective in Kif3a and Mkks show an absence of or shortened kinocilia, kinocilia length appears normal in Mks1 mutant mice (Cui et al., 2011; Sipe and Lu, 2011; Rachel et al., 2012). The siRNA-mediated reduction of Mks1 activity results in blocked centriole migration to the apical membrane, indicating that Mks1is required for centriole positioning (Dawe et al., 2007). In our study, the Ick−/− cochlea showed misorientation of stereociliary bundles and mispositioning of kinocilium relative to the bundle vertex and accumulation of Ift88 at the tip of kinocilium. Previously, we reported that Ick may regulate disassembly between IFT-A and IFT-B subcomplexes in mouse embryonic fibroblasts (MEFs) (Chaya et al., 2014). Accumulation of Ift88 at the tip of the kinocilium indicates that, in HCs, Ick may control transport switching at the tip of the kinocilium. Considering our data together with previous observations suggests that ciliary dysfunction induces the abnormal coupling of the kinocilium and stereociliary bundles, resulting in PCP defects.
In addition, Ick-deficient cochlea displayed elongated cilia in SCs. Why are cilia in Ick-deficient cochlea elongated? Ciliary length is controlled by the precise balance between tubulin assembly and disassembly. These processes require a highly conserved IFT system, which is a bidirectional movement of the protein complex. Tubulin is transported by IFT and simple diffusion. In growing cilia, the concentration of soluble tubulin transported by IFT increases, resulting in the elongation of axonemal microtubules (Craft et al., 2015). Tubulin disassembly is promoted by proteins possessing microtubule-depolymerizing activity (Blaineau et al., 2007; Kobayashi et al., 2011; Niwa et al., 2012). We showed that disruption of Ick leads to Ift88 accumulation at the tip of cilia and cilia elongation in SCs in the cochlea, suggesting that IFT dysfunction causes abnormally elongated cilia in the Ick−/− cochlea. It was reported previously that Ick knockdown increased cilia length and that IFT speed is regulated by Ick in cultured cells (Broekhuis et al., 2014). A loss of IFT speed control by Ick is a possible mechanism underlying the cilia elongation in the Ick−/− cochlea. Although several studies reported elongated cilia in Ick knockdown-cultured cells and in Ick-deficient mouse embryonic limb buds (Broekhuis et al., 2014; Moon et al., 2014; Paige Taylor et al., 2016), we and others observed shortened cilia in the Ick-deficient neural tubes, Ick-deficient MEFs, and primary fibroblasts derived from a human ECO patient (Chaya et al., 2014; Oud et al., 2016). Ick may control ciliary length by different mechanisms in different cell types. Future analysis will be needed to clarify the molecular mechanisms by which Ick controls cilia formation and contributes to PCP establishment in the inner ear.
Footnotes
We thank A.K. Groves (Baylor College of Medicine) for the Pax2-Cre mouse, J.P. Borg (Centre de Recherche en Cancérologie de Marseille) for the anti-Vangl2 antibody, M. Montcouquiol (INSERM Université Bordeaux) for the anti-mPins antibody, and M. Kadowaki, A. Tani, A. Ishimaru, Y. Tojima, K. Hasegawa, H. Abe, and S. Kennedy for technical assistance. This work was supported by Grant-in-Aid for Scientific Research (B) (15H04669) and (C) (16K08583), Young Scientists (B) (15K189550) from Japan Society for the Promotion of Science (JSPS), Nanotechnology Platform (12024046) from The Ministry of Education, Culture, Sports, Science and Technology (MEXT), The Takeda Science Foundation, Senri Life Science Foundation, The Sumitomo Foundation, Koyanagi Foundation, KANAE Foundation for the Promotion of Medical Science, and Terumo Foundation for Life Science and Arts Life Science support program.
The authors declare no competing financial interests.
References
- Belotti E, Puvirajesinghe TM, Audebert S, Baudelet E, Camoin L, Pierres M, Lasvaux L, Ferracci G, Montcouquiol M, Borg JP (2012) Molecular characterisation of endogenous Vangl2/Vangl1 heteromeric protein complexes. PLoS One 7:e46213. 10.1371/journal.pone.0046213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaineau C, Tessier M, Dubessay P, Tasse L, Crobu L, Pagès M, Bastien P (2007) A novel microtubule-depolymerizing kinesin involved in length control of a eukaryotic flagellum. Curr Biol 17:778–782. 10.1016/j.cub.2007.03.048 [DOI] [PubMed] [Google Scholar]
- Broekhuis JR, Verhey KJ, Jansen G (2014) Regulation of cilium length and intraflagellar transport by the RCK-kinases ICK and MOK in renal epithelial cells. PLoS One 9:e108470. 10.1371/journal.pone.0108470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaya T, Omori Y, Kuwahara R, Furukawa T (2014) ICK is essential for cell type-specific ciliogenesis and the regulation of ciliary transport. EMBO J 33:1227–1242. 10.1002/embj.201488175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copley CO, Duncan JS, Liu C, Cheng H, Deans MR (2013) Postnatal refinement of auditory hair cell planar polarity deficits occurs in the absence of Vangl2. J Neurosci 33:14001–14016. 10.1523/JNEUROSCI.1307-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Chen MH, Chuang PT, Reiter JF (2008) Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat Cell Biol 10:70–76. 10.1038/ncb1670 [DOI] [PubMed] [Google Scholar]
- Cotanche DA, Corwin JT (1991) Stereociliary bundles reorient during hair cell development and regeneration in the chick cochlea. Hear Res 52:379–402. 10.1016/0378-5955(91)90027-7 [DOI] [PubMed] [Google Scholar]
- Craft JM, Harris JA, Hyman S, Kner P, Lechtreck KF (2015) Tubulin transport by IFT is upregulated during ciliary growth by a cilium-autonomous mechanism. J Cell Biol 208:223–237. 10.1083/jcb.201409036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C, Chatterjee B, Francis D, Yu Q, SanAgustin JT, Francis R, Tansey T, Henry C, Wang B, Lemley B, Pazour GJ, Lo CW (2011) Disruption of Mks1 localization to the mother centriole causes cilia defects and developmental malformations in Meckel-Gruber syndrome. Dis Model Mech 4:43–56. 10.1242/dmm.006262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabdoub A, Kelley MW (2005) Planar cell polarity and a potential role for a Wnt morphogen gradient in stereociliary bundle orientation in the mammalian inner ear. J Neurobiol 64:446–457. 10.1002/neu.20171 [DOI] [PubMed] [Google Scholar]
- Dawe HR, Smith UM, Cullinane AR, Gerrelli D, Cox P, Badano JL, Blair-Reid S, Sriram N, Katsanis N, Attie-Bitach T, Afford SC, Copp AJ, Kelly DA, Gull K, Johnson CA (2007) The Meckel-Gruber syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet 16:173–186. [DOI] [PubMed] [Google Scholar]
- Ezan J, Lasvaux L, Gezer A, Novakovic A, May-Simera H, Belotti E, Lhoumeau AC, Birnbaumer L, Beer-Hammer S, Borg JP, Le Bivic A, Nürnberg B, Sans N, Montcouquiol M (2013) Primary cilium migration depends on G-protein signalling control of subapical cytoskeleton. Nat Cell Biol 15:1107–1115. 10.1038/ncb2819 [DOI] [PubMed] [Google Scholar]
- Jagger D, Collin G, Kelly J, Towers E, Nevill G, Longo-Guess C, Benson J, Halsey K, Dolan D, Marshall J, Naggert J, Forge A (2011) Alstrom syndrome protein ALMS1 localizes to basal bodies of cochlear hair cells and regulates cilium-dependent planar cell polarity. Hum Mol Genet 20:466–481. 10.1093/hmg/ddq493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C, Roper VC, Foucher I, Qian D, Banizs B, Petit C, Yoder BK, Chen P (2008) Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat Genet 40:69–77. 10.1038/ng.2007.54 [DOI] [PubMed] [Google Scholar]
- Kelly M, Chen P (2007) Shaping the mammalian auditory sensory organ by the planar cell polarity pathway. Int J Dev Biol 51:535–547. 10.1387/ijdb.072344mk [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Tsang WY, Li J, Lane W, Dynlacht BD (2011) Centriolar kinesin Kif24 interacts with CP110 to remodel microtubules and regulate ciliogenesis. Cell 145:914–925. 10.1016/j.cell.2011.04.028 [DOI] [PubMed] [Google Scholar]
- Lahiry P, Wang J, Robinson JF, Turowec JP, Litchfield DW, Lanktree MB, Gloor GB, Puffenberger EG, Strauss KA, Martens MB, Ramsay DA, Rupar CA, Siu V, Hegele RA (2009) A multiplex human syndrome implicates a key role for intestinal cell kinase in development of central nervous, skeletal, and endocrine systems. Am J Hum Genet 84:134–147. 10.1016/j.ajhg.2008.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman JM, Michaud EJ, Schoeb TR, Aydin-Son Y, Miller M, Yoder BK (2008) The Oak Ridge Polycystic Kidney mouse: modeling ciliopathies of mice and men. Dev Dyn 237:1960–1971. 10.1002/dvdy.21515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DJ, Anniko M (1985) Developmental morphology of the mouse inner ear: a scanning electron microscopic observation. Acta Otolaryngol Suppl 422:1–69. [PubMed] [Google Scholar]
- Miyata Y, Nishida E (1999) Distantly related cousins of MAP kinase: biochemical properties and possible physiological functions. Biochem Biophys Res Commun 266:291–295. 10.1006/bbrc.1999.1705 [DOI] [PubMed] [Google Scholar]
- Montcouquiol M, Rachel RA, Lanford PJ, Copeland NG, Jenkins NA, Kelley MW (2003) Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature 423:173–177. 10.1038/nature01618 [DOI] [PubMed] [Google Scholar]
- Moon H, Song J, Shin JO, Lee H, Kim HK, Eggenschwiller JT, Bok J, Ko HW (2014) Intestinal cell kinase, a protein associated with endocrine-cerebro-osteodysplasia syndrome, is a key regulator of cilia length and Hedgehog signaling. Proc Natl Acad Sci U S A 111:8541–8546. 10.1073/pnas.1323161111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, von Hünerbein K, Hoidis S, Smolders JW (2005) A physiological place-frequency map of the cochlea in the CBA/J mouse. Hear Res 202:63–73. 10.1016/j.heares.2004.08.011 [DOI] [PubMed] [Google Scholar]
- Niwa S, Nakajima K, Miki H, Minato Y, Wang D, Hirokawa N (2012) KIF19A is a microtubule-depolymerizing kinesin for ciliary length control. Dev Cell 23:1167–1175. 10.1016/j.devcel.2012.10.016 [DOI] [PubMed] [Google Scholar]
- Ohyama T, Groves AK (2004) Generation of Pax2-Cre mice by modification of a Pax2 bacterial artificial chromosome. Genesis 38:195–199. 10.1002/gene.20017 [DOI] [PubMed] [Google Scholar]
- Omori Y, Chaya T, Katoh K, Kajimura N, Sato S, Muraoka K, Ueno S, Koyasu T, Kondo M, Furukawa T (2010) Negative regulation of ciliary length by ciliary male germ cell-associated kinase (Mak) is required for retinal photoreceptor survival. Proc Natl Acad Sci U S A 107:22671–22676. 10.1073/pnas.1009437108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oud MM, et al. (2016) A novel ICK mutation causes ciliary disruption and lethal endocrine-cerebro-osteodysplasia syndrome. Cilia 5:8. 10.1186/s13630-016-0029-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige Taylor S, Kunova Bosakova M, Varecha M, Balek L, Barta T, Trantirek L, Jelinkova I, Duran I, Vesela I, Forlenza KN, Martin JH, Hampl A, Bamshad M, Nickerson D, Jaworski ML, Song J, Wan Ko H, Cohn DH, Krakow D, Krejci P (2016) An inactivating mutation in intestinal cell kinase, ICK, impairs hedgehog signalling and causes short rib-polydactyly syndrome. Hum Mol Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TJ, Mitchell BJ, Abitua PB, Kintner C, Wallingford JB (2008) Dishevelled controls apical docking and planar polarization of basal bodies in ciliated epithelial cells. Nat Genet 40:871–879. 10.1038/ng.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Baker SA, Deane JA, Cole DG, Dickert BL, Rosenbaum JL, Witman GB, Besharse JC (2002) The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol 157:103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachel RA, et al. (2012) Combining Cep290 and Mkks ciliopathy alleles in mice rescues sensory defects and restores ciliogenesis. J Clin Invest 122:1233–1245. 10.1172/JCI60981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rida PC, Chen P (2009) Line up and listen: planar cell polarity regulation in the mammalian inner ear. Semin Cell Dev Biol 20:978–985. 10.1016/j.semcdb.2009.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross AJ, et al. (2005) Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet 37:1135–1140. 10.1038/ng1644 [DOI] [PubMed] [Google Scholar]
- Schwander M, Kachar B, Müller U (2010) Review series: the cell biology of hearing. J Cell Biol 190:9–20. 10.1083/jcb.201001138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai Y, Satoh H, Takeda N, Fukuda M, Chiba E, Kato T, Kuramochi T, Araki Y (2002) A testicular germ cell-associated serine-threonine kinase, MAK, is dispensable for sperm formation. Mol Cell Biol 22:3276–3280. 10.1128/MCB.22.10.3276-3280.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Krönig C, Schermer B, Benzing T, Cabello OA, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G (2005) Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet 37:537–543. 10.1038/ng1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe CW, Lu X (2011) Kif3a regulates planar polarization of auditory hair cells through both ciliary and non-ciliary mechanisms. Development 138:3441–3449. 10.1242/dev.065961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobkowicz HM, Slapnick SM, August BK (1995) The kinocilium of auditory hair cells and evidence for its morphogenetic role during the regeneration of stereocilia and cuticular plates. J Neurocytol 24:633–653. 10.1007/BF01179815 [DOI] [PubMed] [Google Scholar]
- Soriano P. (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21:70–71. 10.1038/5007 [DOI] [PubMed] [Google Scholar]
- Tilney LG, Tilney MS, DeRosier DJ (1992) Actin filaments, stereocilia, and hair cells: how cells count and measure. Annu Rev Cell Biol 8:257–274. 10.1146/annurev.cb.08.110192.001353 [DOI] [PubMed] [Google Scholar]
- Togawa K, Yan YX, Inomoto T, Slaugenhaupt S, Rustgi AK (2000) Intestinal cell kinase (ICK) localizes to the crypt region and requires a dual phosphorylation site found in map kinases. J Cell Physiol 183:129–139. [DOI] [PubMed] [Google Scholar]
- Wang Y, Guo N, Nathans J (2006) The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J Neurosci 26:2147–2156. 10.1523/JNEUROSCI.4698-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]