

Abstract
Dystonia is a movement disorder characterized by sustained or intermittent muscle contractions and its pathophysiological mechanisms are still poorly understood. Dominant mutations of the GNAL gene are a cause of isolated dystonia (DYT25) in patients. Some mutations result in a complete loss of function of the encoded protein, Gαolf, an adenylyl-cyclase-stimulatory G-protein highly enriched in striatal projection neurons, where it mediates the actions of dopamine and adenosine. We used male and female heterozygous Gnal knock-out mice (Gnal+/−) to study how GNAL haplodeficiency is implicated in dystonia. In basal conditions, no overt dystonic movements or postures or change in locomotor activity were observed. However, Gnal haploinsufficiency altered self-grooming, motor coordination, and apparent motivation in operant conditioning, as well as spine morphology and phospho-CaMKIIβ in the striatum. After systemic administration of oxotremorine, an unselective cholinergic agonist, Gnal+/− mice developed more abnormal postures and movements than WT mice. These effects were not caused by seizures as indicated by EEG recordings. They were prevented by the M1-preferring muscarinic antagonists, telenzepine, pirenzepine, and trihexyphenidyl, which alleviate dystonic symptoms in patients. The motor defects were worsened by mecamylamine, a selective nicotinic antagonist. These oxotremorine-induced abnormalities in Gnal+/− mice were replicated by oxotremorine infusion into the striatum, but not into the cerebellum, indicating that defects in striatal neurons favor the appearance of dystonia-like movement alterations after oxotremorine. Untreated and oxotremorine-treated Gnal+/− mice provide a model of presymptomic and symptomatic stages of DYT25-associated dystonia, respectively, and clues about the mechanisms underlying dystonia pathogenesis.
SIGNIFICANCE STATEMENT Adult-onset dystonia DYT25 is caused by dominant loss-of-function mutations of GNAL, a gene encoding the stimulatory G-protein Gαolf, which is critical for activation of the cAMP pathway in the striatal projection neurons. Here, we demonstrate that Gnal-haplodeficient mice have a mild neurological phenotype and display vulnerability to developing dystonic movements after systemic or intrastriatal injection of the cholinergic agonist oxotremorine. Therefore, impairment of the cAMP pathway in association with an increased cholinergic tone creates alterations in striatal neuron functions that can promote the onset of dystonia. Our results also provide evidence that untreated and oxotremorine-treated Gnal-haplodeficient mice are powerful models with which to study presymptomic and symptomatic stages of DYT25-associated dystonia, respectively.
Keywords: cAMP, dystonia, G-protein, genetic mouse model, muscarinic drugs, striatum
Introduction
Dystonia is defined as a movement disorder that is “characterized by sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures, or both” (Albanese et al., 2013). In most forms of inherited dystonia, the absence of overt neuropathological modifications has made difficult the elucidation of their pathogenesis. The identification of genes causing dystonia may help to reveal their underpinnings. Among the various genes identified so far, TOR1A (DYT1) and THAP1 (DYT6) are the best characterized (Ozelius et al., 2011; Oleas et al., 2013; Ruiz et al., 2015; Weisheit and Dauer, 2015). GNAL (DYT25) was recently identified as a cause of dystonia (Fuchs et al., 2013) and was the first gene linked to adult-onset focal/segmental inherited dystonia (Fuchs et al., 2013; Vemula et al., 2013; Kumar et al., 2014).
The GNAL gene encodes the α subunit of the heterotrimeric G-protein Gαolf, an isoform of Gαs that positively couples various 7-transmembrane domain receptors to adenylyl cyclase (AC) (Jones and Reed, 1989; Zhuang et al., 2000; Corvol et al., 2001; Hervé et al., 2001). Initially identified in the sensory neurons of the olfactory epithelium (Jones and Reed, 1989; Belluscio et al., 1998), Gαolf is enriched in the GABAergic striatal projection neurons (SPNs), in which it replaces the more widely expressed and highly homologous Gαs (Drinnan et al., 1991; Hervé et al., 1993; Hervé et al., 2001). Gαolf is evenly distributed in both categories of SPNs, belonging to either the direct pathway (dSPN) and expressing dopamine D1 receptor (D1R) or the indirect pathway (iSPN) and expressing D2 receptor (D2R) (Hervé et al., 2001). iSPNs also express adenosine A2A receptors (A2ARs) that stimulate AC activity (Schiffmann et al., 2007). In association with Gγ7 (Schwindinger et al., 2003; Schwindinger et al., 2010; Xie et al., 2015), Gαolf couples with D1R in dSPNs and A2AR in iSPNs to AC, which is mostly type 5 AC (AC5) in these neurons (Zhuang et al., 2000; Corvol et al., 2001; Lee et al., 2002). In the striatal cholinergic interneurons, Gαolf is coexpressed with Gαs (Hervé et al., 2001) and could contribute to the signaling of dopamine D5 receptor and A2AR (Yan and Surmeier, 1997; Tozzi et al., 2011). This key role of Gαolf in striatal signaling could be determinant in the GNAL-related dystonia (Fuchs et al., 2013). However, Gαolf is also expressed at low levels in cerebellar Purkinje cells (Belluscio et al., 1998; Vemula et al., 2013) and its mutation could potentially cause dystonic movements by disrupting cerebellar output (Campbell and Hess, 1998; Vemula et al., 2013; Fremont et al., 2014).
Several pathological mutations of GNAL are nonsense mutations or frameshift deletions leading to truncated proteins that are unstable and completely devoid of functional activity (Fuchs et al., 2013). Therefore, heterozygous mice carrying a Gnal-null mutation (Gnal+/−), which express ∼50% of the protein normal levels (Corvol et al., 2007), could be a good model for mimicking the genetic status of dystonic patients. We have shown previously that Gnal+/− mice display a significant phenotype when they are pharmacologically challenged (Corvol et al., 2001; Hervé et al., 2001; Corvol et al., 2007; Hervé, 2011). However, motor behavior was not investigated in the Gnal+/− mice. Here, we show that Gαolf haplodeficiency does not produce any detectable dystonic movement in basal conditions in the mouse, but alters motor coordination, self-grooming, and instrumental task responses as well as spine length in SPNs. Because cholinergic antagonists alleviate dystonia and striatal cholinergic dysfunction may be critical in this condition (Eskow Jaunarajs et al., 2015), we assessed the effects of the muscarinic agonist oxotremorine. We observed that oxotremorine induced more abnormal postures and uncoordinated movements in Gnal+/− mice than in WT mice. These abnormalities displayed some similarities with human dystonia, suggesting that oxotremorine-treated Gnal+/− mice constitute a novel and valuable animal model with which to study dystonia mechanisms.
Materials and Methods
Animals
Gnal+/− mice were mated with C57BL/6J mice to produce male and female Gnal+/− and Gnal+/+ littermates. The mice were kept four per cage on a 12 h light/dark cycle at a constant temperature of 22°C with access to food and water ad libitum unless otherwise indicated. All experiments were in accordance with the guidelines of the French Agriculture and Forestry Ministry for handling animals (decree 87-848). The laboratory animal facility was approved to carry out animal experiments by the Sous-Direction de la Protection Sanitaire et de l'Environnement de la Préfecture de Police (arrêté préfectoral DTPP 2014-724 C75-05-22). The experimental protocols were approved by the Institut du Fer à Moulin local review board. The principal investigators had a personal agreement (D.H., license C-75-828; J.-A.G., license 75-877).
Behavioral tests
Behavioral experiments were conducted using mutant and WT littermates (73 males and 107 females 3–12 months of age) between 1:00 and 7:00 P.M.
Open-field test.
Three-month-old animals of either sex were placed in one of the four squares of open field boxes (50 × 50 × 30 cm) made of white Plexiglas and allowed to explore it freely for 30 min. Locomotion was tracked using a ceiling-mounted camera (WV BP332; Panasonic) and ViewPoint videotracking. Data were analyzed as distance traveled (in centimeters) and number of rearings.
Locomotor activity.
Spontaneous locomotor activity was recorded by video and analyzed using the HomeCageScan (CleverSys), a software that detects rodent movements and behaviors automatically based on video-tracking. Each animal was habituated to the test cage for 2 h and overall motor activity was recorded for 24 h.
Rotarod.
Three- and 12-month-old Gnal+/− and Gnal+/+ littermates of either sex were trained for 3 consecutive days in a rotarod (LE8200; BIOSEB) with 4 trials per day spaced by 20 min intervals. The trial started with 4–40 rpm accelerating rod rotation over 5 min and ended when either the mice fell off the rod or 300 s elapsed. On the following day (test day), mice were placed on the rotarod at three different fixed speeds (16, 24, and 32 rpm), with three trials per speed and 30 min intertrial intervals. The trial ended when the mouse fell off the rod or 60 s elapsed. The rotarod was cleaned using 30% ethanol (v/v) between subjects.
Clasping.
One-year-old mice were tested for clasping by tail suspension (Guyenet et al., 2010).
Grooming.
Three-month-old Gnal+/− and Gnal+/+ littermates of either sex were placed individually in a clear observation cylinder (13 cm diameter, 16 cm height). Grooming was induced by three 25°C water sprays at a distance of 15 cm and mice were observed for 10 min. Analysis of the grooming phases and duration were performed manually by an observer blind to the genotype (Pelosi et al., 2015).
Operant conditioning.
Experiments were done in eight automatic operant chambers (Imetronics) with two holes, a food magazine connected with a food pellet dispenser, and a cue light located above the holes. Three- to 4-month-old mice of either sex were food deprived (3.5 g/d, water ad libitum) for 5 d before the experiment. The session started with the introduction of the animal into the operant chamber, where the light stayed on during the entire session. Once the animal poked in the active hole, the cue light above it turned on until the animal took the pellet in the magazine. During this time, both holes were inactive for 10 s. Animals were trained with a continuous reinforcement schedule, starting with a fixed ratio 1 (FR1) schedule for 5 d, followed by a 5 d FR5 schedule and a 5 d reverse FR5 schedule (rFR5), in which the previously active hole became inactive and vice versa. After this training, the animals were tested in a single 2 h progressive ratio (PR) session, in which the requirement to earn a pellet increased stepwise according to the following series: 1, 2, 3, 5, 12, 18, 27, 40, 60, 90, 135, 200, 300, 450, 675, 1000, and 1500 (Stipanovich et al., 2008). Three animals (two WT and one mutant) that did not reach the criteria were excluded from the analysis. Criteria were a stable response with <20% deviation from mean of the total number of rewards earned in three consecutive sessions (80% stability).
Ledge test.
Mice were placed on the cage ledge. The time and way that the animals walked along the cage were scored according to a previously published method (Guyenet et al., 2010).
Histology
Golgi staining.
Fresh brains from three male animals per genotype (12 months of age) were processed following the Golgi–Cox method (Ricobaraza et al., 2012). After 3 weeks of impregnation in the Golgi–Cox solution, the brains were cut in 200-μm-thick slices using a microtome (VT1000S; Leica). The slices were incubated in 16% (v/v) ammonia, fixed in 10 g/L sodium thiosulfate, dehydrated, and mounted on randomly coded glass slides so that the experimenter was blind to genotype during image acquisition and analysis. Bright-field images of Golgi-impregnated SPNs were acquired with a Micromax CCD camera attached to a Leica DM6000 fluorescent microscope (100× oil objective). Only fully impregnated neurons with their soma entirely within the thickness of section and with at least four orders of dendrites visible were considered for analysis. Image z stacks were taken every 0.5 μm and analyzed with ImageJ software. Dendritic segments were traced through different layers of the stack and spine number and length were measured. Spine density was expressed as the number of spines per micrometer of dendrite.
Immunofluorescence.
Immunostaining was performed on 7- to 9-month-old Gnal+/− and WT littermates of either sex. Under deep pentobarbital anesthesia (500 mg/kg, i.p.), mice were perfused transcardially with 100 ml of a 4% (w/v) paraformaldehyde solution in potassium-free PBS and postfixed overnight in the same solution at 4°C. Brains were cut with a microtome (VT1000S; Leica) in 40-μm-thick coronal sections and stored at −20°C in a buffered solution containing 30% (v/v) ethylene glycol, 30% (v/v) glycerol, and 0.1 m sodium phosphate, pH 7.4, until they were processed for immunofluorescence. After overnight incubation with primary antibodies (Table 1), sections were rinsed and incubated with secondary antibodies (Table 1). Finally, sections were rinsed for 10 min twice in TBS and twice in Tris buffer (0.25 m Tris, pH 7.4) before being mounted in Vectashield (Vector Laboratories). Imaging was performed with a fluorescence microscope (DM 6002; Leica) equipped with a CCD camera (Micromax) and interneurons were counted manually on images in the regions of interest at 10× magnification by an experimenter blind to group identity.
Table 1.
List and dilution of primary and secondary antibodies used for immunoblotting and immunofluorescence
| Source | Dilution | |
|---|---|---|
| Primary antibodies for immunoblotting | ||
| CaMKIIα/β | Abcam | 1:250 |
| pCaMKIIα/β | Abcam | 1:500 |
| Gephyrin | Synaptic Systems | 1:500 |
| GluN1 | BD Pharmingen | 1:500 |
| GluN2A | Millipore | 1:5000 |
| GluN2B | Millipore | 1:1000 |
| GluN3A | Millipore | 1:1000 |
| PSD-95 | Thermo Scientific | 1:2000 |
| vGlut1 | Synaptic Systems | 1:1000 |
| vGlut2 | Synaptic Systems | 1:2000 |
| Synapsin 1 | Millipore | 1:3000 |
| Synaptophysin 1 | Sigma-Aldrich | 1:2000 |
| Homer-1 | Synaptic Systems | 1:2000 |
| Tubulin | Sigma-Aldrich | 1:10,000 |
| Actin | Sigma-Aldrich | 1:5000 |
| Primary antibodies for immunofluorescence | ||
| Choline acetyltransferase (ChAT) | Millipore | 1:200 |
| Parvalbumin | Swant | 1:10,000 |
| Secondary antibodies for immunoblotting | ||
| IRDye 800CW-conjugated anti-mouse IgG | Rockland Immunochemical | 1:5000 |
| IRDye 800 CW-conjugated anti-rabbit IgG | Rockland Immunochemical | 1:5000 |
| Secondary antibodies for immunofluorescence | ||
| Alexa Fluor 483-conjugated anti-goat | Invitrogen | 1:400 |
| Alexa Fluor 546-conjugated anti-mouse | Invitrogen | 1:400 |
Nissl staining.
After perfusion, 50-μm-thick sections were cut with a microtome (VT1000S; Leica) and stained using thionine. The sections were examined under a light microscope (Evos) and the location of injection cannula tips within the striatum and cerebellum was determined.
Immunoblotting
Mice of either sex were killed by decapitation and the heads were immediately frozen in liquid nitrogen (12 s) (Pascoli et al., 2005). The frozen heads were then sliced with a cryostat (210-μm-thick sections) and 6 microdisks (1.4 mm in diameter) were punched out bilaterally from the dorsal striatum, sonicated in 10 g/L SDS, and placed at 100°C for 5 min. Equal amounts of total protein (20 μg) were separated by SDS-PAGE on 4–15% precast gels (Bio-Rad) and transferred electrophoretically to nitrocellulose membranes (GE Healthcare). The membranes were then incubated in primary and secondary antibodies, as indicated in Table 1. Bound antibodies were visualized using an Odyssey infrared fluorescence detection system (LI-COR), followed by quantitation by Odyssey version 1.2 software. Fluorescence intensity values were normalized to actin or tubulin values for variations in loading and transfer.
Tests of oxotremorine on motor behavior
Motor behavior after systemic injection.
Three-month-old Gnal+/− and Gnal+/+ littermates of either sex were allowed to habituate in a clear observation cylinder for 10 min. They were then treated intraperitoneally with either 0.05 or 0.1 mg/kg of oxotremorine methiodide (Sigma-Aldrich), dissolved in saline (NaCl 0.9 g/L). In some experiments, oxotremorine injections (0.1 mg/kg) were preceded by pretreatment with trihexyphenydil (THX, Sigma-Aldrich, 1 or 20 mg/kg, i.p., 70 min before), telenzepine (Tocris Bioscience, 3 mg/kg, i.p., 15 min before), pirenzepine (Tocris Bioscience, 10 mg/kg, i.p., 15 min before), or mecamylamine (Sigma-Aldrich, 3 mg/kg, i.p., 10 min before). After oxotremorine injections, animals were video-recorded for 1 h using a Pacific Electro-optics model PA-290 camera (CleverSys). One or two observers blind to the genotypes analyzed videos and attributed dystonia scores using a modified version of a previously suggested scoring scale (Jinnah et al., 2000) (Table 2) every 10 min for 40 min of observation (starting 20 min after injection).
Table 2.
Description of abnormal postures and movements observed in oxotremorine-treated mice
| Abnormal postures |
| (a) The mouse often extends one or both hind limbs abnormally from the body axis (splayed posture) for a sustained amount of time (>10 s) |
| (b) The mouse stands vertically on hind limbs for short periods motionless, agitating both forelimb paws, or occasionally with a single raised paw |
| (c) The mouse often has a hunched posture with little movement, then attempts to make movements, often making small head jerks |
| Abnormal movements |
| (a) One or both forelimbs move rapidly up and down resembling the first phase of the grooming chain as if the mouse is about to start elliptic movements around the nose, but no elliptic movement is produced; rubbing together of forelimb paws also observed |
| (b) Repetitive jerking of the snout or head |
| (c) Frequent twitching of the body when the animal is in a resting position |
| (d) Slow walking, occasionally with extended body and/or with abnormal hind limb gait |
| Scoring scale |
| 0. Normal motor behavior |
| 1. No impairment, but slightly slowed movements |
| 2. Mild impairment: occasional abnormal postures and movements; ambulation with slow walk |
| 3. Moderate impairment: frequent abnormal postures and movements with limited ambulation |
| 4. Severe impairment: sustained abnormal postures without any ambulation or upright position |
Scoring scale used to evaluate abnormal postures and movements in mice is modified from Jinnah et al. (2000).
Motor behavior after intracranial infusion.
Animals of either sex were anesthetized with a mixture of xylazine (10 mg/ml) and ketamine (25 mg/ml) (Centravet) and mounted in a digitalized stereotactic frame (Stoelting) equipped with a mouse adaptor. Bilateral guide cannulas (6.5 mm long, 26 gauge) were implanted into the striatum (coordinates: anteroposterior, 0.7 mm from bregma; mediolateral, ±1.7; dorsoventral, −3.6) or deep nuclei of cerebellum (coordinates: anteroposterior, −6.24 mm from bregma; mediolateral, ±1.7; dorsoventral, −3.25) according to a mouse brain atlas (Paxinos and Franklin, 2001). The bilateral guides were attached to the skull by dental cement and three anchoring screws. A dummy was inserted into each guide to maintain patency and was removed only for drug infusion. After surgery, the mice received a subcutaneous injection of a nonsteroidal anti-inflammatory drug (flunixin meglumine, 4 mg/kg; Sigma-Aldrich) and were placed on a warm plate for ∼24 h after surgery to avoid hypothermia. Mice were allowed to recover for 8 d before drug infusion. Oxotremorine methiodide was dissolved in PBS and infused into the striatum or cerebellum under a brief isoflurane anesthesia. The injection cannula was inserted into the guide and protruded 2 mm beyond the tip of the guide. Drug (0.1 μg) or vehicle was infused at a rate of 0.5 μl/min for a total volume of 0.5 μl in the striatum and 1 μl in the cerebellum per hemisphere using a 5 μl Hamilton microsyringe connected to the infusion cannula by polyethylene (PE-50) tubing and an automated micro-infusion pump (310; KD Scientific). The injection cannula was held in place for an additional minute before removing to maximize drug diffusion. Animals were video-recorded (see above) for 1 h after infusion and abnormal motor behavior was scored as reported in Table 2 every 10 min starting 30 and 20 min after the infusion into the striatum and cerebellum, respectively.
EEG recording
Surgery.
Gnal+/− mice (n = 5) were anesthetized with a mixture of isoflurane and O2, injected with buprenorphine (0.05 mg/kg, s.c.) to control pain, and maintained at physiological core temperature (37°C). After a local anesthetic injection (lidocaine, s.c.), the scalp was incised and small burr holes were made in the skull above the motor cortex M1 (from the bregma in mm: anteroposterior +2, mediolateral 2), where electrodes were inserted under stereotaxic guidance. The electrodes were fixed with dental cement. The animals were allowed a minimum of 3 d to recover from the surgery.
In vivo EEG recordings.
Before and after oxotremorine administration, EEG activity from the motor cortex was recorded with a sampling rate of 1 kHz (System 3; Tucker-Davis Technologies) while the animal freely explored the recording arena. The animal was filmed with a monochrome video camera (50 Hz, F033; AVT Marlin). The dystonic attack periods were scored offline. Epileptiform activity was probed before and after oxotremorine administration or during dystonic attacks.
Statistical analysis
All data were subjected to tests for normal distribution (Kolmogorov–Smirnov test). They were analyzed with the unpaired Student's t test with Welch's modification, repeated-measures ANOVA, or two-way ANOVA followed by Sidak's post hoc comparisons. All statistical analyses were performed using GraphPad Prism 6.0. A critical value for significance of p < 0.05 was used throughout the study.
Results
Gnal+/− mice have a normal general activity
To test the effect of Gnal haplodeficiency on locomotor activity, we compared 3-month-old Gnal+/− and Gnal+/+ littermates in an open field or in their home cage. In agreement with previous data on locomotor activity in circular corridor (Corvol et al., 2007), we found no difference between genotypes in horizontal activity (Fig. 1A) or vertical rearing (Fig. 1B) in the open field. The measure of traveled distance in the home cage over the entire 24 h light/dark cycle showed that both Gnal+/− and Gnal+/+ mice displayed a normal circadian pattern with a higher activity during the night and no significant difference at any time of 24 h cycle (Fig. 1C). These results showed that Gnal+/− mice have normal spontaneous locomotor activity.
Figure 1.

Similar locomotor activity in Gnal+/− and WT mice. A, No difference was found in the distance traveled in an open field (n = 15–20 mice per group). Two-way ANOVA: genotype, F(1,210) = 0.38, p = 0.54; time, F(1,210) = 0.95, p = 0.4; interaction, F(1,210) = 0.09, p = 0.9. B, Similar number of vertical rears were counted during the open-field test (30 min) in the two genotypes. Student's t test, t = 0.62, p = 0.5. C, Monitoring of distance traveled in housing cage during a 24 h-cycle (12 h of night and day). A circadian variation of locomotor activity was observed, but was not significantly different in Gnal+/− mice compared with WT littermates (n = 7 per group). Two-way ANOVA: genotype, F(1,10) = 0.03; p = 0.8: time, F(1,10) = 174.7; p < 0.0001; interaction, F(1,10) = 3.08; p = 0.1. Data are shown as means ± SEM.
Gnal+/− mice display motor coordination impairments
Motor coordination is one of the most affected functions in dystonia animal models (Sasaki et al., 2013; Richter and Richter, 2014; Rose et al., 2015). To test whether motor coordination was impaired in Gnal+/− mice, we compared their performance with that of Gnal+/+ littermates in the rotarod test at the age of 3 and 12 months. Animals were first trained on an accelerating rotarod for 3 d and their motor coordination improvement was evaluated by measuring the time that they were able to stay on the rod during each trial (Fig. 2A,B). On the test day, the time before falling was measured at three different fixed speeds (16, 24, and 32 rpm; Fig. 2C,D). Three-month-old Gnal+/− mice displayed similar performance as Gnal+/+ during the learning phase, improving their ability to stay on the rod (Fig. 2A). However, on the test day, their latency to fall at 16 rpm was shorter (Fig. 2C). One-year-old Gnal+/− mice displayed a reduced motor learning ability during the training phase (Fig. 2B) and a strong tendency to fall on the test day compared with WT littermates (Fig. 2D). We used another batch of 3-month-old mice to observe motor coordination by performing a ledge test in which mice had to walk along the ledge of their home cage. Gnal+/− mice have a decreased ability to walk without losing their balance as compared with Gnal+/+ mice (Fig. 2E). In contrast, the mutant mice did not display any clasping of the paws when they were suspended by the tail (Fig. 2F), an abnormal reaction reported in DYT1 mutants (Liang et al., 2014; Pappas et al., 2015). Altogether, these results showed that Gnal+/− animals have poor motor coordination, which appears to worsen with age.
Figure 2.
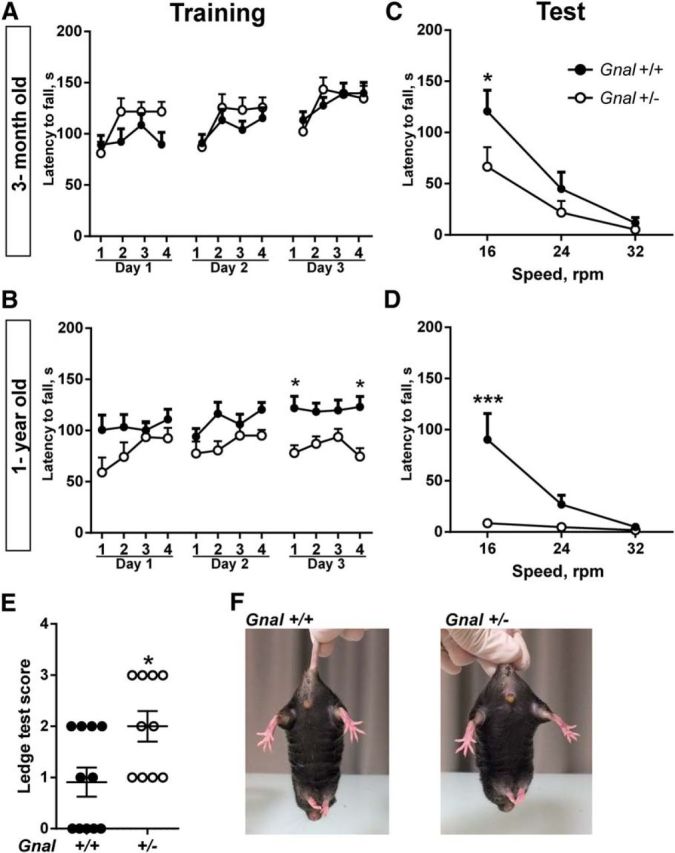
Motor coordination impairment in Gnal+/− and WT mice. A, Training of 3-month-old mice (n = 13–19 animals per group) with accelerating rotarod (4–40 rotations per min, rpm) for 3 consecutive days. Mice performance improved with training, but no significant difference was found between Gnal+/− and Gnal+/+ mice. Two-way ANOVA: sessions, F(11,341) = 9.55, p < 0.0001; genotype, F(1,31) = 0.63, p = 0.43; interaction, F(11,314) = 1.64, p = 0.08. B, The same animals were tested again as in A at the age of 1 year. Gnal+/− animals showed learning impairment. Two-way ANOVA: genotype, F(1,22) = 6.5, p < 0.05; session, F(11,242) = 2.6, p < 0.01; interaction, F(11,242) = 1.6, p = 0.1. One day after the last training session, 3-month-old (C) and 1-year-old mice (D) were tested with rod rotation at 3 fixed speeds. Gnal+/− animals stayed less time on the rod than WT controls at 16 rpm speed. C, Two-way ANOVA: genotype, F(1,30) = 2.7, p = 0.11; speed, F(2,60) = 29, p < 0.0001; interaction, F(2.60) = 2.2, p = 0.12. D, The deficit of 1-year-old Gnal+/− mice was more pronounced than at 3 months of age at 16 rpm speed. Two-way ANOVA: genotype, F(1,22) = 9.3, p < 0.01; speed, F(2,44) = 9.6, p < 0.001; interaction, F(2,44) = 7.1, p < 0.01. A–D, Data are shown as means ± SEM. Post hoc comparisons were done with Sidak's multiple-comparisons test. E, Reduced score of motor coordination of Gnal+/− mice in ledge test compared with WT controls (n = 10–11 animals per group, means ± SEM. are indicated, Student's t test with Welch's correction, t = 2.6, p < 0.05). F, No clasping of the paws was observed in WT and Gnal+/− mice when suspended by the tail. *p < 0.05; **p < 0.01; ***p < 0.001.
Innate sequential behaviors and motivation are altered in Gnal+/− mice
We analyzed self-grooming, an innate rodent behavior characterized by a precise motor sequence execution divided in four phases (Aldridge and Berridge, 1998; Pelosi et al., 2015). Water-spray-elicited grooming was evaluated for a period of 10 min (Fig. 3A–D). The time spent grooming was decreased significantly in Gnal+/− mice compared with WT littermates (Fig. 3A). This decrease resulted from a combination of decreases in the number of bouts (number of times a grooming sequence was started; Fig. 3B) and average duration of grooming bouts (Fig. 3C), which did not reach statistical significance separately. As was the case with the overall duration, the number of transitions during the observation period was reduced in Gnal+/− compared with Gnal+/+ animals (Fig. 3D).
Figure 3.

Self-grooming and operant conditioning alterations in Gnal+/− and WT mice. A–D, Water-spray-induced grooming was analyzed in Gnal+/− and WT littermates (n = 8–12 per group) for 10 min. A, Duration of grooming in the session (t = 3.62, p < 0.01). B, Number of grooming bouts (Student's t test, t = 1.34, p = 0.21). C, Mean duration per bout (Student's t test, t = 1.71, p = 0.11). D, Number of transitions counted during the grooming activity (t = 3.14, p < 0.05). E, Food-restricted Gnal+/− mice and WT littermates (n = 11–12 per group) had to nose-poke in an active hole to obtain food pellets during 5-day of daily sessions under, successively, FR1, FR5, and reversed FR5 (rFR5, active and inactive holes were inverted) schedules. No significant difference was detected between Gnal+/− and WT mice. Two-way ANOVA: session, F(14,210) = 47.35, p < 0.0001; genotype, F(1,15) = 0.003, p = 0.9; interaction, F(14,210) = 0.9, p = 0.55. F, The day after the last rFR5 session, the mice were tested under the PR schedule. In the Gnal+/− animals, the breaking point (number of pokes for the last pellet earned) is decreased compared with WT (t = 2.4, p < 0.05). G, Time course of cumulative nose pokes in the progressive ratio sessions. Two-way ANOVA: genotype, F(1,15) = 3.45, p = 0.08; time, F(23,345) = 63.21, p < 0.0001; interaction, F(23,345) = 5.3, p < 0.0001. Data are shown as means ± SEM. *p < 0.05; **p < 0.01.
We then explored how the Gnal+/− mice learned and executed behavioral sequences in an operant conditioning task. Three-month-old animals were trained to nose-poke in an active hole to get a food reward under FR1 and FR5 schedules. No significant difference was detected between Gnal+/− and Gnal+/+ animals in the acquisition of the task (Fig. 3E). When the active and inactive holes were reversed, Gnal+/− and Gnal+/+ mice learned the new rule similarly, showing a comparable behavioral flexibility. However, in the PR session, in which they had to nose-poke an increasing number of times to get the same reward, Gnal+/− mice poked less than WT littermates and earned significantly fewer rewards (Fig. 3F,G).
Together, these results suggest that partial loss of Gαolf affects innate sequence behaviors. Gnal+/− animals also displayed a significant deficit when an operant conditioning task required an increasing effort to earn reward.
Gnal haplodeficiency reduces spine length in the dorsal striatum
Gnal haplodeficiency was shown to reduce cAMP production in SPNs (Corvol et al., 2001), an effect that could affect spine development in these neurons (Kozorovitskiy et al., 2015). To test this possibility, Golgi–Cox staining was performed in Gnal+/− and Gnal+/+ brains to measure spine density and length. No difference in spine density was found in the dorsal striatum (Fig. 4A,B). In contrast, spine length was slightly decreased in Gnal+/− mice compared with Gnal+/+ mice (Fig. 4C). We investigated whether this change was associated with alterations in synaptic proteins. None of the tested presynaptic markers, vesicular glutamate transporters 1 and 2 (vGlut1 and vGlut2) and synaptophysin, or postsynaptic markers, PSD-95, GluN1, GluN2A, GluN2B, GluN3A, and Homer-1, were altered in the mutant mice (Table 3). Gephyrin, a marker of inhibitory synapses, was also unaltered (Table 3). In contrast, we found that autophosphorylation of CaMKIIβ (Fig. 4D,E), the isoform associated with spine stability (Okamoto et al., 2007), but not CaMKIIα (Fig. 4F), was decreased. These data ruled out a major deficit in synapse organization and stability in the striatal neurons of Gnal+/− mice, but suggested a possible alteration in CaMKIIβ signaling.
Figure 4.

Spine analyses in dorsal striatum. A, Examples of Golgi-stained dendrites of SPNs in Gnal+/+ and Gnal+/− mice. Scale bar, 7 μm. B, Spine density quantification showed no significant difference between genotypes (t = 1, p = 0.3, with a total of ∼50 dendrites measured in three mice per group). C, Distribution of spine lengths in SPN dendrites showed a significant reduction in Gnal+/− mice compared with WT littermates (Kolmogorov–Smirnov test, d = 0.12; **p < 0.01). D, Immunoblots of total CaMKIIα and β and their phosphorylated forms (pCaMKIIα and β). Actin was measured as loading control. E, F, Quantification of pCaMKIIβ/CaMKIIβ (t = 2.6, p < 0.05) (E) and pCaMKIIα/CaMKIIα (t = 0.2, p = 0.8) (F) ratios. Data are shown as means ± SEM. *p < 0.01, two-tailed unpaired t test with Welch's correction.
Table 3.
Quantification of synaptic markers in the striatum
| Protein | Mean ± SEM |
|
|---|---|---|
| Gnal+/+ | Gnal+/− | |
| Synaptophysin | 100 ± 22.4 | 86.5 ± 16.7 |
| Gephyrin | 100 ± 18.1 | 91.9 ± 22.1 |
| Homer | 100 ± 7.4 | 102.5 ± 7.3 |
| PSD-95 | 100 ± 12.7 | 102.8 ± 19.9 |
| GluN1 | 100 ± 17.6 | 100.6 ± 18.4 |
| GluN2A | 100 ± 10.8 | 95.6 ± 9.7 |
| GluN2B | 100 ± 14.3 | 97.2 ± 15.4 |
| GluN3A | 100 ± 20.2 | 98.2 ± 12.2 |
| vGlut1 | 100 ± 6.0 | 98.8 ± 9.3 |
| vGlut2 | 100 ± 6.2 | 115.0 ± 8.9 |
Striatal samples from Gnal+/− and WT mice were analyzed by immunoblotting using antibodies against presynaptic and postsynaptic markers as indicated in Table 1. Actin or tubulin immunolabeling was used as a loading control. Data were normalized by actin or tubulin immunofluorescence for each sample and are expressed as a percentage of the mean obtained in WT animals (n = 6–10 per group).
The number of interneurons is not altered in the striatum of Gnal+/− mice
Gαolf is coexpressed with Gαs in the striatal cholinergic interneurons and possibly in GABAergic interneurons (Hervé et al., 2001). In dystonia animal models, cholinergic and GABAergic interneurons numbers can be altered (Pisani et al., 2006; Hamann et al., 2007; Song et al., 2013; Pappas et al., 2015). To detect such alterations in Gnal+/− mice, we evaluated the number of cholinergic and GABAergic interneurons immunostained by specific antibodies against choline acetyltransferase (ChAT) and parvalbumin (PV), respectively. The number of ChAT- or PV-positive cells in the dorsomedial and dorsolateral striatum was similar in the Gnal+/− and Gnal+/+ mice (Fig. 5). These results indicated that Gαolf haplodeficiency did not result in any gross alteration in the number of striatal interneurons.
Figure 5.

Lack of alteration of cholinergic and parvalbumin-positive interneuron density in the dorsomedial (DMS) and dorsolateral (DLS) striatum of Gnal+/− mice. A–D, ChAT immunostaining (A, C) and quantification (B, D) of cholinergic interneurons in the DMS (A, B) and DLS (C, D) of Gnal+/+ and Gnal+/− animals (n = 5 per group, DMS, t = 0.9, p = 0.4; DLS, t = 0.85, p = 0.4). E–H, PV immunostaining (E, G) and quantification (F, H) of parvalbumin-positive interneurons in the DMS (E, F) and DLS (G, H) of Gnal+/+ and Gnal+/− mice (DMS, t = 0.9, p = 0.2; DLS, t = 0.6, p = 0.5). Data are shown as means ± SEM and statistical analysis was performed using two-tailed t test with Welch's correction.
Activation of muscarinic receptor induces abnormal postures and movements in Gnal+/− mice
In basal conditions, Gnal+/− animals did not display any overt abnormality in movements and postures (see above). Because muscarinic receptor antagonists can ameliorate the motor symptoms in dystonic patients (Ozelius et al., 2011), we reasoned that stimulation of these receptors might trigger the appearance of abnormal movements. To test this hypothesis, Gnal+/− and Gnal+/+ mice were treated with oxotremorine, a nonselective cholinergic agonist. In Gnal+/+ mice, 0.1 mg/kg intraperitoneal oxotremorine induced rare postural changes and abnormal movements. The classical peripheral effects of muscarinic agonists, salivation and lacrimation, were observed, but very few whole-body tremors that were reported at higher doses of oxotremorine (Bymaster et al., 2003). WT mice displayed some abnormalities in their behavior during the first 30 min after injection, with reduced locomotor activity and abnormally persistent resting position, but, afterward, most of them returned to normal activity and few had a slow locomotor activity, abnormal movements, or unusual postures (Movie 1). Grooming chains were often altered, being unusually stopped at the first elliptic phase. In contrast, in Gnal+/− animals, oxotremorine (0.1 mg/kg) often induced abnormal postures and movements during the entire observation period (Fig. 6A–C, Movie 2). The phenotype is mainly characterized by intermittent but sustained abnormal hunched postures. The animals frequently extend hindlimbs from the body axis when in a rest position and the body or head are tilted (Fig. 6B). Several types of abnormal movements were also observed sporadically. They were characterized with rapid up and down movements of one or both forelimb paws. In some cases, during abnormal resting states, the Gnal+/− mice also showed twitching of the body and jerks of the head and snout. Moreover, their locomotion was slow with extended body and abnormal hindlimb gait. To quantify these abnormalities, we used a modified version (Table 2) of a previously published scoring scale (Jinnah et al., 2000). Scoring showed oxotremorine-induced motor abnormalities in both Gnal+/− and Gnal+/+ mice, but their intensity was significantly lower in WT than in mutant animals throughout the observation period 20–60 min after oxotremorine treatment (Fig. 6C, top). Because motor behavior was variable from one animal to the other and the evaluation of motor abnormalities was partially subjective, we performed the experiments on a second independent batch of Gnal+/− and WT littermates and the motor effects of 0.1 mg/kg oxotremorine were evaluated by two raters blind to the genotype. We confirmed that the abnormal motor scores were higher in Gnal+/− mice than in WT controls (Fig. 6C, bottom).
Figure 6.
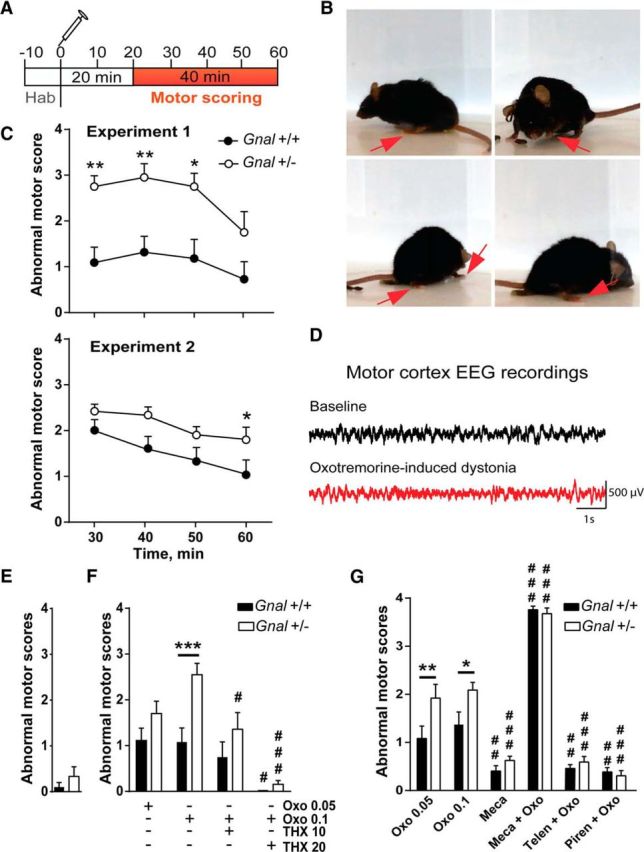
Induction of abnormal movements and postures by systemic oxotremorine in Gnal+/− mice. A, Schedule of behavioral monitoring of oxotremorine injections. Behavior was scored after 20 min to avoid interference with the initial predominant peripheral muscarinic effects. B, Examples of abnormal postures found in Gnal+/− mice 20 min after oxotremorine treatment (0.1 mg/kg). The red arrows indicate abnormal positions of paws. C, Time course of abnormal motor scores after oxotremorine treatment (0.1 mg/kg) in Gnal+/− and Gnal+/+ mice in 2 different experiments rated by 1 (Experiment 1) or 2 (Experiment 2) evaluators blind to the genotype. Motor defects were scored (Table 2) in 10 min intervals and plotted for the end time point. Top, Experiment 1, n = 9 animals per group, two-way ANOVA: time, F(3,57) = 5.04, p < 0.01; genotype, F(1,19) = 13.54, p < 0.01; interaction, F(3,56) = 0.7, p = 0.5. Bottom, Experiment 2, n = 12 animals per group, scores are the mean of the 2 evaluations, two-way ANOVA: time, F(3,33) = 16.82, p < 0.0001; genotype, F(1,11) = 4.97, p < 0.05; interaction, F(3,33) = 1.58, p = 0.2. D, Absence of epileptiform activity in the motor cortex of Gnal+/− mice treated with oxotremorine (0.1 mg/kg). Examples of EEG recording before oxotremorine administration (baseline, top) and during a phase of abnormal posture induced by oxotremorine (bottom). E, Abnormal movement scores during 10 min in the absence of any treatment. Two-tailed t test, t = 1.13, p = 0.3. F, Effects of THX (1 or 20 mg/kg as indicated) pretreatment on oxotremorine (Oxo)-induced responses. Motor scores were measured on four consecutive 10 min intervals and their mean plotted. Two-way ANOVA: treatment, F(3,65) = 13.9, p < 0.0001; genotype, F(1,65) = 12.9, p < 0.001; interaction, F(3,65) = 2.2, p = 0.1. Data are shown as means ± SEM. Sidak's multiple-comparisons test: Gnal+/− versus Gnal+/+; ***p < 0.001; Oxo versus Oxo+THX, #p < 0.05, ###p < 0.001. G, Effects of the nicotinic antagonist, mecamylamine (Meca; 3 mg/kg) and the M1 preferential antagonists telenzepine (Telen, 3 mg/kg) and pirenzepine (Piren, 10 mg/kg) on Oxo-induced responses in Gnal+/− and Gnal+/+ mice. Motor scores were measured and plotted as in F. Two-way ANOVA: treatment, F(5,129) = 158.7, p < 0.0001; genotype, F(1,129) = 11.45, p < 0.001; interaction, F(5,129) = 3.2, p < 0.05. Data are shown as means ± SEM. Sidak's multiple-comparisons test after two-way ANOVA: Gnal+/− versus Gnal+/+; **p < 0.01; **p < 0.01; Oxo (0.1 mg/kg) versus other treatments, ###p < 0.001. Data for Oxo 0.1 mg/kg alone in F and G are those of Experiments 1 and 2 in C, respectively.
Movie 1.
Postures and movements in WT mice after oxotremorine systemic injection. The movie shows the behavior of the WT animal 20 min after oxotremorine (0.1 mg/kg) injection. No abnormal movement was detected.
Movie 2.
Postures and movements in Gnal+/− mice after systemic oxotremorine injection. The movie shows examples of abnormal postures of A and B types as well as abnormal movements of C and D types, as described in Table 2. The videos are taken between 20 and 60 min after oxotremorine injection (0.1 mg/kg).
Systemic treatments with pilocarpine, a muscarinic agonist, can produce epilepsy (Turski et al., 1984) and oxotremorine induces burst firing when applied on hippocampal slices (Williams and Kauer, 1997). We therefore wanted to rule out the possibility that oxotremorine-induced motor alterations in Gnal+/− mice were caused by epileptic seizures. To test this possibility, EEG recordings from the motor cortex of Gnal+/− mice were collected before and after oxotremorine administration (0.1 mg/kg) to determine whether the motor abnormalities were associated with epileptic seizures (Fig. 6D). EEGs during dystonic-like postures were not associated with epileptiform activity and could not be distinguished from baseline EEGs.
We applied the scoring strategy in the absence of oxotremorine and found no alteration in either genotype (Fig. 6E). We also compared two doses, 0.05 and 0. 1 mg/kg, of oxotremorine and found that the higher dose altered the motor score in a more reliable manner than the lower dose (Fig. 6F,G). We then assessed the contribution of muscarinic receptors in eliciting the abnormal behavior by pretreating the mice with three antagonists of these receptors, THX, telenzepine, and pirenzepine. Injection of 20 mg/kg THX 70 min before oxotremorine prevented the appearance of abnormal postures in both Gnal+/− and WT mice, whereas 1 mg/kg had only a partial effect (although it reduced the difference between Gnal+/− and WT mice; Fig. 6F). Pretreatment with telenzepine (3 mg/kg) or pirenzepine (10 mg/kg), two M1 muscarinic receptor-preferring antagonists, significantly reduced the scores of motor defects in both Gnal+/− and WT mice (Fig. 6G). Because oxotremorine reduces acetylcholine release by striatal cholinergic interneurons via activation of M2 and M4 muscarinic autoreceptors (Lapchak et al., 1989, Yan and Surmeier, 1996) and thereby leads to reduced stimulation of nicotinic receptors in the striatum, we tested the role of nicotinic receptors. We treated Gnal+/− and WT mice with the nicotinic antagonist mecamylamine (3 mg/kg) alone or 10 min before oxotremorine administration (Fig. 6G). Mecamylamine alone generated no detectable motor abnormalities. In contrast, when associated with oxotremorine, mecamylamine produced severe motor impairment in both Gnal+/− and WT mice characterized by sustained abnormal postures with sporadic small movements and no ambulation. These motor abnormalities were almost systematically rated with the highest score for each observation period (Fig. 6G). These results showed that blockade of nicotinic receptors enhanced the abnormal movements and postures induced by oxotremorine and suggested that the reduced stimulation of nicotinic receptors resulting from the oxotremorine-induced inhibition of acetylcholine interneurons contributed to the motor defects produced by oxotremorine. The lack of effect of mecamylamine alone showed the necessary role of muscarinic receptor activation in triggering motor defects. Because THX is an effective medication in dystonic patients (Jankovic, 2013), the preventive effect of THX indicated that the oxotremorine-induced motor symptoms in Gnal+/− presented some homologies with human dystonia.
Microinjection of oxotremorine into the striatum, but not into the cerebellum, induces abnormal postures and movements in Gnal+/− mice
To determine whether the striatum or cerebellum could be the site in which oxotremorine induced the dystonic-like abnormalities observed in Gnal+/− mice, we tested the effects of local infusion of oxotremorine into these brain structures. Oxotremorine (0.1 μg) or vehicle was locally infused through cannulas into the right and left striata of Gnal+/− and Gnal+/+ animals (Fig. 7A). Compared with vehicle-treated mice, Gnal+/− animals infused with oxotremorine showed abnormal postures and movements resembling those observed after systemic injections of oxotremorine (Fig. 7B). Although some WT animals displayed abnormal movements, the dystonia scores in these mice were significantly lower than in their Gnal+/− littermates (Movies 3 and 4). Statistical analysis of abnormal motor scores by two-way ANOVA indicated significant effects of oxotremorine administration and genotype (see legend to Fig. 7) and post hoc tests indicated significantly higher abnormal motor scores in oxotremotrine-injected Gnal+/− mice than in WT littermates.
Figure 7.

Microinjections of oxotremorine into the striatum and cerebellum induce abnormal movements. A, Nissl staining showing the bilateral cannula placement in the striatum. White arrows indicate the tip of cannulas. B, Abnormal motor scores (mean 10 min scores) 30–60 min after microinjection of vehicle or oxotremorine (0.1 μg) into the striatum in Gnal+/+ and Gnal+/− mice. Two-way ANOVA: treatment, F(1,13) = 11.9, p < 0.01; genotype, F(1,13) = 6.7, p < 0.05; interaction, F(1,13) = 0.8, p = 0.4. C, Nissl staining showing the bilateral cannula placement into the cerebellum. D, Abnormal motor scores (mean 10 min scores) 20–60 min after microinjection of vehicle or oxotremorine (0.1 μg) into the cerebellum in Gnal+/+ and Gnal+/− mice. Two-way ANOVA: treatment, F(1,26) = 17.0, p < 0.001; genotype, F(1,26) = 0.62, p = 0.4; interaction, F(1,26) = 0.82, p = 0.4. Data are shown as means ± SEM. *p < 0.05; **p < 0.01, ns not significant, Sidak's multiple-comparisons test after two-way ANOVA.
Movie 3.
Postures and movements in WT mice after vehicle or oxotremorine (0.1 μg) infusions into the striatum.
Movie 4.
Postures and movements in Gnal+/− mice after vehicle or oxotremorine (0.1 μg) infusions into the striatum.
We also infused 1 μl of oxotremorine (0.1 μg) into the cerebellum. The cannulas were placed in the deep nuclei of the cerebellum (Fig. 7C), but oxotremorine probably largely spread in other cerebellar areas. After intracerebellar oxotremorine injection, motor defects were observed in both Gnal+/− and WT mice (Fig. 7D). Intense motor disorders were observed immediately after the oxotremorine infusion. These were characterized by highly uncoordinated movements, making walking difficult, which suggested ataxia. Attacks of whole-body tremors were often observed. Such motor abnormalities were not observed after oxotremorine infusion into the striatum. Ten to 15 min after the oxotremorine injections into the cerebellum, the ambulation stopped and numerous animal remained motionless in generally normal but sometimes anomalous postures. The motor abnormalities during this second phase (20–60 min after injection) were scored using the same criteria as used previously for intrastriatal injection. Two-way ANOVA indicated a significant effect of oxotremorine treatment, but the genotype effect and interaction were not significant (see legend to Fig. 7). Post hoc tests confirmed the absence of significant difference between oxotremotrine-injected Gnal+/− and WT mice. These results suggested that the higher severity of motor impairment induced by systemic treatment with oxotremorine in Gnal+/− mice was more probably caused by an action in the striatum rather than in the cerebellum.
Discussion
Heterozygous loss-of-function mutations of the GNAL (DYT25) gene, encoding the protein Gαolf, is a recently identified cause of dystonia (Fuchs et al., 2013; Vemula et al., 2013). Here, we show that Gαolf haplodeficiency in mice impairs self-grooming, motor coordination, and apparent motivation in an instrumental task without affecting spontaneous locomotor activity. However, no overt dystonia-like motor dysfunction was detected in basal conditions. In contrast, Gnal+/− mice displayed significantly more abnormal postures and movements than WT mice after systemic administration of the cholinergic agonist oxotremorine or its local infusion into the striatum. These effects were prevented by THX, a muscarinic antagonist that reduces dystonia in patients.
Genetic evidence indicates that Gnal+/− mice are an animal model with excellent etiological validity for studying the pathophysiology of GNAL-induced dystonia. The pathogenic GNAL mutations reported in patients include a variety of dominant mutations, producing Gαolf forms with no or poor functional activity (Fuchs et al., 2013; Vemula et al., 2013; Kumar et al., 2014; Ziegan et al., 2014; Dos Santos et al., 2016). Therefore, the genetic status of patients with complete loss of function in one GNAL allele is perfectly mimicked by the Gnal+/− mice, which express half of the normal levels of Gαolf, without a compensatory change in Gαs (Corvol et al., 2001; Hervé et al., 2001; Corvol et al., 2007).
Significant physiological and behavioral changes in Gnal+/− mice showed that the partial loss of Gαolf expression was not fully compensated. Gαolf is highly expressed in the striatum, where it replaces the Gαs isoform, the G-protein responsible for adenylyl cyclase stimulation in most cell types (Hervé et al., 2001). In the striatum, Gnal haplodeficiency reduces the levels of AC5 (Xie et al., 2015) and decreases AC responses to D1R or A2AR agonists without affecting the striatal densities of these receptors (Corvol et al., 2001; Hervé et al., 2001). Reduced Gαolf expression markedly alters biochemical and behavioral responses linked to the activation of D1R in the striatum, indicating that Gαolf levels are a limiting factor for the action of D1R (Corvol et al., 2007).
The subtle modifications of SPN spine length in Gnal+/− mice could be related to cAMP signaling alteration because synaptogenesis in young mice depends on the activation of cAMP pathway and its modulation by LRRK2 (Parisiadou et al., 2014; Kozorovitskiy et al., 2015). We also detected a reduced autophosphorylation of CaMKIIβ that might be related to a lack of inhibition of its main phosphatase, protein phosphatase 1, due to an impairment of phosphorylation of its striatal-enriched inhibitor, DARPP-32, in Gnal+/− mice (Alcacer et al., 2012). However, the link between reduced phosphorylation of CaMKIIβ and changes in spine morphology is elusive because CaMKIIβ acts on actin and possibly spine morphology independently of its kinase activity (Okamoto et al., 2007; Lin and Redmond, 2008).
An impairment of motor coordination and learning was observed in the rotarod test, particularly in 1-year-old animals, and was confirmed by the ledge test. Interestingly, mutant mice lacking AC5 or the G-protein subunit Gγ7, two proteins closely associated with Gαolf in the striatum (Schwindinger et al., 2003; Iwamoto et al., 2004; Xie et al., 2015), also exhibit important deficits in the rotarod test (Kheirbek et al., 2009; Sasaki et al., 2013). Several animal models of dystonia show deficits of variable severity in motor coordination in the rotarod test or beam-walking test (which is very similar to the ledge test), although they do not exhibit overt dystonic movements and postures (Oleas et al., 2013; Liang et al., 2014; Richter and Richter, 2014). Therefore, deficits in these tests are suggested as indirect indications of dystonia in genetic rodent models (Oleas et al., 2013; Richter and Richter, 2014).
Despite these behavioral alterations, Gnal+/− mice exhibited no abnormal spontaneous postures or movements reminiscent of dystonia. In contrast, Gnal+/− mice developed significant motor disorders when they were treated with oxotremorine. In WT mice, oxotremorine produced slowness in movement and few abnormal postures. In contrast, oxotremorine-treated Gnal+/− mice displayed significantly more intense motor defects, with limited ambulation, repetitive abnormal movements, and unusual paw or body positions. This phenotype is similar to those reported in symptomatic rodent models of dystonia (Pizoli et al., 2002; Calderon et al., 2011; Fan et al., 2012; Rose et al., 2015). The motor abnormalities were prevented by THX, a drug able to reduce dystonia in patients (Jankovic, 2013). Muscarinic M1 receptor may contribute to the dystonic-like effects of oxotremorine in Gnal+/− mice because these effects were prevented by THX, pirenzepine, or telenzepine, good antagonists of these receptors (Dörje et al., 1991). However, the selectivity of these compounds with respect to the M1 receptors is not very high (<100-fold; Eveleigh et al., 1989; Dörje et al., 1991) and our experiments did not exclude the contribution of other types of muscarinic receptors. Nicotinic mechanisms could also play a role because mecamylamine, a selective nicotinic receptor antagonist, worsened the motor abnormalities induced by oxotremorine. Because opposite nicotinic and muscarinic mechanisms contributed to oxotremorine-induced motor abnormalities, it is unclear whether the higher impairment of Gnal+/− mice results from alterations of nicotinic or muscarinic signaling.
Intrastriatal injection of oxotremorine in Gnal+/− mice produced motor dysfunctions similar to systemic administration, suggesting that the striatum is responsible for their generation. Therefore, we propose that oxotremorine intensifies the striatal dysfunction resulting from decreased Gαolf levels and precipitates the occurrence of dystonic-like movements and postures. However, several studies have suggested that alteration of cerebellar activity can be a cause of dystonia (Jinnah and Hess, 2006; Neychev et al., 2008; LeDoux, 2011). Gαolf is expressed in cerebellar Purkinje cells (Belluscio et al., 1998; Vemula et al., 2013) and the deep nuclei of cerebellum (see the Allen Brain Atlas, http://mouse.brain-map.org/experiment/show?id=69734482). Although, in these neurons, Gαolf is found at lower levels than in SPNs and is coexpressed with Gαs, we cannot rule out that partial Gαolf deficiency in cerebellum contributes to dystonia pathogenesis. However, oxotremorine infusion into the cerebellum induces distinct motor defects, including ataxia and whole-body tremors, which are different from those observed after intrastriatal injections and of similar severity in Gnal+/− and WT mice, suggesting that the vulnerability of Gnal+/− mice to develop movement disorders in response to oxotremorine does not take place in the cerebellum. Nevertheless, the cerebellum may participate in the GNAL phenotype because it has been proposed that dystonia does not depend on specific dysfunction of either basal ganglia or cerebellum, but rather results from defective interactions between the two motor circuits (Calderon et al., 2011; Prudente et al., 2014).
Convergent evidence indicates that alterations in striatal dopamine transmission play a role in various types of dystonia. Dystonia can be caused by mutations in genes critical for the synthesis of dopamine (Ichinose et al., 1999; Rose et al., 2015) or by degeneration of dopamine neurons in Parkinson's disease or MPTP-treated monkeys (Tabbal et al., 2006; Fabbrini et al., 2007; Wichmann, 2008). Data in dystonic patients and DYT1 mutant mice also suggest disruption of dopamine transmission (Playford et al., 1993; Perlmutter et al., 1997; Naumann et al., 1998; Bao et al., 2010; Page et al., 2010). In a recent model of l-DOPA-responsive dystonia, D1R blockade exacerbated dystonic movements, whereas its activation reduced them (Rose et al., 2015). D1R and Gαolf levels are reduced in the striatum of DYT1 mouse models (Yokoi et al., 2015; Zhang et al., 2015). Homozygous Gγ7 knock-out mice, which have a severe impairment of Gαolf signaling in the striatum, display age-dependent dystonia (Sasaki et al., 2013). Finally, mutations of ADCY5, the human gene coding for AC5, the main target of Gαolf in striatal neurons, are responsible for movement disorders, including dystonia (Chen et al., 2014; Carapito et al., 2015; Chen et al., 2015). Collectively, these findings in patients and in mouse models indicate a strong implication of disrupted Gαolf/Gγ7/AC5 signaling in SPNs in dystonia-related symptoms. Impairment of this pathway is expected not only to decrease D1R/cAMP signaling in dSPNs, but also to mimic D2R inhibition of AC in iSPNs while preserving other actions of D2R such as inhibition of K+ currents (Gerfen and Surmeier, 2011). The anticipated result is an imbalance between the direct and indirect pathways, which may be a critical factor favoring the appearance of dystonia in response to other triggering stimuli. The various alterations of dopamine signaling in Gnal+/− mice and their particular sensitivity to oxotremorine are consistent with the proposed crucial role of the dopamine–acetylcholine interaction in the development of dystonic symptoms (Pisani et al., 2007; Pappas et al., 2015).
The particular susceptibility of Gnal+/− mice to oxotremorine-induced motor abnormalities suggests that GNAL mutation carriers could be vulnerable to other minor brain dysfunctions that could precipitate in pathological dystonia. DYT25 onset occurs relatively late, at ∼30 years of age (Fuchs et al., 2013; Vemula et al., 2013). Gnal+/− mice could thus be considered as a presymptomatic model of GNAL-induced dystonia. At this stage, no dystonia is observed, but our results in mice suggest that motor or nonmotor dysfunctions may be present without reaching a clinical threshold. Therefore, Gnal+/− mice and oxotremorine-induced movement alterations provide an excellent model with which to further elucidate the mechanisms of dystonia generation and explore novel therapeutic approaches.
Notes
Supplemental material for this article is available at https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/GOVOUB. Included are videos showing the effects of oxotremorine, injected systematically or locally into the striatum in Gnal+/− and WT mice. This material has not been peer reviewed.
Footnotes
This work was supported in part by Inserm, the Université Pierre et Marie Curie (UPMC, Paris 6, Sorbonne-Universités), the Agence Nationale de la Recherche (Grants ANR09-MNPS-014 to DH, ANR-16-CE37-0003-01 AMEDYST to D.H. and D.P., ANR-12-JSV4-0004 CEREDYSTIM to D.P., and ANR-10-LABX-54 MEMOLIFE and ANR-10-IDEX-0001-02 to PSL Research University), and a European Research Council (Advanced Investigator Grant ERC-AI-2010-250349 to J.-A.G.). Equipment at the Institut du Fer à Moulin was also supported by DIM NeRF from Région Ile-de-France and by the Féfération pour la recherche sur le Cerveau (FRC)/Rotary Espoir en tête. We thank Albert Giralt for the Golgi–Cox staining protocol, Leonardo Belluscio for providing Gnal+/− mice, Domenico Guarino for help in the blind analysis of morphological data, Fiona Francis for language editing of Table 1, the Institut du Fer à Moulin Cell and Tissue Imaging Facility for image acquisition, and the staff of the Institut du Fer à Moulin animal facilities. The Girault and Hervé group is affiliated with the Paris School of Neuroscience (Ecole des Neurosciences de Paris, ENP) and the Bio-Psy laboratory of excellence.
The authors declare no competing financial interests.
References
- Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, Hallett M, Jankovic J, Jinnah HA, Klein C, Lang AE, Mink JW, Teller JK (2013) Phenomenology and classification of dystonia: a consensus update. Mov Disord 28:863–873. 10.1002/mds.25475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcacer C, Santini E, Valjent E, Gaven F, Girault JA, Hervé D (2012) G alpha(olf) Mutation allows parsing the role of cAMP-dependent and extracellular signal-regulated kinase-dependent signaling in l-3,4-dihydroxyphenylalanine-induced dyskinesia. J Neurosci 32:5900–5910. 10.1523/JNEUROSCI.0837-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge JW, Berridge KC (1998) Coding of serial order by neostriatal neurons: a “natural action” approach to movement sequence. J Neurosci 18:2777–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Patel JC, Walker RH, Shashidharan P, Rice ME (2010) Dysregulation of striatal dopamine release in a mouse model of dystonia. J Neurochem 114:1781–1791. 10.1111/j.1471-4159.2010.06890.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belluscio L, Gold GH, Nemes A, Axel R (1998) Mice deficient in G(olf) are anosmic. Neuron 20:69–81. 10.1016/S0896-6273(00)80435-3 [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Carter PA, Yamada M, Gomeza J, Wess J, Hamilton SE, Nathanson NM, McKinzie DL, Felder CC (2003) Role of specific muscarinic receptor subtypes in cholinergic parasympathomimetic responses, in vivo phosphoinositide hydrolysis, and pilocarpine-induced seizure activity. Eur J Neurosci 17:1403–1410. 10.1046/j.1460-9568.2003.02588.x [DOI] [PubMed] [Google Scholar]
- Calderon DP, Fremont R, Kraenzlin F, Khodakhah K (2011) The neural substrates of rapid-onset Dystonia-Parkinsonism. Nat Neurosci 14:357–365. 10.1038/nn.2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DB, Hess EJ (1998) Cerebellar circuitry is activated during convulsive episodes in the tottering (tg/tg) mutant mouse. Neuroscience 85:773–783. 10.1016/S0306-4522(97)00672-6 [DOI] [PubMed] [Google Scholar]
- Carapito R, Paul N, Untrau M, Le Gentil M, Ott L, Alsaleh G, Jochem P, Radosavljevic M, Le Caignec C, David A, Damier P, Isidor B, Bahram S (2015) A de novo ADCY5 mutation causes early-onset autosomal dominant chorea and dystonia. Mov Disord 30:423–427. 10.1002/mds.26115 [DOI] [PubMed] [Google Scholar]
- Chen DH, et al. (2015) ADCY5-related dyskinesia: broader spectrum and genotype-phenotype correlations. Neurology 85:2026–2035. 10.1212/WNL.0000000000002058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YZ, et al. (2014) Gain-of-function ADCY5 mutations in familial dyskinesia with facial myokymia. Ann Neurol 75:542–549. 10.1002/ana.24119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvol JC, Studler JM, Schonn JS, Girault JA, Hervé D (2001) G alpha(olf) is necessary for coupling D1 and A2a receptors to adenylyl cyclase in the striatum. J Neurochem 76:1585–1588. 10.1046/j.1471-4159.2001.00201.x [DOI] [PubMed] [Google Scholar]
- Corvol JC, Valjent E, Pascoli V, Robin A, Stipanovich A, Luedtke RR, Belluscio L, Girault JA, Hervé D (2007) Quantitative changes in G alpha olf protein levels, but not D1 receptor, alter specifically acute responses to psychostimulants. Neuropsychopharmacology 32:1109–1121. 10.1038/sj.npp.1301230 [DOI] [PubMed] [Google Scholar]
- Dörje F, Wess J, Lambrecht G, Tacke R, Mutschler E, Brann MR (1991) Antagonist binding profiles of five cloned human muscarinic receptor subtypes. J Pharmacol Exp Ther 256:727–733. [PubMed] [Google Scholar]
- Dos Santos CO, Masuho I, da Silva-Júnior FP, Barbosa ER, Silva SM, Borges V, Ferraz HB, Rocha MS, Limongi JC, Martemyanov KA, de Carvalho Aguiar P (2016) Screening of GNAL variants in Brazilian patients with isolated dystonia reveals a novel mutation with partial loss of function. J Neurol 263:665–668. 10.1007/s00415-016-8026-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinnan SL, Hope BT, Snutch TP, Vincent SR (1991) G(olf) in the basal ganglia. Mol Cell Neurosci 2:66–70. 10.1016/1044-7431(91)90040-U [DOI] [PubMed] [Google Scholar]
- Eskow Jaunarajs KL, Bonsi P, Chesselet MF, Standaert DG, Pisani A (2015) Striatal cholinergic dysfunction as a unifying theme in the pathophysiology of dystonia. Prog Neurobiol 127–128:91–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eveleigh P, Hulme EC, Schudt C, Birdsall NJ (1989) The existence of stable enantiomers of telenzepine and their stereoselective interaction with muscarinic receptor subtypes. Mol Pharmacol 35:477–483. [PubMed] [Google Scholar]
- Fabbrini G, Brotchie JM, Grandas F, Nomoto M, Goetz CG (2007) Levodopa-induced dyskinesias. Mov Disord 22:1379–1389; quiz 1523. 10.1002/mds.21475 [DOI] [PubMed] [Google Scholar]
- Fan X, Hughes KE, Jinnah HA, Hess EJ (2012) Selective and sustained α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor activation in cerebellum induces dystonia in mice. J Pharmacol Exp Ther 340:733–741. 10.1124/jpet.111.190082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremont R, Calderon DP, Maleki S, Khodakhah K (2014) Abnormal high-frequency burst firing of cerebellar neurons in rapid-onset dystonia-parkinsonism. J Neurosci 34:11723–11732. 10.1523/JNEUROSCI.1409-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs T, Saunders-Pullman R, Masuho I, Luciano MS, Raymond D, Factor S, Lang AE, Liang TW, Trosch RM, White S, Ainehsazan E, Hervé D, Sharma N, Ehrlich ME, Martemyanov KA, Bressman SB, Ozelius LJ (2013) Mutations in GNAL cause primary torsion dystonia. Nat Genet 45:88–92. 10.1038/ng.2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ (2011) Modulation of striatal projection systems by dopamine. Annu Rev Neurosci 34:441–466. 10.1146/annurev-neuro-061010-113641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR, Garden GA (2010) A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp 39: pii: 1787. 10.3791/1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann M, Richter A, Meillasson FV, Nitsch C, Ebert U (2007) Age-related changes in parvalbumin-positive interneurons in the striatum, but not in the sensorimotor cortex in dystonic brains of the dt(sz) mutant hamster. Brain Res 1150:190–199. 10.1016/j.brainres.2007.02.074 [DOI] [PubMed] [Google Scholar]
- Hervé D. (2011) Identification of a specific assembly of the G protein Golf as a critical and regulated module of dopamine and adenosine-activated cAMP pathways in the striatum. Front Neuroanat 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervé D, Lévi-Strauss M, Marey-Semper I, Verney C, Tassin JP, Glowinski J, Girault JA (1993) G(olf) and Gs in rat basal ganglia: possible involvement of G(olf) in the coupling of dopamine D1 receptor with adenylyl cyclase. J Neurosci 13:2237–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervé D, Le Moine C, Corvol JC, Belluscio L, Ledent C, Fienberg AA, Jaber M, Studler JM, Girault JA (2001) G alpha(olf) levels are regulated by receptor usage and control dopamine and adenosine action in the striatum. J Neurosci 21:4390–4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinose H, Suzuki T, Inagaki H, Ohye T, Nagatsu T (1999) Molecular genetics of dopa-responsive dystonia. Biol Chem 380:1355–1364. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Iwatsubo K, Okumura S, Hashimoto Y, Tsunematsu T, Toya Y, Hervé D, Umemura S, Ishikawa Y (2004) Disruption of type 5 adenylyl cyclase negates the developmental increase in G alpha olf expression in the striatum. FEBS Lett 564:153–156. 10.1016/S0014-5793(04)00333-3 [DOI] [PubMed] [Google Scholar]
- Jankovic J. (2013) Medical treatment of dystonia. Mov Disord 28:1001–1012. 10.1002/mds.25552 [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Hess EJ (2006) A new twist on the anatomy of dystonia: the basal ganglia and the cerebellum? Neurology 67:1740–1741. 10.1212/01.wnl.0000246112.19504.61 [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Sepkuty JP, Ho T, Yitta S, Drew T, Rothstein JD, Hess EJ (2000) Calcium channel agonists and dystonia in the mouse. Mov Disord 15:542–551. [DOI] [PubMed] [Google Scholar]
- Jones DT, Reed RR (1989) Golf: an olfactory neuron specific-G protein involved in odorant signal transduction. Science 244:790–795. 10.1126/science.2499043 [DOI] [PubMed] [Google Scholar]
- Kheirbek MA, Britt JP, Beeler JA, Ishikawa Y, McGehee DS, Zhuang X (2009) Adenylyl cyclase type 5 contributes to corticostriatal plasticity and striatum-dependent learning. J Neurosci 29:12115–12124. 10.1523/JNEUROSCI.3343-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozorovitskiy Y, Peixoto R, Wang W, Saunders A, Sabatini BL (2015) Neuromodulation of excitatory synaptogenesis in striatal development. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar KR, et al. (2014) Mutations in GNAL: a novel cause of craniocervical dystonia. JAMA Neurol 71:490–494. 10.1001/jamaneurol.2013.4677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapchak PA, Araujo DM, Quirion R, Collier B (1989) Binding sites for [H-3]AF-DX-116 and effect of AF-DX-116 on endogenous acetylcholine release from rat brain slices. Brain Res 496:285–294. 10.1016/0006-8993(89)91075-5 [DOI] [PubMed] [Google Scholar]
- LeDoux MS. (2011) Animal models of dystonia: Lessons from a mutant rat. Neurobiol Dis 42:152–161. 10.1016/j.nbd.2010.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Hong JH, Choi IY, Che Y, Lee JK, Yang SD, Song CW, Kang HS, Lee JH, Noh JS, Shin HS, Han PL (2002) Impaired D2 dopamine receptor function in mice lacking type 5 adenylyl cyclase. J Neurosci 22:7931–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CC, Tanabe LM, Jou S, Chi F, Dauer WT (2014) TorsinA hypofunction causes abnormal twisting movements and sensorimotor circuit neurodegeneration. J Clin Invest 124:3080–3092. 10.1172/JCI72830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Redmond L (2008) CaMKIIbeta binding to stable F-actin in vivo regulates F-actin filament stability. Proc Natl Acad Sci U S A 105:15791–15796. 10.1073/pnas.0804399105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann M, Pirker W, Reiners K, Lange KW, Becker G, Brücke T (1998) Imaging the pre- and postsynaptic side of striatal dopaminergic synapses in idiopathic cervical dystonia: a SPECT study using [123I] epidepride and [123I] beta-CIT. Mov Disord 13:319–323. 10.1002/mds.870130219 [DOI] [PubMed] [Google Scholar]
- Neychev VK, Fan X, Mitev VI, Hess EJ, Jinnah HA (2008) The basal ganglia and cerebellum interact in the expression of dystonic movement. Brain 131:2499–2509. 10.1093/brain/awn168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Narayanan R, Lee SH, Murata K, Hayashi Y (2007) The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc Natl Acad Sci U S A 104:6418–6423. 10.1073/pnas.0701656104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleas J, Yokoi F, DeAndrade MP, Pisani A, Li Y (2013) Engineering animal models of dystonia. Mov Disord 28:990–1000. 10.1002/mds.25583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius LJ, Lubarr N, Bressman SB (2011) Milestones in dystonia. Mov Disord 26:1106–1126. 10.1002/mds.23775 [DOI] [PubMed] [Google Scholar]
- Page ME, Bao L, Andre P, Pelta-Heller J, Sluzas E, Gonzalez-Alegre P, Bogush A, Khan LE, Iacovitti L, Rice ME, Ehrlich ME (2010) Cell-autonomous alteration of dopaminergic transmission by wild type and mutant (DeltaE) TorsinA in transgenic mice. Neurobiol Dis 39:318–326. 10.1016/j.nbd.2010.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas SS, Darr K, Holley SM, Cepeda C, Mabrouk OS, Wong JM, LeWitt TM, Paudel R, Houlden H, Kennedy RT, Levine MS, Dauer WT (2015) Forebrain deletion of the dystonia protein torsinA causes dystonic-like movements and loss of striatal cholinergic neurons. Elife 4:e08352. 10.7554/eLife.08352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou L, Yu J, Sgobio C, Xie C, Liu G, Sun L, Gu XL, Lin X, Crowley NA, Lovinger DM, Cai H (2014) LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat Neurosci 17:367–376. 10.1038/nn.3636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoli V, Valjent E, Corbillé AG, Corvol JC, Tassin JP, Girault JA, Hervé D (2005) cAMP and extracellular signal-regulated kinase signaling in response to d-amphetamine and methylphenidate in the prefrontal cortex in vivo: role of beta 1-adrenoceptors. Mol Pharmacol 68:421–429. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2001) The mouse brain in stereotaxic coordinates, Ed 2 San Diego: Academic. [Google Scholar]
- Pelosi A, Girault JA, Hervé D (2015) Unilateral lesion of dopamine neurons induces grooming asymmetry in the mouse. PLoS One 10:e0137185. 10.1371/journal.pone.0137185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter JS, Stambuk MK, Markham J, Black KJ, McGee-Minnich L, Jankovic J, Moerlein SM (1997) Decreased [18F]spiperone binding in putamen in idiopathic focal dystonia. J Neurosci 17:843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Martella G, Tscherter A, Bonsi P, Sharma N, Bernardi G, Standaert DG (2006) Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia. Neurobiol Dis 24:318–325. 10.1016/j.nbd.2006.07.006 [DOI] [PubMed] [Google Scholar]
- Pisani A, Bernardi G, Ding J, Surmeier DJ (2007) Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci 30:545–553. 10.1016/j.tins.2007.07.008 [DOI] [PubMed] [Google Scholar]
- Pizoli CE, Jinnah HA, Billingsley ML, Hess EJ (2002) Abnormal cerebellar signaling induces dystonia in mice. J Neurosci 22:7825–7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Playford ED, Fletcher NA, Sawle GV, Marsden CD, Brooks DJ (1993) Striatal [18F]dopa uptake in familial idiopathic dystonia. Brain 116:1191–1199. 10.1093/brain/116.5.1191 [DOI] [PubMed] [Google Scholar]
- Prudente CN, Hess EJ, Jinnah HA (2014) Dystonia as a network disorder: what is the role of the cerebellum? Neuroscience 260:23–35. 10.1016/j.neuroscience.2013.11.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter F, Richter A (2014) Genetic animal models of dystonia: common features and diversities. Prog Neurobiol 121:91–113. 10.1016/j.pneurobio.2014.07.002 [DOI] [PubMed] [Google Scholar]
- Ricobaraza A, Cuadrado-Tejedor M, Marco S, Pérez-Otaño I, García-Osta A (2012) Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus 22:1040–1050. 10.1002/hipo.20883 [DOI] [PubMed] [Google Scholar]
- Rose SJ, Yu XY, Heinzer AK, Harrast P, Fan X, Raike RS, Thompson VB, Pare JF, Weinshenker D, Smith Y, Jinnah HA, Hess EJ (2015) A new knock-in mouse model of l-DOPA-responsive dystonia. Brain 138:2987–3002. 10.1093/brain/awv212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz M, Perez-Garcia G, Ortiz-Virumbrales M, Méneret A, Morant A, Kottwitz J, Fuchs T, Bonet J, Gonzalez-Alegre P, Hof PR, Ozelius LJ, Ehrlich ME (2015) Abnormalities of motor function, transcription and cerebellar structure in mouse models of THAP1 dystonia. Hum Mol Genet 24:7159–7170. 10.1093/hmg/ddv384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Yamasaki T, Omotuyi IO, Mishina M, Ueda H (2013) Age-dependent dystonia in striatal Gγ7 deficient mice is reversed by the dopamine D2 receptor agonist pramipexole. J Neurochem 124:844–854. 10.1111/jnc.12149 [DOI] [PubMed] [Google Scholar]
- Schiffmann SN, Fisone G, Moresco R, Cunha RA, Ferré S (2007) Adenosine A2A receptors and basal ganglia physiology. Prog Neurobiol 83:277–292. 10.1016/j.pneurobio.2007.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwindinger WF, Betz KS, Giger KE, Sabol A, Bronson SK, Robishaw JD (2003) Loss of G protein gamma 7 alters behavior and reduces striatal alpha(olf) level and cAMP production. J Biol Chem 278:6575–6579. 10.1074/jbc.M211132200 [DOI] [PubMed] [Google Scholar]
- Schwindinger WF, Mihalcik LJ, Giger KE, Betz KS, Stauffer AM, Linden J, Hervé D, Robishaw JD (2010) Adenosine A(2A) receptor signaling and G(olf) assembly show a specific requirement for the gamma(7) subtype in the striatum. J Biol Chem 285:29787–29796. 10.1074/jbc.M110.142620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CH, Bernhard D, Bolarinwa C, Hess EJ, Smith Y, Jinnah HA (2013) Subtle microstructural changes of the striatum in a DYT1 knock-in mouse model of dystonia. Neurobiol Dis 54:362–371. 10.1016/j.nbd.2013.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipanovich A, Valjent E, Matamales M, Nishi A, Ahn JH, Maroteaux M, Bertran-Gonzalez J, Brami-Cherrier K, Enslen H, Corbillé AG, Filhol O, Nairn AC, Greengard P, Hervé D, Girault JA (2008) A phosphatase cascade by which rewarding stimuli control nucleosomal response. Nature 453:879–U823. 10.1038/nature06994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabbal SD, Mink JW, Antenor JA, Carl JL, Moerlein SM, Perlmutter JS (2006) 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced acute transient dystonia in monkeys associated with low striatal dopamine. Neuroscience 141:1281–1287. 10.1016/j.neuroscience.2006.04.072 [DOI] [PubMed] [Google Scholar]
- Tozzi A, de Iure A, Di Filippo M, Tantucci M, Costa C, Borsini F, Ghiglieri V, Giampà C, Fusco FR, Picconi B, Calabresi P (2011) The distinct role of medium spiny neurons and cholinergic interneurons in the D/A2A receptor interaction in the striatum: implications for Parkinson's disease. J Neurosci 31:1850–1862. 10.1523/JNEUROSCI.4082-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turski WA, Cavalheiro EA, Bortolotto ZA, Mello LM, Schwarz M, Turski L (1984) Seizures produced by pilocarpine in mice: a behavioral, electroencephalographic and morphological analysis. Brain Res 321:237–253. 10.1016/0006-8993(84)90177-X [DOI] [PubMed] [Google Scholar]
- Vemula SR, Puschmann A, Xiao J, Zhao Y, Rudzińska M, Frei KP, Truong DD, Wszolek ZK, LeDoux MS (2013) Role of Gα(olf) in familial and sporadic adult-onset primary dystonia. Hum Mol Genet 22:2510–2519. 10.1093/hmg/ddt102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisheit CE, Dauer WT (2015) A novel conditional knock-in approach defines molecular and circuit effects of the DYT1 dystonia mutation. Hum Mol Genet 24:6459–6472. 10.1093/hmg/ddv355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann T. (2008) Commentary: dopaminergic dysfunction in DYT1 dystonia. Exp Neurol 212:242–246. 10.1016/j.expneurol.2008.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JH, Kauer JA (1997) Properties of carbachol-induced oscillatory activity in rat hippocampus. J Neurophysiol 78:2631–2640. [DOI] [PubMed] [Google Scholar]
- Xie K, Masuho I, Shih CC, Cao Y, Sasaki K, Lai CW, Han PL, Ueda H, Dessauer CW, Ehrlich ME, Xu B, Willardson BM, Martemyanov KA (2015) Stable G protein-effector complexes in striatal neurons: mechanism of assembly and role in neurotransmitter signaling. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Surmeier DJ (1996) Muscarinic (m2/m4) receptors reduce N- and P-type Ca2+ currents in rat neostriatal cholinergic interneurons through a fast, membrane-delimited, G-protein pathway. J Neurosci 16:2592–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Surmeier DJ (1997) D5 dopamine receptors enhance Zn2+-sensitive GABA(A) currents in striatal cholinergic interneurons through a PKA/PP1 cascade. Neuron 19:1115–1126. 10.1016/S0896-6273(00)80402-X [DOI] [PubMed] [Google Scholar]
- Yokoi F, Dang MT, Liu J, Gandre JR, Kwon K, Yuen R, Li Y (2015) Decreased dopamine receptor 1 activity and impaired motor-skill transfer in Dyt1 ΔGAG heterozygous knock-in mice. Behav Brain Res 279:202–210. 10.1016/j.bbr.2014.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, McCarthy DM, Sharma N, Bhide PG (2015) Dopamine receptor and Gα(olf) expression in DYT1 dystonia mouse models during postnatal development. PLoS One 10:e0123104. 10.1371/journal.pone.0123104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X, Belluscio L, Hen R (2000) G(olf)alpha mediates dopamine D1 receptor signaling. J Neurosci 20:RC91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegan J, Wittstock M, Westenberger A, Dobričić V, Wolters A, Benecke R, Klein C, Kamm C (2014) Novel GNAL mutations in two German patients with sporadic dystonia. Mov Disord 29:1833–1834. 10.1002/mds.26066 [DOI] [PubMed] [Google Scholar]