

Abstract
Energy-dense, yet nutritionally poor food is a high-risk factor for mental health disorders. This is of particular concern during adolescence, a period often associated with increased consumption of low nutritional content food and higher prevalence of mental health disorders. Indeed, there is an urgent need to understand the mechanisms linking unhealthy diet and mental disorders. Deficiency in n-3 polyunsaturated fatty acids (PUFAs) is a hallmark of poor nutrition and mood disorders. Here, we developed a mouse model of n-3 PUFA deficiency lasting from adolescence into adulthood. Starting nutritional deficits in dietary n-3 PUFAs during adolescence decreased n-3 PUFAs in both medial prefrontal cortex (mPFC) and nucleus accumbens, increased anxiety-like behavior, and decreased cognitive function in adulthood. Importantly, we discovered that endocannabinoid/mGlu5-mediated LTD in the mPFC and accumbens was abolished in adult n-3-deficient mice. Additionally, mPFC NMDAR-dependent LTP was also lacking in the n-3-deficient group. Pharmacological enhancement of the mGlu5/eCB signaling complex, by positive allosteric modulation of mGlu5 or inhibition of endocannabinoid 2-arachidonylglycerol degradation, fully restored synaptic plasticity and normalized emotional and cognitive behaviors in malnourished adult mice. Our data support a model where nutrition is a key environmental factor influencing the working synaptic range into adulthood, long after the end of the perinatal period. These findings have important implications for the identification of nutritional risk factors for disease and design of new treatments for the behavioral deficits associated with nutritional n-3 PUFA deficiency.
SIGNIFICANCE STATEMENT In a mouse model mimicking n-3 PUFA dietary deficiency during adolescence and adulthood, we found strong increases in anxiety and anhedonia which lead to decreases in specific cognitive functions in adulthood. We found that endocannabinoid/mGlu5-mediated LTD and NMDAR-dependent LTP were lacking in adult n-3-deficient mice. Acute positive allosteric modulation of mGlu5 or inhibition of endocannabinoid degradation normalized behaviors and synaptic functions in n-3 PUFA-deficient adult mice. These findings have important implications for the identification of nutritional risk for disease and the design of new treatments for the behavioral deficits associated with nutritional n-3 PUFAs' imbalance.
Keywords: accumbens, endocannabinoid, LTD, LTP, mgluR5, prefrontal cortex
Introduction
Poverty and food insecurity are associated with lower food expenditures, low fruit and vegetable consumption, and overall lower-quality diets. Such diets are more affordable than are prudent diets based on lean meats, fish, fresh vegetables, and fruit. The dietary status of adults in Western countries is a serious rising public health concern. This is particularly preoccupying in the low-income segment of the population that is widely exposed to low-nutritional content food containing insufficient quantities of long-chain ω-3 polyunsaturated fatty acids (n-3 PUFAs).
Perinatal and adolescent nutritional n-3 PUFA deficiency is a major environmental risk factor associated with neuropsychiatric disorders, including major depression, bipolar disorder, and schizophrenia (Simopoulos, 2009; Bazinet and Layé, 2014; Bosch-Bouju et al., 2016). ω-3 deficiencies during adolescence may interfere with normal late development and increase the vulnerability to behavioral deficits during adulthood (Drewnowski, 2004). For example, in adolescents, multigenerational dietary n-3 PUFA deficiency causes neurobehavioral deficits that are distinct from those developed in adulthood (Bondi et al., 2014). Thus, n-3 PUFA deficiency during early adolescence may modify the normal developmental trajectory and cause alterations emerging in adulthood. However, the behavioral and a fortiori synaptic consequences of dietary n-3 PUFA deficiency in adults resulting from a PUFA-imbalanced diet during adolescence have seldom been studied (Lalancette-Hébert et al., 2011; Joffre et al., 2014; Delpech et al., 2015).
Developing brains rely solely on, and thus are exquisitely sensitive to, maternal PUFA levels (Bazinet and Layé, 2014). Following lifelong dietary imbalance, our previous work revealed alterations of the endocannabinoid (eCB) system that were paralleled by heightened expression of anxiety and depression-like behaviors in mice (Lafourcade et al., 2011; Larrieu et al., 2012, 2014; Thomazeau et al., 2017) and highlighted the involvement of the nucleus accumbens and the medial prefrontal cortex (mPFC). However, no data are available as to whether the observed emotional deficits are principally related to the effects of n-3 PUFA deficiency during perinatal and/or postnatal periods. This is an important question because nutritional independence from parental supervision often starts at adolescence, a period of ongoing structural and functional refinements of the frontostriatal circuitry implicated in behavioral control (Galvan et al., 2006) and higher cognitive functions (Caballero and Tseng, 2016; Caballero et al., 2016). As a consequence, the full understanding of the pathophysiological underpinnings of dietary n-3 deficiency-induced behavioral and synaptic alterations requires more specific distinction between these two temporal stages.
To clarify the role of nutritional PUFAs in the refinement of adult synaptic circuitry and behavior, we developed a model of n-3 PUFA deficiency starting at adolescence. To this end, mice that had received balanced food until this point in life were exposed to an n-3 PUFA-deficient diet from early adolescence through adulthood.
We discovered severe alterations of the emotional and cognitive repertoire in adult mice exposed to n-3 PUFA deficiency from early adolescence that were paralleled by specific deficits in mGlu5/eCB synaptic plasticity of the mPFC and the accumbens. We found that pharmacological enhancement of the mGlu5/eCB-signaling macromolecular complex similarly normalized behavioral and synaptic deficits.
Materials and Methods
Animals
Animals were treated in compliance with the European Communities Council Directive (86/609/EEC) and the United States National Institutes of Health Guide for the care and use of laboratory animals. All mice arrived at P21 and were group-housed and acclimated to laboratory conditions before experiments with 12 h light/dark cycles and ad libitum access to food and water. All behavioral and synaptic plasticity experiments were performed on male C57BL/6J mice between P90 and P120.
Diets.
Male C57BL/6J mice (Janvier) were fed standard laboratory chow through 1 week after weaning: at P28, male C57BL/6J mice were divided into two groups that received a diet containing 6% of mixed oils containing rapeseed oil (rich in α-linolenic acid, 18:3n-3; “n-3”) or 6% fat in the form of sunflower oil (rich in linolenic acid, 18:2n-6; “n-3-deficient”) (Lafourcade et al., 2011; Madore et al., 2014; Mingam et al., 2008). Diets were isocaloric and differed only in the oil composition. Pellets were prepared by UPAE-Institut national de la recherche agronomique and stored at 4°C. Fatty acid composition was regularly controlled via gas chromatography analyses of organic extracts from manufactured food pellets as previously described (Lafourcade et al., 2011; Moranis et al., 2012; Madore et al., 2014).
Body weight and food intake.
To evaluate the possible effects of the different diets on body weight, mice fed with n-3 and n-3 PUFA-deficient diets were weighed weekly from P28 until commencement of the experiments at P90. Daily food intake was also calculated as average daily food consumption in grams per day for each week.
Fatty acid analysis.
Fatty acid compositions were analyzed in PFC and nucleus accumbens as previously described and expressed as the percentage of total fatty acids (Labrousse et al., 2012; Madore et al., 2014; Delpech et al., 2015; Joffre et al., 2016). Briefly, total lipids were extracted according to the method developed by Folch et al. (1957) and were submitted to fatty acid methylation using 7% boron trifluoride in methanol according to Morrison and Smith (1964). Fatty acid methyl esters were analyzed using gas chromatography as described by Joffre et al. (2016). They were identified by comparison with commercial standards. The proportion of each fatty acid was expressed as a percentage of total fatty acids.
Behavior
All mice were male, experimentally naïve, and used only once.
Social interaction.
The test was performed as previously published (Lafourcade et al., 2011; Larrieu et al., 2012). Mice were transferred to the experimental arena (40 × 40 cm). A social exploration session comprised 5 min without target where the arena contained an empty wire mesh cage in the corner followed by 5 min exposure of an unfamiliar adult CD-1 male enclosed in the wire mesh cage placed in the corner of the arena. The frequency of active investigatory behavior (mainly sniffing the anogenital region, mouth, ears, trunk, and tail) was manually counted by an experimenter blind to the conditions. A video tracking system (Ethovision XT, Noldus Information Technology) was used to measure the time spent in the interaction zone surrounding the target box (Lafourcade et al., 2011; Larrieu et al., 2012).
Temporal order.
Mice were habituated to the experimental arena (40 × 40 cm) without objects for 10 min daily for 2 d before testing. This task consisted of two sample phases and one test trial (Barker et al., 2007). In each sample phase, mice were allowed to explore two copies of an identical object for a total of 4 min. Different objects were used for sample Phases 1 and 2, with a delay between the sample phases of 1 h. After 3 h from sample Phase 2, mice performed the test trial (4 min duration) where a third copy of the objects from sample Phase 1 and a third copy of the objects from sample Phase 2 were used. The positions of the objects in the test and the objects used in sample Phase 1 and sample Phase 2 were counterbalanced between the mice. An intact temporal order memory requested the subjects to spend more time exploring the object from Sample 1 (i.e., the object presented less recently) compared with the object from Sample 2 (i.e., the “new” object). The discrimination ratio was calculated as the difference in time spent by each animal exploring the object from sample Phase 1 compared with the object from sample Phase 2 divided by the total time spent exploring both objects in the test phase.
Novel object location.
In this test, mice ability to recognize that a previously known object had changed location was assessed (Barker et al., 2007). After 2 consecutive days of 10 min habituation/d to the experimental arena (40 × 40 cm) without objects, mice were exposed to objects A1 and A2, which were placed in the far corners of the arena (i.e., sample phase 4 min duration). After a delay of 5 min (test phase), a copy of object A1 was placed in the same position as the original object A1, whereas a copy of object A2 was placed in a different corner than the original A2, diagonally across from A1. Thus, both objects in the test phase were equally familiar, but one was in a new location. The position of the moved object was counterbalanced between mice. The discrimination ratio was calculated as the difference in time spent by each animal exploring the object that changed position compared with the object that remained in the same position divided by the total time spent exploring all objects.
Novel object recognition.
After 2 d of 10 min habituation to the experimental arena, mice were exposed to the procedure of an acquisition or sample phase, followed by a preference test after a delay of either 5 min or 2 h (Barker et al., 2007). In the sample phase, duplicate copies (A1 and A2) of an object were placed near the two corners at either end of one side of the arena (8 cm from each adjacent wall). Mice were placed into the arena facing the center of the opposite wall and allowed a total of 4 min in the arena. At test (4 min duration), mice were placed in the arena, presented with two objects in the same positions: one object (A3) was a third copy of the set of the objects used in the sample phase, and the other object was a novel object (B). The positions of the objects in the test and the objects used as novel or familiar were counterbalanced between the mice. The discrimination ratio was calculated as the difference in time spent by each animal exploring the novel compared with the familiar object divided by the total time spent exploring both objects.
Open-field.
The test was performed as we previously published (Lafourcade et al., 2011; Jung et al., 2012). The apparatus consisted of a Plexiglas arena (40 × 40 cm) illuminated by fluorescent bulbs at a height of 2 m above the floor of the open field apparatus (light intensity of 30 Lux). The floor was cleaned between each trial to avoid olfactory clues. Each mouse was transferred to the open-field facing a corner and was allowed to freely explore the experimental area for 10 min. A video tracking system (Ethovision XT, Noldus Information Technology) recorded the exact track of each mouse as well as total distance traveled (Lafourcade et al., 2011; Larrieu et al., 2012).
Elevated plus maze.
The elevated plus maze apparatus comprised two open arms (30 × 8 cm2) and two closed arms (30 × 8 × 15 cm3) that extended from a common central platform (8 × 8 cm2). The test was performed as previously described (Jung et al., 2012; Manduca et al., 2015). Mice were individually placed on the central platform of the maze facing an open arm and allowed to freely explore the apparatus for 5 min. The 5 min test period was recorded by the video tracking system Ethovision XT (Noldus Information Technology). The following parameters were analyzed: percentage of time spent in the open arms expressed as seconds spent on the open arms of the maze/seconds spent on the open + closed arms × 100); number of entries into the risk-zone reflected by open arm extremities; number of head dipping events (i.e., the animal protruded the head over the ledge of an open arm and down toward the floor while its body is in the closed arms, central platform or in the open arms).
Marble burying.
To quantify behavioral traits of obsessive-like behavior (Woods-Kettelberger et al., 1997) and anxiety (Njung'e and Handley, 1991), 20 clean blue glass marbles were spaced in five rows of four, on 5 cm layer of sawdust bedding, which was lightly pressed down to make a flat even surface, in a plastic cage (30 × 20 cm) (light intensity of 30 Lux). Mice were individually placed facing a corner for a 20 min session. The number of marbles buried was counted at the end of the session. The number of marbles buried was plotted. Buried was defined by having over two-thirds of the total top surface of the marble covered by bedding.
Sucrose preference.
Anhedonia was assessed in the sucrose consumption test (Monleon et al., 1995; Bessa et al., 2009). Mice were individually housed in a laboratory cage and exposed for 72 h to a bottle containing a sucrose solution (1% in tap water, Sigma), placed into the middle of the cage wire top, followed by 18 h of water deprivation and a 2 h exposure to two identical bottles (one filled with 1% sucrose solution and the other with water). Bottles were placed at opposite ends of the cage and counterbalanced across groups to avoid side bias. Sucrose preference was calculated as the ratio of the volume of sucrose versus total volume (sucrose + water) × 100 consumed during the 2 h test.
Drug treatments.
Six hours before behavioral testing, mice were given intraperitoneal injections of vehicle (22.5:77.5 2-hydroxylpropyl-β-cyclodextrin [HBC] or Tween 80: H2O = 1:9), the monoacylglycerol lipase inhibitor 4-nitrophenyl-4-[bis(1,3-benzodioxol-5-yl) (hydroxy)methyl]piperidine-1-carboxylate (JZL184) (16 mg per kg in HBC) (Jung et al., 2012) or the positive allosteric modulator of mGlu5 receptors, 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl) benzamide (CDPPB) (10 mg per kg in Tween 80: H2O) (Won et al., 2012). JZL184 was freshly dissolved in HBC vehicle and mixed thoroughly during 30 min. CDPPB was dissolved in Tween 80: H2O to a final concentration of 1 mg/ml.
Statistical analysis.
All the behavioral parameters were expressed as mean ± SEM. Group comparisons used t test and two-way ANOVA (for different diets and pharmacological treatments), followed by Student-Newman-Keuls post hoc tests where appropriate.
Physiology
Slice preparation.
Adult male mice (P90-P120) were anesthetized with isoflurane and killed as previously described (Robbe et al., 2002; Lafourcade et al., 2011; Jung et al., 2012; Martin et al., 2016a). The brain was sliced (300 μm) in the coronal plane with a vibratome (Integraslice, Campden Instruments) in a sucrose-based solution at 4°C (in mm as follows: 87 NaCl, 75 sucrose, 25 glucose, 2.5 KCl, 4 MgCl2, 0.5 CaCl2, 23 NaHCO3 and 1.25 NaH2PO4). Immediately after cutting, slices containing the mPFC or accumbens were stored for 1 h at 32°C in a low calcium ACSF that contained (in mm) as follows: 130 NaCl, 11 glucose, 2.5 KCl, 2.4 MgCl2, 1.2 CaCl2, 23 NaHCO3, 1.2 NaH2PO4, and were equilibrated with 95% O2/5% CO2 and then at room temperature until the time of recording.
Electrophysiology.
Whole-cell patch-clamp of visualized layer 5 mPFC pyramidal neurons and accumbens medium spiny neurons (MSNs) and field potential recordings were made in coronal slices containing the mPFC or the accumbens as previously described (Robbe et al., 2002; Lafourcade et al., 2011; Jung et al., 2012; Martin et al., 2016a). During the recording, slices were placed in the recording chamber and superfused at 2 ml/min with either low Ca2+ ACSF for mPFC or normal ACSF for the accumbens. All experiments were done at 32°C or room temperature for mPFC and accumbens, respectively. The superfusion medium contained picrotoxin (100 μm) to block GABA Type A (GABA-A) receptors. All drugs were added at the final concentration to the superfusion medium.
For whole-cell patch-clamp experiments, neurons were visualized using an upright microscope with infrared illumination. The intracellular solution was based on K+ gluconate (in mm as follows: 145 K+ gluconate, 3 NaCl, 1 MgCl2, 1 EGTA, 0.3 CaCl2, 2 Na2+ATP, and 0.3 Na+ GTP, 0.2 cAMP, buffered with 10 HEPES). To quantify the AMPA/NMDA ratio, we used a CH3O3SCs-based solution (in mm as follows: 128 CH3O3SCs, 20 NaCl, 1 MgCl2, 1 EGTA, 0.3 CaCl2, 2 Na2+ATP, and 0.3 Na+ GTP, 0.2 cAMP, buffered with 10 HEPES, pH 7.2, osmolarity 290–300 mOsm). The pH was adjusted to 7.2 and osmolarity to 290–300 mOsm. Electrode resistance was 4–6 MOhm.
Whole-cell patch-clamp recordings were performed with an Axopatch-200B amplifier as previously described (Robbe et al., 2002; Kasanetz and Manzoni, 2009; Lafourcade et al., 2011; Jung et al., 2012; Thomazeau et al., 2014, 2017; Martin et al., 2016b). Data were low pass filtered at 2 kHz, digitized (10 kHz, DigiData 1440A, Axon Instruments), collected using Clampex 10.2 and analyzed using Clampfit 10.2 (all from Molecular Device).
A −2 mV hyperpolarizing pulse was applied before each evoked EPSC to evaluate the access resistance, and those experiments in which this parameter changed >25% were rejected. Access resistance compensation was not used, and acceptable access resistance was <30 MOhm. The potential reference of the amplifier was adjusted to zero before breaking into the cell. Cells were held at −76 mV.
Current-voltage (I–V) curves were made by a series of hyperpolarizing to depolarizing current steps immediately after breaking into the cell. Membrane resistance was estimated from the I–V curve around resting membrane potential (Kasanetz et al., 2010; Thomazeau et al., 2014; Martin et al., 2016a).
For extracellular field experiments, the recording pipette was filled with ACSF. The glutamatergic nature of the field EPSP (fEPSP) was systematically confirmed at the end of the experiments using the ionotropic glutamate receptor antagonist CNQX (20 μm), which specifically blocked the synaptic component without altering the nonsynaptic component (data not shown).
Both EPSC/fEPSP area and amplitude were analyzed. Stimulation was performed with a glass electrode filled with ACSF and placed ∼200 μm in the dorsal-medial direction of the recorded cell. The stimulus intensity was adjusted ∼60% of maximal intensity after performing an input–output curve (baseline EPSC amplitudes ranged between 50 and 150 pA). Stimulation frequency was set at 0.1 Hz.
To determine the AMPA/NMDA ratio, the cells were voltage-clamped to 40 mV, and a stable dual response (AMPA+NMDA current) to afferent stimulation was recorded. The EPSC was isolated after bath application of the AMPAR antagonist NBQX (10 μm). The AMPAR EPSC was obtained by digital subtraction of the NMDAR EPSC from the dual response.
Spontaneous AMPAR-mediated EPSC (sEPSCs) were recorded at −76 mV in whole-cell voltage-clamp configuration using Axoscope 10 (Molecular Devices). sEPSCs were filtered at 2 kHz and digitized at 20 kHz. sEPSC amplitude and interinterval time were analyzed with Axograph X using a double exponential template: f(t) = exp(−t/rise) + exp(−t/decay), rise = 0.5 ms. The threshold of amplitude detection was set at 5 pA.
Data acquisition and analysis.
The magnitude of plasticity was calculated 38–40 min after induction as percentage of baseline responses. sEPSCs were analyzed with Axograph X (Axograph). Statistical analysis of data was performed with Prism (GraphPad Software) using tests indicated in the main text after outlier subtraction. All values are given as mean ± SEM, and statistical significance was set at p < 0.05.
Results
Body weight and food intake
Body weight was measured weekly in n-3 and n-3 PUFA-deficient mice from P28 until commencement of the experiments at P90. Average body weight of n-3 PUFA-deficient mice did not differ from diet-matched n-3 controls at any age (F(weeks × diet)8,72 = 1.168, p = 0.330, two-way ANOVA). Daily food intake was calculated as average daily food consumption in grams per day for each week. In n-3 PUFA-deficient mice, daily food consumption did not differ from mice fed with an n-3 diet (F(weeks × diet)8,72 = 1.677, p = 0.119, two-way ANOVA). Collectively, these data indicate that the n-3 PUFA-deficient diet per se did not affect body weight or food consumption in mice.
Starting n-3 PUFA deficiency at adolescence selectively impairs cognition at adulthood
We found that reducing dietary n-3 PUFAs starting at adolescence modified selected behaviors at adulthood. We previously showed the detrimental consequences of perinatal malnutrition on sociability (Lafourcade et al., 2011). Accordingly, we first evaluated social interactions in the two dietary conditions. We observed that n-3-deficient adult mice spent less time in social interactions (Fig. 1A) and had less contact with their congeners than mice fed an n-3 diet.
Figure 1.

Nutritional n-3 deficiency starting in adolescence reduces sociability and temporal order memory at adulthood. A, Social interaction test: n-3 PUFA-deficient group mice spent less time to interact with a congener (n-3-balanced: 121.60 ± 5.94, n = 8; n-3-deficient: 87.34 ± 6.59, n = 10; t(16) = 3.77, p < 0.01, t test) and had less contact with their congeners than mice fed with an n-3 diet (51.88 ± 2.40, n = 8; and 29.50 ± 1.68, n = 10; t(16) = 7.85, p < 0.01, t test; n-3-balanced and n-3-deficient mice, respectively; data not shown). B, Temporal order: there was a marked reduction of discrimination in the temporal order memory task in the n-3-deficient group (n-3-balanced: 0.49 ± 0.06, n = 8; n-3-deficient: 0.14 ± 0.08, n = 8; t(14) = 3.25; p < 0.05, t test). C, Novel object location test: Discrimination between the object that had changed position, and the object that had remained in a constant position was similar in n-3-balanced and n-3 PUFA-deficient groups (n-3-balanced: 0.29 ± 0.04, n = 7; n-3-deficient: 0.31 ± 0.04, n = 5; t(10) = 0.47, p > 0.05, t test). D, Novel object recognition test: n-3 PUFAs deficient adult mice discriminated similarly between the novel and familiar objects after 5 min delay (n-3-balanced: 0.33 ± 0.05, n = 8; n-3-deficient: 0.36 ± 0.04, n = 6; t(12) = 0.52, p > 0.05, t test). Furthermore, n-3-deficient adult mice were also able to discriminate between novel and familiar objects after 2 h delay (0.253 ± 0.167, n = 10; and 0.273 ± 0.196, n = 9; t(17) = 0.82, p > 0.05, t test; n-3-balanced and n-3-deficient mice, respectively, data not shown). Scatter dot plot represents each animal. Error bars indicate SEM. *p < 0.05.
To explore whether the social deficits extended to the cognitive domain, we measured temporal order memory, a task that implicates both PFC and hippocampal circuits (Barker et al., 2007). Mice from the n-3 PUFA-deficient group presented a marked reduction in temporal order memory illustrated by a lack of discrimination in favor of the old object (normally perceived as new) over the most recent object (Fig. 1B). To further indicate impairment in hippocampal function, we next measured the consequences of n-3 deficiency on two tasks strongly associated with hippocampal integrity: location of novel object and novel object recognition (Barker et al., 2007). In marked contrast to temporal order memory and social interaction results, the hippocampal-dependent tasks were conserved in n-3 PUFA-deficient adults. Thus, in the novel object location test, n-3 and n-3 PUFA-deficient groups were indistinguishable in their ability to discriminate the object which had changed position from the object which had not (Fig. 1C). In the novel object recognition test, n-3-deficient adult mice were able to discriminate between novel and familiar objects either after 5 min or 2 h delay (Fig. 1D).
n-3 PUFA deficiency is anxiogenic in adulthood
Based on previous work from our group showing that lifelong dietary n-3 PUFA deficiency augmented anxiety and depression-related behaviors (Lafourcade et al., 2011; Larrieu et al., 2012, 2014), we next compared the n-3 and n-3 PUFA-deficient mice for anxiety-like behaviors.
We found that dietary n-3 PUFA deficiency decreased the percentage of time spent exploring the center of an open-field compared with mice fed with the n-3 PUFA diet (Fig. 2A). The increased anxiety-like behavior was not linked to impaired total motor locomotion, and there was no significant change in the distance traveled between n-3 and n-3-deficient mice (data not shown).
Figure 2.

Nutritional n-3 deficiency starting at adolescence blunts emotional behaviors. A, Open field test: there was a marked reduction of the time spent in the center of the open field arena in the n-3 PUFA-deficient group that indicated increased anxiety (n-3-balanced: 17.76 ± 2.59, n = 8; n-3-deficient: 9.67 ± 1.91, n = 9; t(15) = 2.51, p < 0.05, t test). We controlled that the increased anxiety-like behavior was not linked to impaired total motor locomotion, and found no significant change in the distance traveled between n-3-balanced and n-3-deficient mice (2780 ± 117.8 cm, n = 8; and 2779 ± 228.8 cm, n = 9; t(15) = 0.02, p > 0.05, t test; n-3-balanced and n-3-deficient mice, respectively). B–D, n-3 deficiency augmented all traits of anxiety in the elevated plus maze test. There was a marked reduction of the percentage of time spent in the open arm (B; n-3-balanced: 23.74 ± 2.32, n = 8; n-3-deficient: 16.53 ± 1.04, n = 7; t(13) = 2.80, p < 0.05, t test), the frequency of entries in the risk zone in the open arms (C; n-3-balanced: 3.38 ± 0.38, n = 8; n-3-deficient: 1.71 ± 0.29, n = 7; t(13) = 3.44, p < 0.01, t test) and the frequency of head dipping (D; n-3-balanced: 13.75 ± 1.53, n = 8; n-3-deficient: 5.43 ± 0.75, n = 7; t(13) = 4.65, p < 0.001, t test). E, Dietary n-3 PUFA deprivation in adulthood increased anxiety-like behavior: in the marble burying test, the n-3-deficient group buried twice as many marbles in bedding than the n-3-balanced mice (n-3-balanced: 3.20 ± 0.61, n = 10; n-3-deficient: 8.89 ± 0.99, n = 9; t(17) = 4.99, p < 0.001, t test). F, The marked reduction in sucrose preference of the n-3-deficient group is compatible with increased anhedonia in these mice (n-3-balanced: 52.24 ± 8.55, n = 11; n-3-deficient: 25.00 ± 7.72, n = 8; t(17) = 2.27, p < 0.05, t test). Basal fluid intake was similar in both groups (during the 2 h test period: 5.55 ± 0.72 ml, n = 11; and 5.63 ± 0.53 ml, n = 8; t(17) = 0.08, p > 0.05, t test; n-3-balanced and n-3-deficient mice, respectively; data not shown). Scatter dot plot represents each animal. Error bars indicate SEM. *p < 0.05.
Reminiscent of our previous report with the lifelong n-3 PUFA deficiency model (Lafourcade et al., 2011), this finding suggests that nutritional n-3 PUFA status is closely linked to an anxious state in mice. To confirm this idea, mice groups were compared in an elevated plus maze (Fig. 2B–D). The n-3-deficient mice displayed a clear aversion to the open arms of the elevated plus maze compared with n-3-balanced diet mice (Fig. 2B). Furthermore, both the number of entries in the risk zone (i.e., edge of the open arms; Fig. 2C) and head dippings were strongly reduced (Fig. 2D), suggesting increased risk-aversion in n-3-deficient mice.
Another behavioral marker of anxiety sometime used to identify behavioral traits of obsessive-like behavior (Woods-Kettelberger et al., 1997) is the marble-burying task (Njung'e and Handley, 1991). Mice from the n-3-deficient group buried substantially more marbles than n-3-balanced mice (Fig. 2E). Next, we investigated whether heightened anxiety was accompanied by decreased preference for sucrose, a hallmark of depression-like behavior (Monleon et al., 1995). As previously noted, motor locomotion was unchanged in n-3-deficient mice, and we verified that basal fluid intake was similar in both groups (data not shown). However, there was a massive reduction in the preference for sweet solution, a putative sign of anhedonia, in the n-3-deficient mice compared with mice fed with the n-3-balanced diet (Fig. 2F).
n-3 PUFA deficiency alters fatty acid levels in the mPFC and accumbens
Dietary intake of PUFAs is necessary for maintaining a healthy n-3 PUFAs status in the form of docosahexaenoic acid (DHA) in the brain (Bazinet and Layé, 2014). We and others have reported that perinatal n-3 PUFA deficiency strongly affects brain DHA content (Lafourcade et al., 2011; Joffre et al., 2014); however, the effect of a n-3 PUFA-deficient diet starting at adolescence on brain PUFA content at adulthood has been poorly evaluated (Delpech et al., 2015). Thus, we measured the impact of early life n-3 PUFA dietary manipulation on PUFA content in PFC and accumbens in adulthood. Exposure to the n-3 PUFA-deficient diet from adolescence to adulthood is sufficient to increase n-6 and decrease n-3 PUFA content in the PFC (Table 1) and accumbens (Table 2). In particular, DHA (22:6 n-3) and docosapentaenoic acid (n-3 DPA, 22:5 n-3) levels were significantly decreased in the accumbens and PFC of n-3-deficient mice (Table 2). In addition, n-6 DPA (22:5 n-6) and LA (18:2 n-6) levels were significantly increased, whereas arachidonic acid (AA, 20:4 n-6) levels were similar (Table 2) as previously described in the hippocampus (Delpech et al., 2015). Thus, adolescent malnutrition modified lipid composition in our structures of interest. Correct perinatal nutrition does not protect from dietary deficits later in life, reinforcing the idea that it is necessary to maintain appropriate n-3 PUFA intake after weaning.
Table 1.
Prefrontal cortex fatty acid composition in adult C57Bl6/J mice fed n-3-balanced (n-3) or deficient (n-3-deficient) diet expressed as % of total fatty acid (n = 4 per group)
| Diet fatty acids | n-3 | n-3-deficient | Statistical effect |
|---|---|---|---|
| 14:0 | 0.2 ± 0.01 | 0.2 ± 0.01 | NS |
| 15:0 | 0.05 ± 0.002 | 0.06 ± 0.005 | NS |
| 16:0 | 22.5 ± 0.37 | 22.6 ± 0.61 | NS |
| 17:0 | 0.1 ± 0.01 | 0.1 ± 0.01 | NS |
| 18:0 | 22.1 ± 0.33 | 22.1 ± 0.42 | NS |
| 20:0 | 0.2 ± 0.01 | 0.2 ± 0.02 | NS |
| 22:0 | 0.2 ± 0.02 | 0.2 ± 0.04 | NS |
| 24:0 | 0.2 ± 0.03 | 0.3 ± 0.06 | NS |
| Saturated | 45.6 ± 0.10 | 45.7 ± 0.49 | NS |
| 16:1n-9 | 0.2 ± 0.01 | 0.2 ± 0.01 | NS |
| 16:1n-7 | 0.6 ± 0.02 | 0.6 ± 0.03 | NS |
| 17:1n-9 | 0.04 ± 0.016 | 0.03 ± 0.005 | NS |
| 18:1trans | 0.06 ± 0.009 | 0.06 ± 0.006 | NS |
| 18:1n-9 | 11.9 ± 0.20 | 11.8 ± 0.33 | NS |
| 18:1n-7 | 3.1 ± 0.04 | 3.2 ± 0.02 | NS |
| 20:1n-9 | 0.6 ± 0.03 | 0.6 ± 0.09 | NS |
| 20:1n-7 | 0.14 ± 0.009 | 0.18 ± 0.019 | NS |
| 22:1n-9 | 0.06 ± 0.005 | 0.06 ± 0.012 | NS |
| 24:1n-9 | 0.4 ± 0.07 | 0.6 ± 0.01 | NS |
| Monounsaturated | 17.1 ± 0.28 | 17.4 ± 0.57 | NS |
| 18:2n-6 | 0.3 ± 0.01 | 0.5 ± 0.02 | <0.0001 |
| 20:2n-6 | 0.1 ± 0.01 | 0.1 ± 0.01 | NS |
| 20:3n-6 | 0.3 ± 0.01 | 0.3 ± 0.01 | <0.0001 |
| 20:4n-6 | 9.8 ± 0.15 | 10.1 ± 0.19 | NS |
| 22:4n-6 | 2.1 ± 0.08 | 2.4 ± 0.05 | <0.01 |
| 22:5n-6 | 0.6 ± 0.02 | 2.6 ± 0.21 | <0.0001 |
| n-6 | 13.2 ± 0.17 | 16.0 ± 0.29 | <0.0001 |
| 22:5n-3 | 0.2 ± 0.01 | 0.1 ± 0.01 | <0.01 |
| 22:6n-3 | 17.4 ± 0.09 | 14.0 ± 0.42 | <0.0001 |
| n-3 | 17.6 ± 0.09 | 14.1 ± 0.42 | <0.0001 |
| 20:3n-9 | 0.1 ± 0.00 | 0.1 ± 0.01 | <0.001 |
| Polyunsaturated | 30.9 ± 0.21 | 30.2 ± 0.60 | NS |
| DMA16:0 | 1.7 ± 0.05 | 1.7 ± 0.10 | NS |
| DMA18:0 | 3.3 ± 0.01 | 3.2 ± 0.08 | NS |
| DMA18:1n-9 | 0.7 ± 0.03 | 0.8 ± 0.10 | NS |
| DMA18:1n-7 | 0.7 ± 0.03 | 0.9 ± 0.10 | NS |
| DMA | 6.4 ± 0.07 | 6.6 ± 0.36 | NS |
| n-6/n-3 | 0.7 ± 0.01 | 1.1 ± 0.03 | <0.0001 |
| AA/DHA | 0.6 ± 0.01 | 0.7 ± 0.01 | <0.0001 |
| u.i. | 174.8 ± 0.77 | 167.7 ± 2.71 | <0.05 |
DMA, Dimethylacetal; AA, arachidonic acid, 20:4 n-6; DHA, docosahexaenoic acid, 22:6; SFAs, saturated fatty acids; MUFAs, monounsaturated fatty acids; LA, linoleic acid, 18:2 n-6; n-6 DPA, docosapentaenoic acid, 22:5 n-6; n-3 DPA, docosapentaenoic acid, 22:5 n-3. Adolescence to adulthood n-3 deprivation modifies mPFC fatty acid composition. Fatty acid composition in mice fed with n-3-balanced or n-3-deficient diet from PND 28 to PND 90. Results are expressed as percentage of total fatty acids (n = 4).
Table 2.
Nucleus accumbens fatty acid composition in adult C57Bl6/J mice fed n-3-balanced or n-3-deficient diet expressed as percentage of total fatty acid (n = 4 per group)
| Diet fatty acids | n-3 | n-3-deficient | Statistical effect |
|---|---|---|---|
| 14:0 | 0.2 ± 0.02 | 0.2 ± 0.02 | NS |
| 15:0 | 0.03 ± 0.012 | 0.06 ± 0.07 | NS |
| 16:0 | 19.2 ± 0.61 | 21.0 ± 0.79 | NS |
| 17:0 | 0.12 ± 0.005 | 0.13 ± 0.000 | NS |
| 18:0 | 23.1 ± 0.55 | 22.2 ± 0.39 | NS |
| 20:0 | 0.3 ± 0.03 | 0.3 ± 0.01 | NS |
| 22:0 | 0.3 ± 0.04 | 0.3 ± 0.02 | NS |
| 24:0 | 0.6 ± 0.13 | 0.3 ± 0.02 | NS |
| Saturated | 43.8 ± 0.44 | 44.5 ± 0.44 | NS |
| 16:1n-9 | 0.15 ± 0.002 | 0.15 ± 0.005 | NS |
| 16:1n-7 | 0.6 ± 0.03 | 0.6 ± 0.01 | NS |
| 17:1n-9 | 0.11 ± 0.008 | 0.07 ± 0.026 | NS |
| 18:1trans | 0.09 ± 0.013 | 0.07 ± 0.007 | NS |
| 18:1n-9 | 13.2 ± 0.03 | 13.1 ± 0.10 | NS |
| 18:1n-7 | 3.3 ± 0.04 | 3.4 ± 0.03 | 0.05 |
| 20:1n-9 | 1.1 ± 0.06 | 1.0 ± 0.07 | NS |
| 20:1n-7 | 0.2 ± 0.018 | 0.2 ± 0.010 | NS |
| 22:1n-9 | 0.18 ± 0.048 | 0.09 ± 0.012 | NS |
| 24:1n-9 | 1.0 ± 0.11 | 0.8 ± 0.05 | NS |
| Monounsaturated | 20.0 ± 0.18 | 19.5 ± 0.23 | NS |
| 18:2n-6 | 0.3 ± 0.02 | 0.5 ± 0.02 | <0.01 |
| 20:2n-6 | 0.13 ± 0.023 | 0.14 ± 0.018 | NS |
| 20:3n-6 | 0.4 ± 0.03 | 0.3 ± 0.01 | <0.05 |
| 20:4n-6 | 10.4 ± 0.17 | 10.6 ± 0.23 | NS |
| 22:4n-6 | 2.7 ± 0.06 | 2.7 ± 0.04 | NS |
| 22:5n-6 | 0.5 ± 0.09 | 1.8 ± 0.05 | <0.0001 |
| n-6 | 14.4 ± 0.23 | 16.0 ± 0.30 | <0.01 |
| 22:5n-3 | 0.4 ± 0.06 | 0.1 ± 0.04 | <0.05 |
| 22:6n-3 | 14.3 ± 0.31 | 12.1 ± 0.22 | <0.01 |
| n-3 | 14.7 ± 0.32 | 12.2 ± 0.19 | <0.001 |
| 20:3n-9 | 0.16 ± 0.032 | 0.07 ± 0.015 | <0.05 |
| Polyunsaturated | 29.2 ± 1.15 | 28.3 ± 0.45 | NS |
| DMA16:0 | 1.9 ± 0.22 | 2.3 ± 0.17 | NS |
| DMA18:0 | 3.3 ± 0.11 | 3.4 ± 0.06 | NS |
| DMA18:1n-9 | 0.8 ± 0.05 | 0.9 ± 0.03 | NS |
| DMA18:1n-7 | 1.0 ± 0.06 | 1.1 ± 0.03 | NS |
| DMA | 7.0 ± 0.41 | 7.7 ± 0.23 | NS |
| n-6/n-3 | 1.0 ± 0.01 | 1.3 ± 0.02 | <0.0001 |
| AA/DHA | 0.72 ± 0.01 | 0.88 ± 0.01 | <0.0001 |
| u.i. | 174.8 ± 0.77 | 167.7 ± 2.71 | <0.05 |
DMA, Dimethylacetal; AA, arachidonic acid, 20:4 n-6; DHA, docosahexaenoic acid, 22:6; SFAs, saturated fatty acids; MUFAs, monounsaturated fatty acids; LA, linoleic acid, 18:2 n-6; n-6 DPA, docosapentaenoic acid, 22:5 n-6; n-3 DPA, docosapentaenoic acid, 22:5 n-3. Adolescence to adulthood n-3 deprivation modifies mPFC fatty acid composition. Fatty acid composition in mice fed with n-3-balanced or n-3-deficient diet from PND 28 to PND 90. Results are expressed as percentage of total fatty acids (n = 4).
n-3 PUFA deficiency from adolescence to adulthood reduces the working synaptic range in adult mPFC
A maximum working synaptic range is indispensable to the expression of behavioral flexibility in response to an ever-changing world (Kasanetz et al., 2013). Does nutrition affect the maximum synaptic working range at prefrontal excitatory synapses? In view of our results showing a reduction in prefrontal-dependent cognitive behaviors, we decided to interrogate two critical actors of bidirectional plasticity and reliable prefrontal endophenotypes of neuropsychiatric diseases: NMDAR-dependent LTP and endocannabinoid/mGlu5-dependent LTD (Lafourcade et al., 2011; Kasanetz et al., 2013; Iafrati et al., 2014; Thomazeau et al., 2014; Martin et al., 2016a).
We found that, in mPFC slices prepared from n-3-balanced mice, tetanic stimulation induced a robust, eCB-mediated LTD (eCB-LTD) of excitatory synapses onto layer 5 mPFC pyramidal neurons that matched what we previously reported in mice fed a standard laboratory chow (Lafourcade et al., 2007; Thomazeau et al., 2014). This eCB-LTD was absent in n-3-deficient mice (Fig. 3A).
Figure 3.

n-3 PUFA deficiency starting at adolescence reduces the working synaptic range in adult mPFC. A, Average time courses of mean EPSPs showing that low-frequency stimulation (10 min at 10 Hertz, arrow) of excitatory inputs induced an LTD of evoked EPSPs recorded in the mPFC in n-3-balanced (n = 15; black circles), but not in n-3-deficient (n = 21; white circles), mice. Top, Example traces from baseline and 40 min after LTD induction (n-3-balanced; 10 ms, 0.2 mV n-3-deficient). Calibration: 10 ms, 0.1 mV. B, Nutritional n-3 deficiency did not modify synaptic CB1R potency or efficiency in the mPFC. Dose–response curve for the cannabimimetic CP55,940 in n-3-balanced (black symbols, n = 3–7 animals, EC50 = 0.18, top value 44.33%, 95% CI for EC50 = 0.03167–1.068) and n-3-deficient mice (white symbols, n = 3–5 animals, EC50 = 0.19, top value 48.35%, 95% CI for EC50 = 0.05193–0.6623). fEPSP amplitudes were measured 30 min after application of CP55,940. Each point is expressed as the percentage of inhibition of its basal value. Error bars indicate SEM. C, Average time courses of mean EPSPs showing that, after bath perfusion with the mGluR2/3-specific agonist LY379268 (30 nm), n-3 PUFA deficiency did not alter mGlu2–3-dependent LTD in the mPFC of n-3-deficient diet mice (n = 3; black and white circles). D, Average time courses of mean EPSPs showing that TBS induces LTP at layer V/VI mPFC synapses in n-3-balanced diet mice (black circles), but not n-3 PUFA-deficient mice (white circles) (n = 16 animals in n-3-balanced group; n = 11 animals in n-3-deficient mice).
We next searched for functional alterations that could explain the lack of endocannabinoid/mGlu5-LTD. Neither the potency nor the efficacy of presynaptic CB1R was modified in the deficient group (Fig. 3B). The consequences of starting n-3 PUFA deficiency in adolescence and adulthood were different from those of gestational/perinatal deficiency where a marked reduction in CB1R functions was reported (Lafourcade et al., 2011; Larrieu et al., 2012). Of note, the effects of n-3 PUFA depletion did not extend to all types of synaptic plasticity: mGlu2/3-mediated LTD, a specific biomarker of cocaine addiction (Kasanetz et al., 2013), was indistinguishable in n-3-balanced and n-3-deficient mice (Fig. 3C).
Theta-burst stimulation (TBS) induces an NMDAR-dependent LTP at mPFC pyramidal synapses. The absence of TBS-LTP in the mPFC is an endophenotype common to several models of neuropsychiatric diseases, including schizophrenia, autism, and mental retardation (Iafrati et al., 2014; Thomazeau et al., 2014; Martin et al., 2016a). TBS-LTP was readily induced in n-3-balanced mice similar to that observed in mice fed standard chow (Iafrati et al., 2014; Thomazeau et al., 2014; Martin et al., 2016a). In marked contrast, TBS-induced LTP was absent in n-3-deficient mice (Fig. 3D).
In search of mechanistic insights, we compared intrinsic firing properties of pyramidal neurons in the two dietary conditions. Independent of the dietary treatment, all recorded mPFC neurons showed similar membrane reaction profiles in response to a series of somatic current steps, illustrated by superimposable I–V plots (Fig. 4A). The resting membrane potential (Fig. 4B) and the rheobase (Fig. 4C) were similar in all groups. The number of action potentials in response to somatic current steps was also similar in n-3-balanced and n-3-deficient mice (Fig. 4D). Thus, n-3 PUFA deficiency from adolescence did not impact the intrinsic properties of mPFC pyramidal cells in adulthood. This result is compatible with our observations following lifelong n-3 PUFA deficiency.
Figure 4.

Intrinsic properties of layer 5 mPFC pyramidal neurons in n-3 and n-3 PUFA-deficient mice. A, Left, Summary of all the I–V curves recorded in the two dietary groups showing no difference between n-3-balanced group (n = 18 cells/10 mice, black symbols) and n-3-deficient mice (n = 24 cells/13 mice, white symbols; F(26,1072) = 0.438, p > 0.05, two-way ANOVA). Right, Typical membrane responses to somatic current steps of mPFC neurons from n-3-balanced or n-3-deficient mice. Sample spike trains in response to 300 pA depolarizing current step from n-3-balanced and n-3-deficient mice. B, The resting membrane potential from n-3-balanced mice (black bar; −70.48 ± 1.19, n = 18 cells/10 mice) and n-3-deficient mice (white bar; −70.68 ± 1.04, n = 24 cells/13 mice) and (C) the rheobase (59.44 ± 11.45, n = 18 cells/10 mice from n-3-balanced mice, black bar; 69 ± 96.95, n = 24 cells/13 mice from n-3-deficient mice, white bar) was similar in both groups (p > 0.05, Mann–Whitney U test). D, The number of evoked action potentials (number of spikes) in response to 300 pA depolarizing current step (n-3-balanced: 117.17 ± 0.83, n = 18 cells/10 mice, black bar symbols; n-3-deficient: 19.83 ± 0.62, n = 24 cells/13 mice black bar symbols) was similar in both groups (p > 0.05, Mann–Whitney U test). Error bars indicate SEM.
Circuit properties were next compared in the two dietary conditions. Input–output profiles were measured in both groups: fEPSPs evoked by electrical stimulation showed a consistent profile distribution in response to increasing stimulation intensity across different slices and mice: “input–output” curves from n-3-balanced and n-3-deficient mice were similar (Fig. 5A). The relative contribution of AMPAR and NMDAR to synaptic currents is an indicator of synaptic plasticity; in particular, those dependent upon the incorporation or removal of AMPAR in the postsynaptic density (Kasanetz and Manzoni, 2009; Neuhofer et al., 2015). The ratio between AMPAR- and NMDAR-evoked EPSCs was similar in both dietary conditions, suggesting that n-3 PUFA deficiency did not modify the overall gain of prefrontal synapses (Fig. 5B). We next compared quantal events by recording spontaneous AMPAR-mediated EPSCs (sEPSCs) in mPFC neurons from both n-3-balanced and n-3-deficient mice. The mean amplitudes and distribution of sEPSC events (Fig. 5C) were similar in the two groups. We also compared the frequency of sEPSCs by comparing the interval between AMPAR events (Fig. 5D). No difference was found between the two dietary conditions. These results are consistent with what we observed in mice mPFC following lifelong n-3 PUFA deficiency (Lafourcade et al., 2011).
Figure 5.

Nutritional n-3 deficiency does not change the basic synaptic properties in mPFC pyramidal neurons. A, Input–output profile from n-3-balanced and n-3 PUFA-deficient mice from mPFC layer 5 fEPSPs. Averaged fEPSP area measured as a factor of stimulus intensity (F(6,63) = 0.154, p > 0.05, two-way ANOVA; n-3-balanced group n = 5 mice, n-3-deficient group n = 6 mice). B, AMPAR/NMDAR ratios measured in the n-3-balanced were similar to that measured in n-3 PUFA-deficient mice (n-3-balanced: 0.5725 ± 0.09, n = 9 cells from 9 mice; n-3-deficient: 0.739 ± 0.17, n = 9 cells from 9 mice; p > 0.05, Mann–Whitney U test). C, Sample traces from layer 5 mPFC pyramidal neurons clamped at −70 mV from n-3-balanced and n-3-deficient mice. Calibration: 0.25 s, 20 pA. sEPSCs were recorded in the whole-cell configuration in accumbens MSNs clamped at −70 mV. AMPAR-mediated mEPSC amplitudes were not changed in n-3-deficient mice (black symbols, n = 14 cells/12 mice; white symbols, n = 14 cells/9 mice). The cumulative distribution (Kolmogorov–Smirnov test; not significant) and mean amplitude (inset) of the sEPSC were similar in the two dietary conditions (p > 0.05; paired t test). D, The frequency of spontaneous AMPAR-mediated events was indistinguishable in the two dietary conditions. Cumulative probability distributions of intervals between events were similar in the two dietary conditions (Kolmogorov–Smirnov test, not significant); mean frequency (inset) of the mEPSC was similar in both dietary conditions (p > 0.05; paired t test). Error bars indicate SEM.
The data show that starting malnutrition at adolescence reduced the working synaptic range in adult mice from the n-3-deficient group. These synaptic aberrations in LTP or LTD were not caused by alterations of basal synaptic gain, neuronal excitability, postsynaptic AMPAR and NMDAR, or presynaptic CB1R coupling.
n-3 PUFA deficiency impairs eCB-mediated plasticity in the accumbens
We recently showed that eCB-mediated plasticity in the accumbens is instrumental in underlying depression-like behavior (Bosch-Bouju et al., 2016). How n-3 PUFA deficiency impacts accumbens synapses and synaptic plasticity is currently not known.
The vast majority of accumbens neurons are so-called MSNs. Independent of the dietary treatment, all recorded MSNs showed similar membrane response profiles to a series of somatic current steps, illustrated by superimposable I–V plots (Fig. 6A). The resting membrane potential (Fig. 6B) and the rheobase (Fig. 6C) were similar in all groups. The number of action potentials in response to somatic current steps was also similar in n-3-balanced and n-3-deficient mice (Fig. 6D). Thus, n-3 PUFA deficiency from adolescence did not impact the intrinsic properties of MSNs in adulthood.
Figure 6.

Intrinsic properties of accumbens MSNs in n-3 and n-3-deficient mice. A, Right, Summary of all the I–V curves recorded in the two dietary groups showing no difference between n-3-balanced (n = 10 cells/8 mice, black symbols) and n-3-deficient mice (n = 10 cells/6 mice, white symbols; F(17,306) = 0.697, p > 0.05, two-way ANOVA). Left, Typical membrane responses to somatic current steps of accumbens MSNs from n-3-balanced or n-3-deficient mice. Sample spike trains in response to 300 pA depolarizing current step from n-3-balanced and n-3-deficient mice. B, The resting membrane potential from n-3-balanced mice (black bar; −75.18 ± 2.54, n = 10 cells/8 mice), n-3-deficient mice (white bar; −78.73 ± 2.65, n = 10 cells/6 mice), and (C) the rheobase (149.0 ± 22.58, n = 10 cells from n-3-balanced mice, black bar; 139.0 ± 20.25, n = 10 cells from n-3-deficient mice, white bar) were similar in both groups (p > 0.05, Mann–Whitney U test). D, The number of evoked action potentials in response to somatic current steps was similar in both dietary conditions (n-3-balanced: n = 10 cells/8 mice, black bar symbols; n-3-deficient: n = 10 cells/6 mice white bar symbols; p > 0.05, Mann–Whitney U test).
We next measured fEPSPs of accumbens MSNs to build input–output profiles in the two dietary conditions. fEPSPs evoked by electrical stimulation showed a consistent profile distribution in response to increasing stimulation intensity across different slices and mice; thus, “input–output” curves from n-3-balanced and n-3-deficient mice were similar (Fig. 7A). The ratio between AMPAR- and NMDAR-evoked EPSCs was similar in both groups, suggesting that n-3 PUFA deficiency did not modify the overall gain of accumbens excitatory synapses (Fig. 7B). We next compared quantal events by recording sEPSCs in accumbens MSNs from both n-3-balanced and n-3-deficient mice. The mean amplitude of spontaneous events was similar in the two dietary conditions (Fig. 7C) at the resting membrane potential (−70 mV), where these events are principally mediated by AMPAR. Therefore, adult n-3 deficiency does not appear to affect AMPAR currents in accumbens MSNs. We also compared the frequency of sEPSCs by comparing the cumulative distribution of the interval between events (Fig. 7D). Both dietary conditions had a similar distribution and average interevent intervals.
Figure 7.

Nutritional n-3 deficiency does not change the basic synaptic properties in MSNs from the accumbens core. A, There was no change in synaptic excitability in the accumbens of n-3-deficient mice. Input–output curves of evoked fEPSP in accumbens neurons in n-3-balanced (n = 9 animals, black symbols) and in n-3-deficient mice (n = 3 animals, white symbols) for increasing stimulation intensities. Average mean fEPSPs are represented (F(6,91) = 0.182, p > 0.05, two-way ANOVA). B, Summary histograms with superimposed scatterplots showing that the AMPAR-mediated EPSC/NMDAR-mediated EPSC ratio was similar in the two dietary groups (0.86 ± 0.22, n = 7 cells from 7 n-3-balanced mice vs 0.98 ± 0.13, n = 8 cells from 8 n-3-deficient mice; p > 0.05, Mann–Whitney U test). C, D, Sample traces from MSN clamped at −70 mV from n-3-balanced and n-3-deficient mice. Calibration: 0.25 s, 20 pA. Quantal synaptic parameters were similar in both dietary conditions. Cumulative sEPSC amplitude (C) and interevent (D) distributions showed no difference between n-3-balanced (black symbols, n = 9 cells/4 mice) and n-3-deficient mice (white symbols, n = 9 cells/5 mice) (p > 0.05, Kolmogorov-Smirnov test). Insets, Summary histograms with superimposed scatterplots showing the average amplitudes and interevent values. Error bars indicate SEM.
In accumbens slices prepared from mice fed with a n-3-balanced diet, tetanic stimulation induced a robust eCB-LTD of excitatory synapses onto MSNs (Fig. 8A), which matched that observed in mice fed with normal chow (Robbe et al., 2002). In marked contrast, we found that this form of long-term synaptic depression was totally absent in n-3-deficient mice (Fig. 8A,B). Next, we estimated CB1R presynaptic functions by building dose–response curves for the cannabinoid agonist CP55,940. In this model of n-3 deficiency from adolescence, maximal inhibitory effect of CP55,940 and its EC50 were comparable in both dietary conditions (Fig. 8C) and similar to what we previously reported in mice fed with standard laboratory chow (Mato et al., 2005). Of note, lifelong n-3 PUFA depletion strongly lowered the maximal efficacy of CB1R in the mPFC (Puente et al., 2011). To determine whether n-3 deficiency impairments generalized to a second form of presynaptic LTD, we compared mGlu2/3-triggered LTD in the two dietary conditions (Mato et al., 2005; Lafourcade et al., 2011; Kasanetz et al., 2013). The mGlu2/3-LTD was strongly expressed in n-3-balanced and n-3-deficient mice (Fig. 8D), leaving little doubt that n-3 deficiency had a minimal effect on the downstream presynaptic signaling cascades common to mGlu2/3 and CB1R in the accumbens (Mato et al., 2005).
Figure 8.

n-3 deficiency during adolescence ablates eCB-dependent plasticity. A, Example traces from baseline and 40 min after LTD induction. Calibration: 5 ms, 0.1 mV. Average time courses of mean EPSPs showing that low-frequency stimulation induced an LTD of evoked EPSPs recorded in accumbens MSNs in n-3-balanced (black circles), but not in n-3-deficient (white circles), mice. B, Individual experiments (gray) and group average (black represents n-3-balanced; white represents n-3-deficient), before (baseline) and after (40 min) LTD induction showing the lack of LTD in the n-3-deficient group. In n-3-balanced mice: 101.08 ± 1.22, n = 10 before LTD, versus 80.81 ± 3.70, n = 10, after LTD induction. In n-3-deficient mice: 100.31 ± 0.91, n = 16 before LTD versus 98.38 ± 3.87, n = 16, after LTD. C, Nutritional n-3 deficiency did not modify synaptic CB1R potency or efficiency in the accumbens. Dose–response curve for the cannabimimetic CP55,940 in n-3-balanced (black symbols, n = 3–8 animals, EC50 = 0.03 μm, top value 49.54%, 95% CI for EC50 = 0.006510–0.1496) and n-3-deficient (white symbols, n = 3–5 animals, EC50 = 0.23 μm, top value 59.99%, 95% CI for EC50 = 0.05782–0.9020) mice. fEPSP amplitudes were measured 30 min after application of CP55,940. Each point is expressed as the percentage of inhibition of its basal value. Error bars indicate SEM. D, n-3 PUFA deficiency does not alter mGlu2–3-dependent LTD in n-3-deficient mice. Individual experiments (gray) and group average (black: n-3-balanced; white: n-3-deficient), before (baseline) and after (40 min) bath perfusion with the mGlu2/3-specific agonist LY379268 (LY, 30 nm). In n-3-balanced mice: 100.40 ± 1.16, n = 5 before LTD versus 39.79 ± 2.20, n = 5, after LTD induction. In n-3-deficient mice: 100.70 ± 1.03, n = 4 before LTD versus 50.07 ± 5.60, n = 4, after LTD (n-3-balanced and n-3-deficient, p < 0.05; paired t test). Error bars indicate SEM. *p < 0.05.
These data show that deprivation of dietary n-3 PUFAs from adolescence until adult life is sufficient to selectively abolish the development of mGlu5-eCB-mediated synaptic plasticity in the mice accumbens.
Amplification of the mGlu5-eCB signaling restores the working synaptic range in accumbens and mPFC of n-3-deficient mice
Pharmacological exploration of synaptic plasticity deficits in the brain slice preparation is a valuable approach to identify new therapeutic targets. Recent findings show that mGlu5- and/or eCB-mediated signaling is a prime target for such intervention (Jung et al., 2012; Won et al., 2012; Martin et al., 2016a, b; Bosch-Bouju et al., 2016; Wei et al., 2016; Thomazeau et al., 2017).
The lack of eCB-mediated LTD in the mPFC and accumbens of mice deprived of dietary n-3 PUFAs prompted us to test the new hypothesis that amplification of some of its key molecular elements could restore synaptic plasticity (Fig. 9A, schematic).
Figure 9.

Acute pharmacological amplification of the mGlu5-eCB signalosome restores eCB synaptic plasticity in the accumbens of n-3-deficient mice. A, Schematic representation of the mGlu5-eCB signalosome and of the targets of the pharmacological agents used in the study. B, Time course of eCB-LTD in n-3-deficient accumbens slices preincubated for 45 min in 1 μm JZL184 (orange circles). C, Time course of normalized fEPSP responses from n-3-deficient accumbens slices preincubated with 10 μm CDPPB (green circles). D, Bar chart of fEPSP percentage change from baseline 38–40 min after 10 Hz stimulation in n-3 and n-3-deficient mice before and after treatment with JZL184 ± SR141716A (5 μm) (orange bars, F(4,37) = 4.960, p < 0.05, one-way ANOVA) and CDPPB ± SR141716A (green bars, F(4,34) = 7.357, p < 0.05, one-way ANOVA). Error bars indicate SEM. *p < 0.05.
We first tested whether inhibition of 2-arachidonylglycerol (2-AG)'s main degrading enzyme monoacylglycerol lipase (MGL) would allow 2-AG levels to reach the threshold for LTD induction in n-3-deficient mice (Fig. 9A). Accumbens slices were incubated with the MGL inhibitor JZL184 (1 μm) for 45–90 min before LTD induction. This treatment was sufficient to restore eCB-LTD in the accumbens of n-3-deficient mice (Fig. 9B–D). JZL184 had no effect in mice fed with a n-3-balanced diet (Fig. 9D). The CB1R antagonist SR141716A (5 μm) blocked the JZL184 rescue, showing that CB1R activation mediates the effects of the MGL inhibitor in the accumbens (Fig. 9D). Next, we reasoned that amplification of the most upstream element of the eCB signaling complex (i.e., mGlu5) might also restore LTD. To this aim, we used CDPPB (10 μm), a well-described positive allosteric modulator (PAM) of mGlu5 (Nickols and Conn, 2014) (Fig. 9A). CDPPB fully restored the ability of excitatory accumbens synapses to express LTD in the n-3 PUFA-deficient group (Fig. 9C) while having no effect in the n-3 PUFA-balanced group (Fig. 9D). Importantly, we verified that CB1R ultimately mediated the effects of CDPPB; preincubation with SR141716A prevented the CDPPB rescue (Fig. 9D).
We found that JZL184 allowed the expression of normal LTD in the mPFC of n-3-deficient mice (Fig. 10A) while having no effect in slices prepared from the n-3-balanced-group (Fig. 10B). The JZL184 rescue was fully prevented by the CB1R antagonist (Fig. 10B). mPFC LTD was also restored in slices acutely exposed to the mGlu5 PAM and CDPPB (Fig. 10C). Antagonizing CB1R similarly prevented this rescue of LTD (Fig. 10D), suggesting that, as in the accumbens, CDPPB acted on mGlu5 to increase eCB production. Finally, we tested the response of LTP to the two LTD rescuing compounds, JZL184 and CDPPB. Enhancing 2-AG levels also restored LTP (Fig. 11A), an effect that was not blocked by the CB1R antagonist (Fig. 11B). Postsynaptic mGlu5 positively modulate NMDAR and mGlu5 PAMs have been shown to rescue NMDAR-dependent synaptic plasticity and behavior in autism and Huntington models (Won et al., 2012; Doria et al., 2015). We found that CDPPB restored LTP in the n-3-deficient group (Fig. 11C,D). CB1R antagonism did not prevent the recovery effect of CDPPB on LTP (Fig. 11D; see Discussion). An integrated interpretation of these results is that CDPPB potentiated mGlu5, which in turn enhanced NMDAR responses, allowing the expression of full-blown LTP in n3-deficient mice.
Figure 10.

Pharmacological amplification mGlu5-eCB signaling restores eCB synaptic plasticity in the mPFC of n-3-deficient mice. A, Time course of eCB-LTD in n-3-deficient mPFC slices in control conditions (white circles) or preincubated for 45 min in 1 μm JZL184 (orange circles). B, Summary bar chart of fEPSP percentage change from baseline 38–40 min after 10 Hz stimulation in n-3-balanced and n-3-deficient mice before and after treatment with JZL184 (orange bars) ± SR141716A (5 μm) (F(4,48) = 8.062, p < 0.05, one-way ANOVA). C, Time course of normalized fEPSP responses from mPFC slices in control conditions (white circles) or preincubated with CDPPB (green circles). D, Summary bar chart of fEPSP percentage change from baseline 38–40 min after 10 Hz stimulation in n-3-balanced and n-3-deficient mice with or without treatment with CDPPB ± SR141716A (F(4,44) = 12.30, p < 0.05, one-way ANOVA). Error bars indicate SEM. *p < 0.05.
Figure 11.

Pharmacological amplification mGlu5-eCB signaling restores mPFC LTP in n-3-deficient mice. A, Time course of LTP in n-3-deficient mPFC slices in control conditions (white circles) or preincubated with JZL184 (45 min, 1 μm, orange circles). B, Summary bar chart of fEPSP percentage change from baseline 38–40 min after LTP induction in n-3-balanced and n-3-deficient mice before and after treatment with JZL184 (orange bars) ± SR141716A (5 μm) (F(4,37) = 4.834, p < 0.05, one-way ANOVA). C, Time course of LTP in mPFC slices in control conditions (white circles) or preincubated with CDPPB (green circles). D, Summary bar chart of fEPSP percentage change from baseline 38–40 min after LTP induction in n-3-balanced and n-3-deficient mice with or without treatment with CDPPB ± SR141716A (F(4,37) = 9.346, p < 0.05, one-way ANOVA). Error bars indicate SEM. *p < 0.05.
Amplification of the mGlu5-eCB signaling complex rescues the behavioral deficits associated with n-3 PUFA deficiency
We next tested whether pharmacological enhancements of the mGlu5-eCB signaling complex could normalize behavioral deficits.
Inhibition of 2-AG degradation corrects cognitive and emotional deficits in n-3-deficient mice
Acute pharmacological enhancement of 2-AG with JZL184 (16 mg/kg, i.p.) ameliorated cognitive deficits in n-3-deficient mice. Social interaction (Fig. 12A) and temporal order memory deficits (Fig. 12B) were normalized, suggesting that the rescue effects of JZL184 extended to the cognitive domain. Furthermore, in the n-3-deficient group, time spent in the center of an open field (Fig. 12C), time spent in the open arm of an elevated plus maze (Fig. 12D), number of entries in the risk zone (Fig. 12E), head dipping in an elevated plus maze (Fig. 12F), number of marbles buried (Fig. 12G), and sucrose preference (Fig. 12H) were normalized. Of note, JZL184 had no effect on behaviors in the n-3-balanced group, indicating selectivity of the drug's effects to the disease state at the dose used. Thus, blocking 2-AG's degradation normalized emotional and cognitive deficits induced by the n-3 PUFAs deficient diet.
Figure 12.
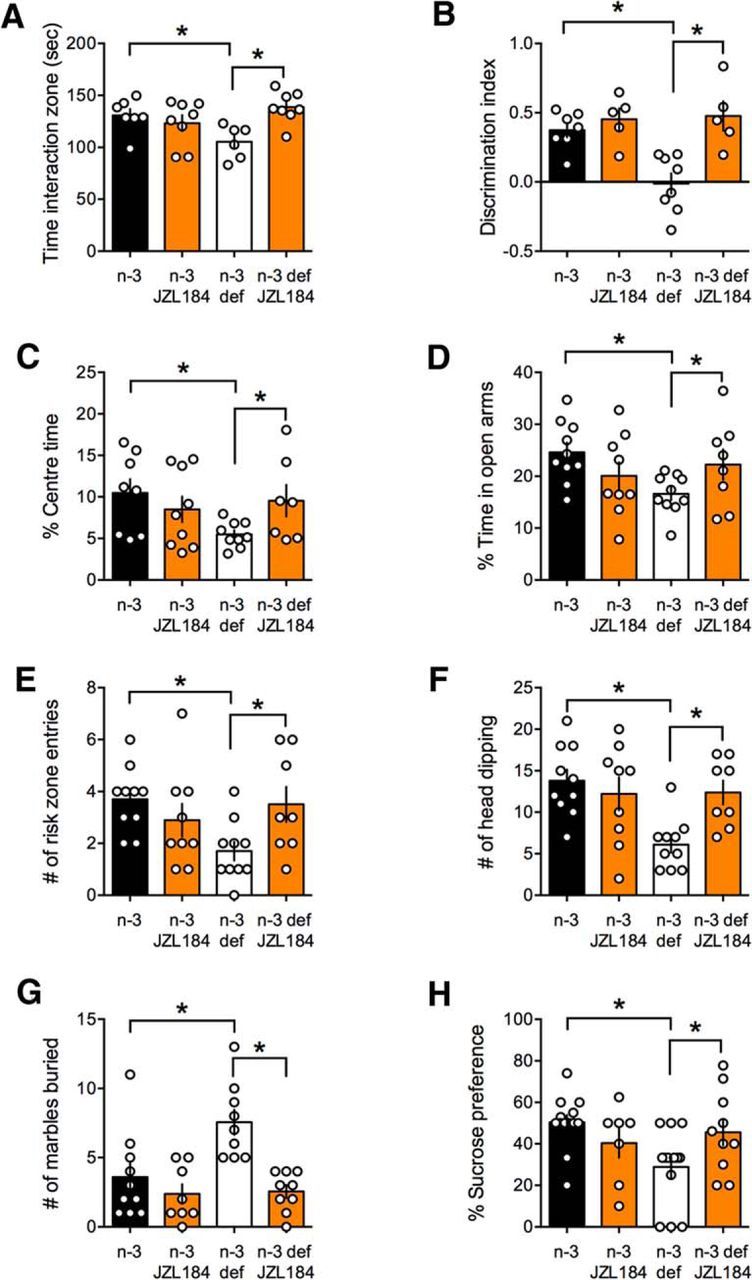
Inhibition of 2-AG degradation corrects cognitive and emotional deficits in n-3-deficient mice. JZL184 (16 mg/kg, i.p.) rescued cognitive deficits observed in the social interaction (A; F(dietxtreat) = 9.596, p < 0.05) and temporal order memory (B; F(dietxtreat) = 6.948, p < 0.05) tasks in n-3 PUFA-deficient mice. Enhancing 2-AG levels also normalized anxiety-related behaviors as measured by (C; F(dietxtreat) = 4.201, p < 0.05) the time spent in the center of the open field; (D; F(dietxtreat) = 5.569, p < 0.05) the percentage of time spent in the open arm; (E; F(dietxtreat) = 6.362, p < 0.05) the frequency of entries in the risk zone in the open arms of the elevated plus maze test; and (F; F(dietxtreat) = 7.136, p < 0.05) the frequency of head dipping. JZL184 corrected the deficits in the marble burying test (G; F(dietxtreat) = 5.291, p < 0.05) and reduced anhedonia in sucrose preference of the n-3-deficient group (H; F(dietxtreat) = 4.306, p < 0.05). All measures were performed 6 h after JZL184 treatment. Scatter dot plot represents each animal. Error bars indicate SEM. *p < 0.05.
Potentiation of mGlu5 rescues cognitive and emotional behaviors in n-3-deficient mice
Modulators of mGlu5 receptors may be useful in the treatment of neuropsychiatric disease (Belzung, 2001; Won et al., 2012; Doria et al., 2015). In the n-3-deficient group, social cognition and temporal order memory (Fig. 13A,B), time spent in the center of an open field (Fig. 13C), time spent in the open arm (Fig. 13D), number of entries in the risk zone and head dipping in an elevated plus maze (Fig. 13E,F), number of marbles buried (Fig. 13G), and sucrose preference (Fig. 13H) were normalized following injection of CDPPB (10 mg/kg, i.p.). We determined that, in the same conditions, CDPPB had no effect on these behaviors in the n-3-balanced group, indicating selectivity of the drug's effects to the disease-state at the dose used.
Figure 13.
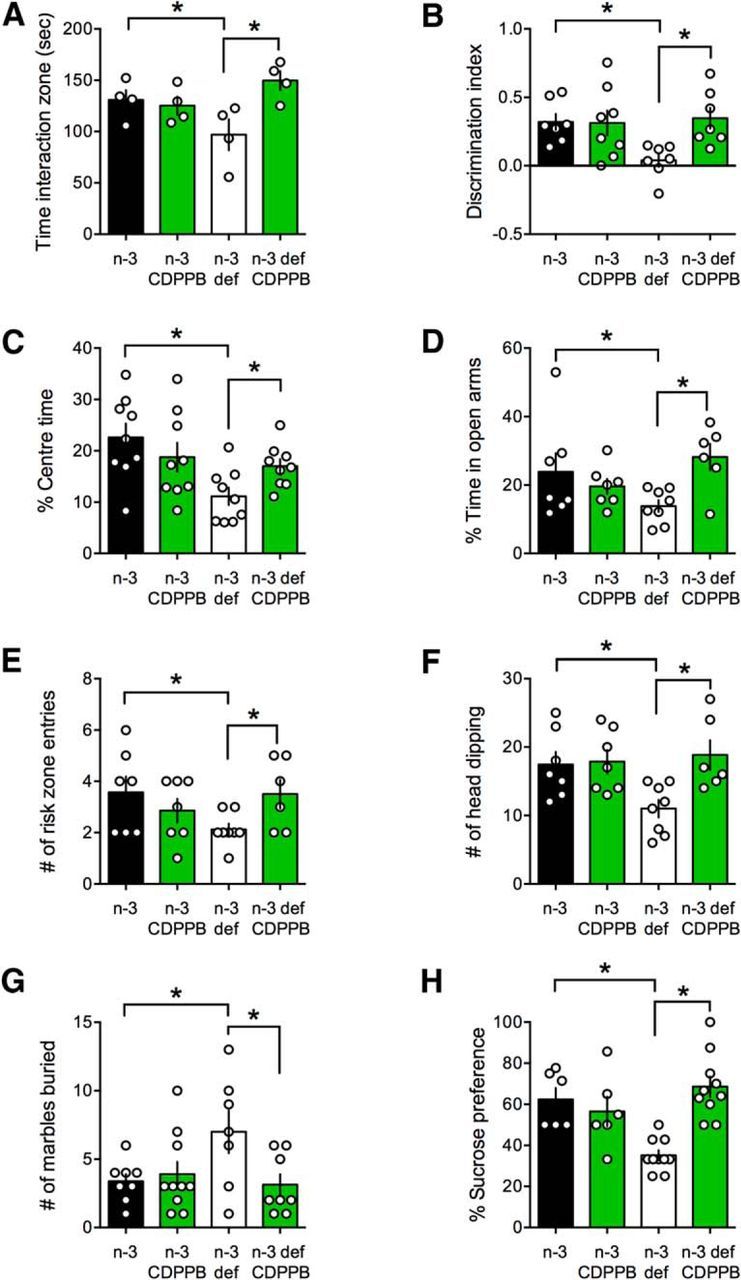
Positive allosteric modulation of mGlu5 normalized behavioral deficits in n-3-deficient mice. CDPPB rescued cognitive deficits observed in the social interaction (A; F(dietxtreat) = 7.066, p < 0.05) and temporal order memory (B; F(dietxtreat) = 4.818, p < 0.05) tasks in n-3 PUFA-deficient mice. CDPPB also normalized anxiety-related behaviors as measured by (C; F(dietxtreat) = 5.496, p < 0.05) the time spent in the center of the open field; (D; F(dietxtreat) = 6.925, p < 0.05) the percentage of time spent in the open arm; and (E; F(dietxtreat) = 4.911, p < 0.05) the frequency of entries in the risk zone in the open arms of the elevated plus maze test as well as the frequency of head dipping (F; F(dietxtreat) = 4.5127, p < 0.05). CDPPB corrected the deficits in the marble burying test (G; F(dietxtreat) = 5.291, p < 0.05) and reduced anhedonia in sucrose preference of the n-3-deficient group (H; F(dietxtreat) = 15.360, p < 0.01). All measures were performed 6 h after CDPPB treatment. Scatter dot plot represents each animal. Error bars indicate SEM. *p < 0.05.
Discussion
Here, we developed a new mouse model of n-3 PUFA deficiency commencing in adolescence and continuing through adulthood to better understand the consequences of nutritional imbalance during a period of ongoing structural and functional refinements in frontostriatal circuitry. The results are threefold: (1) we found that starting n-3 PUFA-deficient diet at adolescence increases anxiety-related behavior and anhedonia in adults in agreement with the idea that malnutrition is accompanied by enhanced susceptibility to emotional distress. We also discovered that cognitive functions are impeded by nutritional deficits after the neurodevelopmental period. (2) The behavioral deficits were paralleled by a reduction of the working range at excitatory synapses in the mPFC and accumbens where mGlu5/eCB-LTD and NMDAR-dependent LTP were abolished. (3) Pharmacological enhancement of the mGlu5/eCB signaling complex following positive allosteric modulation of mGlu5 or inhibition of 2-AG degradation normalized the behavioral repertoire and synaptic plasticity in n-3-deficient mice. These findings strongly suggest that an n-3 PUFA-balanced diet is necessary throughout life to maintain optimal frontostriatal function, cognition and emotional behavior.
Nutrition influences cognition. Adolescent exposure to dietary n-3 PUFA deficiency does not influence adult spatial memory, despite reducing DHA in the hippocampus (Delpech et al., 2015). In contrast, perinatal exposure to a deficient n-3 PUFA diet triggers adult memory deficits (Moranis et al., 2012; Lépinay et al., 2015). Nutritional n-3 PUFA deficits during adolescence impact mPFC development (Bondi et al., 2014). Here we show, for the first time, that nutritional n-3 deficiency impairs mPFC-dependent cognition, suggesting that adolescent nutrition is a determinant of adult behaviors and that balanced nutrition during the perinatal period does not protect from later dietary deficits. Furthermore, we also observed excessive anxiety and reduced sucrose preference in adult n-3-deficient mice, showing that, when dietary n-3 PUFA deficiency starts in adolescence, it impedes emotional behaviors later in life. These findings support the idea that malnutrition is accompanied by enhanced susceptibility to emotional distress and that adolescent nutritional n-3 PUFA deficiency is a major environmental risk factor associated with neuropsychiatric disorders (Simopoulos, 2009; Bazinet and Layé, 2014; Bosch-Bouju et al., 2016).
In mammals, the mPFC is one of the most evolved brain regions, and its malfunctions are a common denominator in several neuropsychiatric diseases, including (but not limited to) mental retardation, autism, schizophrenia, depression, and addiction (Goto et al., 2010). Previously, our laboratories showed that lifelong n-3 deficiency impairs eCB-mediated synaptic plasticity in the mPFC (Lafourcade et al., 2007; Larrieu et al., 2012). The idea that dietary PUFAs strongly control mPFC functions was reinforced by the results that n-3 deficiency starting at adolescence led to specific alterations of cognitive tasks classically associated with mPFC but not hippocampus. The lack of mPFC eCB-LTD cannot be readily explained by a massive reduction of CB1R function as previously observed after an entire life of n-3 PUFA deficiency (Lafourcade et al., 2011). In marked contrast, presynaptic CB1R potency was normal in the mPFC, suggesting that perinatal n-3 PUFA deficiency was instrumental in reducing CB1R functions in our previous study (Lafourcade et al., 2011).
Dietary n-3 PUFAs protect LTP and cognition from aging and environmental insults (McGahon et al., 1999; Kotani et al., 2006; Lynch et al., 2007; Cao et al., 2010; Kelly et al., 2011; Kavraal et al., 2012; Patten et al., 2013). Consistent with this idea, we report that NMDAR-LTP in the mPFC was absent in the n-3-deficient group. LTP was still expressed in the hippocampus of mice that had received the same dietary treatment (Delpech et al., 2015), suggesting a higher sensitivity of the mPFC to PUFA deficiency. The latter hypothesis is supported by our finding that mPFC-dependent, but not hippocampal-dependent cognition was affected in n-3 PUFA-deficient mice.
The nucleus accumbens, widely implicated in reward and motivation (Carelli, 2002; Carlezon and Thomas, 2009), has also recently drawn attention for its participation in the emotional repertoire, including anxiety and compulsivity (Sturm et al., 2003; Galvan et al., 2006; Millet et al., 2014; Bagot et al., 2015; Floresco, 2015). While deregulated neuronal and synaptic activity in the accumbens is also observed in naturalistic rodent models of addiction (Kasanetz et al., 2010) or depression (Bosch-Bouju et al., 2016), the accumbens by and large was not identified as a primary target of dietary PUFA deficiency. Here, we found that n-3 deficiency beginning during adolescence is sufficient to abolish eCB-LTD in adult mice. The fact that basic intrinsic and synaptic properties of accumbens medium spiny or mPFC pyramidal neurons were unchanged in n-3-deficient mice made the lack of eCB-mediated LTD even more remarkable. The lack of eCB-LTD cannot be readily explained by a massive reduction of CB1R functions: accumbens presynaptic CB1R efficacy and potency were not affected. Thus, it is safe to conclude that reduction of CB1R potency is not a general response to n-3 PUFA deficiency, an idea supported by our previous report of normal CB1R presynaptic functions in lifelong n-3-deficient hippocampi (Thomazeau et al., 2017). Moreover, based on our mGlu2/3 LTD experiments, the presynaptic LTD machinery is presumed intact. It only makes sense that the central eCB system shows such sensitivity to dietary PUFAs. Not only are PUFAs precursors of eCBs which share common biosynthetic pathways (Katona and Freund, 2012), but nanoscale modifications of the mGlu5/eCB signalosome in absence of CB1R desensitization or other gross alterations impede mGlu5 plasticity (Jung et al., 2012). Further work will be needed to understand how n-3 PUFA deficiency alters eCB-LTD in the mPFC and accumbens.
In the CNS, eCBs are produced on-demand by a multimolecular signaling complex made of a spatially organized network of postsynaptic G-protein-coupled receptors of various neurotransmitters, scaffolding proteins, ion channels, intracellular calcium stores, as well as synthesizing and degrading enzymes (Katona and Freund, 2012). The lack of eCB-LTD in both mPFC and accumbens in n-3-deficient adult mice was reminiscent of what we reported in unrelated synaptopathies: the fmr1−/y model of Fragile X (Jung et al., 2012), mice overexpressing the Down-syndrome gene candidate Dyrk1A (Thomazeau et al., 2014), mice submitted to social stress (Bosch-Bouju et al., 2016), and, more recently, lifelong n-3 deprivation (Thomazeau et al., 2017). In all cases, inhibition of the eCB-deactivating enzyme MGL normalized prefrontal and hippocampal eCB-LTD and in fmr1−/y acutely corrected behavioral deficits, including anxiety traits (Jung et al., 2012). Here, we found that JZL184, a MGL inhibitor, restored LTD in n-3-deficient mice. It is important to emphasize that the JZL184 effect on eCB-LTD was mediated by CB1R, excluding off-target effects of the molecule. Surprisingly, enhancing 2-AG levels also restored LTP. In contrast to LTD, the rescue of LTP did not require CB1R. It is not clear how 2-AG elevation restores LTP. A rescue of LTP through normalization of heterosynaptic plasticity, as we observed in the hippocampus (Thomazeau et al., 2017), is excluded here where GABA-A receptors were blocked. Another possibility is that 2-AG potentiates NMDAR in a CB1R-independent manner as recently reported (Yang et al., 2014). MGL inhibition decreases AA levels (Kerr et al., 2013) and AA potentiates NMDAR (Miller et al., 1992). Thus, 2-AG elevation is able to modulate NMDAR independently of CB1R.
NMDAR and mGlu1/5 are the principal postsynaptic effectors of LTD and LTP (Emes et al., 2008; Coba et al., 2009; Melis and Pistis, 2012; Grant, 2016). In the accumbens and mPFC, postsynaptic mGlu5 receptors are indispensable to eCB-LTD (Robbe et al., 2002; Lafourcade et al., 2007). The pharmacology of the family of mGlu receptors offers great therapeutic promise, notably because of the development of subtype-selective allosteric modulators. We reasoned that amplification of mGlu5 may enhance eCB-LTD in n-3-deficient mice. We found that a positive allosteric mGlu5 modulator normalized n-3-deficient LTD in mPFC and accumbens. A possible confound in this interpretation is that mGlu5 positively interacts with NMDAR (Coba et al., 2009). Indeed, allosteric modulation of mGlu5 has proven beneficial in restoring NMDAR functions in a mouse model of autism (Won et al., 2012). In contrast, we found here that CB1R was necessary for the CDPPB effects on LTD, in line with our working hypothesis that the mGlu5 modulator CDPPB enhances eCB signaling.
We found that acute in vivo treatment with either an inhibitor of 2-AG degradation or a positive allosteric modulator of mGlu5 corrected excessive anxiety/anhedonia and cognitive deficits in adult n-3 PUFAs' deficient mice. These results are important for two reasons: (1) they confirm the key roles of the mGlu5-eCB signaling complex; and (2) they shed light on potential therapeutic routes to compensate for dietary n-3 PUFA deficiency. These new data indicate that an n-3 PUFA balanced diet is necessary throughout life to maintain optimal cognition, emotional control, and synaptic plasticity. By demonstrating that enhancement of mGlu5 activity or 2-AG levels corrects synaptic and behavioral deficits, this work reveals two new therapeutic routes to treat malnutrition-induced cognitive and emotional impairments. These results warrant a thorough assessment of the behavioral and synaptic consequences of adolescent and adult nutritional deficiencies. In conclusion, these data add to the growing evidence that the mGlu5-eCB signalosome is a key postsynaptic hub whose functional integrity guarantees a maximal working synaptic range and associated behavioral flexibility.
Footnotes
This work was supported by Institut National de la Santé et de la Recherche Médicale, Institut national de la recherche agronomique, and ANR-Blanc MoodFood to O.J.M. and S.L., T.L. was the recipient of Société Française de Nutrition and Labex BRAIN postdoctoral fellowship. We thank members of our laboratories for discussions; Dr. Andrew F. Scheyer for proofreading the manuscript; and the National Institute of Mental Health's Chemical Synthesis and Drug Supply Program for providing CNQX, CDPPB, JZL-184, and NBQX.
The authors declare no competing financial interests.
References
- Bagot RC, Parise EM, Peña CJ, Zhang HX, Maze I, Chaudhury D, Persaud B, Cachope R, Bolaños-Guzmán CA, Cheer JF, Cheer J, Deisseroth K, Han MH, Nestler EJ (2015) Ventral hippocampal afferents to the nucleus accumbens regulate susceptibility to depression. Nat Commun 6:7062. 10.1038/ncomms8062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker GR, Bird F, Alexander V, Warburton EC (2007) Recognition memory for objects, place, and temporal order: a disconnection analysis of the role of the medial prefrontal cortex and perirhinal cortex. J Neurosci 27:2948–2957. 10.1523/JNEUROSCI.5289-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazinet RP, Layé S (2014) Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat Rev Neurosci 15:771–785. 10.1038/nrn3820 [DOI] [PubMed] [Google Scholar]
- Belzung C. (2001) Rodent models of anxiety-like behaviors: are they predictive for compounds acting via non-benzodiazepine mechanisms? Curr Opin Investig Drugs 2:1108–1111. [PubMed] [Google Scholar]
- Bessa JM, Mesquita AR, Oliveira M, Pêgo JM, Cerqueira JJ, Palha JA, Almeida OF, Sousa N (2009) A trans-dimensional approach to the behavioral aspects of depression. Front Behav Neurosci 3:1. 10.3389/neuro.08.001.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondi CO, Taha AY, Tock JL, Totah NK, Cheon Y, Torres GE, Rapoport SI, Moghaddam B (2014) Adolescent behavior and dopamine availability are uniquely sensitive to dietary omega-3 fatty acid deficiency. Biol Psychiatry 75:38–46. 10.1016/j.biopsych.2013.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch-Bouju C, Larrieu T, Linders L, Manzoni OJ, Layé S (2016) Endocannabinoid-mediated plasticity in nucleus accumbens controls vulnerability to anxiety after social defeat stress. Cell Rep 16:1237–1242. 10.1016/j.celrep.2016.06.082 [DOI] [PubMed] [Google Scholar]
- Caballero A, Tseng KY (2016) GABAergic function as a limiting factor for prefrontal maturation during adolescence. Trends Neurosci 39:441–448. 10.1016/j.tins.2016.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero A, Granberg R, Tseng KY (2016) Mechanisms contributing to prefrontal cortex maturation during adolescence. Neurosci Biobehav Rev 70:4–12. 10.1016/j.neubiorev.2016.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao XJ, Cao JJ, Chen TT, Chen WH, Ruan DY (2010) Protective effects of omega-3 fish oil on lead-induced impairment of long-term potentiation in rat dentate gyrus in vivo. Sheng Li Xue Bao 62:225–230. [PubMed] [Google Scholar]
- Carelli RM. (2002) The nucleus accumbens and reward: neurophysiological investigations in behaving animals. Behav Cogn Neurosci Rev 1:281–296. 10.1177/1534582302238338 [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Thomas MJ (2009) Biological substrates of reward and aversion: a nucleus accumbens activity hypothesis. Neuropharmacology 56 [Suppl 1]:122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coba MP, Pocklington AJ, Collins MO, Kopanitsa MV, Uren RT, Swamy S, Croning MD, Choudhary JS, Grant SG (2009) Neurotransmitters drive combinatorial multistate postsynaptic density networks. Sci Signal 2:ra19. 10.1126/scisignal.2000102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpech JC, Thomazeau A, Madore C, Bosch-Bouju C, Larrieu T, Lacabanne C, Remus-Borel J, Aubert A, Joffre C, Nadjar A, Layé S (2015) Dietary n-3 PUFA deficiency increases vulnerability to inflammation-induced spatial memory impairment. Neuropsychopharmacology 40:2774–2787. 10.1038/npp.2015.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doria JG, de Souza JM, Andrade JN, Rodrigues HA, Guimaraes IM, Carvalho TG, Guatimosim C, Dobransky T, Ribeiro FM (2015) The mGluR5 positive allosteric modulator, CDPPB, ameliorates pathology and phenotypic signs of a mouse model of Huntington's disease. Neurobiol Dis 73:163–173. 10.1016/j.nbd.2014.08.021 [DOI] [PubMed] [Google Scholar]
- Drewnowski A. (2004) Obesity and the food environment: dietary energy density and diet costs. Am J Prev Med 27:154–162. 10.1016/j.amepre.2004.06.011 [DOI] [PubMed] [Google Scholar]
- Emes RD, Pocklington AJ, Anderson CN, Bayes A, Collins MO, Vickers CA, Croning MD, Malik BR, Choudhary JS, Armstrong JD, Grant SG (2008) Evolutionary expansion and anatomical specialization of synapse proteome complexity. Nat Neurosci 11:799–806. 10.1038/nn.2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB. (2015) The nucleus accumbens: an interface between cognition, emotion, and action. Annu Rev Psychol 66:25–52. 10.1146/annurev-psych-010213-115159 [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH (1957) A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226:497–509. [PubMed] [Google Scholar]
- Galvan A, Hare TA, Parra CE, Penn J, Voss H, Glover G, Casey BJ (2006) Earlier development of the accumbens relative to orbitofrontal cortex might underlie risk-taking behavior in adolescents. J Neurosci 26:6885–6892. 10.1523/JNEUROSCI.1062-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Yang CR, Otani S (2010) Functional and dysfunctional synaptic plasticity in prefrontal cortex: roles in psychiatric disorders. Biol Psychiatry 67:199–207. 10.1016/j.biopsych.2009.08.026 [DOI] [PubMed] [Google Scholar]
- Grant SG. (2016) The molecular evolution of the vertebrate behavioural repertoire. Philos Trans R Soc Lond B Biol Sci 371:20150051. 10.1098/rstb.2015.0051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrati J, Orejarena MJ, Lassalle O, Bouamrane L, Gonzalez-Campo C, Chavis P (2014) Reelin, an extracellular matrix protein linked to early onset psychiatric diseases, drives postnatal development of the prefrontal cortex via GluN2B-NMDARs and the mTOR pathway. Mol Psychiatry 19:417–426. 10.1038/mp.2013.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joffre C, Nadjar A, Lebbadi M, Calon F, Laye S (2014) n-3 LCPUFA improves cognition: the young, the old and the sick. Prostaglandins Leukot Essent Fatty Acids 91:1–20. 10.1016/j.plefa.2014.05.001 [DOI] [PubMed] [Google Scholar]
- Joffre C, Grégoire S, De Smedt V, Acar N, Bretillon L, Nadjar A, Layé S (2016) Modulation of brain PUFA content in different experimental models of mice. Prostaglandins Leukot Essent Fatty Acids 114:1–10. 10.1016/j.plefa.2016.09.003 [DOI] [PubMed] [Google Scholar]
- Jung KM, Sepers M, Henstridge CM, Lassalle O, Neuhofer D, Martin H, Ginger M, Frick A, DiPatrizio NV, Mackie K, Katona I, Piomelli D, Manzoni OJ (2012) Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nat Commun 3:1080. 10.1038/ncomms2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasanetz F, Manzoni OJ (2009) Maturation of excitatory synaptic transmission of the rat nucleus accumbens from juvenile to adult. J Neurophysiol 101:2516–2527. [DOI] [PubMed] [Google Scholar]
- Kasanetz F, Deroche-Gamonet V, Berson N, Balado E, Lafourcade M, Manzoni O, Piazza PV (2010) Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science 328:1709–1712. 10.1126/science.1187801 [DOI] [PubMed] [Google Scholar]
- Kasanetz F, Lafourcade M, Deroche-Gamonet V, Revest JM, Berson N, Balado E, Fiancette JF, Renault P, Piazza PV, Manzoni OJ (2013) Prefrontal synaptic markers of cocaine addiction-like behavior in rats. Mol Psychiatry 18:729–737. 10.1038/mp.2012.59 [DOI] [PubMed] [Google Scholar]
- Katona I, Freund TF (2012) Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci 35:529–558. 10.1146/annurev-neuro-062111-150420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavraal S, Oncu SK, Bitiktas S, Artis AS, Dolu N, Gunes T, Suer C (2012) Maternal intake of Omega-3 essential fatty acids improves long term potentiation in the dentate gyrus and Morris water maze performance in rats. Brain Res 1482:32–39. 10.1016/j.brainres.2012.09.002 [DOI] [PubMed] [Google Scholar]
- Kelly L, Grehan B, Chiesa AD, O'Mara SM, Downer E, Sahyoun G, Massey KA, Nicolaou A, Lynch MA (2011) The polyunsaturated fatty acids, EPA and DPA exert a protective effect in the hippocampus of the aged rat. Neurobiol Aging 32:2318.e1–15. 10.1016/j.neurobiolaging.2010.04.001 [DOI] [PubMed] [Google Scholar]
- Kerr DM, Harhen B, Okine BN, Egan LJ, Finn DP, Roche M (2013) The monoacylglycerol lipase inhibitor JZL184 attenuates LPS-induced increases in cytokine expression in the rat frontal cortex and plasma: differential mechanisms of action. Br J Pharmacol 169:808–819. 10.1111/j.1476-5381.2012.02237.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotani S, Sakaguchi E, Warashina S, Matsukawa N, Ishikura Y, Kiso Y, Sakakibara M, Yoshimoto T, Guo J, Yamashima T (2006) Dietary supplementation of arachidonic and docosahexaenoic acids improves cognitive dysfunction. Neurosci Res 56:159–164. 10.1016/j.neures.2006.06.010 [DOI] [PubMed] [Google Scholar]
- Labrousse VF, Nadjar A, Joffre C, Costes L, Aubert A, Grégoire S, Bretillon L, Layé S (2012) Short-term long chain omega3 diet protects from neuroinflammatory processes and memory impairment in aged mice. PLoS One 7:e36861. 10.1371/journal.pone.0036861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafourcade M, Elezgarai I, Mato S, Bakiri Y, Grandes P, Manzoni OJ (2007) Molecular components and functions of the endocannabinoid system in mouse prefrontal cortex. PLoS One 2:e709. 10.1371/journal.pone.0000709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafourcade M, Larrieu T, Mato S, Duffaud A, Sepers M, Matias I, De Smedt-Peyrusse V, Labrousse VF, Bretillon L, Matute C, Rodríguez-Puertas R, Layé S, Manzoni OJ (2011) Nutritional omega-3 deficiency abolishes endocannabinoid-mediated neuronal functions. Nat Neurosci 14:345–350. 10.1038/nn.2736 [DOI] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Julien C, Cordeau P, Bohacek I, Weng YC, Calon F, Kriz J (2011) Accumulation of dietary docosahexaenoic acid in the brain attenuates acute immune response and development of postischemic neuronal damage. Stroke 42:2903–2909. 10.1161/STROKEAHA.111.620856 [DOI] [PubMed] [Google Scholar]
- Larrieu T, Madore C, Joffre C, Layé S (2012) Nutritional n-3 polyunsaturated fatty acids deficiency alters cannabinoid receptor signaling pathway in the brain and associated anxiety-like behavior in mice. J Physiol Biochem 68:671–681. 10.1007/s13105-012-0179-6 [DOI] [PubMed] [Google Scholar]
- Larrieu T, Hilal ML, Hilal LM, Fourrier C, De Smedt-Peyrusse V, Sans N, Capuron L, Layé S (2014) Nutritional omega-3 modulates neuronal morphology in the prefrontal cortex along with depression-related behaviour through corticosterone secretion. Transl Psychiatry 4:e437. 10.1038/tp.2014.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lépinay AL, Larrieu T, Joffre C, Acar N, Gárate I, Castanon N, Ferreira G, Langelier B, Guesnet P, Brétillon L, Parnet P, Layé S, Darnaudéry M (2015) Perinatal high-fat diet increases hippocampal vulnerability to the adverse effects of subsequent high-fat feeding. Psychoneuroendocrinology 53:82–93. 10.1016/j.psyneuen.2014.12.008 [DOI] [PubMed] [Google Scholar]
- Lynch AM, Loane DJ, Minogue AM, Clarke RM, Kilroy D, Nally RE, Roche OJ, O'Connell F, Lynch MA (2007) Eicosapentaenoic acid confers neuroprotection in the amyloid-beta challenged aged hippocampus. Neurobiol Aging 28:845–855. 10.1016/j.neurobiolaging.2006.04.006 [DOI] [PubMed] [Google Scholar]
- Madore C, Nadjar A, Delpech JC, Sere A, Aubert A, Portal C, Joffre C, Layé S (2014) Nutritional n-3 PUFA deficiency during perinatal periods alters brain innate immune system and neuronal plasticity-associated genes. Brain Behav Immun 41:22–31. 10.1016/j.bbi.2014.03.021 [DOI] [PubMed] [Google Scholar]
- Manduca A, Morena M, Campolongo P, Servadio M, Palmery M, Trabace L, Hill MN, Vanderschuren LJ, Cuomo V, Trezza V (2015) Distinct roles of the endocannabinoids anandamide and 2-arachidonoylglycerol in social behavior and emotionality at different developmental ages in rats. Eur Neuropsychopharmacol 25:1362–1374. 10.1016/j.euroneuro.2015.04.005 [DOI] [PubMed] [Google Scholar]
- Martin HG, Lassalle O, Brown JT, Manzoni OJ (2016a) Age-dependent long-term potentiation deficits in the prefrontal cortex of the Fmr1 knockout mouse model of Fragile X syndrome. Cereb Cortex 26:2084–2092. [DOI] [PubMed] [Google Scholar]
- Martin HG, Lassalle O, Manzoni OJ (2016b) Differential adulthood onset mGlu5 signaling saves prefrontal function in the Fragile X mouse. Cereb Cortex. Advance online publication. Retrieved Oct. 23, 2016. doi: 10.1093/cercor/bhw328. 10.1093/cercor/bhw328 [DOI] [PubMed] [Google Scholar]
- Mato S, Robbe D, Puente N, Grandes P, Manzoni OJ (2005) Presynaptic homeostatic plasticity rescues long-term depression after chronic Delta 9-tetrahydrocannabinol exposure. J Neurosci 25:11619–11627. 10.1523/JNEUROSCI.2294-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGahon BM, Murray CA, Horrobin DF, Lynch MA (1999) Age-related changes in oxidative mechanisms and LTP are reversed by dietary manipulation. Neurobiol Aging 20:643–653. 10.1016/S0197-4580(99)00027-5 [DOI] [PubMed] [Google Scholar]
- Melis M, Pistis M (2012) Hub and switches: endocannabinoid signalling in midbrain dopamine neurons. Philos Trans R Soc Lond B Biol Sci 367:3276–3285. 10.1098/rstb.2011.0383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B, Sarantis M, Traynelis SF, Attwell D (1992) Potentiation of NMDA receptor currents by arachidonic acid. Nature 355:722–725. 10.1038/355722a0 [DOI] [PubMed] [Google Scholar]
- Millet B, Jaafari N, Polosan M, Baup N, Giordana B, Haegelen C, Chabardes S, Fontaine D, Devaux B, Yelnik J, Fossati P, Aouizerate B, Krebs MO, Robert G, Jay T, Cornu P, Vérin M, Drapier S, Drapier D, Sauleau P, et al. (2014) Limbic versus cognitive target for deep brain stimulation in treatment-resistant depression: accumbens more promising than caudate. Eur Neuropsychopharmacol 24:1229–1239. 10.1016/j.euroneuro.2014.05.006 [DOI] [PubMed] [Google Scholar]
- Monleon S, D'Aquila P, Parra A, Simon VM, Brain PF, Willner P (1995) Attenuation of sucrose consumption in mice by chronic mild stress and its restoration by imipramine. Psychopharmacology (Berl) 117:453–457. 10.1007/BF02246218 [DOI] [PubMed] [Google Scholar]
- Moranis A, Delpech JC, De Smedt-Peyrusse V, Aubert A, Guesnet P, Lavialle M, Joffre C, Layé S (2012) Long term adequate n-3 polyunsaturated fatty acid diet protects from depressive-like behavior but not from working memory disruption and brain cytokine expression in aged mice. Brain Behav Immun 26:721–731. 10.1016/j.bbi.2011.11.001 [DOI] [PubMed] [Google Scholar]
- Morrison WR, Smith LM (1964) Preparation of fatty acid methyl esters and dimethylacetals from lipids with boron fluoride-methanol. J Lipid Res 5:600–608. [PubMed] [Google Scholar]
- Neuhofer D, Henstridge CM, Dudok B, Sepers M, Lassalle O, Katona I, Manzoni OJ (2015) Functional and structural deficits at accumbens synapses in a mouse model of Fragile X. Front Cell Neurosci 9:100. 10.3389/fncel.2015.00100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickols HH, Conn PJ (2014) Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol Dis 61:55–71. 10.1016/j.nbd.2013.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njung'e K, Handley SL (1991) Evaluation of marble-burying behavior as a model of anxiety. Pharmacol Biochem Behav 38:63–67. 10.1016/0091-3057(91)90590-X [DOI] [PubMed] [Google Scholar]
- Patten AR, Sickmann HM, Dyer RA, Innis SM, Christie BR (2013) Omega-3 fatty acids can reverse the long-term deficits in hippocampal synaptic plasticity caused by prenatal ethanol exposure. Neurosci Lett 551:7–11. 10.1016/j.neulet.2013.05.051 [DOI] [PubMed] [Google Scholar]
- Puente N, Cui Y, Lassalle O, Lafourcade M, Georges F, Venance L, Grandes P, Manzoni OJ (2011) Polymodal activation of the endocannabinoid system in the extended amygdala. Nat Neurosci 14:1542–1547. 10.1038/nn.2974 [DOI] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ (2002) Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A 99:8384–8388. 10.1073/pnas.122149199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simopoulos AP. (2009) Evolutionary aspects of the dietary omega-6:omega-3 fatty acid ratio: medical implications. World Rev Nutr Diet 100:1–21. 10.1159/000235706 [DOI] [PubMed] [Google Scholar]
- Sturm V, Lenartz D, Koulousakis A, Treuer H, Herholz K, Klein JC, Klosterkötter J (2003) The nucleus accumbens: a target for deep brain stimulation in obsessive-compulsive- and anxiety-disorders. J Chem Neuroanat 26:293–299. 10.1016/j.jchemneu.2003.09.003 [DOI] [PubMed] [Google Scholar]
- Thomazeau A, Lassalle O, Iafrati J, Souchet B, Guedj F, Janel N, Chavis P, Delabar J, Manzoni OJ (2014) Prefrontal deficits in a murine model overexpressing the Down syndrome candidate gene dyrk1a. J Neurosci 34:1138–1147. 10.1523/JNEUROSCI.2852-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomazeau A, Bosch-Bouju C, Manzoni O, Layé S (2017) Nutritional n-3 PUFA deficiency abolishes endocannabinoid gating of hippocampal long-term potentiation. Cereb Cortex 27:2571–2579. 10.1093/cercor/bhw052 [DOI] [PubMed] [Google Scholar]
- Wei D, Dinh D, Lee D, Li D, Anguren A, Moreno-Sanz G, Gall CM, Piomelli D (2016) Enhancement of anandamide-mediated endocannabinoid signaling corrects autism-related social impairment. Cannabis Cannabinoid Res 1:81–89. 10.1089/can.2015.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won H, Lee HR, Gee HY, Mah W, Kim JI, Lee J, Ha S, Chung C, Jung ES, Cho YS, Park SG, Lee JS, Lee K, Kim D, Bae YC, Kaang BK, Lee MG, Kim E (2012) Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486:261–265. 10.1038/nature11208 [DOI] [PubMed] [Google Scholar]
- Woods-Kettelberger A, Kongsamut S, Smith CP, Winslow JT, Corbett R (1997) Animal models with potential applications for screening compounds for the treatment of obsessive-compulsive disorder. Expert Opin Investig Drugs 6:1369–1381. 10.1517/13543784.6.10.1369 [DOI] [PubMed] [Google Scholar]
- Yang K, Lei G, Xie YF, MacDonald JF, Jackson MF (2014) Differential regulation of NMDAR and NMDAR-mediated metaplasticity by anandamide and 2-AG in the hippocampus. Hippocampus 24:1601–1614. 10.1002/hipo.22339 [DOI] [PubMed] [Google Scholar]