

Abstract
The glucagon-like peptide-1 (GLP-1) receptor agonist exenatide stimulates microglial β-endorphin expression and subsequently produces neuroprotection and antinociception. This study illustrated an unrecognized autocrine role of IL-10 in mediation of exenatide-induced β-endorphin expression. Treatment with exenatide in cultured primary spinal microglia concentration dependently stimulated the expression of the M2 microglial markers IL-10, IL-4, Arg 1, and CD206, but not the M1 microglial markers TNF-α, IL-1β, IL-6, or CD68. Intrathecal exenatide injection also significantly upregulated spinal microglial expression of IL-10, IL-4, Arg 1, and CD206, but not TNF-α, IL-1β, IL-6, or CD68. Intrathecal injection of exenatide stimulated spinal microglial expression of IL-10 and β-endorphin in neuropathic rats. Furthermore, treatment with IL-10 (but not IL-4) stimulated β-endorphin expression in cultured primary microglia, whereas treatment with β-endorphin failed to increase IL-10 expression. The IL-10-neutralizing antibody entirely blocked exenatide-induced spinal microglial expression of β-endorphin in vitro and in vivo and fully blocked exenatide mechanical antiallodynia in neuropathic rats. Moreover, specific cAMP/PKA/p38 signal inhibitors and siRNA/p38β, but not siRNA/p38α, completely blocked exenatide-induced IL-10 expression in cultured primary microglia. Knock-down of IL-10 receptor-α mRNA using siRNA fully inhibited exenatide-induced spinal microglial β-endorphin expression and mechanical antiallodynia in neuropathy. Exenatide also markedly stimulated phosphorylation of the transcription factor STAT3 in cultured primary microglia and β-endorphin stimulation was completely inhibited by the specific STAT3 activation inhibitor. These results revealed that IL-10 in microglia mediated β-endorphin expression after GLP-1 receptor activation through the autocrine cAMP/PKA/p38β/CREB and subsequent IL-10 receptor/STAT3 signal pathways.
SIGNIFICANCE STATEMENT Activation of GLP-1 receptors specifically and simultaneously stimulates the expression of anti-inflammatory cytokines IL-10 and IL-4, as well as the neuroprotective factor β-endorphin from microglia. GLP-1 receptor agonism induces β-endorphin expression and antinociception through autocrine release of IL-10. Activation of GLP-1 receptors stimulates IL-10 and β-endorphin expression subsequently through the Gs-cAMP/PKA/p38β/CREB and IL-10/IL-10 receptor-α/STAT3 signal transduction pathways.
Keywords: β-endorphin, autocrine, GLP-1 receptor, IL-10, microglia, spinal cord
Introduction
Neuroprotection and antinociception have been increasingly documented after activation of glucagon-like peptide-1 (GLP-1) receptors in multiple animal models. GLP-1 receptor agonists such as GLP-1, exenatide, and liraglutide were able to rescue synaptic degradation and reduce the brain levels of β-amyloid in the mouse Alzheimer's disease model (Perry et al., 2002; Perry et al., 2003). Mice with overexpressed GLP-1 receptors in the hippocampus exhibited increased neurite growth and improved learning capacity (During et al., 2003). Exenatide also promoted neurogenesis, normalized dopamine imbalance, and increased the amount of dopaminergic neurons in the substantia nigra in a rat 6-hydroxydopamine model of Parkinson's disease (Bertilsson et al., 2008; Li et al., 2009). Exenatide treatment normalized motor activity and rescued spinal motor neurons in an amyotrophic lateral sclerosis model in the SOD1 G93A-mutant mice (Li et al., 2012). Moreover, in the mouse and rat ischemia and stroke model, treatment with exenatide also reduced brain damage after stroke and improved functional motor outcomes (Li et al., 2009). Exenatide also showed neuroprotection in a mouse model of multiple sclerosis (Aharoni et al., 2011; Hölscher, 2012, 2014). In addition, we recently reported that activation of spinal GLP-1 receptors produced marked antinociception in a variety of rodent models of chronic pain, including neuropathic, inflammatory, bone cancer, and diabetic neuropathic pain (Gong et al., 2014a; Gong et al., 2014b; Zhu et al., 2014; Fan et al., 2015; Fan et al., 2016; Xu et al., 2017). Interestingly, GLP-1 receptor agonists have been in clinical trials to assess their neuroprotective effects on patients with Parkinson's disease (www.clinicaltrials.gov identifier: NCT01174810; https://clinicaltrials.gov/ct2/show/NCT01174810?term=NCT01174810&rank=1) and Alzheimer's disease (www.clinicaltrials.gov identifier: NCT01255163; https://clinicaltrials.gov/ct2/show/NCT01255163?term=NCT01255163&rank=1).
The underlying mechanisms for GLP-1 receptors producing neuroprotection and antinociception are complicated and debatable (Athauda and Foltynie, 2016). Most neurodegenerative diseases are accompanied with chronic inflammatory responses in the brain and spinal cord (Ayasolla et al., 2004), which may mainly involve microglial activation, expression, and release of proinflammatory cytokines and an increase in monocyte adhesion and microvascular permeability (Dozier et al., 2009; Arakawa et al., 2010). However, a recent study reported that exenatide treatment in mice failed to inhibit the expression of proinflammatory cytokines such as TNF-α, IL-6, and IL-1β in brains that were induced by ischemia and reperfusion, although it was able to reduce brain damage significantly (Darsalia et al., 2014). Consistently, our studies have also confirmed that exenatide treatment in rats markedly reduced complete Freund adjuvant (CFA)-induced inflammatory nociception without decreasing spinal expression of proinflammatory cytokines (Fan et al., 2015). Exenatide treatment also failed to inhibit lipopolysaccharide (LPS)-induced overproduction of proinflammatory cytokines in cultured primary microglia (Fan et al., 2015).
In addition to destructive and proinflammatory effects, peripheral macrophages and central microglia also exhibit anti-inflammatory and neuroprotective properties associated with the expression and release of anti-inflammatory cytokines, including IL-10, IL-4, Arg 1, and TGF-β (Franco and Fernandez-Suarez, 2015; Carniglia et al., 2017). IL-4 and particularly IL-10, two secretory cytokines, exhibited wide-ranging anti-inflammatory functions by inhibition of immune mediator release, antigen presentation, and phagocytosis (Sabat et al., 2010; Ul-Haq et al., 2016). They also exhibited marked neuroprotective and antinociceptive properties (Licinio and Wong, 1999; Vale et al., 2003). GLP-1 receptor agonists were also able to induce expression of these anti-inflammatory cytokines (IL-10 and IL-4) in peripheral macrophages (Shiraishi et al., 2012). The herbal iridoid GLP-1 receptor agonist shanzhiside methylester (Zhu et al., 2014; Fan et al., 2016) upregulated the spinal IL-10 expression in neuropathic rats (Zheng et al., 2015). We have also demonstrated that the GLP-1 receptor agonists, including the peptidic agonists exenatide and GLP-1, nonpeptidic agonist WB4–24, and small-molecule agonists shanzhiside methylester and catalpol, stimulated microglial expression and secretion of β-endorphin in the hippocampus and spinal cord to produce antinociception and neuroprotection (Gong et al., 2014b; Fan et al., 2015; Jia et al., 2015; Fan et al., 2016). Therefore, we hypothesized that GLP-1 receptor-induced neuroprotection and antinociception in the CNS are predominately mediated by microglial expression and secretion of anti-inflammatory cytokines and neuroprotective factors (IL-10, IL-4, and β-endorphin) rather than by inhibition of overproduction of proinflammatory cytokines.
This study first assessed the stimulatory effects of exenatide on gene and protein expression of M2 microglial markers IL-10, IL-4, Arg 1, and CD206, as well as β-endorphin and its precursor proopiomelanocortin (POMC) in cultured primary microglia and in the spinal cord of neuropathic rats. The cross talks between cytokines and opioid peptides have been investigated (Kelley, 1988; Kamphuis et al., 1997; Awad et al., 2012). We further explored the parallel or sequential relationship among IL-10, IL-4, and β-endorphin expression by assaying the stimulatory effect of IL-4 and IL-10 on β-endorphin directly in microglial and, vice versa, and by applying the IL-10-neutralizing antibody. Moreover, by measuring phosphorylation and using specific signaling inhibitors and gene silencers, we characterized two subsequent signal transduction pathways involved in the microglial cells: GLP-1 receptor/cAMP/PKA/p38β/CREB and IL-10/IL-10 receptor-α/STAT3/β-endorphin signals. We provide novel information regarding the neuroprotective and antinociceptive role of the GLP-1 receptor/IL-10/β-endorphin through an autocrine mechanism in microglia in the CNS.
Materials and Methods
Chemicals and reagents.
Exenatide was from Kaijie Bio-Pharmaceuticals and IL-10 and IL-4 were from PeproTech. The CREB inhibitor KG501 and naloxone were from Sigma-Aldrich. The adenylate cyclase inhibitor 2′,5′-dideoxyadenosine (DDA) and the PKA activation inhibitor H-89 were from Santa Cruz Biotechnologies. The p38 activation inhibitor SB203580 and STAT3 activation inhibitor NSC74859 were from Selleck Chemicals and Medchem Express, respectively.
Animals.
Male 1-d-old neonatal and adult (8- to 10-week-old) Wistar rats were from the Shanghai Experimental Animal Institute for Biological Sciences (Shanghai, China). The adult rats, housed in a temperature- and humidity-controlled environment on a 12 h light/dark cycle with food and water ad libitum, were acclimatized to the laboratory environment for 3–5 d before our experiments. The animal protocols were approved by the Animal Care and Welfare Committee of the Shanghai Jiao Tong University (Shanghai, China) and followed the regulatory animal care guidelines of the U.S. National Institutes of Health.
Primary cell cultures.
The spinal cord from the 1-d-old neonatal rats was isolated and minced and then incubated in 0.05% trypsin. The dissociated cells were thereafter suspended in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (100 μg/ml). For glial cell cultures, suspended cells were plated into 75 cm2 tissue culture flasks (1 × 107 cells/flask) that were precoated with poly-l-lysine (100 μg/ml) and cultured at 37°C in a 5% carbon dioxide incubator. After 8 d of cell culture, microglial cells were prepared as floating cell suspensions by shaking the flasks at 260 rpm for 2 h. The aliquots were transferred into new plates and unattached cells were removed by washing with serum-free DMEM. Harvested microglial cells exhibited a purity >95%, as determined by Iba-1 immunoreactivity (Wu et al., 2017). After 11 d of cell culture, astrocytes were prepared by shaking the flasks for 2 h and then incubating with 10 ml of 0.05% trypsin-EDTA (Invitrogen) in a cell incubator for 15 min to separate the oligodendrocytes. After neutralization of trypsin with 10 ml of complete DMEM, the floating cell suspensions were discarded. A nearly intact layer of astrocytes at the bottom of the flasks was then trypsinized and subcultured conventionally. Prepared astrocytes exhibited a purity >90% as determined by the GFAP immunoreactivity (Gong et al., 2014b).
For the neuronal culture, the cell suspension was plated into poly-l-lysine (100 μg/ml)-coated plates. After incubation in DMEM with 10% FBS for 4 h, the medium was changed to the Neurobasal (Invitrogen) containing B27 supplement and 0.5 mm glutamine. Experiments were initiated 5–6 d after cell plating. Harvested neurons exhibited a purity >85% as determined by the NeuN immunoreactivity (Gong et al., 2014b).
RNA isolation and quantitative reverse transcriptase-PCR (qRT-PCR).
For the spinal cords, contralateral and ipsilateral spinal lumbar enlargements (L3–L5) were isolated from neuropathic rats and mechanically homogenized in TRIzol reagent (Invitrogen) on ice. Total RNA from spinal homogenates and primary microglia were isolated using TRIzol and reverse transcribed into cDNA using the ReverTraAce qRT-PCR RT-kit (Toyobo) according to the manufacturers' protocols. qRT-PCR amplification was conducted in a Mastercycler ep realplex (Eppendorf) using the RealmasterMix (SYBR Green I; Toyobo). The primers used were as follows: 5′-CCTATCGGGTGGAGCACTTC-3′ and 5′-TGGCTCTTCTCG GAGGTCAT-3′ (POMC exon 2–3), 5′-CCAAGGTCATCCATGACGAC-3′ and 5′-TC CACAGTCTTCTGAGTGGC-3′ (GAPDH) (Gong et al., 2014b), 5′-GGCTCAGCA CTGCTATGTTGCC-3′ and 5′-AGCATGTGGGTCTGGCTGACTG-3′ (IL-10; NM_012854.2), 5′-GGTCTCAG CCCCCACCTTGC-3′ and 5′-CCGTGGTGTTCCT TGTTGCCGT-3′ (IL-4; NM_201270.1), 5′-CCTATGCGTCATTTGGGTGG-3′ and 5′-TACACGATGTCCTTGGCAGA-3′ (Arg 1; NM_017134.3), 5′-CGGGGACTTGG GCTGTATTC-3′ and 5′-GCCGTGAGTCCAAGAGTTGA-3′ (CD206; NM_001106123.2), 5′-CCCCGACTATGTGCCCTCAC-3′ and 5′-AGG GCTCTTGATGGCGGA-3′ (TNF-α) (Fan et al., 2015), 5′-GGAAGGCAG TGTCACTCATTGTG-3′ (IL-1β forward), 5′-GGT CCT CAT CCT GGA AGCTCC-3′ (IL-1β reverse) (Fan et al., 2015) and 5′-GGGACTGA TGTTGTTGACAGCC-3′ and 5′-CATATGTAATTAAGCCTCCGACTTGTG-3′ (IL-6) (Fan et al., 2015). qRT-PCR was first identified to be specific using the melting curves and the relative expression of each mRNA level was calculated with the 2−▵▵CT method after normalizing Ct (cycle threshold) values with GAPDH Ct (Gong et al., 2014b).
Western blot.
Protein supernatants were from homogenized spinal lumbar enlargements (L3–L5) and cultured microglia and extracted in immunoprecipitation analysis buffer containing the protease inhibitor phenylmethylsulfonyl fluoride. The protein was denatured at 100°C for 10 min and separated in 10% SDS-PAGE gel and then transferred to a polyvinylidene fluoride membrane using the electrophoretic method. The membrane was then blocked in 5% skim milk dissolved in 1× TBS containing 0.1% Tween 20 (TBS-T) and then incubated with a primary antibody against IL-10 receptor-α (1:500; Santa Cruz Biotechnology), phospho-Stat3 (Ser727) (1:1000; Cell Signaling Technology), and GAPDH (1:5000; Protein Tech Group) at 4°C overnight with slight shaking. The Odyssey Infrared Imaging system (Li-Cor Biosciences) was used to detect the protein bands after 1 h of incubation at 37°C with Dylight 680-conjugated anti-mouse IgG (1:10,000) and Dylight 800-conjugated anti-rabbit IgG (1:10,000) (Rockland Immunochemicals). The protein band intensity was analyzed and quantified using ImageJ. The relative expression of each target protein was obtained after normalization to the GAPDH level. The experiments were repeated at least three times (Gong et al., 2014b; Fan et al., 2016).
Immunofluorescence staining.
IL-10 or β-endorphin and microglia, astrocytes, or neurons in the spinal cord were doubly labeled with immunofluorescence and visualized under a TCS SP8 confocal microscope (Leica Microsystems) as described previously (Huang et al., 2016). Briefly, rats were subjected to the intracardial perfusion with 100 ml of normal saline, followed by 60 ml of 4% paraformaldehyde (w/v) in PBS under pentobarbital anesthesia (40 mg/kg). Spinal lumbar enlargements (L3–L5) were isolated and fixed in 4% buffered paraformaldehyde for 12 h and dehydrated in gradient sucrose solutions (10–30%) at 4°C. Tissues were entrapped in the optimal cutting temperature embedding agent (Leica Microsystems) and cut into 20 μm frozen sections. The frozen sections were incubated in 10% goat serum (v/v) and 0.5% Triton X-100 (v/v) in PBS for 1 h and then incubated with the rat IL-10 antibody (1:100; goat polyclonal; R&D Systems), rat β-endorphin antibody (1:100; rabbit polyclonal; Phoenix Pharmaceuticals), and primary cellular biomarker antibodies at 4°C for 24 h. Spinal neuronal and glial cells were identified by the following biomarkers: Iba-1 (1:100; mouse monoclonal; Millipore) for microglia, GFAP (1:100; mouse polyclonal; Millipore) for astrocytes, and NeuN (1:60; mouse polyclonal; Millipore) for neurons. IL-10 and β-endorphin staining was visualized with the Alexa Fluor-555-conjugated donkey anti-goat secondary antibody (1:200; Invitrogen). The Alexa Fluor-488-conjugated goat anti-mouse secondary antibody (1:200; Invitrogen) was used to detect other antibodies.
For quantification of the intensity of IL-10-, β-endorphin-, Iba-1-, GFAP- and NeuN-immunopositive cells, photomicrographs of the medial three-fourths of the dorsal horn (laminas I–V) were taken under a confocal microscope with a 10× or 40× magnification. An investigator blinded to the experimental groups measured the positively stained surface area using a computer-assisted image analysis program (ImageJ). The background fluorescence was excluded and only immunofluorescence intensity measurements from positive staining areas were included by low- and high-threshold setup. For colocalization analysis, the program of colocalization finder from ImageJ software was used to generate merged images in which colocalized pixels appeared as white. All surface areas in each group were measured following the same configuration setup at the same time. The average percentage immunolabeled surface area was calculated as the fraction of the positive immunofluorescence surface area of the total measured picture area from three nonadjacent sections of each spinal cord (Huang et al., 2016). Data were then calculated from five to six rats of each group.
β-Endorphin and IL-10 measurements.
For the spinal cord homogenates, contralateral and ipsilateral spinal lumbar enlargements were isolated from neuropathic rats 1 h after intrathecal injection of exenatide and homogenized at 4000 rpm for 15 s with a homogenizer (Fluko Equipment) in 10 mm Tris-HCl (1 g of tissue/5 ml) and centrifuged at 4000 rpm in 4°C for 15 min. The cultured primary microglia were stimulated with 10 nm exenatide for 2 h and then placed in 24-well plates (1 × 105 cells/well) and washed twice with 1 ml of warm DMEM containing 2 mg/ml BSA and 15 mmol/L N-(2-hydroxyethyl) piperazine-N′-2-ethanesulfonic acid. The total protein concentrations in the spinal cord homogenates and cultured primary microglia were measured using the standard bicinchoninic acid protein assay (Beyotime Institute of Biotechnology, Jiangsu, China), in which the levels of IL-10 (eBioscience) and β-endorphin (Phoenix Pharmaceuticals) were measured using enzyme-linked fluorescent immunoassay kits. A fluorescence microplate reader (Thermo Labsystems) was used to measure the relative fluorescence values and the concentration of IL-10 or β-endorphin was calculated by a calibration curve performed at the same time. The assays were validated with the linear range of 1–1000 pg/ml for IL-10 and 1–100 pg/ml for β-endorphin. According to the manufacturer's information, there was cross-reactivity of the β-endorphin with α-endorphin (100%) and γ-endorphin (60%), but not with met-enkephalin (0%) or leu-enkephalin (0%).
RNA interference.
The siRNAs targeting IL-10 receptor-α and the nonspecific oligonucleotides (oligos) were designed and synthesized by Shanghai GenePharma to target the following sequences: IL-10 receptor-α, 5′-CCCUGAGGAGAAAGACCAATT-3′ and 5′-UUGGUCUUUCUCCUCAGGGTT-3′; p38β/MAPK11, 5′-GCACGAGAACGUCAUAGGATT-3′ and 5′-UCCUAUGACGUUCUCGUGCTT-3′ and the nonspecific oligos, 5′-UUCUCCGAACGUGUCACGUTT-3′ and 5′-ACGUGACACGUUCGGAGAATT-3′. The complex of siRNA and the Lipofectin DOTAP (1,2-dioleoyl-3-trimethylammonium-propane; Avanti Polar Lipids) was prepared with a mass ratio of 1:8 following manufacturer's instructions. For in vitro transfection, primary microglial cells were seeded into 24-well/6-well plates. The siRNA-DOTAP complex was added into the plates with supplement of 300 μl/500 μl of basic DMEM to make the final siRNA concentration of 5 μg/ml and cells then incubated for 5 h. After transfection, cells were further cultured for 24 h routinely. For in vivo transfection, the siRNA-DOTAP complex was injected intrathecally into rats successively for 6 d (Wu et al., 2017).
Intrathecal catheterization and injection in rats.
A catheter (PE-10: 0.28 mm inner diameter and 0.61 mm outer diameter; Clay Adams) was administered into the rat lumbar level of the spinal cord under inhaled isoflurane anesthesia as described previously (Huang et al., 2012; Lu et al., 2012). Only rats that had no motor impairment after intrathecal catheter were selected for the experiments. For intrathecal administration, 10 μl of drug solution was administered in a 50 μl microinjector followed by a 20 μl saline flush.
Rat model of neuropathic pain and mechanical allodynia assessment.
The left L5 and L6 spinal nerves of rats were carefully isolated and tightly ligated with 6–0 silk sutures under inhaled isoflurane anesthesia following the methodologies described previously (Kim and Chung, 1992; Zhang et al., 2013). After the nerve ligation, the lumbar fascia and skin were sutured by 4–0 resorbable polyglactin suture and the rats were allowed to recover. The intrathecal catheterization was performed at the same time just before spinal nerve ligation. After recovery, only rats with significant unilateral allodynia to mechanical stimulation (hindlimb withdrawal thresholds in the operated side <8 g) and with no major motor impairment were used for further studies. Neuropathic rats, starting the drug testing 1 week after spinal nerve ligation, were assigned randomly to experimental groups.
To evaluate mechanical allodynia, the animals were acclimatized for at least 30 min to the testing environment: a Plexiglas box on a metal grid. An examiner blinded to the treatment groups performed the behavior testing using a 2290 CE electrical von Frey hair (IITC Life Science). The monofilament generated a force that ranged from 0.1 to 90 g. The withdrawal thresholds evoked in contralateral and ipsilateral were determined by stimulation of the hindlimb while the rat stood on a metal grid. The increasing increments of force were applied to stimulate the footpad until the rat suddenly withdrew its hindlimb. The lowest force evoking a withdrawal response was considered the threshold, which was averaged from triplicate measurements at an ∼1 min interval.
Experimental design and statistical analysis.
For animal studies, five to six male rats were assigned randomly into each group and their behavioral tests were undertaken at different time points. Data are summarized as means ± SEM. Two-tailed and unpaired Student's t test and one-way or repeated-measures two-way ANOVA were used to generate the statistical significance values using Prism (version 5.01; GraphPad Software). The post hoc Student–Newman–Keuls test was performed when the effect of the drug (dose) (for the one-way ANOVA, the factor was drug [dose]; for the two-way ANOVA, the factors were drug [dose], time, and their interaction) was statistically significant. Probability values were two-tailed and the statistical significance criterion p-value was 0.05.
For analysis of concentration–response curves, the following parameters were calculated by fitting nonlinear least-squares curves to the relation Y = a + bx, where x = [C]n/(EC50n + [C]n); that is, the minimum effect, maximum effect (Emax), half-effective concentration (EC50), and Hill coefficient (n). The values of EC50 and b (Emax) were projected by yielding a minimum residual sum of squares of deviations from the theoretical curve (Wang and Pang, 1993).
Results
Exenatide stimulated IL-10 and β-endorphin expression specifically in spinal microglia
We have demonstrated previously that GLP-1 receptor agonism stimulated β-endorphin expression specifically in spinal and hippocampal microglia (Gong et al., 2014b; Fan et al., 2015; Jia et al., 2015; Fan et al., 2016). In this study, we assessed whether GLP-1 receptor agonism also altered the expression of M2 microglial markers such as IL-10, IL-4, Arg 1, and CD206 in spinal microglia. Treatment of cultured primary spinal microglial cells with the GLP-1 receptor agonist exenatide (0.1, 1, 10, 100, and 1000 nm) for 2 h was able to increase the expression of IL-10, IL-4, Arg 1, and CD206 mRNA concentration dependently as measured by using qRT-PCR, with EC50 values of 1.1, 1.4, 5.0, and 6.0 nm, respectively (Fig. 1A–D). In contrast, treatment with exenatide up to 100 nm did not alter the expression of M1 microglial markers TNF-α, IL-1β, IL-6, and CD68 significantly (Fig. 1E–H).
Figure 1.

Effects of exenatide on the expression of IL-10 (A), IL-4 (B), Arg 1 (C), CD206 (D), TNF-α (E), IL-1β (F), IL-6 (G), and CD68 mRNA (H) and IL-10 protein (I, J) in cultured primary microglia from the spinal cord. Data are shown as means ± SEM (n = 4–5 in each group). *p < 0.05 compared with the control group (analyzed by one-way ANOVA followed by the post hoc Student–Newman–Keuls test).
The stimulatory effect of exenatide on IL-10 protein expression was further measured in spinal microglial cells using a commercial fluorescent immunoassay kit. Treatment with exenatide for 2 h stimulated the expression of IL-10 protein concentration dependently in the culture medium, with an EC50 value of 1.9 nm (Fig. 1I). The stimulatory effect of exenatide on IL-10 expression in microglia was also compared with that in astrocytes and neurons. As shown in Figure 1J, the baseline level of IL-10 protein in microglia was 10.9 ± 5.0 pg/ml, which was significantly higher than those in astrocytes (F = 3.218, p = 0.04, one-way ANOVA followed by the post hoc Student–Newman–Keuls test) and neurons (F = 11.21, p = 0.04). Treatment with exenatide (10 nm) for 2 h increased the level of IL-10 protein markedly by 3.2-fold in cultured microglia (F = 2.621, p = 0.005, one-way ANOVA followed by the post hoc Student–Newman–Keuls test). However, exenatide treatment failed to enhance the expression of IL-10 protein significantly in astrocytes or neurons.
The expression of M1 and M2 microglial markers was further measured in both contralateral and ipsilateral spinal lumbar enlargements of neuropathy rats 1 h after intrathecal injection of exenatide. There was no significant difference in IL-10, IL-4, Arg 1, CD206, TNF-α, IL-1, IL-6, and CD68 mRNA levels between contralateral and ipsilateral spinal lumbar enlargements. Compared with the normal saline control, intrathecal injection of exenatide (100 ng) increased the expression of IL-10, IL-4, Arg 1, and CD206 mRNA significantly in both contralateral/ipsilateral spinal cords by 2.4-/2.2-fold (F = 4.995/1.596, p = 0.016/0.024, one-way ANOVA followed by the post hoc Student–Newman–Keuls test; Fig. 2A), 1.8-/1.5-fold (F = 34.26/1.444, p = 0.0133/0.0041; Fig. 2B), 1.5-/3.0-fold (p = 3.179/315, p = 0.0474/0.022; Fig. 2C), and 2.7-/2.1-fold (p = 156.1/27.58, p = 0.0239/0.0292; Fig. 2D), respectively. However, intrathecal exenatide injection did not alter the expression of TNF-α, IL-6, and CD68 mRNA remarkably in the either contralateral or ipsilateral spinal cord (Fig. 2E–G). IL-10 protein levels were further measured in spinal homogenates. Our preliminary spike test showed that the spike recoveries of IL-10 in spinal homogenates were within 80–120%, validating the specificity of the IL-10 ELISA assay in spinal homogenates. There was no significant difference in the baseline levels of IL-10 protein expression between contralateral and ipsilateral spinal cords. As shown in Figure 2I, intrathecal exenatide significantly upregulated IL-10 protein levels by 3.2-fold (F = 2.991, p = 0.0154, one-way ANOVA followed by the post hoc Student–Newman–Keuls test) and 4.7-fold (F = 20.49, p = 0.0421), respectively.
Figure 2.

Stimulatory effects of intrathecal injection of exenatide on spinal expression of IL-10 (A), IL-4 (B), Arg 1 (C), CD206 (D), TNF-α (E), IL-1β(F), IL-6 (G), and CD68 mRNA (H) and IL-10 protein (I) in neuropathic rats induced by tight ligation of L5/L6 spinal nerves. Data are shown as means ± SEM (n = 5–6 in each group). *p < 0.05 compared with the control group (analyzed by one-way ANOVA followed by the post hoc Student–Newman–Keuls test and two-tailed and unpaired Student's t test, respectively).
After the specific IL-10 immunostaining was validated by negative control and antigen absorption test in the spinal cord (data not shown), we identified cell types that promoted IL-10 expression specifically after exenatide treatment through double immunofluorescence labeling of IL-10 with intracellular biomarkers of microglia, astrocytes, and neurons in the ipsilateral dorsal horn. IL-10 colocalized extensively with the microglial biomarker IBA-1 in the spinal dorsal horn (Fig. 3A) and its laminase I–V (Fig. 3B), but much less with the astrocytic biomarker GFAP (Fig. 3C,D) or the neuronal biomarker NeuN (Fig. 3E,F). Compared with saline treatment, intrathecal injection of exenatide (10 ng) increased the immunostaining of IL-10 with IBA-1 markedly (Fig. 3G,H), but failed to stimulate the immunostaining of IL-10 with GFAP (Fig. 3I,J) or NeuN (Fig. 3K,L). To measure quantitatively the double labeling of IL-10 and cellular biomarkers, a computer-assisted image analysis program was used to calculate immunolabeled area ratios in the dorsal horn laminase I–V. Intrathecal exenatide, compared with the saline control, increased the double immunofluorescence intensity of IL-10 with IBA-1 significantly by 3.0-fold (p = 0.0154, by two-tailed and unpaired Student's t test; Fig. 3M), but not with GFAP (Fig. 3N) or NeuN (Fig. 3O).
Figure 3.

Specific stimulatory effects of exenatide on spinal microglial IL-10 expression in neuropathic rats. IL-10 was colabeled using double fluorescence staining using the IL-10 antibody and the microglial marker IBA-1 antibody, the IL-10 antibody and the astrocytic marker GFAP antibody, and the IL-10 antibody and the neuronal marker NeuN antibody in the spinal dorsal horn (A, C, E, G, I, K, 250 μm) and the indicated dorsal horn I–V laminate (B, D, F, H, J, L, 50 μm). Double labeling of IL-10 and biomarkers was indicated by arrows. The immunolabeled surface area ratios of IL-10/IBA-1 (M), IL-10/GFAP (N), and IL-10/NeuN (O) from the indicated spinal dorsal horn laminae I–V were quantified using ImageJ. Data are shown as means ± SEM (n = 5–6 in each group). *p < 0.05 compared with the saline control group (analyzed by unpaired and two-tailed Student's t test).
To further assess the cell types that also promoted the expression of β-endorphin in parallel after exenatide treatment, double immunofluorescence labeling of β-endorphin with microglial, astrocytic, and neuronal cellular markers was also performed in the same spinal slides from the above two groups of neuropathic rats. As shown in Figure 4, A and B, β-endorphin immunofluorescence was colabeled with Iba-1 in the I–V laminae of the dorsal horn in saline-treated rats. However, the β-endorphin immunofluorescence was less colabeled with GFAP (Fig. 4C,D) and NeuN (Fig. 4E,FF). Intrathecal injection of exenatide enhanced the double immunofluorescence of β-endorphin and Iba-1 markedly (Fig. 4G,H). However, it did not increase coexpression of β-endorphin and GFAP (Fig. 4I,J) or β-endorphin and NeuN (Fig. 4K,L) significantly. The computer-assisted image analysis confirmed quantitatively that exenatide treatment enhanced the double immunofluorescence of β-endorphin and Iba-1 by 4.5-fold (p = 0.022, two-tailed and unpaired Student's t test; Fig. 4M), but not of β-endorphin and GFAP (Fig. 4N) or β-endorphin and NeuN (Fig. 4O).
Figure 4.

Specific stimulatory effects of exenatide on spinal microglial β-endorphin expression in neuropathic rats. β-endorphin was colabeled using double fluorescence staining using the β-endorphin antibody and the microglial marker IBA-1 antibody, the β-endorphin antibody and the astrocytic marker GFAP antibody, and the β-endorphin antibody and the neuronal marker NeuN antibody in the spinal dorsal horn (A, C, E, G, I, K, 250 μm) and the indicated dorsal horn I–V laminate (B, D, F, H, J, L, 50 μm). Double labeling of β-endorphin and biomarkers is indicated by arrows. The immunolabeled surface area ratios of β-endorphin/IBA-1 (M), β-endorphin/GFAP (N), and β-endorphin/NeuN (O) from the indicated spinal dorsal horn laminae I–V were quantified using ImageJ. Data are shown as means ± SEM (n = 4–6 in each group). *p < 0.05 compared with the saline control group (analyzed by unpaired and two-tailed Student's t test).
IL-10 mediated exenatide-stimulated β-endorphin expression in spinal microglia
The above results indicated that exenatide stimulated spinal microglial expression of IL-10, IL-4, Arg 1, and CD206, in addition to β-endorphin as reported previously (Gong et al., 2014b). We further assessed whether exenatide stimulated the release of IL-10 and β-endorphin in sequence or in parallel. The directly stimulatory effect of IL-10 on β-endorphin expression was confirmed in cultured primary microglia. Treatment with IL-10 (30 ng/ml) for 2 h increased levels of β-endorphin mRNA significantly (F = 336.6, p = 0.0321, one-way ANOVA followed by the post hoc Student–Newman–Keuls test; Fig. 5A) and protein (F = 1.082, p = 0.0124; Fig. 5B). In contrast, treatment with IL-4 at the same concentration of 30 ng/ml failed to stimulate the expression of microglial β-endorphin mRNA (Fig. 5C). In addition, treatment with β-endorphin in a gradient of concentrations (0.3, 1, and 3 μg/ml) for 2 h failed to enhance the expression of IL-10 mRNA (Fig. 5D). Furthermore, treatment with the IL-10-neutralizing antibody (4 μg/ml) did not alter the baseline POMC expression significantly in cultured primary microglia, but 1 h of pretreatment totally blocked exenatide-stimulated POMC expression (F = 1.445, p = 0.0299, one-way ANOVA followed by the post hoc Student–Newman–Keuls test; Fig. 5E).
Figure 5.

Effects of IL-10 (A, B), IL-4 (C), and β-endorphin (D) on the mRNA and protein expression of β-endorphin and IL-10 and blocking effects of the IL-10-neutralizing antibody on exenatide-stimulated POMC expression (E) in cultured primary microglia. Data are shown as means ± SEM (n = 3 in each group). *#p < 0.05 compared with the control and exenatide-only treatment groups, respectively (analyzed by one-way ANOVA followed by the post hoc Student–Newman–Keuls test).
The indirect stimulatory effect of exenatide on spinal β-endorphin expression through IL-10 release was confirmed in neuropathic rats. Four groups of spinal nerve ligation-induced neuropathic rats received intrathecal injection of either saline (10 μl) or the IL-10-neutralizing antibody (2 μg), followed by a second injection of exenatide (100 ng) 30 min later. The hindpaw withdrawal thresholds to mechanical stimuli were measured in both contralateral and ipsilateral paws before and 0.5, 1, and 2 h after the second intrathecal administration. As shown in Figure 6A, intrathecal injection of the IL-10 antibody did not affect the mechanical thresholds in either the contralateral or ipsilateral hindpaws compared with normal saline treatment. Intrathecal exenatide in the ipsilateral paws produced a marked mechanical antiallodynia, which was nearly entirely prevented by IL-10 antibody pretreatment (F(3,100) = 46.55, p < 0.0001, repeated-measures two-way ANOVA followed by the post hoc Student–Newman–Keuls test).
Figure 6.

Blockade effects of the IL-10-neutralizing antibody on exenatide-induced spinal mechanical antiallodynia (A) and expression of POMC (B) and β-endorphin (C) in neuropathic rats induced by spinal nerve ligation. Data are shown as means ± SEM (n = 6 in each group). *#p < 0.05 compared with the control and exenatide-only treatment groups, respectively (analyzed by one-way and repeated-measures two-way ANOVA followed by the post hoc Student–Newman–Keuls test).
Spinal cord enlargements were obtained immediately upon completion of behavioral assessment and the levels of β-endorphin mRNA and protein were assayed using qRT-PCR and the commercial fluorescent immunoassay kit, respectively. Intrathecal injection of the IL-10 antibody did not affect the baseline POMC expression significantly in the spinal cord but suppressed exenatide-stimulated POMC expression significantly by 47% (F = 2.428, p = 0.0177, one-way ANOVA followed by the post hoc Student–Newman–Keuls test; Fig. 6B). Similarly, intrathecal injection of the IL-10 antibody did not affect the baseline level of β-endorphin peptide significantly, but completely blocked exenatide-stimulated spinal expression of β-endorphin (F = 8.375, p = 0.0471, one-way ANOVA followed by the post hoc Student–Newman–Keuls test; Fig. 6C).
GLP-1 receptor/cAMP/PKA/p38β/CREB signaling pathway mediated exenatide-stimulated IL-10 expression in spinal microglia
To confirm that exenatide-stimulated microglial expression of IL-10 was mediated by direct GLP-1 receptor activation, the specific GLP-1 receptor antagonist exendin(9–39) was used. Treatment with 10 nm exendin(9–39) did not affect the baseline IL-10 level significantly, but 1 h of pretreatment completely suppressed exenatide-stimulated IL-10 expression (F = 5.411, p = 0.0421, one-way ANOVA followed by the post hoc Student–Newman–Keuls test; Fig. 7A).
Figure 7.

Blockade effects of the specific GLP-1 receptor antagonist exendin(9–39) (A), the adenylyl cyclase inhibitor DDA and the PKA inhibitor H-89 (B), the p38 inhibitor rSB203580 (C), the CREB inhibitor KG501 (D), and siRNA/p38β (E) on exenatide-stimulated IL-10 expression in cultured primary microglia. Data are shown as mean ± SEM (n = 4 in each group). *#p < 0.05 compared with the control and exenatide-only treatment groups, respectively (analyzed by one-way ANOVA followed by the post hoc Student–Newman–Keuls test).
It was shown recently that the Gs/cAMP/PKA/p38β/CREB signal pathway mediated exenatide-induced microglial expression of β-endorphin, in which exenatide stimulated adenylyl cyclase activity and phosphorylation of PKA, p38 and selective p38β, and CREB specifically (Wu et al., 2017). To further verify the causal role of these signaling molecules in IL-10 expression, the specific adenylyl cyclase inhibitor DDA, the PKA inhibitor H-89, the p38 inhibitor SB203580, and the CREB inhibitor KG501 were used in exenatide-induced IL-10 expression. Treatment with 100 μm DDA (F = 1.884, p = 0.0387; Fig. 7B), 10 μm H-89 (F = 5.048, p = 0.0147; Fig. 7B), 50 μm SB203580 (F = 4.663, p < 0.0001; Fig. 7C), and 25 μm KG501 (F = 243.6, p = 0.047; Fig. 7D) did not alter baseline IL-10 expression significantly in cultured primary microglia, but 1 h of pretreatment totally suppressed exenatide-increased IL-10 mRNA expression (one-way ANOVA followed by the post hoc Student–Newman–Keuls test).
We demonstrated recently that the gene silencers siRNA/p38α and siRNA/p38β specifically attenuated their respective mRNA expression (Huang et al., 2017; Li et al., 2017; Wu et al., 2017). In this study, transfection (24 h before) with siRNA/p38β (5 μg/ml), but not the same concentrations of siRNA/p38α, totally blocked exenatide-increased IL-10 expression in cultured primary microglia (F = 2.562, p = 0.0026, one-way ANOVA followed by the post hoc Student–Newman–Keuls test; Fig. 7E).
IL-10 receptor-α/STAT3 signaling pathway mediated exenatide-induced β-endorphin expression in spinal microglia
The biological functions of IL-10, such as inhibition of inflammation and antigen presentation, are known to be mediated by IL-10 receptors (Sabat et al., 2010). Due to the lack of specific IL-10 receptor antagonists, the autocrine mediation of the IL-10 receptor in exenatide-stimulated spinal microglial β-endorphin expression was assessed using RNA interference technology to knock down IL-10 receptor expression. The effectiveness of siRNA/IL-10 receptor-α was first determined using Western blot in cultured primary microglial cells. Compared with the nonspecific oligo control (5 μg/ml), transfection with siRNA/IL-10 receptor-α (5 μg/ml) for 24 h reduced the expression of IL-10 receptor-α protein markedly by 72% (F = 5.805, p = 0.0016, one-way ANOVA followed by post hoc Student–Newman–Keuls test; Fig. 8A). Furthermore, treatment with exenatide (10 nm) for 2 h increased POMC expression significantly by 1.4-fold in cultured microglial cells. Although transfection with siRNA/IL-10 receptor-α for 24 h did not affect baseline POMC expression significantly compared with the nonspecific oligo control, it completely blocked exenatide-increased POMC expression in cultured microglial cells (F = 1.128, p = 0.0039, one-way ANOVA followed by the post hoc Student–Newman–Keuls test; Fig. 8B).
Figure 8.
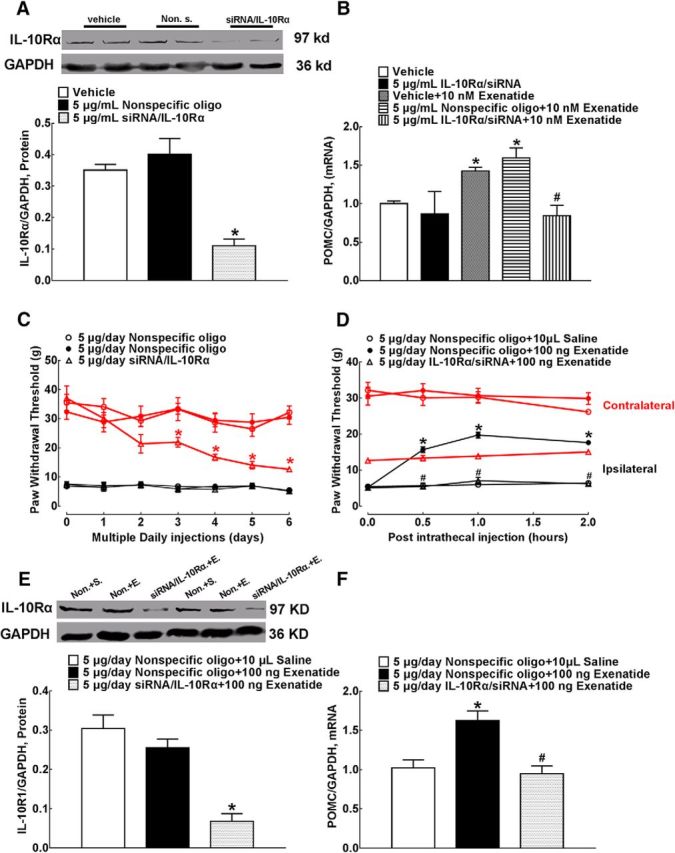
Blockade effects of siRNA/IL-10 receptor-α on IL-10 receptor-α expression (A) and exenatide-increased POMC mRNA level (B) in primary cultures of microglia, baseline paw withdrawal thresholds (C), intrathecal exenatide-induced mechanical antiallodynia (D), and spinal expression of IL-10 receptor-α mRNA (E) and protein (F) in neuropathic rats. Representative gels are inserted in their respective figures. Data are shown as means ± SEM (n = 4 in each group in the cell culture and n = 6 in each group in neuropathic rats). *#p < 0.05 compared with the control and exenatide-only treatment groups, respectively (analyzed by one-way or repeated-measures two-way ANOVA followed by the post hoc Student–Newman–Keuls test).
We further assessed the effects of siRNA/IL-10 receptor-α in neuropathic rats. For 6 d, three groups of neuropathic rats received multiple daily intrathecal injections of nonspecific oligos (5 μg/d, the first 2 groups) or siRNA/IL-10 receptor-α (5 μg/d). The withdrawal thresholds to mechanical stimuli were measured daily in both contralateral and ipsilateral paws before each siRNA injection. As shown in Figure 8C, compared with the nonspecific oligo control (5 μg/d), multiple daily intrathecal injections of siRNA/IL-10 receptor-α decreased baseline withdrawal responses significantly in the contralateral paws in a time-dependent manner (F(2,105) = 25.84, p < 0.0001, repeated-measures two-way ANOVA followed by the post hoc Student–Newman–Keuls test), but did not affect the reduced withdrawal responses significantly in the ipsilateral paws.
On the seventh day, a single dose of normal saline (10 μl, the first group) or exenatide (100 ng, the last two groups) was injected intrathecally into rats and the paw withdrawal thresholds were tested before and 0.5, 1, and 2 h after injection. As shown in Figure 8D, intrathecal exenatide injection did not affect the withdrawal thresholds significantly in the contralateral paws of either nonspecific oligo- or siRNA/IL-10 receptor-α-pretreated rats, although the latter had lower baseline mechanical thresholds. However, intrathecal exenatide produced a time-dependent mechanical antiallodynia in the ipsilateral paws of the nonspecific oligo-treated rats. Pretransfection with siRNA/IL-10 receptor-α completely blocked exenatide-induced mechanical antiallodynia (F(2,60) = 230.1, p < 0.0001, repeated-measures two-way ANOVA followed by the post hoc Student–Newman–Keuls test).
The spinal cord enlargements were obtained immediately after completion of behavioral assessment and the effect of siRNA/IL-10 receptor-α on spinal IL-10 receptor-α knock-down was assessed by Western blot. Compared with the nonspecific oligo treatment group, multiple daily intrathecal injections of siRNA/IL-10 receptor-α reduced the expression of IL-10 receptor-α protein significantly by 73% in the spinal homogenates (F = 1.272, p = 0.0002, one-way ANOVA followed by the post hoc Student–Newman–Keuls test; Fig. 8E). In addition, intrathecal injection of exenatide in the nonspecific oligo-treated rats stimulated spinal POMC expression markedly, which was entirely attenuated by siRNA/IL-10 receptor-α (F = 1.536, p = 0.0026, one-way ANOVA followed by the post hoc Student–Newman–Keuls test; Fig. 8F).
STAT3 is the main transcription factor after IL-10 receptor activation (Sabat et al., 2010). We explored the causal relationship between STAT3 activation and exenatide-stimulated β-endorphin expression. Exenatide incubation (10 nm) for 30 min upregulated STAT3 phosphorylation markedly by 3-fold in cultured microglial cells (F = 5.095, p = 0.0144, by 2-tailed and unpaired Student's t test; Fig. 9A). To further explore the casual role of the STAT3 signaling in exenatide-increased expression of β-endorphin, gradient concentrations (10, 30, and 100 μm) of the specific STAT3 activation inhibitor NSC74859 were incubated with primary microglia 1 h before exenatide treatment. Exenatide treatment (10 nm) for 2 h increased the level of POMC mRNA markedly. Although NSC74859 did not alter baseline POMC levels significantly, it completely suppressed exenatide-induced POMC overexpression (F = 26.53, p < 0.0001, one-way ANOVA followed by the post hoc Student–Newman–Keuls test; Fig. 9B).
Figure 9.

Stimulatory effects of exenatide on STAT3 phosphorylation (A) and the blockage of the specific STAT3 inhibitor NSC74859 on exenatide-induced POMC mRNA expression (B) in primary microglial cells. A representative gel is inserted in the figure. Data are shown as means ± SEM (n = 4 in each group). *#p < 0.05 compared with the control and exenatide-only treatment groups, respectively (analyzed by one-way ANOVA followed by the post hoc Student–Newman–Keuls test).
Discussion
Microglial cells are resident macrophages in the CNS (David and Kroner, 2011). Peripheral and central nerve injury and insult or pathogenic agents can induce M1 type polarization of microglial cells by upregulating the expression of surface proteins such as CD68, CD86, and CD32. M1 microglia also release proinflammatory mediators such as TNF-α, IL-1β, IL-6, nitric oxide, and chemokines, which are also considered to be M1 markers, but functionally deteriorate neuronal activities (David and Kroner, 2011). Conversely, during the neuronal injury recovery process, microglial cells can be polarized to an alternative (M2) type by increasing expression of the cellular surface proteins CD206 and CD163 and cytoplasmic protein Arg 1 (Yang and Ming, 2014; Sasaki, 2017; Tang and Le, 2016). M2 microglial cells further secrete anti-inflammatory or neuroprotective factors such as IL-10, IL-4, and TGF-β, which are also regarded as M2 markers and functionally promote tissue regeneration and facilitate the neuronal homeostasis balance (Franco and Fernandez-Suarez, 2015).
In consistent with a recent finding that activation of GLP-1 receptors could promote the expression of M2 microglial genes in the cortex (Darsalia et al., 2014), in the current study, we revealed that exenatide treatment stimulated the expression of Arg 1 and CD206 specifically in cultured primary spinal microglia and the spinal cord of neuropathic rats. More importantly, we found that exenatide stimulated the expression and secretion of the anti-inflammatory cytokines IL-10 and IL-4 and neuroprotective factor β-endorphin specifically and simultaneously from microglia (but not in neurons or astrocytes) in primary cultures and in the spinal cord of neuropathic rats. We also found that exenatide induced β-endorphin overexpression and antinociception through autocrine release of IL-10, but not IL-4, and that exenatide stimulated IL-10 and β-endorphin expression subsequently through the Gs-cAMP/PKA/p38β/CREB and IL-10/IL-10 receptor-α/STAT3 signal transduction pathways. Activated central microglia and peripheral macrophages basically share the same cellular antigens, including Iba-1 and morphology (David and Kroner, 2011). It is thus possible that our identified role of Iba-1-positive cells in IL-10/β-endorphin expression is due to peripheral microphage infiltration. Indeed, infiltration of macrophages to the injured central and peripheral nerve has been described previously (David and Kroner, 2011; Kim and Moalem-Taylor, 2011; Jin and Yamashita, 2016). However, there is no significant infiltration of macrophages found in the CNS, including the spinal cord, after peripheral nerve injuries (Ajami et al., 2007; Cao and DeLeo, 2008; Kim and Moalem-Taylor, 2011; Guan et al., 2016). It was reported that circulating macrophages were not recruited to the dorsal portion of the brainstem under normal physiological and facial nerve damage in chimeric animals obtained by parabiosis (Ajami et al., 2007). In addition, RNA-sequencing and antigen immunochemistry analysis showed that monocytes did not infiltrate the spinal cord substantially after spinal nerve ligation (Guan et al., 2016) or partial ligation of the sciatic nerve (Kim and Moalem-Taylor, 2011) in rodents. Therefore, we postulate that spinal microglial expression of IL-10/β-endorphin plays a dominate role in exenatide-induced antinociception in neuropathy.
Although it was reported previously that exenatide inhibited the expression of proinflammatory cytokines in macrophage and β-cells in diabetic animals (Pugazhenthi et al., 2010; Darsalia et al., 2014; Varin et al., 2016), we and others have demonstrated that activation of GLP-1 receptors by exenatide and nonpeptide agonist WB4–24 did not inhibit LPS-induced microglial and CFA- or ischemia-induced spinal or brain expression of proinflammatory cytokines, TNF-α, IL-1β, and IL-6 significantly (Darsalia et al., 2014; Fan et al., 2015). Furthermore, our current study showed that exenatide treatment increased the expression of IL-10 and IL-4 specifically in primary spinal microglia, but not in astrocytes and neurons, similar to the previous studies in which exenatide stimulated IL-10 and IL-4 expression in human monocytes/macrophage and regulatory T cells (Ayasolla et al., 2004; Xue et al., 2008; Arakawa et al., 2010; McClean et al., 2011; Bułdak et al., 2016). We also found that intrathecal exenatide injection stimulated the expression of IL-10 and IL-4 in both the contralateral and ipsilateral spinal cord of neuropathic rats. Together, these results suggest that GLP-1 receptor-mediated neuroprotection and antinociception are associated with the expression and release of anti-inflammatory cytokines, rather than inhibition of the expression and release of proinflammatory cytokines.
IL-10 has been considered the most important anti-inflammatory cytokine so far studied, and acts by binding to the IL-10 receptor-α followed by activation of IL-10 receptor-β localized on the cell membrane (Sabat et al., 2010; Glocker et al., 2011). Exogenous application of IL-10 exhibited antinociceptive activities in sciatic nerve chronic constriction injury- and paclitaxel-induced neuropathic pain, formalin- and CFA-induced inflammatory pain, bone cancer pain, and diabetic neuropathic pain (Milligan et al., 2005; Ledeboer et al., 2007; Zhou et al., 2008; Kim et al., 2011; Thakur et al., 2016). In the current study, multiple daily intrathecal injections of siRNA/IL-10 receptor-α progressively induced mechanical allodynia in the contralateral hindpaws, although it did not further attenuate the reduced withdrawal thresholds in the ipsilateral paws, which may be due to the ceiling effect. However, intrathecal injection of the IL-10-neutralizing antibody for 2 h failed to affect the baseline mechanical thresholds significantly in the contralateral paws, consistent with the previous finding that daily intrathecal injections of the IL-10-neutralizing antibody for 5 d had no nociceptive effects in rats (Tai et al., 2009). The reason for different outcomes of the IL-10-neutralizing antibody and siRNA/IL-10 receptor-α on normal nociceptive thresholds is not known, but may be due to the fact that other endogenous IL-10-related anti-inflammatory cytokines such as IL-19, IL-20, IL-22, IL-24, and IL-26 also bind to IL-10 receptors (Sheppard et al., 2003; Oral et al., 2006; Scrivo et al., 2015).
Nevertheless, the IL-10 and siRNA/IL-10 receptor-α were used to validate the causal relationship between IL-10 and β-endorphin expression after GLP-1 receptor activation. Our current results demonstrated that intrathecal injection of exenatide stimulated spinal microglial expression of IL-10 and β-endorphin markedly and in parallel in neuropathic rats as measured by the immunofluorescence staining technique. We further showed that treatment with IL-10, but not IL-4, increased the expression of β-endorphin markedly in microglia, consistent with a previous study in which the β-endorphin expression was stimulated by IL-10 in human promyelocytic cells (Awad et al., 2012), although it was also reported that IL-4 induced POMC expression in lymphocytes (Busch-Dienstfertig et al., 2012). In contrast, treatment with β-endorphin did not alter IL-10 expression significantly. More importantly, the IL-10-neutralizing antibody completely blocked exenatide-induced β-endorphin overexpression in primary microglia. Furthermore, intrathecal injection of the IL-10-neutralizing antibody in neuropathic rats fully inhibited exenatide-induced spinal β-endorphin overexpression and antinociception, the latter of which was entirely blocked by intrathecal naloxone administration. These results indicate a sequential expression of IL-10 before β-endorphin in microglia. We have demonstrated previously that different GLP-1 agonists in primary microglia and in the spinal cord and hippocampus upregulated the expression of β-endorphin, which mediated their neuroprotection and antinociception (Gong et al., 2014b; Fan et al., 2015; Jia et al., 2015). Therefore, GLP-1 receptor activation-induced β-endorphin expression and antinociception occur through autocrine IL-10 expression. More broadly, the autocrine regulation of POMC expression by leukemia inhibitory factor has been demonstrated in AtT-20 murine cells (Akita et al., 1995). It has also been reported that dendritic cell-derived IL-10 blocked dendritic cell maturation effectively (Corinti et al., 2001) and macrophage-derived IL-10 inhibited LPS-induced prostaglandin production via a feedback loop in macrophages (MacKenzie et al., 2013).
IL-10 expression has been well characterized in monocytes, T-cell subsets, and macrophages (Glocker et al., 2011). Activation of TLR by LPS stimulated IL-10 expression via the MYD88/MAPKs/NF-κB signals in macrophages (Saraiva and O'Garra, 2010). The TLR-independent signals, including CREB, STAT, MAF, and Jun, regulated IL-10 expression in immune cells (Saraiva and O'Garra, 2010). More specifically, it was demonstrated recently that the cAMP/PKA/p38β/CREB signaling pathway in microglia was activated by GLP-1 receptor agonism (Wu et al., 2017), in which p38 and the transcription factor CREB are known to regulate IL-10 expression (Saraiva and O'Garra, 2010; Lobo-Silva et al., 2016). In this study, we further demonstrated that pharmacological inhibition of cAMP production by DDA, PKA activation by H-89, p38 activation by SB203580, and CREB activation by KG501 completely blocked exenatide-induced IL-10 expression, indicating that the cAMP/PKA/p38β/CREB signaling transduction cascade casually mediated GLP-1 receptor-induced IL-10 overexpression in microglia (Fig. 9). p38β, but not p38α, was also involved in bulleyaconitine A- and cynandione A-induced microglial expression of dynorphin A and β-endorphin (Huang et al., 2017; Li et al., 2017).
After binding and activation of membrane-bound IL-10 receptors, the Janus tyrosine kinases were activated, resulting in recruitment and activation of the transcription factor STAT3 (Sabat et al., 2010), which has been speculated to be a strong inducer encoding the β-endorphin gene POMC (Scully and Rosenfeld, 2002; Bates et al., 2003). In agreement with a previous study showing that GLP-1 activated STAT3 significantly in human macrophages (Shiraishi et al., 2012), our current study showed that exenatide treatment stimulated STAT3 phosphorylation significantly in microglia. Furthermore, the specific STAT3 inhibitor NSC74859 completely suppressed exenatide-induced β-endorphin expression. Conversely, knock-down of IL-10 receptor-α expression completely blocked exenatide-induced β-endorphin overexpression in microglia and spinal knock-down of IL-10 receptor-α expression entirely attenuated intrathecal exenatide-induced spinal β-endorphin expression and mechanical antiallodynia. Together, our current data suggest that the IL-10/IL-10 receptor-α/STAT3 signaling mediated exenatide-induced microglial expression of β-endorphin in an autocrine mechanism (Fig. 10). Our results also rule out the direct cross talk between transcription factors of CREB and STAT3. In contrast, the cross talk between STAT3 and NF-κB, in which STAT3 stimulated the expression of Abin-3, Dusp-1, Bcl-3, and IkBNS to inhibit the NF-κB transcription activity, has been studied extensively (Kuwata et al., 2003; Hirotani et al., 2005; Weaver et al., 2007; Sabat et al., 2010).
Figure 10.

Illustration of the proposed IL-10 autocrine mechanisms underlying GLP-1 receptor activation-induced β-endorphin expression in microglia and spinal antinociception in neuropathy. After activation of GLP-1 receptors localized on the microglial cell membrane, the cAMP/PKA/p38β signal cascade is activated, leading to phosphorylation of the transcription factor CREB, which subsequently mediates IL-10 expression and secretion. The secreted IL-10 then binds to and activates IL-10 receptors localized on microglial cell membrane to stimulate phosphorylation of the transcription factor STAT3 and in turn to induce expression and secretion of β-endorphin. The released β-endorphin thereafter passes the microglial–neuronal synapse to activate postsynaptic neuronal μ-opioid receptors, leading to antinociception in neuropathy.
In conclusion, our study uncovered for the first time the antinociceptive and neuroprotective pathway of GLP-1 receptor/IL-10/β-endorphin in microglia in the CNS. The autocrine mechanisms are involved in two signal transduction pathways: the cAMP/PKA/p38β/CREB signals and subsequent IL-10/IL-10 receptor-α/STAT3 signals.
Footnotes
This work was supported in part by the National Natural Science Foundation of China (Grants 81374000 and 81673403) and the Shanghai Industrial Translational Project (Grant 15401901300).
The authors declare no competing financial interests.
References
- Aharoni R, Vainshtein A, Stock A, Eilam R, From R, Shinder V, Arnon R (2011) Distinct pathological patterns in relapsing-remitting and chronic models of experimental autoimmune enchephalomyelitis and the neuroprotective effect of glatiramer acetate. J Autoimmun 37:228–241. 10.1016/j.jaut.2011.06.003 [DOI] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM (2007) Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10:1538–1543. 10.1038/nn2014 [DOI] [PubMed] [Google Scholar]
- Akita S, Webster J, Ren SG, Takino H, Said J, Zand O, Melmed S (1995) Human and murine pituitary expression of leukemia inhibitory factor: novel intrapituitary regulation of adrenocorticotropin hormone synthesis and secretion. J Clin Invest 95:1288–1298. 10.1172/JCI117779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa M, Mita T, Azuma K, Ebato C, Goto H, Nomiyama T, Fujitani Y, Hirose T, Kawamori R, Watada H (2010) Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes 59:1030–1037. 10.2337/db09-1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athauda D, Foltynie T (2016) The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson's disease: mechanisms of action. Drug Discov Today 21:802–818. 10.1016/j.drudis.2016.01.013 [DOI] [PubMed] [Google Scholar]
- Awad H, Abas M, Elgharably H, Tripathi R, Theofilos T, Bhandary S, Sai-Sudhakar C, Sen CK, Roy S (2012) Endogenous opioids in wound-site neutrophils of sternotomy patients. PloS One 7:e47569. 10.1371/journal.pone.0047569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayasolla K, Khan M, Singh AK, Singh I (2004) Inflammatory mediator and beta-amyloid (25–35)-induced ceramide generation and iNOS expression are inhibited by vitamin E. Free Radic Biol Med 37:325–338. 10.1016/j.freeradbiomed.2004.04.007 [DOI] [PubMed] [Google Scholar]
- Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG Jr (2003) STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature 421:856–859. 10.1038/nature01388 [DOI] [PubMed] [Google Scholar]
- Bertilsson G, Patrone C, Zachrisson O, Andersson A, Dannaeus K, Heidrich J, Kortesmaa J, Mercer A, Nielsen E, Rönnholm H, Wikström L (2008) Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson's disease. J Neurosci Res 86:326–338. 10.1002/jnr.21483 [DOI] [PubMed] [Google Scholar]
- Bułdak Ł, Machnik G, Bułdak RJ, Łabuzek K, Bołdys A, Belowski D, Basiak M, Okopień B (2016) Exenatide (a GLP-1 agonist) expresses anti-inflammatory properties in cultured human monocytes/macrophages in a protein kinase A and B/Akt manner. Pharmacol Rep 68:329–337. 10.1016/j.pharep.2015.10.008 [DOI] [PubMed] [Google Scholar]
- Busch-Dienstfertig M, Labuz D, Wolfram T, Vogel NN, Stein C (2012) JAK-STAT1/3-induced expression of signal sequence-encoding proopiomelanocortin mRNA in lymphocytes reduces inflammatory pain in rats. Mol Pain 8:83. 10.1186/1744-8069-8-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, DeLeo JA (2008) CNS-infiltrating CD4+ T lymphocytes contribute to murine spinal nerve transection-induced neuropathic pain. Eur J Immunol 38:448–458. 10.1002/eji.200737485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carniglia L, Ramírez D, Durand D, Saba J, Turati J, Caruso C, Scimonelli TN, Lasaga M (2017) Neuropeptides and microglial activation in inflammation, pain, and neurodegenerative diseases. Mediators Inflamm 2017:5048616. 10.1155/2017/5048616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corinti S, Albanesi C, la Sala A, Pastore S, Girolomoni G (2001) Regulatory activity of autocrine IL-10 on dendritic cell functions. J Immunol 166:4312–4318. 10.4049/jimmunol.166.7.4312 [DOI] [PubMed] [Google Scholar]
- Darsalia V, Hua S, Larsson M, Mallard C, Nathanson D, Nyström T, Sjöholm A, Johansson ME, Patrone C (2014) Exendin-4 reduces ischemic brain injury in normal and aged type 2 diabetic mice and promotes microglial M2 polarization. PLoS One 9:e103114. 10.1371/journal.pone.0103114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S, Kroner A (2011) Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci 12:388–399. 10.1038/nrn3053 [DOI] [PubMed] [Google Scholar]
- Dozier KC, Cureton EL, Kwan RO, Curran B, Sadjadi J, Victorino GP (2009) Glucagon-like peptide-1 protects mesenteric endothelium from injury during inflammation. Peptides 30:1735–1741. 10.1016/j.peptides.2009.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- During MJ, Cao L, Zuzga DS, Francis JS, Fitzsimons HL, Jiao X, Bland RJ, Klugmann M, Banks WA, Drucker DJ, Haile CN (2003) Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med 9:1173–1179. 10.1038/nm919 [DOI] [PubMed] [Google Scholar]
- Fan H, Gong N, Li TF, Ma AN, Wu XY, Wang MW, Wang YX (2015) The non-peptide GLP-1 receptor agonist WB4–24 blocks inflammatory nociception by stimulating beta-endorphin release from spinal microglia. Br J Pharmacol 172:64–79. 10.1111/bph.12895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Li TF, Gong N, Wang YX (2016) Shanzhiside methylester, the principle effective iridoid glycoside from the analgesic herb Lamiophlomis rotata, reduces neuropathic pain by stimulating spinal microglial beta-endorphin expression. Neuropharmacology 101:98–109. 10.1016/j.neuropharm.2015.09.010 [DOI] [PubMed] [Google Scholar]
- Franco R, Fernández-Suárez D (2015) Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol 131:65–86. 10.1016/j.pneurobio.2015.05.003 [DOI] [PubMed] [Google Scholar]
- Glocker EO, Kotlarz D, Klein C, Shah N, Grimbacher B (2011) IL-10 and IL-10 receptor defects in humans. Ann NY Acad Sci 1246:102–107. 10.1111/j.1749-6632.2011.06339.x [DOI] [PubMed] [Google Scholar]
- Gong N, Fan H, Ma AN, Xiao Q, Wang YX (2014a) Geniposide and its iridoid analogs exhibit antinociception by acting at the spinal GLP-1 receptors. Neuropharmacology 84:31–45. 10.1016/j.neuropharm.2014.04.007 [DOI] [PubMed] [Google Scholar]
- Gong N, Xiao Q, Zhu B, Zhang CY, Wang YC, Fan H, Ma AN, Wang YX (2014b) Activation of spinal glucagon-like peptide-1 receptors specifically suppresses pain hypersensitivity. J Neurosci 34:5322–5334. 10.1523/JNEUROSCI.4703-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, Guan AK, Evans-Reinsch Z, Braz J, Devor M, Abboud-Werner SL, Lanier LL, Lomvardas S, Basbaum AI (2016) Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci 19:94–101. 10.1038/nn.4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirotani T, Lee PY, Kuwata H, Yamamoto M, Matsumoto M, Kawase I, Akira S, Takeda K (2005) The nuclear IkappaB protein IkappaBNS selectively inhibits lipopolysaccharide-induced IL-6 production in macrophages of the colonic lamina propria. J Immunol 174:3650–3657. 10.4049/jimmunol.174.6.3650 [DOI] [PubMed] [Google Scholar]
- Hölscher C. (2012) Potential role of glucagon-like peptide-1 (GLP-1) in neuroprotection. CNS Drugs 26:871–882. 10.2165/11635890-000000000-00000 [DOI] [PubMed] [Google Scholar]
- Hölscher C. (2014) Central effects of GLP-1: new opportunities for treatments of neurodegenerative diseases. J Endocrinol 221:T31–41. 10.1530/JOE-13-0221 [DOI] [PubMed] [Google Scholar]
- Huang JL, Chen XL, Guo C, Wang YX (2012) Contributions of spinal D-amino acid oxidase to bone cancer pain. Amino Acids 43:1905–1918. 10.1007/s00726-012-1390-z [DOI] [PubMed] [Google Scholar]
- Huang Q, Mao XF, Wu HY, Li TF, Sun ML, Liu H, Wang YX (2016) Bullatine A stimulates spinal microglial dynorphin A expression to produce anti-hypersensitivity in a variety of rat pain models. J Neuroinflammation 13:214. 10.1186/s12974-016-0696-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Mao XF, Wu HY, Liu H, Sun ML, Wang X, Wang YX (2017) Cynandione A attenuates neuropathic pain through p38beta MAPK-mediated spinal microglial expression of beta-endorphin. Brain Behav Immun 62:64–77. 10.1016/j.bbi.2017.02.005 [DOI] [PubMed] [Google Scholar]
- Jia Y, Gong N, Li TF, Zhu B, Wang YX (2015) Peptidic exenatide and herbal catalpol mediate neuroprotection via the hippocampal GLP-1 receptor/beta-endorphin pathway. Pharmacol Res 102:276–285. 10.1016/j.phrs.2015.10.008 [DOI] [PubMed] [Google Scholar]
- Jin X, Yamashita T (2016) Microglia in central nervous system repair after injury. J Biochem 159:491–496. 10.1093/jb/mvw009 [DOI] [PubMed] [Google Scholar]
- Kamphuis S, Kavelaars A, Brooimans R, Kuis W, Zegers BJ, Heijnen CJ (1997) T helper 2 cytokines induce preproenkephalin mRNA expression and proenkephalin A in human peripheral blood mononuclear cells. J Neuroimmunol 79:91–99. 10.1016/S0165-5728(97)00113-6 [DOI] [PubMed] [Google Scholar]
- Kelley KW. (1988) Cross-talk between the immune and endocrine systems. J Anim Sci 66:2095–2108. 10.2527/jas1988.6682095x [DOI] [PubMed] [Google Scholar]
- Kim CF, Moalem-Taylor G (2011) Detailed characterization of neuro-immune responses following neuropathic injury in mice. Brain Res 1405:95–108. 10.1016/j.brainres.2011.06.022 [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM (1992) An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50:355–363. 10.1016/0304-3959(92)90041-9 [DOI] [PubMed] [Google Scholar]
- Kim WM, Jeong CW, Lee SH, Kim YO, Cui JH, Yoon MH (2011) The intrathecally administered kappa-2 opioid agonist GR89696 and interleukin-10 attenuate bone cancer-induced pain through synergistic interaction. Anesth Analg 113:934–940. 10.1213/ANE.0b013e318227824e [DOI] [PubMed] [Google Scholar]
- Kuwata H, Watanabe Y, Miyoshi H, Yamamoto M, Kaisho T, Takeda K, Akira S (2003) IL-10-inducible Bcl-3 negatively regulates LPS-induced TNF-alpha production in macrophages. Blood 102:4123–4129. 10.1182/blood-2003-04-1228 [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Jekich BM, Sloane EM, Mahoney JH, Langer SJ, Milligan ED, Martin D, Maier SF, Johnson KW, Leinwand LA, Chavez RA, Watkins LR (2007) Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain Behav Immun 21:686–698. 10.1016/j.bbi.2006.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li TF, Wu HY, Wang YR, Li XY, Wang YX (2017) Molecular signaling underlying bulleyaconitine A (BAA)-induced microglial expression of prodynorphin. Sci Rep 7:45056. 10.1038/srep45056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, Holloway HW, Powers K, Shen H, Egan JM, Sambamurti K, Brossi A, Lahiri DK, Mattson MP, Hoffer BJ, Wang Y, Greig NH (2009) GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci U S A 106:1285–1290. 10.1073/pnas.0806720106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chigurupati S, Holloway HW, Mughal M, Tweedie D, Bruestle DA, Mattson MP, Wang Y, Harvey BK, Ray B, Lahiri DK, Greig NH (2012) Exendin-4 ameliorates motor neuron degeneration in cellular and animal models of amyotrophic lateral sclerosis. PLoS One 7:e32008. 10.1371/journal.pone.0032008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licinio J, Wong ML (1999) The role of inflammatory mediators in the biology of major depression: central nervous system cytokines modulate the biological substrate of depressive symptoms, regulate stress-responsive systems, and contribute to neurotoxicity and neuroprotection. Mol Psychiatry 4:317–327. 10.1038/sj.mp.4000586 [DOI] [PubMed] [Google Scholar]
- Lobo-Silva D, Carriche GM, Castro AG, Roque S, Saraiva M (2016) Balancing the immune response in the brain: IL-10 and its regulation. J Neuroinflammation 13:297. 10.1186/s12974-016-0763-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JM, Gong N, Wang YC, Wang YX (2012) D-Amino acid oxidase-mediated increase in spinal hydrogen peroxide is mainly responsible for formalin-induced tonic pain. Br J Pharmacol 165:1941–1955. 10.1111/j.1476-5381.2011.01680.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie KF, Van Den Bosch MW, Naqvi S, Elcombe SE, McGuire VA, Reith AD, Blackshear PJ, Dean JL, Arthur JS (2013) MSK1 and MSK2 inhibit lipopolysaccharide-induced prostaglandin production via an interleukin-10 feedback loop. Mol Cell Biol 33:1456–1467. 10.1128/MCB.01690-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClean PL, Parthsarathy V, Faivre E, Hölscher C (2011) The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer's disease. J Neurosci 31:6587–6594. 10.1523/JNEUROSCI.0529-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Sloane EM, Langer SJ, Cruz PE, Chacur M, Spataro L, Wieseler-Frank J, Hammack SE, Maier SF, Flotte TR, Forsayeth JR, Leinwand LA, Chavez R, Watkins LR (2005) Controlling neuropathic pain by adeno-associated virus driven production of the anti-inflammatory cytokine, interleukin-10. Mol Pain 1:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oral HB, Kotenko SV, Yilmaz M, Mani O, Zumkehr J, Blaser K, Akdis CA, Akdis M (2006) Regulation of T cells and cytokines by the interleukin-10 (IL-10)-family cytokines IL-19, IL-20, IL-22, IL-24 and IL-26. Eur J Immunol 36:380–388. 10.1002/eji.200425523 [DOI] [PubMed] [Google Scholar]
- Perry T, Haughey NJ, Mattson MP, Egan JM, Greig NH (2002) Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther 302:881–888. 10.1124/jpet.102.037481 [DOI] [PubMed] [Google Scholar]
- Perry T, Lahiri DK, Sambamurti K, Chen D, Mattson MP, Egan JM, Greig NH (2003) Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (Abeta) levels and protects hippocampal neurons from death induced by Abeta and iron. J Neurosci Res 72:603–612. 10.1002/jnr.10611 [DOI] [PubMed] [Google Scholar]
- Pugazhenthi U, Velmurugan K, Tran A, Mahaffey G, Pugazhenthi S (2010) Anti-inflammatory action of exendin-4 in human islets is enhanced by phosphodiesterase inhibitors: potential therapeutic benefits in diabetic patients. Diabetologia 53:2357–2368. 10.1007/s00125-010-1849-y [DOI] [PubMed] [Google Scholar]
- Sabat R, Grütz G, Warszawska K, Kirsch S, Witte E, Wolk K, Geginat J (2010) Biology of interleukin-10. Cytokine Growth Factor Rev 21:331–344. 10.1016/j.cytogfr.2010.09.002 [DOI] [PubMed] [Google Scholar]
- Saraiva M, O'Garra A (2010) The regulation of IL-10 production by immune cells. Nat Rev Immunol 10:170–181. 10.1038/nri2711 [DOI] [PubMed] [Google Scholar]
- Sasaki A. (2017) Microglia and brain macrophages: an update. Neuropathology 37:452–464. 10.1111/neup.12354 [DOI] [PubMed] [Google Scholar]
- Scrivo R, Conigliaro P, Riccieri V, Di Franco M, Alessandri C, Spadaro A, Perricone R, Valesini G (2015) Distribution of interleukin-10 family cytokines in serum and synovial fluid of patients with inflammatory arthritis reveals different contribution to systemic and joint inflammation. Clin Exp Immunol 179:300–308. 10.1111/cei.12449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully KM, Rosenfeld MG (2002) Pituitary development: regulatory codes in mammalian organogenesis. Science 295:2231–2235. 10.1126/science.1062736 [DOI] [PubMed] [Google Scholar]
- Sheppard P, et al. (2003) IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol 4:63–68. 10.1038/ni873 [DOI] [PubMed] [Google Scholar]
- Shiraishi D, Fujiwara Y, Komohara Y, Mizuta H, Takeya M (2012) Glucagon-like peptide-1 (GLP-1) induces M2 polarization of human macrophages via STAT3 activation. Biochem Biophys Res Commun 425:304–308. 10.1016/j.bbrc.2012.07.086 [DOI] [PubMed] [Google Scholar]
- Tai YH, Tsai RY, Lin SL, Yeh CC, Wang JJ, Tao PL, Wong CS (2009) Amitriptyline suppresses neuroinflammation-dependent interleukin-10-p38 mitogen-activated protein kinase-heme oxygenase-1 signaling pathway in chronic morphine-infused rats. Anesthesiology 110:1379–1389. 10.1097/ALN.0b013e31819fccd5 [DOI] [PubMed] [Google Scholar]
- Tang Y, Le W (2016) Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol 53:1181–1194. 10.1007/s12035-014-9070-5 [DOI] [PubMed] [Google Scholar]
- Thakur V, Gonzalez M, Pennington K, Chattopadhyay M (2016) Viral vector mediated continuous expression of interleukin-10 in DRG alleviates pain in type 1 diabetic animals. Mol Cell Neurosci 72:46–53. 10.1016/j.mcn.2016.01.006 [DOI] [PubMed] [Google Scholar]
- Ul-Haq Z, Naz S, Mesaik MA (2016) Interleukin-4 receptor signaling and its binding mechanism: a therapeutic insight from inhibitors tool box. Cytokine growth Factor Rev 32:3–15. 10.1016/j.cytogfr.2016.04.002 [DOI] [PubMed] [Google Scholar]
- Vale ML, Marques JB, Moreira CA, Rocha FA, Ferreira SH, Poole S, Cunha FQ, Ribeiro RA (2003) Antinociceptive effects of interleukin-4, -10, and -13 on the writhing response in mice and zymosan-induced knee joint incapacitation in rats. J Pharmacol Exp Ther 304:102–108. 10.1124/jpet.102.038703 [DOI] [PubMed] [Google Scholar]
- Varin EM, Wojtusciszyn A, Broca C, Muller D, Ravier MA, Ceppo F, Renard E, Tanti JF, Dalle S (2016) Inhibition of the MAP3 kinase Tpl2 protects rodent and human beta-cells from apoptosis and dysfunction induced by cytokines and enhances anti-inflammatory actions of exendin-4. Cell Death Dis 7:e2065. 10.1038/cddis.2015.399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Pang CC (1993) Halothane inhibits the pressor effect of diphenyleneiodonium. Br J Pharmacol 109:1186–1191. 10.1111/j.1476-5381.1993.tb13747.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BK, Bohn E, Judd BA, Gil MP, Schreiber RD (2007) ABIN-3: a molecular basis for species divergence in interleukin-10-induced anti-inflammatory actions. Mol Cell Biol 27:4603–4616. 10.1128/MCB.00223-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Mao XF, Fan H, Wang YX (2017) p38beta MAPK signaling mediates exenatide-stimulated microglial beta-endorphin expression. Mol Pharmacol 91:451–463. 10.1124/mol.116.107102 [DOI] [PubMed] [Google Scholar]
- Xu M, Wu HY, Liu H, Gong N, Wang YR, Wang YX (2017) Morroniside, a secoiridoid glycoside from Cornus officinalis, attenuates neuropathic pain by activation of spinal glucagon-like peptide-1 receptors. Br J Pharmacol 174:580–590. 10.1111/bph.13720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S, Wasserfall CH, Parker M, Brusko TM, McGrail S, McGrail K, Moore M, Campbell-Thompson M, Schatz DA, Atkinson MA, Haller MJ (2008) Exendin-4 therapy in NOD mice with new-onset diabetes increases regulatory T cell frequency. Ann NY Acad Sci 1150:152–156. 10.1196/annals.1447.049 [DOI] [PubMed] [Google Scholar]
- Yang Z, Ming XF (2014) Functions of arginase isoforms in macrophage inflammatory responses: impact on cardiovascular diseases and metabolic disorders. Front Immunol 5:533. 10.3389/fimmu.2014.00533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JY, Gong N, Huang JL, Guo LC, Wang YX (2013) Gelsemine, a principal alkaloid from Gelsemium sempervirens Ait., exhibits potent and specific antinociception in chronic pain by acting at spinal alpha3 glycine receptors. Pain 154:2452–2462. 10.1016/j.pain.2013.07.027 [DOI] [PubMed] [Google Scholar]
- Zheng Y, Yin X, Huo F, Xiong H, Mei Z (2015) Analgesic effects and possible mechanisms of iridoid glycosides from Lamiophlomis rotata (Benth.) Kudo in rats with spared nerve injury. J Ethnopharmacol 173:204–211. 10.1016/j.jep.2015.06.045 [DOI] [PubMed] [Google Scholar]
- Zhou Z, Peng X, Hao S, Fink DJ, Mata M (2008) HSV-mediated transfer of interleukin-10 reduces inflammatory pain through modulation of membrane tumor necrosis factor alpha in spinal cord microglia. Gene Ther 15:183–190. 10.1038/sj.gt.3303054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Gong N, Fan H, Peng CS, Ding XJ, Jiang Y, Wang YX (2014) Lamiophlomis rotata, an orally available Tibetan herbal painkiller, specifically reduces pain hypersensitivity states through the activation of spinal glucagon-like peptide-1 receptors. Anesthesiology 121:835–851. 10.1097/ALN.0000000000000320 [DOI] [PubMed] [Google Scholar]