

Abstract
Kv2.1 is a major delayed-rectifier voltage-gated potassium channel widely expressed in neurons of the CNS. Kv2.1 localizes in high-density cell-surface clusters in the soma and proximal dendrites as well as in the axon initial segment (AIS). Given the crucial roles of both of these compartments in integrating signal input and then generating output, this localization of Kv2.1 is ideal for regulating the overall excitability of neurons. Here we used fluorescence recovery after photobleaching imaging, mutagenesis, and pharmacological interventions to investigate the molecular mechanisms that control the localization of Kv2.1 in these two different membrane compartments in cultured rat hippocampal neurons of mixed sex. Our data uncover a unique ability of Kv2.1 channels to use two molecularly distinct trafficking pathways to accomplish this. Somatodendritic Kv2.1 channels are targeted by the conventional secretory pathway, whereas axonal Kv2.1 channels are targeted by a nonconventional trafficking pathway independent of the Golgi apparatus. We further identified a new AIS trafficking motif in the C-terminus of Kv2.1, and show that putative phosphorylation sites in this region are critical for the restricted and clustered localization in the AIS. These results indicate that neurons can regulate the expression and clustering of Kv2.1 in different membrane domains independently by using two distinct localization mechanisms, which would allow neurons to precisely control local membrane excitability.
SIGNIFICANCE STATEMENT Our study uncovered a novel mechanism that targets the Kv2.1 voltage-gated potassium channel to two distinct trafficking pathways and two distinct subcellular destinations: the somatodendritic plasma membrane and that of the axon initial segment. We also identified a distinct motif, including putative phosphorylation sites, that is important for the AIS localization. This raises the possibility that the destination of a channel protein can be dynamically regulated via changes in post-translational modification, which would impact the excitability of specific membrane compartments.
Keywords: excitability, phosphorylation, potassium channel, sorting, targeting
Introduction
The axon initial segment (AIS) is an essential neuronal subdomain controlling action potential generation and the maintenance of neuronal polarity (Buffington and Rasband, 2011). These physiological and biological functions are accomplished by a large assembly of diverse molecules that localize specifically to the AIS upon development of a neuronal cell. Two classes of voltage-gated ion channels, voltage-gated sodium (Nav) and potassium (Kv) channels, are highly enriched in the AIS plasma membrane where they regulate the firing of action potentials. The significance of the clustering of these ion channels at specific sites in the AIS is underscored by the fact that disruption of their localization by mutation, disease, or injury has a major impact on neuronal excitability. For instance, mutations in the genes encoding AIS-specific Kv7 channels (Pan et al., 2006; Rasmussen et al., 2007) have been found in newborns with the epileptic disorder benign familial neonatal convulsions (Lerche et al., 1999). Also, alterations in the localization of AIS ion channels occur in epileptic (Harty et al., 2013) and injured (Schafer et al., 2009) brain, and in response to stroke (Hinman et al., 2013).
The Kv2.1 delayed rectifier channel is a Kv channel isoform highly expressed in the AIS (Sarmiere et al., 2008; King et al., 2014), and it has recently been shown that de novo mutations in the KCNB1 gene encoding Kv2.1 are associated with congenital encephalopathic epilepsy (Torkamani et al., 2014; Saitsu et al., 2015; Thiffault et al., 2015). Furthermore, a recent study implied that certain polymorphisms KCNB1 comprise a major risk factor of schizophrenia (Peltola et al., 2016). Kv2.1 is unique in that it is expressed in both the somatodendritic membrane (Trimmer, 1991; Scannevin et al., 1996; Du et al., 1998) and in the AIS (Sarmiere et al., 2008; King et al., 2014), contrasting the general tendency of voltage-gated ion channels to exhibit specific localization in either the somatodendritic or axonal membranes (Jensen et al., 2011; Trimmer, 2015). Because Kv2.1 is localized in both the proximal dendrites and soma, where synaptic potentials are integrated, and at the AIS, precise regulation of the localization of Kv2.1 at these sites likely contributes to its role in controlling overall neuronal excitability (Misonou, 2010). In fact, Kv2.1 has been shown to impact the firing frequencies in a number neuronal cell types including sympathetic neurons (Malin and Nerbonne, 2000), neocortical neurons (Guan et al., 2013; Pathak et al., 2016), substantia nigra dopaminergic neurons (Kimm et al., 2015), superior cervical ganglion cells, and hippocampal pyramidal neurons (Liu and Bean, 2014), and Kv2.1 knock-out mice exhibit a hyperexcitable phenotype (Speca et al., 2014). However, the specific role of Kv2.1 in the AIS is not fully understood, primarily due to the difficulty of isolating the function of Kv2.1 in the AIS from that in the adjacent somatodendritic membrane.
The somewhat atypical dual localization of Kv2.1 raises questions as to the molecular mechanisms underlying the ability of the neuron to populate these distinct polarized compartments with Kv2.1. Here, we investigate the determinants on Kv2.1 responsible for these distinct subcellular localizations, and the different trafficking pathways underlying the targeting to these sites. Previous studies have identified a peptide motif (PRC motif) in the cytoplasmic C-terminus of Kv2.1 that determines the restricted and clustered localization on the soma and proximal dendrites (Lim et al., 2000; Jensen et al., 2014). Here we demonstrate that upon disruption of this motif, Kv2.1 channel clustering in the AIS is still apparent, suggesting that the distinct localizations in the somatodendritic and AIS membranes are differentially regulated, presumably by different mechanisms. Using mutagenesis, quantitative live-cell imaging, and pharmacological interventions, we provide evidence for a previously unappreciated ability of neurons to sort a specific ion channel protein into two separate independent trafficking pathways with distinct polarity, resulting in different compartment-specific localizations.
Materials and Methods
Hippocampal neuronal culture.
Primary dissociated rat hippocampal neurons from embryos of either sex were plated on 25 mm round coverslips and grown in coculture with astroglial cells as described in detail previously (Jensen and Misonou, 2015). All animal use was approved by the institutional animal care and use committee. Neurons were transiently transfected at 8–10 d in vitro (DIV) using Lipofectamine 2000 transfection reagent (ThermoFisher Scientific). Neurons were transfected in separate dishes with 0.5 μg cDNA (mixed with 0.4 μg noncoding vector DNA) per coverslip.
Molecular biology.
GFP-Kv2.1 constructs were made from cloning of the 853 aa rat Kv2.1 (Frech et al., 1989; NM_013186) into pEGFP-C1 (Clontech) using EcoRI and SalI restriction sites. The previously described Kv2.1 S586A point mutant (Lim et al., 2000) was subcloned into pEFGP-C1. The GFP-Kv2.1 C-terminal deletion constructs were generated from the GFP-Kv2.1 wild-type (WT) plasmid by introduction of a stop codon using QuikChange mutagenesis (Agilent). SEP-Kv2.1 WT was made by inserting the super-ecliptic pHluorin (SEP) into the first extracellular (S1–S2) loop of Kv2.1 using standard overlap PCR. From the SEP-Kv2.1 WT plasmid, deletion constructs (see Fig. 8) and the T728A/S732A mutant were generated by QuikChange mutagenesis with primers designed to eliminate different regions of the Kv2.1 C-terminus. The GFP Kv4.2/Kv2.1 chimera was constructed by overhang PCR to generate Kv4.2 (NM_031730) aa 1-477/Kv2.1 (NM_013186) aa 535–853. HaloTag (Los et al., 2008) was amplified from pFN21K (Promega) using PCR, and cloned into ApaI and SalI site of pDisplay (Invitrogen) to generate Halo-platelet-derived growth factor receptor (PDGFR). Halo-tagged Kv2.1 (Halo-Kv2.1) was generated by inserting the Halo tag into the first extracellular loop. Both were then cloned into a tetracyclin-inducible expression vector based on the pTet-On vector (Clontech).
Figure 8.

Critical role of conserved putative phosphorylation sites in the clustered localization of Kv2.1 in the AIS. A, Sequence alignment of the AIS motif across different species. Vertical lines between sequences indicate location of identical amino acid residues, and colons indicate similar residues. Two putative phosphorylation sites (T728, S732) in the motif are colored in red. B, Axonal localization of Kv2.1 Δ583–599 lacking the core sequence of the PRC motif. Neurons were immunostained for surface SEP and ankyrin G. Scale bar, 10 μm. C, Uniform localization of Kv2.1 Δ583–599 with T728A and S732A mutations in the axon. Scale bar, 10 μm. D, Quantification of CV of fluorescence intensity in the AIS. The difference between the values from transfected with Kv2.1 Δ583–599 or Kv2.1 Δ583–599 T728A/S732A was statistically significant (*t(16) = 7.839, p < 0.0001) using Student's t test. E, Line-scan analysis of the fluorescence signals in the neurons shown in B and C for Ankyrin G (light gray line) and Kv2.1 (black line). Segmented lines were drawn over the somatic membrane and into the axon.
Immunofluorescence labeling.
Neurons were fixed in 4% paraformaldehyde/PBS for 20 min. Blocking and permeabilization was done in 0.2% fish skin gelatin/0.1% Triton X-100/TBS. Primary and secondary antibodies were diluted in the same buffer and applied in separate incubation steps of 1 h and 45 min, respectively. Coverslips were mounted on glass microscope slides using ProLong Gold anti-fade reagent (ThermoFisher Scientific).
Brefeldin A experiments.
Neurons at 9 DIV were transfected with the Halo-PDGFR or Halo-Kv2.1 vector using Lipofectamine 2000. After a 4 d incubation, transfected neurons (at 13 DIV) were pretreated with 0.1 μg/ml Brefeldin A (BFA; Nacalai Tesque) for 2 h at 37°C. Doxycycline (Sigma-Aldrich) was added to the final concentration of 1 μg/ml, and then incubated for 16 h at 37°C in the presence or absence of BFA. Cells were labeled with membrane-impermeable AlexaFluor 488-conjugated HaloTag ligand (Promega) at 500 nm for 15 min at 37°C, washed with fresh culture medium, and then labeled with membrane-permeable tetramethylrhodamine-conjugated HaloTag ligand (Promega) at 500 nm for 15 min at 37°C. Cells were fixed as for the immunofluorescence labeling (without permeabilization).
Antibodies.
All Kv2.1 constructs tagged with GFP or SEP were immunolabeled with an anti-GFP rabbit polyclonal antibody (ThermoFisher Scientific, A-11122, AB_2576216; 2.5 μg/ml). The AIS was identified by ankyrin G immunolabeling using an anti-ankyrin G mouse monoclonal IgG2b antibody (NeuroMab, clone N106/65 supernatant, RRID:AB_10675130; 1:5 dilution). The monoclonal antibody N106/65 was developed by and/or obtained from the UC Davis/NIH NeuroMab Facility (supported by NIH Grant U24NS050606 and maintained by the Department of Neurobiology, Physiology, and Behavior, College of Biological Sciences, University of California, Davis, CA). The somatodendritic compartment was identified by immunolabeling for microtubule-associated protein-2 (MAP2) with an anti-MAP2 rabbit polyclonal antibody (Santa Cruz Biotechnology, H-300, RRID:AB_2250101; 0.25 μg/ml). All secondary antibodies were AlexaFluor 488-, 555-, or 647-conjugated secondary antibodies used at 2 μg/ml (ThermoFisher Scientific).
Imaging.
The steady-state localization of WT and mutant Kv2.1 isoforms were documented using a Olympus IX73 microscope with a 60×/1.42 NA objective lens and Andor Zyla5.5 camera, a Carl Zeiss inverted laser scanning microscope (LSM 780) with a 63×/1.4 NA lens, or a Carl Zeiss ApoTome system with a 63×/1.4 NA lens. Line-scan analysis of fluorescence intensities was performed using ImageJ. Temperature-block experiments were performed as previously reported (Jensen and Misonou, 2015).
Fluorescence recovery after photobleaching.
Neurons expressing GFP-Kv2.1 S586A were imaged using an Olympus FV-1000 microscope equipped with a 450 nm diode laser. Images were taken every 5 s. Photobleaching was induced by applying the 450 nm laser to a circular spot of a fixed diameter (5 μm) at 100% power for 200 ms. Fluorescence intensity was measured using ImageJ, corrected for background fluorescence and the overall bleaching due to imaging, and normalized for the maximum and minimum fluorescence intensities.
Electrophysiology.
Human embryonic kidney (HEK) 293T cells were transiently transfected with the SEP-tagged Kv2.1 WT or deletion constructs, using Lipofectamine2000 (ThermoFisher Scientific). The constructs were evaluated with the whole-cell patch-clamp technique, 24 h post-transfection, as previously described (David et al., 2015). HEK293T cells were continuously perfused with extracellular solution containing the following (in mm): 145 NaCl, 4 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, and 10 glucose, with the pH adjusted to 7.35 with NaOH. The intracellular solution contained the following (in mm): 110 KCl, 5 K2ATP, 5 K4BAPTA, 2 MgCl2, and 10 HEPES, with the pH adjusted to 7.2 with KOH.
Experimental design and statistical analysis.
All statistical analyses were performed on GraphPad Prism software. Power analysis was performed using G*Power (Faul et al., 2007, 2009) with parameters taken from previous reports or similar experiments.
To quantify the degree of clustering at the AIS, we measured the coefficient of variation (CV) of fluorescence signals (Bishop et al., 2015) in the first 20 μm of the axon in transfected neurons (4 cells each from 2 independent cultures) using ImageJ. Axons were identified by its morphology with the cell-fill mKate2 or as MAP2-negative neurites. For statistical comparison of the mean CV, we designed to use either Student's t test for two groups or one-way ANOVA with Sidak post hoc test for three or more groups.
Fluorescence recovery after photobleaching (FRAP) data were obtained from neurons expressing Kv2.1 S586A in the soma (8 cells) and AIS (10 cells from 4 independent cultures), background subtracted, compensated for bleaching from imaging, and normalized to make the minimum and maximum 0 and 1, respectively. The recovery phase from the minimum were fitted with the one-phase association model [Y= Y0 + (Plateau - Y0) × (1 − 10−k × X)]. The difference between the fitted curves in the soma and AIS were analyzed with F test for the null-hypothesis that a curve with shared parameters (slope k and Plateau) fits better than those with different parameters for each dataset. To test whether the recovery in the AIS is negligible, the individual datasets were also fitted with the linear function to obtain the mean slope. This was compared with the mean slope obtained from simulated random values in the same range as the real data using Student's t test.
To quantify the peak fluorescence intensity in neurons treated without or with BFA (5 cells each from 3 independent cultures), we first identify the axon in images, of which signals were intensified to reveal the entire morphology. Then, we performed the line-scan analysis over the axon, the soma, and a dendrite to measure signal intensity. The values were normalized to either that in the AIS or in the soma. The mean values were compared using Student's t test.
To identify the motif involved in AIS localization, 24–33 transfected neurons from ≥3 independent cultures were analyzed per deletion condition. We distinguished specific axonal clustering from a rather nonspecific clustering by the criterion that neurons should have at least four clusters in the AIS that were readily identifiable. From this criterion, we calculated the percentage of the transfected neurons with Kv2.1 AIS clusters. We designed the results to be analyzed with one-way ANOVA with Sidak post hoc test.
Results
Different molecular mechanisms traffic Kv2.1 to axonal and somatodendritic membranes
The somatodendritic localization and clustering of Kv2.1 is determined by the PRC motif (573–598 aa) within the cytoplasmic C-terminal tail (411–853 aa) of Kv2.1 (Lim et al., 2000). Point mutations in four critical residues within the PRC motif, such as S586A, were found to completely disrupt the restricted localization in proximal dendrites, as well as the clustering of Kv2.1, although they are still efficiently expressed on the surface (Lim et al., 2000). We confirmed these findings in neurons expressing GFP-tagged WT and mutant Kv2.1 isoforms, as shown in Figure 1A. In neurons expressing GFP-tagged WT Kv2.1, line-scan analysis of fluorescence intensity revealed somatodendritic and axonal clusters as discrete peaks (Fig. 1C). These peaks were absent in the somata of neurons expressing the GFP-tagged Kv2.1 S586A mutant. However, GFP-Kv2.1 S586A still exhibited intact AIS localization, as defined by overlap with immunolabeling for ankyrin G (Fig. 1B). We also evaluated the degree of AIS clustering by quantifying the CV of fluorescence intensity as a measure of the extent of nonuniformity of the fluorescent signal (Bishop et al., 2015). Figure 1D shows the intact clustering of Kv2.1 S586A in the AIS, as evidenced by the CV of fluorescence intensity comparable to that of WT (Fig. 1C,D), suggesting that AIS localization is determined by a peptide motif(s) outside of the PRC motif. Interestingly, we also found that truncation of the Kv2.1 C-terminus from aa 536–853 (GFP-Kv2.1 ΔC318) resulted in disruption of AIS clustering, as evidenced by the significantly decreased CV of AIS fluorescence signals (Fig. 1D). These results indicate that two different trafficking mechanisms, possibly dictated by different C-terminal peptide motifs, underlie the distinct localization of Kv2.1 channels in the somatodendritic and axonal compartment.
Figure 1.

Distinct molecular mechanisms underlie the somatodendritic and AIS localizations of Kv2.1. A, Localization of GFP-tagged WT Kv2.1, the Kv2.1 S586A point mutant, and the Kv2.1 ΔC318 deletion mutant, lacking the last 318 aa of Kv2.1 (536–853) in cultured hippocampal neurons at 14 DIV. Arrowheads indicate the AIS. Scale bar, 20 μm. B, Colocalization of Kv2.1 S586A with endogenous ankyrin G. C, Line-scan analysis of the fluorescence signals in the neurons shown in A. Segmented lines were drawn over the somatic membrane and into the axon. The border between these two regions was defined by ankyrin G immunolabeling and is indicated by a gray line. D, Quantification of CV of fluorescence intensity in the AIS. Data are the mean ± SEM. *F(2,9) = 6.405, p = 0.0186, ANOVA.
Inefficient lateral diffusion of Kv2.1 from the somatic membrane to the AIS
Polarized localization of membrane proteins such as ion channels and receptors can be achieved by different neuronal targeting mechanisms (Jensen et al., 2011). To understand how Kv2.1 is trafficked and localized in the AIS, we first performed FRAP experiments to probe lateral diffusion. We considered two possible scenarios for the trafficking of Kv2.1 to the AIS. Either Kv2.1 obtains its AIS localization by (1) lateral diffusion from the somatic membrane to the AIS, or (2) intracellular vesicular trafficking to the AIS membrane. To test whether lateral diffusion occurs between somatic and axonal membrane, we performed FRAP experiments with Kv2.1 S586A. Unlike the relatively immobile Kv2.1 WT in the somatic membrane (O'Connell and Tamkun, 2005), we assumed that this mutant could be used to probe lateral membrane diffusion between these two compartments. The first scenario would predict that fluorescence recovery occurs within the AIS for Kv2.1 S586A by recruiting unbleached molecules from the adjacent somatic membrane, whereas the second scenario predicts no recovery in the AIS adjacent to the soma.
To test this, GFP was bleached in two adjacent areas, one in the soma close to the AIS and the other at the edge of Kv2.1 AIS clusters (Fig. 2A). As expected from its uniform localization in somata, Kv2.1 S586A in the somatic membrane exhibited robust recovery after photobleaching (Fig. 2B). In contrast, there was virtually no recovery in the AIS (Fig. 2B). Regression analysis with the exponential one-phase association model suggested that the recovery curves for the soma and AIS are significantly different (F(2,835) = 88.3, p < 0.0001, F test). When fitted with the linear function, the slope of the AIS recovery (0.0000265 ± 0.0000039) was not statistically different (t(7) = 1.571, p = 0.1602, t test) from that of simulated random signals (0.0000063 ± 0.0000109), which is virtually zero.
Figure 2.

Restricted diffusion of Kv2.1 S586A in the AIS. A, Neurons expressing GFP-Kv2.1 S586A were imaged live and subjected to FRAP. FRAP laser was applied to the AIS and nearby somatic membrane as indicated by gray circles. Scale bar, 5 μm. B, FRAP of GFP-Kv2.1 S586A. The signals were fit with a curve defined by a single exponential. The data are the mean ± SEM from 8 (soma) or 10 (AIS) cells from four independent cultures. C, Neurons were subjected to simultaneous FRAP in the AIS and in the somatic membrane, at the sites indicated by the arrow and arrowhead, respectively. Images of a representative cell are shown before bleaching, immediately after bleaching (Bleach), and 100 s after bleaching. Scale bar, 10 μm. D, Line-scan analysis of fluorescence intensity before and after photobleaching. A line was drawn over the two bleached areas. The arrow and arrowhead indicate the locations of the AIS and somatic bleached areas, respectively. The dotted line denotes the level of recovery in the somatic membrane FRAP site 100 s after bleaching. Note that there was no detectable recovery in the AIS.
This is not due to the large difference in fluorescence intensities of GFP signals in these membrane domains, since there was no recovery even to the level of somatic GFP signals in the AIS (Fig. 2C,D). Simultaneous FRAP in the soma and AIS (Fig. 2C) showed that a negligible level of recovery (from 49% of the somatic level after photobleaching to 50% at 100 s) occurred in the AIS during the same time period as it was occurring rapidly in the somatic membrane (38–69%). These results indicate that there is no significant lateral diffusion of GFP-Kv2.1 S586A from the somatic membrane to the AIS, and suggest that the AIS localization of Kv2.1 is established at the level of intracellular vesicular trafficking.
A unique nonconventional trafficking route for AIS-localized Kv2.1
We have previously shown, using a temperature-block and release protocol, that Kv2.1 destined for somatodendritic localization is sorted into a specific population of transport vesicles in the Golgi apparatus; a population distinct from those carrying proteins destined for distal dendrites (Jensen et al., 2014). In this protocol, GFP-Kv2.1 transfected neurons (3 h post-transfection) were subjected to a 19.5°C temperature block, which leads to trapping of most of the newly synthesized GFP-Kv2.1 in the Golgi apparatus (Jensen and Misonou, 2015), as shown here in Figure 3A. However, we found that a subpopulation of GFP-Kv2.1 escaped from this temperature-block and formed clusters in the AIS (Fig. 3A). These distinct Kv2.1 populations were also evident when neurons were cotransfected with the ARF1 I71Q mutant (Dascher and Balch, 1994), which blocks the formation of transport vesicles at the Golgi apparatus. Expression of this mutant blocked the expression of GFP-Kv2.1 in the somatodendritic membrane, but a subpopulation of GFP-Kv2.1 was still able to traffic to and from clusters in the AIS (Fig. 3A). From these results, we hypothesized that Kv2.1 destined for the AIS bypasses the Golgi apparatus (Fig. 3B), a noncanonical trafficking pathway that has been implicated for a small subset of other transmembrane proteins (Grieve and Rabouille, 2011).
Figure 3.
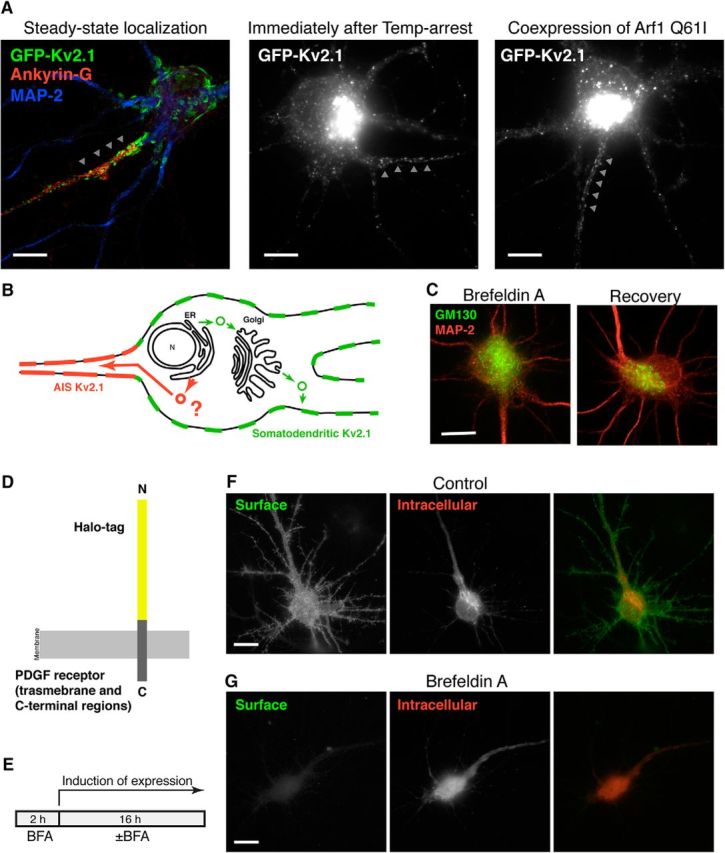
Golgi-independent trafficking pathway of Kv2.1 to the AIS. A, Localization and clustering of GFP-Kv2.1 in the AIS when the secretory trafficking pathway is inhibited. Left, The steady-state localization of GFP-Kv2.1, particularly in the Ankyrin G-positive AIS indicated by arrowheads. Middle, The neuron was transfected with GFP-Kv2.1 for 3 h and then subjected to temperature-block at 19.5°C for 3 h. This procedure trapped most of GFP-Kv2.1 in the perinuclear Golgi apparatus in the soma and prevented its expression in somatodendritic clusters. GFP-Kv2.1 can still be found clustered in the AIS. Right, The ARF1 I71Q mutant, which inhibits the formation of transport vesicles at the Golgi apparatus, was coexpressed with GFP-Kv2.1 for 3 h. GFP-Kv2.1 was still detected in the AIS. Scale bars, 10 μm. B, Schematic diagram of the model whereby Kv2.1 destined to the AIS takes an alternative intracellular trafficking route bypassing the Golgi apparatus. C, Effect of BFA on the Golgi apparatus in neurons. Neurons were incubated for 2 h with BFA at 0.1 μg/ml and immediately fixed, or fixed following a 16 h recovery period without BFA, and immunolabeled for MAP-2 (red) and a Golgi marker, GM130 (green), to assess the neuronal morphology and the structure of the Golgi apparatus, respectively. Scale bar, 10 μm. D, Diagram of the Halo-PDGFR reporter construct used to monitor secretory pathway trafficking in neurons. HaloTag and PDGFR regions are not shown to scale. E, Schematic of the BFA experiments. Neurons were preincubated with 0.1 μg/ml BFA for 2 h, and then incubated with 1 μg/ml doxycycline for 16 h in the absence (Control) or presence of BFA. F, Trafficking of Halo-PDGFR in control as reported by membrane impermeant (green) and membrane permeant (red) HaloTag ligands. Scale bar, 10 μm. G, Effect of BFA on the trafficking of Halo-PDGFR. The images in E and F were captured using the same exposure time for each fluorophore. Scale bar, 10 μm.
Because Kv2.1 is not subjected to N-linked glycosylation (Shi and Trimmer, 1999), processing of a glycan chain cannot be used as readout of Kv2.1 trafficking though ER and the Golgi apparatus. Instead, we used pharmacological block of the Golgi-dependent trafficking pathway using BFA (Helms and Rothman, 1992). This fungal toxin has been widely used to disrupt the Golgi apparatus, because of its ability to suppress ARF1 activity by inhibiting GTP-exchange factors. We verified that BFA (0.1 μg/ml) exhibited the expected (Wisco et al., 2003; Leterrier et al., 2006) disruption of the neuronal Golgi apparatus within 2 h (Fig. 3C). The disruption was reversible, such that neurons recovered normal Golgi structures 16 h after BFA washout (Fig. 3C). We then used an in-house generated probe, Halo-PDGFR, which allows us to monitor the conventional secretory pathway (Fig. 3D). Halo-PDGFR comprises an N-terminal extracellular domain HaloTag fused to the transmembrane region and the C-terminal tail of the PDGFR. The 34 kDa HaloTag (Los et al., 2008) is fused with the leader sequence of IgK, which directs the probe into the secretory pathway (Coloma et al., 1992). Therefore, Halo-PDGFR should be trafficked through the secretory pathway and then expressed in the plasma membrane, as detected using a membrane-impermeable HaloTag ligand. The intracellular Halo-PDGFR pool could subsequently be labeled using a membrane-permeable HaloTag ligand. Halo-PDGFR was constructed in a doxycycline-inducible plasmid vector to precisely control the timing of expression. To ensure that the Golgi apparatus was fully disrupted before the expression of Halo-PDGFR, we preincubated neurons with BFA for 2 h and then induced the expression of the probe in the absence (Control) or the presence of BFA for 16 h (Fig. 3E). In control neurons, Halo-PDGFR was detected on the cell surface, as well as in perinuclear structures, presumably the Golgi apparatus (Fig. 3F), indicating that the Golgi apparatus restores its function during the recovery period. However, in the presence of BFA, there was no detectable Halo-PDGFR on the cell surface, whereas the intracellular pool was readily detected (Fig. 3F). These results suggest that in our hands BFA treatment blocks the neuronal secretory pathway.
Next, we tested whether Kv2.1 can traffic to the AIS in the presence of BFA. We again used the HaloTag, but this time inserted into the first extracellular (S1–S2) loop of Kv2.1 to generate Halo-Kv2.1 (Fig. 4A). We verified that under control conditions Halo-Kv2.1 is expressed on the cell surface of neurons in large discrete clusters, similar to WT Kv2.1 (Fig. 4B). BFA greatly reduced the level of cell surface Halo-Kv2.1 in the somatodendritic region (Fig. 4C). However, the cell-surface expression of Halo-Kv2.1 in the AIS remained relatively intact in BFA-treated neurons (Fig. 4C). As shown in the line scans in Figure 4D, although somatodendritic and AIS surface clusters of Halo-Kv2.1 were present in control cells, in neurons treated with BFA clusters were only apparent in the AIS. The expression level of Halo-Kv2.1 in the somatodendritic region relative to that in the AIS was significantly reduced in BFA-treated cells (t(6) = 12.86, p < 0.0001, t test; Fig. 4E). Also, the degree of clustering in BFA-treated neurons, as quantified by the CV of fluorescence intensity, was comparable to that in control neurons (t(6) = 1.613, p = 0.1578, t test; Fig. 4E). Together with the results from the FRAP experiments with Kv2.1 S586A, and from the temperature-arrest experiments, these results collectively suggest that Kv2.1 channels that are localized in the AIS can traffic independently of somatodendritic Kv2.1 channels, through a Golgi-independent pathway used for AIS but not somatodendritic Kv2.1.
Figure 4.

Effect of inhibiting the secretory pathway on the expression of Kv2.1 in the AIS. A, Diagram of the Halo-Kv2.1 reporter construct used to monitor Kv2.1 trafficking in neurons. HaloTag and Kv2.1 are not shown to scale. B, Surface expression of Halo-Kv2.1 in the absence of BFA, as reported by membrane impermeant (green) and membrane-permeant (red) HaloTag ligands. Arrowheads indicate the AIS, identified by the morphology. Scale bar, 10 μm. C, Effect of BFA on the trafficking of Halo-Kv2.1. The images in B and C were captured using the same exposure time for each fluorophore. Scale bar, 10 μm. D, Line-scan analysis of the fluorescence signals in the neurons shown in B and C. Segmented lines were drawn over the somatic membrane and into the axon. The border between these two regions is indicated by a gray line. E, Quantification of the relative peak fluorescence intensity in the somatodendritic region (right bars and y-axis) and CV of fluorescence intensity in the AIS (left bars and y-axis). The relative somatodendritic intensity was significantly reduced in BFA-treated neurons (*t(6) = 12.86, p < 0.0001, t test). However, the CV values from control and BFA-treated cells were not significantly different (n.s.) using Student's t test (t(6) = 1.613, p = 0.16). Data are the mean ± SEM.
Identification of the peptide motif for the AIS localization of Kv2.1
The data showing the distinct AIS localization of the Kv2.1 S586A and ΔC318 mutants indicated that the localization and clustering of Kv2.1 in the AIS is determined by a region in the cytoplasmic C-terminal tail outside of the PRC motif. To test whether the C-terminal tail of Kv2.1 is sufficient for the AIS localization, we generated a chimeric protein (Kv4.2-Kv2.1C), with another Kv channel, Kv4.2, which has a subcellular localization in neurons distinct from Kv2.1 (Fig. 5B; Jensen et al., 2014). Kv4.2-Kv2.1C has the cytoplasmic N-terminus, transmembrane segments and proximal C-terminus of Kv4.2 followed by the region of the Kv2.1 C-terminus deleted in the ΔC318 mutant (536–853aa). The Kv4.2-Kv2.1C chimera exhibited a Kv2.1-like subcellular localization, including AIS localization and clustering, further supporting the notion that the determinant for AIS localization is located within the C-terminal tail of Kv2.1 (Fig. 5A).
Figure 5.

Determinants for AIS localization and clustering in the cytoplasmic C-terminal tail of Kv2.1. A, Localization of a chimeric Kv subunit containing the cytoplasmic N-terminal tail, membrane spanning regions and proximal C-terminus of Kv4.2, and a cytoplasmic C-terminal tail fragment (aa 536–853) of Kv2.1. This GFP-tagged chimera exhibited a subcellular localization typical of WT Kv2.1 (Fig. 4A) including clustering in the AIS (arrowheads). Scale bar, 10 μm. B, Localization of GFP-Kv4.2 in the same culture as A. KChIP2 was also exogenously expressed in neurons in A and B. Scale bar, 10 μm. C, Diagram of the reporter construct generated to report the surface expression of Kv2.1 and its deletion mutants, with a cytoplasmic mCherry tag, and SEP inserted in the first extracellular loop of Kv2.1. D, SEP fluorescence in neurons transfected for 16 h. Fluorescence signals are intensified to show the overall distribution of SEP-Kv2.1 and the morphology of the neuron. Scale bar, 20 μm. E, Same neuron as in C. The images were taken in the soma in a single z-plane where more centrally located intracellular signals can be separated from the plasma membrane-delimited signals. At this early time point after transfection, a large portion of SEP-Kv2.1 is intracellular, as evidenced by mCherry signals in the absence of SEP fluorescence in the more central regions of the soma, and SEP fluorescence specific to the peripheral edge of the soma. Scale bar, 10 μm. F, Line-scan analysis of the images in E. G, Functional channels formed by SEP-Kv2.1. Whole-cell patch-clamp recordings from HEK293T cells expressing either GFP-Kv2.1 or SEP-Kv2.1. Representative whole-cell currents from cells in which the membrane potential was held at −80 mV and increased to +80 mV in 10 mV increments for 200 ms to evaluate activation, followed by a step to +30 mV to evaluate deactivation, of Kv2.1 channels. H, Cell-surface expression of SEP-Kv2.1 in the AIS membrane. Neurons transfected with SEP-Kv2.1 were immunolabeled with an anti-GFP antibody, which recognizes SEP, without permeabilization, followed by permeabilization and immunolabeling for Ankyrin G. Scale bar, 5 μm.
To identify the putative AIS localization motif, we generated a series of internal deletion mutants that lacked parts of the cytoplasmic C-terminal tail. We generated these mutants in a SEP-Kv2.1 construct, in which SEP was inserted into the first extracellular (S1–S2) loop of Kv2.1, and mCherry fused to the cytoplasmic N-terminus (Fig. 5C). This construct allowed us to distinguish Kv2.1 WT and mutant Kv2.1 isoforms delivered to the cell surface by either the intrinsic pH-sensitive fluorescence of SEP (Sankaranarayanan et al., 2000) or through cell-surface immunolabeling of SEP. Figure 5D–F shows a neuron expressing SEP-Kv2.1 WT in which the extracellular SEP fluorescence reveals cell-surface localized Kv2.1, whereas pools positive for mCherry fluorescence and negative for SEP represent intracellular Kv2.1 channels. Patch-clamp analysis showed that the SEP-Kv2.1 channel generated robust outward currents when heterologously expressed in HEK293T cells, similar to those obtained from expression of GFP-Kv2.1, which carries GFP on the N-terminal cytoplasmic domain. The macroscopic voltage-dependent activation, and the activation and deactivation kinetics of SEP-Kv2.1 were similar to GFP-Kv2.1 (Fig. 5G), suggesting that SEP-Kv2.1 is efficiently expressed at the cell surface and forms functional channels. Last, we confirmed the cell-surface expression of SEP-Kv2.1 in the AIS (Fig. 5H).
We then tested a series of C-terminal deletion mutants generated in the SEP-Kv2.1 background for their localization and clustering in the AIS. Because deletion of the bulk of the 443 aa C-terminal tail (aa 411–853) and the ΔC318 mutant (deletion of aa 536–853) disrupted the AIS localization of Kv2.1 (Fig. 1A), we dissected this region of the C-terminus by a series of internal deletion mutants. We found that a large deletion of SEP-Kv2.1 from aa 521 to 745 significantly disrupted AIS localization (Fig. 6A,B). Because a point mutation (S586A) in the PRC motif itself (aa 573–598) did not affect AIS localization (Fig. 1A), we speculated that an internal deletion (Δ601–745) removing the Kv2.1 region from just beyond the PRC domain to aa 745 would exhibit a loss of clustering phenotype similar to the Δ521–745 mutant. Surprisingly, this mutant exhibited normal AIS localization, as well as somatodendritic clustering typical of WT Kv2.1, as expected (Lim et al., 2000) for any internal deletion and truncation mutants containing the PRC domain (Fig. 6A,B). We therefore hypothesized that the region containing the PRC motif acts in conjunction with an additional downstream motif to establish the AIS localization of Kv2.1. As the Δ521–745 mutant showed a slight, but significant reduction of AIS clustering, and because the Δ521–720 mutant retained the robust AIS localization typical of WT Kv2.1, we concluded that a motif located within aa 720–745 is important to establish the AIS localization of Kv2.1. To address this possibility, we generated a double internal deletion mutant lacking both the bulk of the PRC motif (aa 583–599) as well as aa 720–745. This double-deletion mutant exhibited uniform cel-surface localization throughout the axon, but was not clustered in the AIS (Fig. 6A–C). Consistent with the SEP fluorescence signal that suggested robust surface expression of the double-deletion mutant, expression of this mutant in heterologous HEK293T cells yielded functional channels (Fig. 6D). Together, these results reveal that a novel motif in the C-terminus of Kv2.1, downstream of the previously characterized PRC motif, is involved in the localization and clustering of Kv2.1 in the AIS but not the somatodendritic compartment.
Figure 6.

Identification of the peptide motif important for the AIS localization of Kv2.1. A, Schematic diagrams on the left illustrate the internal deletion mutants used in the study. The PRC motif is indicated by the black circle. Note that the sizes of deletions are not to scale in the diagrams. Neurons were transfected with wild-type Kv2.1 and these deletion mutants and immunolabeled for SEP without permeabilization, and scored for AIS clustering. The overall difference was statistically significant (F(6,179) = 49.19, p < 0.0001, ANOVA) with multiple comparisons (*p < 0.05, Sidak post hoc tests). B, Representative images of neurons expressing Kv2.1 Δ521–745, Δ521–720, Δ601–745, and the double PRC and AIS motif mutant, as indicated. Arrowheads denote the AIS as defined by Ankyrin G immunolabeling (data not shown). Scale bar, 5 μm. C, Line-scan analysis of the images shown in B. Gray bars indicate the border between the soma and the axon. D, Whole-cell patch-clamp recordings from HEK293T cells expressing the double PRC and AIS motif mutant, using the protocol described in Figure 5G (legend).
We then tested whether the double mutant fails to enter the Golgi-independent trafficking pathway to the AIS and rather takes the default secretory pathway to the surface. We generated a double deletion construct with the Halo-tag inserted in the first extracellular loop of the channel subunit as done for the WT Kv2.1 (Fig. 7A) and used the BFA approach as in Figure 3. This Halo-tag mutant exhibited a uniform distribution similar to the SEP-tagged channel, when the expression was induced in the absence of BFA (Fig. 7B,C). When the expression was induced in the presence of BFA, which blocked the expression of Halo-Kv2.1 in the soma but not the AIS (Fig. 7D), the double mutant failed to be expressed on surface in most transfected neurons (Fig. 7E). Even in a few neurons exhibiting surface labeling (Fig. 7F), the signal levels were low, and the distribution was uniform, as evidenced by the line-scan analysis (Fig. 7G). This is presumably because the mutant is trafficked by promiscuous trafficking pathways largely through the Golgi apparatus. We also measured the peak fluorescence intensity in the AIS relative to that in the somatodendritic area in cells treated without or with BFA. The relative intensity was not statistically different between control and BFA-treated neurons (0.83 ± 0.09 in control vs 0.91 ± 0.16 in BFA; t(8) = 0.4619, p = 0.66, t test), suggesting that the surface expression of the double mutant was inhibited equally in the AIS and the somatodendritic region by BFA. This is distinct from the WT channel, for which expression in the AIS was refractory to BFA (Fig. 4E). These results indicate that the identified motifs are important to route the channel protein to the Golgi-independent trafficking pathway.
Figure 7.

Golgi-dependent trafficking of a mutant lacking the PRC and AIS motifs. A, Diagram of the Halo-Kv2.1 double mutant lacking aa 583–599 and 720–745. B, Uniform surface localization of the Halo-Kv2.1 double mutant in the AIS in the absence of BFA. Arrowheads indicate the AIS. C, Line-scan analysis of the fluorescence signals in the neurons shown in B. Segmented lines were drawn over the somatic membrane and into the axon. The border between these two regions is indicated by a gray line. D, Surface expression of Halo-Kv2.1 in the presence of BFA. Note that only signals in the AIS are apparent as shown in Figure 4. E, Effect of BFA on the trafficking of Halo-Kv2.1 double mutant. The arrow indicates a neuron without the transgene. F, A neuron exhibiting detectable surface expression of the Halo-Kv2.1 double mutant in the presence of BFA. Arrowhead indicate the AIS. G, Line-scan analysis of the fluorescence signals in the neurons shown in F.
Putative phosphorylation sites critical for the clustered localization of Kv2.1 at the AIS
Finally, in the identified AIS localization motif (aa 720–745), we noticed the presence of two conserved putative phosphorylation sites (T728 and S732), which scored highly in the phosphorylation site prediction algorithms ExPASy and NetPhos 2.0 (Fig. 8A). We therefore considered the possibility that sorting of Kv2.1 into the two distinct trafficking pathways, presumably at the endoplasmic reticulum, is regulated by the phosphorylation state of the AIS motif. To test whether these sites are critical for the clustered localization of Kv2.1 in the AIS, we mutated the threonine and serine residues to alanine (expected to be dephosphorylation mimetic), in SEP-Kv2.1 Δ583–599 lacking the PRC motif, a mutant background that exhibits an AIS localization similar to Kv2.1 S586A (Fig. 1A) and Kv2.1 Δ521–720 (Fig. 8B). We chose this mutant as a backbone for the mutagenesis because the presence of the PRC motif would complicate interpretation due to its cooperative action on the AIS localization of Kv2.1.
We found that, in contrast to the clustered and restricted localization of Kv2.1 Δ583–599 in the AIS, Kv2.1 Δ583–599 T728A/S732A exhibited a uniform distribution and nonrestricted localization in the axon (Fig. 8C), which is similar to that of the double PRC and AIS motif mutant (Fig. 6B). Quantitative analysis of the CV of fluorescence intensity and line-scan analysis also revealed that Kv2.1 Δ583–599 T728A/S732A does not form discrete clusters (Fig. 8D), and that it distributes uniformly in the axon beyond the ankyrin G-positive AIS (Fig. 8E). These results indicate that the putative phosphorylation sites in the AIS motif are critical for the clustered localization of Kv2.1 in the AIS.
Discussion
The localization of Kv2.1 in both the proximal somatodendritic membrane and in the AIS is an exceptional feature, because ion channels have almost exclusively been found to exhibit compartment-specific localization (i.e., either a somatodendritic or axonal localization; Lai and Jan, 2006; Trimmer, 2015). Growing evidence points to the notion that the AIS acts as a filter to separate axonal from somatodendritic proteins (Rasband, 2010). Kv2.1 is a prime exception to this model. It can enter both dendrites and the axon and form clusters in both neuronal cell-surface membranes. Our results suggest that the mechanisms dictating these specific localizations of Kv2.1 are distinct, because mutations in the motif responsible for its somatodendritic localization and clustering (the PRC motif) alone do not affect the localization and clustering of the Kv2.1 in the AIS. In this study, we defined a novel peptide motif (aa 720–745) located in the cytoplasmic C-terminal tail of Kv2.1 distal to the PRC motif, which works in conjunction with the PRC motif to ensure the AIS localization of Kv2.1 channels. Deletion of both of the PRC motif and this new motif, which we term “the AIS motif”, completely disrupts the AIS specific localization and clustering of Kv2.1, without impairing its cell-surface expression. We do not know why Kv2.1 needs two separate localization signals for its AIS localization, but speculate that having two distinct motifs would provide an opportunity for an additional layer of independent regulation of the specific population of Kv2.1 in the AIS, relative to the population in the somatodendritic membrane. In fact, we have previously shown that Kv2.1 clusters in the AIS are refractory to Ca2+-dependent modulation of localization, whereas somatodendritic clusters of Kv2.1 disperse, in both cultured neurons (Misonou et al., 2004) and intact brains (King et al., 2014). The AIS motif is highly conserved (Fig. 8A) across species from human (96% identity) to Xenopus (78% identity), similar to the extent of the conservation of the PRC motif. This underscores its importance in the physiological role of Kv2.1, and is consistent with the AIS localization of Kv2.1 observed in diverse mammalian species (King et al., 2014).
To understand how Kv2.1 traffics to the AIS, we performed FRAP experiments with the Kv2.1 localization mutant S586A. Although this mutant is localized and clustered in the AIS, it also exhibits a uniform somatodendritic distribution and significant diffusion in the somatodendritic membrane. However, the diffusible somatic Kv2.1 S586A did not diffuse into the AIS membrane, although it could diffuse within the adjacent somatic membrane. These results indicate that Kv2.1 is trafficked directly to the AIS through an intracellular trafficking mechanism, as opposed to arriving there from the somatodendritic domain via lateral diffusion. It has been proposed that Kv2.1 is directly trafficked into the somatic surface clusters via an intracellular vesicular trafficking pathway (Deutsch et al., 2012). Our results are consistent with such a model as proposed for somatic Kv2.1 channels, and extend it to suggest that axonal Kv2.1 channels cluster via a mechanism that involves directed intracellular trafficking.
We also provided evidence that Kv2.1 destined to the AIS uses a nonconventional trafficking pathway largely independent of the Golgi apparatus, whereas that targeted to the somatodendritic membrane takes the canonical secretory pathway. This model is supported by evidence obtained in experiments using temperature block of protein export from the Golgi apparatus, blockade of COP I-dependent vesicle budding from the Golgi apparatus by the Arf1 Q61I mutant, and pharmacological disruption of the Golgi apparatus by BFA treatment, which yielded altogether consistent results. Under all of these conditions, Kv2.1 was still readily localized and clustered in the AIS membrane. Furthermore, the Kv2.1 mutant lacking both the PRC and AIS signals failed to localize in the AIS and became sensitive to BFA treatment for cell-surface expression. Collectively, these results suggest that AIS Kv2.1 is trafficked through a Golgi bypass route, distinct from the pathway taken by somatodendritic Kv2.1, and that trafficking via this alternate route requires both the PRC and AIS dual signals.
Golgi bypass trafficking routes have been described in a number of cell types, including polarized cells such as MDCK cells (Grieve and Rabouille, 2011). A number of cargo proteins have been reported to be trafficked though a Golgi bypass route, including the receptor tyrosine phosphatase CD45 (Baldwin and Ostergaard, 2002), and the cystic fibrosis transmembrane conductance regulator (Yoo et al., 2002). Although the role and significance of Golgi bypass routes have not been fully addressed, especially in neurons, these alternative “secretory” pathways have been shown to provide distinct glycosylation characteristics on these other transmembrane N-linked glycoproteins (Baldwin and Ostergaard, 2002; Yoo et al., 2002). However, Kv2.1 lacks N-linked glycosylation (Shi and Trimmer, 1999), and as such should not be similarly affected, although other Golgi specific posttranslational modifications, such as phosphorylation and tyrosine sulfation, would likely be impacted. Kv2.1 is extensively phosphorylated at up to 31 distinct sites (Trimmer and Misonou, 2015), primarily on the C-terminus, although whether any of these phosphorylation events occur in the Golgi apparatus is not known. Grieve and Rabouille, (2011) suggested that Golgi bypass routes may provide faster and more efficient trafficking of proteins to the cell surface. We did observe that the surface expression of Kv2.1 destined for the AIS is relatively fast and efficient compared with that targeted to the somatodendritic membrane, consistent with the previous finding by Sarmiere et al. (2008). Precise timing of ion channel expression in distinct membrane compartment might be of importance to plasticity of intrinsic excitability, especially during neuronal development, where neuronal excitability and Ca2+ transients play crucial roles (Spitzer, 2006). Therefore, sorting Kv2.1 into the two distinct trafficking pathways defined here may allow neurons to independently regulate the cell-surface expression of Kv2.1 channels in these two distinct membrane compartments. A recently published report suggested a role for a Golgi bypass route in the trafficking of another class of ion channels, ionotropic glutamate receptors, to dendritic spines and excitatory synapses of neurons (Hanus et al., 2016).
In addition to the finding that the AIS trafficking of Kv2.1 can occur independently of the Golgi apparatus, the mechanism also appears to be different from the “selective-retention” mechanism reported for axonal Nav1.2 channels (Garrido et al., 2003). Although this is still largely unexplored, it is interesting that the complete machinery for protein translation and cell-surface expression has been found in axons (Brittis et al., 2002; González et al., 2016). Together with our data, this suggests that local synthesis and plasma membrane expression of Kv2.1 on the AIS could occur without the need of the axonal Golgi apparatus. In fact, it was recently proposed that Kv2.1 cell-surface clusters, both in the somatodendritic and AIS membranes, are associated with intracellular ER membranes, at sites where the ER comes into close proximity with the plasma membrane to form specialized ER–plasma membrane junctions (Fox et al., 2015). Neuronal ER–plasma membrane junctions are present in somata (Rosenbluth, 1962), and on the AIS, where the specialized ER of the AIS, termed the cisternal organelle, comes into close apposition to the plasma membrane (Peters et al., 1968). Kv2.1 clusters in the AIS are found at these sites, which are also highly enriched in ryanodine receptors and synaptopodin, and that occur at sites deficient in the specialized ankyrin G-associated cytoskeleton (King et al., 2014), which plays a prominent role in localizing Nav and Kv7 channels to the AIS (Zhang and Rasband, 2016). Sites of Kv2.1 clustering have also been proposed as an insertion platform for plasma membrane proteins, including Kv2.1 (Deutsch et al., 2012). Therefore, the possibility that Kv2.1 is locally synthesized in the AIS can at this point not be excluded, nor can the possibility that Kv2.1 sorting to somatodendritic or axonal compartments involves the smooth ER membranes associated with the ER–plasma membrane junctions at which Kv2.1 is clustered.
Our results demonstrate that there are two distinct pools of Kv2.1 that are sorted into the two different trafficking pathways and regulated independently thereafter. These pools are presumably identical in primary structure. However, posttranslational modifications such as phosphorylation may act as a molecular tag to determine the fate of Kv2.1. We found two putative highly conserved phosphorylation sites in the AIS signal, and mutating these sites to alanine (in conjunction with deletion of the PRC motif), completely disrupted the restricted and clustered localization of Kv2.1 in the AIS. Recent studies have shown that there exist distinct mechanisms to confer activity-dependent plasticity to AIS structure and function (Grubb and Burrone, 2010; Kuba et al., 2010; Evans et al., 2015). It is intriguing to speculate that differences in phosphorylation could provide a tag for directing Kv2.1 to one or the other pathway during its biosynthetic trafficking. Such a mechanism would allow neurons to deliver Kv2.1 and perhaps other protein species into the AIS in a regulated manner, and provide precise compartment-specific regulation of AIS structure and function, and, for ion channels such as Kv2.1, control of local membrane excitability.
Footnotes
This work was supported by the Lundbeck Foundation and Carlsberg Foundation to C.S.J., the Research Foundation Flanders (J.I.S.), JGC-S Scholarship Foundation (S.W.), Core-to-Core Program A Advanced Research Networks, the Strategic Research Foundation at Private Universities (S1201009), Mitsubishi Life Science Foundation, Naito Foundation, and MEXT/JSPS KAKENHI Grants 26640017 and 16K07006 (H.M.).We thank Minori Iwata for her help preparing cultured neurons, Dr. Nobuyuki Nukina for assistance on FRAP experiments, and the Core Facility for Integrated Microscopy at the University of Copenhagen, Denmark.
The authors declare no competing financial interests.
References
- Baldwin TA, Ostergaard HL (2002) The protein-tyrosine phosphatase CD45 reaches the cell surface via golgi-dependent and -independent pathways. J Biol Chem 277:50333–50340. 10.1074/jbc.M209075200 [DOI] [PubMed] [Google Scholar]
- Bishop HI, Guan D, Bocksteins E, Parajuli LK, Murray KD, Cobb MM, Misonou H, Zito K, Foehring RC, Trimmer JS (2015) Distinct cell- and layer-specific expression patterns and independent regulation of Kv2 channel subtypes in cortical pyramidal neurons. J Neurosci 35:14922–14942. 10.1523/JNEUROSCI.1897-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittis PA, Lu Q, Flanagan JG (2002) Axonal protein synthesis provides a mechanism for localized regulation at an intermediate target. Cell 110:223–235. 10.1016/S0092-8674(02)00813-9 [DOI] [PubMed] [Google Scholar]
- Buffington SA, Rasband MN (2011) The axon initial segment in nervous system disease and injury. Eur J Neurosci 34:1609–1619. 10.1111/j.1460-9568.2011.07875.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coloma MJ, Hastings A, Wims LA, Morrison SL (1992) Novel vectors for the expression of antibody molecules using variable regions generated by polymerase chain reaction. J Immunol Methods 152:89–104. 10.1016/0022-1759(92)90092-8 [DOI] [PubMed] [Google Scholar]
- Dascher C, Balch WE (1994) Dominant inhibitory mutants of ARF1 block endoplasmic reticulum to Golgi transport and trigger disassembly of the Golgi apparatus. J Biol Chem 269:1437–1448. [PubMed] [Google Scholar]
- David JP, Stas JI, Schmitt N, Bocksteins E (2015) Auxiliary KCNE subunits modulate both homotetrameric Kv2.1 and heterotetrameric Kv2.1/Kv6.4 channels. Sci Rep 5:12813. 10.1038/srep12813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch E, Weigel AV, Akin EJ, Fox P, Hansen G, Haberkorn CJ, Loftus R, Krapf D, Tamkun MM (2012) Kv2.1 cell surface clusters are insertion platforms for ion channel delivery to the plasma membrane. Mol Biol Cell 23:2917–2929. 10.1091/mbc.E12-01-0047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Tao-Chang J-H, Zerfas P, McBain CJ (1998) The K+ channel, Kv2.1, is apposed to astrocytic processes and is associated with inhibitory postsynaptic membranes in hippocampal and cortical principal neurons and inhibitory interneurons. Neuroscience 8439:37–48. 10.1016/s0306-4522(97)00519-8 [DOI] [PubMed] [Google Scholar]
- Evans MD, Dumitrescu AS, Kruijssen DL, Taylor SE, Grubb MS (2015) Rapid modulation of axon initial segment length influences repetitive spike firing. Cell Rep 13:1233–1245. 10.1016/j.celrep.2015.09.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul F, Erdfelder E, Lang AG, Buchner A (2007) G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 39:175–191. 10.3758/BF03193146 [DOI] [PubMed] [Google Scholar]
- Faul F, Erdfelder E, Buchner A, Lang AG (2009) Statistical power analyses using G*Power 3.1: tests for correlation and regression analyses. Behav Res Methods 41:1149–1160. 10.3758/BRM.41.4.1149 [DOI] [PubMed] [Google Scholar]
- Fox PD, Haberkorn CJ, Akin EJ, Seel PJ, Krapf D, Tamkun MM (2015) Induction of stable ER-plasma-membrane junctions by Kv2.1 potassium channels. J Cell Sci 128:2096–2105. 10.1242/jcs.166009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frech GC, VanDongen AM, Schuster G, Brown AM, Joho RH (1989) A novel potassium channel with delayed rectifier properties isolated from rat brain by expression cloning. Nature 340:642–645. 10.1038/340642a0 [DOI] [PubMed] [Google Scholar]
- Garrido JJ, Giraud P, Carlier E, Fernandes F, Moussif A, Fache MP, Debanne D, Dargent B (2003) A targeting motif involved in sodium channel clustering at the axonal initial segment. Science 300:2091–2094. 10.1126/science.1085167 [DOI] [PubMed] [Google Scholar]
- González C, Cánovas J, Fresno J, Couve E, Court FA, Couve A (2016) Axons provide the secretory machinery for trafficking of voltage-gated sodium channels in peripheral nerve. Proc Natl Acad Sci U S A 113:1823–1828. 10.1073/pnas.1514943113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieve AG, Rabouille C (2011) Golgi bypass: skirting around the heart of classical secretion. Cold Spring Harb Perspect Biol 3:a005298. 10.1101/cshperspect.a005298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Burrone J (2010) Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature 465:1070–1074. 10.1038/nature09160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan D, Armstrong WE, Foehring RC (2013) Kv2 channels regulate firing rate in pyramidal neurons from rat sensorimotor cortex. J Physiol 591:4807–4825. 10.1113/jphysiol.2013.257253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanus C, Geptin H, Tushev G, Garg S, Alvarez-Castelao B, Sambandan S, Kochen L, Hafner AS, Langer JD, Schuman EM (2016) Unconventional secretory processing diversifies neuronal ion channel properties. Elife 5:e20609. 10.7554/eLife.20609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty RC, Kim TH, Thomas EA, Cardamone L, Jones NC, Petrou S, Wimmer VC (2013) Axon initial segment structural plasticity in animal models of genetic and acquired epilepsy. Epilepsy Res 105:272–279. 10.1016/j.eplepsyres.2013.03.004 [DOI] [PubMed] [Google Scholar]
- Helms JB, Rothman JE (1992) Inhibition by brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature 360:352–354. 10.1038/360352a0 [DOI] [PubMed] [Google Scholar]
- Hinman JD, Rasband MN, Carmichael ST (2013) Remodeling of the axon initial segment after focal cortical and white matter stroke. Stroke 44:182–189. 10.1161/STROKEAHA.112.668749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen CS, Misonou H (2015) Live-cell imaging of post-Golgi transport vesicles in cultured hippocampal neurons. Cell Mol Neurobiol 35:123–135. 10.1007/s10571-014-0103-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen CS, Rasmussen HB, Misonou H (2011) Neuronal trafficking of voltage-gated potassium channels. Mol Cell Neurosci 48:288–297. 10.1016/j.mcn.2011.05.007 [DOI] [PubMed] [Google Scholar]
- Jensen CS, Watanabe S, Rasmussen HB, Schmitt N, Olesen SP, Frost NA, Blanpied TA, Misonou H (2014) Specific sorting and post-Golgi trafficking of dendritic potassium channels in living neurons. J Biol Chem 289:10566–10581. 10.1074/jbc.M113.534495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimm T, Khaliq ZM, Bean BP (2015) Differential regulation of action potential shape and burst-frequency firing by BK and Kv2 channels in substantia nigra dopaminergic neurons. J Neurosci 35:16404–16417. 10.1523/JNEUROSCI.5291-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AN, Manning CF, Trimmer JS (2014) A unique ion channel clustering domain on the axon initial segment of mammalian neurons. J Comp Neurol 522:2594–2608. 10.1002/cne.23551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba H, Oichi Y, Ohmori H (2010) Presynaptic activity regulates Na(+) channel distribution at the axon initial segment. Nature 465:1075–1078. 10.1038/nature09087 [DOI] [PubMed] [Google Scholar]
- Lai HC, Jan LY (2006) The distribution and targeting of neuronal voltage-gated ion channels. Nat Rev Neurosci 7:548–562. 10.1038/nrn1938 [DOI] [PubMed] [Google Scholar]
- Lerche H, Biervert C, Alekov AK, Schleithoff L, Lindner M, Klinger W, Bretschneider F, Mitrovic N, Jurkat-Rott K, Bode H, Lehmann-Horn F, Steinlein OK (1999) A reduced K+ current due to a novel mutation in KCNQ2 causes neonatal convulsions. Ann Neurol 46:305–312. [DOI] [PubMed] [Google Scholar]
- Leterrier C, Lainé J, Darmon M, Boudin H, Rossier J, Lenkei Z (2006) Constitutive activation drives compartment-selective endocytosis and axonal targeting of type 1 cannabinoid receptors. J Neurosci 26:3141–3153. 10.1523/JNEUROSCI.5437-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ST, Antonucci DE, Scannevin RH, Trimmer JS (2000) A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron 25:385–397. 10.1016/S0896-6273(00)80902-2 [DOI] [PubMed] [Google Scholar]
- Liu PW, Bean BP (2014) Kv2 channel regulation of action potential repolarization and firing patterns in superior cervical ganglion neurons and hippocampal CA1 pyramidal neurons. J Neurosci 34:4991–5002. 10.1523/JNEUROSCI.1925-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV (2008) HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 3:373–382. 10.1021/cb800025k [DOI] [PubMed] [Google Scholar]
- Malin SA, Nerbonne JM (2000) Elimination of the fast transient in superior cervical ganglion neurons with expression of KV4.2W362F: molecular dissection of IA. J Neurosci 20:5191–5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H. (2010) Homeostatic regulation of neuronal excitability by K+ channels in normal and diseased brains. Neuroscientist 16:51–64. 10.1177/1073858409341085 [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS (2004) Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci 7:711–718. 10.1038/nn1260 [DOI] [PubMed] [Google Scholar]
- O'Connell KM, Tamkun MM (2005) Targeting of voltage-gated potassium channel isoforms to distinct cell surface microdomains. J Cell Sci 118:2155–2166. 10.1242/jcs.02348 [DOI] [PubMed] [Google Scholar]
- Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, Bennett V, Scherer SS, Cooper EC (2006) A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci 26:2599–2613. 10.1523/JNEUROSCI.4314-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak D, Guan D, Foehring RC (2016) Roles of specific Kv channel types in repolarization of the action potential in genetically identified subclasses of pyramidal neurons in mouse neocortex. J Neurophysiol 115:2317–2329. 10.1152/jn.01028.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltola MA, Kuja-Panula J, Liuhanen J, Võikar V, Piepponen P, Hiekkalinna T, Taira T, Lauri SE, Suvisaari J, Kulesskaya N, Paunio T, Rauvala H (2016) AMIGO-Kv2.1 potassium channel complex is associated with Schizophrenia-related phenotypes. Schizophr Bull 42:191–201. 10.1093/schbul/sbv105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Proskauer CC, Kaiserman-Abramof IR (1968) The small pyramidal neuron of the rat cerebral cortex: the axon hillock and initial segment. J Cell Biol 39:604–619. 10.1083/jcb.39.3.604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN. (2010) The axon initial segment and the maintenance of neuronal polarity. Nat Rev Neurosci 11:552–562. 10.1038/nrn2852 [DOI] [PubMed] [Google Scholar]
- Rasmussen HB, Frøkjaer-Jensen C, Jensen CS, Jensen HS, Jørgensen NK, Misonou H, Trimmer JS, Olesen SP, Schmitt N (2007) Requirement of subunit co-assembly and ankyrin-G for M-channel localization at the axon initial segment. J Cell Sci 120:953–963. 10.1242/jcs.03396 [DOI] [PubMed] [Google Scholar]
- Rosenbluth J. (1962) Subsurface cisterns and their relationship to the neuronal plasma membrane. J Cell Biol 13:405–421. 10.1083/jcb.13.3.405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitsu H, Akita T, Tohyama J, Goldberg-Stern H, Kobayashi Y, Cohen R, Kato M, Ohba C, Miyatake S, Tsurusaki Y, Nakashima M, Miyake N, Fukuda A, Matsumoto N (2015) De novo KCNB1 mutations in infantile epilepsy inhibit repetitive neuronal firing. Sci Rep 5:15199. 10.1038/srep15199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, De Angelis D, Rothman JE, Ryan TA (2000) The use of pHluorins for optical measurements of presynaptic activity. Biophys J 79:2199–2208. 10.1016/S0006-3495(00)76468-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmiere PD, Weigle CM, Tamkun MM (2008) The Kv2.1 K+ channel targets to the axon initial segment of hippocampal and cortical neurons in culture and in situ. BMC Neurosci 9:112. 10.1186/1471-2202-9-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannevin RH, Murakoshi H, Rhodes KJ, Trimmer JS (1996) Identification of a cytoplasmic domain important in the polarized expression and clustering of the Kv2.1 K+ channel. J Cell Biol 135:1619–1632. 10.1083/jcb.135.6.1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Jha S, Liu F, Akella T, McCullough LD, Rasband MN (2009) Disruption of the axon initial segment cytoskeleton is a new mechanism for neuronal injury. J Neurosci 29:13242–13254. 10.1523/JNEUROSCI.3376-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G, Trimmer JS (1999) Differential asparagine-linked glycosylation of voltage-gated K+ channels in mammalian brain and in transfected cells. J Membr Biol 168:265–273. 10.1007/s002329900515 [DOI] [PubMed] [Google Scholar]
- Speca DJ, Ogata G, Mandikian D, Bishop HI, Wiler SW, Eum K, Wenzel HJ, Doisy ET, Matt L, Campi KL, Golub MS, Nerbonne JM, Hell JW, Trainor BC, Sack JT, Schwartzkroin PA, Trimmer JS (2014) Deletion of the Kv2.1 delayed rectifier potassium channel leads to neuronal and behavioral hyperexcitability. Genes Brain Behav 13:394–408. 10.1111/gbb.12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer NC. (2006) Electrical activity in early neuronal development. Nature 444:707–712. 10.1038/nature05300 [DOI] [PubMed] [Google Scholar]
- Thiffault I, Speca DJ, Austin DC, Cobb MM, Eum KS, Safina NP, Grote L, Farrow EG, Miller N, Soden S, Kingsmore SF, Trimmer JS, Saunders CJ, Sack JT (2015) A novel epileptic encephalopathy mutation in KCNB1 disrupts Kv2.1 ion selectivity, expression, and localization. J Gen Physiol 146:399–410. 10.1085/jgp.201511444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torkamani A, Bersell K, Jorge BS, Bjork RL Jr, Friedman JR, Bloss CS, Cohen J, Gupta S, Naidu S, Vanoye CG, George AL Jr, Kearney JA (2014) De novo KCNB1 mutations in epileptic encephalopathy. Ann Neurol 76:529–540. 10.1002/ana.24263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS. (1991) Immunological identification and characterization of a delayed rectifier K+ channel polypeptide in rat brain. Proc Natl Acad Sci U S A 88:10764–10768. 10.1073/pnas.88.23.10764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS. (2015) Subcellular localization of K channels in mammalian brain neurons: remarkable precision in the midst of extraordinary complexity. Neuron 85:238–256. 10.1016/j.neuron.2014.12.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS, Misonou H (2015) Phosphorylation of voltage-gated ion channels. In: Handbook of ion channels (Trudeau MC, Zheng J, eds). Boca Raton, FL: CRC. [Google Scholar]
- Wisco D, Anderson ED, Chang MC, Norden C, Boiko T, Fölsch H, Winckler B (2003) Uncovering multiple axonal targeting pathways in hippocampal neurons. J Cell Biol 162:1317–1328. 10.1083/jcb.200307069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo JS, Moyer BD, Bannykh S, Yoo HM, Riordan JR, Balch WE (2002) Non-conventional trafficking of the cystic fibrosis transmembrane conductance regulator through the early secretory pathway. J Biol Chem 277:11401–11409. 10.1074/jbc.M110263200 [DOI] [PubMed] [Google Scholar]
- Zhang C, Rasband MN (2016) Cytoskeletal control of axon domain assembly and function. Curr Opin Neurobiol 39:116–121. 10.1016/j.conb.2016.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]