

Abstract
The exact function of the polybasic juxtamembrane region (5RK) of the plasma membrane neuronal SNARE, syntaxin 1A (Syx), in vesicle exocytosis, although widely studied, is currently not clear. Here, we addressed the role of 5RK in Ca2+-triggered release, using our Syx-based intramolecular fluorescence resonance energy transfer (FRET) probe, which previously allowed us to resolve a depolarization-induced Ca2+-dependent close-to-open transition (CDO) of Syx that occurs concomitant with evoked release, both in PC12 cells and hippocampal neurons and was abolished upon charge neutralization of 5RK. First, using dynamic FRET analysis in PC12 cells, we show that CDO occurs following assembly of SNARE complexes that include the vesicular SNARE, synaptobrevin 2, and that the participation of 5RK in CDO goes beyond its participation in the final zippering of the complex, because mutations of residues adjacent to 5RK, believed to be crucial for final zippering, do not abolish this transition. In addition, we show that CDO is contingent on membrane phosphatidylinositol 4,5-bisphosphate (PIP2), which is fundamental for maintaining regulated exocytosis, as depletion of membranal PIP2 abolishes CDO. Prompted by these results, which underscore a potentially significant role of 5RK in exocytosis, we next amperometrically analyzed catecholamine release from PC12 cells, revealing that charge neutralization of 5RK promotes spontaneous and inhibits Ca2+-triggered release events. Namely, 5RK acts as a fusion clamp, making release dependent on stimulation by Ca2+.
SIGNIFICANCE STATEMENT Syntaxin 1A (Syx) is a central protein component of the SNARE complex, which underlies neurotransmitter release. Although widely studied in relation to its participation in SNARE complex formation and its interaction with phosphoinositides, the function of Syx's polybasic juxtamembrane region (5RK) remains unclear. Previously, we showed that a conformational transition of Syx, related to calcium-triggered release, reported by a Syx-based FRET probe, is abolished upon charge neutralization of 5RK (5RK/A). Here we show that this conformational transition is dependent on phosphatidylinositol 4,5-bisphosphate (PIP2) and is related to SNARE complex formation. Subsequently, we show that the 5RK/A mutation enhances spontaneous release and inhibits calcium-triggered release in neuroendocrine cells, indicating a previously unrecognized role of 5RK in neurotransmitter release.
Keywords: amperometry, exocytosis, FRET, fusion clamp, neuroendocrine cells, syntaxin-1A
Introduction
The Soluble N-ethylmaleimide-sensitive factor activating protein receptor (SNARE) complex, composed of the plasma membrane (PM)-anchored syntaxin-1 (Syx) and SNAP-25, along with the vesicle membrane-anchored synaptobrevin-2 (Syb2), plays a central role in neurosecretory dense-core vesicles release (Rothman, 1996; Jahn and Fasshauer, 2012; Fang and Lindau, 2014; Rizo and Xu, 2015). At a prefusion trans state (Fasshauer et al., 1998), the SNARE domains of the proteins form a coiled-coil structure with helical extension through the linkers of Syx and Syb2 into their transmembrane (TM) domains, at a postfusion cis state (Stein et al., 2009).
Accumulating evidence suggests that in its primed state, the SNARE complex is assembled in the membrane-distal part of the SNARE domains, whereas membrane-proximal zippering is inhibited (Sørensen et al., 2006; Gao et al., 2012; Liang et al., 2013; Lou and Shin, 2016), possibly through a clamping interaction of the SNARE complex with complexin, until the arrival of a trigger mediated by Ca2+-bound synaptotagmin1 (Syt1) to fully zipper and complete fusion (Chen et al., 2002; Tang et al., 2006; Krishnakumar et al., 2011). Syt1, in addition to its role as the Ca2+ sensor for Ca2+-triggered release, has also been shown to clamp the frequency of miniature spontaneous events (Yoshihara and Littleton, 2002; Maximov and Südhof, 2005; Kochubey and Schneggenburger, 2011). However, the origin of Syt1 clamping of spontaneous release, as well as the impact of complexin on spontaneous release, remains unresolved (Trimbuch and Rosenmund, 2016). Although Syt1-SNARE interaction at physiological ionic strength has been questioned (Park et al., 2015), recent crystal structures of the Syt1-SNARE complex (Zhou et al., 2015) revealed a unit thought to promote Ca2+-triggered fusion via remodeling of PM. Notably, the Syt1-SNARE structures did not include the juxtamembrane linkers of Syx and Syb2.
The polybasic stretch of five lysines and arginines, 5RK, in the juxtamembrane linker of Syx, the focus of our study, was shown to interact electrostatically with phosphatidylinositol 4,5-bisphosphate (PIP2) and phosphatidylinositol 3,4,5 trisphosphate (PIP3), promoting Syx clustering (Aoyagi et al., 2005; James et al., 2008; Lam et al., 2008; Murray and Tamm, 2011; van den Bogaart et al., 2011; Honigmann et al., 2013; Khuong et al., 2013). However, the exact function of these interactions in vesicle exocytosis is currently unclear (for review, see van den Bogaart et al., 2013), especially in view of the only partially reduced evoked release reported for 5RK charge neutralization mutants (Lam et al., 2008; Khuong et al., 2013).
To gain insights into the domain-specific functioning of Syx associated with exocytosis in live cells, we previously constructed a novel Syx-based intramolecular fluorescence resonance energy transfer (FRET) reporter probe, termed CSYS, which reports a conformational close-to-open transition (opening) of Syx (Greitzer-Antes et al., 2013), which allows Syx to enter SNARE complexes (Sutton et al., 1998; Margittai et al., 2003; Jahn and Scheller, 2006). CSYS, shown to substitute for native Syx and to support secretion in PC12 cells (Greitzer-Antes et al., 2013) and in hippocampal neurons (Vertkin et al., 2015), enabled to resolve in PC12 cells a discrete opening of Syx in response to high K+ depolarization, which is dependent on Ca2+ entry through voltage-gated Ca2+ channels that stimulate exocytosis. Importantly, this Ca2+-dependent opening (CDO) was found to be absolutely dependent on intact 5RK, because it does not occur upon 5RK charge neutralization (Greitzer-Antes et al., 2013).
Here, we further demonstrate that CDO is specifically linked to 5RK and not to other residues in the juxtamembrane linker; we also establish a role for PIP2 as a key regulator of CDO. To get a clue as to the physiological relevance of CDO to secretion, we determined the secretion phenotype of a 5RK charge neutralization mutant. Our results suggest a significant role for 5RK in making transmitter release in PC12 cells dependent on stimulation by Ca2+.
Materials and Methods
Plasmid construction.
Double-labeled Syx (CSYS) and CSYS-5RK/A cDNA were generated as described by Greitzer-Antes et al. (2013). CSYS-YQ/AA was generated by introducing two point mutations, Y257A and Q258A, at the linker region domain of CSYS. The BoNT-C1-resistant mutation (CSYS-R) was generated by introducing one point mutation, K253I, at the BoNT-C1 recognizing sequence (Lam et al., 2008). For PC12 transfection, the constructs were cloned into pcDNA3 vector using EcoRI and XbaI restriction sites. The PLCη2 construct was kindly provided by T. F. J. Martin (University of Wisconsin). The 5-phosphatase Inp54p system constructs were purchased from Addgene. BoNT-C1α51 was kindly provided by M. B. Jackson (University of Wisconsin). BoNT-C1α51 and both PIP2 depletion constructs, described above, were subcloned into mRFP pcDNA3 vector containing IRES (internal ribosome entry site).
PC12 cells preparation and transfection for FRET experiments.
PC12 cells were maintained at 37°C/5% CO2 in DMEM with high glucose (Sigma-Aldrich) supplemented with 10% bovine serum, 5% l-glutamin, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. For imaging, cells were replated to a ∼60% confluence onto poly-l-lysine (Sigma-Aldrich)-coated 35 mm glass bottom culture dishes and allowed to adhere overnight. Cells were transfected with 1.5 μg of the CSYS mutation probes, using Lipofectamine 2000 (Invitrogen). Imaging experiments were conducted at room temperature, 24 h after transfection. During the experiment the transfected cells were superfused through a 2 ml bath, with physiological (2.8 mm K+) and high K+ (105 mm K+) solutions as described in by An and Almers (2004).
Dynamic FRET assay in PC12.
PC12 cells were imaged using a C-Apochromat 40×/1.2 NA water objective and excited with a 405 nm laser every 5 s for a total of 400 s. During the sequential imaging, the cells were imaged in a control, 2.8 mm K+ solution for 100 s before and after 200 s of 105 mm high K+ solution, for a total imaging time of 400 s. Fluorescent signals were collected with a Zeiss 510META confocal microscope using its “channel mode”. Cells were excited with a 405 nm laser band and the emission was filtered through the main beam splitter HFT405/514/633 nm and further separated by a secondary beam splitter, NFT515 nm. CFP and YFP fluorescence were collected by 470–500 and 505–550 nm bandpass filters, respectively, and directed into two separate photomultipliers. Under these settings, the leak of YFP into the CFP recording window is purely optical, very low (<1%; Okamoto et al., 2004), and remained constant regardless of changes in FRET, thus not requiring any corrections. YFP and CFP intensities at a region-of-interest (ROI) on the cell's membrane were calculated, and background fluorescence was quantified from an ROI in each image defined in an area containing no fluorescent cells. The background-subtracted fluorescence intensity at each exposure time point was normalized to the average of the initial measurements in each cell (before high K+ solution was added). The FRET ratio of normalized intensities was denoted as FYFP/FCFP. Changes in FRET are reflected as changes in the FRET ratio.
Static FRET assay in PC12.
PC12 cells were imaged in physiological solution (2.8 mm K+) in a 0.7 mm glass-bottom dish. Fluorescence emissions from CFP and/or YFP-tagged proteins were collected from the PM of the cells with a Zeiss inverted confocal microscope (Axiovert LSM 510META), using a 20 × 0.75 NA air objective and laser excitations of 405 and 514 nm, respectively. We used a spectrum-based method to remove contamination caused by donor emission and for direct excitation of the acceptor. The FRET assay was performed as described by Zheng and Zagotta (2004). Briefly, two emission spectra were collected from each cell, one with 405 nm excitation and the other with 514 nm excitation. A scaled CFP spectrum, collected from control cells expressing CFP-tagged proteins only, was used to normalize the CFP emissions from the spectrum taken from cells expressing both fluorophores at 405 nm excitation. This procedure allows one to dissect the YFP emission spectrum, termed F405, into two components: one is due to the direct excitation of YFP (F405direct) and the other is due to FRET (F405FRET). F405 is normalized to the total YFP emission with 514 nm excitation at the same cell, F514. The resulting ratio, termed RatioA, can be expressed as RatioA = . The direct excitation component in the calculated RatioA, termed RatioA0, was experimentally determined from a large population of oocytes expressing only YFP-tagged proteins. This allows the bleed through of the direct excitation of YFP to be precisely calculated by the 405 nm laser. The difference between RatioA and RatioA0 (RatioA − RatioA0) is directly proportional to FRET efficiency: RatioA − RatioA0 = . The apparent FRET efficiency from an individual cell, Eapp, can be calculated as Eapp = , where εD and εA are molar extinction coefficients for the donor and acceptor, respectively, at the donor excitation wavelength (Takanishi et al., 2006; Gao et al., 2007). To avoid autofluorescence contamination in the CFP spectrum, the autofluorescence of native PC12 cells in the CFP spectrum was subtracted from each cell.
5′ phosphatase PIP2 depletion assay in PC12.
PIP2 depletion was conducted as described previously (Suh et al., 2006). Briefly, this dimerizer strategy uses heterodimerization of protein domains from FKBP and from FRB by the immunosuppressant Rapamycin. FRB is anchored to PM via Lyn11. Inp54p, a yeast inositol polyphosphate 5-phosphatase that specifically cleaves the phosphate at the 5 position of PIP2 fused to FKBP, is recruited from the cytosol upon addition of Rapamycin, forming the tripartite complex Lyn11 FRB-Rapamycin-FKBP Inp54p. Membrane recruitment of Inp54p rapidly induces specific dephosphorylation at the 5 position of PIP2. In our experiments, PIP2 was depleted from PC12 cells transfected with plasmids encoding FRB (0.75 μg) and mRFP FKBP Inp54p (0.75 μg), by the addition of the dimerizing agent, rapamycin (5 μm), to the physiological and high K+ solutions. To verify PIP2 depletion from PM, PC12 cells were transfected with plasmids encoding the 5′ phosphatase system, described above, together with GFP-PH PLCδ1 (0.5 μg), and imaged by confocal microscopy.
Cell culture for the amperometry experiments.
PC12 cells were obtained from American Type Culture Collection and maintained in RPMI-1640 media supplemented with 10% heat-inactivated horse serum and 5% fetal bovine serum (Sigma-Aldrich), in 7% CO2 atmosphere at 37°C. The cells were grown on mouse collagen IV-coated flasks (Becton Dickinson) and were subcultured approximately every 7 d. Cells were transfected using Lipofectamine-2000 (Invitrogen).
Amperometry measurements.
Amperometric currents were recorded with a VA-10 amplifier (npi Electronics GmbH). A constant voltage of +700 mV was applied to the 5 μm OD carbon electrode (ALA Scientific). The currents were filtered at 1 kHz and digitized at 10 kHz using a Digidata 1322A analog-to-digital converter and the Clampex 9 software package (Axon CNS, Molecular Devices). Data were analyzed by the customized macro for Igor Pro software (Mosharov, 2008). Spikes smaller than 5 pA were considered as noise and were discarded. The bath solution contained the following (in mm): 150 NaCl, 5 KCl, 1.2 MgCl2, 5 glucose, 10 HEPES, 2 CaCl2, pH 7.4, and the high K+ solution contained the following (in mm): 100 KCl, 50 CaCl2, 0.7 MgCl2, HEPES 10, pH 7.4. Similarly to what was shown in chromaffin cells (Shang et al., 2014), in PC12 cells we observed reduced spontaneous release in the presence of elevated [Ca2+] (data not shown); therefore, spontaneous events were monitored in physiological [Ca2+] (2 mm).
Experimental design and statistical analysis.
Data were summarized as mean ± SEM. The number of samples (n) indicates the number of cells per group. Statistical analysis was performed in either SPSS v24 (IBM) or Sigma-AldrichPlot v11. For time course FRET (dynamic FRET) experiments, one-way ANOVA for repeated measures was conducted. For amperometry experiments, where events were monitored before and after stimulation in each cell, paired t test analysis was used; multiple groups were compared by ANOVA followed by post hoc Tukey's test. Asterisks in the figures indicate statistically significant differences as follows: *p < 0.05, **p < 0.001.
3D alignment and electrostatic surface calculation.
Swiss PDB Viewer 4.1 was used for: (1) 3D alignments, using a built-in magic fit algorithm with subsequent RMS deviation calculation, and (2) electrostatic surface calculations using the Poisson–Boltzmann method on atomic partial charges, setting the solvent ionic strength to 0.3 mol/L, the protein dielectric constant to 4, and the solvent dielectric constant to 80. Proteins were visualized in MolSoft MolBrowser 3.8.
Results
The juxtamembrane basic residues, 5RK, in Syx are absolutely required for the CDO of CSYS induced by high K+ depolarization
Previously, we generated a Syx-based FRET probe, CSYS (Fig. 1A), which reports close-to-open transitions (openings) of Syx during exocytosis (Greitzer-Antes et al., 2013). Importantly, CSYS could rescue the inhibition of exocytosis by botulinum neurotoxin C1 (which cleaves native Syx) in both PC12 cells (Greitzer-Antes et al., 2013) and hippocampal neurons (Vertkin et al., 2015), demonstrating that it is functional and supports fusion. Next, using dynamic FRET analysis in PC12 cells expressing CSYS, we monitored a significant reduction in the FYFP/FCFP ratio (FRET ratio), reporting opening of CSYS, in response to high K+ depolarization. The opening consisted of two distinct conformational transitions, a partial, Ca2+-independent opening [resistant to blockade by cadmium (Cd)] and a CDO that is blocked by Cd (Greitzer-Antes et al., 2013; Fig. 1B; F(79,15800) = 22.68, p < 0.0001, ANOVA with repeated measures). Importantly, the same concentration of Cd blocked Ca2+ entry through voltage-gated Ca2+ channels and blocked secretion stimulated by high K+ depolarization (Greitzer-Antes et al., 2013), suggesting that CDO may reflect a secretion-related conformational transition of Syx. Starting a quest to better understand the nature of this transition, we examined the involvement of the five positive charges, termed 5RK, in the juxtamembrane linker of Syx. Dynamic FRET analysis in cells expressing a 5RK charge neutralization mutant of CSYS, CSYS-5RK/A (in which the positive charges were replaced with alanines; Fig. 1A), revealed that CDO (blocked by Cd) did not occur in response to high K+ depolarization (Greitzer-Antes et al., 2013; Fig. 1C). Subsequently, we verified that the apparent elimination of this transition by 5RK neutralization was not due to a lower initial FRET signal that would indicate an inherent opening imposed by the mutation itself. Indeed, static FRET analysis showed that the initial FRET efficiencies, measured under resting conditions, of CSYS and CSYS-5RK/A were similar (Fig. 1D), indicating that the 5RK/A mutation abolished CDO.
Figure 1.
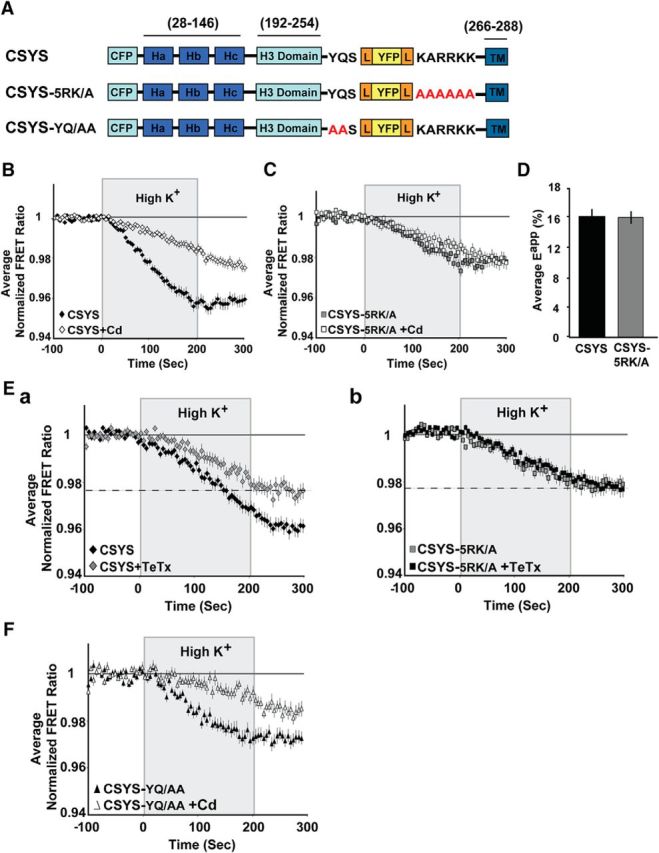
5RK is crucial for the CDO of CSYS. A, Domain structure of CSYS (Greitzer-Antes et al., 2013), and its mutants, CSYS-5RK/A and CSYS-YQ/AA. Two fluorescent molecules, CFP and YFP, are fused to Syx via flexible linkers (L). B, Changes in the average normalized FRET ratio in PC12 cells expressing CSYS in response to high K+ depolarization. Addition of 200 μm Cd to both physiological and high K+ solutions results in a smaller decrease in the FRET ratio compared with its absence (white diamonds, n = 109, and black diamonds, n = 122, respectively; 9 experiments; F(79,15800) = 22.68, p < 0.0001 ANOVA with repeated measures). C, No significant difference in the FRET ratio reduction between cells expressing CSYS-5RK/A in the presence and absence of Cd (white squares, n = 67, and gray squares, n = 51, respectively; 5 experiments). D, The initial FRET efficiencies (Eapp) of PC12 cells expressing CSYS and CSYS-5RK/A are similar (black bar, n = 31, and gray bar, n = 25, respectively; 2 experiments). E, CDO involves ternary SNARE complex assembly Ea, The reduction in the FRET ratio of CSYS (black diamond, n = 47; 4 experiments) was statistically smaller (F(79,6636) = 5.232, p < 0.0001, ANOVA with repeated measures) upon Syb2 cleavage by 30 nm TeTx-LC (gray diamond, n = 39; 4 experiments). Eb, On the contrary, the FRET ratio of CSYS-5RK/A (gray square, n = 31; 4 experiments) is not affected by the Syb2 cleavage (black square, n = 35; 4 experiments). Dashed lines indicate a similar FRET ratio reduction of CSYS-5RK/A compared with CSYS+TeTx-LC. F, Addition of Cd to CSYS-YQ/AA-expressing cells results in a smaller decrease in the FRET ratio compared with its absence (white triangles, n = 33 and black triangles, n = 41, respectively; 3 experiments; F(79,3239) = 5.621, p < 0.0001 ANOVA with repeated measures).
To better understand the role of 5RK in Syx conformational transition, we had to determine the characteristics of CDO and whether it reports Syx conformational transition before or following SNARE complex assembly. To determine whether CDO occurs in the context of the ternary SNARE complex, which includes the vesicular SNARE Syb2, we used the light chain of tetanus toxin (TeTx-LC) to cleave Syb2, previously shown to inhibit exocytosis (Huang et al., 2001). Importantly, we showed that the opening of CSYS was significantly reduced following 4 h of incubation with the toxin (Fig. 1Ea; F(79,6636) = 5.232, p < 0.0001, ANOVA with repeated measures), whereas the opening of CSYS-5RK/A, which lacks CDO (see above), was not affected by the toxin (Fig. 1Eb). Notably, the opening of CSYS in the presence of the toxin was similar to that of CSYS-5RK/A (Fig. 1E). Together, these data show that CDO involves the ternary SNARE complex, underscoring a critical role for 5RK in Syx conformational transition following ternary SNARE complex assembly.
The next question that arose was whether the role of 5RK is related to the final zippering of the SNARE complex, which is mediated by the formation of continuous helices throughout the linker regions and the TM domains of Syx and Syb2 (Stein et al., 2009). In such a case, mutation of any residue in the Syx linker that makes contact with the Syb2 linker will result in impaired CDO. Thus, we targeted the YQ motif, which forms an aromatic layer with residues in the Syb2 linker, thought to be crucial for the linker contacts (Stein et al., 2009). Notably, dynamic FRET analysis in response to high K+ depolarization of cells expressing the mutant CSYS-YQ/AA (Fig. 1A) revealed a conformational opening that did consist of CDO (sensitive to Cd blockade; Fig. 1F; (F(79,3239) = 5.621, p < 0.0001, ANOVA with repeated measures), underscoring a unique role for 5RK in CDO, which is not related to the final zippering.
PIP2 is absolutely and specifically required for CDO
In continuing our quest to better understand the nature of CDO, we considered the role of Syx-phospholipid interactions, implicated in exocytosis regulation (Lam et al., 2008; Murray and Tamm, 2009; Honigmann et al., 2013; Khuong et al., 2013), focusing specifically on the most characterized candidate underlying these interactions, PIP2 (for review, see Martin, 2015). To assess the involvement of PIP2 in CDO regulation, two different experimental approaches of PIP2 depletion from PM were taken. First, we used the strongly Ca2+-activated phospholipase, Cη2 (PLCη2), shown to hydrolyze PIP2 from the PM of PC12 cells upon activation by high K+ depolarization (Kabachinski et al., 2016). PC12 cells transfected with CSYS, with or without mRFP-tagged PLCη2 were subjected to dynamic FRET analysis during high K+ depolarization. The opening of CSYS in the presence of PLCη2 was significantly smaller than in its absence (Fig. 2A; F(79,7110) = 7.192, p < 0.0001 ANOVA with repeated measures), insensitive to Cd and similar in size to the opening in the absence of the phospholipase but in the presence of Cd (Fig. 2B), indicating that the phospholipase eliminated CDO. To further substantiate this notion, the opening of CSYS-5RK/A, which completely lacks CDO (Greitzer-Antes et al., 2013; Fig. 1C), was compared in the absence and presence of coexpressed PLCη2 and indeed was found to be insensitive to the presence of the phospholipase (Fig. 2C), thus confirming that the phospholipase eliminates CDO in CSYS.
Figure 2.
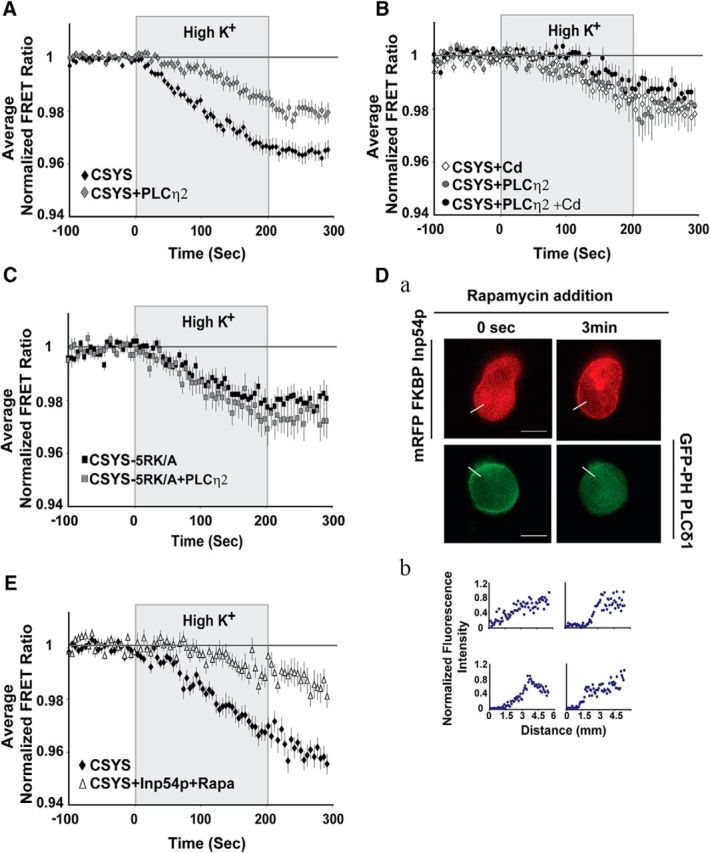
PIP2 depletion abolishes CDO of CSYS. A–C, PIP2 hydrolysis by PLCη2 (tagged with m-Kate fluorophore) in PC12 cells. A, PLCη2 coexpression with CSYS resulted in a significantly smaller decrease in the FRET ratio in response to high K+ depolarization compared with no treatment (black diamonds, n = 49, and gray diamonds, n = 50, respectively; 3 experiments; F(79,7110) = 7.192, p < 0.0001, ANOVA with repeated measures). B, Addition of Cd had no effect on the FRET ratio reduction of CSYS coexpressed with PLCη2 (black circles, n = 9; gray circles, n = 8), which was similar to that of CSYS alone but in the presence of Cd (white diamonds, n = 11); all cells were assayed in a single experiment. C, Hydrolysis of PIP2 by PLCη2 did not have any effect on the FRET reduction of CSYS-5RK/A (gray squares, n = 17; black squares, n = 17; 2 experiments). D, E, PIP2 depletion induced by the 5-phosphatase Inp54p system in PC12 cells. Da, Rapamycin addition induced heterodimerization between FRB and mRFP FKBP Inp54p, resulting in mRFP tagging of PM (top) and a decrease in the PM tagging of GFP-PH PLCδ, which binds PIP2 (bottom). Scale bar, 5 μm. Db, Normalized fluorescence intensity profiles of the above cells, indicating PM expression, determined from line scans (Da, white lines) taken from the outside to the middle of each cell. E, PIP2 depletion by the 5-phosphatase Inp54p system resulted in a significantly smaller decrease in the FRET ratio of CSYS in response to high K+ depolarization (white triangles, n = 16; black diamonds, n = 20; 2 experiments; F(79,2686) = 7.632, p < 0.0001, ANOVA with repeated measures).
Bearing in mind that hydrolysis of PIP2 by PLCη2 generates two second messengers, inositol 1,4,5-trisphosphate (IP3), which releases Ca2+ from intracellular stores (Nakahara et al., 2005), and diacylglycerol (DAG), which mediates the activation of protein kinase C, it was essential to demonstrate that the effect of PLCη2 is directly related to PIP2 depletion and not to the generation of second messengers. Thus, we used an additional PIP2 depletion system, the 5-phosphatase Inp54p, with the aim of replicating the above results obtained by PLCη2 expression. Activation by rapamycin of the Inp54p system, previously shown to deplete, within seconds, PM-associated PIP2 in living cells without producing DAG, IP3, or calcium signals (Suh et al., 2006; see Materials and Methods), resulted in recruitment of the phosphatase to PM and subsequent PIP2 depletion (Fig. 2D), significantly reducing the opening of CSYS in PC12 cells (Fig. 2E; F(79,2686) = 7.63, p < 0.0001, ANOVA with repeated measures; compare Fig. 2A), as expected. Together, we concluded that PIP2 depletion directly abolishes CDO, indicating that PIP2 is essential for this conformational transition of Syx.
Next, in an attempt to assess the unique role of PIP2 in CDO, we tested the role of another acidic phospholipid, phosphatidic acid (PA), previously shown to form functional interactions with Syx to facilitate membrane fusion (Lam et al., 2008). Additionally, it has been shown that overexpression of Phospholipase D1 (PLD1), a stimulus-activated enzyme that cleaves phosphatidylcholine to generate PA, enhances regulated exocytosis, whereas knockdown of PLD1 activity results in decreased secretion (Humeau et al., 2001; Vitale et al., 2001; Hughes et al., 2004; Huang et al., 2005; Zeniou-Meyer et al., 2007). Here, inhibition of PA production by PLD1 upon a 1 h incubation in 6 μm of the PLD-inhibitor VU0155056 (Fig. 3A) had no effect on CDO (Fig. 3B), underscoring the unique role of PIP2 in CDO.
Figure 3.

Inhibition of PA production in the PM has no effect on CSYS opening. A, Left, Confocal images, taken with a 488 excitation laser, demonstrating the cellular distribution of a wild-type PA-binding probe coupled to EGFP (wt-PABD) in PC12 cells. The cells were incubated for 1 h with (Ac, Ad) or without (Aa, Ab) 6 μm PLD-inhibitor, VU0155056. In both groups the cells were maintained under resting conditions (Aa, Ac) and stimulated for 5 min with high K+ solution (Ab, Ad). Middle, Normalized fluorescence intensity profiles, as determined by line scans (5 μm; white line) taken from the outside to the middle of each cell, corresponding to PA distribution on PM shown on the left. Line scans were normalized and the peaks were aligned, for each condition. Right, Changes in membrane fluorescence before and after high K+ depolarization from cells expressing wt-PABD, in the presence (right) or absence (left) of the PLD-inhibitor (**p < 0.001). B, No significant difference between the changes of the FRET ratio of CSYS in response to high K+ depolarization in the absence (n = 22) and presence (n = 21) of the PLD-inhibitor.
Neutralization of the basic residues (5RK) in the juxtamembrane region of Syx increases spontaneous release but inhibits evoked release
At this point we were convinced that CDO reports one (or more) specific conformational transition of Syx following SNARE complex assembly, triggered by the elevation of intracellular [Ca2+] in response to high K+ depolarization; this transition is absolutely dependent on both PIP2 levels and intact 5RK. To gain insights into possible secretion-related correlates of the transition, we decided to pursue the 5RK dependence, rather than the PIP2 dependence, because 5RK is specific to Syx functioning, whereas PIP2 is involved in a growing number of processes, including synaptic growth as well as synaptic vesicle cycling and formation (Cremona et al., 1999; Wenk et al., 2001; Verstreken et al., 2009; Khuong et al., 2010; Martin, 2012). Thus, to address the secretion correlates of CDO, we used CSYS-5RK/A and compared its secretion phenotype with that of CSYS, using carbon fiber amperometry in PC12 cells. Specifically, we were interested in evaluating the impact of 5RK neutralization on both spontaneous and evoked releases, since previous studies that substituted native Syx with 5RK charge neutralization mutants reported an impact on evoked release only (Lam et al., 2008; Khuong et al., 2013). To reduce potentially confounding effects from endogenous Syx, we used cells transfected with the light chain of BoNT-C1α51 (BoNT-C1), which spares SNAP-25 cleavage and cleaves only endogenous Syx (Wang et al., 2011), precluding it from mediating membrane fusion (Schiavo et al., 1995). Previously, we showed that CSYS-R, a CSYS mutant bearing a K253I mutation, which confers resistance to BoNT-C1 (Lam et al., 2008), coexpressed with BoNT-C1 in PC12 cells, is resistant to the toxin and retains its PM expression (Greitzer-Antes et al., 2013). Importantly, CSYS-R was shown to rescue the inhibition of exocytosis by the toxin, both in PC12 cells (Greitzer-Antes et al., 2013) and in hippocampal neurons (Vertkin et al., 2015). Accordingly, here we showed that also the toxin-resistant CSYS-5RK/A-R retains its PM expression in cells coexpressing the toxin, in contrast with its toxin-nonresistant version (Fig. 4A). Thus, the secretion phenotype of CSYS was examined in cells coexpressing CSYS-R with the toxin by amperometric measurements, 1 min before (to detect spontaneous events) and 1 min following a 10 s application of the high K+ solution (to detect evoked events). Whereas no release, measured as amperometric spikes, was detected in cells expressing the toxin alone (mRFP-tagged; data not shown), in cells coexpressing the toxin with the toxin-resistant version of CSYS a small spontaneous release and a significant evoked release were detected, both in terms of charge release and number of spikes per cell (Fig. 4C; t(19) = 2.22, p = 0.038 and: t(19) = 2.1, p = 0.05, respectively; paired t test). Importantly, this pattern of secretion with a few spontaneous events and a significant evoked release, driven by the expressed CSYS, was quite similar to that driven by the endogenous Syx in native (not treated with the toxin; control) cells, both in terms of charge release and the number of events per cell (Fig. 4B; t(19) = 2.72, p = 0.012 and t(19) = 3.98, p = 0.001, respectively; paired t test). In addition, the single-spike characteristics, quantal size, half-width, and peak amplitude were similar between the two groups (Fig. 4E).
Figure 4.

Neutralization of 5RK increases the frequency of spontaneous events and diminishes evoked events. A, CSYS-5RK/A-R is resistant to cleavage by BoNT-C1 and targets to PM regions in PC12 cells. Top, Confocal images of PC12 cells demonstrating that CSYS-5RK/A-R expression with (Aiv) or without (Aiii) BoNT-C1 is distributed to the PM, in contrast with the cytosolic expression of CSYS-5RK/A in the presence of BoNT-C1 (compare Aii with Ai). Scale bar, 5 mm. Bottom, Normalized fluorescence intensity profiles of the above cells indicating PM or cytosolic expression. The fluorescence profiles were determined from line scans (red lines, top) taken from the outside to the middle of each cell. B–E, Representative amperometric recordings of catecholamine release (left), total charge release (middle), and average number of events (right) for spontaneous and evoked release from either native PC12 cells (control, B; t(19) = 2.72, *p = 0.012 and t(19) = 3.98, **p = 0.001, for total charge release and average number of events, respectively; paired t test) or those transfected with BoNT-C1 and cotransfected with either BoNT-C1-resistant CSYS (C; t(19) = 2.22, *p = 0.038 and: t(19) = 2.1, *p = 0.05, for total charge release and average number of events, respectively; paired t test) or BoNT-C1-resistant CSYS-5RK/A (D), before and after application of high K+ depolarization by a 10 s pressure pulse (horizontal line) through a micropipette positioned close to the cell. Ea, An amperometric spike example. Eb–Ed, Single-spike analysis of the cells in A–C: mean half-width (Eb), mean peak amplitude (Ec), and mean quantal size (Ed) of individual events.
Strikingly, the pattern of secretion of the 5RK neutralization mutant CSYS-5RK/A, examined in cells coexpressing the toxin with CSYS-5RK/A-R (Fig. 4D), was markedly different from that of CSYS (Fig. 4C), assayed in parallel in the same experiments. First, spontaneous release in CSYS-5RK/A-expressing cells was sixfold higher in the number of spikes per cell (F(2,52) = 9.763, p < 0.0001) and approximately sevenfold higher in the charge release per cell (F(2,52) = 7.517, p < 0.001; ANOVA followed by post hoc Tukey's test). Second, whereas in CSYS-expressing cells the evoked release was approximately fourfold higher than the spontaneous release, both in the number of spikes and in the charge release per cell (Fig. 4C), in CSYS-5RK/A-expressing cells there was no significant difference between the spontaneous and evoked releases (Fig. 4D), suggesting that the evoked release was inhibited by the 5RK/A mutation. To substantiate this notion, we performed additional kinetic analysis, looking at the time course of the amperometric events by plotting the normalized cumulative spike frequency distribution and the normalized cumulative charge release distribution (Fig. 5A and B, respectively). Remarkably, in contrast to those of CSYS, the plots of CSYS-5RK/A could be well fitted to one-line function throughout periods before and after the high K+ depolarization, confirming the absence of evoked release.
Figure 5.

5RK neutralization mutation diminishes evoked release. Cumulative distribution of spike frequency (A) or quantal release (B) normalized to the total number of spikes or to the total quantal release, respectively, in all cells in Figure 4B and C. Each point was normalized to the total number of spikes or to the total quantal release in each group before and after application of high K+ solution (horizontal line). The cumulative distribution of each plot was fitted with a linear function; r2 (A) = 0.98 and 0.99, r2 (B) = 0.97 and 0.98 for CSYS and CSYS-5RKA, respectively.
Although the secretion phenotype of CSYS was completely different from that of CSYS-5RK/A, the single-spike characteristics, quantal size, half-width, and peak amplitude were not appreciably different between the two groups and were similar to those of control cells (Fig. 4E). Together, these results strongly suggest that 5RK neutralization enhances spontaneous release and inhibits evoked release, suggesting that 5RK plays a role in clamping spontaneous release and in promoting Ca2+-triggered release.
Neutralization of residues adjacent to 5RK in the juxtamembrane region of Syx has no apparent effect on secretion
Correlation of the FRET analyses of CSYS and CSYS-5RK/A (Fig. 1B,C) with the corresponding amperometric analyses (Fig. 4C,D) suggested a link between the absence of CDO in CSYS-5RK/A in the FRET analysis and its unique secretion phenotype, with enhanced spontaneous release and inhibited evoked release. To substantiate this link, we characterized the secretion phenotype of CSYS-YQ/AA, which exhibited CDO in the FRET analysis (Fig. 1E). Indeed, in contrast to CSYS-5RK/A, the secretion phenotype of CSYS-YQ/AA, with few spontaneous events and a significant evoked release, was not appreciably different from that of CSYS, assayed in parallel in the same experiments (Fig. 6; CSYS: t(16) = 2.89, p = 0.011; CSYS-YQ/AA: t(13) = 2.17, p = 0.048; paired t test). The single-spike characteristics were not appreciably different between the two groups (data not shown).
Figure 6.

YQ neutralization mutant, CSYS-YQ/AA, does not affect the secretion phenotype. A, Average number of events for spontaneous and evoked release per cells transfected with BoNT-C and either CSYS or CSYS-YQ/AA, both of which are resistant to BoNT-C cleavage (CSYS t(16) = 2.89, *p = 0.011; CSYS-YQ/AA t(13) = 2.17, *p = 0.048; paired t test:). B, Cumulative distribution of spike frequency in the cells of A, normalized to the total number of spikes in each group after 120 s of recording; 60 s in low K+ (5 mm K+) and 60 s after 10 s application of high K+ (100 mm) solutions (indicated by a line).
Discussion
Using our Syx-based FRET probe, CSYS, we captured a CDO of Syx in response to high K+ depolarization (Greitzer-Antes et al., 2013), which occurs following SNARE complex assembly (Fig. 1E), and is absolutely dependent on the juxtamembrane 5RK stretch (Greitzer-Antes et al., 2013); Fig. 1B,C), but not on the adjacent YQ motif, believed to be crucial for final zippering of the complex (Fig. 1G). This indicated that the participation of 5RK in CDO goes beyond its participation in the final zippering and that CDO may reflect a highly important functional property of Syx, as indicated by the conservation of juxtamembrane charge clusters and the essential role of the juxtamembrane domain of the yeast syntaxin proteins Sso1/2p in SNARE function in vivo (Van Komen et al., 2005). The finding that PIP2, fundamental for maintaining regulated exocytosis (for review, see Martin, 2012), is specifically essential for CDO (Figs. 2, 3), underscored the notion of an important, secretion-related, structural transition of Syx in which 5RK plays a significant role. Guided by this notion, we conducted amperometric analysis to assess the role of 5RK in catecholamine release from PC12 cells. Remarkably, we found that the charge neutralization mutation 5RK/A that abolished CDO (Figs. 4, 5), but not the YQ/AA mutation that retained CDO (Fig. 6), induced the enhancement of spontaneous release, with a sixfold increased number of events and a sevenfold increased charge per cell, and the concomitant inhibition of Ca2+-triggered release. In fact, studies of the molecular basis of vesicle fusion conducted at various synapses have shown that Ca2+-triggered and spontaneous release are often concomitantly regulated, in either opposing or equal directions (Schneggenburger and Rosenmund, 2015). Our data suggest that 5RK is involved in the concomitant regulation of both release processes, but in opposing directions. Although the regulation of these two processes do not necessarily share a single underlying mechanism, the convergence at 5RK enables this stretch to act as a fusion clamp that makes catecholamine release from PC12 cells dependent on stimulation that elevates the intracellular Ca2+ concentration.
The role of 5RK in Ca2+ triggering
To elucidate the mechanism underlying the role of 5RK in Ca2+-triggered dense-core vesicle release one should consider the groups of proteins that, upon binding to Ca2+, trigger the merger of vesicles with the PM. The fast kinetics of Ca2+ sensors belonging to the synaptotagmin family, especially Syt1,2 play a direct role in triggering membrane fusion. Other Ca2+ sensing proteins having intrinsically slower kinetics, such as Munc13 proteins, act on Ca2+-dependent processes upstream of fusion (Bittner and Holz, 1992; von Rüden and Neher, 1993) such as mobilization and priming of vesicles (Pinheiro et al., 2016). The slow time scale of CDO, which may reflect one or more Ca2+-dependent transitions of Syx along the sustained high K+ depolarization that are related to the replenishment of the readily-releasable pool (RRP) of vesicles, corresponds to the slow Ca2+ sensors' longer temporal scale. Specifically, the well documented role of Munc13s in mediating the opening of Syx, by accelerating the transition from the closed Syx-Munc18-1 complex to the SNARE complex (Rizo and Xu, 2015), supports their involvement in CDO. Furthermore, a possible link between the function of Munc13s as effectors for PIP2 in evoked release (Martin, 2015) and the well documented PIP2-binding ability of 5RK (see Introduction), may underlie a crucial role of 5RK in RRP replenishment.
Nevertheless, by acknowledging the role of Syt1 as the main Ca2+ sensor for Ca2+-triggered fusion (Xu et al., 2007), we reasoned that Syt1, despite its fast kinetics, could still be relevant to CDO. According to this scenario CDO may reflect one or more Ca2+-dependent transitions of Syx directly related to fast Syt-1-driven Ca2+-triggered fusion events. However, these events involve a nonhomogenous population of vesicles undergoing a series of maturation steps and reaching the RRP, poised for Ca2+-triggered fusion, at different times throughout the sustained high K+ depolarization. Along these lines, we considered the suggested model for Syx's role in Ca2+-triggered release, based on the recently resolved crystal structures of the Syt1-SNARE complex (Zhou et al., 2015). This model assigns an important functional role to a flat face of the Syt1-SNARE complex, with an extensive pattern of positive charges comprising the polybasic Syt1 region and the C-terminal end of the SNARE complex, which upon Ca2+ elevation, forms simultaneous PM interactions that deform the PM and promote fusion. Noting that this model relies on SNARE structures up to the juxtamembrane linkers of Syx and Syb2, we reasoned that an extended model that includes the linkers may reveal that the positive charges of 5RK extend and enhance the positively charged surface of the C-terminal end of the SNARE complex, providing substantial leverage for more efficient PM deformation. To test this notion, we attempted to extend the model so that it includes the linkers. First, we conducted a structural alignment by superimposing on the structure of the Syt1-SNARE complex (Zhou et al., 2015; 5ccg.pdb) the previously resolved structure of the SNARE complex in its cis state, which includes the linkers and the TM domains of Syx and Syb2 (Stein et al., 2009; 3ipd.pdb). Successful structural alignment with RMS = 0.95Å (involving 65 atoms) generated a combined complex (Fig. 7A) that could potentially serve as a basis for an extended model that incorporates 5RK. We focused only on the linker regions in the combined complex. A plot of the electrostatic surface of the linker region of Syx (AA 259–265: SKARRKKI), alone (Fig. 7Ba) or together with that of Syb2 (AA 91–95: KNLKM; Fig. 7Bb) shows a positive charge density, as was previously shown for the resolved cis-SNARE complex (Stein et al., 2009). Zooming in Figure 7, Ca and Cb, it becomes clear that the 5RK residues K260, R263, and R262 create a positive surface that points in the same direction as that of the Syt1 polybasic region; the latter is thought to point toward the PM (Zhou et al., 2015). Together, we suggest that, upon entry of Ca2+ and helical extension of the SNARE complex, the positively charged surface created by K260, R262, and R263 extends and enhances the positively charged surface of the C-terminal end of the SNARE complex. Thus, it acts in concert with the positive surface created by the Syt1 polybasic region and forms simultaneous PM interactions, resulting in membrane deformation that promotes fusion. In this context, 5RK plays a crucial role in Ca2+-triggered PM deformation that promotes fusion.
Figure 7.

3D-alignment-based reconstitution of Syt1-SNARE model. A, SNARE complex protein alignment based on magic fit (Swiss-prot viewer) derived from the pdb files 5ccg.pdb (containing the Syt-SNARE complex without the linker and transmembrane regions of Syx and Syb2) and 3ipd.pdb (containing the cis-SNARE complex with linker and transmembrane regions of Syx and Syb2). Aa, Superimposition of SNARE complex proteins (red/purple, Syx; gray/yellow, SNAP25; dark/light blue, Syb2) together with the C2B domain of Syt1 (green). Ab, Underlined alignment of Syx helices from 5ccg.pdb (purple) and 3ipd.pdb (red) files. Amino acid stretches of Syx used in 5ccg.pdb (purple) and 3ipd.pdb (red) files are denoted. The root mean square deviation of the alignment was 0.95Å. B, A reconstituted Syt1-SNARE complex with an electrostatic surface for the Syx linker region, alone (Ba) and together with the Syb2 linker region (Bb). Stick model representing amino acids of the linker regions of Syx and Syb2 (red and blue, respectively). C, Close-up views of the electrostatic surface of the linker region (the electrostatic potential was visualized within a −5 to 5 kT/e range) composed of Syx, alone (Ca) and together with Syb2 (Cb), in longitudinal projection along the helices (left) and projection through a cross-section (black bars) at the Syx-K264 level, looking toward the N-termini of Syx and Syb2 (right). D, Electrostatic surface relevant for synaptotagmin-SNARE complex (reconstituted based on the cis-SNARE complex model; A) interaction with presynaptic PM and a vesicular membrane, in longitudinal (Da) and cross sectional (Da, black bar; Db) projections. The linker region of Syx and the polybasic region of Syt1 are denoted to emphasize the similar orientations of their positive charges with respect to the PM.
The role of 5RK in spontaneous release
In considering the mechanism underlying the clamping of spontaneous release by 5RK, we envision two possible scenarios, not mutually exclusive. The first scenario relies on the well accepted role of Syt1 in suppression of spontaneous release that is separated from its function in evoked release (Schupp et al., 2016). This function of Syt1 was suggested to possibly involve a specific configuration of the C2B lob, which differs from that involved in its function in evoked release (Bai et al., 2016), and to possibly arrest the SNARE complex in a partially assembled trans state (Chicka et al., 2008) for long enough that complexin can bind and stabilize clamping (Schupp et al., 2016). The fusion clamp property of 5RK can be incorporated into this scenario, assuming that its interaction with the PM, possibly via the well documented interaction with phospholipids (see Introduction), stabilizes the specific configuration of the Syt1-SNARE complex that clamps spontaneous fusion.
In a second, more minimalistic scenario, the interaction of 5RK with phospholipids before the Ca2+ stimulus acts as a membrane anchor that clamps spontaneous release by preventing spontaneous fluctuations of the loose Syx linker (in the partially zippered SNARE complex). Consequently, this phospholipid-5RK interaction prevents Syx's linker from interacting with that of Syb2 to finalize zippering of the complex, which will draw the plasma and vesicular membranes closely together to promote the formation of the fusion pore. This scenario is along the lines of the fusion clamp role suggested for the tryptophans, W89 and W90, in the Syb2 linker (Lindau, 2012; Lindau et al., 2012) and suggests for the 5RK stretch a complementary role, on the PM side, in clamping spontaneous release.
Our results support the notion (Schneggenburger and Rosenmund, 2015) that at various instances both evoked and spontaneous releases are highly regulated processes that use the same pool of vesicles (as suggested in our study by the similar single-spike parameters for spontaneous and evoked events). Moreover, in line with studies that have shown that proteins involved in regulating the final steps of vesicle fusion often concomitantly regulate Ca2+-triggered and spontaneous releases (Schneggenburger and Rosenmund, 2015), we put forward the notion that the 5RK stretch acts as a molecular target for these regulations.
Noteworthy, our Syx-based FRET probe enabled us to gain insights about novel Syx functions in vivo, lending credence to the use this probe as a tool to gain insights into mechanistic questions linked to Syx functionality in secretion.
Footnotes
This work was supported by the Israel Academy of Sciences (Grant 234/14 to I.L.).
The authors declare no competing financial interests.
References
- An SJ, Almers W (2004) Tracking SNARE complex formation in live endocrine cells. Science 306:1042–1046. 10.1126/science.1102559 [DOI] [PubMed] [Google Scholar]
- Aoyagi K, Sugaya T, Umeda M, Yamamoto S, Terakawa S, Takahashi M (2005) The activation of exocytotic sites by the formation of phosphatidylinositol 4,5-bisphosphate microdomains at syntaxin clusters. J Biol Chem 280:17346–17352. 10.1074/jbc.M413307200 [DOI] [PubMed] [Google Scholar]
- Bai H, Xue R, Bao H, Zhang L, Yethiraj A, Cui Q, Chapman ER (2016) Different states of synaptotagmin regulate evoked versus spontaneous release. Nat Commun 7:10971. 10.1038/ncomms10971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner MA, Holz RW (1992) Kinetic analysis of secretion from permeabilized adrenal chromaffin cells reveals distinct components. J Biol Chem 267:16219–16225. [PubMed] [Google Scholar]
- Chen X, Tomchick DR, Kovrigin E, Araç D, Machius M, Südhof TC, Rizo J (2002) Three-dimensional structure of the complexin/SNARE complex. Neuron 33:397–409. 10.1016/S0896-6273(02)00583-4 [DOI] [PubMed] [Google Scholar]
- Chicka MC, Hui E, Liu H, Chapman ER (2008) Synaptotagmin arrests the SNARE complex before triggering fast, efficient membrane fusion in response to Ca2+. Nat Struct Mol Biol 15:827–835. 10.1038/nsmb.1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona O, Di Paolo G, Wenk MR, Lüthi A, Kim WT, Takei K, Daniell L, Nemoto Y, Shears SB, Flavell RA, McCormick DA, De Camilli P (1999) Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99:179–188. 10.1016/S0092-8674(00)81649-9 [DOI] [PubMed] [Google Scholar]
- Fang Q, Lindau M (2014) How could SNARE proteins open a fusion pore? Physiology 29:278–285. 10.1152/physiol.00026.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer D, Sutton RB, Brunger AT, Jahn R (1998) Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc Natl Acad Sci U S A 95:15781–15786. 10.1073/pnas.95.26.15781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Liu SS, Qiu S, Cheng W, Zheng J, Luo JH (2007) Fluorescence resonance energy transfer analysis of subunit assembly of the ASIC channel. Biochem Biophys Res Commun 359:143–150. 10.1016/j.bbrc.2007.05.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Zorman S, Gundersen G, Xi Z, Ma L, Sirinakis G, Rothman JE, Zhang Y (2012) Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science 337:1340–1343. 10.1126/science.1224492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greitzer-Antes D, Barak-Broner N, Berlin S, Oron Y, Chikvashvili D, Lotan I (2013) Tracking Ca2+-dependent and Ca2+-independent conformational transitions in syntaxin 1A during exocytosis in neuroendocrine cells. J Cell Sci 126:2914–2923. 10.1242/jcs.124743 [DOI] [PubMed] [Google Scholar]
- Honigmann A, van den Bogaart G, Iraheta E, Risselada HJ, Milovanovic D, Mueller V, Müllar S, Diederichsen U, Fasshauer D, Grubmüller H, Hell SW, Eggeling C, Kühnel K, Jahn R (2013) Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment. Nat Struct Mol Biol 20:679–686. 10.1038/nsmb.2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Altshuller YM, Hou JC, Pessin JE, Frohman MA (2005) Insulin-stimulated plasma membrane fusion of Glut4 glucose transporter-containing vesicles is regulated by phospholipase D1. Mol Biol Cell 16:2614–2623. 10.1091/mbc.E04-12-1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Kang YH, Pasyk EA, Sheu L, Wheeler MB, Trimble WS, Salapatek A, Gaisano HY (2001) Ca2+ influx and cAMP elevation overcame botulinum toxin A but not tetanus toxin inhibition of insulin exocytosis. Am J Physiol Cell Physiol 281:C740–750. [DOI] [PubMed] [Google Scholar]
- Hughes WE, Elgundi Z, Huang P, Frohman MA, Biden TJ (2004) Phospholipase D1 regulates secretagogue-stimulated insulin release in pancreatic beta-cells. J Biol Chem 279:27534–27541. 10.1074/jbc.M403012200 [DOI] [PubMed] [Google Scholar]
- Humeau Y, Vitale N, Chasserot-Golaz S, Dupont JL, Du G, Frohman MA, Bader MF, Poulain B (2001) A role for phospholipase D1 in neurotransmitter release. Proc Natl Acad Sci U S A 98:15300–15305. 10.1073/pnas.261358698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D (2012) Molecular machines governing exocytosis of synaptic vesicles. Nature 490:201–207. 10.1038/nature11320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Scheller RH (2006) SNAREs: engines for membrane fusion. Nat Rev Mol Cell Biol 7:631–643. 10.1038/nrm2002 [DOI] [PubMed] [Google Scholar]
- James DJ, Khodthong C, Kowalchyk JA, Martin TF (2008) Phosphatidylinositol 4,5-bisphosphate regulates SNARE-dependent membrane fusion. J Cell Biol 182:355–366. 10.1083/jcb.200801056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabachinski G, Kielar-Grevstad DM, Zhang X, James DJ, Martin TF (2016) Resident CAPS on dense-core vesicles docks and primes vesicles for fusion. Mol Biol Cell 27:654–668. 10.1091/mbc.E15-07-0509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khuong TM, Habets RL, Slabbaert JR, Verstreken P (2010) WASP is activated by phosphatidylinositol-4,5-bisphosphate to restrict synapse growth in a pathway parallel to bone morphogenetic protein signaling. Proc Natl Acad Sci U S A 107:17379–17384. 10.1073/pnas.1001794107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khuong TM, Habets RL, Kuenen S, Witkowska A, Kasprowicz J, Swerts J, Jahn R, van den Bogaart G, Verstreken P (2013) Synaptic PI(3,4,5)P3 is required for Syntaxin1A clustering and neurotransmitter release. Neuron 77:1097–1108. 10.1016/j.neuron.2013.01.025 [DOI] [PubMed] [Google Scholar]
- Kochubey O, Schneggenburger R (2011) Synaptotagmin increases the dynamic range of synapses by driving Ca(2)+-evoked release and by clamping a near-linear remaining Ca(2)+ sensor. Neuron 69:736–748. 10.1016/j.neuron.2011.01.013 [DOI] [PubMed] [Google Scholar]
- Krishnakumar SS, Radoff DT, Kümmel D, Giraudo CG, Li F, Khandan L, Baguley SW, Coleman J, Reinisch KM, Pincet F, Rothman JE (2011) A conformational switch in complexin is required for synaptotagmin to trigger synaptic fusion. Nat Struct Mol Biol 18:934–940. 10.1038/nsmb.2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam AD, Tryoen-Toth P, Tsai B, Vitale N, Stuenkel EL (2008) SNARE-catalyzed fusion events are regulated by Syntaxin1A-lipid interactions. Mol Biol Cell 19:485–497. 10.1091/mbc.E07-02-0148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang B, Kiessling V, Tamm LK (2013) Prefusion structure of syntaxin-1A suggests pathway for folding into neuronal trans-SNARE complex fusion intermediate. Proc Natl Acad Sci U S A 110:19384–19389. 10.1073/pnas.1314699110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindau M. (2012) High resolution electrophysiological techniques for the study of calcium-activated exocytosis. Biochim Biophys Acta 1820:1234–1242. 10.1016/j.bbagen.2011.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindau M, Hall BA, Chetwynd A, Beckstein O, Sansom MS (2012) Coarse-grain simulations reveal movement of the synaptobrevin C-terminus in response to piconewton forces. Biophys J 103:959–969. 10.1016/j.bpj.2012.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Shin YK (2016) SNARE zippering. Biosci Rep 36:e00327. 10.1042/BSR20160004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margittai M, Fasshauer D, Jahn R, Langen R (2003) The Habc domain and the SNARE core complex are connected by a highly flexible linker. Biochemistry 42:4009–4014. 10.1021/bi027437z [DOI] [PubMed] [Google Scholar]
- Martin TF. (2012) Role of PI(4,5)P(2) in vesicle exocytosis and membrane fusion. Subcell Biochem 59:111–130. 10.1007/978-94-007-3015-1_4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TF. (2015) PI(4,5)P(2)-binding effector proteins for vesicle exocytosis. Biochim Biophys Acta 1851:785–793. 10.1016/j.bbalip.2014.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Südhof TC (2005) Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron 48:547–554. 10.1016/j.neuron.2005.09.006 [DOI] [PubMed] [Google Scholar]
- Mosharov EV. (2008) Analysis of single vesicle exocytosis events recorded by amperometry. Methods Mol Biol 440:315–327. 10.1007/978-1-59745-178-9_24 [DOI] [PubMed] [Google Scholar]
- Murray DH, Tamm LK (2009) Clustering of syntaxin-1A in model membranes is modulated by phosphatidylinositol 4,5-bisphosphate and cholesterol. Biochemistry 48:4617–4625. 10.1021/bi9003217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray DH, Tamm LK (2011) Molecular mechanism of cholesterol- and polyphosphoinositide-mediated syntaxin clustering. Biochemistry 50:9014–9022. 10.1021/bi201307u [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara M, Shimozawa M, Nakamura Y, Irino Y, Morita M, Kudo Y, Fukami K (2005) A novel phospholipase C, PLC(eta)2, is a neuron-specific isozyme. J Biol Chem 280:29128–29134. 10.1074/jbc.M503817200 [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y (2004) Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci 7:1104–1112. 10.1038/nn1311 [DOI] [PubMed] [Google Scholar]
- Park Y, Seo JB, Fraind A, Pérez-Lara A, Yavuz H, Han K, Jung SR, Kattan I, Walla PJ, Choi M, Cafiso DS, Koh DS, Jahn R (2015) Synaptotagmin-1 binds to PIP(2)-containing membrane but not to SNAREs at physiological ionic strength. Nat Struct Mol Biol 22:815–823. 10.1038/nsmb.3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro PS, Houy S, Sørensen JB (2016) C2-domain containing calcium sensors in neuroendocrine secretion. J Neurochem 139:943–958. 10.1111/jnc.13865 [DOI] [PubMed] [Google Scholar]
- Rizo J, Xu J (2015) The synaptic vesicle release machinery. Annu Rev Biophys 44:339–367. 10.1146/annurev-biophys-060414-034057 [DOI] [PubMed] [Google Scholar]
- Rothman JE. (1996) The protein machinery of vesicle budding and fusion. Protein Sci 5:185–194. 10.1002/pro.5560050201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo G, Shone CC, Bennett MK, Scheller RH, Montecucco C (1995) Botulinum neurotoxin type C cleaves a single Lys-Ala bond within the carboxyl-terminal region of syntaxins. J Biol Chem 270:10566–10570. 10.1074/jbc.270.18.10566 [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Rosenmund C (2015) Molecular mechanisms governing Ca(2+) regulation of evoked and spontaneous release. Nat Neurosci 18:935–941. 10.1038/nn.4044 [DOI] [PubMed] [Google Scholar]
- Schupp M, Malsam J, Ruiter M, Scheutzow A, Wierda KD, Söllner TH, Sørensen JB (2016) Interactions between SNAP-25 and synaptotagmin-1 are involved in vesicle priming, clamping spontaneous and stimulating evoked neurotransmission. J Neurosci 36:11865–11880. 10.1523/JNEUROSCI.1011-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang S, Wang C, Liu B, Wu Q, Zhang Q, Liu W, Zheng L, Xu H, Kang X, Zhang X, Wang Y, Zheng H, Wang S, Xiong W, Liu T, Zhou Z (2014) Extracellular Ca(2)(+) per se inhibits quantal size of catecholamine release in adrenal slice chromaffin cells. Cell Calcium 56:202–207. 10.1016/j.ceca.2014.07.006 [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Wiederhold K, Müller EM, Milosevic I, Nagy G, de Groot BL, Grubmüller H, Fasshauer D (2006) Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. EMBO J 25:955–966. 10.1038/sj.emboj.7601003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A, Weber G, Wahl MC, Jahn R (2009) Helical extension of the neuronal SNARE complex into the membrane. Nature 460:525–528. 10.1038/nature08156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Inoue T, Meyer T, Hille B (2006) Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science 314:1454–1457. 10.1126/science.1131163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT (1998) Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395:347–353. 10.1038/26412 [DOI] [PubMed] [Google Scholar]
- Takanishi CL, Bykova EA, Cheng W, Zheng J (2006) GFP-based FRET analysis in live cells. Brain Res 1091:132–139. 10.1016/j.brainres.2006.01.119 [DOI] [PubMed] [Google Scholar]
- Tang J, Maximov A, Shin OH, Dai H, Rizo J, Südhof TC (2006) A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126:1175–1187. 10.1016/j.cell.2006.08.030 [DOI] [PubMed] [Google Scholar]
- Trimbuch T, Rosenmund C (2016) Should I stop or should I go? The role of complexin in neurotransmitter release. Nat Rev Neurosci 17:118–125. 10.1038/nrn.2015.16 [DOI] [PubMed] [Google Scholar]
- van den Bogaart G, Meyenberg K, Risselada HJ, Amin H, Willig KI, Hubrich BE, Dier M, Hell SW, Grubmüller H, Diederichsen U, Jahn R (2011) Membrane protein sequestering by ionic protein-lipid interactions. Nature 479:552–555. 10.1038/nature10545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G, Lang T, Jahn R (2013) Microdomains of SNARE proteins in the plasma membrane. Curr Top Membr 72:193–230. 10.1016/B978-0-12-417027-8.00006-4 [DOI] [PubMed] [Google Scholar]
- Van Komen JS, Bai X, Rodkey TL, Schaub J, McNew JA (2005) The polybasic juxtamembrane region of Sso1p is required for SNARE function in vivo. Eukaryot Cell 4:2017–2028. 10.1128/EC.4.12.2017-2028.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Ohyama T, Haueter C, Habets RL, Lin YQ, Swan LE, Ly CV, Venken KJ, De Camilli P, Bellen HJ (2009) Tweek, an evolutionarily conserved protein, is required for synaptic vesicle recycling. Neuron 63:203–215. 10.1016/j.neuron.2009.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertkin I, Styr B, Slomowitz E, Ofir N, Shapira I, Berner D, Fedorova T, Laviv T, Barak-Broner N, Greitzer-Antes D, Gassmann M, Bettler B, Lotan I, Slutsky I (2015) GABAB receptor deficiency causes failure of neuronal homeostasis in hippocampal networks. Proc Natl Acad Sci U S A 112:E3291–3299. 10.1073/pnas.1424810112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale N, Caumont AS, Chasserot-Golaz S, Du G, Wu S, Sciorra VA, Morris AJ, Frohman MA, Bader MF (2001) Phospholipase D1: a key factor for the exocytotic machinery in neuroendocrine cells. EMBO J 20:2424–2434. 10.1093/emboj/20.10.2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Rüden L, Neher E (1993) A Ca-dependent early step in the release of catecholamines from adrenal chromaffin cells. Science 262:1061–1065. 10.1126/science.8235626 [DOI] [PubMed] [Google Scholar]
- Wang D, Zhang Z, Dong M, Sun S, Chapman ER, Jackson MB (2011) Syntaxin requirement for Ca2+-triggered exocytosis in neurons and endocrine cells demonstrated with an engineered neurotoxin. Biochemistry 50:2711–2713. 10.1021/bi200290p [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk MR, Pellegrini L, Klenchin VA, Di Paolo G, Chang S, Daniell L, Arioka M, Martin TF, De Camilli P (2001) PIP kinase Iγ is the major PI(4,5)P(2) synthesizing enzyme at the synapse. Neuron 32:79–88. 10.1016/S0896-6273(01)00456-1 [DOI] [PubMed] [Google Scholar]
- Xu J, Mashimo T, Südhof TC (2007) Synaptotagmin-1, −2, and -9: Ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron 54:567–581. 10.1016/j.neuron.2007.05.004 [DOI] [PubMed] [Google Scholar]
- Yoshihara M, Littleton JT (2002) Synaptotagmin I functions as a calcium sensor to synchronize neurotransmitter release. Neuron 36:897–908. 10.1016/S0896-6273(02)01065-6 [DOI] [PubMed] [Google Scholar]
- Zeniou-Meyer M, Zabari N, Ashery U, Chasserot-Golaz S, Haeberlé AM, Demais V, Bailly Y, Gottfried I, Nakanishi H, Neiman AM, Du G, Frohman MA, Bader MF, Vitale N (2007) Phospholipase D1 production of phosphatidic acid at the plasma membrane promotes exocytosis of large dense-core granules at a late stage. J Biol Chem 282:21746–21757. 10.1074/jbc.M702968200 [DOI] [PubMed] [Google Scholar]
- Zheng J, Zagotta WN (2004) Stoichiometry and assembly of olfactory cyclic nucleotide-gated channels. Neuron 42:411–421. 10.1016/S0896-6273(04)00253-3 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Lai Y, Bacaj T, Zhao M, Lyubimov AY, Uervirojnangkoorn M, Zeldin OB, Brewster AS, Sauter NK, Cohen AE, Soltis SM, Alonso-Mori R, Chollet M, Lemke HT, Pfuetzner RA, Choi UB, Weis WI, Diao J, Südhof TC, Brunger AT (2015) Architecture of the synaptotagmin-SNARE machinery for neuronal exocytosis. Nature 525:62–67. 10.1038/nature14975 [DOI] [PMC free article] [PubMed] [Google Scholar]