

Abstract
Functional synapse formation is critical for the wiring of neural circuits in the developing brain. The cell adhesion molecule N-cadherin plays important roles in target recognition and synaptogenesis. However, the molecular mechanisms that regulate the localization of N-cadherin and the subsequent effects remain poorly understood. Here, we show that protein kinase D1 (PKD1) directly binds to N-cadherin at amino acid residues 836–871 and phosphorylates it at Ser 869, 871, and 872, thereby increasing the surface localization of N-cadherin and promoting functional synapse formation in primary cultured hippocampal neurons obtained from embryonic day 18 rat embryos of either sex. Intriguingly, neuronal activity enhances the interactions between N-cadherin and PKD1, which are critical for the activity-dependent growth of dendritic spines. Accordingly, either disruption the binding between N-cadherin and PKD1 or preventing the phosphorylation of N-cadherin by PKD1 in the hippocampal CA1 region of male rat leads to the reduction in synapse number and impairment of LTP. Together, this study demonstrates a novel mechanism of PKD1 regulating the surface localization of N-cadherin and suggests that the PKD1-N-cadherin interaction is critical for synapse formation and function.
SIGNIFICANCE STATEMENT Defects in synapse formation and function lead to various neurological diseases, although the mechanisms underlying the regulation of synapse development are far from clear. Our results suggest that protein kinase D1 (PKD1) functions upstream of N-cadherin, a classical synaptic adhesion molecule, to promote functional synapse formation. Notably, we identified a crucial binding fragment to PKD1 at C terminus of N-cadherin, and this fragment also contains PKD1 phosphorylation sites. Through this interaction, PKD1 enhances the stability of N-cadherin on cell membrane and promotes synapse morphogenesis and synaptic plasticity in an activity-dependent manner. Our study reveals the role of PKD1 and the potential downstream mechanism in synapse development, and contributes to the research for neurodevelopment and the therapy for neurological diseases.
Keywords: activity-dependent, N-cadherin, protein kinase D1, synapse formation, synaptic plasticity
Introduction
Brain function relies on the establishment and organization of proper neuronal circuitry consisting of a vast but sophisticated interconnected network of synapses. Synapses are specialized asymmetrical connections between neurons that function in information processing and integration. Synapse formation, elimination, and remodeling are essential for the development of neural circuits and cognitive functions, such as learning and memory (Kandel et al., 2014). Cell adhesion molecules, which can align the presynaptic active zone and the PSD across the synaptic cleft, have been intensively studied for their important roles in neurite outgrowth, neuronal polarity, neuronal migration, synapse formation, and synaptic plasticity both in the developing and mature brain (Togashi et al., 2002; Arikkath and Reichardt, 2008; Seong et al., 2015).
N-cadherin, one of the important cell adhesion molecules in the nervous system, is a Type I classical cadherin that is widely expressed in both presynaptic and postsynaptic membranes of excitatory neurons in mammals (Arikkath and Reichardt, 2008). N-cadherin mediates calcium-dependent, homophilic interactions across the synaptic cleft and plays crucial roles in dendrite morphogenesis, synapse formation, synaptic plasticity, and neural disorders associated with autism, bipolar disease, schizophrenia, and Alzheimer's disease (Schrick et al., 2007; Bacchelli et al., 2014; Tucci et al., 2014; Uribe-Arias et al., 2016). Although the functions of N-cadherin in neurons have been extensively investigated, the post-translational modification and regulation of the membrane localization of N-cadherin have been rarely studied.
Protein kinase D (PKD) is a serine/threonine protein kinase family that includes three kinase isoforms: PKD1, PKD2, and PKD3 (Johannes et al., 1994). PKDs are activated by a novel PKC family and are recruited to the plasma membrane or to intracellular membranes through binding DAG to achieve full activation (Valverde et al., 1994; Iglesias and Rozengurt, 1998; Iglesias et al., 1998). PKDs have been studied for their roles in diverse cellular functions, including cell proliferation and migration, gene expression, cell motility, and adhesion (Rozengurt, 2011). In the nervous system, PKDs have been shown to regulate Golgi functions, neuronal polarity, dendrite development, synaptic plasticity, and memory formation (Bisbal et al., 2008; Yin et al., 2008; Czöndör et al., 2009; Avriyanti et al., 2015; Bencsik et al., 2015). It has been reported that PKD1 can bind to and phosphorylate epithelial-type cadherin (E-cadherin) and thus regulate the adhesion of cancer cells (Jaggi et al., 2005). Based on the observations that N-cadherin shares a similar structure and a highly conserved cytoplasmic domain with E-cadherin (Gumbiner, 2005), we proposed that N-cadherin might contribute to the “cell–cell adhesion” between neurons under regulation of PKD1.
In this work, we used morphological and electrophysiological studies of cultured hippocampal neurons to demonstrate that PKD1 promotes functional synapse formation by acting upstream of N-cadherin. We found that PKD1 directly binds to N-cadherin at amino acid residues 836–871 and phosphorylates N-cadherin at Ser 869, 871, and 872, leading to increased membrane localization of N-cadherin. Furthermore, we demonstrated that disruption of the interaction between PKD1 and N-cadherin reduces the surface localization of N-cadherin and thus inhibits functional synapse formation and LTP. We also demonstrated that neural activity enhances the interaction between N-cadherin and PKD1 and that neural activity-driven spine growth requires the kinase activity of PKD1. This study identifies N-cadherin as a novel synaptic substrate of PKD1 and demonstrates the pivotal roles of these proteins in synaptogenesis and in synaptic plasticity.
Materials and Methods
Animals.
Sprague Dawley rats were housed in a temperature-controlled (23 ± 2°C) and humidity-controlled (50 ± 5%) environment. The animals were maintained on a 12 h light and dark cycle with food and water ad libitum. All animal studies were approved by the Animal Center of the Peking University Health Science Center, and the experiments were performed in accordance with the relevant guidelines, including any relevant details.
DNA constructs and chemicals.
Human PKD1 (h-PKD1) cDNA was cloned into the mammalian expression vector pcDNA3.1 to produce a his-myc-tagged expression plasmid. DN-hPKD1 (D727A-hPKD1) and rat-hPKD1 were generated by site-directed mutagenesis using PCR. Rat N-cadherin plasmid with myc tag (N-cad) was kindly provided by Prof. Tanaka (Osaka University, Osaka, Japan). N-cadΔ836–871 (N-cadherin with a deletion of amino acids of 836–871) and N-cad mut (N-cadherin with S869A, S871A, and S872A) were generated by PCR. GFP-hPKD1 and related mutants were constructed as previously described (Wang et al., 2004; Yin et al., 2008). shRNAs coexpressing EGFP against rat PKD1 and N-cadherin (shPKD1 and shN-cad) were purchased from Shanghai Genechem. The target mRNA sequences were as follows: for PKD1, 5′-GGUUCUGGACAGUUCGGAA-3′; and for N-cadherin, 5′-GACUGGAUUUCCUGAAGAU-3′ (Zhang et al., 2010; Lewallen et al., 2011). shRNA targeting a nonspecific sequence (shGFP) was used as a control. Endogenous rat PKD1 and transfected rat-hPKD1 can be knocked down by shPKD1, whereas transfected hPKD1 resists shPKD1.
To prepare constructs for protein purification, PKD1, cytoplasmic N-cadherin, and various constructs were cloned into the pGEX-5X-1 vector with an N-terminal GST tag or into the pET-28a(+) vector with an N-terminal His6 tag.
Two pairs of interfering peptides were created and used in the experiments. Fusion with the TAT protein sequence (RKKRRQRRR) renders the peptides cell-permeable. TAT-836–871, which was used to disrupt the binding of PKD1 to N-cadherin, consists of the TAT sequence and amino acids 836–871 of N-cadherin, the region through which N-cadherin binds to PKD1 (the sequence was RKKRRQRRR-INEGLKAADNDPTAPPYDSLLVFDYEGSGSTAGSLS). As a control peptide, amino acids 836–871 of N-cadherin were replaced with a scrambled sequence of the same 36 amino acids (termed TAT-scramble, the sequence was RKKRRQRRR-LPTALGLSLYKSFPGEYDGSVISDGEDAPDAASNNT). TAT-S3, which was used to disrupt the phosphorylation of N-cadherin by PKD1, consists of the TAT sequence and amino acids 865–878 of N-cadherin, including the three phosphorylation sites, Ser 869, 871, and 872 (the sequence was RKKRRQRRR-STAGSLSSLNSSSS). As a control peptide, Ser 869, 871, and 872 were replaced with alanine in the peptide TAT-S3A (the sequence was RKKRRQRRR-STAGALAALNSSSS). For neuronal morphology analysis, 3 μm peptide was added to the medium at DIV 10 after GFP transfection at DIV 8. For electrophysiological recording, 3 μm peptides were added to the medium without transfection. The TAT fusion peptides were synthesized by GL Biochem. The mass and purity of the peptides were verified by HPLC.
To study the role of PKD1 in neural activity-induced spine growth, 12 mm KCl (Sigma-Aldrich) was added to the culture medium to increase neural activity. For biochemical experiments, KCl was added at DIV 6 or 7, and cortical neurons were harvested 12 h later. For morphological experiments, KCl was added at DIV 10 (2 d after transfection) and maintained until hippocampal neurons were fixed at DIV 15.
Gö6976 (Calbiochem), Bim (Calbiochem), and PMA (Sigma-Aldrich) were diluted in DMSO (Sigma-Aldrich) for use.
Cell culture and transfection.
Hippocampal and cortical neurons were obtained from embryonic day 18 rat embryos of either sex and plated onto 35 mm dishes coated with poly-D-lysine (Sigma-Aldrich) at an appropriate density. After 4 h in plating media (10% FBS in DMEM), the cultures were transferred to neurobasal medium supplemented with 2% B27 and 0.5 mm GlutaMAX-I (Invitrogen). Half of the medium was replaced with fresh medium every 3 d. At DIV 3, cytosine arabinoside was added to the maintenance medium at a final concentration of 10 μm to inhibit glial proliferation. For most morphological and electrophysiological experiments, hippocampal neurons at DIV 8 were transfected with 3 μg of the indicated PKD1/N-cadherin constructs together with 1 μg GFP-expressing plasmid (pEGFP-N1) or shRNAs coexpressing EGFP to label entire neurons using Lipofectamine-2000 (Invitrogen) following the manufacturer's guidelines. For the biochemical experiments, cortical neurons were primarily used.
Mouse N2a and rat C6 cells were maintained in DMEM supplemented with 10% FBS. The cells were transfected with Lipofectamine-2000 (Invitrogen) according to the manufacturer's instructions.
Western blots.
Western blotting experiments were performed according to previously described protocols (Xing et al., 2012). The antibodies used in Western blots were rabbit polyclonal anti-PKD1 (sc-639; Santa Cruz Biotechnology), rat monoclonal anti-N-cadherin MNCD2 (Developmental Studies Hybridoma Bank), mouse monoclonal anti-N-cadherin 3B9 (Invitrogen), mouse monoclonal anti-myc (TA-01; Origene), mouse monoclonal anti-β-catenin (BD Biosciences), mouse monoclonal anti-GST (Applygen), mouse monoclonal anti-His (Applygen), mouse monoclonal anti-human Tfr (Invitrogen), mouse monoclonal anti-GFP (Santa Cruz Biotechnology), mouse monoclonal anti-β-actin (TA-09; Origene) followed by HRP-conjugated secondary antibodies (Sigma-Aldrich and Origene), mouse anti-Synaptophysin (BD Biosciences), and mouse anti-PSD95 (BD Biosciences).
Pulldown assay.
GST-fusion proteins were expressed in Escherichia coli BL21 (DE3) cells and purified using Glutathione Sepharose 4 Fast Flow (GE Healthcare Pharmacia) according to the manufacturer's instructions; His6-fusion proteins were purified using Ni-NTA agarose beads (QIAGEN) following the manufacturer's instructions. For binding assays, eluted His6-fusion protein was incubated with immobilized GST (as a control) or with GST-fusion protein for 2 h at 4°C. The mixture was then washed, eluted, and subjected to Western blot analysis.
Immunoprecipitation.
Protein extracts from transfected cells, neurons, or rat hippocampal tissues were prepared as for Western blots. Extracts containing 400–500 μg of protein were incubated with antibodies against PKD1 (1:50) or N-cadherin 3B9 (1:50) at 4°C for 3 h before incubation with protein A-Sepharose CL-4B resin (GE Healthcare) overnight. The immunoprecipitates were washed six times with 0.1% Triton X-100 in TBS. The final pellets were boiled in SDS-PAGE sample buffer and subjected to Western blot analysis.
Cell surface biotinylation assay.
Cell cultures were washed with ice-cold PBS, pH 8.0, and then incubated for 45 min at 4°C with EZ-Link Sulfo-NHS-SS-biotin (Pierce) to biotinylate surface proteins. After quenching with PBS containing 100 mm glycine and washing with PBS, pH 7.4, the cells were lysed with RIPA lysis buffer (1% Triton X-100 and 0.1% SDS in TBS). The supernatants from the cell lysates were incubated with Ultralink Plus immobilized streptavidin beads (Pierce) overnight at 4°C to capture biotinylated surface proteins. After washing the beads six times with RIPA buffer, the bound proteins were eluted by boiling for 5 min with SDS-PAGE sample buffer and were analyzed by Western blot.
In vitro kinase assay.
The phosphorylation of N-cadherin by PKD1 under various treatments was assessed following immunoprecipitation. Extracts containing 150 μg of protein obtained from lysates of young hippocampal neurons treated with Gö6976, Bim, PMA, or control solvent (DMSO) were incubated with an antibody against PKD1 (1:100) or with normal rabbit IgG (sc-2027, Santa Cruz Biotechnology) as a control to immunoprecipitate PKD1 at 4°C for 3 h before overnight incubation with protein A-Sepharose CL-4B resin. After successively washing with lysis buffer and assay buffer (30 mm Tris, pH 7.6, 10 mm MgCl2, and 1 mm dithiothreitol), the final pellets were resuspended to a final volume of 25 μl in assay buffer. To initiate the phosphorylation reaction, 10 μl of phosphorylation mix (assay buffer containing 5 μCi [γ-32P]ATP) was added. The mixture was incubated at 30°C for 30 min, and the reaction was terminated by the addition of SDS-PAGE sample buffer. After boiling for 5 min, the samples were subjected to SDS-PAGE. The gels were stained with Coomassie Brilliant Blue, dried, and exposed to x-ray film for autoradiography.
To detect the potential sites of phosphorylation in N-cadherin by PKD1, purified His6-N-cadherin c-ter protein and various constructs were respectively mixed with commercial purified PKD1 (Merck Millipore) in assay buffer in a final volume of 25 μl, and kinase assays were conducted as described above.
Immunostaining.
For analysis of PKD1 distribution in neurons, cultured hippocampal neurons were fixed at DIV 15 by 4% PFA and 4% sucrose in PBS for 20 min at room temperature, followed by 1 h blocking solution with 3% BSA and 0.3% Triton X-100 in PBS. Incubation with primary antibodies and secondary antibodies was done in the blocking buffer (1% BSA and 0.3% Triton X-100 in PBS) at 4°C overnight, respectively. Finally, the cells were mounted on slides, and the stained sections were examined using a Leica SP8 confocal laser scanning microscope at 63× (NA 1.4) objective at 0.75× and 4× zoom. Antibodies used in immunostaining are rabbit polyclonal anti-PKD1 (sc-639; Santa Cruz Biotechnology) and mouse monoclonal anti-N-cadherin 3B9 (Invitrogen) followed by AlexaFluor-488 goat anti-rabbit IgG (Invitrogen) and AlexaFluor-594 donkey anti-mouse IgG (Invitrogen).
PSD preparations.
Cytosol, synaptosome, synaptosomal membrane, and PSD fractions from the hippocampal CA1 region of rat brain were prepared using the previously described procedure with slight modifications (Carlin et al., 1980; Cho et al., 1992; Yang et al., 2015). In brief, the CA1 region of hippocampus was homogenized on ice using 20 strokes of a Teflon-glass homogenizer in 1 ml of HEPES-buffered sucrose (0.32 m sucrose, 4 mm HEPES, pH 7.4) containing freshly added protease inhibitors, then centrifuged at 800–1000 × g at 4°C to remove the pelleted nuclear fraction (P1). Supernatant (S1) was centrifuged at 10,000 × g for 15 min to yield the crude synaptosomal pellet (P2), and the pellet was washed once in 1 ml HEPES-buffered sucrose. P2 was lysed by hypoosmotic shock in 900 μl ice-cold 4 mm HEPES, pH 7.4, plus protease inhibitors, homogenized by pipetting and mixed for 30 min at 4°C. The lysate was centrifuged at 25,000 × g for 20 min to yield supernatant (S3, crude synaptic vesicle fraction) and pellet (P3, lysed synaptosomal membrane fraction). To prepare the PSD fraction, P3 was resuspended in 900 μl of ice-cold 50 mm HEPES, pH 7.4, 2 mm EDTA, plus protease inhibitors and 0.5% Triton X-100, rotated for 15 min at 4°C and centrifuged at 32,000 × g for 20 min to obtain the PSD pellet. PSD pellets were resuspended in 50 μl ice-cold 50 mm HEPES, pH 7.4, 2 mm EDTA plus protease inhibitors.
Analysis of neuronal morphology.
Dissociated hippocampal neurons grown at low density were used to determine the morphological characteristics of the neurons. Hippocampal neurons were fixed at DIV 15 in 4% PFA and 4% sucrose in PBS for 20 min at room temperature, mounted on slides, and imaged using an Olympus confocal laser scanning microscope (FV1000) with a 60× (NA 1.42) objective at 3× zoom. Spine stacks were acquired at 0.35 μm z intervals to image the entire thickness of the dendrite. Measurement and analysis of the images were performed using Image Pro Plus (Media Cybernetics). Protrusions whose length exceeds twice the value of its width are considered as filopodia, and the rest are spines. Spine density, protrusion density, and filopodia density are presented as average numbers per 10 μm of dendrites. For spine area, protrusion area, protrusion width, and protrusion length, the experimental groups were normalized to the control groups. Representative images were imaged using a Leica SP8 confocal laser scanning microscope with a 63× (NA 1.4) objective at 0.75× and 4× zoom.
Whole-cell recordings on cultured neurons.
E18 rat hippocampal neurons were cultured in vitro for 10–12 d. Whole-cell patch-clamp recordings were performed at room temperature using an EPC9 amplifier (HEKA Elektronik) and low-resistance pipettes (3–5 mΩ). The pipettes for whole-cell recordings were pulled (P-97, Sutter Instruments) using borosilicate glass capillaries (1.5 mm OD, 0.84 mm ID; VitalSense Scientific Instruments). The intracellular solutions contained the following (in mm): 110 K-gluconate, 20 KCl, 5 MgCl2, 20 HEPES, 0.6 EGTA, 2 Mg-ATP, and 0.2 Na-GTP, pH 7.3, 300 mOsm. The extracellular solution contained the following (in mm): 129 NaCl, 5 KCl, 30 glucose, 25 HEPES, 2 CaCl2, 1 MgCl2, 0.001 TTX, 0.001 strychnine, and 0.02 bicuculline, pH 7.3, 315 mOsm. For mEPSC recordings, neurons were voltage-clamped at −60 mV, and data were sampled at 10 kHz and filtered at 2 kHz. Neurons with a resting potential of at least −60 mV and an Rseries that fluctuated within 15% of the initial value (<20 mΩ) were analyzed. The total recording duration for each neuron was 250 s. Data analysis was performed blind using MiniAnalysis software (Synaptosoft) offline. The frequency and amplitude of mEPSCs were compared using Student's t test or one-way ANOVA with Bonferroni's post hoc test. p < 0.05 was considered to be statistically significant. Data are presented as mean ± SEM.
Stereotactic injection (intrahippocampal injection).
Stereotaxic surgery was performed under anesthesia with 10% chloral hydrate (m/v, i.p.). Peptides were bilaterally injected into the hippocampal CA1 region of rats using a microinfusion pump (CMA 100; CMA Microdialysis). The coordinates used to target the CA1 region of hippocampus were based on the stereotaxic atlas of Paxinos and Watson (1986): anteroposterior −3.8 mm, mediolateral ±2.5 mm, and dorsoventral −2.8 mm. Peptides were dissolved in saline to a concentration of 20 μg/μl, and 0.5 μl peptide per side was infused over 3 min followed by 2 additional min to allow diffusion before withdrawal. The injection of peptides was performed 12 h before slice preparation and electrophysiological recording.
Brain slice preparation and electrophysiological recording.
Three- to four-week-old rats were anesthetized with 10% chloral hydrate (m/v, i.p.). Brains were quickly removed and submerged in ice-cold sucrose-replaced ACSF dissection solution containing the following (in mm): 10 glucose, 213 sucrose, 3 KCl, 1 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 5 MgCl2, pH 7.4, 315 mOsm (saturated with 95% O2-5% CO2). Acute hippocampal slices (300 μm) were prepared using a Leica VT1000S vibratome. After dissection, slices were incubated at 33°C for at least 1 h in ACSF containing the following (in mm): 10 glucose, 125 NaCl, 5 KCl, 1.2 NaH2PO4, 26 NaHCO3, 2.6 CaCl2, 1.3 MgCl2 (saturated with 95% O2-5% CO2) and 100 μm picrotoxin, pH 7.4, 315 mOsm. A single slice was then transferred to a recording chamber and submerged beneath continuously perfusing oxygenated ACSF. The chamber was perfused at a speed of 2 ml/min.
fEPSPs were recorded in the stratum radiatum of CA1 with a patch pipette (1–2 mΩ) filled with ACSF. A bipolar tungsten stimulating electrode (FHC) was placed in the Schaffer collaterals to deliver test and conditioning stimuli. Test stimuli were delivered at 0.05 Hz with an intensity adjusted to elicit a fEPSP amplitude 40%–50% of maximum as the baseline. After obtaining a stable baseline for 20 min, LTP was induced by 4 consecutive trains (1 s) of stimuli at 100 Hz (high-frequency stimulation) separated by 20 s using the same stimulus strength as in baseline recording. LTP values were recorded for another 60 min at 0.05 Hz after high-frequency stimulation induction. The initial slope of the response was used to assess changes in synaptic strength. Paired-pulse responses were recorded at 50, 100, 150, 200, 250, and 300 ms interstimulus intervals. The facilitation ratio was calculated as fEPSP2 slope/fEPSP1 slope.
The fEPSP was recorded using an EPC10 amplifier (HEKA Elektronik) and PatchMaster software (HEKA Elektronik). The data were sampled at 10 kHz and filtered at 2 kHz. Statistical analysis was performed using Igor software (Wavemetrics) and Prism 5.0 (GraphPad Software). Two-way ANOVA with Bonferroni's post hoc test and Student's t test were used to determine the statistical significance of the data. p < 0.05 was considered to be statistically significant. Data are presented as mean ± SEM.
Experimental design and statistical analysis.
Morphological and electrophysiological studies on primary cultured hippocampal neurons or brain slices were conducted to explore the function of PKD1 and PKD1-N-cadherin interaction. Biochemical studies on primary cultured cortical neurons, etc., were conducted to explore the detailed mechanism.
Statistical analysis was performed using Prism 5.0 (GraphPad Software). Comparisons between groups were performed using Student's t test, one-way ANOVA followed by Bonferroni's post hoc test, or two-way ANOVA followed by Bonferroni's post hoc test. All data are presented as mean ± SEM.
Results
N-cadherin directly interacts with PKD1
Considering that PKD1 binds to and phosphorylates E-cadherin in LNCaP prostate cancer cells (Jaggi et al., 2005), we speculated that there might be a comparable link between PKD1 and N-cadherin in the nervous system. The GST pulldown assay verified our speculation that PKD1 could directly interact with N-cadherin (Fig. 1A). The immunoprecipitated PKD1 from the rat brain with PKD1 antibody was used in the in vitro kinase assay, and N-cad-C-ter was phosphorylated by the immunoprecipitated PKD1 (Fig. 1B, left). This phosphorylation level of N-cad-C-ter was reduced in the presence of the PKD1 inhibitor Gö6976 and the PKC inhibitor BIM, and increased by the application of the PKC activator PMA (Fig. 1B, right). The amount of the immunoprecipitated PKD1 was represented by IgG bands (the Coomassie staining IgG bands; Fig. 1B, right). These results support our hypothesis that PKD1 can bind to and phosphorylate N-cadherin. To determine the subcellular distribution of PKD1, we performed the PSD preparation experiments on hippocampal CA1 region of postnatal 3- to 4-week-old rats. Subcellular fractionation assays revealed that a significant amount of PKD1 was distributed in the PSD fraction (Fig. 1C), which suggested that PKD1 is mainly presented and interacts with N-cadherin in the postsynaptic compartment. Moreover, immunofluorescence staining showed that PKD1 colocalized with N-cadherin partially at dendritic spines of hippocampal neurons (Fig. 1D). These results demonstrated that PKD1 colocalizes with N-cadherin at dendritic spines mainly in the postsynaptic compartment and might regulate N-cadherin in the nervous system.
Figure 1.
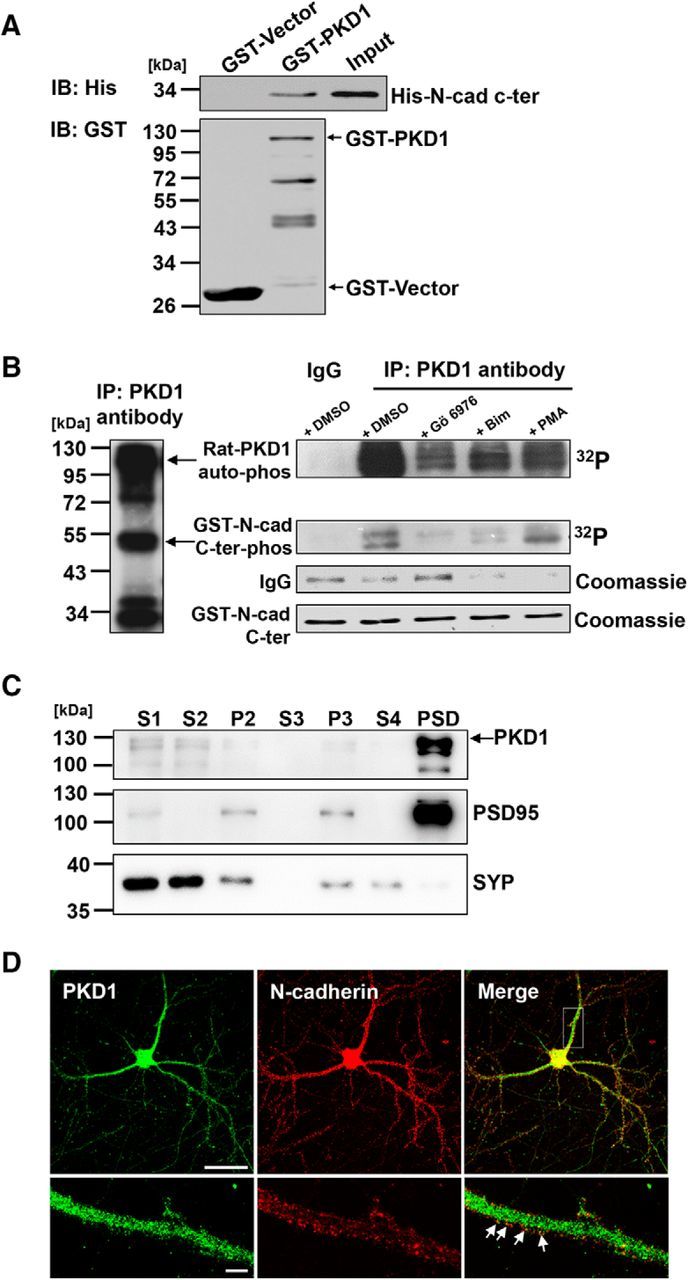
N-cadherin is directly associated with PKD1. A, IB of His-N-cad-c-ter pulldown by GST-PKD1 in vitro. Top, Probed for His-N-cad-c-ter binding to GST-PKD1. Bottom, Probed for total GST fusion protein present within each pulldown reaction. IB, Immunoblot. B, In vitro kinase assay showed that N-cadherin c-ter (N-cad 747–906) was phosphorylated by immunoprecipitated PKD1 (left) from the rat brain. The amount of the immunoprecipitated PKD1 was represented by the Coomassie staining IgG bands and indicated treatments were shown (right). Gö6976, a PKD1 inhibitor; Bim, a PKC inhibitor; and PMA, a PKC activator. C, Hippocampal CA1 region of rat brain lysates were fractionated by differential centrifugation, and subcellular fractions were analyzed by immunoblotting with antibodies to PKD1. S1, homogenates; S2, supernatant after P2 precipitation; P2, crude synaptosomes; S3, cytosol; P3, light membranes; S4, supernatant after PSD precipitation. SYP, synaptophysin; PSD95, post-synaptic density protein 95. D, Immunofluorescence staining for showing the colocalization of endogenous PKD1 and N-cadherin in DIV 15 hippocampal neurons. Arrows indicate the PKD1 puncta overlapping with N-cadherin. Scale bars: Top, 50 μm; Bottom, 5 μm.
PKD1 promotes synapse formation and function by acting upstream of N-cadherin
N-cadherin has been shown to play important roles in synapse formation and synaptic plasticity (Bozdagi et al., 2000; Friedman et al., 2015; Seong et al., 2015). Combined with the observations above, we first used in vitro cultured hippocampal neurons to examine the potential functions of PKD1 in synapse formation and function. The cultured neurons were transfected at DIV 8 with various PKD1 constructs along with GFP to show the morphology of dendritic spines where most excitatory synapses are formed. The density and morphology of spines were analyzed at DIV 15, the time at which the majority of the protrusions have developed into mature spines (Yoshimura et al., 2006; Tahirovic and Bradke, 2009). Overexpression of wild-type human PKD1 (hPKD1) increased the spine density (p < 0.0001, F(2,49) = 47.25, one-way ANOVA) and spine area (p < 0.0001, F(2,49) = 149.8, one-way ANOVA) compared with the empty vector, suggesting that PKD1 promotes spine formation (Fig. 2A). To confirm whether a correlation exists between the observed changes in spine morphology and synaptic transmission, we performed whole-cell patch-clamp recordings of mEPSCs in neurons to measure their basal synaptic properties. We found that overexpression of hPKD1 significantly increased mEPSC frequency (p < 0.0001, F(2,28) = 20.53, one-way ANOVA) but did not affect mEPSC amplitude (p = 0.6167, F(2,28) = 0.4918, one-way ANOVA) (Fig. 2B), indicating that a greater number of functional synapses were formed after hPKD1 overexpression.
Figure 2.
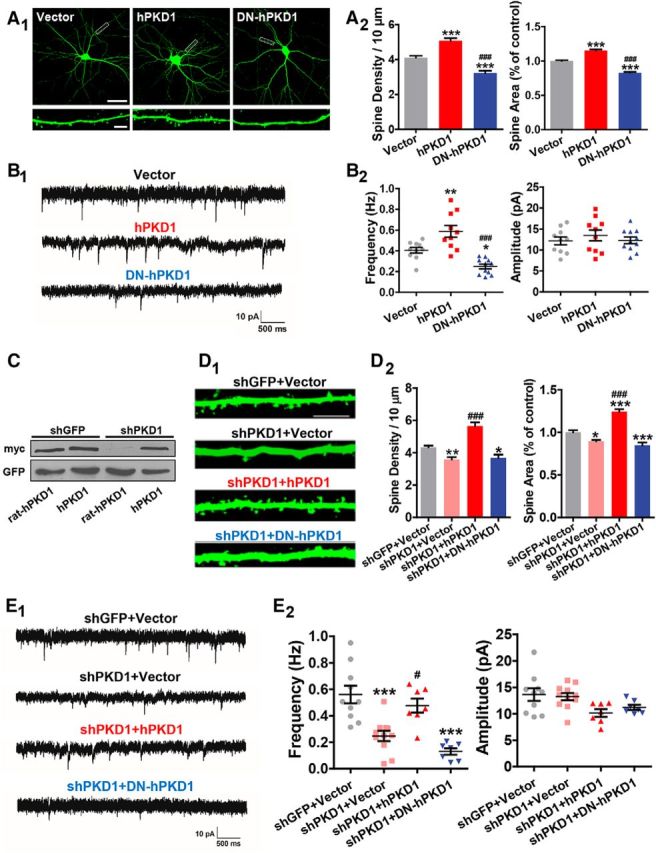
PKD1 is essential for synapse formation and function. A, Representative images and quantification of spine density and area in DIV 15 hippocampal neurons cotransfected GFP with his-myc-tagged Vector, hPKD1, or DN-hPKD1 at DIV 8 (n = 20, 16, 16 cells, respectively, in columns shown in the graphs). Scale bars: Top, 50 μm; Bottom, 5 μm. One-way ANOVA with Bonferroni's post hoc test: ***p < 0.001, compared with Vector. ###p < 0.001, compared with hPKD1. B, Representative traces (B1) and plots (B2) of mEPSC frequencies and amplitudes in hippocampal neurons cotransfected GFP with Vector, hPKD1, or DN-hPKD1 (n = 10, 10, 11 cells, respectively). Calibration: 10 pA, 500 ms. One-way ANOVA with Bonferroni's post hoc test: *p < 0.05, compared with Vector. **p < 0.01, compared with Vector. ###p < 0.001, compared with hPKD1. No significant differences in mEPSC amplitude were detected. C, Knockdown effect of myc-tagged rat PKD1 (rat-hPKD1) by shPKD1 in N2a cells. Rat-hPKD1 is an analog of hPKD1 with the rat target sequence of shPKD1 replacing the homologous domain in hPKD1 by site-directed mutagenesis. D, Representative images and quantification of spine density and area of DIV 15 hippocampal neurons transfected with shGFP+Vector, shPKD1+Vector, shPKD1+hPKD1, or shPKD1+DN-hPKD1 at DIV 8 (n = 18, 17, 15, and 16 cells, respectively). Scale bar, 2.5 μm. One-way ANOVA with Bonferroni's post hoc test: *p < 0.05, compared with shGFP+Vector. **p < 0.01, compared with shGFP+Vector. ***p < 0.001 compared with shGFP+Vector. ###p < 0.001, compared with shPKD1+Vector. E, Representative traces (E1) and plots (E2) of the mEPSCs in hippocampal neurons transfected with shGFP+Vector, shPKD1+Vector, shPKD1+hPKD1, or shPKD1+DN-hPKD1 (n = 10, 11, 7, and 7 cells, respectively). Calibration: 10 pA, 500 ms. One-way ANOVA with Bonferroni's post hoc test: **p < 0.01, compared with shGFP+Vector. ***p < 0.001, compared with shGFP+Vector. #p < 0.05 compared with shPKD1+Vector. No significant differences in mEPSC amplitude were detected among the groups. Data are mean ± SEM.
As a serine/threonine kinase, the activity of endogenous PKD1 changes in parallel with neuronal maturation. By using an antibody recognizing autophosphorylated human PKD1, Czöndör et al. (2009) reported that the relative amount of activated PKD1 decreases since DIV 2 and maintains at a certain level since DIV 7 in the mouse hippocampal cultures. To examine whether the effects of PKD1 on spine development rely on its kinase activity or not, a kinase-dead version of hPKD1 (dominant-negative hPKD1, DN-hPKD1) was overexpressed and it decreased the spine density, spine area, and mEPSC frequency compared with the empty vector (Fig. 2A,B), suggesting that the kinase activity is indispensable for synapse formation and function.
To investigate whether endogenous PKD1 is required for spine development, we constructed a short hairpin RNA of rat PKD1 (hereafter referred to as shPKD1) and examined its silencing efficiency by Western blot. The expression of myc-tagged rat PKD1 (rat-hPKD1) was significantly decreased in shPKD1-transfected N2a cells compared with that of control shRNA (shGFP)-transfected cells. In addition, shPKD1 had no effect on hPKD1, and hPKD1 rescued the knockdown effects of shPKD1, excluding off-target effects of the shRNA (Fig. 2C). Knockdown of PKD1 significantly decreased the spine density, spine area, and mEPSC frequency in cultured neurons, and these could be rescued by overexpression of hPKD1. However, DN-hPKD1 failed to rescue the spine phenotypes and synaptic transmission defects caused by PKD1 knockdown (Fig. 2D,E; for spine density, p < 0.0001, F(3,62) = 31.52, one-way ANOVA; for spine area, p < 0.0001, F(3,57) = 51.76, one-way ANOVA; for mEPSC frequency, p < 0.0001, F(3,31) = 14.88, one-way ANOVA; for mEPSC amplitude, p = 0.0307, F(3,31) = 3.374, one-way ANOVA). These results indicate that PKD1 is both necessary and sufficient to promote functional synapses formation in cultured hippocampal neurons.
Because membrane N-cadherin promotes synapse formation during development (Benson and Tanaka, 1998; Togashi et al., 2002), PKD1 might exert its effects by regulating the localization of N-cadherin on neuronal membrane. We did biotinylation assays on N-cadherin in C6 cells transfected with wild-type or dominant-negative hPKD1 to evaluate their effects on the membrane distribution of N-cadherin. The results showed that overexpression of GFP-hPKD1 promoted the surface localization of N-cadherin, whereas GFP-DN-hPKD1 reduced the amount of N-cadherin on the cell surface (Fig. 3A; p = 0.0002; F(2,9) = 25.77; one-way ANOVA), demonstrating that the kinase activity of PKD1 is responsible for its regulation of the cell surface localization of N-cadherin.
Figure 3.
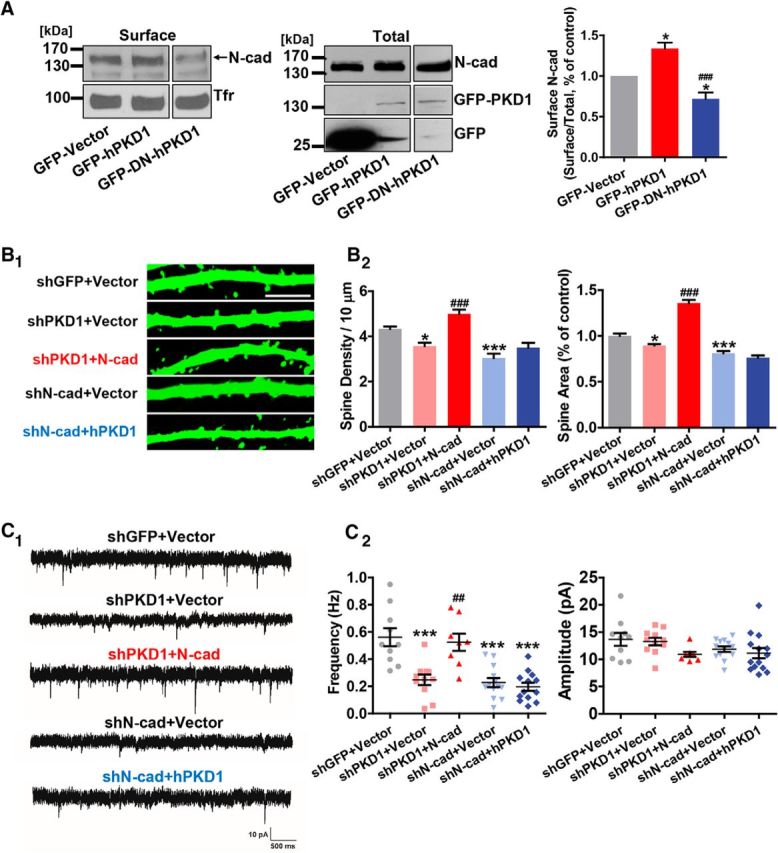
PKD1 promotes synapse formation and function by acting upstream of N-cadherin. A, Surface biotinylation assay for membrane location of N-cadherin in C6 cells overexpressing GFP-Vector, GFP-hPKD1, or GFP-DN-hPKD1; n = 4. Tfr, Transferrin receptor. One-way ANOVA with Bonferroni's post hoc test: *p < 0.05, compared with GFP-Vector. ###p < 0.001, compared with hPKD1. B, Representative images and quantification of spine density and area of DIV 15 hippocampal neurons transfected with shGFP+Vector, shPKD1+Vector, shPKD1+N-cad (myc-N-cadherin), shN-cad+Vector, or shN-cad+hPKD1 at DIV 8 (n = 18, 17, 20, 16, 16 cells, respectively). Scale bar, 2.5 μm. Same shGFP+Vector and shPKD1+Vector data as in Figure 2D. One-way ANOVA with Bonferroni's post hoc test: *p < 0.05, compared with shGFP+Vector. ***p < 0.001, compared with shGFP+Vector. ###p < 0.001, compared with shPKD1+Vector. C, Representative traces (C1) and plots (C2) of mEPSC frequencies and amplitudes in hippocampal neurons transfected with shGFP+Vector, shPKD1+Vector, shPKD1+N-cad, shN-cad+Vector, or shN-cad+hPKD1 (n = 10, 11, 8, 13, 13 cells, respectively). Calibration: 10 pA, 500 ms. Same shGFP+Vector and shPKD1+Vector data as in Figure 2E. One-way ANOVA with Bonferroni's post hoc test: ***p < 0.001, compared with shGFP+Vector. ##p < 0.01, compared with shPKD1+Vector. No significant differences in mEPSC amplitude were detected. Data are mean ± SEM.
The results above suggest that PKD1 very likely functions upstream of N-cadherin. If so, the reduced spine density, spine area, and synaptic strength caused by PKD1 knockdown should be rescued by overexpression of N-cadherin. Indeed, morphological and electrophysiological studies showed that overexpression of N-cadherin rescued the phenotypes caused by knocking down PKD1, whereas overexpression of PKD1 failed to rescue the knockdown effect of N-cadherin (Fig. 3B,C; for spine density, p < 0.0001, F(4,82) = 21.29, one-way ANOVA; for spine area, p < 0.0001, F(4,67) = 93.26, one-way ANOVA; for mEPSC frequency, p < 0.0001, F(4,50) = 14.72, one-way ANOVA; for mEPSC amplitude, p = 0.0915, F(4,51) = 2.122, one-way ANOVA). Together, these results show that PKD1 regulates the membrane localization of N-cadherin to promote synapse formation and function.
PKD1 binds directly to the C terminus of N-cadherin at amino acid residues 836–871
The results described above demonstrate that PKD1 interacts with N-cadherin in vitro and in cultured hippocampal neurons (Fig. 1A,C). To identify the critical amino acids in N-cadherin that are responsible for its binding with PKD1, we generated a series of GST-tagged constructs expressing various portions of the C terminus of N-cadherin (N-cad-C-ter) and tested them with His-PKD1. We found that the fragment of 836–871 amino acids had the highest affinity for binding with PKD1 (Fig. 4A, lane 5 compared with lanes 4 and 6), suggesting that amino acid residues 836–871 of N-cadherin are necessary and sufficient for its binding with PKD1.
Figure 4.
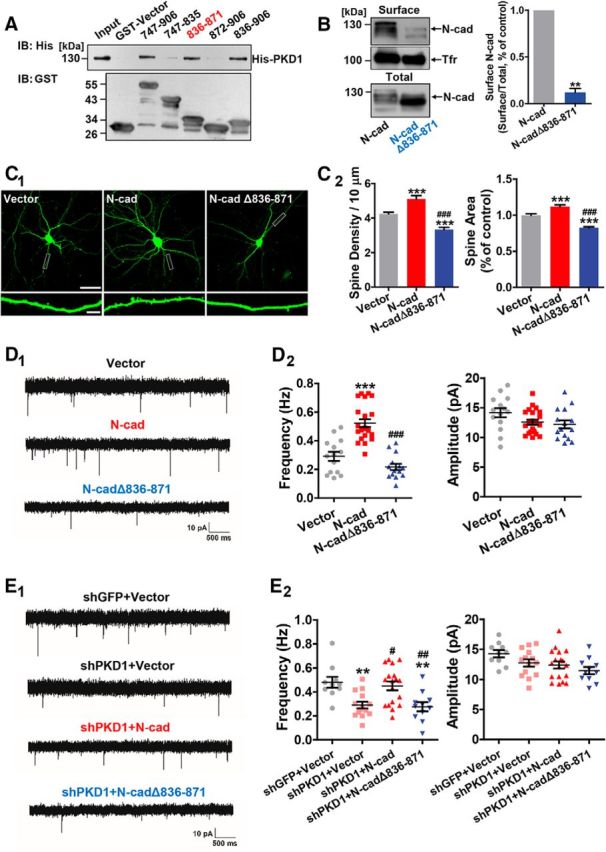
PKD1 binds to N-cadherin at amino acid residues 836–871 and promotes functional synapse formation. A, Immunoblot of His-PKD1 pulldown by the indicated constructs of cytoplasmic N-cadherin. Top, Probed for His-PKD1 binding to cytoplasmic N-cadherin fused to GST. Bottom, Probed for total GST fusion protein present within each pulldown reaction. IB, Immunoblot. B, Membrane location of N-cadherin in N2a cells transfected with N-cad or N-cadΔ836–871 (N-cadherin with a deletion of amino acids 836–871); n = 3. **p < 0.01 (paired t test). C, Representative images and quantification of spine density and area of DIV 15 hippocampal neurons cotransfected GFP with Vector, N-cad, or N-cadΔ836–871 at DIV 8 (n = 16, 15, and 17 cells, respectively). Scale bars: Top, 50 μm; Bottom, 5 μm. One-way ANOVA with Bonferroni's post hoc test: ***p < 0.001, compared with Vector. ###p < 0.001, compared with N-cad. D, Representative traces (D1) and plots (D2) of mEPSC frequencies and amplitudes in hippocampal neurons cotransfected GFP with Vector, N-cad, or N-cadΔ836–871 (n = 14, 22, and 15 cells, respectively). Calibration: 10 pA, 500 ms. One-way ANOVA with Bonferroni's post hoc test: ***p < 0.001, compared with Vector. ###p < 0.001, compared with N-cad. E, Representative traces (E1) and plots (E2) of frequencies and amplitudes of the mEPSCs in hippocampal neurons transfected with shGFP+Vector, shPKD1+Vector, shPKD1+N-cad, or shPKD1+N-cadΔ836–871 (n = 10, 14, 18, and 11 cells, respectively). Calibration: 10 pA, 500 ms. One-way ANOVA with Bonferroni's post hoc test: **p < 0.01, compared with shGFP+Vector. #p < 0.05, compared with shPKD1+Vector. ##p < 0.01, compared with shPKD1+Vector. No significant differences in mEPSC amplitude were detected among the groups. Data are mean ± SEM.
We further explored the function of PKD1 binding with N-cadherin through an analog of N-cadherin with a deletion of the PKD1 binding motif (N-cadΔ836–871). The results of surface biotinylation assays showed that a much lower proportion of N-cadΔ836–871 was present on the cell membrane compared with full-length N-cadherin (Fig. 4B; p = 0.0022, t(2) = 21.40, paired t test). Overexpression of full-length N-cadherin increased the spine density, spine area, and mEPSC frequency, whereas overexpression of N-cadΔ836–871 caused a reduction in these parameters (Fig. 4C,D; for spine density, p < 0.0001, F(2,45) = 38.83, one-way ANOVA; for spine area, p < 0.0001, F(2,41) = 56.16, one-way ANOVA; for mEPSC frequency, p < 0.0001, F(2,48) = 36.78, one-way ANOVA; for mEPSC amplitude, p = 0.0808, F(2,48) = 2.653, one-way ANOVA). Furthermore, N-cadΔ836–871 failed to rescue the reduced mEPSC frequency caused by PKD1 knockdown (Fig. 4E; for mEPSC frequency, p = 0.0003, F(3,49) = 7.695, one-way ANOVA; for mEPSC amplitude, p = 0.0461, F(3,49) = 2.865, one-way ANOVA). These results suggest that deletion of amino acids 836–871 of N-cadherin blocks the effects of PKD1 on synapse formation and function.
To disrupt the binding of endogenous PKD1 and N-cadherin, we created an interfering peptide of which the sequence corresponds to amino acids 836–871 of N-cadherin, and made it cell-permeable by fusing it to the HIV TAT protein (RKKRRQRRR) (Brooks et al., 2005; Wang et al., 2014). This peptide is referred to as TAT-836–871. A peptide corresponding to a randomly scrambled version of residues 836–871 of N-cadherin fused to the TAT protein was used as a control (TAT-scramble). TAT-836–871 efficiently disrupted the binding of PKD1 and N-cadherin in cultured neurons (see Fig. 6A; p = 0.0006, t(5) = 7.602, paired t test). It also led to a significant reduction in the surface localization of N-cadherin (see Fig. 6B; p = 0.0075, t(2) = 11.44, paired t test). Moreover, TAT-836–871 treatment of cultured neurons caused a reduction in the spine density and spine area, as well as an inhibition of synaptic transmission, compared with treatment with TAT-scramble (see Fig. 6C,D; for spine density, p = 0.0016, t(23) = 3.576, unpaired t test; for spine area, p < 0.0001, t(24) = 6.243, unpaired t test; for mEPSC frequency, p = 0.0077, t(18) = 3.002, unpaired t test; for mEPSC amplitude, p = 0.3826, t(18) = 0.8949, unpaired t test). These results confirm that the direct binding of PKD1 to amino acid residues 836–871 of N-cadherin is critical for functional synapse formation.
Figure 6.
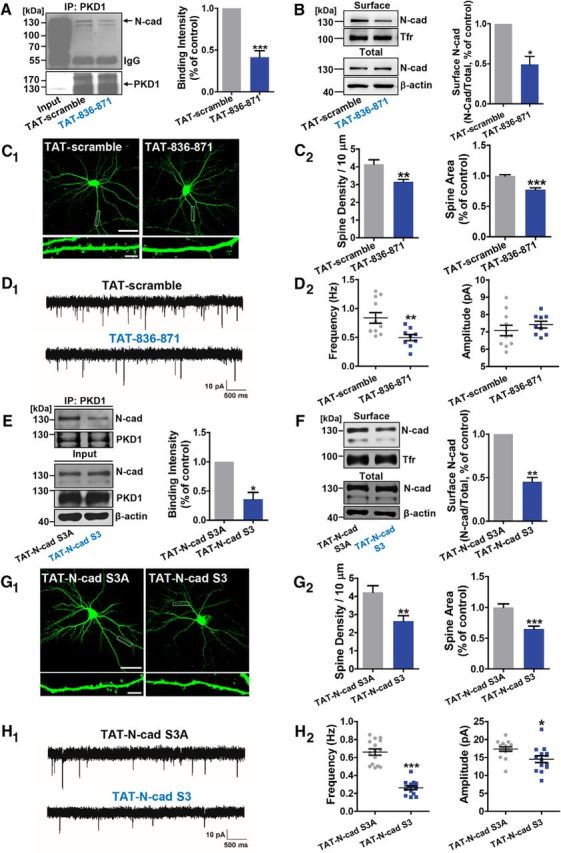
The effects of interfering peptides disrupting PKD1-N-cadherin interactions in cultured neurons. A, Effect of 3 μm TAT-836–871 on the binding of endogenous PKD1 and N-cadherin in cortical neurons shown by IP; n = 6. ***p < 0.001 (paired t test). B, Surface N-cadherin in cortical neurons treated with TAT-scramble (3 μm) or TAT-836–871 (3 μm); n = 3. *p < 0.05 (paired t test). C, Representative images and quantification of spine density and area in DIV 15 hippocampal neurons transfected with GFP at DIV 8 and treated with TAT-scramble (3 μm) or TAT-836–871 (3 μm) at DIV 10 (n = 16, 17 cells). Scale bars: Top, 50 μm; Bottom, 5 μm. **p < 0.01 (unpaired t test). ***p < 0.001 (unpaired t test). D, Representative mEPSC traces (D1) and plots (D2) of the frequencies and amplitudes of hippocampal neurons treated with TAT-scramble (3 μm) or TAT-836–871 (3 μm) (n = 11, 9 cells). Calibration: 10 pA, 500 ms. **p < 0.01 (unpaired t test). E, Coimmunoprecipitation of PKD1 with N-cadherin from cortical neurons under TAT-N-cad S3A (3 μm) or TAT-N-cad S3 (3 μm) treatments; n = 4. *p < 0.05 (paired t test). F, Surface N-cadherin in cortical neurons treated with TAT-N-cad S3A (3 μm) or TAT-N-cad S3 (3 μm); n = 3. **p < 0.01 (paired t test). G, Representative images and quantification of spine density and area of DIV 15 hippocampal neurons transfected with GFP at DIV 8 and treated with TAT-N-cad S3A (3 μm) or TAT-N-cad S3 (3 μm) at DIV 10 (n = 17, 17 cells). Scale bars: Top, 50 μm; Bottom, 5 μm. **p < 0.01 (unpaired t test). ***p < 0.001 (unpaired t test). H, Representative mEPSC traces (H1) and plots (H2) of the frequencies and amplitudes of hippocampal neurons treated with TAT-N-cad S3A (3 μm) or TAT-N-cad S3 (3 μm) (n = 15, 14 cells). Calibration: 10 pA, 500 ms. *p < 0.05 (unpaired t test). ***p < 0.001 (unpaired t test). Data are mean ± SEM.
N-cadherin is a novel substrate of PKD1
PKD1 participates in a variety of cellular processes through its phosphorylation of downstream substrates (Cabrera-Poch et al., 2004; Döppler et al., 2005; Krueger et al., 2010; Rozengurt, 2011). Based on our discoveries that PKD1 phosphorylates N-cadherin (Fig. 1B) and that the kinase activity of PKD1 is essential for functional synapse formation and the membrane distribution of N-cadherin (Figs. 2, 3), we sought to identify the critical sites at which PKD1 phosphorylates N-cadherin and explore the subsequent effects of the phosphorylation.
First, we generated a series of His-tagged constructs encoding successively longer fragments of N-cad-C-ter; each longer construct encodes several additional serines or threonines that might possibly be phosphorylated (Fig. 5A). Phosphorylation assays showed that PKD1 phosphorylated His-747–872 (Fig. 5B), which contains Ser 869, 871, and 872. Because these three serines have leucines in their 5 positions, which match the consensus phosphorylation sequence of PKD1 (Nishikawa et al., 1998), we speculated that Ser 869, 871, and 872 in N-cadherin might be PKD1 phosphorylation sites. The mutation of these three serine residues to alanines decreased the phosphorylation of N-cadherin by PKD1 (Fig. 5C). These results suggest that N-cadherin is a novel substrate of PKD1 in the nervous system and can be phosphorylated at Ser 869, 871, and 872.
Figure 5.
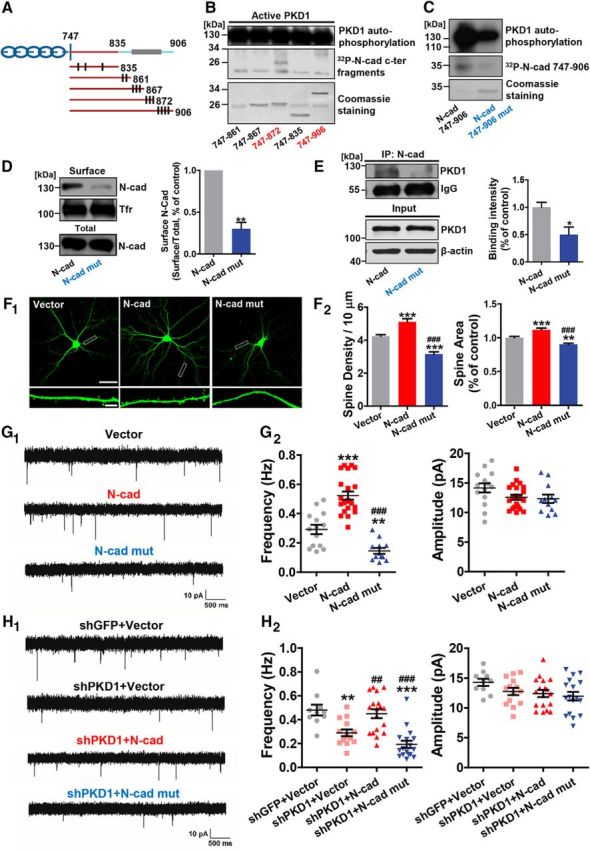
PKD1 phosphorylates N-cadherin at Ser 869, 871, and 872 and promotes functional synapse formation. A, Schematic depiction of cytoplasmic N-cadherin constructs illustrating the approximate localization of serine and threonine residues (black lines) that represent potential sites of phosphorylation by PKD1. B, His-747–906 and His-747–872 containing the potential phosphorylation sites Ser 869, 871, and 872 were phosphorylated by PKD1. C, N-cadherin c-ter (His-N-cad-747–906) phosphorylation by purified PKD1 was reduced by the triple mutation (His-N-cad-747–906 mut) as shown by in vitro kinase assay. D, Membrane location of N-cadherin in N2a cells transfected with N-cad or Ν-cad mut (N-cadherin with Ser 869, 871, and 872 mutated to alanine); n = 4. **p < 0.01 (paired t test). E, Coimmunoprecipitation of N-cadherin with PKD1 in N2a cells transfected with N-cad or N-cad mut; n = 3. *p < 0.05 (unpaired t test). IP, Immunoprecipitation. F, Representative images and quantification of spine density and area of DIV 15 hippocampal neurons cotransfected GFP with Vector, N-cad, or N-cad mut at DIV 8 (n = 16, 15, 18 cells, respectively). Same Vector and N-cad data as in Figure 4C. Scale bars: Top, 50 μm; Bottom, 5 μm. One-way ANOVA with Bonferroni's post hoc test: **p < 0.01, compared with Vector. ***p < 0.001, compared with Vector. ###p < 0.001, compared with N-cad. G, Representative mEPSC traces (G1) and plots (G2) of the frequencies and amplitudes of hippocampal neurons cotransfected GFP with Vector, N-cad, or Ν-cad mut (n = 14, 22, 12 cells, respectively). Same Vector and N-cad data as in Figure 4D. Calibration: 10 pA, 500 ms. One-way ANOVA with Bonferroni's post hoc test: **p < 0.01, compared with Vector. ***p < 0.001, compared with Vector. ###p < 0.001, compared with N-cad. H, Representative traces (H1) and plots (H2) of frequencies and amplitudes of the mEPSCs in hippocampal neurons transfected with shGFP+Vector, shPKD1+Vector, shPKD1+N-cad, or shPKD1+N-cad mut (n = 10, 14, 18, and 17 cells, respectively). Same shGFP+Vector, shPKD1+Vector, and shPKD1+N-cad data as in Figure 4E. Calibration: 10 pA, 500 ms. One-way ANOVA with Bonferroni's post hoc test: **p < 0.01, compared with shGFP+Vector. ***p < 0.001, compared with shGFP+Vector. ##p < 0.01, compared with shPKD1+Vector. ###p < 0.001, compared with shPKD1+Vector. No significant differences in mEPSC amplitude were detected among the groups. Data are mean ± SEM.
We next mutated the three serines to alanines to generate an analog of N-cadherin (N-cad mut) for further study. Biotinylation assays showed that much less N-cad mut was distributed on the membrane compared with wild-type N-cadherin (Fig. 5D; p = 0.0026, t(3) = 9.283, paired t test). Overexpression of N-cadherin increased the spine density, spine area, and mEPSC frequency, whereas N-cad mut lost the ability to promote synapse formation or function (Fig. 5F,G; for spine density, p < 0.0001, F(2,46) = 46.09, one-way ANOVA; for spine area, p < 0.0001, F(2,40) = 28.12, one-way ANOVA; for mEPSC frequency, p < 0.0001, F(2,45) = 46.86, one-way ANOVA; for mEPSC amplitude, p = 0.0968, F(2,45) = 2.461, one-way ANOVA). Like N-cadΔ836–871, N-cad mut also failed to rescue the reduced mEPSC frequency caused by PKD1 knockdown (Fig. 5H; for mEPSC frequency, p < 0.0001, F(3,55) = 15.09, one-way ANOVA; for mEPSC amplitude, p = 0.1346, F(3,55) = 1.935, one-way ANOVA). These results suggest that phosphorylation of N-cadherin by PKD1 is necessary for functional synapse formation.
To further investigate the roles of N-cadherin phosphorylation by endogenous PKD1, we created an interfering peptide, TAT-N-cad S3, containing the three identified serine phosphorylation sites, to compete with endogenous N-cadherin for the phosphorylation by PKD1. TAT-N-cad S3A, in which the three serines were mutated to alanines, was used as a control. TAT-N-cad S3 reduced the amount of N-cadherin on cell surface as expected (Fig. 6F; p = 0.0066, t(2) = 12.25, paired t test). Likewise, TAT-N-cad S3 led to a significant reduction in the spine density, spine area, mEPSC frequency, and amplitude (Fig. 6G,H; for spine density, p = 0.0017, t(32) = 3.425, unpaired t test; for spine area, p < 0.0001, t(32) = 4.837, unpaired t test; for mEPSC frequency, p < 0.0001, t(27) = 9.636, unpaired t test; for mEPSC amplitude, p = 0.0172, t(27) = 2.538, unpaired t test). Additionally, we found that interference with PKD1 phosphorylating N-cadherin also affected the physical association between them (Figs. 5E, 6E; in Fig. 5E, p = 0.0365, t(4) = 3.093, unpaired t test; in Fig. 6E, p = 0.0120, t(3) = 5.472, paired t test).
Thus far, we have demonstrated that N-cadherin is a novel substrate of PKD1, and we have shown that PKD1 phosphorylates N-cadherin at Ser 869, 871, and 872, which are essential for the effects of PKD1 on synapse formation and function.
Interaction of PKD1 and N-cadherin promotes the binding of N-cadherin to β-catenin
It has been reported that the C-terminal domain of N-cadherin interacts with β-catenin, which interacts with α-catenin and F-actin to stabilize the membrane localization of N-cadherin (Arikkath and Reichardt, 2008). This prompted the idea that the increased cell surface abundance of N-cadherin regulated by PKD1 might be due to enhanced binding of N-cadherin to β-catenin. Overexpression of hPKD1 increased the binding of N-cadherin to β-catenin, whereas overexpression of DN-hPKD1 curtailed their interaction (Fig. 7A;p < 0.0001, F(2,12) = 35.64, one-way ANOVA), suggesting that the kinase activity of PKD1 is critical for the binding of N-cadherin to β-catenin. Furthermore, overexpression of N-cadΔ836–871 or N-cad mut decreased the binding of N-cadherin to β-catenin compared with wild-type N-cadherin (Fig. 7B,C; in Fig. 7B, p = 0.0181, t(4) = 3.865, unpaired t test; in Fig. 7C; p = 0.0017, t(3) = 10.90, paired t test). These results provide a possible mechanism for the regulation of the membrane localization of N-cadherin by PKD1: the interactions between PKD1 and N-cadherin, including the binding and phosphorylation of N-cadherin by PKD1, promote the association between N-cadherin and β-catenin, leading to increased membrane localization of N-cadherin.
Figure 7.
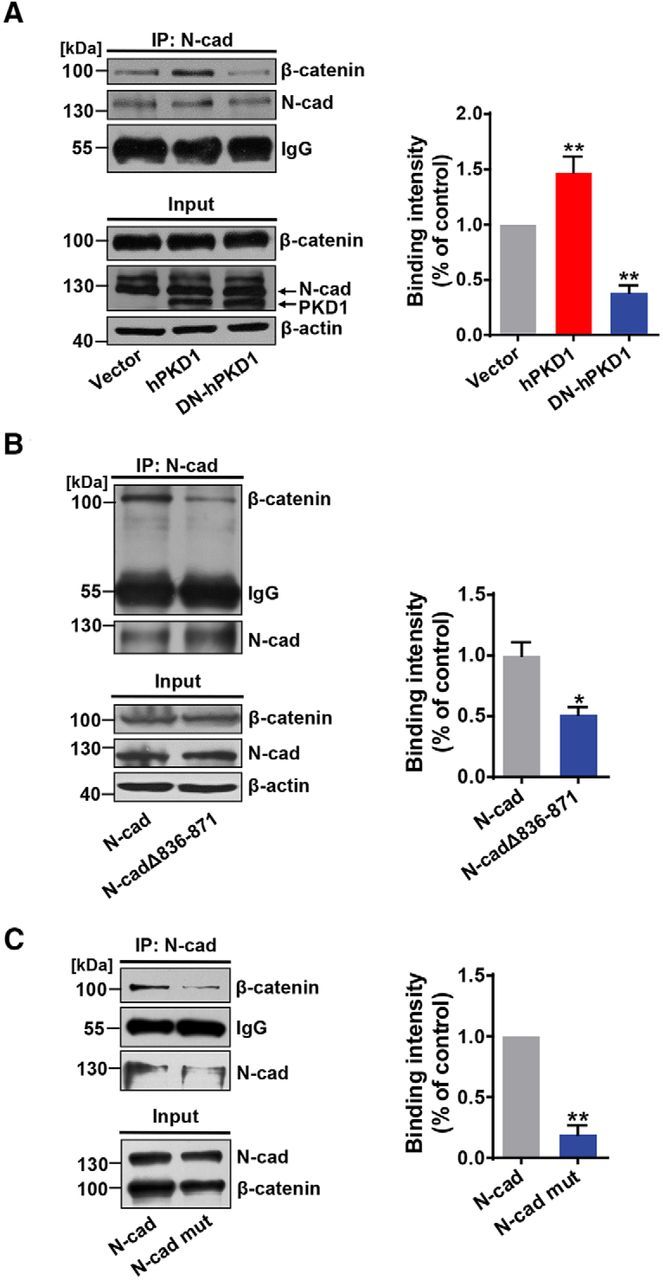
Interaction of PKD1 and N-cadherin promotes the binding of N-cadherin and β-catenin. A, Coimmunoprecipitation of N-cadherin with β-catenin in N2a cells cotransfected N-cadherin with vector, hPKD1, or DN-hPKD1. n = 5. **p < 0.01 (one-way ANOVA with Bonferroni's post hoc). B, C, N-cadherin 836–871aa deletion or Ser869, 871, and 872Ala mutation decreased its binding intensity with β-catenin in N2a cells. B, n = 3. C, n = 4. B. *p < 0.05 (unpaired t test). **p < 0.01 (unpaired t test). C. *p < 0.05 (paired t test). **p < 0.01 (paired t test). Data are mean ± SEM.
PKD1 is a mediator of the activity-dependent dendritic spine development
External environments can reshape neural circuits through the dynamic changes of synapses (Bozdagi et al., 2000; Johansson and Belichenko, 2002). Additionally, PKD1 has been demonstrated to translocate to dendritic spines during synaptic development (Czöndör et al., 2009). For these reasons, we postulated that PKD1 might regulate the effects of neural activity on synapse formation. KCl treatment has been widely used to mimic increased neural activity during maturation and to examine the activity-dependent effects on neuronal morphology (Sin et al., 2002; Peng et al., 2009; Qi et al., 2014). In our experiments, the extracellular K+ concentration was increased to 16 mm. Immunoprecipitation showed an activity-driven increase in the binding of PKD1 to N-cadherin (Fig. 8A; p = 0.0076, t(6) = 3.940, unpaired t test). In agreement with previous reports that the function of N-cadherin in synapse formation is regulated by neural activity (Sugiura et al., 2009; Mendez et al., 2010), the surface localization of N-cadherin also increased after KCl stimulation (Fig. 8B; p = 0.0090, t(4) = 4.747, unpaired t test). These results reveal a neuronal activity-dependent association between PKD1 and N-cadherin, suggesting that the activity-dependent spine growth may partially rely on the PKD1-N-cadherin signaling pathway. As for the effects on neuronal morphology, increased neural activity led to significant increase in protrusion density, spine density, filopodia density (Fig. 8C2; left, group effect: KCl, F(1,184) = 21.64, p < 0.0001, group effect: hPKD1, F(2,184) = 38.63, p < 0.0001; middle, group effect: KCl, F(1,184) = 9.331, p = 0.0026, group effect: hPKD1, F(2,184) = 39.60, p < 0.0001; right, group effect: KCl, F(1,184) = 43.07, p < 0.0001, group effect: hPKD1, F(2,184) = 15.87, p < 0.0001), protrusion area and protrusion length, along with mild enlargement of protrusion width (Fig. 8C3; left, group effect: KCl, F(1,124) = 146.2, p < 0.0001, group effect: hPKD1, F(2,124) = 85.10, p < 0.0001; middle, group effect: KCl, F(1,124) = 13.21, p = 0.0004, group effect: hPKD1, F(2,124) = 66.31, p < 0.0001; right, group effect: KCl, F(1,124) = 128.7, p < 0.0001, group effect: hPKD1, F(2,124) = 43.58, p < 0.0001), and these changes were maintained by overexpression of hPKD1 but prevented by overexpression of DN-hPKD1. We also noted that KCl stimulation did not cause significant changes in protrusion density, spine density, and filopodia density for neurons transfected with hPKD1 (Fig. 8C2), which might be due to the saturation effects of hPKD1 overexpressing leading to a limited extent of protrusion growth. These data demonstrate that the kinase activity of PKD1 is indispensable for activity-dependent spine growth in cultured neurons and that the interaction between PKD1 and N-cadherin partially regulates the spine growth induced by KCl stimulation in vitro.
Figure 8.
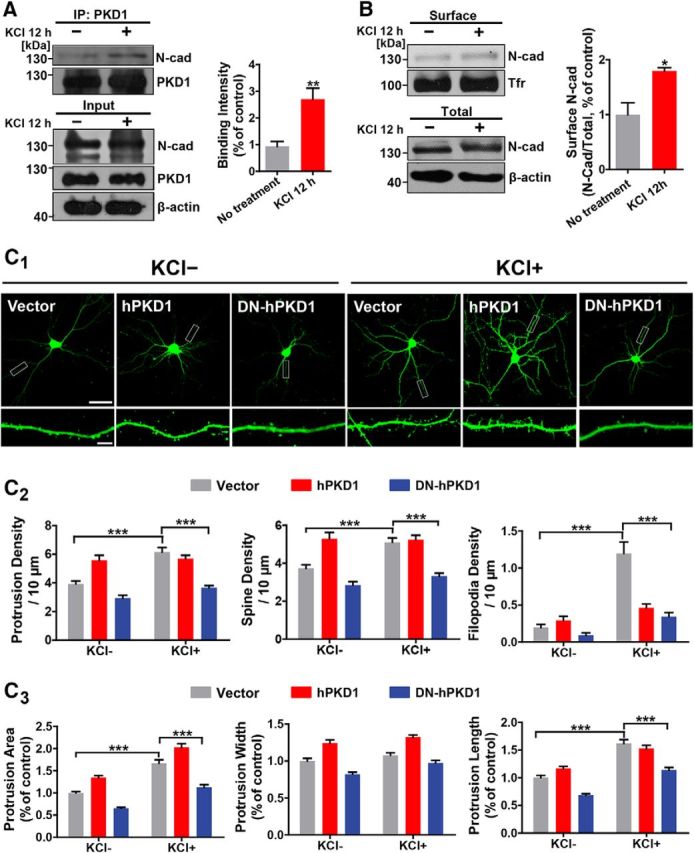
Neural activity promotes spine growth in a manner that depends on the kinase activity of PKD1. A, Activity-driven increase in N-cadherin binding to PKD1 in cultured cortical neurons treated with KCl (12 mm) for 12 h; n = 4. **p < 0.01 (unpaired t test). B, Activity-driven increase in the amount of membrane-associated N-cadherin in cortical neurons treated with KCl (12 mm) for 12 h; n = 3. *p < 0.05 (unpaired t test). C1, Representative images of DIV 15 hippocampal neurons transfected with GFP plus Vector, hPKD1, or DN-hPKD1 at DIV 8 and treated without or with KCl (12 mm) at DIV 10. Scale bars: Top, 50 μm; Bottom, 5 μm. C2, Quantification of protrusion density, spine density, and filopodia density in neurons from the experiment shown in C1 (n = 15, 15, 15, 14, 13, and 18 cells, respectively). ***p < 0.001 (two-way ANOVA with Bonferroni's post hoc test). C3, Quantification of protrusion area, protrusion width, and protrusion length in neurons from the experiment shown in C1 (n = 15, 15, 15, 14, 13, and 18 cells, respectively). ***p < 0.001 (two-way ANOVA with Bonferroni's post hoc test). Data are mean ± SEM.
PKD1-N-cadherin interaction is critical for LTP induction and regulates synaptic plasticity
To investigate the effects of interfering peptides on synaptic plasticity, electrophysiological recordings were performed in rat hippocampal slices to measure LTP after a bilateral injection of TAT-836–871, TAT-N-cad S3, or respective control peptides into the CA1 region of the hippocampus (Fig. 9A,B). Immunoprecipitation assays showed that TAT-836–871 injection did decrease PKD1 binding to N-cadherin in the hippocampus in vivo (Fig. 9D; p = 0.0299, t(2) = 5.649, paired t test). LTP is critically involved not only in learning and memory but also in the activity-dependent development of neural circuits and synaptic plasticity (Bliss and Collingridge, 1993; Engert and Bonhoeffer, 1999). We obtained hippocampal slices 12 h after peptide injection and induced LTP in Schaffer collaterals using high-frequency stimulation (Fig. 9C). In slices from rats injected with interfering peptides, the initial strength of LTP was decreased and the fEPSP slope diminished to baseline by ∼30 min after induction, whereas in slices from control animals significant and robust LTP was maintained for the entire recording period (Fig. 9E,F).
Figure 9.
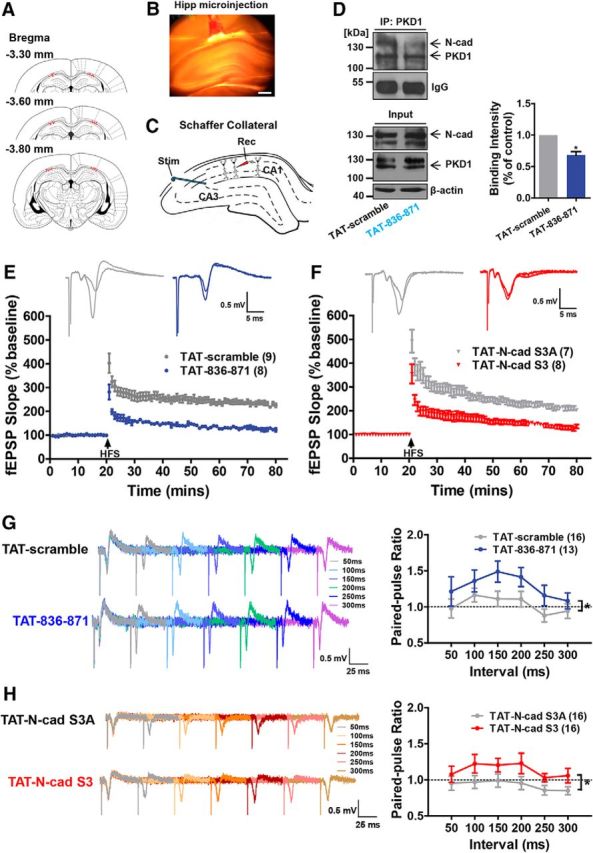
Disruption of the interaction of PKD1 and N-cadherin inhibits LTP and increases the PPR in the stratum radiatum of CA1. A, Column showing cannula tip placement in rats that received hippocampal injection of interfering peptides or control peptides. B, Representative coronal section showing the hippocampal injection site. Scale bar, 500 μm. C, Schematic of field recording configuration in CA1. The stimulating electrode was placed in the CA3 region, and the recording electrode was placed in the stratum radiatum of the CA1 region to measure the field response of the Schaffer collaterals. D, Effect of TAT-836–871 on the binding of PKD1 to N-cadherin in vivo measured by IP. Hippocampal tissues obtained from rats 12 h after peptide injection, respectively (10 μg); n = 3. *p < 0.05 (paired t test). E, Injection of TAT-836–871 (10 μg) to CA1 region of hippocampus suppressed LTP induction (blue circles, n = 8 slices/from 7 rats) compared with the injection of TAT-scramble (gray circles, n = 9/8). F, Injection of TAT-N-cad S3 (10 μg) to CA1 region of hippocampus suppressed LTP induction (red diamonds, n = 8 slices/from 7 rats) compared with TAT-N-cad S3A (gray diamonds, n = 7/7). G, H, Representative traces of fEPSPs evoked by paired pulses at six interstimulus intervals (interstimulation interval; 50, 100, 150, 200, 250, and 300 ms). The PPR in the hippocampal CA1 region was increased by TAT-836–871 (10 μg) and TAT-N-cad S3 (10 μg) injections compared with the injections of their control peptides, respectively. *p < 0.05 (unpaired t test). Data are mean ± SEM.
The paired-pulse ratio (PPR) is primarily associated with changes in presynaptic transmitter release (Hessler et al., 1993). We measured the PPR after peptide injection and calculated it at every interstimulation interval (50–300 ms) (Fig. 9G,H; in Fig. 9G, p = 0.0498, t(23) = 2.071, unpaired t test; in Fig. 9H, p = 0.0481, t(30) = 2.061, unpaired t test). The increased PPR observed in the slices injected with the interfering peptides indicated a reduced probability of transmitter release, although no significant differences at each interstimulation interval time point between groups were detected. These results demonstrate that synaptic plasticity is directly modulated by the interaction of PKD1 and N-cadherin and that their interaction may have potential effects on learning and memory.
Discussion
The results of this study indicate a direct functional interaction between PKD1 and N-cadherin and identify N-cadherin as a novel substrate of PKD1 in the nervous system. Our work demonstrates that the interaction between PKD1 and N-cadherin increases the surface localization of N-cadherin, leading to increases in spine density, spine area, and transmission efficacy that are correlated with synaptic potentiation (Fig. 10). These activity-dependent PKD1-N-cadherin interactions provide new molecular mechanisms for synapse formation and function.
Figure 10.
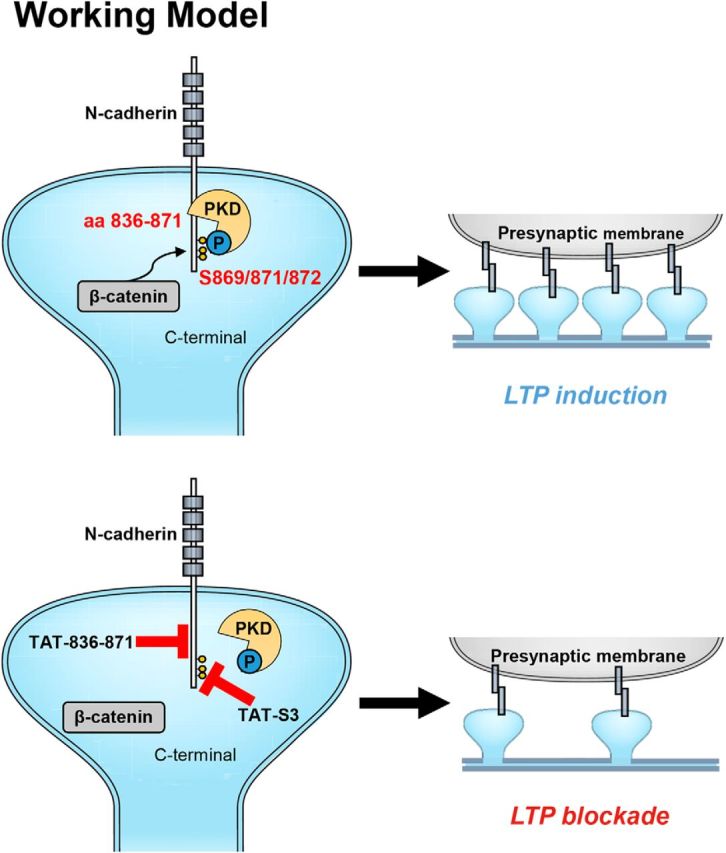
Working model. PKD1 binds to N-cadherin at amino acid residues 836–871 and phosphorylates it at Ser 869, 871, and 872. Disruption of the modification of N-cadherin by PKD1 impairs the binding of N-cadherin to β-catenin and the membrane localization of N-cadherin, thereby inhibiting synapse formation and synaptic plasticity.
Kinase activity and possible substrates are often first taken into account when exploring how a protein kinase works. Previous study showed that PKD1 phosphorylates E-cadherin, but it remains unknown which amino acid residues in cadherins are phosphorylated by PKD1 (Jaggi et al., 2005). In this study, we identified the sites in N-cadherin phosphorylated by PKD1 as Ser 869, 871, and 872. Further functional studies revealed that the three phosphorylation sites are crucial for the membrane localization of N-cadherin and synapse development, indicating that PKD1 promotes synapse formation and function not only through its physical binding to N-cadherin but also by phosphorylation of N-cadherin. It is of interest that two of the observed phosphorylation sites, Ser 869 and Ser 871, are located within the PKD1 binding motif (amino acids 836–871) of N-cadherin. This overlap may underlie the decreased binding of PKD1 to N-cadherin that occurs when the phosphorylation of N-cadherin by PKD1 is disrupted (Figs. 5E, 6E). We also found that increasing neuronal activity promoted the binding of PKD1 and N-cadherin, and the surface localization of N-cadherin (Fig. 8A,B). From the morphological studies, KCl treatment failed to promote spine formation in DN-hPKD1-expressing neurons (Fig. 8C). Combined with these results, we speculated that neuronal activity induced surface localization of N-cadherin is dependent on PKD1 kinase activity.
Subcellular fractionation assay of hippocampal CA1 region of postnatal 3- to 4-week-old rats showed that PKD1 was significantly enriched in the PSD fraction (Fig. 1C), which suggested that PKD1 is mainly presented in the postsynaptic compartment. Our results clearly demonstrated the localization of PKD1 in adolescent rat CA1 region during the late phase of neural development. Consequently, PKD1 interacts with N-cadherin postsynaptically in dendritic spines. In the electrophysiological studies, we observed a significant increase in mEPSC frequency but not in mEPSC amplitude after overexpression of PKD1 (Fig. 2B) or N-cadherin (Figs. 4D, 5G). Alterations in mEPSC frequency generally reflect changes in the number of synapses and the probability of presynaptic neurotransmitter release. Combined with the subcellular fractionation assay that PKD1 was presented in postsynaptic compartment, the increase of mEPSC frequency was much more likely caused by the increase in synapse number rather than the presynaptic changes. However, we cannot rule out the possibility that these manipulations might also change the presynaptic neurotransmitter release probability at existing synapses. Previous studies have demonstrated that N-cadherin trans-synaptically regulates presynaptic function in a retrograde manner and that knockdown of N-cadherin results in impairment of vesicle exocytosis and replenishment of the readily releasable vesicle pool (Bamji et al., 2003; Saglietti et al., 2007). We also observed increased PPR after disrupting PKD1-N-cadherin interaction in CA1 regions (Fig. 9G,H), which suggested that this impaired transmitter release was regulated by postsynaptic manipulations on PKD1-N-cadherin interactions. Therefore, postsynaptic manipulation of PKD1 may also regulate presynaptic vesicle release through retrograde control of presynaptic N-cadherin.
PKD has been implicated in salt taste-induced learning in Caenorhabditis elegans (Fu et al., 2009) and in cocaine-induced locomotor hyperactivity (Wang et al., 2014). A study has reported that actin stabilization needed for the enlargement of dendritic spines is dependent on PKD activity; thus, impaired PKD functions attenuate LTP formation and spatial memory formation (Bencsik et al., 2015). The authors performed the Morris water maze using the kdPKD-EGFP-expressing mice during the 9–10th weeks of DOX treatment. In our experiments, we specifically conducted the electrophysiological studies on adolescent animals at the age of postnatal 3–4 weeks, which corresponds to the late phase of neuronal development. We observed that disrupting PKD1-N-cadherin interaction in rat hippocampus 12 h before obtaining the acute brain slices inhibited LTP induction and maintenance (Fig. 9E,F), confirming the correlated changes in spine morphology and in synaptic plasticity in vitro.
Overall, our study demonstrates one of the multiregulatory mechanisms of PKD1 in the late phase of neuronal development: the precise regulation of membrane N-cadherin by PKD1 is critical for synapse formation and synaptic plasticity, as shown in our working hypothesis (Fig. 10). Our study also provides a possible therapeutic target for the clinical treatment of neurodevelopmental diseases, such as schizophrenia, and neurodegenerative diseases, such as Alzheimer's disease.
Footnotes
This work was supported by National Natural Science Foundation of China Grant 81600989 to C.C., and Grants 31530028 and 31720103908 to Y.W., Ministry of Science and Technology of China Grants 2014CB542204 and 2015BAI08B02 to Y.W., and China Postdoctoral Science Foundation Grant 2015M580025 to C.C. C.C. was supported in part by Peking-Tsinghua Center for Life Sciences Postdoctoral Fellowship. We thank Prof. Qihua He (Cell Analytical Laboratory, Medical and Healthy Analytical Center, Peking University) for technical support for imaging.
The authors declare no competing financial interests.
References
- Arikkath J, Reichardt LF (2008) Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends Neurosci 31:487–494. 10.1016/j.tins.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avriyanti E, Atik N, Kunii M, Furumoto N, Iwano T, Yoshimura S, Harada R, Harada A (2015) Functional redundancy of protein kinase D1 and protein kinase D2 in neuronal polarity. Neurosci Res 95:12–20. 10.1016/j.neures.2015.01.007 [DOI] [PubMed] [Google Scholar]
- Bacchelli E, Ceroni F, Pinto D, Lomartire S, Giannandrea M, D'Adamo P, Bonora E, Parchi P, Tancredi R, Battaglia A, Maestrini E (2014) A CTNNA3 compound heterozygous deletion implicates a role for alphaT-catenin in susceptibility to autism spectrum disorder. J Neurodev Disord 6:17. 10.1186/1866-1955-6-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamji SX, Shimazu K, Kimes N, Huelsken J, Birchmeier W, Lu B, Reichardt LF (2003) Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron 40:719–731. 10.1016/S0896-6273(03)00718-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bencsik N, Szíber Z, Liliom H, Tárnok K, Borbély S, Gulyás M, Rátkai A, Szűcs A, Hazai-Novák D, Ellwanger K, Rácz B, Pfizenmaier K, Hausser A, Schlett K (2015) Protein kinase D promotes plasticity-induced F-actin stabilization in dendritic spines and regulates memory formation. J Cell Biol 210:771–783. 10.1083/jcb.201501114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DL, Tanaka H (1998) N-cadherin redistribution during synaptogenesis in hippocampal neurons. J Neurosci 18:6892–6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisbal M, Conde C, Donoso M, Bollati F, Sesma J, Quiroga S, Díaz Añel A, Malhotra V, Marzolo MP, Cáceres A (2008) Protein kinase D regulates trafficking of dendritic membrane proteins in developing neurons. J Neurosci 28:9297–9308. 10.1523/JNEUROSCI.1879-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31–39. 10.1038/361031a0 [DOI] [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW (2000) Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron 28:245–259. 10.1016/S0896-6273(00)00100-8 [DOI] [PubMed] [Google Scholar]
- Brooks H, Lebleu B, Vivès E (2005) Tat peptide-mediated cellular delivery: back to basics. Adv Drug Deliv Rev 57:559–577. 10.1016/j.addr.2004.12.001 [DOI] [PubMed] [Google Scholar]
- Cabrera-Poch N, Sánchez-Ruiloba L, Rodríguez-Martínez M, Iglesias T (2004) Lipid raft disruption triggers protein kinase C and Src-dependent protein kinase D activation and Kidins220 phosphorylation in neuronal cells. J Biol Chem 279:28592–28602. 10.1074/jbc.M312242200 [DOI] [PubMed] [Google Scholar]
- Carlin RK, Grab DJ, Cohen RS, Siekevitz P (1980) Isolation and characterization of postsynaptic densities from various brain regions: enrichment of different types of postsynaptic densities. J Cell Biol 86:831–845. 10.1083/jcb.86.3.831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KO, Hunt CA, Kennedy MB (1992) The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron 9:929–942. 10.1016/0896-6273(92)90245-9 [DOI] [PubMed] [Google Scholar]
- Czöndör K, Ellwanger K, Fuchs YF, Lutz S, Gulyás M, Mansuy IM, Hausser A, Pfizenmaier K, Schlett K (2009) Protein kinase D controls the integrity of Golgi apparatus and the maintenance of dendritic arborization in hippocampal neurons. Mol Biol Cell 20:2108–2120. 10.1091/mbc.E08-09-0957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döppler H, Storz P, Li J, Comb MJ, Toker A (2005) A phosphorylation state-specific antibody recognizes Hsp27, a novel substrate of protein kinase D. J Biol Chem 280:15013–15019. 10.1074/jbc.C400575200 [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T (1999) Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399:66–70. 10.1038/19978 [DOI] [PubMed] [Google Scholar]
- Friedman LG, Benson DL, Huntley GW (2015) Cadherin-based transsynaptic networks in establishing and modifying neural connectivity. Curr Top Dev Biol 112:415–465. 10.1016/bs.ctdb.2014.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Ren M, Feng H, Chen L, Altun ZF, Rubin CS (2009) Neuronal and intestinal protein kinase d isoforms mediate Na+ (salt taste)-induced learning. Sci Signal 2:ra42. 10.1126/scisignal.2000224 [DOI] [PubMed] [Google Scholar]
- Gumbiner BM. (2005) Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol 6:622–634. 10.1038/nrm1699 [DOI] [PubMed] [Google Scholar]
- Hessler NA, Shirke AM, Malinow R (1993) The probability of transmitter release at a mammalian central synapse. Nature 366:569–572. 10.1038/366569a0 [DOI] [PubMed] [Google Scholar]
- Iglesias T, Rozengurt E (1998) Protein kinase D activation by mutations within its pleckstrin homology domain. J Biol Chem 273:410–416. 10.1074/jbc.273.1.410 [DOI] [PubMed] [Google Scholar]
- Iglesias T, Waldron RT, Rozengurt E (1998) Identification of in vivo phosphorylation sites required for protein kinase D activation. J Biol Chem 273:27662–27667. 10.1074/jbc.273.42.27662 [DOI] [PubMed] [Google Scholar]
- Jaggi M, Rao PS, Smith DJ, Wheelock MJ, Johnson KR, Hemstreet GP, Balaji KC (2005) E-cadherin phosphorylation by protein kinase D1/protein kinase Cμ is associated with altered cellular aggregation and motility in prostate cancer. Cancer Res 65:483–492. [PubMed] [Google Scholar]
- Johannes FJ, Prestle J, Eis S, Oberhagemann P, Pfizenmaier K (1994) PKCu is a novel, atypical member of the protein kinase C family. J Biol Chem 269:6140–6148. [PubMed] [Google Scholar]
- Johansson BB, Belichenko PV (2002) Neuronal plasticity and dendritic spines: effect of environmental enrichment on intact and postischemic rat brain. J Cereb Blood Flow Metab 22:89–96. 10.1097/00004647-200201000-00011 [DOI] [PubMed] [Google Scholar]
- Kandel ER, Dudai Y, Mayford MR (2014) The molecular and systems biology of memory. Cell 157:163–186. 10.1016/j.cell.2014.03.001 [DOI] [PubMed] [Google Scholar]
- Krueger DD, Osterweil EK, Bear MF (2010) Activation of mGluR5 induces rapid and long-lasting protein kinase D phosphorylation in hippocampal neurons. J Mol Neurosci 42:1–8. 10.1007/s12031-010-9338-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewallen KA, Shen YA, De la Torre AR, Ng BK, Meijer D, Chan JR (2011) Assessing the role of the cadherin/catenin complex at the Schwann cell-axon interface and in the initiation of myelination. J Neurosci 31:3032–3043. 10.1523/JNEUROSCI.4345-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez P, De Roo M, Poglia L, Klauser P, Muller D (2010) N-cadherin mediates plasticity-induced long-term spine stabilization. J Cell Biol 189:589–600. 10.1083/jcb.201003007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa K, Toker A, Wong K, Marignani PA, Johannes FJ, Cantley LC (1998) Association of protein kinase Cmu with type II phosphatidylinositol 4-kinase and type I phosphatidylinositol-4-phosphate 5-kinase. J Biol Chem 273:23126–23133. 10.1074/jbc.273.36.23126 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1986) The rat brain in stereotaxic coordinates. Cambridge, MA: Academic Press. [DOI] [PubMed] [Google Scholar]
- Peng YR, He S, Marie H, Zeng SY, Ma J, Tan ZJ, Lee SY, Malenka RC, Yu X (2009) Coordinated changes in dendritic arborization and synaptic strength during neural circuit development. Neuron 61:71–84. 10.1016/j.neuron.2008.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi C, Liu S, Qin R, Zhang Y, Wang G, Shang Y, Wang Y, Liang J (2014) Coordinated regulation of dendrite arborization by epigenetic factors CDYL and EZH2. J Neurosci 34:4494–4508. 10.1523/JNEUROSCI.3647-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozengurt E. (2011) Protein kinase D signaling: multiple biological functions in health and disease. Physiology (Bethesda) 26:23–33. 10.1152/physiol.00037.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saglietti L, Dequidt C, Kamieniarz K, Rousset MC, Valnegri P, Thoumine O, Beretta F, Fagni L, Choquet D, Sala C, Sheng M, Passafaro M (2007) Extracellular interactions between GluR2 and N-cadherin in spine regulation. Neuron 54:461–477. 10.1016/j.neuron.2007.04.012 [DOI] [PubMed] [Google Scholar]
- Schrick C, Fischer A, Srivastava DP, Tronson NC, Penzes P, Radulovic J (2007) N-cadherin regulates cytoskeletally associated IQGAP1/ERK signaling and memory formation. Neuron 55:786–798. 10.1016/j.neuron.2007.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong E, Yuan L, Arikkath J (2015) Cadherins and catenins in dendrite and synapse morphogenesis. Cell Adh Migr 9:202–213. 10.4161/19336918.2014.994919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin WC, Haas K, Ruthazer ES, Cline HT (2002) Dendrite growth increased by visual activity requires NMDA receptor and Rho GTPases. Nature 419:475–480. 10.1038/nature00987 [DOI] [PubMed] [Google Scholar]
- Sugiura H, Tanaka H, Yasuda S, Takemiya T, Yamagata K (2009) Transducing neuronal activity into dendritic spine morphology: new roles for p38 MAP kinase and N-cadherin. Neuroscientist 15:90–104. 10.1177/1073858408324024 [DOI] [PubMed] [Google Scholar]
- Tahirovic S, Bradke F (2009) Neuronal polarity. Cold Spring Harb Perspect Biol 1:a001644. 10.1101/cshperspect.a001644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, Takeichi M (2002) Cadherin regulates dendritic spine morphogenesis. Neuron 35:77–89. 10.1016/S0896-6273(02)00748-1 [DOI] [PubMed] [Google Scholar]
- Tucci V, Kleefstra T, Hardy A, Heise I, Maggi S, Willemsen MH, Hilton H, Esapa C, Simon M, Buenavista MT, McGuffin LJ, Vizor L, Dodero L, Tsaftaris S, Romero R, Nillesen WN, Vissers LE, Kempers MJ, Vulto-van Silfhout AT, Iqbal Z, et al. (2014) Dominant beta-catenin mutations cause intellectual disability with recognizable syndromic features. J Clin Invest 124:1468–1482. 10.1172/JCI70372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uribe-Arias A, Posada-Duque RA, González-Billault C, Villegas A, Lopera F, Cardona-Gómez GP (2016) p120-catenin is necessary for neuroprotection induced by CDK5 silencing in models of Alzheimer's disease. J Neurochem 138:624–639. 10.1111/jnc.13697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde AM, Sinnett-Smith J, Van Lint J, Rozengurt E (1994) Molecular cloning and characterization of protein kinase D: a target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc Natl Acad Sci U S A 91:8572–8576. 10.1073/pnas.91.18.8572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Su P, Zhang Y, Lu J, Xing B, Kang K, Li W, Wang Y (2014) Protein kinase D1-dependent phosphorylation of dopamine D1 receptor regulates cocaine-induced behavioral responses. Neuropsychopharmacology 39:1290–1301. 10.1038/npp.2013.341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kedei N, Wang M, Wang QJ, Huppler AR, Toth A, Tran R, Blumberg PM (2004) Interaction between protein kinase Cmu and the vanilloid receptor type 1. J Biol Chem 279:53674–53682. 10.1074/jbc.M410331200 [DOI] [PubMed] [Google Scholar]
- Xing BM, Yang YR, Du JX, Chen HJ, Qi C, Huang ZH, Zhang Y, Wang Y (2012) Cyclin-dependent kinase 5 controls TRPV1 membrane trafficking and the heat sensitivity of nociceptors through KIF13B. J Neurosci 32:14709–14721. 10.1523/JNEUROSCI.1634-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wei M, Xiong Y, Du X, Zhu S, Yang L, Zhang C, Liu JJ (2015) Endophilin A1 regulates dendritic spine morphogenesis and stability through interaction with p140Cap. Cell Res 25:496–516. 10.1038/cr.2015.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin DM, Huang YH, Zhu YB, Wang Y (2008) Both the establishment and maintenance of neuronal polarity require the activity of protein kinase D in the Golgi apparatus. J Neurosci 28:8832–8843. 10.1523/JNEUROSCI.1291-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Arimura N, Kaibuchi K (2006) Signaling networks in neuronal polarization. J Neurosci 26:10626–10630. 10.1523/JNEUROSCI.3824-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Woodhead GJ, Swaminathan SK, Noles SR, McQuinn ER, Pisarek AJ, Stocker AM, Mutch CA, Funatsu N, Chenn A (2010) Cortical neural precursors inhibit their own differentiation via N-cadherin maintenance of beta-catenin signaling. Dev Cell 18:472–479. 10.1016/j.devcel.2009.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]