

Abstract
Potassium voltage-gated channel interacting protein 3 (KChIP3), also termed downstream regulatory element antagonist modulator (DREAM) and calsenilin, is a multifunctional protein belonging to the neuronal calcium sensor (NCS) family. Recent studies revealed the expression of KChIP3 in dorsal root ganglion (DRG) neurons, suggesting the potential role of KChIP3 in peripheral sensory processing. Herein, we show that KChIP3 colocalizes with transient receptor potential ion channel V1 (TRPV1), a critical molecule involved in peripheral sensitization during inflammatory pain. Furthermore, the N-terminal 31-50 fragment of KChIP3 is capable of binding both the intracellular N and C termini of TRPV1, which substantially decreases the surface localization of TRPV1 and the subsequent Ca2+ influx through the channel. Importantly, intrathecal administration of the transmembrane peptide transactivator of transcription (TAT)-31-50 remarkably reduces Ca2+ influx via TRPV1 in DRG neurons and alleviates thermal hyperalgesia and gait alterations in a complete Freund's adjuvant-induced inflammatory pain model in male rats. Moreover, intraplantar injection of TAT-31-50 attenuated the capsaicin-evoked spontaneous pain behavior and thermal hyperalgesia, which further strengthened the regulatory role of TAT-31-50 on TRPV1 channel. In addition, TAT-31-50 could also alleviate inflammatory thermal hyperalgesia in kcnip3−/− rats generated in our study, suggesting that the analgesic effect mediated by TAT-31-50 is independent of endogenous KChIP3. Our study reveals a novel peripheral mechanism for the analgesic function of KChIP3 and provides a potential analgesic agent, TAT-31-50, for the treatment of inflammatory pain.
SIGNIFICANCE STATEMENT Inflammatory pain arising from inflamed or injured tissues significantly compromises the quality of life in patients. This study aims to elucidate the role of peripheral potassium channel interacting protein 3 (KChIP3) in inflammatory pain. Direct interaction of the KChIP3 N-terminal 31-50 fragment with transient receptor potential ion channel V1 (TRPV1) was demonstrated. The KChIP3–TRPV1 interaction reduces the surface localization of TRPV1 and thus alleviates heat hyperalgesia and gait alterations induced by peripheral inflammation. Furthermore, the transmembrane transactivator of transcription (TAT)-31-50 peptide showed analgesic effects on inflammatory hyperalgesia independently of endogenous KChIP3. This work reveals a novel mechanism of peripheral KChIP3 in inflammatory hyperalgesia that is distinct from its classical role as a transcriptional repressor in pain modulation.
Keywords: DRG, hyperalgesia, inflammation, interaction, KChIP3, TRPV1
Introduction
Calcium signaling is involved in a range of cellular processes, including action potential generation, neurotransmitter release, gene transcriptional regulation, and synaptic plasticity. Changes in intracellular [Ca2+]i can be detected precisely by Ca2+-binding proteins such as the ubiquitous calcium sensor calmodulin (CaM), calcium-binding protein (CaBP) family members, and neuronal calcium sensor (NCS) proteins (Burgoyne, 2007; Burgoyne and Haynes, 2015). Downstream regulatory element antagonist modulator (DREAM), a member of the NCS family, was first reported as a critical transcriptional repressor in pain processing (Carrión et al., 1999; Cheng et al., 2002). Numerous genes have been shown to be regulated by DREAM, such as preprodynorphin, c-fos, bdnf (brain-derived neurotrophic factor), the sodium/calcium exchanger NCX3 gene, the circadian AA-NAT (arylalkylamine N-acetyltransferase) gene, IL-2 (interleukin-2), the deubiquitinase TNFAIP3 (tumor necrosis factor α-induced protein 3), MGLL (monoglyceride lipase), and the protease CTSL (cathepsin L) (Carrión et al., 1999; Link et al., 2004; Gomez-Villafuertes et al., 2005; Savignac et al., 2005; Rivera-Arconada et al., 2010; Tiruppathi et al., 2014; Benedet et al., 2017). Genetic DREAM ablation results in elevated prodynorphin levels in the spinal cord, ultimately markedly attenuating pain response (Cheng et al., 2002). In contrast, transgenic mice expressing a constitutively active DREAM mutant displayed hyperalgesia in the basal state (Rivera-Arconada et al., 2010). Unexpectedly, these mice showed reduced nociceptive behaviors and impaired spinal sensitization after the intraplantar injection of complete Freund's adjuvant (CFA) that was attributable to reduced BDNF content in the spinal cord. These contradictory findings indicate the complex regulatory roles of DREAM in nociceptive processing.
DREAM is also termed calsenilin for its interaction with the C terminus of presenilin (Buxbaum et al., 1998) and potassium channel-interacting protein 3 (KChIP3) for its binding to the cytoplasmic N terminus of the Kv4 α-subunit (An et al., 2000). Hereafter, the name KChIP3 is used for consistency with the gene nomenclature. In total, four KChIP proteins (KChIP1-4), sharing expression patterns and functions constitute the KChIP subfamily of NCS proteins. Coexpression of the KChIP proteins remarkably promotes the membrane localization of Kv4 channels, increases current density, slows current inactivation, and speeds recovery from inactivation (An et al., 2000; Takimoto et al., 2002; Jerng et al., 2005; Hu et al., 2006). Reciprocally, the membrane localization of KChIP3, particularly when coexpressed with the Kv4.2 or Kv4.3 channels, was demonstrated in previous studies by our group and others (Takimoto et al., 2002; Ruiz-Gomez et al., 2007; Zhang et al., 2010). In addition to Kv4 channels, direct interactions of KChIP3 with ion channels or anchoring proteins, including Cav3.2 or Cav3.3, the GluN1 subunit of NMDA receptors, and PSD95, were indicated (Anderson et al., 2010; Wu et al., 2010; Zhang et al., 2010). Altogether, these findings provide further evidence for the multifunctional properties of KChIP3.
Transient receptor potential ion channel V1 (TRPV1), a nonselective cation channel enriched in primary sensory neurons, responds to a variety of noxious stimuli, including heat (>43°C), capsaicin, low pH, and arachidonic acid metabolites (Caterina et al., 1997; Julius, 2013). Sensitization and desensitization are two functional features of TRPV1. During inflammatory pain, TRPV1 undergoes sensitization and contributes to the development of peripheral sensitization. The underlying mechanism is related to the phosphorylation of TRPV1 intracellular domains by multiple protein kinases, which leads to the increased surface localization of the receptor and altered channel kinetics. In contrast, Ca2+ effectors activated after Ca2+ influx via TRPV1, including CaM and phosphatase calcineurin, mediate TRPV1 desensitization (Numazaki et al., 2003; Jung et al., 2004; Rosenbaum et al., 2004; Mohapatra and Nau, 2005; Lishko et al., 2007). Direct associations of CaM with both the intracellular N and C termini of TRPV1 were demonstrated (Rosenbaum et al., 2004; Lishko et al., 2007). Here, we demonstrate that the KChIP3 N-terminal 31-50 fragment mediates its association with TRPV1 and alleviates inflammatory thermal hyperalgesia. This study reveals a novel mechanism of peripheral KChIP3 in inflammatory hyperalgesia that is distinct from its classical role as a transcriptional repressor in pain modulation.
Materials and Methods
General experimental design.
In this study, multiple behavioral tests (e.g., radiant heat measurement) and nonbehavioral tests (e.g., Ca2+ imaging) were used. Behavioral tests were performed in a double-blinded manner and data were collected from more than five animals of each group. Nonbehavioral tests include data from at least three duplications. The between-subject factors include genotype (i.e., wild-type vs kcnip3 KO), peptide [i.e., transactivator of transcription (TAT)-scramble vs TAT-31-50] or plasmids (i.e., EGFP vs EGFP-KChIP3) administration, time points after CFA intraplantar injection (i.e., 1, 3, 6, 12, and 24 h), and dorsal root ganglion (DRG) neuron size (small, medium, large).
Animals.
Male Sprague Dawley (SD) rats (130–180 g) were used. All experiments were performed according to the guidelines of the Animal Care and Use Committee of Peking University. Kcnip3−/− rats were generated by the Nanjing Biomedical Research Institute of Nanjing University. In kcnip3−/− rats, deletion of exon 2 of the kcnip3 gene using the CRISPR/Cas9 system induces a frameshift mutation. Null KChIP3 expression in the KO rats was verified by PCR and Western blot. The animals were housed in temperature-controlled rooms on a 12/12 h light-dark cycle with ad libitum access to food and water.
CFA-induced inflammatory pain model.
In the CFA-induced inflammatory pain model, 100% CFA (Sigma-Aldrich) was diluted to 25% in incomplete Freund's adjuvant (Sigma-Aldrich) to avoid excessive inflammation and spontaneous pain behavior. Nociceptive responses after the intrathecal injection or delivery of the plasmids or peptides were measured in a blinded manner.
Plasmids.
The EGFP-KChIP3 plasmids were constructed by PCR amplification of rat brain cDNA and subcloned into the XhoI-EcoRI site of the pEGFP-C2 vector (Clontech). The prokaryotic expression constructs containing different KChIP3 N termini fragments were generated by PCR and subcloned into the PGEX-5x-1 (Clontech) and pet28a (+) (Novagen) vectors, respectively. The prokaryotic expression constructs encoding the intracellular TRPV1 domains were generated in a similar manner. The EGFP-KChIP3Δ31–50 plasmid was constructed by deletion of amino acids 1–50 followed by insertion of amino acids 1–30 via the BamHI site.
Peptides.
The TAT-31-50 peptide (RKKRRQRRR-KWQRPRFTRQALMRCCLIKW) and the control peptides TAT-1-20 (RKKRRQRRR-MQRTKEAMKASDGSLLGDPG) and TAT-scramble (RKKRRQRRR-TKMWWQRARLPFLRKCRCIQ) were synthesized and purified by Shanghai Gil Biochemical. The peptides were dissolved in sterile, double-distilled water.
Intrathecal catheterization and injection.
For the intrathecal delivery of drugs, a PE-10 polyethylene catheter was implanted into the intrathecal space, reaching the lumbar enlargement of the spinal cord (Li et al., 2014). Surgeries were performed under 10% chloral hydrate anesthesia (0.3 g/kg, i.p.), and rats were allowed 4–5 d for recovery. Any animals showing motor impairment from the intrathecal cannula placement were excluded from the study.
After 4 d of surgical recovery from the intrathecal catheter placement, the rats were randomly assigned to different experimental groups: EGFP, EGFP-KChIP3, or EGFP-KChIP3Δ31–50. Their basal paw withdrawal latency (PWL) was measured before subjection to the intrathecal injection of the plasmids. DNA and Lipofectamine 2000 (Invitrogen) complexes were prepared as described below. DNA (10 μg, 1 μg/μl) was diluted in saline (10 μl) and mixed gently. Next, Lipofectamine 2000 (20 μl) was added to saline (10 μl) and mixed gently. After 5 min of incubation at room temperature, the diluted DNA was combined and mixed with the diluted Lipofectamine 2000 and the mixture was incubated for 20 min at room temperature to allow DNA–Lipofectamine complexes to form. The mixture was then injected intrathecally. The ratio of DNA (in micrograms) to Lipofectamine 2000 (in microliters) was 1:2. After the injection, the needle remained in situ for 2 min before withdrawal. The basal PWL was then measured 4 d after gene delivery and 25% CFA was subsequently injected into the plantar surface of the left hindpaw. Nociceptive responses were then measured 1, 3, 6, 12, and 24 h after CFA administration.
For in vivo application of the peptide, after 4 d of recovery from placing the intrathecal catheter surgically, the rats were randomly divided into the different groups. Their basal PWLs were measured and the rats were then subjected to an intrathecal injection of the peptides at different dosages: 3 μg (1 mg/ml), 10 μg (1 mg/ml), and 30 μg (3.33 mg/ml). After injection, the needle remained in situ for 2 min before withdrawal. Nociceptive responses were measured 1, 3, 6, and 12 h and 1 d after the subcutaneous administration of 25% CFA into their left hindpaws.
Immunofluorescence staining.
Rats were anesthetized by 10% chloral hydrate and briefly perfused transcardially with warm saline, followed by cold 4% paraformaldehyde (PFA) in PBS (137 mm NaCl, 2.7 mm KCl, 10 mm NaH2PO4, 2 mm K2HPO4, pH 7.4). After perfusion, lumbar 4/5 DRG neurons were isolated and immersed in 4% PFA at 4°C for 4 h. After dehydration for cryoprotection in 30% (w/v) sucrose in 0.1 m PBS, all samples were kept at 4°C until sectioning.
Sections of DRG neurons were cut with a cryostat at 10 μm thickness and mounted directly onto gelatin-coated slides. Sections were washed with PBS once for 10 min and then once with PBS containing 0.1% Triton X-100 and 3% bovine serum albumin (BSA) for 40 min at room temperature. Sections were then incubated at 4°C for 24 h with primary antibodies (KChIP3, Abcam, ab3474, 1:100; TRPV1, Merck-Millipore, AB5566, 1:100; NF200, Sigma-Aldrich, N0142, 1:200; CGRP, Abcam, ab81887, 1:100; GFP, Abcam, ab5450, 1:400) diluted in PBS containing 1% BSA. After incubation with the primary antibodies, sections were washed with PBS 3 times for 5 min each and then incubated at 4°C overnight with secondary antibodies (Alexa Fluor 594-conjugated donkey anti-rabbit, Invitrogen, A-21207, 1: 500; Alexa Fluor 488-conjugated donkey anti-mouse, Invitrogen, A-21202, 1: 500; Alexa Fluor 680-conjuagted donkey anti-guinea pig, Jackson Immuno Research, 706-625-148, 1: 500; Alexa Fluor 488-conjugated donkey anti-goat, Invitrogen, A-11055, 1: 500), IB4-FITC (Sigma-Aldrich, L2895, 1:100) or Hoechst 33342 (Abcam, ab145597, 1:3000) diluted in PBS containing 1% BSA. After incubation with the secondary antibodies, the sections were washed 3 times for 5 min each and air-dried before being placed on coverslips with mounting medium (Invitrogen, P36961)
Images were acquired by a confocal laser scanning microscope (Leica, TCS SP8 STED) at a 512 × 512 pixel resolution. The neuronal areas and staining densities of all the images were analyzed using Image-Pro Plus version 6.0 software. Only DRG neurons with clear nuclear morphology were subjected to quantification analysis.
Tissue preparation and Western blot.
At 1, 6, and 24 h after CFA injection, treated rats and naive controls were deeply anesthetized with 10% chloral hydrate and their L4–L5 DRG neurons were quickly removed and immediately homogenized in ice-cold lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1.5 mm MgCl2, 10% glycerol, 1% Triton X-100, 5 mm EGTA, 1 mm phenylmethylsulfonyl fluoride, 1 mm Na3VO4, 10 mm NaF, and a proteinase inhibitor mixture purchased from AMRESCO that included 0.5 mm AEBSF, 0.3 mm aprotinin, 10 mm bestatin, 10 mm E-64, 10 mm leupeptin). The homogenates were centrifuged at 12,000 × g for 5 min at 4°C and the supernatants were analyzed. Protein concentrations were measured using a BCA assay kit (Pierce). Next, 50 μg of each sample was boiled for 5 min with SDS-PAGE sample buffer, subjected to SDS-PAGE using 10% or 12% running gels, and transferred onto nitrocellulose membranes. The membranes were blocked with 5% nonfat milk in TBST (50 mm tris-HCl, pH 7.5, 150 mm NaCl, and 0.05% Tween 20) for 1 h at room temperature and then incubated overnight at 4°C with the appropriate primary antibody (KChIP3, Santa Cruz Biotechnology, sc-166916, 1:100; KChIP3, Abcam, ab3474, 1:100; TRPV1, Santa Cruz Biotechnology, sc-12498, 1:100; β-actin, Santa Cruz Biotechnology, sc-47778, 1:500). The blots were then washed with TBST three times for 10 min each time. Next, the membranes were incubated with the appropriate horseradish peroxidase-conjugated secondary antibody (goat anti-mouse, Santa Cruz Biotechnology, sc-2005, 1:2000; goat anti-rabbit, Santa Cruz Biotechnology, sc-2004, 1:2000; Santa Cruz Biotechnology, sc-2768, rabbit-anti goat, 1: 2000) for 1 h at room temperature. Finally, the blots were developed with a lightening chemiluminescence kit (Santa Cruz Biotechnology, sc-2048).
Films were analyzed by Quantity One software and the densities of specific bands were measured and normalized to that of a loading control band.
Cell culture, plasmids transfection, and incubation with TAT fusion peptides.
HEK293 cells and CHO-TRPV1 cells were cultured in 35 mm dishes (Fisher) in a humidified atmosphere of 95% air and 5% CO2 at 37°C. The culture media were DMEM (Invitrogen) and F12 medium (Invitrogen), respectively, which were supplemented with 10% FBS (HyClone). Cells were transfected with Lipofectamine 2000 24–36 h after passage according to the methods described above. At 24–48 h after transfection, the cells were used for further analyses. For peptide applications, CHO-TRPV1 cells were incubated with either 3 μm TAT-scramble, TAT-1-20, or TAT-31-50 for 3 h until further analysis.
Coimmunoprecipitation (co-IP).
For co-IP, protein extracts containing 400–500 μg of protein from rat DRG tissues were incubated overnight at 4°C with a goat anti-TRPV1 antibody (Santa Cruz Biotechnology) or normal goat IgG (Santa Cruz Biotechnology, sc-2028) and a protein A-Sepharose CL-4B resin (GE Healthcare) in a volume of 500 μl. The samples were then washed six times with TBS/0.1% Triton X-100 to solubilize the bound proteins. Proteins in the TRPV1 precipitates were separated by 10% SDS-PAGE electrophoresis and KChIP3 was detected by Western blot analysis (Santa Cruz Biotechnology).
GST pull-down assay.
GST or His6 fusion proteins were expressed in Escherichia coli BL21 bacteria and purified according to the manufacturer's instructions. For binding assays, GST or GST fusion proteins were immobilized on a glutathione-Sepharose resin (GE Healthcare). The His6 fusion protein was purified using Ni-nitrilotriacetic acid magnetic beads (QIAGEN). The purified His6 fusion protein was then incubated with the immobilized GST or GST fusion proteins for 1 h at 4°C, washed, and subjected to SDS-PAGE electrophoresis.
Cell surface biotinylation assay.
Cell cultures were washed twice with ice-cold PBS and incubated for 30 min at 4°C with EZ-Link Sulfo-NHS-SS-biotin (1 mg/ml; Pierce) to biotinylate surface proteins. Excess biotin reagent was quenched and removed by washing the cells with PBS containing 100 mm glycine. Cells were lysed with lysis buffer (137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 2 mm KH2PO4, 1% Triton X-100, 0.1% SDS, and proteinase inhibitor mixture, pH 7.4). The lysates were centrifuged at 12,000 × g for 5 min to yield protein extracts in the supernatant and then the protein extracts were incubated with UltraLink Plus immobilized streptavidin beads (Pierce) for 2 h at 4°C to capture biotinylated surface proteins. After being washed with lysis buffer 5 times, bound proteins were eluted by boiling for 5 min with loading buffer and subjected to SDS-PAGE electrophoresis.
RT-PCR.
Total RNA was extracted using TRIzol reagent (Invitrogen). RNA concentration and purity were measured using a NanoDrop 2000c spectrophotometer (Thermo Scientific). Total RNA was reverse-transcribed for 1 h at 42°C in buffer containing 1 m oligo (dT) 18 primer, 2 mm dNTPs, 40 U of rRNasin (Promega) and 25 U of AMV reverse transcriptase (Roche). The primers pairs used for RT-PCR were designed to span introns, and the sequences were as follows: 5′-CCGAGGCTTCAAGAACGAATG-3′ and 5′-TGCTCCAGAGGTGCGTCCTT-3′ (351 bp fragment, spanning exon 3–7); 5′-AGCCTCGAGGCCACCATGCAGAGGACCAAGGAA-3′ or 5′-AGCAAGCTTGCCACCACCATGCAGAGGACCAAGGAA-3′ and 5′-AGCGAATTCTAGATGACGTTCTCAAACAG-3′ (full-length). The PCR products were separated by agarose gel electrophoresis with 0.5 μg/ml ethidium bromide, which was then observed under an ultraviolet light system.
Dissociation of DRG neurons and Ca2+ imaging.
The DRG neuron dissociation and Fura-2 AM-based Ca2+ imaging experiments were performed as described previously (Liu et al., 2015). After 4–6 d of recovery from the surgical placement of the intrathecal catheter, plasmids (EGFP, EGFP-KChIP3, EGFP-KChIP3Δ31–50) or peptides (TAT-scramble, TAT-31-50, TAT-1-20 of incremental dosages of 3, 10, and 30 μg) were delivered to the DRGs. One hour after CFA injection, the rats were anesthetized with 10% chloral hydrate and their left L4–L5 DRG neurons were removed and digested sequentially with collagenase type IA (1.5 mg/ml; Sigma-Aldrich) for 40–50 min and 0.125% trypsin (Sigma-Aldrich) for 8–10 min at 37°C. Enzymatic treatment was terminated by the addition of DMEM followed by gentle trituration of the ganglia with a flame-polished Pasteur pipette and centrifugation at 500 rpm for 5 min. The cell pellet was then resuspended in DMEM with 10% FBS. The dissociated cells were plated on poly-d-lysine-coated (100 mg/ml; Sigma-Aldrich) glass coverslips inside 35 mm culture dishes with a 10 mm bottom well and incubated for 1.5 h at 37°C with 5% CO2 and 95% air.
DRG cells were washed with extracellular solution (ES) containing the following (in mm): 130 NaCl, 5 KCl, 2 KH2PO4, 1.5 CaCl2, 6 MgCl2, 10 glucose, and 10 HEPES, pH 7.2, twice and then incubated with Fura-2 AM (Invitrogen, 5 μm) in ES at room temperature for 1 h. The cells were then washed with ES and incubated in ES at room temperature in the dark for a recovery period of 1 h. For calcium imaging, an inverted fluorescence microscope (Olympus) equipped with 340 and 380 nm excitation filters and a computer with MetaFluor software were used. Fluorescence images and F340/F380 ratios were acquired every 10 s. TRPV1 activation was evoked by the addition of 5 μm capsaicin.
Radiant heat measurement.
Thermal hyperalgesia was evaluated by following a thermal stimulus paradigm adapted from Hargreave's published reports (Hargreaves et al., 1988). Before testing, the animals were allowed to acclimatize to their environment for 10 min. The radiant heat source was adjusted to a range of 12–15 s for rats as the baseline latency with a cutoff time of 30 s to prevent tissue damage. To record the PWL in response to the heat stimulus, each hindpaw was measured 4 times at 5 min intervals and the mean value was recorded.
Hot plate.
The rat was placed on a hot plate set at 50°C or 54°C (IITC) and the latencies to flinching and licking of hindpaws were measured. All animals were tested sequentially with a minimum interval of 5 min between tests. To avoid tissue injury, a cutoff time was set at 60 and 30 s for assays at 50°C and 54°C, respectively (Duan et al., 2014).
Intraplantar injection of TAT peptide/capsaicin.
30 μg TAT-scramble or TAT-31-50 was injected into left hindpaw 3 h before 3 μg capsaicin intraplantar injection. All intraplantar injection was performed via microsyringe (Hamilton) in awake SD rats and the needle remained in situ for 10 s before withdrawal. After intraplantar injection of capsaicin, spontaneous pain behavior was tested by counting the total lifting time in the first 10 min. After that, evoked nociceptive responses were measured by radiant heat (as described above) 15, 30, 60, 90, and 120 min after capsaicin injection.
Von Frey filament measurement.
The paw withdrawal threshold in response to mechanical stimulus was determined using a series of von Frey filaments (Stoelting) ranging from 1.08 to 21.09 g. Animals were placed in a plastic cage with a metal mesh floor and allowed to move freely. They were allowed to acclimate to this environment for ∼10 min before testing to allow for behavioral accommodation. Von Frey filaments were applied to the midplantar surface of the operated hindpaw through the mesh floor. Probing was only performed when the animal's paw was in contact with the floor. Each probe was applied to the foot until it bent and was kept in this position for 6–8 s (Chaplan et al., 1994). The interval between consecutive filaments was at least 5 s. Filaments were started in 2 g increments and the mechanical threshold was calculated by the formula provided in previous studies (Chaplan et al., 1994).
Light touch sensitivity.
To assess light touch sensitivity, rats were habituated on an elevated wire grid for 15 min. The plantar hindpaw was stroked lightly with a paintbrush in the direction from heel to toe. This stimulation was repeated three times with interval of 10 s. For each test, no evoked movement was scored as 0 and walking away or occasionally brief paw lifting (∼1 s or less) was scored as 1. For each rat, the accumulative scores of three tests were used to indicate touch score (Duan et al., 2014).
Sticky tape assay.
For the sticky tape assay, we put a 1 × 2 cm rectangular adhesive sticky tape (Comix) on the hindpaw plantar of each rat and measured the latency of biting or licking to remove the tape (Duan et al., 2014).
Pinprick test.
For the pinprick test, we stroked the plantar surface of the hindpaw with a pin without skin penetration and counted the quantity of withdrawal response per 10 tries with 1 min interval (Duan et al., 2014).
Catwalk XT analysis.
The core of the CatWalk system (Noldus Information Technology) consists of an enclosed walkway on a glass plate that is traversed by a rodent from one side of the walkway to the other (Chen et al., 2014). Green light enters at the long edge of the plate and is completely internally reflected. Light can escape only at areas in which an animal's paw contacts the glass plate, resulting in scattered light. Using Illuminated Footprint technology, paws are captured by a high-speed video camera that is positioned underneath the walkway. The CatWalk XT system includes a high-speed digital camera (GP-3360; Gevicam) that acquires images at 100 frames/s. The CatWalk XT system's lens is 8.5 mm in size and 65 degrees in curvature (Fujinon). The pixel brightness depends on the amount of light received from such an area by the camera. The Illuminated Footprint enables intensity differences between an animal's paws to be detected.
The 3D footprint intensity tab plots the print intensity of the four paws for each individual frame in which the paws contact the glass plate in a 3D chart. The intensities vary from 0 to 225 and are represented by different colors. The 3D chart can be rotated in all directions.
For our experiment, 1 week before surgery, animals were trained daily on the CatWalk XT system until they could consistently make uninterrupted runs. Training was considered complete when the animals could make 3 consecutive uninterrupted runs within 3.0 s. Measurements were taken at 6 h after CFA plantar injection. Each rat was placed in the CatWalk XT system and runs were performed repeatedly until three uninterrupted runs were completed.
Analysis of these recordings yields many parameters, of which the following are of the most interest in the CFA-induced inflammatory pain model (Chen et al., 2014).
Intensity data include STAND intensity, CATWALK max intensity, and CATWALK max intensity of the top 15 pixels. STAND intensity is the mean intensity of the contact area of the hindpaw at the moment of maximal paw–floor contact during standing. CATWALK max intensity and CATWALK max intensity of the top 15 pixels are the mean intensity of the contact area and the 15 most intensive pixels of the hindpaw at the moment of maximal paw–floor contact during the catwalk, respectively. This parameter is expressed in arbitrary units (AU).
Coordination data include CATWALK swing duration. This parameter is the duration of the swing phase during catwalk. Because the duration of the stance or swing phase depends on the animal's walking speed and degree of dysfunction, these parameters are transformed to a fraction of the total step duration according to the following formula: fraction swing phase = [time in swing phase/(time in single step)] × 100%. Duration of the swing phase and total steps are expressed in seconds.
Print area data include STAND contact area, CATWALK max contact area, and CATWALK print area. The STAND contact area is the total floor area contacted by the paw during standing in pixels. The CATWALK max contact area is the maximum of the total floor area in pixels from single pictures of the paw in the stance phase during the catwalk. The CATWALK print area is the total floor area in pixels summated from all the pictures of the paw in the stance phase during the catwalk (compared with the max contact area, this parameter reflects the entire paw area from the third toe to the heel).
To illustrate alterations of the intensity and print area data of the inflamed left hindpaw clearly, we used the formula (RH − LH)/RH to highlight difference between two hindpaws (where RH is the right hindpaw and LH is the left hindpaw) and to remove confounding factor influences such as the weight and baseline levels of the print area.
Statistical analysis.
All of the data are represented as the mean ± SEM. Statistical analyses were performed using Prism version 7.0 software. Comparisons between two groups were performed by either Student's t test (interval and ratio scale data) or χ2 test (nominal scale data). Comparisons among groups were made by one-way ANOVA followed by Tukey's multiple-comparisons test for comparing the mean of each group with the mean of every other group or Dunnett's multiple-comparisons test for comparing the mean of each group with the mean of a control group. Comparisons among groups with different time points were made by two-way ANOVA followed by Sidak's multiple-comparisons test. Statistical significance was set at p < 0.05. In cases of heterogeneity, Welch's correction (for Student's t test) or the Greenhouse–Geisser correction (for ANOVA) was applied to adjust the degree of freedom.
Results
KChIP3 is widely expressed in different subpopulations of DRG neurons
Recent immunostaining studies revealed the distribution of KChIP3 in myelinated Aβ and Aδ sensory neurons as well as that of peptidergic nociceptive neurons in rats (Cheng et al., 2016). Considering the high homology of KchIP1–4 in their C-terminal regions, we investigated the expression pattern of KChIP3 in rat DRG neurons using a commercial antibody generated against the N-terminal 31-45 fragment of KChIP3. The specificity of the antibody was proven in kcnip3 KO rats (Fig. 1-1, and Fig. 8B). The immunostaining results indicated that the KChIP3 protein is present in different subpopulations of DRG neurons, including small-, medium- and large-diameter neurons (Fig. 1A), which is consistent with previous studies (Pruunsild and Timmusk, 2005; Palczewska et al., 2011; Matsuyoshi et al., 2012; Cheng et al., 2016). For quantification analysis, 2754 neurons (small: n = 1389; medium: n = 852; large: n = 513) from rat L4 or L5 DRGs were counted. In total, 42.34% of the DRG neurons were KChIP3 immunoreactive. Among these KChIP3-positive neurons, 33.53% were small, 35.85% were medium, and the remaining 30.62% were large (Fig. 1B). Moreover, the percentage of KChIP3-positive neurons increased as the cell diameter increased (Fig. 1C). The quantification results indicated that 28.15% of the small neurons, 49.06% of the medium neurons, and 69.59% of the large neurons were KChIP3 positive, respectively (χ2 test: small vs medium: χ(2)2 = 100.1; p < 0.001; small vs large: χ(2)2 = 269.6, p < 0.001; medium vs large: χ(2)2 = 54.99, p < 0.001; Fig. 1D). In addition, staining densitometry was performed to measure the staining intensity of KChIP3 in the different sizes of neurons (Fig. 1E). As seen in Figure 1A, compared with KChIP3-negative DRG neurons (marked by white arrows), cell diameter was positively correlated with KChIP3 staining intensity (marked by colored arrows). The KChIP3 staining intensity increased with the cell diameter increment (small: n = 389; medium: n = 352; large: n = 225, one-way ANOVA with Greenhouse–Geisser correction: F(2,963) = 46.44, p < 0.001; Tukey's multiple-comparisons test: small vs medium, p < 0.001; small vs large: p < 0.001; medium vs large, p < 0.001; Fig. 1F).
Figure 8.
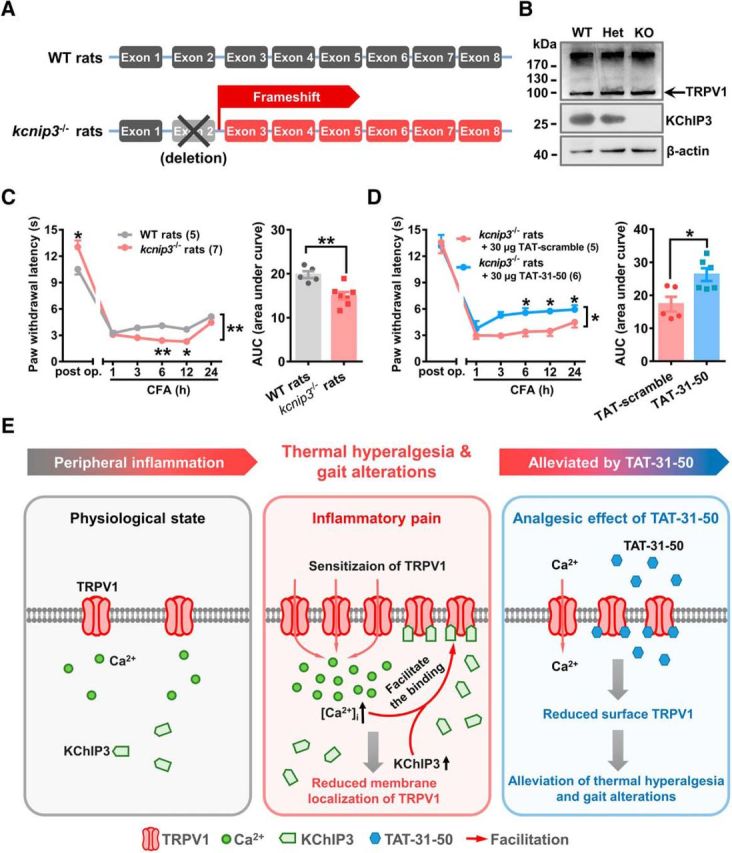
TAT-31-50 alleviates CFA-induced thermal hyperalgesia in kcnip3 KO rats. A, Schematic diagram of the genetic editing in kcnip3−/− rats. B, Representative Western blots of KChIP3 and TRPV1 in wild-type (WT), heterozygous (Het), and kcnip3 KO rats. C, D, Time course of the PWL of the ipsilateral hindpaw before and after CFA injection in WT and kcnip3 KO rats (C) or kcnip3 KO rats pretreated with an intrathecal administration of 30 μg of TAT-31-50 or TAT-scramble (D). C, D, Left, Two-way ANOVA, group effect: *p < 0.05, shown at the ends of the lines; post test: Sidak's multiple-comparisons test, *p < 0.05, **p < 0.01, shown above the lines. Right, AUC values of the PWL time courses were compared between the WT and kcnip3 KO rats or the TAT-31-50- and TAT-scramble-injected rats. Unpaired t test, *p < 0.05, **p < 0.01. E, Schematic diagram of the inhibitory effect of KChIP3 and TAT-31-50 on TRPV1. Under physiological conditions, a low amount of TRPV1 is expressed on the plasma membrane. During inflammatory pain, TRPV1 undergoes functional sensitization and the intracellular [Ca2+]i is increased. Concurrently, expression of the KChIP3 protein is upregulated at 6 h after CFA injection. Therefore, binding between KChIP3 and the intracellular domains of TRPV1 is enhanced, which leads to a reduction in the surface localization of TRPV1 and the subsequent inhibition of TRPV1-mediated Ca2+ influx by KChIP3. Importantly, TAT-31-50, constructed based on the critical binding site of KChIP3 mediating its interaction with TRPV1, alleviates thermal hyperalgesia and gait alterations induced by intraplantar CFA injection significantly.
Figure 1.
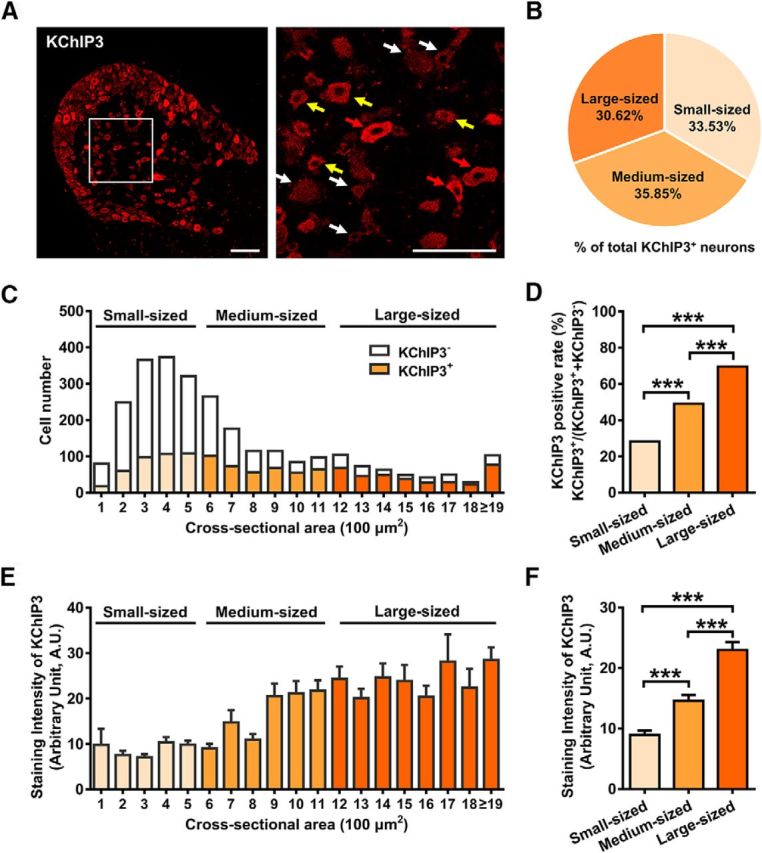
KChIP3 is widely expressed in different-sized DRG neurons, including small-, medium-, and large-diameter neurons. A, Representative image of immunostaining of KChIP3 in rat DRG. Enlarged image of the boxed area in the left image is shown on the right. Red, yellow, and white arrows indicate the strongly positive, weakly positive, and negative neurons, respectively. Scale bar, 200 μm. B, Quantification analysis of the proportion of small, medium, or large neurons among KChIP3+ neurons (KChIP3+ neurons of each subtype/total KChIP3+ neurons). C, Cell counting of KChIP3− and KChIP3+ neurons in DRG neurons with cross-sectional area of <600 (small), 600–1200 (medium), and >1200 mm2 (large), respectively. D, Quantification analysis of the percentage of KChIP3+ neurons in small, medium, and large neurons, respectively [KChIP3+/(KChIP3+ plus KChIP3−)]. χ2 test, ***p < 0.001. E, Staining intensity of KChIP3 in DRG neurons with cross-sectional area of <600 (small), 600–1200 (medium), and >1200 mm2 (large), respectively. F, Quantification analysis of the staining intensity of KChIP3 in small, medium, and large sized DRG neurons. One-way ANOVA followed by Tukey's multiple-comparisons test, ***p < 0.001. For more information, refer to Figures 1-1 and 1-2, respectively.
This extended data figure supports for Figure 1. Representative images of the immunostaining of KChIP3 (green) and Hoechst 33342 (blue) in DRG neurons of WT (left image) and kcnip3 KO rats (right image), respectively. A commercial antibody (Abcam) against the N-terminal 30-45 fragment of KChIP3 was used. Scale bar = 200 μm. Download Figure 1-1, TIF file (4.9MB, tif)
This extended data figure supports for Figure 1. KChIP3 mRNA detection in DRG tissue by RT-PCR. A, Detection of KChIP3 mRNA in L4-5 DRG extracts by PCR using the primer pairs 1 (351 bp fragment), 2 and 3 (full-length), respectively. A no primer group was used as the negative control. B, Detection of KChIP3 mRNA in DRG neurons and spinal cord extracts, using the full-length primer pairs, respectively. A no cDNA group was used as the negative control. Download Figure 1-2, TIF file (1MB, tif)
In addition to immunofluorescence, RT-PCR experiments were performed to detect KChIP3 mRNA expression in the rat DRG tissue. Three pairs of primers were used to amplify fragmented or full-length KChIP3 mRNA. PCR products with the expected sizes were acquired (Fig. 1-2A). The PCR product from spinal cord tissue was simultaneously used as a positive control (Fig. 1-2B). Our studies confirm the broad expression of KChIP3 in different subpopulations of DRG neurons, supporting its potential role in peripheral sensory processing.
KChIP3 alleviates the thermal hyperalgesia in the rat inflammatory pain model
Considering the distribution of KChIP3 in small nociceptive neurons, we speculated whether KChIP3 is involved in nociceptive processing. First, an inflammatory pain model was induced in rats via an intraplantar injection of 25% CFA (100 μl/paw). The time course of KChIP3 protein expression in L4/5 (ipsilateral) DRG tissue after CFA injection was examined. Expression of the KChIP3 protein peaked at 6 h (∼2.1-fold that of the naive rat) and returned to the basal level at 24 h after CFA injection (n = 3; one-way ANOVA: F(3,8) = 23.23, p < 0.001; Tukey's multiple-comparisons test: naive vs 6 h, p = 0.004; Fig. 2A).
Figure 2.
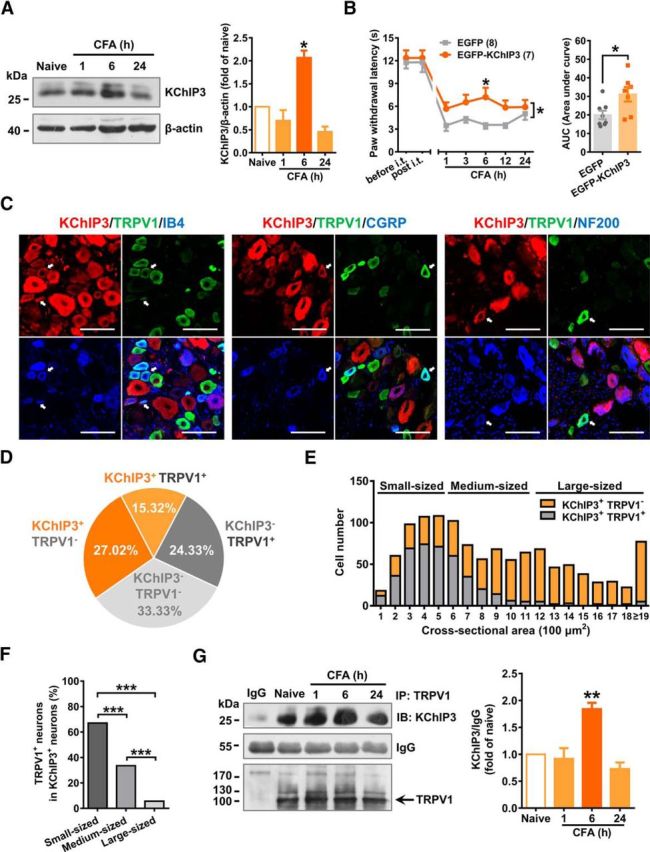
Introduction of exogeneous KChIP3 expression alleviates CFA-induced thermal hyperalgesia behavior and an association between KChIP3 and TRPV1 was observed in DRG tissue. A, Representative image of KChIP3 Western blots in ipsilateral L4–5 DRG lysates at 1, 6, 24 h after the intraplantar injection of CFA (left). Quantification analysis of the KChIP3 protein expression in the left image (right). One-way ANOVA followed by Tukey's multiple-comparisons test, *p < 0.05. B, Time course of PWL of the ipsilateral hindpaw before and after CFA injection in EGFP or EGFP-KChIP3 plasmid-injected rats (left). Two-way ANOVA, group effect: *p < 0.05, shown at the end of the lines; post test: Sidak's multiple-comparisons test, *p < 0.05, shown above the lines. AUC values from 1–24 h after CFA injection were compared between the EGFP- and EGFP-KChIP3-injected groups, which is shown on the right (right). Unpaired t test, *p < 0.05. C, Representative images of the immunostaining of KChIP3 (red), TRPV1 (green), and IB4 (blue, left)/CGRP (blue, middle)/NF200 (blue, right) in DRG neurons. Scale bar, 100 μm. D, Quantification analysis of the percentage of KChIP3+-TRPV1+, KChIP3+-TRPV1−, KChIP3−-TRPV1+, and KChIP3−-TRPV1− neurons in DRG neurons. E, Cell counting of KChIP3+-TRPV1− and KChIP3+-TRPV1+ neurons in DRG neurons with cross-sectional areas of 100–500 (small), 600–1100 (medium), and >1200 mm2 (large), respectively. F, Quantification analysis of the percentage of TRPV1+ neurons in KChIP3+ neurons classified as small, medium, or large DRG neurons. χ2 test, ***p < 0.001. G, Representative result of TRPV1 and KChIP3 co-IP studies in ipsilateral DRG lysates at 1, 6, and 24 h after CFA injection. One-way ANOVA followed by Tukey's multiple-comparisons test, **p < 0.01. For more information, see Figure 2-1.
This extended data figure supports for Figure 2. Expression of EGFP-KChIP3 plasmids in HEK293 cell line and DRG tissues. A, Representative images of the expression of EGFP-KChIP3 in HEK293 cells. B, Representative images of the expression of EGFP-KChIP3 in DRG lysates. C, Representative images of immunostaining of EGFP (green), KChIP3 (red), Hoechst 33342 (blue) in DRG slices after intrathecal delivery of EGFP-KChIP3 or EGFP plasmids and the DRG slices of naïve rats. Enlarged images of the boxed area in the upper rows are shown in the lower rows. Yellow arrows indicate the DRG neurons labelled by green and red immunofluorescence concurrently. Red and green arrows indicate the neurons labelled by red or green immunofluorescence respectively. Scale bar = 100 μm. Download Figure 2-1, TIF file (38.3MB, tif)
To investigate the potential role of KChIP3 upregulation in inflammatory pain behaviors, we injected the EGFP-KChIP3 plasmids intrathecally to induce exogenous KChIP3 expression in DRG neurons 7 d before the CFA injection. Efficient expression of the plasmids was verified by the detection of an ∼55 kDa EGFP-KChIP3 protein band in both in vitro and in vivo studies (Fig. 2-1A,B) and the EGFP fluorescence in the majority of DRG neurons (Fig. 2-1C). The radiant heat test indicated that exogenous EGFP-KChIP3 expression did not alter the PWL of the hindpaw in the basal state; however, EGFP-KChIP3 expression significantly alleviated CFA-induced thermal hyperalgesia at 6 h after CFA injection (n = 7 or 8, two-way repeated-measures ANOVA: group effect F(1,13) = 6.466, p = 0.0245; Sidak's multiple-comparisons test: p(6) = 0.0105; Fig. 2B), which was consistent with the peak KChIP3 protein expression after CFA injection. In addition, the area under the curve (AUC) from 1–24 h was calculated and significant differences were detected between the control and EGFP-KChIP3 groups (n = 7 or 8, unpaired Student's t test: t(13) = 2.543, p = 0.0245; Fig. 2B). These findings suggest that increased KChIP3 levels in DRG neurons after plantar inflammation might reduce thermal hyperalgesia.
Association of KChIP3 and TRPV1 is enhanced after peripheral inflammation
Given that KChIP3 is a calcium sensor protein that regulates inflammatory thermal hyperalgesia negatively, we tested whether the calcium-permeable channel TRPV1, a key molecule responsible for thermal hyperalgesia behavior, could be modulated by KChIP3.
First, triple immunostaining in rat DRG tissue slices confirmed the colocalization of KChIP3 and TRPV1 in multiple subpopulations of DRG neurons (Fig. 2C). In total, 2754 DRG neurons were counted, among which 15.32% were double labeled by KChIP3 and TRPV1, 27.02% were KChIP3+/TRPV1−, 24.33% were KChIP3−/TRPV1+, and the remaining 33.33% were KChIP3−/TRPV1− (Fig. 2D). Further quantification analysis revealed that the colocalization of KChIP3 and TRPV1 was more prominent in small KChIP3-positive DRG neurons than in the medium and large neurons (χ2 test: small vs medium: χ(2)2 = 90.77; p < 0.001; small vs large: χ(2)2 = 299.59, p < 0.001; medium vs large: χ(2)2 = 91.43, p < 0.001; Fig. 2E,F). Of these KChIP3+/TRPV1+ neurons, 44.76% were colabeled with IB4 (64/143, n = 1028), 26.15% were coexpressed with CGRP (51/195, n = 1167), and 21.18% were merged with NF200 (18/85, n = 567), suggesting that the interaction between KChIP3 and TRPV1 occurs mainly in both nonpeptidergic and peptidergic nociceptive DRG neurons (Fig. 2C).
Next, co-IP studies in DRG lysates showed that the TRPV1 antibody could precipitate KChIP3 and this interaction peaked 6 h after CFA injection (n = 3; one-way ANOVA: F(3,8) = 15.00, p = 0.0012; Tukey's multiple-comparisons test: naive vs 6 h, p = 0.0069; Fig. 2G). The peak time of interaction between TRPV1 and KChIP3 was consistent with that of elevated KChIP3 protein expression and the analgesic effect exhibited by exogenous KChIP3 after CFA injection. Altogether, these data imply that the interaction between KChIP3 and TRPV1 might be involved in the analgesic effect afforded by exogenous EGFP-KChIP3 expression.
KChIP3 binds to both the intracellular N and C termini of TRPV1
Interaction between TRPV1 and KChIP3 was further investigated with co-IP studies in HEK293 cells cotransfected with TRPV1 and KChIP3 plasmids. The TRPV1 protein can be precipitated together with EGFP-KChIP3 by a GFP antibody (Fig. 3A). Reciprocally, the KChIP3 protein can be precipitated together with GFP-TRPV1 by a GFP antibody (Fig. 3B). Because co-IP studies could not exclude the possibility of indirect protein–protein interaction involving an accessory protein, we performed GST pull-down assays to investigate direct binding between KChIP3 and TRPV1. As expected, GST-KChIP3 precipitated both the N and C terminus of His6-TRPV1 (Fig. 3C,D). Conversely, both GST-TRPV1 N and C terminus precipitated His6-KChIP3 (Fig. 3E). Moreover, the binding of KChIP3 and TRPV1 showed calcium sensitivity. The addition of 100 μm Ca2+ enhanced the binding between GST-KChIP3 and His6-TRPV1 C terminus, whereas Ca2+ chelation by 1 mm EGTA reduced this effect (Fig. 3F). The above in vitro studies revealed direct binding between KChIP3 and the intracellular N and C termini of TRPV1.
Figure 3.

KChIP3 binds with both the intracellular N and C termini of TRPV1. A, B, Representative results of KChIP3 and TRPV1 co-IP studies in HEK293 cells transfected with the EGFP-KChIP3 and GST-TRPV1 plasmids (A) or the EGFP-TRPV1 and KChIP3 plasmids (B). C–E, Representative results of GST pull-down assays. GST-KChIP3 precipitated the His6-TRPV1 N terminus (Nt; C) and the His6-TRPV1 C terminus (Ct; D); His6-KChIP3 precipitated the GST-TRPV1 Nt and GST-TRPV1 Ct (E). F, Representative results of the GST pull-down assays showing the Ca2+ sensitivity of the association between GST-KChIP3 and His6-TRPV1 Ct. The addition of 100 μm Ca2+ in the reaction buffer increased the binding of KChIP3 and TRPV1-Ct, whereas the chelation of intracellular Ca2+ by 1 mm EGTA reduced this effect. G, H, Representative results of biotinylation studies on the surface of TRPV1 (G) in CHO-TRPV1 cells transfected with EGFP or EGFP-KChIP3 plasmids. Statistical analysis of the surface level of TRPV1 is shown on the right. Unpaired t test, *p < 0.05. The total level of TRPV1 was also examined, and quantification analysis of the results is shown on the right (unpaired t test).
To investigate the potential effect of KChIP3 binding on TRPV1 function, we examined surface TRPV1 using a biotinylation assay in CHO-TRPV1 cells (CHO cells stably expressing TRPV1) transfected with either the EGFP or EGFP-KChIP3 plasmids. A significant decrease (∼50%) in the surface TRPV1 level was detected (n = 4, unpaired Student's t test: t(6) = 2.849, p = 0.0292; Fig. 3G). However, the total amount of TRPV1 protein was not affected by the transfection of EGFP-KChIP3 (n = 5 or 6, unpaired Student's t test with Welch's correction: t(9) = 1.377, p = 0.2266; Fig. 3H). This finding suggests that KChIP3 modulates the surface localization of TRPV1 negatively by binding its intracellular domains.
KChIP3 N-terminal 31-50 fragment is responsible for the binding with TRPV1 N and C termini
To identify the critical domains of KChIP3 responsible for its binding with TRPV1, GST fusion proteins containing different fragments of KChIP3 were generated, including GST-1-50, GST-51-100, GST-101-256, GST-Δ1-50, and GST-KChIP3 (Fig. 4A). GST pull-down assays showed that GST-1-50 strongly precipitated both His6-TRPV1 N terminus and His6-TRPV1 C terminus (Fig. 4C). In contrast, deletion of the N-terminal 1-50 fragment of KChIP3 remarkably reduced the binding between KChIP3 and the TRPV1 N or C terminus (Fig. 4C). These data suggest that the KChIP3 1–50 fragment is required for its binding with TRPV1.
Figure 4.
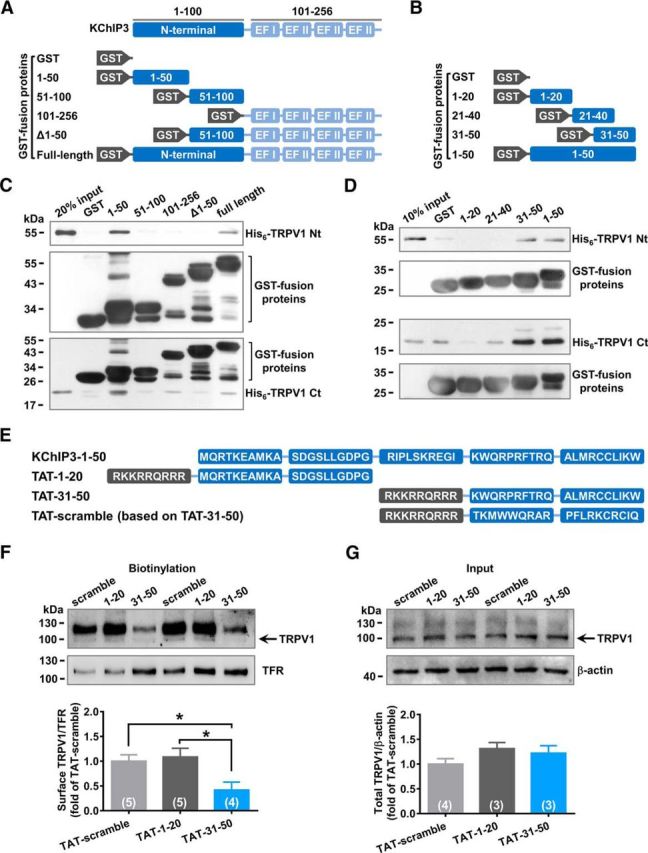
KChIP3 N-terminal 31-50 fragment binds with both the N and C termini of TRPV1. A, B, Schematic diagram of the GST fusion proteins containing different fragments of KChIP3 (A) or KChIP3–1-50 (B). C, D, Representative results of GST-pull down assays. GST-1-50 precipitated both His6-TRPV1 N and C termini (C). More specificly, GST-31–50 precipitated both His6-TRPV1 N and C termini (D). E, Schematic diagram of TAT fusion peptides containing different fragments of KChIP3-1-50 and a scramble control of TAT-31-50. F, G, Representative results of biotinylation studies on the TRPV1 surface level in CHO-TRPV1 cells incubated with 3 μm TAT fusion peptides for 3 h. F, Bottom, Quantification analysis of the surface level of TRPV1. One-way ANOVA followed by Tukey's multiple-comparisons test, *p < 0.05. Total protein level of TRPV1 was also examined as well (G) and quantification analysis of the results is shown below (one-way ANOVA).
To further map critical binding sites in the KChIP3 1-50 fragment, GST-1-20, GST-21-40, and GST-31-50 fusion proteins were generated (Fig. 4B). GST pull-down assays indicated that GST-31-50 precipitated both His6-TRPV1 N terminus and His6-TRPV1 C terminus in a manner similar to that of GST-1-50 (Fig. 4D). Therefore, the 31-50 fragment constitutes the core domain mediating KChIP3 binding with the intracellular domains of TRPV1.
Next, we investigated whether the 31-50 fragment of KChIP3 was sufficient for mediating the effect of KChIP3 on TRPV1. The protein transduction domain of the HIV-1 TAT protein was fused with the fragments of KChIP3 1-20, 31-50, and the scramble control of 31-50 to introduce these fragments into the cytoplasm (Fig. 4E). Incubation with the TAT-31-50 peptide (3 μm, 3 h) reduced the surface TRPV1 significantly in CHO-TRPV1 cells compared with groups treated with TAT-1-20 or TAT-scramble (n = 4 or 5, one-way ANOVA: F(2,11) = 4.956, p = 0.0292; Dunnett's multiple-comparisons test: TAT-31-50 vs TAT-1-20, p = 0.0471; TAT-31-50 vs TAT-scramble, p = 0.0244; Fig. 4F). However, the total TRPV1 protein levels were similar among the groups after treatment with the TAT fusion peptide (Fig. 4G). Together, these data suggest that TAT-31-50 inhibits the surface localization of TRPV1 dramatically.
Transmembrane TAT-31-50 peptide exhibits the analgesic effect in the rat inflammatory pain model
Given the inhibitory effect of TAT-31-50 on the surface localization of TRPV1 in CHO-TRPV1 cells, we next investigated whether intrathecal administration of this peptide could alleviate CFA-induced thermal hyperalgesia by downregulating the function of TRPV1 (see procedure diagram in Fig. 5A).
Figure 5.
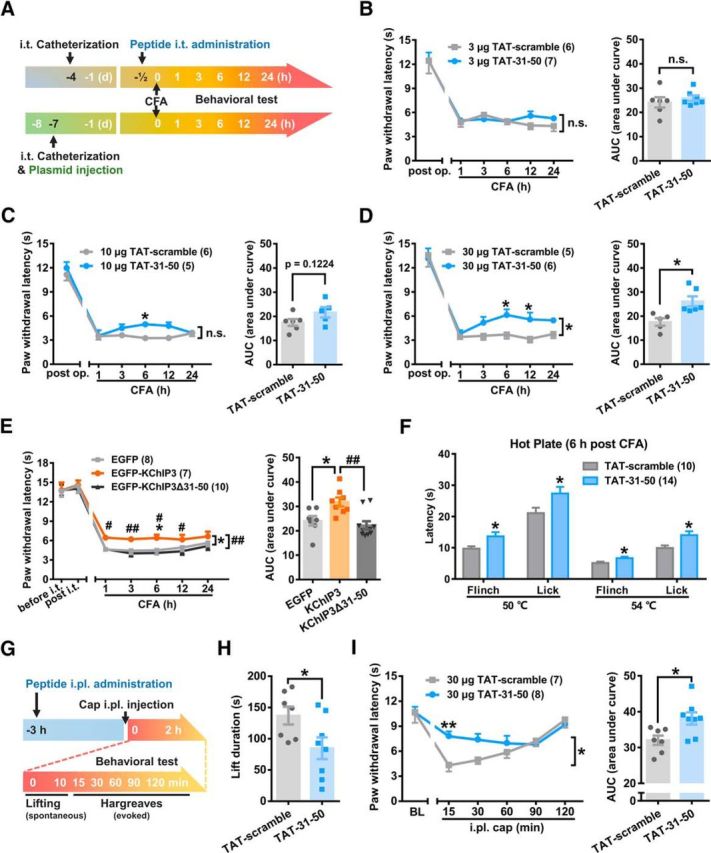
Intrathecal injection of TAT-31-50 peptide alleviates CFA-induced thermal hyperalgesia. A, Schematic diagram of the protocol for the measurement of thermal pain threshold after intrathecal administration of peptides. B–D, Time course of PWL of the ipsilateral hindpaw before and after CFA injection from 1–24 h in the TAT-31-50- or TAT-scramble-injected (i.t.) rats. A dose of 3 (B), 10 (C), or 30 μg (D) TAT-31-50 or TAT-scramble peptide was injected intrathecally. B–D, Left, Two-way ANOVA, group effect: *p < 0.05, shown in the end of the lines; post test: Sidak's multiple-comparisons test, *p < 0.05, shown above the lines. Right, AUC from 1–24 h after CFA injection was compared between the TAT-31-50- and TAT-scramble-injected groups. Unpaired t test, *p < 0.05. E, Time course of PWL of the ipsilateral hindpaw before and after CFA injection (left) in the EGFP, EGFP-KChIP3, or EGFP-KChIP3Δ31–50 plasmid-injected rats. Two-way ANOVA, post test: Tukey's multiple-comparisons test, *p < 0.05, compared with the EGFP group, ##p < 0.01, compared with the EGFP-KChIP3 group, shown in the end of the lines; post test: Sidak's multiple-comparisons test, *p < 0.05 compared with the EGFP group, #p < 0.05, and ##p < 0.01 compared with the EGFP-KChIP3 group, shown above the lines. Right, AUC from 1–24 h after CFA injection was compared among the three groups. One-way ANOVA, *p < 0.05, compared with the EGFP group, ##p < 0.01, compared with the EGFP-KChIP3 group. F, Latency of flinching and licking on hot plate (50°C and 54°C) was compared between the 30 μg TAT-31-50- and TAT-scramble-injected (i.t.) groups 6 h after CFA injection. Multiple t test, *p < 0.05. G, Schematic diagram of the protocol for the measurement of spontaneous and evoked pain after intraplantar (i.pl.) injection of the peptide and capsaicin (cap). H, Lifting time in the first 10 min after capsaicin injection was compared between the TAT-31-50- and TAT-scramble-injected groups. Unpaired t test, *p < 0.05. I, Time course of PWL of the left hindpaw before and after capsaicin injection from 15–120 min in the TAT-31-50- or TAT-scramble-injected rats. Left, Two-way ANOVA, group effect: *p < 0.05, shown in the end of the lines; post test: Sidak's multiple-comparisons test, **p < 0.01, shown above the lines. Right, AUC from 15–120 min after capsaicin injection was compared between the TAT-31-50- and TAT-scramble-injected groups. Unpaired t test, *p < 0.05. For more information, see Figures 5-1 and 5-2, respectively.
This extended data figure supports for Figure 5. Intrathecal injection of TAT-31-50 alleviates CFA-induced thermal hyperalgesia compared to TAT-1-20. A–B, Time course of the PWL of the ipsilateral hind paw before and after CFA injection from 1 to 24 h in TAT-1-20- or TAT-31-50-injected rats. The rats were pre-treated with an intrathecal administration of 10 or 30 μg of the TAT-1-20 or TAT-31-50 (n = 5) peptides (B). The left panel of A–B shows two-way ANOVA, group effect: *p < 0.05, shown at the ends of the lines; post-test: Sidak's multiple comparisons test, **p < 0.01, *p < 0.05, shown above the lines. In the right panel of A–B, area under the curve values of the paw withdrawal latencies from 1 to 24 h after CFA injection were compared between the TAT-31-50- and TAT-scramble-injected groups. Unpaired t-test, *p < 0.05. Download Figure 5-1, TIF file (733.2KB, tif)
This extended data figure supports for Figure 5. Intrathecal injection of TAT-31-50 exerts no influence on the mechanosensation of A-fibers. A–B, Time course of PWL (A) or mechanical threshold (B) of the ipsilateral hindpaw before and after CFA injection from 6 to 24 h in the 30 μg TAT-scramble- or TAT-31-50-injected (i.t.) rats. Two-way ANOVA, group effect: ***p < 0.001, shown in the end of the lines; post-test: Sidak's multiple comparisons test, ***p < 0.001, **p < 0.01, *p < 0.05, shown above the lines. C, Correlation analysis between PWL and mechanical threshold in the 30 μg TAT-scramble- or TAT-31-50-injected (i.t.) rats. Linear regression, n = 10 or 14. D, E, F, Light touch sensitivity (D), latency to detect the sticky tape (E) and pinprick reactivity (F) of the ipsilateral hindpaws are compared between the 30 μg TAT-scramble- or TAT-31-50-injected (i.t.) rats 6 h post CFA injection. Unpaired t test. Download Figure 5-2, TIF file (1.6MB, tif)
The thermal pain thresholds were comparable between the groups in which TAT-31-50 and TAT-scramble were injected after catheterization. However, the intrathecal injection of TAT-31-50 (10 μg) prolonged the PWL significantly 6 h after CFA injection compared with that of the TAT-scramble group (n = 5 or 6, two-way repeated-measures ANOVA: group effect F(1,9) = 2.907, p = 0.1224; Sidak's multiple-comparisons test: p(6) = 0.0292; Fig. 5C, left). The analgesic effect was remarkable when the peptide dosage was increased to 30 μg (n = 5 or 6, two-way repeated-measures ANOVA: group effect F(1,9) = 10.26, p = 0.0108; Sidak's multiple-comparisons test: p(6) = 0.0251, p(12) = 0.0256; Fig. 5D, left). Simultaneously, the AUC (1–24 h) values were compared between the TAT-31-50- and TAT-scramble-injected groups and a significant difference was detected (n = 5–6, unpaired Student's t test: t(9) = 3.204, p = 0.0108; Fig. 5D, right). In addition, due to its inability to bind TRPV1, TAT-1-20 was also used as a negative control. Compared with TAT-1-20, TAT-31-50 increased the PWL and AUC (1–24 h) values significantly after -CFA injection (Fig. 5-1). These data demonstrate the analgesic effect of intrathecally administered TAT-31-50 on thermal hyperalgesia.
To further prove that the 31–50 fragment was indispensable for the analgesic effect of KChIP3, we constructed the EGFP-KChIP3Δ31–50 plasmid. In contrast to the analgesia elicited by EGFP-KChIP3, intrathecal injection of EGFP-KChIP3Δ31–50 showed no effect on PWL after CFA injection (n = 7, 8, or 10, PWL: two-way repeated-measures ANOVA: group effect F(2,22) = 7.980, p = 0.0025; Tukey's multiple-comparisons test of group effects: EGFP vs EGFP-KChIP3, p = 0.0239; EGFP vs EGFP-KChIP3Δ31–50, p = 0.7586; EGFP-KChIP3 vs EGFP-KChIP3Δ31–50, p = 0.0024; Tukey's multiple-comparisons test of each time point: EGFP vs EGFP-KChIP3, p(6) = 0.0435; EGFP-KChIP3 vs EGFP-KChIP3Δ31–50, p(1) = 0.0397, p(3) = 0.008, p(6) = 0.0045, p(12) = 0.0354; Fig. 5E, left). AUC (1–24 h) analysis also showed no difference between the EGFP and EGFP-KChIP3Δ31–50 groups (AUC: one-way ANOVA: F(2,22) = 7.980, p = 0.0025; Tukey's multiple-comparisons test: EGFP vs EGFP-KChIP3, p = 0.0239; EGFP vs EGFP-KChIP3Δ31–50, p = 0.7586; EGFP-KChIP3 vs EGFP-KChIP3Δ31–50, p = 0.0024; Fig. 5E, right). However, consistent with the results in Figure 2B, the analgesic effect exerted by the intrathecal injection of EGFP-KChIP3 was remarkable. These findings demonstrated that deletion of the 31–50 fragment abolished the analgesic effect of KChIP3, suggesting the necessity of this fragment for the functional effect of full-length KChIP3.
In addition to radiant heat, a hot plate test with the plate set at 50°C or 54°C was performed 6 h after CFA injection. Intrathecal administration of 30 μg of TAT-31-50 prolonged the latency of flinching or licking significantly compared with scramble control (multiple t tests, TAT-scramble vs TAT-31-50: 50°C flinching: p = 0.0338; 50°C licking: p = 0.0398; 54°C flinching: p = 0.0400, 54°C licking: p = 0.0223; Fig. 5F).
In addition to the attenuation of thermal hyperalgesia by TAT-31-50 both 6 and 24 h post CFA injection (n = 10 or 14, two-way repeated-measures ANOVA: group effect F(1,22) = 48.25, p < 0.0001; Sidak's multiple-comparisons test: p(6) < 0.0001, p(12) < 0.0001; Fig. 5-2A), mechanical nociceptive threshold was slightly increased 6 h after CFA injection (n = 10 or 14, two-way repeated-measures ANOVA, Sidak's multiple-comparisons test: p(6) = 0.0433; Fig. 5-2B). Moreover, the correlation between the thermal hyperalgesia and mechanical nociceptive threshold was detected (n = 10 or 14, linear regression, p(TAT-scramble) = 0.0244, p(TAT-31-50) = 0.0486; Fig. 5-2C).
In consideration of the wide expression of KChIP3 in different-sized DRG neurons and thereby the possible effect of TAT-31-50 on A-fibers where TRPV1 is scarcely expressed, multiple somatosensory assays dependent on A-fibers are performed, including: (1) the light touch sensitivity test, (2) the pinprick test, and (3) the sticky tape assay. TAT-31-50 exerted no effect on these A-fiber-related somatosensation 6 h after CFA injection (Fig. 5-2D–F).
To further confirm the TRPV1-dependent in vivo analgesic effect in the periphery, we administered 30 μg of TAT-scramble or TAT-31-50 through intraplantar injection 3 h before capsaicin (3 μg) injection (Fig. 5G). Spontaneous pain behavior was assayed by counting the total lifting time in the first 10 min. Intraplantar injection of TAT-31-50 significantly shortened the lifting duration (n = 7 or 8, unpaired t test, p = 0.0401; Fig. 5H). In addition, evoked nociceptive responses were measured by radiant heat at 15, 30, 60, 90, and 120 min after capsaicin injection. The PWL at 15 min (n = 7 or 8, two-way repeated-measures ANOVA: group effect F(1,13) = 7.686, p = 0.0158; Sidak's multiple-comparisons test: p(15) = 0.0047; Fig. 5I, left) and total AUC (n = 7 or 8, unpaired t test, p = 0.0158; Fig. 5I, right) were increased significantly by TAT-31-50. In summary, both central and peripheral application of TAT-31-50 had an analgesic effect.
TAT-31-50 alleviates the gait alterations elicited by plantar inflammation
CatWalk gait analysis, a computer-assisted locomotor analysis, has been used to detect gait alterations and has been proposed as an objective tool for evaluating mechanical allodynia induced by nerve injury (Vrinten and Hamers, 2003). It allows the rapid quantification of individual paw parameters and parameters related to interlimb coordination. To evaluate the effect of TAT-31-50 on inflammation-induced mechanical allodynia, the Catwalk XT gait analysis system was used to assess changes in both static and dynamic gaits (Fig. 6A) at 6 h after CFA injection. The intensity difference of paw prints between the RH (contralateral side) and the LH (ipsilateral side) was calculated. To rule out body weight and pedal size influences on the paw print intensities, the RH − LH/RH ratio was used in the following studies.
Figure 6.
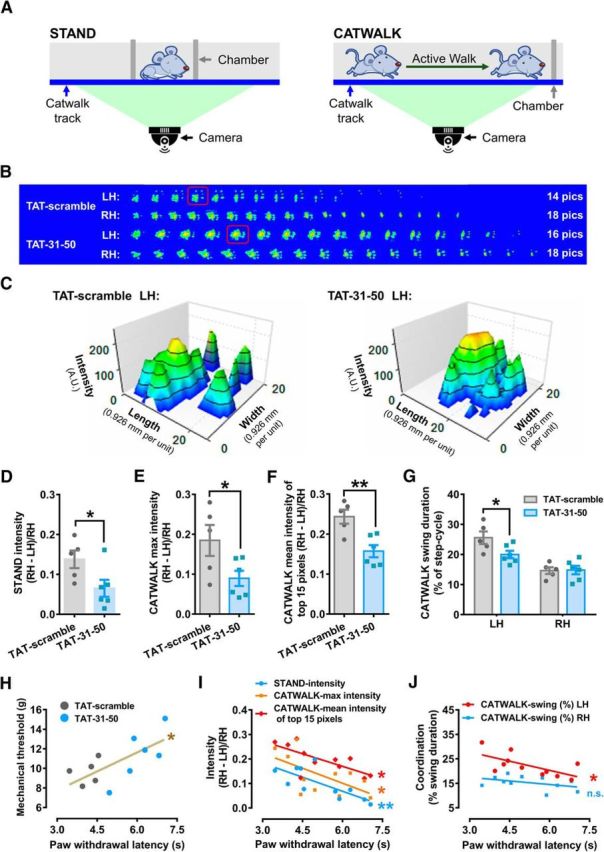
TAT-31-50 administration attenuates the gait alterations induced by CFA injection. A, Schematic diagram of the procedure evaluating gait alterations during STAND (left) or CATWALK (right). B, Consecutive graphs within a duty cycle of the LH and RH 6 h after CFA injection in the LH. C, 3D graphs of the LH print boxed by the red line in B. x-axis: length (0.926 mm per unit), y-axis: width (0.926 mm per unit), z-axis: intensity (A.U.). D–F, STAND intensity (D), CATWALK max intensity (E), and CATWALK mean intensity of the top 15 pixels (F) in TAT-31-50- or TAT-scramble-injected rats at 6 h after CFA injection. Unpaired t test, n = 5 or 6, *p < 0.05, **p < 0.01. G, CATWALK swing duration (% of step-cycle) of LH or RH at 6 h after CFA injection in the LH. Two-way ANOVA, interaction effect: p < 0.05; post test: Sidak's multiple-comparisons test, *p < 0.05. H–J, Correlation analysis between PWL and mechanical threshold (H), intensity data (I), and coordination data (J). Linear regression, n = 11, *p < 0.05, **p < 0.01. For more information, see Figure 6-1.
This extended data figure supports for Figure 6. TAT-31-50 administration did not affect the contact area or hind paw print area after CFA injection. A, Three-dimensional graphs of the right hind paw. x-axis: length (0.926 mm per unit), y-axis: width (0.926 mm per unit), z-axis: intensity (A.U.). B–D, STAND contact area (B), CATWALK max contact area (C), and CATWALK paw print area (D) in rats 6 h after CFA injection. Unpaired t- test, n = 5 or 6. E–F, Ipsilateral paw withdrawal latencies (E) and mechanical thresholds (F) in TAT-31-50- or TAT-scramble-injected rats before and 6 h after CFA injection. Two-way ANOVA, post-test: Sidak's multiple comparisons test, *p < 0.05, **p < 0.01. G, Correlation analysis between PWL and print area data. Download Figure 6-1, TIF file (3.3MB, tif)
For static gait, the STAND intensity ratio was 13.8% in the TAT-scramble group, whereas administration of the TAT-31-50 peptide lowered this value to 6.5% (n = 5 or 6, unpaired Student's t test: t(9) = 2.359, p = 0.0427; Fig. 6D), suggesting that the difference in STAND intensity between the two limbs was reduced by TAT-31-50. For dynamic gait, gait-related parameters were collected during walking. The representative paw print graphs showed that the rats initially contacted the floor with their palm, then transferred to their toes, and ended with their heel (Fig. 6B). Footprints with the largest contact area were highlighted in red frames and further displayed in 3D graphs (Fig. 6C). The yellow or red regions in the 3D graphs, representing regions with high intensity within one step cycle, were enlarged in the TAT-31-50-injected group compared with those in the TAT-scramble control group. However, no significant differences in the contralateral hindpaws were detected between the two groups (Fig. 6-1A).
In addition to the STAND intensity, the maximal and mean intensities of the most intense 15 pixels during walking were compared between the TAT-scramble- and TAT-31-50-injected groups. The results showed that both parameters were reduced by the administration of TAT-31-50 (max intensity: n = 5 or 6, unpaired Student's t test: t(9) = 2.359, p = 0.0454; mean intensity of top 15 pixels: n = 5 or 6, unpaired Student's t test: t(9) = 3.753, p = 0.0045; Fig. 6E,F), suggesting the alleviation of inflammation-induced gait alterations by TAT-31-50.
In humans, individuals suffering from unilateral knee pain tend to avoid bearing weight on their injured leg. Therefore, the percentage of swing duration, or the time of noncontact with the floor during one step cycle, was calculated. As expected, intraplantar inflammation increased the swing duration of the ipsilateral hindpaw to ∼25%, whereas administration of the TAT-31-50 peptide reduced this time to ∼20% (n = 5 or 6, two-way repeated-measures ANOVA: interaction effect F(1,9) = 7.108, p = 0.0476; Sidak's multiple-comparisons test: p(LH) = 0.0318, p(RH) = 0.9952; Fig. 6G). In addition, the STAND contact area (Fig. 6-1B), the CATWALK maximal contact area (Fig. 6-1C), and the paw print area (calculated according to the accumulation of all the footprints; Fig. 6-1D) were compared between the TAT-scramble- and TAT-31-50-injected groups and no significant differences were detected, suggesting that the paw area and its contact with the floor were not affected by the analgesic peptide.
In the following studies, correlation analysis of the nociceptive behaviors and parameters of CatWalk was performed. The developments of both thermal hyperalgesia (n = 5 or 6, two-way repeated-measures ANOVA: interaction effect F(1,9) = 6.093, p = 0.0200; Sidak's multiple-comparisons test: p(6) = 0.0077; Fig. 6-1E) and mechanical allodynia behaviors (n = 5 or 6, two-way repeated-measures ANOVA: interaction effect F(1,9) = 6.093, p = 0.0196; Sidak's multiple-comparisons test: p(6) = 0.0448; Fig. 6-1F) were attenuated by administration of the TAT-31-50 peptide. A significant positive correlation between the thermal and mechanical thresholds was detected (n = 11, linear regression: R2 = 0.4881, F(1,9) = 8.582, p = 0.0168; Fig. 6H). In contrast, a significant negative correlation between intensity (including STAND intensity, CATWALK max intensity, and CATWALK mean intensity of the top 15 pixels) and the thermal pain threshold was detected (n = 11, linear regression, STAND intensity: R2 = 0.5595, F(1,9) = 11.43, p = 0.0081; CATWALK max intensity: R2 = 0.3629, F(1,9) = 5.127, p = 0.0498; CATWALK mean intensity of the top 15 pixels: R2 = 0.5076, F(1,9) = 9.279, p = 0.0139; Fig. 6I). In addition, a significant negative correlation between the coordination (percent swing duration of the ipsilateral hindpaw) and the thermal pain threshold was detected (n = 11, linear regression, CATWALK swing (%) LH: R2 = 0.4199, F(1,9) = 6.515, p = 0.0311; CATWALK swing (%) RH: R2 = 0.1928, F(1,9) = 2.149, p = 0.1767; Fig. 6J). However, no correlation between the print area data (including the STAND contact area, the CATWALK max contact area, and the CATWALK print area) and the thermal pain threshold was detected (Fig. 6-1G). The above data indicate that TAT-31-50 contributed to the attenuation of gait alterations elicited by plantar inflammation and that this improvement could be correlated (∼40%, estimated by the R2 in the statistical analysis of linear regression in Fig. 5I,J) to the alleviation of thermal hyperalgesia.
KChIP3–31-50 fragment inhibits the TRPV1-mediated Ca2+ influx
As shown in the above data (Fig. 4F), application of TAT-31-50 induced a significant reduction of surface TRPV1, mimicking the function of the KChIP3 31-50 fragment. To further clarify the effect of TAT-31-50 on the function of TRPV1, Ca2+ imaging assays were performed in freshly dissociated ipsilateral DRG neurons 6 h after CFA injection.
Compared with TAT-scramble, 10 μg TAT-31-50 slightly reduced the Ca2+ influx evoked by capsaicin (5 μm), the most specific known TRPV1 agonist (n = 12, two-way repeated-measures ANOVA: group effect F(1,22) = 5.649, p = 0.0266; Fig. 7A,B). Furthermore, tripling the dose of TAT-31-50 induced a much greater decrease (∼60%) of TRPV1-mediated Ca2+ influx (n = 5 or 7, two-way repeated-measures ANOVA: group effect F(1,10) = 20.01, p = 0.0012; Fig. 7C,D). Likewise, diminishment of capsaicin-induced Ca2+ influx by TAT-31-50 was also observed compared with that by the TAT-1-20 peptide, which was unable to bind TRPV1 (Fig. 7-1). Therefore, the KChIP3-31-50 fragment is sufficient to inhibit TRPV1-mediated Ca2+ influx.
Figure 7.
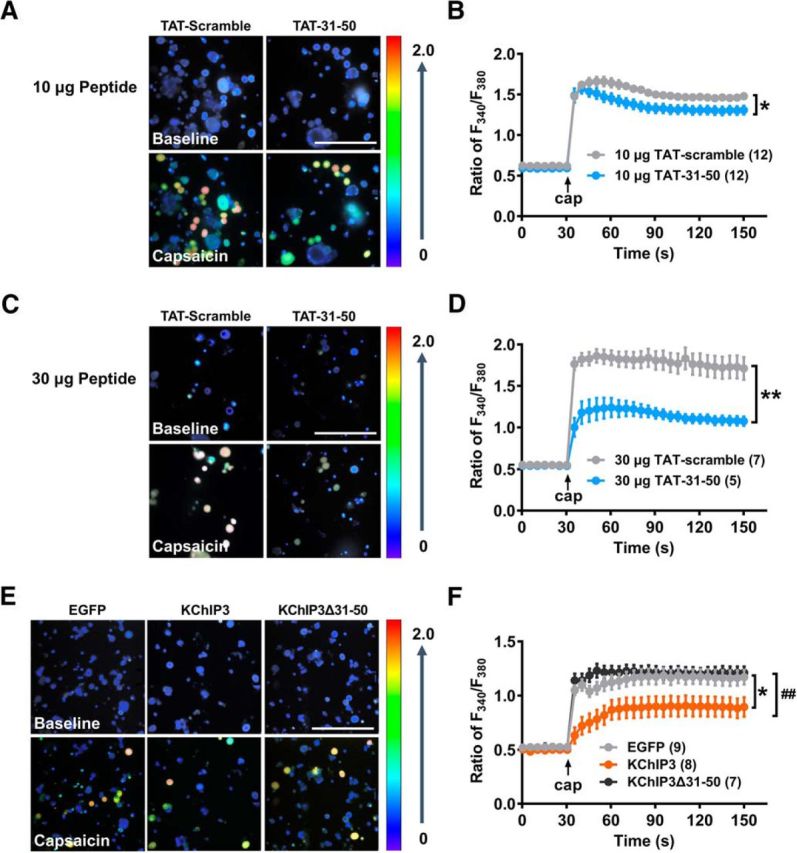
The KChIP3 N-terminal 31-50 fragment is necessary and sufficient for the inhibition of TRPV1-mediated Ca2+ influx by KChIP3. A–F, Ca2+ imaging assays in dissociated ipsilateral L4–5 DRG neurons 6 h after CFA injection. The rats were pretreated with an intrathecal administration of 10 (A, B) or 30 μg of the TAT-scramble or TAT-31-50 peptides (C, D) or the EGFP, EGFP-KChIP3 or EGFP-KChIP3Δ31–50 plasmids (E, F). A, C, E, Images of dissociated DRG neurons at the baseline are shown above and images after capsaicin treatment are shown below. Scale bar, 200 μm. B, D, F, Quantification analysis of the capsaicin (5 μm)-evoked Ca2+ influx in A, C, and E, respectively. B, D, Group effect: *p < 0.05, **p < 0.01. F, two-way ANOVA, post test: Sidak's multiple-comparisons test, *p < 0.05, compared with the EGFP group, ##p < 0.01, compared with the EGFP-KChIP3 group. For more information, see Figure 7-1.
This extended data figure supports for Figure 7. TAT-31-50 decreases TRPV1-mediated Ca2+ influx compared to TAT-1-20. A, B, Ca2+ imaging assay in dissociated ipsilateral L4-5 DRG neurons of rats 6 h after CFA injection. The rats were pre-treated with an intrathecal administration of 10 μg of TAT-1-20 or TAT-31-50. A, Images of dissociated DRG neurons at the baseline are shown above, and images after capsaicin treatment are shown below. Scale bar = 200 μm. B, Quantification analysis of the capsaicin (5 mM)-evoked Ca2+ influx in A. Two-way ANOVA, group effect: **p < 0.01. Download Figure 7-1, TIF file (2.1MB, tif)
Next, we investigated whether the 31-50 fragment was required for the inhibitory effect of KChIP3 on TRPV1-mediated Ca2+ influx. Consistent with the results shown in Figure 5F, exogenous EGFP-KChIP3 expression via intrathecal injection, but not EGFP-KChIP3Δ31–50 expression, decreased the capsaicin-evoked Ca2+ influx (n = 7, 8, or 9, two-way repeated-measures ANOVA: group effect F(2,21) = 6.580, p = 0.006; Tukey's multiple-comparisons test of group effects: EGFP vs EGFP-KChIP3, p = 0.0213; EGFP vs EGFP-KChIP3Δ31–50, p = 0.8264; EGFP-KChIP3 vs EGFP-KChIP3Δ31–50, p = 0.0088; Fig. 7E,F). In other words, deletion of the 31–50 fragment abolished the inhibition of Ca2+ influx through TRPV1 by full-length KChIP3. The above data indicate that the 31–50 fragment is sufficient and necessary for the inhibitory effect of KChIP3 on TRPV1-mediated Ca2+ influx.
TAT-31-50 alleviated the CFA-induced thermal hyperalgesia in kcnip3 KO rats
To further investigate the analgesic mechanisms of TAT-31-50, we constructed kcnip3 KO rats (Fig. 8A). Efficient deletion of the kcnip3 gene was verified by immunoblotting analysis of DRG lysates. At the same time, expression of the TRPV1 protein was not affected by kcnip3 deletion (Fig. 8B). Consistent with previous studies (Cheng et al., 2002; Rivera-Arconada et al., 2010), kcnip3 deletion led to an increased thermal pain threshold in the basal state (n = 5 or 7, unpaired Student's t test: t(10) = 2.456, p = 0.0339; Fig. 8C, left). However, kcnip3 KO rats showed reduced PWL values in the radiant heat test at 6 and 12 h after intraplanter CFA injection (n = 5 or 7, two-way repeated-measures ANOVA: group effect F(1,10) = 15.06, p = 0.0031; Sidak's multiple-comparisons test: p(6) = 0.0049, p(12) = 0.0295; Fig. 8C, right). Quantification analysis of the AUC (1–24 h) also showed the increased sensitivity of the kcnip3 KO rats to thermal pain in the inflammatory pain state. The opposite roles of KChIP3 in the basal and inflammatory states might be attributed to its distinct roles in the nucleus and plasma membrane. In the nucleus, Ca2+-bound KChIP3 was shown to dimerize to block its DNA-binding ability (Carrión et al., 1999; Lusin et al., 2008) and depress the expression of genes such as bdnf, thus strengthening the role of BDNF and facilitating spinal sensitization in inflammation-induced thermal hyperalgesia. Although localized on the plasma membrane, binding between KChIP3 and TRPV1 receptors may result in decreased surface level of TRPV1, which may lead to reduced thermal hypersensitivity.
To exclude the possibility that TAT-31-50 induces analgesia by inhibiting the binding of KChIP3 and TRPV1 competitively, the analgesic effect of the TAT-31-50 peptide was tested in the kcnip3 KO rats. The results showed that TAT-31-50 retained its ability to alleviate thermal hyperalgesia relative to the scramble control in the kcnip3 KO rats (n = 5 or 6, PWL: two-way repeated-measures ANOVA: group effect F(1,9) = 9.382, p = 0.0135; Sidak's multiple-comparisons test: p(3) = 0.0176; p(6) = 0.0265; p(12) = 0.0192; AUC: unpaired Student's t test: t(9) = 3.063, p = 0.0135; Fig. 8D). These results indicate that the kcnip3 deletion had distinct effects on the thermal pain thresholds in the basal and inflammatory states and that the analgesic effect of TAT-31-50 was independent of endogenous KChIP3.
Discussion
In the present study, we revealed the peripheral analgesic effect of KChIP3 in CFA-induced inflammatory pain and identified TRPV1 as a new KChIP3-binding protein. In the basal state, binding between KChIP3 and TRPV1 remains at a low level (Fig. 8E, left). During inflammatory pain, TRPV1 undergoes functional sensitization, which leads to increased Ca2+ influx via TRPV1. Concurrently, the protein expression of KChIP3 is upregulated. Therefore, the binding between KChIP3 and TRPV1 is enhanced, which results in the reduced surface localization of TRPV1 and the subsequent diminishment of Ca2+ influx through the channel (Fig. 8E, middle). Importantly, the transmembrane TAT-31-50 peptide, which was generated based on the critical binding site of KChIP3 mediating its association with TRPV1, has an effect on the surface localization of TRPV1 similar to that of KChIP3. In the behavioral tests, intrathecal injection of TAT-31-50 alleviates inflammation-induced thermal hyperalgesia and gait alterations (Fig. 8E, right). Therefore, TAT-31-50 might be a potential analgesic agent for the treatment of inflammatory pain.
Peripheral KChIP3 contributes to pain modulation
Previous studies have shown that KChIP3 is a critical transcriptional repressor in pain modulation. Kcnip3 KO mice exhibit elevated levels of prodynorphin mRNA in their spinal cords and reduced sensitivity to noxious stimuli in both physiological and acute inflammatory pain states (Cheng et al., 2002). In later studies, transgenic mice expressing DREAM with the EF hands mutant (EFmDREAM or daDREAM), which shows sustained transcriptional repression activity in a Ca2+-independent manner, were generated (Rivera-Arconada et al., 2010). These mice exhibit basal hyperalgesia but impaired response to inflammatory pain. Results in these mice are consistent with our present results in kcnip3 KO rats, which show hypoalgesia in the basal state and exacerbated thermal hyperalgesia after CFA injection (Fig. 8C,D). However, a recent study found the opposite results in the trigeminal neurons of mice overexpressing EFmDREAM. These mice showed enhanced responses to acute inflammatory pain stimuli. Further cDNA microarray analysis revealed two new target genes of KChIP3 related to pain modulation in trigeminal ganglion neurons, including cathepsin L and the monoglyceride lipase, which are involved in the proteolytic processing of dynorphin in secretory vesicles and the hydrolysis of endocannabinoid 2-arachidnonoyl glycerol, respectively (Benedet et al., 2017). In our opinion, the multiple KChIP3 target genes in peripheral sensory neurons and the CNS might contribute to its complex regulatory roles in physiological and pathological pain states. In addition, because the binding of KChIP3 with Kv4.2 or NMDA receptors on the plasma membrane has significant effects on neuronal excitability (Anderson et al., 2010; Zhang et al., 2010), their association might further complicate the profound role of KChIP3 in pain modulation.
Although the expression of KChIP3 in DRG neurons has been reported previously (Matsuyoshi et al., 2012; Cheng et al., 2016), herein, we describe the expression pattern of KChIP3 in different subpopulations of rat DRG neurons in detail (Fig. 1). In DRG, unlike the other Kv4 auxiliary subunits (KChIP1, KChIP2, and dipeptidyl peptidase like protein 10), KChIP3 exhibited no colocalization with Kv4.3, the major contributor of the subthreshold A type K+ current in primary sensory neurons (Kuo et al., 2017), suggesting the unique role of KChIP3 in the periphery.
Furthermore, our studies showed the molecular weight of KChIP3 in DRG neurons to be ∼27 kDa, which is slightly smaller than that reported previously (Carrión et al., 1999; An et al., 2000). Previous studies indicated that the hKChIP3-Ib isoform, which contains 230 amino acids and carries an N-terminal 1-60 amino acid replacement compared with KChIP3-Ia (Pruunsild and Timmusk, 2012), has a similar molecular weight to the peripheral KChIP3 identified in our studies. Therefore, to determine whether KChIP3-Ib is the major isoform of KChIP3 expressed in DRG neurons, we used a commercial antibody generated against the N-terminal 31-45 fragment of KChIP3 during Western blot analysis. A clear band corresponding to ∼27 kDa was detected in the DRG tissues of WT but not kcnip3−/− rats (Fig. 8B). Therefore, it is inferred that a different KChIP3 isoform containing the 31-45 fragment is present in primary sensory neurons and further studies are needed to delineate the characteristics of the KChIP3 isoform in DRG neurons.
The KChIP3–TRPV1 interaction reduces the surface localization of TRPV1
In the present study, the KChIP3–TRPV1 interaction was proven by co-IP studies in both tissue and cell cultures and with GST pull-down studies. Furthermore, the Ca2+ sensitivity of this association, which possibly contributed to the increased binding between KChIP3 and TRPV1 6 h after CFA injection, was revealed in the in vitro study. As previous studies indicated, the surface localization and gating properties of TRPV1 were modulated by the interaction of its intracellular N or C termini with scaffolding proteins, signaling molecules, and protein kinases such as AKAP79/150, CaM, phosphatidylinositol 4,5-bisphosphate (PIP2), and protein kinase D (Numazaki et al., 2003; Wang et al., 2004; Lishko et al., 2007; Zhang et al., 2008; Zhu et al., 2008; Fischer et al., 2013; Julius, 2013; Chung et al., 2015). Therefore, we speculate that the KChIP3–TRPV1 binding might interfere with the interaction between TRPV1 and its intracellular binding partners, ultimately leading to the reduction of surface TRPV1. Because the critical binding sites of the N and C termini of TRPV1 with KChIP3 were not investigated in the present study, the mechanisms underlying the inhibition of TRPV1 surface localization and receptor function by KChIP3 need to be explored in future studies.
KChIP3 N-terminal 31-50 fragment exhibits an analgesic effect on inflammatory pain via negatively regulating the function of TRPV1
Mapping studies indicated that the KChIP3 N-terminal 31-50 fragment had the strongest ability of binding to both the N and C termini of TRPV1 (Fig. 4A–D). Functional studies further indicated that deletion of the 31–50 fragment abolished, not only the inhibitory effect of KChIP3 on TRPV1-mediated Ca2+ influx (Fig. 7E,F), but also its analgesic effect on inflammatory thermal hyperalgesia (Fig. 5E).
Because the TAT fusion peptide construction has been proven an efficient method with which to facilitate the cellular entrance of peptide (Xing et al., 2012; Li et al., 2014; Liu et al., 2015), the TAT-31-50 peptide was generated and applied in the behavioral tests. Previous studies have shown that TRPV1 is essential for the sensation of noxious heat and contributes to mechanical static pressure (von Frey; Martínez-Rojas et al., 2014; Borbiro et al., 2015; Watanabe et al., 2015; Xu et al., 2016), but is not involved in the light touch perception and fast nociceptive reflex (Brenneis et al., 2013). In the present work, behavioral tests of the other somatosensory modalities such as light touch and pinprick perception indicated that the TAT-31-50 did not affect the TRPV1-independent somatosensation (Fig. 5-2); however, application of TAT-31-50 via intrathecal injection alleviated thermal hyperalgesia and gait alterations induced by CFA injection significantly. The mechanism of the analgesic effect of TAT-31-50 is related to the decreased surface localization of TRPV1 and the subsequent reduction of TRPV1-mediated Ca2+ influx. In addition, intraplantar injection of TAT-31-50 decreased the capsaicin-evoked spontaneous and evoked pain behavior, which further strengthen the specificity of the action of TAT-31-50 on TRPV1 (Fig. 5G–I).
TAT-31-50 shows a similar analgesic effect as full-length KChIP3
Generally, TAT fusion peptides compete with endogenous full-length proteins in binding the target proteins and thereby downregulate the function of full-length proteins through competitive inhibition. However, in our results, TAT-31-50 reduced capsaicin-evoked Ca2+ influx and thermal hyperalgesia, exhibiting an effect similar to that of KChIP3. We speculated that the binding of TAT-31-50 to the TRPV1 intracellular termini might block the function of critical domains mediating the interaction of TRPV1 with its binding partners and therefore interfere with the membrane trafficking of TRPV1.
To clarify whether TAT-31-50 exerts its function via inhibiting the function of endogenous KChIP3 competitively, we tested its analgesic effect in kcnip3 KO rats, in which TAT-31-50 showed a similar inhibitory effect on inflammatory thermal hyperalgesia. Therefore, TAT-31-50 mimics the inhibitory effect on TRPV1 and the analgesic effect of full-length KChIP3. Elucidation of the critical role of the N-terminal 31-50 fragment in the functional regulation of TRPV1 by KChIP3 provides further evidence for the importance of the unique KChIP3 N terminus compared with other KChIP subtypes.
In conclusion, our findings reveal a new role of KChIP3 in peripheral nociception that is independent of its transcriptional repressor activity and provide new insights into the multifunctional properties of KChIP3. Inhibitory effects of KChIP3 and the transmembrane peptide TAT-31-50 on the surface localization of TRPV1 and the Ca2+ influx through this channel were demonstrated. Because the TAT-31-50 peptide alleviated inflammatory thermal hyperalgesia and gait alterations significantly, it represents a potential analgesic agent for the treatment of inflammatory pain. A better understanding of the role of KChIP3 in pain modulation may contribute to the development of new pain-relieving drugs.
Footnotes
This work was supported by the National Natural Science Foundation of China (Grants 31371143, 31771295, 31530028, 31720103908, and 81521063), the National Key Technology Support Program (Grant 2015BAI08B01), and the Ministry of Science and Technology of China (973 Program Grant 2014CB542204).
The authors declare no competing financial interests.
References
- An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ (2000) Modulation of A-type potassium channels by a family of calcium sensors. Nature 403:553–556. 10.1038/35000592 [DOI] [PubMed] [Google Scholar]
- Anderson D, Mehaffey WH, Iftinca M, Rehak R, Engbers JD, Hameed S, Zamponi GW, Turner RW (2010) Regulation of neuronal activity by Cav3-Kv4 channel signaling complexes. Nat Neurosci 13:333–337. 10.1038/nn.2493 [DOI] [PubMed] [Google Scholar]
- Benedet T, Gonzalez P, Oliveros JC, Dopazo JM, Ghimire K, Palczewska M, Mellström B, Naranjo JR (2017) Transcriptional repressor DREAM regulates trigeminal noxious perception. J Neurochem 141:544–552. 10.1111/jnc.13584 [DOI] [PubMed] [Google Scholar]
- Borbiro I, Badheka D, Rohacs T (2015) Activation of TRPV1 channels inhibits mechanosensitive Piezo channel activity by depleting membrane phosphoinositides. Sci Signal 8:ra15. 10.1126/scisignal.2005667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneis C, Kistner K, Puopolo M, Segal D, Roberson D, Sisignano M, Labocha S, Ferreirós N, Strominger A, Cobos EJ, Ghasemlou N, Geisslinger G, Reeh PW, Bean BP, Woolf CJ (2013) Phenotyping the function of TRPV1-expressing sensory neurons by targeted axonal silencing. J Neurosci 33:315–326. 10.1523/JNEUROSCI.2804-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne RD. (2007) Neuronal calcium sensor proteins: generating diversity in neuronal Ca(2+) signalling. Nat Rev Neurosci 8:182–193. 10.1038/nrn2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne RD, Haynes LP (2015) Sense and specificity in neuronal calcium signalling. Biochim Biophys Acta 1853:1921–1932. 10.1016/j.bbamcr.2014.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Choi EK, Luo Y, Lilliehook C, Crowley AC, Merriam DE, Wasco W (1998) Calsenilin: a calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat Med 4:1177–1181. 10.1038/2673 [DOI] [PubMed] [Google Scholar]
- Carrión AM, Link WA, Ledo F, Mellström B, Naranjo JR (1999) DREAM is a Ca2+-regulated transcriptional repressor. Nature 398:80–84. 10.1038/18044 [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389:816–824. 10.1038/39807 [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53:55–63. 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]
- Chen YJ, Cheng FC, Sheu ML, Su HL, Chen CJ, Sheehan J, Pan HC (2014) Detection of subtle neurological alterations by the catwalk XT gait analysis system. J Neuroeng Rehabil 11:62. 10.1186/1743-0003-11-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CF, Wang WC, Huang CY, Du PH, Yang JH, Tsaur ML (2016) Coexpression of auxiliary subunits KChIP and DPPL in potassium channel Kv4-positive nociceptors and pain-modulating spinal interneurons. J Comp Neurol 524:846–873. 10.1002/cne.23876 [DOI] [PubMed] [Google Scholar]
- Cheng HY, Pitcher GM, Laviolette SR, Whishaw IQ, Tong KI, Kockeritz LK, Wada T, Joza NA, Crackower M, Goncalves J, Sarosi I, Woodgett JR, Oliveira-dos-Santos AJ, Ikura M, van der Kooy D, Salter MW, Penninger JM (2002) DREAM is a critical transcriptional repressor for pain modulation. Cell 108:31–43. 10.1016/S0092-8674(01)00629-8 [DOI] [PubMed] [Google Scholar]
- Chung MK, Lee J, Joseph J, Saloman J, Ro JY (2015) Peripheral group I metabotropic glutamate receptor activation leads to muscle mechanical hyperalgesia through TRPV1 phosphorylation in the rat. J Pain 16:67–76. 10.1016/j.jpain.2014.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan B, Cheng L, Bourane S, Britz O, Padilla C, Garcia-Campmany L, Krashes M, Knowlton W, Velasquez T, Ren X, Ross S, Lowell BB, Wang Y, Goulding M, Ma Q (2014) Identification of spinal circuits transmitting and gating mechanical pain. Cell 159:1417–1432. 10.1016/j.cell.2014.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer MJ, Btesh J, McNaughton PA (2013) Disrupting sensitization of transient receptor potential vanilloid subtype 1 inhibits inflammatory hyperalgesia. J Neurosci 33:7407–7414. 10.1523/JNEUROSCI.3721-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Villafuertes R, Torres B, Barrio J, Savignac M, Gabellini N, Rizzato F, Pintado B, Gutierrez-Adan A, Mellström B, Carafoli E, Naranjo JR (2005) Downstream regulatory element antagonist modulator regulates Ca2+ homeostasis and viability in cerebellar neurons. J Neurosci 25:10822–10830. 10.1523/JNEUROSCI.3912-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32:77–88. 10.1016/0304-3959(88)90026-7 [DOI] [PubMed] [Google Scholar]
- Hu HJ, Carrasquillo Y, Karim F, Jung WE, Nerbonne JM, Schwarz TL, Gereau RW 4th (2006) The kv4.2 potassium channel subunit is required for pain plasticity. Neuron 50:89–100. 10.1016/j.neuron.2006.03.010 [DOI] [PubMed] [Google Scholar]
- Jerng HH, Kunjilwar K, Pfaffinger PJ (2005) Multiprotein assembly of Kv4.2, KChIP3 and DPP10 produces ternary channel complexes with ISA-like properties. J Physiol 568:767–788. 10.1113/jphysiol.2005.087858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julius D. (2013) TRP channels and pain. Annu Rev Cell Dev Biol 29:355–384. 10.1146/annurev-cellbio-101011-155833 [DOI] [PubMed] [Google Scholar]
- Jung J, Shin JS, Lee SY, Hwang SW, Koo J, Cho H, Oh U (2004) Phosphorylation of vanilloid receptor 1 by Ca2+/calmodulin-dependent kinase II regulates its vanilloid binding. J Biol Chem 279:7048–7054. 10.1074/jbc.M311448200 [DOI] [PubMed] [Google Scholar]
- Kuo YL, Cheng JK, Hou WH, Chang YC, Du PH, Jian JJ, Rau RH, Yang JH, Lien CC, Tsaur ML (2017) K+ channel modulatory subunits KChIP and DPP participate in Kv4-mediated mechanical pain control. J Neurosci 37:4391–4404. 10.1523/JNEUROSCI.1619-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hu F, Chen HJ, Du YJ, Xie ZY, Zhang Y, Wang J, Wang Y (2014) LIMK-dependent actin polymerization in primary sensory neurons promotes the development of inflammatory heat hyperalgesia in rats. Sci Signal 7:ra61. 10.1126/scisignal.2005353 [DOI] [PubMed] [Google Scholar]
- Link WA, Ledo F, Torres B, Palczewska M, Madsen TM, Savignac M, Albar JP, Mellström B, Naranjo JR (2004) Day-night changes in downstream regulatory element antagonist modulator/potassium channel interacting protein activity contribute to circadian gene expression in pineal gland. J Neurosci 24:5346–5355. 10.1523/JNEUROSCI.1460-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lishko PV, Procko E, Jin X, Phelps CB, Gaudet R (2007) The ankyrin repeats of TRPV1 bind multiple ligands and modulate channel sensitivity. Neuron 54:905–918. 10.1016/j.neuron.2007.05.027 [DOI] [PubMed] [Google Scholar]
- Liu J, Du J, Yang Y, Wang Y (2015) Phosphorylation of TRPV1 by cyclin-dependent kinase 5 promotes TRPV1 surface localization, leading to inflammatory thermal hyperalgesia. Exp Neurol 273:253–262. 10.1016/j.expneurol.2015.09.005 [DOI] [PubMed] [Google Scholar]
- Lusin JD, Vanarotti M, Li C, Valiveti A, Ames JB (2008) NMR structure of DREAM: implications for Ca(2+)-dependent DNA binding and protein dimerization. Biochemistry 47:2252–2264. 10.1021/bi7017267 [DOI] [PubMed] [Google Scholar]
- Martínez-Rojas VA, Barragán-Iglesias P, Rocha-González HI, Murbartián J, Granados-Soto V (2014) Role of TRPV1 and ASIC3 in formalin-induced secondary allodynia and hyperalgesia. Pharmacol Rep 66:964–971. 10.1016/j.pharep.2014.06.011 [DOI] [PubMed] [Google Scholar]
- Matsuyoshi H, Takimoto K, Yunoki T, Erickson VL, Tyagi P, Hirao Y, Wanaka A, Yoshimura N (2012) Distinct cellular distributions of Kv4 pore-forming and auxiliary subunits in rat dorsal root ganglion neurons. Life Sci 91:258–263. 10.1016/j.lfs.2012.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Nau C (2005) Regulation of Ca2+-dependent desensitization in the vanilloid receptor TRPV1 by calcineurin and cAMP-dependent protein kinase. J Biol Chem 280:13424–13432. 10.1074/jbc.M410917200 [DOI] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Takeuchi K, Murayama N, Toyooka H, Tominaga M (2003) Structural determinant of TRPV1 desensitization interacts with calmodulin. Proc Natl Acad Sci U S A 100:8002–8006. 10.1073/pnas.1337252100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewska M, Casafont I, Ghimire K, Rojas AM, Valencia A, Lafarga M, Mellström B, Naranjo JR (2011) Sumoylation regulates nuclear localization of repressor DREAM. Biochim Biophys Acta 1813:1050–1058. 10.1016/j.bbamcr.2010.11.001 [DOI] [PubMed] [Google Scholar]
- Pruunsild P, Timmusk T (2005) Structure, alternative splicing, and expression of the human and mouse KCNIP gene family. Genomics 86:581–593. 10.1016/j.ygeno.2005.07.001 [DOI] [PubMed] [Google Scholar]
- Pruunsild P, Timmusk T (2012) Subcellular localization and transcription regulatory potency of KCNIP/Calsenilin/DREAM/KChIP proteins in cultured primary cortical neurons do not provide support for their role in CRE-dependent gene expression. J Neurochem 123:29–43. 10.1111/j.1471-4159.2012.07796.x [DOI] [PubMed] [Google Scholar]
- Rivera-Arconada I, Benedet T, Roza C, Torres B, Barrio J, Krzyzanowska A, Avendaño C, Mellström B, Lopez-Garcia JA, Naranjo JR (2010) DREAM regulates BDNF-dependent spinal sensitization. Mol Pain 6:95. 10.1186/1744-8069-6-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum T, Gordon-Shaag A, Munari M, Gordon SE (2004) Ca2+/calmodulin modulates TRPV1 activation by capsaicin. J Gen Physiol 123:53–62. 10.1085/jgp.200308906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Gomez A, Mellström B, Tornero D, Morato E, Savignac M, Holguín H, Aurrekoetxea K, González P, González-García C, Ceña V, Mayor F Jr, Naranjo JR (2007) G protein-coupled receptor kinase 2-mediated phosphorylation of downstream regulatory element antagonist modulator regulates membrane trafficking of Kv4.2 potassium channel. J Biol Chem 282:1205–1215. 10.1074/jbc.M607166200 [DOI] [PubMed] [Google Scholar]
- Savignac M, Pintado B, Gutierrez-Adan A, Palczewska M, Mellström B, Naranjo JR (2005) Transcriptional repressor DREAM regulates T-lymphocyte proliferation and cytokine gene expression. EMBO J 24:3555–3564. 10.1038/sj.emboj.7600810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto K, Yang EK, Conforti L (2002) Palmitoylation of KChIP splicing variants is required for efficient cell surface expression of Kv4.3 channels. J Biol Chem 277:26904–26911. 10.1074/jbc.M203651200 [DOI] [PubMed] [Google Scholar]
- Tiruppathi C, Soni D, Wang DM, Xue J, Singh V, Thippegowda PB, Cheppudira BP, Mishra RK, Debroy A, Qian Z, Bachmaier K, Zhao YY, Christman JW, Vogel SM, Ma A, Malik AB (2014) The transcription factor DREAM represses the deubiquitinase A20 and mediates inflammation. Nat Immunol 15:239–247. 10.1038/ni.2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrinten DH, Hamers FF (2003) ‘CatWalk’ automated quantitative gait analysis as a novel method to assess mechanical allodynia in the rat; a comparison with von frey testing. Pain 102:203–209. 10.1016/s0304-3959(02)00382-2 [DOI] [PubMed] [Google Scholar]
- Wang Y, Kedei N, Wang M, Wang QJ, Huppler AR, Toth A, Tran R, Blumberg PM (2004) Interaction between protein kinase cmu and the vanilloid receptor type 1. J Biol Chem 279:53674–53682. 10.1074/jbc.M410331200 [DOI] [PubMed] [Google Scholar]
- Watanabe M, Ueda T, Shibata Y, Kumamoto N, Ugawa S (2015) The role of TRPV1 channels in carrageenan-induced mechanical hyperalgesia in mice. Neuroreport 26:173–178. 10.1097/WNR.0000000000000322 [DOI] [PubMed] [Google Scholar]
- Wu LJ, Mellström B, Wang H, Ren M, Domingo S, Kim SS, Li XY, Chen T, Naranjo JR, Zhuo M (2010) DREAM (downstream regulatory element antagonist modulator) contributes to synaptic depression and contextual fear memory. Mol Brain 3:3. 10.1186/1756-6606-3-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing BM, Yang YR, Du JX, Chen HJ, Qi C, Huang ZH, Zhang Y, Wang Y (2012) Cyclin-dependent kinase 5 controls TRPV1 membrane trafficking and the heat sensitivity of nociceptors through KIF13B. J Neurosci 32:14709–14721. 10.1523/JNEUROSCI.1634-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XX, Cao Y, Ding TT, Fu KY, Li Y, Xie QF (2016) Role of TRPV1 and ASIC3 channels in experimental occlusal interference-induced hyperalgesia in rat masseter muscle. Eur J Pain 20:552–563. 10.1002/ejp.758 [DOI] [PubMed] [Google Scholar]
- Zhang X, Li L, McNaughton PA (2008) Proinflammatory mediators modulate the heat-activated ion channel TRPV1 via the scaffolding protein AKAP79/150. Neuron 59:450–461. 10.1016/j.neuron.2008.05.015 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Su P, Liang P, Liu T, Liu X, Liu XY, Zhang B, Han T, Zhu YB, Yin DM, Li J, Zhou Z, Wang KW, Wang Y (2010) The DREAM protein negatively regulates the NMDA receptor through interaction with the NR1 subunit. J Neurosci 30:7575–7586. 10.1523/JNEUROSCI.1312-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Yang Y, Zhang H, Han Y, Li Y, Zhang Y, Yin D, He Q, Zhao Z, Blumberg PM, Han J, Wang Y (2008) Interaction between protein kinase D1 and transient receptor potential V1 in primary sensory neurons is involved in heat hypersensitivity. Pain 137:574–588. 10.1016/j.pain.2007.10.025 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This extended data figure supports for Figure 1. Representative images of the immunostaining of KChIP3 (green) and Hoechst 33342 (blue) in DRG neurons of WT (left image) and kcnip3 KO rats (right image), respectively. A commercial antibody (Abcam) against the N-terminal 30-45 fragment of KChIP3 was used. Scale bar = 200 μm. Download Figure 1-1, TIF file (4.9MB, tif)
This extended data figure supports for Figure 1. KChIP3 mRNA detection in DRG tissue by RT-PCR. A, Detection of KChIP3 mRNA in L4-5 DRG extracts by PCR using the primer pairs 1 (351 bp fragment), 2 and 3 (full-length), respectively. A no primer group was used as the negative control. B, Detection of KChIP3 mRNA in DRG neurons and spinal cord extracts, using the full-length primer pairs, respectively. A no cDNA group was used as the negative control. Download Figure 1-2, TIF file (1MB, tif)
This extended data figure supports for Figure 2. Expression of EGFP-KChIP3 plasmids in HEK293 cell line and DRG tissues. A, Representative images of the expression of EGFP-KChIP3 in HEK293 cells. B, Representative images of the expression of EGFP-KChIP3 in DRG lysates. C, Representative images of immunostaining of EGFP (green), KChIP3 (red), Hoechst 33342 (blue) in DRG slices after intrathecal delivery of EGFP-KChIP3 or EGFP plasmids and the DRG slices of naïve rats. Enlarged images of the boxed area in the upper rows are shown in the lower rows. Yellow arrows indicate the DRG neurons labelled by green and red immunofluorescence concurrently. Red and green arrows indicate the neurons labelled by red or green immunofluorescence respectively. Scale bar = 100 μm. Download Figure 2-1, TIF file (38.3MB, tif)
This extended data figure supports for Figure 5. Intrathecal injection of TAT-31-50 alleviates CFA-induced thermal hyperalgesia compared to TAT-1-20. A–B, Time course of the PWL of the ipsilateral hind paw before and after CFA injection from 1 to 24 h in TAT-1-20- or TAT-31-50-injected rats. The rats were pre-treated with an intrathecal administration of 10 or 30 μg of the TAT-1-20 or TAT-31-50 (n = 5) peptides (B). The left panel of A–B shows two-way ANOVA, group effect: *p < 0.05, shown at the ends of the lines; post-test: Sidak's multiple comparisons test, **p < 0.01, *p < 0.05, shown above the lines. In the right panel of A–B, area under the curve values of the paw withdrawal latencies from 1 to 24 h after CFA injection were compared between the TAT-31-50- and TAT-scramble-injected groups. Unpaired t-test, *p < 0.05. Download Figure 5-1, TIF file (733.2KB, tif)
This extended data figure supports for Figure 5. Intrathecal injection of TAT-31-50 exerts no influence on the mechanosensation of A-fibers. A–B, Time course of PWL (A) or mechanical threshold (B) of the ipsilateral hindpaw before and after CFA injection from 6 to 24 h in the 30 μg TAT-scramble- or TAT-31-50-injected (i.t.) rats. Two-way ANOVA, group effect: ***p < 0.001, shown in the end of the lines; post-test: Sidak's multiple comparisons test, ***p < 0.001, **p < 0.01, *p < 0.05, shown above the lines. C, Correlation analysis between PWL and mechanical threshold in the 30 μg TAT-scramble- or TAT-31-50-injected (i.t.) rats. Linear regression, n = 10 or 14. D, E, F, Light touch sensitivity (D), latency to detect the sticky tape (E) and pinprick reactivity (F) of the ipsilateral hindpaws are compared between the 30 μg TAT-scramble- or TAT-31-50-injected (i.t.) rats 6 h post CFA injection. Unpaired t test. Download Figure 5-2, TIF file (1.6MB, tif)
This extended data figure supports for Figure 6. TAT-31-50 administration did not affect the contact area or hind paw print area after CFA injection. A, Three-dimensional graphs of the right hind paw. x-axis: length (0.926 mm per unit), y-axis: width (0.926 mm per unit), z-axis: intensity (A.U.). B–D, STAND contact area (B), CATWALK max contact area (C), and CATWALK paw print area (D) in rats 6 h after CFA injection. Unpaired t- test, n = 5 or 6. E–F, Ipsilateral paw withdrawal latencies (E) and mechanical thresholds (F) in TAT-31-50- or TAT-scramble-injected rats before and 6 h after CFA injection. Two-way ANOVA, post-test: Sidak's multiple comparisons test, *p < 0.05, **p < 0.01. G, Correlation analysis between PWL and print area data. Download Figure 6-1, TIF file (3.3MB, tif)
This extended data figure supports for Figure 7. TAT-31-50 decreases TRPV1-mediated Ca2+ influx compared to TAT-1-20. A, B, Ca2+ imaging assay in dissociated ipsilateral L4-5 DRG neurons of rats 6 h after CFA injection. The rats were pre-treated with an intrathecal administration of 10 μg of TAT-1-20 or TAT-31-50. A, Images of dissociated DRG neurons at the baseline are shown above, and images after capsaicin treatment are shown below. Scale bar = 200 μm. B, Quantification analysis of the capsaicin (5 mM)-evoked Ca2+ influx in A. Two-way ANOVA, group effect: **p < 0.01. Download Figure 7-1, TIF file (2.1MB, tif)