

Abstract
Consolidation of newly formed fear memories requires a series of molecular events within the basolateral complex of the amygdala (BLA). Once consolidated, new information can be assimilated into these established associative networks to form higher-order associations. Much is known about the molecular events involved in consolidating newly acquired fear memories but little is known about the events that consolidate a secondary fear memory. Here, we show that, within the male rat BLA, DNA methylation and gene transcription are crucial for consolidating both the primary and secondary fear memories. We also show that consolidation of the primary, but not the secondary, fear memory requires de novo protein synthesis in the BLA. These findings show that consolidation of a fear memory and its updating to incorporate new information recruit distinct processes in the BLA, and suggest that DNA methylation in the BLA is fundamental to consolidation of both types of conditioned fear.
SIGNIFICANCE STATEMENT Our data provide clear evidence that a different set of mechanisms mediate consolidation of learning about cues that signal learned sources of danger (i.e., second-order conditioned fear) compared with those involved in consolidation of learning about cues that signal innate sources of danger (i.e., first-order conditioned fear). These findings carry important implications because second-order learning could underlie aberrant fear-related behaviors (e.g., in anxiety disorders) as a consequence of neutral secondary cues being integrated into associative fear networks established through first-order pairings, and thereby becoming potent conditioned reinforcers and predictors of fear. Therefore, our data suggest that targeting such second-order conditioned triggers of fear may require pharmacological intervention different to that typically used for first-order conditioned cues.
Keywords: amygdala, epigenetic regulation, intracellular signalling pathways, memory consolidation, protein synthesis, second-order fear conditioning
Introduction
Fear is an adaptation to selection pressures imposed by environmental sources of danger. These sources can be classified in either of two ways. Innate sources of danger originate in the experiences of our ancestors and have been hard-wired in our brains. They share a capacity to induce pain/discomfort and are common to the individuals of a species, e.g., pain, spiders, snakes, heights, and open spaces. In contrast, learned sources of danger originate in the experiences of an individual. As such, they can be common to the individuals of a social group or culture, e.g., guns, or unique to an individual, e.g., the song on the radio at the time of a car crash.
Cues that signal innate or learned sources of danger evoke a common set of reactions: attention is redirected from the pursuit of current goals toward cues that identify the danger, its location in space and time; ongoing social and other activities are suppressed; and programs are activated to prepare for fight or flight. Such commonalities suggest that the cue-danger (or fear) memory that forms in each case may be encoded and stored in the same way. Consistent with this view, laboratory studies have shown that both types of fear memory require neuronal activity in the basolateral complex of the amygdala (BLA), including activation of NMDA receptors (Campeau et al., 1992; Gewirtz and Davis, 1997; Muller et al., 1997; Fendt and Fanselow, 1999; Wilensky et al., 1999; Maren and Quirk, 2004; Zhang et al., 2008; Parkes and Westbrook, 2010; Holmes et al., 2013). However, there are also reasons to suppose they may be encoded differently. Specifically, cues that signal learned sources of danger retain their fear-eliciting properties even after the learned source of danger has been eliminated (Rizley and Rescorla, 1972; Cheatle and Rudy, 1978; Rescorla, 1982; Barnet et al., 1991; Holmes et al., 2014), thereby raising the possibility that the two types of cue-danger fear memory are consolidated via different signaling cascades and nuclear processes in the BLA.
The present study addressed this gap in our knowledge. It specifically examined whether, within the BLA, the signaling cascades and nuclear processes required for consolidating an association between a cue and an innate source of danger are also required for consolidating an association between a cue and a learned source of danger. In each experiment, rats were first subjected to what Pavlov (1927) termed first-order conditioning. This consisted in pairings of a relatively innocuous conditioned stimulus, S1, and brief but aversive foot shock [the innate or unconditioned source of danger (US)]. Forty-eight hours later, they received what Pavlov (1927) termed second-order conditioning. This consisted in pairings of a second innocuous stimulus, S2, with the already-conditioned S1 (the learned source of danger; for a further demonstration of second-order conditioning using these parameters; Holmes et al., 2014). Immediately after these S2-S1 pairings, rats received a BLA infusion of vehicle or a drug known to disrupt molecular processes involved in consolidation of the first-order conditioned fear memory. Particularly, the major intracellular signaling cascades [Ca2+/calmodulin (CaM)- and cAMP-dependent protein kinase pathways (CaMKII/IV, PKA, PKC) and extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK)], as well as the nuclear changes that are known to be triggered by their activation (epigenetic regulation of gene expression, gene transcription, and synthesis of protein products).
Materials and Methods
Subjects
Subjects were 309 experimentally naive male outbred Sprague-Dawley rats (weighing between 240 and 540 g) obtained from a commercial supplier (Animal Resources Centre). They were housed in groups of eight in an opaque plastic box (22 cm height × 67 cm length × 40 cm width). The boxes were kept in an air-conditioned colony room maintained on a 12 h light/dark cycle (lights on at 7:00 A.M.). Food and water were continuously available in the home cage during all phases of the experiment. All experimental procedures occurred between 8:00 A.M. and 6:00 P.M. All procedures were approved by the Animal Care and Ethics Committee of the University of New South Wales and in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals (revised 2011).
Surgery and drug infusions
Before behavioral training and testing, rats were implanted with guide cannulae directed toward the BLA. Rats were injected intraperitoneally with 1.3 ml/kg of the anesthetic ketamine (Ketapex, Apex Laboratories), at a concentration of 100 mg/ml and 0.3 ml/kg xylazine, a muscle relaxant (Rompun, Bayer), at a concentration of 20 mg/ml. Anesthetized rats were then mounted on a stereotaxic apparatus (David Kopf Instruments), and 26-gauge guide cannulae (Plastics One) were implanted through holes drilled in both hemispheres of the skull. The tips of the guide cannulae were aimed bilaterally at the BLA using the following coordinates: 2.6 mm posterior to bregma, 4.9 mm lateral to the midline, and 7.7–7.9 mm ventral to the skull; or the central nucleus of the amygdala (CeA) using the following coordinates: 2.3 mm posterior to bregma, 4.2 mm lateral to the midline, and 7.5 mm ventral to the skull. The guide cannulae were secured to the skull with four jeweller's screws and dental cement. A dummy cannula was kept in each guide at all times except during microinjections. Immediately after the surgical procedure, rats received an intraperitoneal injection of a prophylactic (0.4 ml) dose of 300 mg/kg solution of procaine penicillin. Rats were allowed 7 d to recover from surgery, during which time they were handled and weighed daily.
Bupivacaine, U0126, H7, KN-62, cycloheximide, actinomycin-d, RG108, 5-AZA, or vehicle was infused bilaterally in the BLA by inserting a 33-gauge internal cannula into the guide cannula. The internal cannula was connected to a 25 μl glass syringe attached to an infusion pump (Harvard Apparatus) and projected an additional 1 mm ventral to the tip of the guide cannula. The internal cannula remained in place for an additional 2 min after the infusion and was then removed.
Drugs
A sterile solution of 0.5% (w/v) bupivacaine hydrochloride (Delta West) was used to produce a reversible inactivation of the BLA at a concentration of 2.5 μg/0.5 μl. Nonpyrogenic saline (0.9% w/v) was used as a vehicle solution. A total volume of 0.5 μl was delivered to both sides at a rate of 0.25 μl/min.
The following set of drugs were used target specific intracellular signaling pathways in the BLA. For instance, a specific inhibitor of MAP kinase kinase (MEK), U0126 (Promega), was dissolved in 100% dimethyl sulfoxide (DMSO; Sigma-Aldrich) to a concentration of 4 μg/μl as described by Schafe et al. (2000). The stock was then diluted 1:1 in artificial CSF (ACSF; Tocris Bioscience) to obtain a final concentration of 2 μg/μl. Vehicle (50% DMSO-ACSF) was prepared by diluting 100% DMSO 1:1 in ACSF. A total volume of 0.5 μl was delivered to both sides at a rate of 0.2 μl/min. A broad-spectrum protein kinase inhibitor, H7-dihydrochloride (Tocris Bioscience), was prepared in the manner described by Goosens et al. (2000). H7 was dissolved in 100 mm PBS, pH 7.4, to a final concentration of 8 μg/μl. Vehicle (100 mm PBS, pH 7.4) was prepared by dissolving 0.276% sodium dihydrogen orthophosphate, 1.094% d-sodium hydrogen orthophosphate, and 0.85% sodium chloride in 1 L of distilled water (dH2O) and adjusted to pH 7.4. A total volume of 0.2 μl was delivered to both sides at a rate of 0.1 μl/min. A CaMKII inhibitor, KN-62 (Sigma-Aldrich), was dissolved in 45% w/v 2-hydropropyl-β-cyclodextrin (45% w/v HBC; Sigma-Aldrich) to a concentration of 680 ng/μl as described by Rodrigues et al. (2004). A 45% w/v solution of HBC was used as the vehicle. A total volume of 0.5 μl was delivered to both sides at a rate of 0.25 μl/min.
Finally, the remaining set of inhibitors were used to assess the role of nuclear processes in consolidation of second-order fear within the BLA. In particular, a protein synthesis inhibitor, cycloheximide (Sigma-Aldrich), was prepared in the manner described by Duvarci et al. (2005). Cycloheximide was dissolved in 70% ethanol (EtOH) to a stock solution of 200 μg/μl concentration. The stock was then diluted 1:4 in ACSF to result in a final concentration of 40 μg/μl. Vehicle (EtOH-ACSF) was prepared by diluting 70% EtOH 1:4 in ACSF. A total volume of 0.5 μl was delivered to both sides at a rate of 0.25 μl/min. A transcription inhibitor, actinomycin D (Sigma-Aldrich), was prepared in the manner described by Bailey et al. (1999). Actinomycin D was diluted in ACSF in a serial manner to result in a final concentration of 5 ng/μl. Vehicle was ACSF. A total volume of 0.5 μl was delivered to both sides at a rate of 0.25 μl/min. The DNA methyltransferase inhibitors, RG108 and 5-AZA-2′-deoxycytidine (Sigma-Aldrich) were prepared in the manner described by Maddox et al. (2014). RG108 and 5-AZA were dissolved in 100% DMSO to a stock solution of 2 μg/μl concentration. The stock was then diluted 1:1 in ACSF to result in a final concentration of 1 μg/μl. Vehicle (50% DMSO-ACSF) was prepared by diluting 100% DMSO 1:1 in ACSF. A total volume of 0.5 μl was delivered to both sides at a rate of 0.125 μl/min.
Where information for the half-life of these drugs was available, they ranged from 20 min to 4 h (Tattersall et al., 1975; Young and Dowling, 1975; Momparler, 2005; Ikeda et al., 2010; Bae et al., 2013; Schneeberger et al., 2016). Moreover, the total volumes used (0.2 and 0.5 μl) in the present experiments were chosen to accurately target specific nuclei in the brain. Studies have shown that infusions of these volume stay within the boundaries of the region of interest, with a radial diffusion of ∼520 μm for both 0.2 and 0.5 μl microinjections (Goosens et al., 2000; Lohamn et al., 2005; Holmes et al., 2013).
Histology
Subsequent to behavioral testing, rats received a lethal dose of sodium pentobarbital (1 ml). The brains were removed and sectioned coronally at 40 μm through the BLA or CeA. Every second section was collected on a slide and stained with cresyl violet. The location of the cannulation tips was determined under a microscope using the boundaries defined by the atlas of Paxinos and Watson (2007; Figs. 1, 2, 8b, 9b). Rats with incorrect placements or damage were excluded from statistical analysis.
Figure 1.

Cannula placements as verified on Nissl-stained sections for (a) bupivacaine, (b) U0126, (c) H7, and (d) KN-62 in the BLA. The symbols represent the most ventral point of the cannula track for each rat on coronal sections based on the atlas of Paxinos and Watson (2007).
Figure 2.

Cannula placements as verified on Nissl-stained sections for (a) cycloheximide, (b) actinomycin-d, (c) RG108, and (d) 5-AZA in the BLA. The symbols represent the most ventral point of the cannula track for each rat on coronal sections based on the atlas of Paxinos and Watson (2007).
Figure 8.

Presence of shock is not sufficient to recruit molecular processes in consolidation of second-order conditioned fear. a, Experimental timeline of second-order conditioning and retraining with first-order conditioning. The procedure was similar to that previously described except that rats received the first three of the four S1-shock pairings on 1 d followed by the fourth S1-shock pairing before second-order conditioning on the next day. This single S1-shock pairing before the S2-S1 pairings thus incorporated the presence of shock across the second-order session but did so without changing the number of S2-S1 pairings. Immediately after this session, rats received an infusion of the same dose and volume of U0126 or vehicle in the BLA. b, Cannula placements as verified on Nissl-stained section. The symbols represent the most ventral point of the cannula track for each rat on coronal sections based on the atlas of Paxinos and Watson (2007). c, Percentage freezing to S1 across first-order conditioning (left) and S2 and S1 across second-order conditioning (right) for U0126- (n = 14) and vehicle- (n = 13) treated rats. Percentage freezing to (d) S2 at test, (e) S2 across first-order acquisition, and (f) S2 at test for U0126- and vehicle-treated rats. Drug-treated rats are shown with filled squares and vehicle-treated rats are shown with open squares. Data are represented as mean ± SEM.
Figure 9.

Protein synthesis in the CeA is not required for the consolidation of second-order conditioned fear. a, Experimental timeline of second-order conditioning and retraining with first-order conditioning. The procedure was similar to that previously described except that rats received an infusion of the same dose and volume of cycloheximide or vehicle in the CeA immediately after second-order conditioning. b, Cannula placements as verified on Nissl-stained section for cycloheximide in the CeA. The symbols represent the most ventral point of the cannula track for each rat on coronal sections based on the atlas of Paxinos and Watson (2007). c, Percentage freezing to S1 across first-order conditioning (left) and S2 and S1 across second-order conditioning (right) for cycloheximide- (n = 13) and vehicle- (n = 13) treated rats. Percentage freezing to (d) S2 at test, (e) S2 across first-order acquisition, and (f) S2 at test for cycloheximide- and vehicle-treated rats. Drug-treated rats are shown with filled squares and vehicle-treated rats are shown with open squares. Data are represented as mean ± SEM.
Behavioral apparatus
Behavioral procedures were conducted in four identical chambers, each measuring 30 cm height × 27 cm length × 30 cm width. Their side walls and ceilings were made of aluminum, and the back and front walls were made of clear plastic. Their floors consisted of stainless steel rods, 2 mm in diameter, spaced 13 mm apart, center to center. A tray below the floor contained bedding material (corncob). Each chamber was enclosed in a sound- and light-attenuating shell. A white fluorescent tube and speaker mounted on the back wall of each shell were used, respectively, for the presentation of a visual CS (∼57 lux measured at the center of the chamber) flashing at a rate of 3.5 Hz, and an auditory CS (620 Hz square-wave tone) measuring ∼70 dB (A scale) against a background noise of ∼45 dB measured by a digital sound level meter (Dick Smith Electronics). The physical identity of the CSs was fully counterbalanced in all experiments. The levels of freezing to the CSs did not differ as a function of their physical identity in any phase of the experiments. A constant-current shock generator, capable of delivering unscrambled AC 50 Hz to the floor of each chamber, was used for the presentation of a 0.5 s duration foot-shock US at 0.8 mA intensity. The floor of each chamber was cleaned with water after removal of each rat. Illumination of each chamber was provided by an infrared light source (940 ± 25 nm). A camera mounted on the back wall of each shell recorded the behavior of each rat. Each camera was connected to a monitor and a DVD recorder located in another room of the laboratory. This room contained the computer that controlled stimulus presentations via appropriate software (MATLAB, MathWorks).
Behavioral procedure
Second-order fear conditioning was performed as previously described (Parkes and Westbrook, 2010; Fig. 3a). Briefly, on days 1 and 2, rats received two 20 min exposures per day to the conditioning chambers, one in the morning and then 2 h later in the afternoon. No stimuli were presented in this phase.
Figure 3.

Consolidation of second-order fear requires BLA activity. a, Experimental timeline of second-order conditioning and retraining with first-order conditioning. Percentage freezing to (b) S1 across first-order conditioning for bupivacaine- (n = 10) and vehicle- (n = 12) treated rats, (c) S2 and S1 across second-order conditioning, and (d) to S2 across test. Drug-treated rats are shown with filled squares and circle and vehicle-treated rats are shown with open squares. Data are represented as mean ± SEM.
First-order conditioning.
On day 3, rats received four pairings of the S1 (10 s) and the US (0.5 s, 0.8 mA foot-shock). Rats were placed into the conditioning context, and after a 5 min adaptation period, S1 was presented. Presentation of S1 coterminated with the US. The intertrial interval (ITI) between paired S1-US presentations was 5 min. Rats remained in the chamber for 1 min following the final stimulus presentation.
Context extinction.
On day 4, rats received two context extinction sessions, one in the morning and the other, 3 h later, in the afternoon. In each session, rats were placed in the conditioning chamber for 30 min and were then returned to their home cage. No stimuli were presented in this phase. The context was extinguished to allow a clearer assessment of the acquisition of freezing to S2 across its pairings with S1.
Second-order conditioning.
On day 5, rats received four pairings of S2 and S1 such that the offset of S2 immediately preceded the onset of S1. The duration of each presentation of S2 was 30 s and that of S1 was 10 s. Rats were placed in the conditioning context, and, after a 2 min adaptation period, S2 was presented and terminated in the presentation of S1. The ITI between the S2-S1 pairings was 5 min. Rats remained in the conditioning chamber for an additional 1 min after the final stimulus presentation. Immediately after second-order conditioning, rats received a bilateral infusion of drug or vehicle into the BLA. Six hours later, rats infused with drug were infused with vehicle while those infused with vehicle now received an infusion of drug.
Rats received control infusions of the drug or vehicle 6–7 h after second-order conditioning to equate all rats for drug exposure so that any differences in test performance, where they are observed, cannot be attributed to differential experience of the drug per se. Where no such difference is observed, it is possible that the delayed (control) drug infusion had some effect on consolidation of second-order fear, thereby reducing the levels of test performance in the control group. However, there are five reasons why we are confident that this was not the case. First, the level of performance in the control group was remarkably consistent across experiments in which a drug effect was or was not detected on consolidation of second-order fear. If the absence of a difference between groups that received immediate (treatment) or delayed (control) drug infusions was because the delayed drug infusion affected consolidation of second-order fear, it follows that the test levels of responding to S2 in the control groups would have been lower in experiments where no group-difference was observed. Second, where we failed to detect an effect of a drug on consolidation of second-order fear, we consistently observed an effect of the same drug on consolidation of first-order conditioned fear, using the same counterbalanced infusion-strategy. Third, the level of performance in the control group is equivalent to that observed in past work from our laboratory using identical parameters and equipment. That is, the level of second-order fear in our behavioral control experiments (Parkes and Westbrook, 2010; Holmes et al., 2013, 2014), where rats had not undergone surgery, is identical to that observed in the control groups in the present study. Fourth, in the case of cycloheximide, which failed to disrupt consolidation of second-order fear when infused into the BLA, we have replicated this finding in several experiments where no counterbalanced drug infusions were given (D. M. Leidl, B. P. P. Lay, C. Chakouch, R. F. Westbrook, & N. M. Holmes, unpublished observations). Last, studies have shown that drugs that affect consolidation of first-order fear immediately after a conditioning session fail to disrupt consolidation when administered from 3 to 6 h after that session (Nader et al., 2000; Schafe and LeDoux, 2000; Schafe et al., 2000; Rodrigues et al., 2004).
Context extinction.
On day 6, rats received a brief context extinction session. Rats were placed in the conditioning chamber for 10 min and were then returned to their home cage. No stimuli were presented in this session.
Tests.
One hour following context extinction on day 6, all rats were tested with S2. On day 7, rats were tested with S1. On each test, rats were placed into the conditioning chamber, and after a 2 min adaptation period, the stimulus was presented. Each test session consisted of eight stimulus alone presentations with an ITI of 2 min. Rats remained in the conditioning chamber for an additional 1 min after the final stimulus presentation.
First-order conditioning to S2 and test.
In the instance where we failed to detect an effect of the drug on consolidation of second-order conditioned fear, S2 was retrained as a first-order stimulus. In such instances, on day 8, rats received four, 30 s presentations of S2, each coterminating with onset of foot-shock (0.5 s, 0.8 mA). Immediately after S2-shock pairings, rats that had previously received an infusion of drug immediately after second-order conditioning now received an immediate infusion of vehicle, and vice versa (i.e., group DRUG becomes group VEH, and group VEH becomes group DRUG). On day 9, rats received a brief 10 min context extinction session followed 1 h later by S2 test. All rats were tested for fear to S2 in the manner described previously.
Data analysis
Freezing was used to assess conditioned fear. It was defined as the absence of all movement except those related to breathing (Fanselow, 1980). Each rat was observed every 2 s and scored as either “freezing” or “not freezing” by two observers, one of whom was blind to group assignment. The correlation between the scores was high (≥0.9) and any discrepancies were resolved in favor of the score by the naive observer. A percentage score was calculated for the proportion of the total observations each rat spent freezing during the first 2 min of the session (baseline) as well as the total duration of each CS presentation. The levels of freezing to each stimulus are reported as the percentage freezing to the CS during each CS alone presentation in the session, and does not include freezing during the ITI. Data were analyzed in SPSS 22.0 (IBM) using repeated-measures ANOVA. In each case, the criterion for rejection of the null hypothesis (α) was set at 0.05. Effects of trial, where reported, were measured with contrasts testing for the presence of a linear trend. Where appropriate, the Greenhouse–Geisser sphericity correction was used when Mauchley's test of sphericity was violated. Confidence intervals (CIs; 95% for the mean difference) and measures of effect size (ηp2 for ANOVA and Cohen's d for contrasts; Cohen, 1988) are reported for each significant comparison. For nonsignificant comparisons, an inverse Bayes factor, BF01, was calculated using JASP 0.7.5.6. (Rouder et al., 2009). The BF01 is the ratio of the likelihood of the null hypothesis (H0; no impairment in consolidation) relative to the alternative (H1; an impairment in consolidation). Hence, the larger the BF01 value, the more support there is in favor of the null hypothesis (i.e., H0).
Results
Consolidation of second-order conditioned fear requires the BLA
Previous studies have demonstrated that silencing neural activity in the BLA or blocking NMDA receptors interferes with the formation of a second-order fear memory (Gewirtz and Davis, 1997; Parkes and Westbrook, 2010; Holmes et al., 2013). To determine whether activity in the BLA is also required to consolidate a second-order fear memory, we functionally inactivated the BLA using a sodium channel blocker, bupivacaine, immediately after pairings of S2 and the already conditioned S1. Subsequent to extinction of any freezing elicited by the context, rats were tested for their levels of fear (freezing) to S2. One day later, rats were also tested with S1 to assess whether silencing the BLA had disrupted reconsolidation of the first-order fear memory.
Levels of freezing before the first stimulus presentation (i.e., the baseline period) were <10% in each training and test session, and did not differ significantly between groups (max F(1,20) = 1.82, p = 0.19; Fig. 3). First- and second-order conditioning were successful (Fig. 3b,c). There was no significant between-group difference (F(1,20) = 2, p = 0.17) or linear trend × group interaction across S1-shock pairings (F(1,20) = 0.47, p = 0.5). Similarly, there was no significant between-group difference in the overall level of freezing to S2 (F(1,20) = 0.19, p = 0.67) or to S1 (F(1,20) = 0.001, p = 0.98) or significant linear × group interactions (max F(1,20) = 0.84, p = 0.37) across the S2-S1 pairings. Figure 3d shows the mean (±SEM) levels of freezing to S2 across testing. Baseline levels of freezing to the context (M = 4.7, SEM = 1.3) was significantly lower compared with freezing during context extinction (M = 15.95, SEM = 2.76; t(21) = 3.58, p = 0.002, paired t test). Bupivacaine disrupted the consolidation of second-order conditioned fear. Rats infused with bupivacaine immediately after the S2-S1 pairings froze significantly less when tested with S2 than rats infused with vehicle [F(1,20) = 17.54, p < 0.001, 95% CI (7.85, 23.43), d = 1.85, ANOVA]. A significant linear × group interaction confirmed that the decline in freezing to S2 was greater in the vehicle group than the drug group [F(1,20) = 10.27, p = 0.004, ηp2 = 0.34, 95% CI (−30.18, −15.85), ANOVA]. There was no detectable effect of the drug on the levels of freezing elicited by S1. Rats infused with bupivacaine after the S2-S1 pairings froze just as much when tested with S1 as rats infused with vehicle (F(1,20) = 0.77, p = 0.39) and the linear × group interaction was not significant (F(1,20) = 1.97, p = 0.18). This was true in each of the remaining experiments: i.e., disruptions of cellular and molecular signaling in the BLA after S2-S1 pairings in stage 2 had no effect on the test levels of freezing to S1. Rats infused with drug after the S2-S1 pairings froze just as much when tested with S1 as rats infused with vehicle (U0126: F(1,21) = 0.2, p = 0.66; H7: F(1,24) = 0.001, p = 0.98; KN-62: F(1,25) = 0.29, p = 0.6; DNMT: F(1,49) = 0.057, p = 0.81; actinomycin-d: F(1,21) = 1.66, p = 0.21; cycloheximide: F(1,20) < 0.001, p = 0.99) and the linear × group interaction was not significant (U0126: F(1,21) = 0.21, p = 0.66; H7: F(1,24) = 0.12, p = 0.74; KN-62: F(1,25) = 0.272, p = 0.41; DNMT: F(1,49) = 2.64, p = 0.11; actinomycin-d: F(1,21) = 1.61, p = 0.22; cycloheximide: F(1,20) = 0.37, p = 0.55).
Consolidation of second-order fear requires activation of CaMKII/IV but not PKA/PKC or ERK/MAPK signaling pathways in the BLA
The next series of experiments examined the intracellular signaling cascades that are necessary for consolidation of second-order conditioned fear. Specifically, they examined whether consolidation of the S2-S1 memory was disrupted by targeting the second-messenger molecules known to be critical for consolidation of the S1-shock memory. For instance, ERK/MAPK and PKA/PKC translocate from the cytoplasm to the nucleus and initiate the transcriptional changes that are required for gene expression (Schafe et al., 1999; Goosens et al., 2000; Schafe and LeDoux, 2000; Moita et al., LeDoux, 2002). CaMKIV is primarily localized in the nuclei of neurons and allows for rapid activation of transcription factors, such as CREB, upon calcium entry to the cell (Jensen et al., 1991; Ho et al., 2000; Wu et al., 2001). Moreover, CaMKII rapidly translocates to postsynaptic dendrites and strengthens recently activated synapses through interactions with glutamate receptors (Lisman et al., 2002; Thiagarajan et al., 2002; Rodrigues et al., 2004). Together, we targeted these molecules via BLA infusions of the CaMKII/CaMKIV inhibitor, KN-62, the highly selective MEK inhibitor, U0126, or the PKA/PKC inhibitor, H7, immediately after the S2-S1 pairings.
Levels of freezing before the first stimulus presentation (i.e., the baseline period) were <15% in each training and test session, and did not differ significantly between groups (KN-62: max F(1,25) = 1.31, p = 0.26; U0126: max F(1,21) = 3.67, p = 0.069; H7: max F(1,24) = 2.21, p = 0.15; Figs. 4, 5). First- and second-order conditioning was successful in each of the treatment and control groups (Fig. 4): there was no significant between-group difference in the overall level of freezing to S1 (KN-62: F(1,25) = 0.27, p = 0.61; U0126: F(1,21) = 0.006, p = 0.94; H7: F(1,24) = 0.083, p = 0.78) and no significant linear × group interaction across the S1-shock pairings (KN-62: F(1,25) = 0.66, p = 0.42; U0126: F(1,21) = 0.19, p = 0.67; H7: F(1,24) = 0.12, p = 0.73), indicating that groups acquired freezing to S1 at the same rate. Similarly, there was no significant between-group difference in the overall level of freezing to S2 (KN-62: F(1,25) = 2.73, p = 0.11; U0126: F(1,21) = 0.044, p = 0.84; H7: F(1,24) = 0.002, p = 0.97) or to S1 (KN-62: F(1,25) = 3.27, p = 0.083; U0126: F(1,21) = 0.26, p = 0.62; H7: F(1,24) = 0.41, p = 0.53) or significant linear × group interactions (KN-62: max F(1,25) = 0.52, p = 0.48; U0126: max F(1,21) = 0.28, p = 0.6; H7: max F(1,24) = 1.37, p = 0.25) across the S2-S1 pairings. These results indicate that, while groups acquired freezing to S2 at the same rate, they also maintained similar levels of freezing to S1, and therefore, that the original fear memory was preserved across the S2-S1 pairings. Figure 5, b, c (left), and d (left), shows the mean (±SEM) levels of freezing to S2 across testing in each of these experiments. Baseline levels of freezing to the context (KN-62: M = 8.27, SEM = 2.24; U0126: M = 4.49, SEM = 1.26) was significantly lower compared with freezing during context extinction (KN-62: M = 16.44, SEM = 2.84, t(26) = 3.11, p = 0.0045, paired t test; U0126: M = 12.49, SEM = 1.66, t(22) < 0.001, p = 5.88, paired t test). Freezing during context extinction was already low for H7 and its control group (M = 12.09, SEM = 2.15, t(25) = 0.88, p = 0.39, paired t test). BLA infusions of KN-62, U0126, and H7 had different effects on consolidation of the second-order fear memory. Infusions of KN-62 immediately after S2-S1 pairings disrupted consolidation of second-order conditioned fear. The control group froze significantly more during test presentations of S2 than Group KN-62 [F(1,25) = 6.036, p = 0.021, 95% CI (2.22, 25.28), d = 0.94, ANOVA]. Moreover, there was no significant linear × group interaction (F(1,25) = 2.03, p = 0.17), confirming that the between-group differences in freezing were similar across the S2 test presentations. In contrast, infusions of U1026 or H7 failed to affect consolidation of second-order conditioned fear. Levels of freezing to S2 at test did not differ between groups (U0126: F(1,21) = 0.014, p = 0.91, BF01 = 2.63; H7: F(1,24) = 1.31, p = 0.26, BF01 = 1.7) and there was no significant linear × group interaction across the S2 test presentations (U0126: F(1,21) = 0.014, p = 0.91; H7: F(1,24) = 0.098, p = 0.76) in either experiment.
Figure 4.
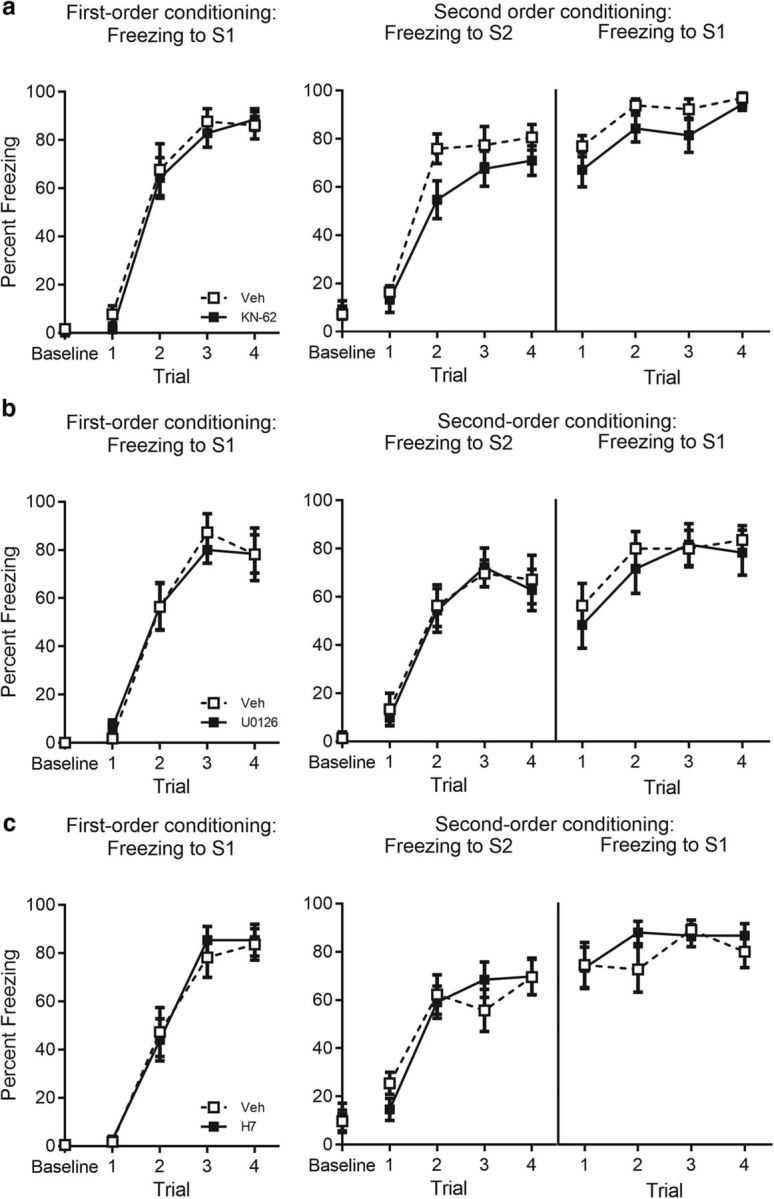
Acquisition data for experiments targeting CaMKII/IV, ERK/MAPK, and PKA/PKC pathways in the BLA. a, Percentage freezing to S1 across first-order conditioning (left) and S2 and S1 across second-order conditioning (right) for KN-62- (n = 14) and vehicle- (n = 13) treated rats. b, Percentage freezing to S1 across first-order conditioning (left) and S2 and S1 across second-order conditioning (right) for U0126- (n = 12) and vehicle- (n = 11) treated rats. c, Percentage freezing to S1 across first-order conditioning (left) and S2 and S1 across second-order conditioning (right) for H7- (n = 15) and vehicle- (n = 11) treated rats. Drug-treated rats are shown with filled squares, and vehicle-treated rats are shown with open squares. Data are represented as mean ± SEM.
Figure 5.

Consolidation of second-order fear requires CaMKII/CaMKIV activation in the BLA but not ERK/MAPK and PKA/PKC activation. a, Experimental timeline of second-order conditioning and retraining with first-order conditioning. b, Percentage freezing to S2 across test for KN-62- and vehicle-treated rats. c, Percentage freezing to S2 across test (left), S2 across first-order acquisition (middle), and S2 across test (right) for U0126- and vehicle-treated rats. d, Percentage freezing to S2 across test (left), S2 across first-order acquisition (middle), and S2 across test (right) for H7- and vehicle-treated rats. Drug-treated rats are shown with filled squares and vehicle-treated rats are shown with open squares. Data are represented as mean ± SEM.
Importantly, the failure of U0126 and H7 to disrupt consolidation of the second-order fear memory was not due to their ineffectiveness when infused into the BLA. We verified the effectiveness of these infusions within each experiment by replicating their established effects on consolidation of a first-order fear memory. Specifically, subsequent to the testing of S2 and S1, rats were reconditioned to fear S2 through its pairings with shock, thereby establishing it as a first-order CS. Immediately after this reconditioning, rats that had previously received vehicle after the S2-S1 pairings now received a BLA infusion of drug (U0126 or H7), whereas rats that had previously received drug after the S2-S1 pairings now received a BLA infusion of vehicle. All rats were then tested with presentations of S2 alone.
Figure 5, c and d, shows the mean (±SEM) levels of freezing to S2 across its pairings with shock (middle) and subsequent testing (right). In each experiment, acquisition to S2 as a first-order CS was successful: there was no significant between-group difference in overall freezing to S2 (U0126: F(1,20) = 0.57, p = 0.46; H7: F(1,23) = 0.15, p = 0.71) or linear × group interaction across the pairings (U0126: F(1,20) = 0.36, p = 0.56; H7: F(1,23) = 0.032, p = 0.86), thereby providing further evidence that the previous drug infusion had failed to affect the consolidation of second-order conditioned fear. Baseline levels of freezing to the context (U0126: M = 8, SEM = 2.58; H7: M = 13.27, SEM = 3.27) was significantly lower compared with freezing during context extinction (U0126: M = 26.68, SEM = 4.43, t(21) < 0.001, p = 4.05, paired t test; H7: M = 32.81, SEM = 5.59, t(24) = 3.89, p < 0.001, paired t test). Critically, infusions of either drug disrupted consolidation of first-order fear, thereby demonstrating their efficacy in the BLA and replicating previous findings (Schafe et al., 1999; Goosens et al., 2000; Schafe and LeDoux, 2000). At test, the control group froze significantly more to S2 than the group treated with drug after S2-shock pairings [U0126: F(1,20) = 5.92, p = 0.024, 95% CI (2.037, 26.49), d = 1.04, ANOVA; H7: F(1,23) = 15.25, p = 0.001, 95% CI (12.80, 41.64), d = 1.57, ANOVA]. There was no significant linear × group interaction, confirming that the size of the difference between the control and treatment groups was maintained across test presentations of S2 (U0126: F(1,20) = 0.49, p = 0.49; H7: F(1,23) = 0.1, p = 0.75).
Together, the results from this set of experiments reveal both commonalities and differences in the signaling pathways through which first- and second-order fear memories are consolidated in the BLA. They permit three major conclusions. First, like consolidation of a first-order fear memory, consolidation of a second-order fear memory requires CaMKII/CaMKIV signaling in the BLA. Second, unlike consolidation of a first-order fear memory, consolidation of a second-order fear memory occurs independently of ERK/MAPK and PKA/PKC activation in the BLA. Third, under the conditions used in these experiments, inhibition of major cell signaling pathways in the BLA has no effect on the maintenance of already-established first-order conditioned fear memories.
Consolidation of second-order fear is conditional on DNA methylation and gene transcription, but not newly synthesized proteins in the BLA
The next set of experiments examined the involvement of nuclear processes in consolidating the second-order fear memory. They again asked whether consolidation of second-order fear requires the nuclear processes known to be required for consolidation of first-order fear, including DNA methylation, transcription of mRNA for various genes, and translation of that mRNA into newly synthesized proteins. Thus, immediately after the S2-S1 pairings, rats received a BLA infusion of a DNMT inhibitor, 5-AZA or RG108, the broad spectrum transcriptional inhibitor, actinomycin-d, or the protein synthesis inhibitor, cycloheximide.
Levels of freezing before the first stimulus presentation (i.e., the baseline period) were <15% in each training and test session, and did not differ significantly between groups (DNMT: max F(1,49) = 2.12, p = 0.15; actinomycin-d: max F(1,21) = 2.71, p = 0.12; cycloheximide: max F(1,20) = 1.30, p = 0.27; Figs. 6, 7). First- and second-order conditioning was successful in each of the experiments (Fig. 6). In each experiment, there was no significant between-group difference in the overall level of freezing to S1 (DNMT: F(1,49) = 0.006, p = 0.94; actinomycin-d: F(1,21) = 0.048, p = 0.83; cycloheximide: F(1,20) = 0.63, p = 0.44) and no significant linear × group interaction across the S1-shock pairings (DNMT: F(1,49) = 0.087, p = 0.77; actinomycin-d: F(1,21) = 0.18, p = 0.68; cycloheximide: F(1,20) = 0.3, p = 0.59), indicating that groups acquired freezing to S1 at the same rate. Similarly, there was no significant between-group difference in the overall level of freezing to S2 (DNMT: F(1,49) = 0.12, p = 0.73; actinomycin-d: F(1,21) = 0.97, p = 0.34; cycloheximide: F(1,20) = 0.19, p = 0.67) or to S1 (DNMT: F(1,49) = 2.35, p = 0.13; actinomycin-d: F(1,21) = 0.28, p = 0.60; cycloheximide: F(1,20) = 0.005, p = 0.94) or significant linear × group interactions (DNMT: max F(1,49) = 0.17, p = 0.69; actinomycin-d: max F(1,21) = 0.76, p = 0.39; cycloheximide: max F(1,20) = 0.033, p = 0.86) across the S2–S1 pairings. Figure 7, b, c, and d (left), shows the mean (±SEM) levels of freezing to S2 across testing in these experiments. Baseline levels of freezing to the context (DNMT: M = 4.87, SEM = 0.86; actinomycin-d: M = 2.25, SEM = 0.59) were significantly lower compared with freezing during context extinction (DNMT: M = 10.81, SEM = 1.84, t(21) = 3.16, p = 0.0027, paired t test; actinomycin-d: M = 13.55, SEM = 3.26, t(22) = 0.0021, p = 3.49, paired t test). Freezing during context extinction was already low for cycloheximide and its control group (M = 17.61, SEM = 2.83, t(21) = 1.99, p = 0.06, paired t test). Inspection of the figures suggests that BLA infusions of RG108, 5-AZA, actinomycin-d, and cycloheximide had different effects on consolidation of the second-order fear memory. The statistical analysis confirmed that infusions of a DNMT inhibitor or actinomycin-d disrupted consolidation: the overall level of freezing to S2 at test was significantly lower in the treatment group relative to its control [DNMT: F(1,49) = 16.387, p < 0.001, 95% CI (7.37, 26.99), d = 1.15, ANOVA; actinomycin-d: F(1,21) = 6.91, p = 0.016, 95% CI (2.79, 23.95), d = 1.11, ANOVA], and the linear × group interaction was not significant in these experiments (DNMT: F(1,49) = 0.14, p = 0.71; actinomycin-d: F(1,21) = 0.97, p = 0.34), showing that the test level of freezing to S2 declined at the same rate in the treatment and control group. In contrast to these effects, the BLA infusion of cycloheximide had no detectable effect on consolidation of the second-order fear memory: test levels of freezing to S2 at test did not differ between groups (F(1,20) = 0.74, p = 0.4, BF01 = 1.99) and there was no significant linear × group interaction across the S2 test presentations (F(1,20) = 0.016, p = 0.90).
Figure 6.
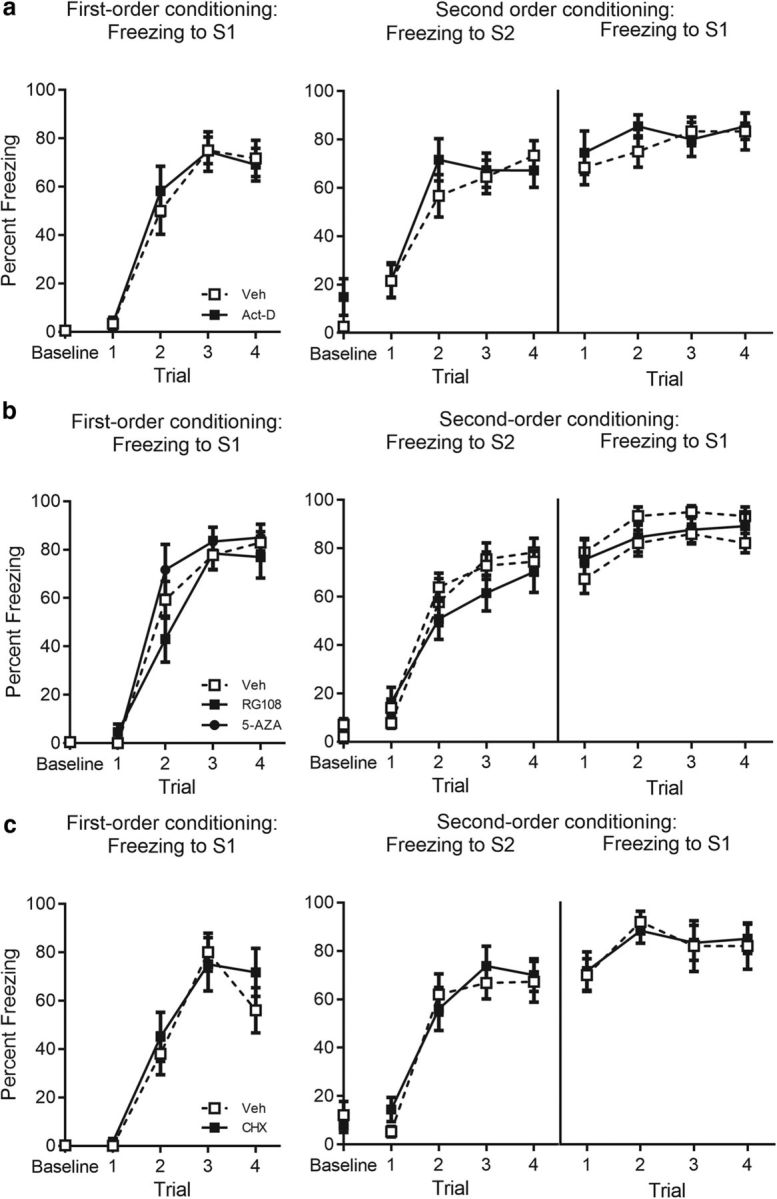
Acquisition data for experiments targeting gene transcription, DNA methylation, and protein synthesis in the BLA. a, Percentage freezing to S1 across first-order conditioning (left) and S2 and S1 across second-order conditioning (right) for actinomycin-d- (n = 11) and vehicle- (n = 12) treated rats. b, Percentage freezing to S1 across first-order conditioning (left) and S2 and S1 across second-order conditioning (right) for RG108- (n = 13), 5-AZA- (n = 12) and vehicle- (n = 27) treated rats. c, Percentage freezing to S1 across first-order conditioning (left) and S2 and S1 across second-order conditioning (right) for cycloheximide- (n = 12) and vehicle- (n = 10) treated rats. Drug-treated rats are shown with filled squares and circle and vehicle-treated rats are shown with open squares. Data are represented as mean ± SEM.
Figure 7.

Consolidation of second-order fear is dependent on DNA methylation and transcription but not de novo protein synthesis in the BLA. a, Experimental timeline of second-order conditioning and retraining with first-order conditioning. b, Percentage freezing to S2 across test for actinomycin-d- and vehicle-treated rats. c, Percentage freezing to S2 across test RG108-, 5-AZA-, and vehicle-treated rats. d, Percentage freezing to S2 across test (left), S2 across first-order acquisition (middle), and S2 across test (right) for cycloheximide- and vehicle-treated rats. Drug-treated rats are shown with filled squares and circle and vehicle-treated rats are shown with open squares. Data are represented as mean ± SEM.
As for the cases of U0126 and H7, the failure of cycloheximide to disrupt consolidation of second-order conditioned fear was not due to the ineffectiveness of the drug when infused into the BLA. We verified the effectiveness of the cycloheximide infusion by replicating its established effects on consolidation of a first-order fear memory. Specifically, S2 was retrained as a first-order CS through its pairings with shock. These S2-shock pairings were followed by a BLA infusion of cycloheximide in the rats infused with vehicle after the S2-S1 pairings and by a BLA infusion of vehicle in the rats that had been infused with the drug after the S2-S1 pairings. Figure 7d (middle and right) shows the mean (±SEM) levels of freezing to S2 across its pairings with shock and subsequent testing, respectively. Acquisition to S2 as a first-order CS was successful: there was no significant between-group difference in freezing to S2 (F(1,19) = 0.46, p = 0.51) or linear × group interaction (F(1,19) = 0.31, p = 0.59), thereby providing further evidence that the previous drug infusion had failed to affect the consolidation of second-order conditioned fear. Baseline levels of freezing to the context (M = 7.46, SEM = 2.80) were significantly lower compared with freezing during context extinction (M = 27.68, SEM = 5.08, t(20) = 3.58, p = 0.0019, paired t test). Critically, infusion of cycloheximide disrupted consolidation of first-order fear: rats infused with vehicle froze significantly more across the test presentation of S2 than rats that received a BLA infusion of cycloheximide after the S2-shock pairings [F(1,19) = 6.02, p = 0.024, 95% CI (3.018, 38.13), d = 1.07, ANOVA]. The linear × group interaction was not significant (F(1,19) = 0.29, p = 0.6), indicating that the between-group differences in freezing were similar across the test presentations.
These results show that, within the BLA, there are both commonalities and differences in the nuclear processes required for consolidation of first- and second-order fear memories. Consolidation of both types of fear memory was disrupted by drugs that inhibit DNA methylation and de novo mRNA synthesis. However, whereas consolidation of the first-order fear memory was disrupted by a drug that inhibits protein synthesis, there was no such disruption by the drug on consolidation of the second-order fear memory. This difference is not due to the absence of shock at the time when rats form the second-order fear memory: even when the second-order conditioning session included an additional pairing of S1 and shock, an immediate post-training infusion of U0126 into the BLA had no effect on consolidation of the second-order fear memory. That is, there was no significant between-group difference in freezing to S2 (F(1,25) = 1.13, p = 0.3, BF01 = 1.84) or linear × group interaction (F(1,25) = 0.38, p = 0.55; Fig. 8b). Moreover, this difference was not due to a shift in the regional requirement for protein synthesis for second-order fear from the BLA to the CeA: infusion of protein synthesis inhibitor cycloheximide into the CeA immediately after second-order conditioning failed to affect consolidation of the second-order fear memory. That is, there was no significant between-group difference in freezing to S2 (F(1,24) = 0.77, p = 0.39, BF01 = 2.08) or linear × group interaction (F(1,24) = 0.13, p = 0.72; Fig. 9b). In each of these experiments, infusion of drug immediately after first-order conditioning (i.e., S2-shock pairings) disrupted consolidation of first-order fear (Figs. 8c, right, 9c, right): rats infused with vehicle froze significantly more across test presentation of S2 than rats that received drug after the S2-shock pairings [U0126: F(1,24) = 4.78, p = 0.039, 95% CI (1.28, 44.65), d = 0.86, ANOVA; CeA: F(1,24) = 8.1, p = 0.009, 95% CI (5.16, 32.41), d = 1.12, ANOVA]. No linear × group interaction was found in the CeA experiment (F(1,24) = 0.069, p = 0.79), however, there was a significant linear × group interaction between U0126- and vehicle-treated rats (F(1,24) = 6.62, p = 0.017, ηp2 = 0.22, 95% CI (−20.20, −3.51), ANOVA], reflecting the greater initial levels of freezing by vehicle-treated rats.
To quantify the level of support for the null hypothesis in these tests of second-order conditioned fear, we reported Bayes Factors which showed that, on average, the observed pattern of results was twice as likely to occur under the null hypothesis. As an additional, and perhaps more sensitive means of assessing this support, we randomly allocated the rats from experiments in which BLA infusions of the drug (U0126, H7, or cycloheximide) failed to disrupt consolidation of second-order fear into four groups: rats that had received drug infusions across the course of the experiment were allocated to two new drug groups, and rats that had received vehicle infusions were allocated to two new vehicle groups. For one of the new drug and new vehicle groups in each experiment, the data committed to the additional statistical analysis was taken from the levels of freezing to S2 when it was tested as a second-order CS; for the remaining two groups, the data were taken from the levels of freezing to S2 when it was tested as a first-order CS (i.e., the test levels of freezing to S2 in the final verification stage). From there, we pooled across groups of rats from the different experiments to yield four final groups: 2ndOrder-Vehicle, 2ndOrder-Drug, 1stOrder-Vehicle, and 1stOrder-Drug, and these data were then analyzed using a two-factor ANOVA (drug × type of conditioning). Across multiple runs of this data construction and analysis, each differing in the random allocation of rats to first-order and second-order groups, there was a significant drug × type of conditioning interaction [min F(1,89) = 7.49, p = 0.007, ηp2 = 0.081, 95% CI (−45.56, −5.27)], supporting our claim that consolidation of first- and second-order fear memories differ at the level of the processes targeted by the three drugs.
Discussion
Recapitulation of the results
The present findings show for the first time that there are both commonalities and differences in the molecular events that are required for consolidation of first- and second-order fear memories in the BLA. Specifically, consolidation of the second-order memory that formed in stage 2 of our fear conditioning protocol was disrupted by post-training infusions of drugs that inhibit neuronal activity in the BLA (bupivacaine), including the phosphorylation of kinases that regulate CaMKII/CaMKIV signaling (KN-62), enzymes that catalyze methylation of DNA (RG108 and 5-AZA), and de novo synthesis of mRNA (actinomycin-d). The disruption of consolidation was evident as low levels of freezing when rats were subsequently tested with the second-order CS, S2. These low levels of freezing were not due to effects of the drug infusions on freezing per se, as rats that received control infusions 6 h after conditioning exhibited high levels of freezing at test. Instead, these results are in-line with those reported for the consolidation of a first-order fear memory, which is also disrupted by inhibition of these same cellular processes in the BLA. Together with findings reported in the literature, we have shown that consolidation of both first- and second-order fear memories requires CaMKII/CaMKIV signaling, DNA methylation, and de novo mRNA synthesis in the BLA, implying a general role for these processes in the consolidation of long-term fear memories in the BLA.
The present findings also showed that the processes required for consolidation of first- and second-order fear memories are partially distinct. Specifically, we replicated past findings that, within the BLA, consolidation of a first-order fear memory requires activation of the ERK/MAPK signaling pathway (Schafe et al., 2000), the PKA/PKC signaling pathway (Goosens et al., 2000), and critically, de novo protein synthesis (Schafe and LeDoux, 2000), which is thought to produce the structural changes in BLA neurons that mediate storage and retrieval of a newly formed memory. For example, auditory fear conditioning has been associated with increases in the size, shape, and number of dendritic spines on BLA neurons (Ostroff et al., 2010); increases that are blocked by post-training infusions of a protein synthesis inhibitor into the BLA (Schafe and LeDoux, 2000; Maren et al., 2003). However, in contrast to their requirement for consolidation of first-order conditioned fear, consolidation of second-order fear occurred independently of these processes: when tested with S2, rats that had received post-training infusions of drugs that inhibited ERK/MAPK, PKA/PKC, or de novo protein synthesis in the BLA displayed just as much freezing as rats that received infusions of vehicle alone. These findings cannot be due to the ineffectiveness of drug infusions. Past studies have shown that the same drug volumes and concentrations as those used in the present study are highly effective in inhibiting targeted processes within the BLA (Goosens et al., 2000; Schafe et al., 2000; Schafe and LeDoux, 2000), and in the present study, the same volumes and concentrations were used to replicate the roles of these processes in consolidation of first-order conditioned fear. That is, even though the pharmacological efficacy of drug infusions was not verified in the present study, in cases where a drug failed to disrupt consolidation of second-order conditioned fear (U0126, H7, and cycloheximide), we verified the behavioral effect of the same infusion on consolidation of first-order fear, thereby demonstrating a dissociable role of the targeted process in consolidation of first- and second-order fear memories.
The differential involvement of these molecular events in consolidation of first- and second-order fear memories was not due to the presence of shock in first-order conditioning and its absence in second-order conditioning; and it was not due to a shift in the regional requirement for protein synthesis for second-order fear from the BLA to the CeA. Specifically, consolidation of second-order conditioned fear was unaffected by a post-training BLA infusion of U0126 (the MEK inhibitor) when the second-order conditioning session contained an additional pairing of the first-order CS and shock or by a post-training CeA infusion of cycloheximide (the protein synthesis inhibitor). Instead, a potential explanation of the results just described is that, within the BLA, the changes that regulate consolidation of a first-order fear memory are used, or exploited, for consolidation of a subsequent second-order fear memory; hence consolidation of a second-order fear memory does not require de novo protein synthesis in the BLA.
Differential requirement for early phases of protein synthesis versus differences in the content of learning
Recent evidence indicates that memory is consolidated in distinct phases of protein synthesis, and that the proteins synthesized at the time of exposure to salient events play an especially critical role in this consolidation (Pearce et al., 2017). In rodent models, early phases of protein synthesis have been identified with activation of immediate early genes, such as Arc, and through this activation, polymerization of the major cytoskeletal protein, actin (Steward et al., 2014; Bahrami and Drabløs, 2016). In turn, actin polymerization regulates structural changes at recently activated synapses (Matus, 2000; Messaoudi et al., 2007) as well as different aspects of memory. For example, in slice electrophysiology studies, the induction of LTP, a cellular correlate of memory, triggers rapid increases in Arc-regulated actin polymerization, and leads to changes in the size, shape, and number of new dendritic spines. Furthermore, in studies of fear conditioning, Arc mRNA is expressed in the nucleus of activated neurons as rapidly as 5 min after the onset of auditory fear conditioning (Guzowski et al., 2005), and knockdown of this expression before auditory CS-shock pairings (by infusion of an Arc antisense oligodeoxynucleotide) disrupts consolidation of the first-order conditioned auditory fear memory (Ploski et al., 2008). Therefore, the present findings leave open the possibility that the plasticity-related protein products needed for this consolidation are synthesized during the second-order conditioning session.
Alternatively, the present results suggest that while activation of a first-order fear memory is necessary for the formation of a second-order fear memory (Parkes and Westbrook, 2010; Holmes et al., 2013, 2014), the two types of memory are encoded and stored independently within the BLA. Consistent with this view, previous research using the exact same stimuli and parameters confirmed that our protocol generates second-order fear conditioning, and additionally showed that the expression of fear to the second-order CS, S2, is not conditional on the current value of the first-order CS, S1: specifically, subsequent to second-order fear conditioning, extinguishing S1 had no effect on expression of conditioned fear to S2. Based on these and other findings in the literature (Rizley and Rescorla, 1972; Cheatle and Rudy, 1978; Rescorla, 1982; Barnet et al., 1991; Holmes et al., 2014), we concluded that the content of the second-order fear memory established in our conditioning protocol is a direct association between S2 and the freezing reactions elicited by S1, i.e., an S2-freeze association. Thus, another explanation for the present findings is that, rather than reflecting a difference in the type of conditioning per se, the differential signaling (ERK/MAPK and PKA/PKC) and protein synthesis requirements for consolidation of first- and second-order fear memories in the BLA may reflect differences in the content of the association that forms in the two types of conditioning. At present, we know nothing about the cellular and molecular events in the BLA (or indeed, anywhere in the brain) that are required for consolidation of a first-order S-R association. A potential implication of the present findings is that this consolidation would require neuronal activity in the BLA, including CaMK signaling, gene expression, and methylation of DNA, but would occur independently of ERK/MAPK, PKA/PKC and de novo protein synthesis in the BLA.
Conclusions
In summary, our results make two major contributions to our understanding of the role of the BLA in consolidation of a fear memory. First, our results show that the processes mediating the storage of primary fear memories do not generalize completely to those that consolidate secondary fear memories. This is particularly informative because the processes underlying first-order fear are typically cited as those which contribute to anxiety disorders such as PTSD, social and specific phobias, as well as the treatments of these disorders (Holmes and Singewald, 2013; VanElzakker et al., 2014). By determining how new information is incorporated into established associative networks, this may improve our understanding of anxiety disorders and their treatments. Second, we demonstrate that, in contrast to the traditional view that protein synthesis is crucial for stabilizing synaptic remodelling, epigenetic regulation and gene transcription may function as fundamental processes in fear memory consolidation and memory storage (Holliday, 1999; Queenan et al., 2017). Future work will examine the role of late dendritic protein synthesis in consolidating long-term fear memories, the transcripts involved in consolidating both first- and second-order fear memories, and the link, if any, between CaMK signaling and DNA methylation, given their importance for consolidation of first- and second-order fear memories. Finally, future work will determine the time course of changes in expression of these molecular processes following first- and second-order conditioning.
Footnotes
This work was supported by an Australian Research Council Discovery Project DP130103687. We thank Kelly Clemens and Neil Youngson for comments on the findings.
The authors declare no competing financial interests.
References
- Bae SH, Hwang JW, Shin SJ, Park GH, Yoon KD, Bae SK (2013) Quantitation and pharmacokinetics of 1,4-diamino-2,3-dicyano-1,4-bis (2-aminophenylthio) butadiene (U0126) in rat plasma by liquid chromatography-tandem mass spectrometry. J Sep Sci 36:239–245. 10.1002/jssc.201200779 [DOI] [PubMed] [Google Scholar]
- Bahrami S, Drabløs F (2016) Gene regulation in the immediate-early response process. Adv Biol Regul 62:37–49. 10.1016/j.jbior.2016.05.001 [DOI] [PubMed] [Google Scholar]
- Bailey DJ, Kim JJ, Sun W, Thompson RF, Helmstetter FJ (1999) Acquisition of fear conditioning in rats requires the synthesis of mRNA in the amygdala. Behav Neurosci 113:276–282. 10.1037/0735-7044.113.2.276 [DOI] [PubMed] [Google Scholar]
- Barnet RC, Arnold M, Miller RR (1991) Simultaneous conditioning demonstrated in second-order conditioning: evidence for similar associative structure in forward and simultaneous conditioning. Learn Motiv 22:253–268. 10.1016/0023-9690(91)90008-V [DOI] [Google Scholar]
- Campeau S, Miserendino MJ, Davis M (1992) Intra-amygdala infusion of the N-methyl-d-aspartate receptor antagonist AP5 blocks acquisition but no expression of fear-potentiated startle to an auditory conditioned stimulus. Behav Neurosci 106:569–574. 10.1037/0735-7044.106.3.569 [DOI] [PubMed] [Google Scholar]
- Cheatle MD, Rudy JW (1978) Analysis of second-order odor-aversion conditioning in neonatal rats: implications for Kamin's blocking effect. J Exp Psychol Anim Behav Process 4:237–249. 10.1037/0097-7403.4.3.237 [DOI] [PubMed] [Google Scholar]
- Cohen J. (1988) Statistical power analysis for the behavioral sciences. New York: Routledge Academic. [Google Scholar]
- Duvarci S, Nader K, LeDoux JE (2005) Activation of extracellular signal-regulated kinase-mitogen-activated protein kinase cascade in the amygdala is required for memory reconsolidation of auditory fear conditioning. Eur J Neurosci 21:283–289. 10.1111/j.1460-9568.2004.03824.x [DOI] [PubMed] [Google Scholar]
- Fanselow MS. (1980) Conditional and unconditional components of post-shock freezing. Pavlov J Biol Sci 15:177–182. [DOI] [PubMed] [Google Scholar]
- Fendt M, Fanselow MS (1999) The neuroanatomical and neurochemical basis of conditioned fear. Neurosci Biobehav Rev 23:743–760. 10.1016/S0149-7634(99)00016-0 [DOI] [PubMed] [Google Scholar]
- Gewirtz JC, Davis M (1997) Second-order fear conditioning prevented by blocking NMDA receptors in amygdala. Nature 388:471–474. 10.1038/41325 [DOI] [PubMed] [Google Scholar]
- Goosens KA, Holt W, Maren S (2000) A role for amygdaloid PKA and PKC in the acquisition of long-term conditional fear memories in rats. Behav Brain Res 114:145–152. 10.1016/S0166-4328(00)00224-2 [DOI] [PubMed] [Google Scholar]
- Guzowski JF, Timlin JA, Roysam B, McNaughton BL, Worley PF, Barnes CA (2005) Mapping behaviorally relevant neural circuits with immediate-early gene expression. Curr Opin Neurobiol 15:599–606. 10.1016/j.conb.2005.08.018 [DOI] [PubMed] [Google Scholar]
- Holliday R. (1999) Is there an epigenetic component in long-term memory? J Theor Biol 200:339–341. 10.1006/jtbi.1999.0995 [DOI] [PubMed] [Google Scholar]
- Holmes A, Singewald N (2013) Individual differences in recovery from traumatic fear. Trends Neurosci 36:23–31. 10.1016/j.tins.2012.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes NM, Parkes SL, Killcross AS, Westbrook RF (2013) The basolateral amygdala is critical for learning about neutral stimuli in the presence of danger, and the perirhinal cortex is critical in the absence of danger. J Neurosci 33:13112–13125. 10.1523/JNEUROSCI.1998-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes NM, Cai SY, Lay BP, Watts NR, Westbrook RF (2014) Extinguished second-order conditioned fear responses are renewed but not reinstated. J Exp Psychol Anim Learn Cogn 40:440–456. 10.1037/xan0000036 [DOI] [PubMed] [Google Scholar]
- Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM, Wozniak DF, Nardi A, Arvin KL, Holtzman DM, Linden DJ, Zhuo M, Muglia LJ, Chatila TA (2000) Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci 20:6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Oda Y, Nakamura T, Takahashi R, Miyake W, Hase I, Asada A (2010) Pharmacokinetics of lidocaine, bupivacaine, and levobupivacaine in plasma and brain in awake rats. Anesthesiology 112:1396–1403. 10.1097/ALN.0b013e3181d9cc54 [DOI] [PubMed] [Google Scholar]
- Jensen KF, Ohmstede CA, Fisher RS, Sahyoun N (1991) Nuclear and axonal localization of Ca2+/calmodulin-dependent protein kinase type gr in rat cerebellar cortex. Proc Natl Acad Sci U S A 88:2850–2853. 10.1073/pnas.88.7.2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H (2002) The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3:175–190. 10.1038/nrn753 [DOI] [PubMed] [Google Scholar]
- Lohamn RJ, Liu L, Morris M, O'Brien TJ (2005) Validation of a method for localised microinjection of drugs into thalamic subregions in rats for epilepsy pharmacological studies. J Neurosci Methods 146:191–197. 10.1016/j.jneumeth.2005.02.008 [DOI] [PubMed] [Google Scholar]
- Maddox SA, Watts CS, Schafe GE (2014) DNA methyltransferase activity is required for memory-related neural plasticity in the lateral amygdala. Neurobiol Learn Mem 107:93–100. 10.1016/j.nlm.2013.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Quirk GJ (2004) Neuronal signalling of fear memory. Nat Rev Neurosci 5:844–852. 10.1038/nrn1535 [DOI] [PubMed] [Google Scholar]
- Maren S, Ferrario CR, Corcoran KA, Desmond TJ, Frey KA (2003) Protein synthesis in the amygdala, but not the auditory thalamus, is required for consolidation of Pavlovian fear conditioning in rats. Eur J Neurosci 18:3080–3088. 10.1111/j.1460-9568.2003.03063.x [DOI] [PubMed] [Google Scholar]
- Matus A. (2000) Actin based plasticity in dendritic spines. Science 290:754–758. 10.1126/science.290.5492.754 [DOI] [PubMed] [Google Scholar]
- Messaoudi E, Kanhema T, Soulé J, Tiron A, Dagyte G, da Silva B, Bramham CR (2007) Sustained Arc/Arg3.1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J Neurosci 27:10445–10455. 10.1523/JNEUROSCI.2883-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moita MA, Lamprecht R, Nader K, LeDoux JE (2002) A-kinase anchoring proteins in amygdala are involved in auditory fear memory. Nat Neurosci 5:837–838. 10.1038/nn901 [DOI] [PubMed] [Google Scholar]
- Momparler RL. (2005) Pharmacolgy of 5-Aza-2′-deoxycytidine (decitabine). Semin Hematol 42:S9–S16. 10.1053/j.seminhematol.2005.05.002 [DOI] [PubMed] [Google Scholar]
- Muller J, Corodimas KP, Fridel Z, LeDoux JE (1997) Functional inactivation of the lateral and basal nuclei of the amygdala by muscimol infusion prevents fear conditioning to an explicit conditioned stimulus and to contextual stimuli. Behav Neurosci 111:683–691. 10.1037/0735-7044.111.4.683 [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, LeDoux JE (2000) Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406:722–726. 10.1038/35021052 [DOI] [PubMed] [Google Scholar]
- Ostroff LE, Cain CK, Bedont J, Monfils MH, LeDoux JE (2010) Fear and safety learning differentially affect synapse size and dendritic translation in the lateral amygdala. Proc Natl Acad Sci U S A 107:9418–9423. 10.1073/pnas.0913384107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes SL, Westbrook RF (2010) The basolateral amygdala is critical for the acquisition and extinction of associations between a neutral stimulus and a learned danger signal but not between two neutral stimuli. J Neurosci 30:12608–12618. 10.1523/JNEUROSCI.2949-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov I. (1927) Conditioned reflexes: an investigation of the physiological activity. Oxford, UK: Oxford UP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C (2007) The rat brain in stereotaxic coordinates. Sydney: Academic. [DOI] [PubMed] [Google Scholar]
- Pearce K, Cai D, Roberts AC, Glanzman DL (2017) Role of protein synthesis and DNA methylation in the consolidation and maintenance of long-term memory in aplysia. eLife 6:e18299. 10.7554/eLife.18299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploski JE, Pierre VJ, Smucny J, Park K, Monsey MS, Overeem KA, Schafe GE (2008) The activity-regulated cytoskeletal-associated protein (Arc/Arg3.1) is required for memory consolidation of Pavlovian fear conditioning in the lateral amygdala. J Neurosci 28:12383–12395. 10.1523/JNEUROSCI.1662-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queenan BN, Ryan TJ, Gazzaniga MS, Gallistel CR (2017) On the research of time past: the hunt for the substrate of memory. Ann N Y Acad Sci 1396:108–125. 10.1111/nyas.13348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescorla RA. (1982) Simultaneous second-order conditioning produces S-S learning in conditioned suppression. J Exp Psychol Anim Behav Process 8:23–32. 10.1037/0097-7403.8.1.23 [DOI] [PubMed] [Google Scholar]
- Rizley RC, Rescorla RA (1972) Associations in second-order conditioning and sensory preconditioning. J Comp Physiol Psychol 81:1–11. 10.1037/h0033333 [DOI] [PubMed] [Google Scholar]
- Rodrigues SM, Farb CR, Bauer EP, LeDoux JE, Schafe GE (2004) Pavlovian fear conditioning regulates Thr286 autophosphorylation of Ca2+/calmodulin-dependent protein kinase II at lateral amygdala synapses. J Neurosci 24:3281–3288. 10.1523/JNEUROSCI.5303-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouder JN, Speckman PL, Sun D, Morey RD, Iverson G (2009) Bayesian t tests for accepting and rejecting the null hypothesis. Psychon Bull Rev 16:225–237. 10.3758/PBR.16.2.225 [DOI] [PubMed] [Google Scholar]
- Schafe GE, Nadel NV, Sullivan GM, Harris A, LeDoux JE (1999) Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn Mem 6:97–110. [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE (2000) Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of Pavlovian fear conditioning. J Neurosci 20:8177–8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE (2000) Memory consolidation of auditory Pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci 20:RC96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger Y, Stenzig J, Hübner F, Schaefer A, Reichenspurner H, Eschenhagen T (2016) Pharmacokinetics of the experimental non-nucleosidic DNA methyl transferase inhibitor N-phthalyl-l-tryptophan (RG108) in rats. Basic Clin Pharmacol Toxicol 118:327–332. 10.1111/bcpt.12514 [DOI] [PubMed] [Google Scholar]
- Steward O, Farris S, Pirbhoy PS, Darnell J, Driesche SJ (2014) Localization and local translation of Arc/Arg3.1 mRNA at synapses: some observations and paradoxes. Front Mol Neurosci 7:101. 10.3389/fnmol.2014.00101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattersall MHN, Sodergren JE, Sengupta SK, Trites DH, Modest EJ, Frei E (1975) Pharmcokinetics of actinomycin 0 in patients with malignant melanoma. Clinl Pharmacol Ther 17:701–708. 10.1002/cpt1975176701 [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Piedras-Renteria ES, Tsien RW (2002) α- and βCaMKII: inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron 36:1103–1114. 10.1016/S0896-6273(02)01049-8 [DOI] [PubMed] [Google Scholar]
- VanElzakker MB, Dahlgren MK, Davis FC, Dubois S, Shin LM (2014) From pavlov to PTSD: the extinction of conditioned fear in rodents, humans, and anxiety disorders. Neurobiol Learn Mem 113:3–18. 10.1016/j.nlm.2013.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilensky AE, Schafe GE, LeDoux JE (1999) Functional inactivation of the amygdala before but not after auditory fear conditioning prevents memory formation. J Neurosci 19:RC48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW (2001) Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogenactivated protein kinase pathway. Proc Natl Acad Sci U S A 98:2808–2813. 10.1073/pnas.051634198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CW, Dowling MD Jr (1975) Antipyretic effect of cycloheximide, and inhibitor of protein synthesis, in patients with Hodgkin's disease or other malignant neoplasms. Cancer Res 35:1218–1224. [PubMed] [Google Scholar]
- Zhang XH, Liu F, Chen Q, Zhang CL, Zhuo M, Xiong ZQ, Li BM (2008) Conditioning-strength dependent involvement of NMDA NR2B subtype receptor in the basolateral nucleus of amygdala in acquisition of auditory fear memory. Neuropharmacology 55:238–246. 10.1016/j.neuropharm.2008.05.030 [DOI] [PubMed] [Google Scholar]