

Abstract
Members of the SCY1-like (SCYL) family of protein kinases are evolutionarily conserved and ubiquitously expressed proteins characterized by an N-terminal pseudokinase domain, centrally located Huntingtin, elongation factor 3, protein phosphatase 2A, yeast kinase TOR1 repeats, and an overall disorganized C-terminal segment. In mammals, three family members encoded by genes Scyl1, Scyl2, and Scyl3 have been described. Studies have pointed to a role for SCYL1 and SCYL2 in regulating neuronal function and viability in mice and humans, but little is known about the biological function of SCYL3. Here, we show that the biochemical and cell biological properties of SCYL3 are similar to those of SCYL1 and both proteins work in conjunction to maintain motor neuron viability. Specifically, although lack of Scyl3 in mice has no apparent effect on embryogenesis and postnatal life, it accelerates the onset of the motor neuron disorder caused by Scyl1 deficiency. Growth abnormalities, motor dysfunction, hindlimb paralysis, muscle wasting, neurogenic atrophy, motor neuron degeneration, and loss of large-caliber axons in peripheral nerves occurred at an earlier age in Scyl1/Scyl3 double-deficient mice than in Scyl1-deficient mice. Disease onset also correlated with the mislocalization of TDP-43 in spinal motor neurons, suggesting that SCYL1 and SCYL3 regulate TDP-43 proteostasis. Together, our results demonstrate an overlapping role for SCYL1 and SCYL3 in vivo and highlight the importance the SCYL family of proteins in regulating neuronal function and survival. Only male mice were used in this study.
SIGNIFICANCE STATEMENT SCYL1 and SCYL2, members of the SCY1-like family of pseudokinases, have well established roles in neuronal function. Herein, we uncover the role of SCYL3 in maintaining motor neuron viability. Although targeted disruption of Scyl3 in mice had little or no effect on embryonic development and postnatal life, it accelerated disease onset associated with the loss of Scyl1, a novel motor neuron disease gene in humans. Scyl1 and Scyl3 double-deficient mice had neuronal defects characteristic of amyotrophic lateral sclerosis, including TDP-43 pathology, at an earlier age than did Scyl1-deficient mice. Thus, we show that SCYL1 and SCYL3 play overlapping roles in maintaining motor neuronal viability in vivo and confirm that SCYL family members are critical regulators of neuronal function and survival.
Keywords: CASP, COPI, motor neuron, SCYL1, SCYL3, TDP-43
Introduction
Members of the SCY1-like family of protein kinases, which comprises SCYL1, SCYL2, and SCYL3, are evolutionarily conserved and ubiquitously expressed proteins characterized by an N-terminal kinase domain; centrally located Huntingtin, elongation factor 3, protein phosphatase 2A, yeast kinase TOR1 (HEAT) repeats; and an overall disorganized C-terminal segment. Unlike functional protein kinases that catalyze the transfer of phosphate groups to protein substrates, kinase domains of SCYL proteins are likely inactive and possibly serve other functions, such as modulation of active kinases or assembly of signaling pathways (Boudeau et al., 2006; Pelletier, 2016).
Genetic studies in humans and mice have begun to elucidate the biological function of SCYL proteins. Disruptive mutations in SCYL1 are associated with a rare syndrome characterized by liver failure, cerebellar atrophy, ataxia and, most importantly peripheral neuropathy (Schmidt et al., 2015). In mice, spontaneous or targeted deletion of Scyl1 causes an early onset motor neuron disorder characterized by progressive loss of motor function, muscle wasting, loss of large-caliber axons in the periphery, and loss of lower motor neurons in the ventral horn of the spinal cord (Blot et al., 1995; Schmidt et al., 2007; Pelletier et al., 2012). The disease is neural cell-autonomous and involves molecular pathways associated with other motor neuron disorders such as amyotrophic lateral sclerosis (ALS; Pelletier et al., 2012). Mislocalization of the RNA-binding protein (RBP) TDP-43 from the nucleus to cytoplasmic inclusions, a hallmark of ALS and frontotemporal lobar degeneration, occurs in lower motor neurons of Scyl1-deficient mice and correlates with disease onset (Pelletier et al., 2012). Similarly, targeted disruption of Scyl2 in mice causes severe neurological disorders, including impaired suckling behavior, which result in premature death of most newborn mice (Gingras et al., 2015). In surviving mice, the absence of Scyl2 results in the degeneration of several neuronal populations, most notably CA3 pyramidal neurons of the hippocampus (Gingras et al., 2015). The degenerative process associated with the loss of SCYL2 is also neural cell-autonomous and caused by excessive excitatory signaling (Gingras et al., 2015).
Although these studies clearly established SCYL1 and SCYL2 as critical regulators of neuronal function and survival, the biological function of SCYL3 has remained elusive. In this study, we use a combination of biochemical, cell biological and genetic approaches to elucidate the function of SCYL3 in mammalian development and physiology. We show that SCYL3 is a widely expressed Golgi membrane-associated protein with biochemical and cell biological properties similar to those of SCYL1 and both proteins play an overlapping role in maintaining motor neuron viability in mice. Although targeted disruption of Scyl3 in mice did not produce overt abnormalities, absence of Scyl3 accelerated the onset of the motor phenotype associated with loss of Scyl1. Disease onset in Scyl1 and Scyl3 double-deficient mice also correlated with TDP-43 pathology in spinal motor neurons suggesting a link between SCYL1, SCYL3, and TDP-43 proteostasis.
Materials and Methods
SCYL3 antibody.
A peptide corresponding to amino acids 7–27 [ALKSYTLRESPFTLPSGLAVY (serum 7688)] of SCYL3 was synthesized by the Hartwell Center for Bioinformatics and Biotechnology at St. Jude Children's Research Hospital (St. Jude) and conjugated to glutaraldehyde-activated keyhole limpet hemocyanin (KLH). The KLH-conjugated peptide was used to immunize rabbits (Rockland Immunochemicals). The serum was enriched for the peptide of interest by affinity chromatography on a matrix coupled to the corresponding peptide. Selectivity of the antibody was tested against other SCY1-like family members by RNA interference (RNAi)-mediated knockdown of SCYL1, SCYL2, and SCYL3. No cross-reactivity with other SCYL proteins was found. Reactivity of the peptide-purified antibody was also tested by Western blot, using protein extracts from Scyl3+/+, Scyl3+/−, and Scyl3−/− mouse embryonic fibroblasts (MEFs). No signal was detected in Scyl3−/− MEFs. The serum detected endogenous or overexpressed protein in Western blot and immunofluorescence studies.
Antibodies.
The following antibodies were used: anti-SCYL1 antibody (7645; Pelletier et al., 2012); anti-SCYL2 (7649; Gingras et al., 2015); anti-FLAG M2 (Sigma-Aldrich, catalog #F3165; RRID:AB_259529); HRP-conjugated anti FLAG (M2) antibody (Sigma-Aldrich, catalog #A8592; RRID:AB_439702); rabbit polyclonal anti-FLAG antibody (Sigma-Aldrich, catalog #F7425; RRID:AB_439687); anti-GM130 clone 35 (BD Biosciences, catalog #610823; RRID:AB_398142); anti-μ2-adaptin clone 31 (BD Biosciences, catalog #611351; RRID:AB_398873); anti-GS28 clone 1 (BD Biosciences, catalog #611185; RRID:AB_398719); anti-COPG (Santa Cruz Biotechnology, catalog #sc-14165; RRID:AB_2081447); anti-COPA (Santa Cruz Biotechnology, catalog #sc-398099); anti-COPB H-300 (Santa Cruz Biotechnology, catalog #sc-30091; RRID:AB_638665); anti-COPB2 (Santa Cruz Biotechnology, catalog #sc-23167; RRID:AB_2081312); anti-Iba1 (BioCore Medical Technologies, catalog #CP290A); anti-GFAP (Dako, catalog #Z0334; RRID:AB_10013382); anti–TDP-43 (Proteintech Group, catalog #10782-2-AP; RRID:AB_615042); anti-caprin1 (Proteintech Group, catalog #15112-1-AP; RRID:AB_2070016); anti-FUS/TLS (Bethyl Laboratories, catalog #IHC00074); anti-hnRNPA2B1 (Sigma-Aldrich, catalog #R4653; RRID:AB_261967); anti-ezrin (Cell Signaling Technology, catalog #3145; RRID:AB_2100309); anti-SCYL3 (Proteintech Group, catalog #11249-1-AP; RRID:AB_2071154); anti-CASP antibody (Invitrogen, catalog #PA5-30003); anti-rabbit IgG, HRP-linked antibody (Cell Signaling Technology, catalog #7074; RRID:AB_2099233); and anti-mouse IgG, HRP-linked antibody (Cell Signaling Technology, catalog #7076; RRID:AB_330924); AlexaFluor 488 donkey anti-rabbit antibody (ThermoFisher Scientific, catalog #R37118; RRID:AB_2556546); AlexaFluor 568 donkey anti-mouse antibody (ThermoFisher Scientific, catalog #A10037; RRID:AB_2534013).
DNA constructs.
All oligonucleotides were produced by the Hartwell Center for Bioinformatics and Biotechnology or Integrated DNA Technologies. Plasmids pBR322-DTA (diphtheria toxin A) and PL451-TK were generated as described previously (Pelletier et al., 2012). Plasmids PL452 and PL451 were obtained from Dr. Neil A. Copeland (Liu et al., 2003). Plasmids encoding CRE recombinases (pMC-Cre) were obtained from Klaus Rajewski (Max Delbrück Center for Molecular Medicine, Berlin, Germany). The Scyl3-targeting construct was engineered by using gap-repair technology as described previously (Liu et al., 2003). Briefly, a 9.6 kb fragment containing genomic sequences of Scyl3 was subcloned by gap repair into pBR322-DTA. The first cassette containing the neomycin-resistance gene flanked by 2 loxP sites (PL452) was inserted in intron 4. After excision of the neomycin cassette by CRE recombinase, a second cassette containing the neomycin-resistance and thymidine kinase cDNAs flanked by 2 Flp recombinase target (Frt) sites and a second loxP site (PL451-TK), was inserted in intron 6 (see Fig. 4). Locations of loxP sites were chosen such that a null allele would be generated upon CRE recombination. Full-length cDNAs encoding SCYL1 (IRAV3672524), SCYL2 (IRAV4037878), and SCYL3 (IRAK30435732) were purchased from Open Biosystems. Sequences were verified by DNA sequencing (Hartwell Center for Bioinformatics and Biotechnology). The FLAG epitope was inserted at the N-terminal end of SCYL1, SCYL2, or SCYL3 of the coding region. The HA epitope was inserted at the C-terminal end of SCYL3. Truncated versions of SCYL3 were generated by using conventional molecular biology techniques. VSVG-tsO45-eGFP was obtained from Dr. Jennifer Lippincott-Schwartz (Presley et al., 1997). VSVG-tsO45-KDELR-Myc was obtained from Dr. Victor W. Hsu (Yang et al., 2006). pSpCas9(BB)-2A-GFP (PX458) was a gift from Dr. Feng Zhang (Addgene plasmid #48138; Ran et al., 2013). px458-CFP was generated by replacing the cDNA encoding GFP with a gBlock encoding CFP by using the Gibson assembly method.
Figure 4.
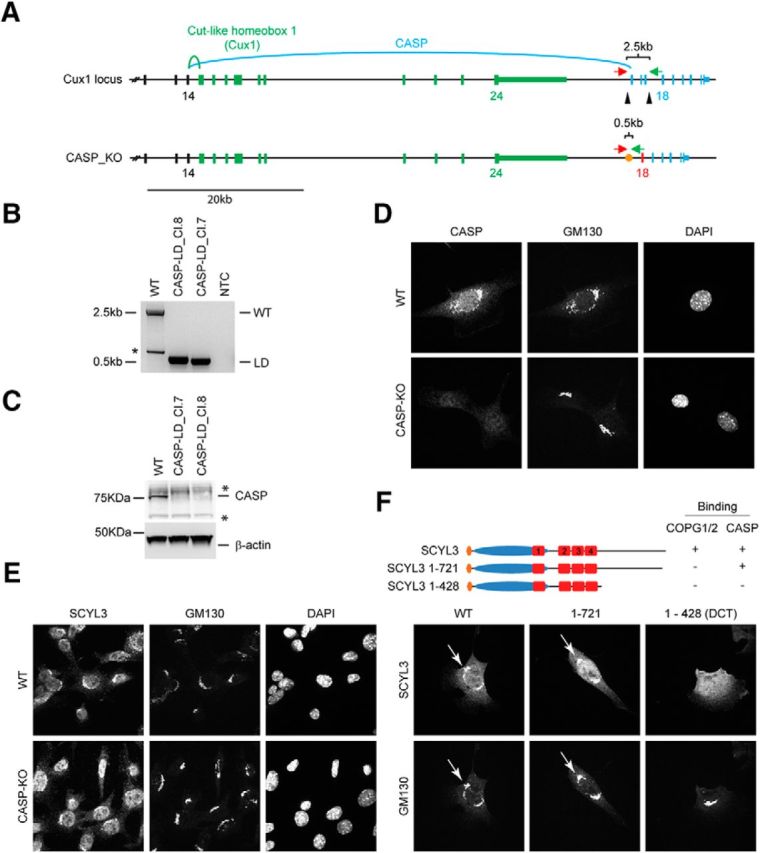
COPI and CASP interactions contribute to localization of SCYL3 to the Golgi apparatus. A, Generation of CASP-deficient MEFs using CRISPR–Cas9 technology. Structure of the Cux1 gene encoding CDP and CASP. Exons common to CDP and CASP are illustrated in black (only exons 12–14 illustrated). Exons encoding CDP are illustrated in green, and those encoding CASP are illustrated in blue. To generate CASP-deficient cells, 2 sgRNAs (black arrowheads) were designed to delete exons 15–17. Splicing of exon 14 into exon 18 causes a frame shift and premature stop codon. Red arrow, Cux1-F51 primer; green arrow, Cux-R32 primer. B, PCR genotyping of the WT and 2 independently derived CASP-deficient MEF lines (Cl.7 and Cl.8). Bands of 2496 and ∼495 bp correspond to WT and null CASP-null alleles, respectively. A smaller band (*) from internal priming sites was also obtained by using these primers. NTC, No template control. Sanger sequencing confirmed proper recombination of the allele in CASP-deficient MEFs. C, Western blot analysis of CASP and β-actin (as loading control) in WT and CASP-deficient MEFs. *Indicates nonspecific bands. D, Immunofluorescence staining of CASP and GM130 in WT and CASP-deficient MEFs. Exponentially growing WT and CASP-deficient MEFs were fixed and stained for CASP and GM130 and imaged by confocal microscopy. E, SCYL3 localization in WT and CASP-deficient MEFs. Exponentially growing WT and CASP-deficient MEFs were fixed and stained for SCYL3 and GM130 and imaged by confocal microscopy. F, Subcellular localization of full-length and key truncated version of SCYL3 in Scyl3-deficient MEFs. Exponentially growing Scyl3−/− MEFs were transfected with full-length or the indicated truncated versions of SCYL3, fixed and stained for SCYL3 and GM130, and imaged by confocal microscopy.
Mouse husbandry.
All animals were housed in an Association for Assessment and Accreditation of Laboratory Animal Care accredited facility and maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All animal experiments were reviewed and approved by the St. Jude Institutional Animal Care and Use Committee.
Generation of mice bearing a null allele of Scyl3.
EmbryoMax mouse embryonic stem (ES) cells (CMTI-1, strain 129/svev, Millipore) were used to target the Scyl3 gene. Positive clones were identified by Southern blot analyses of EcoRI-digested genomic DNA, using 5′ and 3′ external probes. To identify single recombination events, EcoRI-digested genomic DNA obtained from positive clones was also analyzed by Southern blot, using a neomycin probe. ES cell clones bearing the null allele (Scyl3−) were generated in vitro by electroporating ES cell clones with plasmids encoding the CRE recombinase. During this process, 1-(2-deoxy-2-fluoro-β-d-arabinofuranosyl)-5-iodouracil (0.5 μm) was used for negative selection. Properly rearranged clones were identified by Southern blot analysis, using 5′ and 3′ external probes and then PCR assays. Two distinct ES cell clones were injected into blastocysts, which were implanted into BALB/c recipient females to produce chimeras. Chimeras were crossed with C57BL/6J mice (Jackson Laboratories) to obtain heterozygous mice. Heterozygous mice were crossed twice with C57BL/6 background, and their progeny were used for the heterozygous intercross. Scyl1-deficient mice were generated as described previously (Pelletier et al., 2012).
Mouse genotyping.
The Scyl3 locus was genotyped by PCR, using the following primers: S3F01: 5′-CTCAGACCCTCTGATGGGAAGAGGA-3′, S3R51: 5′-CTGTCCTCACAGCGAGACAGCTCAGTA-3′, and S3R02: 5′-CTGGGACATCTCCATCTTGATA-3′. Bands of 521 and 310 bp, corresponding to the WT (Scyl3+) and null allele (Scyl3−), respectively, were obtained. The Scyl1 locus was genotyped as described previously (Pelletier et al., 2012).
SCYL3 localization studies.
Exponentially growing WT or Scyl3−/− MEFs were cultured on gelatin-coated round glass coverslips (10 mm) in DMEM, high glucose (11965092) supplemented with 100 U/ml penicillin (Invitrogen, 15140122), 100 μg/ml streptomycin (Invitrogen, 15140122), 1× GlutaMAX (Invitrogen, 35050061), and 1 mm sodium pyruvate (Invitrogen, 11360070) and 10% animal serum complex (Gemini Bio-Products, 100-602). Cells were transfected with 0.5 μg of plasmid DNA encoding SCYL3 (and variants) using lipofectamine 2000 as described by the manufacturer (Life Technologies, 11668-019). Twenty four to 36 h after transfection, cells were fixed and stained as described in the next section.
For immunofluorescence staining, cells were fixed with 4% paraformaldehyde [in Dulbecco's PBS (DPBS)] for 15 min. Cells were washed twice with DPBS and incubated with 100 mm Glycine (in DPBS) for 5 min. Cells were then incubated with DPBS containing 0.1% Triton X-100 and 1% bovine serum albumin (BSA) for 1 h at room temperature. Coverslips were inverted onto a 50 μl droplet containing primary antibodies diluted in DPBS-BSA (1%) and incubated overnight at 4°C. Cells were washed three times with DPBS and incubated with secondary antibodies for 2 h at room temperature. Cells were then washed three times with DPBS and incubated with 300 nm DAPI (Invitrogen, D1306) for 5 min. Cells were finally washed three times and mounted onto glass slides using Fluoromount-G (SouthernBiotech). Cells were visualized using a Zeiss Observer Z1, and images were captured with Slidebook6 acquisition and analysis software (3i Technologies).
Generation of CASP-deficient fibroblasts.
CASP was deleted in MEFs by using CRISPR-Cas9 technology. Two sg RNAs (sgRNAs) targeting introns 14 and 17 of Cux1 were selected as described previously (Pelletier et al., 2015). Cux1_Guide 01: 5′-CCTCCTTCCTCCGAGTATCC-3′ (chr5:136,258,192–136,258,211; GRCm38/mm10) and Cux1_Guide 02: 5′-ATTTGAAGCCTACGGGGAAC-3′ (chr5:136,256,191–136,256,210; GRCm38/mm10) have no potential off-target sites with <3 mismatches, and the protospacer-adjacent motif-proximal 13 nt seed sequences are unique in the mouse genome. Guide sequences were cloned in PX458 and PX458-CFP to generate px458-Cux1 Guide_01 and px458-CFP-Cux1_Guide 02 plasmids. SV40-transformed WT MEFs were transfected with px458-Cux1_Guide 01 and px458-CFP-Cux1_Guide 02 plasmids, and after 2 d GFP- and CFP-positive cells were single-cell sorted by fluorescence-activated cell sorting into individual wells of 96-well plates. The region surrounding target sequences was PCR amplified with the following primers: Cux1-F51, 5′-CAGGGTTACAGGTCAGGGGA-3′, and Cux1-R32, 5′-TCTGTGGATGGTTGAACCCG-3′. Thirty-seven clones were analyzed at the genomic level by PCR, and fragments of ∼500 and 2496 bp corresponding to the deleted and WT alleles, respectively, were obtained. Twenty clones lost their WT allele and contained the genomic deletion. Sequencing confirmed the proper deletion of CASP in these clones. Two clones (CASP_Cl.7 and CASP_Cl.8) were used for subsequent studies (CASP and SCYL3 immunofluorescence staining).
Retrograde and anterograde transport assay.
Three independently derived WT and Scyl3−/− MEFs were transfected with a plasmid expressing VSVG-ts-O45-eGFP or VSVG-ts-O45-Myc to monitor forward and reverse movement, respectively. To monitor forward movement from the ER to plasma membrane, 24 h after transfection cells were incubated at 40°C for 12 h to promote accumulation of the chimeric protein in the ER. Cells were then transferred to an incubator at 32°C to allow transport of the chimeric protein from the ER to the Golgi apparatus and ultimately the plasma membrane. Cells were taken out at the indicated times, fixed, and stained using an AlexaFluor 488-conjugated anti-GFP antibody (A-21311, Invitrogen) and GM130, a Golgi marker, to follow progression of the chimeric protein along the secretory pathway. To monitor retrograde transport from the Golgi/ERGIC to the ER, transfected cells were incubated at 32°C to allow the chimeric protein to move freely along the secretory pathway. Cells were then transferred to an incubator at 40°C to trap the chimeric protein that returned to the ER. Cells were taken out at indicated times, fixed, and stained using an anti-Myc antibody (A-14, Santa Cruz Biotechnology) and GM130 to follow progression of the chimeric protein along the secretory pathway. The number of cells with predominant staining within the ER, Golgi, or plasma membrane was scored. Values obtained from the 3 individually derived cell lines (∼25 cells/cell line per time point) were averaged and plotted. Values are expressed as the mean ± SEM.
Golgi size.
Exponentially growing WT and Scyl3−/− MEFs were fixed and stained for GM130 and imaged by confocal microscopy. GM130-positive areas (pixels) were determined from 162 WT and 142 Scyl3−/− MEFs (obtained from 3 independently derived cell lines for each genotype). Data are expressed as the mean ± SEM. No significant differences in size or overall morphology were observed between WT and Scyl3−/− MEFs.
Cell migration.
Cell migration was assessed as described previously (Rankin et al., 2006). Briefly, three independently derived WT and Scyl3−/− MEF lines were seeded in quintuplicate on gelatin-coated 10 mm glass coverslips placed at the bottom of the well of a 24-well plate at a density of 50,000 cells per well. Cells were cultured in DMEM, high glucose (Invitrogen, 11965092) supplemented with 100 U/ml penicillin (Invitrogen, 15140122), 100 μg/ml streptomycin (Invitrogen, 15140122), 1× GlutaMAX (Invitrogen, 35050061), 1 mm sodium pyruvate (Invitrogen, 11360070), and 10% animal serum complex (Gemini Bio-Products, 100-602) for a few days until they reached confluence. Cells were then washed three times with DPBS. The glass coverslips were flipped and placed into wells of a 6-well plate and cultured in DMEM high glucose (11965092) supplemented with 100 U/ml penicillin (Invitrogen, 15140122), 100 μg/ml streptomycin (Invitrogen, 15140122), 1× GlutaMAX (Invitrogen, 35050061), 1 mm sodium pyruvate (Invitrogen, 11360070), and 2 or 10% animal serum complex (Gemini Bio-Products, 100-602). Cells were then incubated at 37°C for 24 h. Plates were removed from the incubator and imaged using an inverted microscope fitted with a digital camera. Distances traveled by cells from the edge of the coverslip were measured. Five measurements were made for each cell line and culture condition. Values from three individually derived cell lines were averaged and plotted.
Immunoprecipitation.
Exponentially growing HEK293T cells were seeded at a density of 1.5 × 106 cells per 10 cm dish and transfected the next day with 10 μg (or as indicated in figure legends) of plasmids encoding FLAG-tagged SCYL3 (SCYL3-N-FLAG). After 36 h of transfection, HEK293T cells were washed three times with ice-cold PBS and lysed in NP-40 lysis buffer [50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 50 mm sodium fluoride, 5 mm EDTA, 40 mm β-glycerophosphate, 1 mm sodium orthovanadate, 1% NP-40 (Igepal-CA-630) supplemented with protease inhibitors (Complete, Roche)] at 4°C for 30 min. Lysates were cleared by centrifugation at 20,000 × g for 5 min at 4°C, and protein content was quantified by the BCA assay. Cleared lysates (1 mg of protein) were incubated with 20 μl of EZview Red ANTI-FLAG M2 Affinity Gel for 2 h at 4°C. Beads were washed three times with NP40 lysis buffer and eluted using 100 μl of 200 ng/ml 3X FLAG peptide (Sigma-Aldrich, F4799) for 20 min at 4°C for liquid chromatography-tandem mass spectrometry (LC-MS/MS) or using 100 μl of 1× Laemmli buffer (Bio-Rad) for Western blotting. Proteins in samples were resolved by 10% SDS-PAGE and either transferred to nitrocellulose for Western blotting or stained with SYPRO Ruby for MS analyses.
LC-MS/MS.
Protein bands from immune complexes resolved by SDS-PAGE were cut out, reduced, and alkylated with iodoacetamide before being digested with trypsin. Mass spectrometric analysis was performed using an LTQ Linear Ion Trap Mass Spectrometer (Thermo Electron). Database searches were performed using LTQ.raw files in combination with the Mascot search engine (http://www.matrixscience.com). Protein/peptide assignments were made on the basis of MS/MS spectra. The Swiss-Prot database (http://au.expasy.org) was used for protein identification.
Tissue expression of SCYL proteins.
For Western blotting of mouse tissues, tissues were lysed in RIPA buffer [50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% NP-40 (Igepal-CA-630), 0.5% sodium deoxycholate, 0.1% SDS, PhosSTOP phosphatase inhibitor cocktail tablets (Sigma-Aldrich) and cOmplete protease inhibitors (Sigma-Aldrich)] and then resolved using 10% SDS-PAGE and analyzed by Western blot.
Western blot analyses.
Western blot analyses of MEFs and HEK293T cell extracts were performed as described previously (Pelletier et al., 2006). Briefly, membranes were blocked for 1 h at room temperature in TBST (137 mm sodium chloride, 20 mm Tris, 0.1% Tween 20, pH 7.6) supplemented with 5% nonfat dry milk. Then, they were incubated with antibodies overnight at 4°C. Membranes were incubated with secondary antibodies at room temperature for 1 h. Then, membranes were washed three times with TBST. Bands were detected using the GE Healthcare ECL Western Blotting System (GE Healthcare Life Sciences, RPN2108) according to the manufacturer's recommendations.
Motor dysfunction scoring system and grip test.
The scoring system used to follow disease progression and the grip test was described previously (Pelletier et al., 2012). Disease progression in animals was scored as follows: 1, growth defects; 2, growth defects and abnormal gait; 3, posterior waddle and abnormal gait; 4, growth abnormalities, abnormal gait, and tremor when suspended by their tails; 5, aforementioned phenotypes and partial paralysis; 6, complete paralysis; and 7, fattening of the pelvis and dorsally contracted hindlimbs. For the grip test, mice were placed on top of an elevated cage grid. When mice were holding tightly to the cage grid, the grid was inverted and then the amount of time that they remained suspended was recorded.
Histology and immunohistochemistry.
Immunohistochemistry and histologic studies were performed on tissues collected from 3 males from each age group and genotype. Immediately after the animals were killed, mice were perfused with 10% buffered formalin. Limbs, spines, and brains were postfixed by immersion in 10% buffered formalin for at least 24 h before being decalcified in formic acid (TBD-2 Decalcifier; ThermoFisher Scientific). Tissues were embedded in paraffin, sectioned at 6 μm, mounted on positively charged glass slides (Superfrost Plus; ThermoFisher Scientific), and dried in a 60°C oven for 20 min. Slides were stain with hematoxylin and eosin (H&E) or used for immunohistochemistry. H&E-stained limb cross-sections were acquired with an AT2 Aperio Digital Pathology Slide Scanner and images were analyzed with NIH ImageJ v1.42q (http://rsbweb.nih.gov/ij/). Muscle cross-sectional areas (CSAs) for rectus femoris and bicep brachii were determined from similar cross sections. Mean fiber CSAs were estimated by counting the number of muscle fibers within a given area. The area was then divided by the number of fibers counted. Muscle fiber numbers were estimated by counting the number of fibers within a given area and extrapolated for the whole muscle. The percentage of atrophied muscle fibers was determined on H&E-stained cross-sections under blinded conditions. To calculate the percentage of atrophied muscle fibers, the number of angulated/atrophied fibers was divided by the total number of muscle fibers in a field and then multiplied by 100. The percentage of atrophied/angulated fibers was determined from two images (0.0768 mm2) obtained from three mice per age group and genotype. Similarly, the percentage of fibers with central nucleation was determined under blinded conditions by counting the number of fibers with central nucleation divided by the total number of muscle fibers and multiplied by 100. The number of myelinated axons in the sciatic nerve was determined under blinded conditions by using a light microscope and NIH ImageJ on toluidine blue-stained plastic embedded cross-sections. Axon counts in the sciatic nerve were determined by counting the number of myelinated fibers in 15–20% of the nerve multiplied by the total area of the nerve. Analysis was based on six images from three mice per age group and genotype. Axonal caliber in sciatic nerve was quantified by using a light microscope and ImageJ software on toluidine blue-stained cross-sections. Between 415 and 1785 axons per sciatic nerve were evaluated. The number of healthy ventral horn motor neurons in the lumbar region of the spinal cord was determined under blinded conditions from H&E-stained cross-sections by using a light microscope and ImageJ. Large motor neurons were considered healthy if cells exhibited clearly defined Nissl substance and a large nucleus. The number of neurons showing rarefaction of cytosolic organelles or central chromatolysis was quantified under blinded conditions. Analysis was based on 30 images obtained from three mice per age group and genotype. The number of ventral horn motor neurons exhibiting cytoplasmic TDP-43 was determined under blinded conditions. Analysis was based on 24 images obtained from three mice per age group and genotype. The number of GFAP-positive and Iba1-positive cells in the spinal ventral horn was determined by counting the number of cells with clearly defined nucleus and immunoreactivity/0.04 mm2.
Experimental design and statistical analysis.
Three males for each age group and genotype were used for histological studies. All samples were analyzed in blind manner. Statistical analyses were performed by GraphPad Prism software v6.02. To assess whether Scyl3 deficiency worsened phenotypic changes caused by Scyl1 deficiency, Student's t test was used for comparison between groups and two-tailed p values ≤0.05 were considered significant.
Sequence alignment.
Protein sequence alignments were performed by Vector NTI software 9.0.0 using the BLOSUM62 mt2 matrix.
Results
Biochemical and cell biological properties of SCYL3
SCYL3, like SCYL1 and SCYL2, is characterized by an N-terminal pseudokinase domain, centrally located HEAT repeats, and an overall disorganized C-terminal segment (Fig. 1A). Unlike SCYL1 and SCYL2, SCYL3 has an N-terminal myristoylation consensus sequence that aids its subcellular localization to the Golgi apparatus (Sullivan et al., 2003) and does not possess coiled-coil domains or compositional bias, as seen in the proline-rich region of SCYL1. The overall sequence identity between SCYL3 and SCYL1 or SCYL2 is 19.7% and 10.5%, respectively; thus, the closest analog of SCYL3 is SCYL1 (Fig. 1B).
Figure 1.

SCYL3 domain structure, distribution, and localization. A, Schematic representation of the SCYL family of protein pseudokinases and predicted domains. SCYL3, like SCYL1 and SCYL2, consists of an N-terminal pseudokinase domain, four HEAT repeats, and a C-terminal segment containing no known protein domain. B, Sequence identity among SCYL family members. The overall identity between SCYL1 and SCYL2 is 17%; between SCYL1 and SCYL3 is 19.7%; between SCYL2 and SCYL3 is 14%; and between SCYL2 and SCYL3 is 10.5%. C, Selectivity of antibodies against SCYL1, SCYL2, and SCYL3. NIH 3T3 cells were transfected with or without RNAi targeting Scyl1, Scyl2, or Scyl3 transcripts. Then, 48 h after transfection, protein extracts were analyzed by Western blot using antibodies against SCYL1, SCYL2, SCYL3, or μ2-adaptin as a loading control. *Indicates nonspecific band. D, Tissue distribution of SCYL proteins. Protein extracts prepared from various mouse tissues were resolved by SDS-PAGE and analyzed by Western blot, using antibodies against SCYL1, SCYL2, and SCYL3. E, Exponentially growing WT and Scyl3−/− MEFs were stained with anti-SCYL3 antibody and imaged by confocal microscopy. Note the presence of SCYL3-positive staining in the perinuclear region of WT (arrow) but not Scyl3−/− MEFs. *Indicates nonspecific nuclear staining in WT and Scyl3−/− MEFs. Images are representative of several independent experiments. F, SCYL3 localizes to the Golgi apparatus. Exponentially growing WT MEFs were fixed and stained with antibodies against SCYL3 and GM130, GS28, COPG1/2 or COPA and counterstained with DAPI. Images are representative of several independent experiments. G, Scyl3−/− MEFs were transfected with plasmid encoding SCYL3. Thirty-six hours after transfection, cells were fixed and stained with antibodies against SCYL3 and GM130, and counterstained with DAPI. Images are representative of several independent experiments. H, SCYL3 is a membrane-associated protein. Microsomal (M) and cytosolic (C) fractions from WT mouse liver were resolved by SDS-PAGE and analyzed by Western blot using antibodies against SCYL3, COPG2, the Golgi-associated protein GM130, and the membrane anchored protein Vti1b. Microsomal fractions were also washed once (1×) or twice (2×) with NaHCO3, pH 11.5, to dislodge membrane-associated but not membrane-anchored proteins. Images are representative of two independent experiments. Scale bars, 10 μm
To determine the function of SCYL3, its distribution in mouse tissues and subcellular localization in cultured cells was examined. A rabbit polyclonal antibody against SCYL3 (see Materials and Methods) was generated and its specificity was tested by RNAi-mediated knockdown studies. The antibody against SCYL3 (serum 7688AP) detected a band of ∼75 kDa corresponding to SCYL3 in SCYL1- or SCYL2-depleted cells but not in SCYL3-depleted cells (Fig. 1C). Further, Western blot using this antibody showed that unlike SCYL1 and SCYL2, which were expressed in virtually all tissues examined (Fig. 1D), SCYL3 was predominantly expressed in the forebrain, cerebellum, kidney, liver, lung, lymph nodes, ovary, pancreas, spleen, testis, and thymus (Fig. 1D). There was low or no expression of SCYL3 in the eyes, heart, large intestine, and skeletal muscles.
Next, subcellular localization of SCYL3 was examined in MEFs derived from a wild-type (WT) mouse by using indirect immunofluorescence staining and confocal microscopy. Both endogenously and ectopically expressed SCYL3 localized to vesicular structures in the periphery of the nucleus, which resembled the Golgi apparatus (Fig. 1E,F). Specificity of the anti-SCYL3 antibody for immunofluorescence staining was tested in Scyl3-deficient MEFs derived from Scyl3-deficient embryos (see “SCYL3 is dispensable for the core function of COPI and cellular migration” section for details about the generation of Scyl3-deficient MEFs). The absence of perinuclear staining in Scyl3-deficient MEFs confirmed signal specificity (Fig. 1E). Further, confocal analyses revealed that endogenous and ectopically expressed SCYL3 colocalized with the Golgi markers GM130 (Fig. 1F,G) and GS28 (Fig. 1F). SCYL3 also partially colocalized with the coatomer complex I (COPI) subunits COPG2 and COPA (Fig. 1F).
Given the presence of SCYL3 in the Golgi apparatus, we tested whether SCYL3 exclusively associated with membranes and acted like a membrane-associated or membrane-anchored protein. Cytosolic and microsomal membrane fractions were isolated from the mouse liver and the presence of SCYL3 and other membrane and cytosolic markers was determined in these fractions. Similar to GM130 (a membrane-associated protein) and Vti1b (a membrane-anchored protein), SCYL3 cofractionated almost exclusively with the microsomal membrane fraction. Also, SCYL3 was present in small amounts in the cytosolic fraction, suggesting that it cycles between the two compartments. To test whether SCYL3 acted like a membrane-associated or membrane-anchored protein, the membrane fraction was washed once or twice with a high-pH solution (sodium carbonate buffer, pH 11.5), which removes membrane-associated but not membrane-anchored proteins. Like GM130 and COPG1/2, but unlike Vti1b, SCYL3 readily dissociated from microsomal membranes under high-pH conditions (Fig. 1H). Together, these results indicate that SCYL3 is a widely expressed Golgi membrane-associated protein and, like other SCYL family members, may regulate intracellular trafficking.
To further characterize the properties of SCYL3, we identified SCYL3-interacting proteins using affinity enrichment and LC-MS/MS analysis. A FLAG-tagged version of SCYL3 was expressed in Hek293T cells and enriched by affinity capture using anti-FLAG M2 beads. Resolution of the immune complex by SDS-PAGE and SYPRO Ruby gel staining revealed several potential interacting proteins (Fig. 2A). Nine clearly identifiable bands were excised and analyzed by MS (Fig. 2A and Fig. 2-1). Several peptides corresponding to COPA, COPB, COPB2, COPD, COPE, COPG1, and COPG2 were identified. These proteins are components of COPI, a heptameric protein complex involved in Golgi-to-ER and intra-Golgi transport of cargo molecules (Szul and Sztul, 2011). We also identified CASP, a Golgi membrane protein related to giantin thought to regulate intra-Golgi and Golgi–ER retrograde transport (Gillingham et al., 2002) and kinesin-1 heavy chain (KINH), a motor protein involved in transporting organelles along microtubules (Verhey and Hammond, 2009). Several peptides ascribed to various proteins were also identified, most being involved in protein folding and likely associated with SCYL3 as the polypeptide is synthesized and folded. Several RBPs, including caprin1, were also identified. Fig. 2-1 lists all proteins identified by MS analysis. Although SCYL3 was originally identified as a putative interacting partner for erzin [a member of the ERM (ezrin, radixin, moesin) family of cytoplasmic peripheral membrane proteins linking the plasma membrane to the cytoskeleton; Sullivan et al., 2003], no peptides ascribed to ezrin were identified in the MS analysis.
Figure 2.
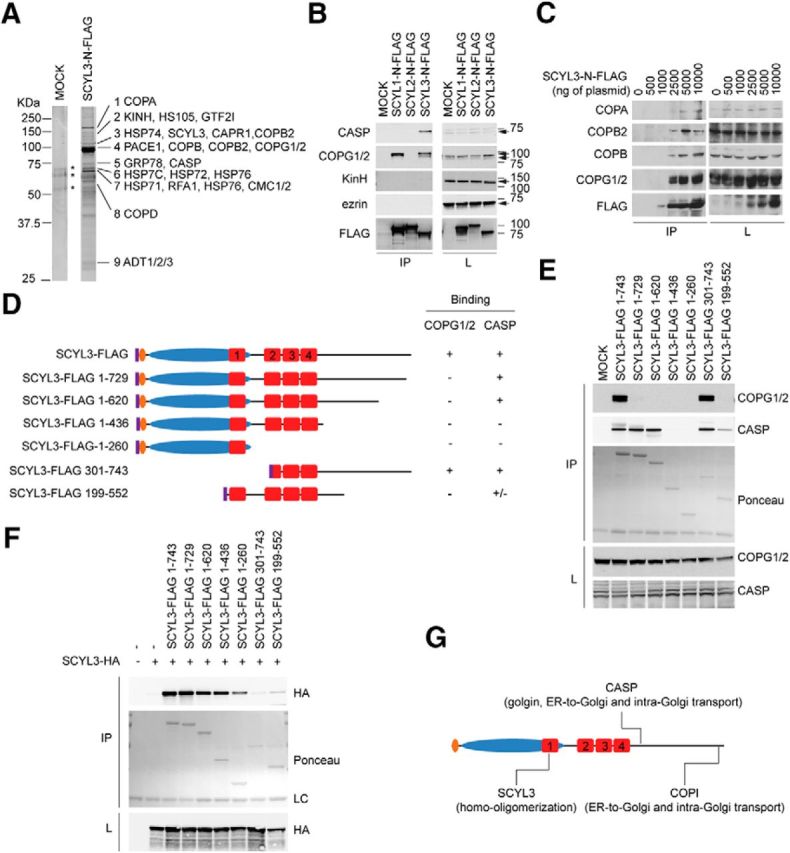
Identification of SCYL3-interacting partners. A, Identification of CASP and components of the COPI complex by affinity enrichment and LC-MS/MS. HEK293T cells were transfected with a plasmid encoding a FLAG-tagged version of SCYL3. Then, 36 h after transfection, lysates from MOCK-transfected or SCYL3-transfected cells were prepared and immunoprecipitated using the M2 affinity matrix. Immune complexes were resolved by SDS-PAGE, and the gel was stained with SYPRO Ruby protein gel stain. Nine bands (1–9) were excised and analyzed by LC-MS/MS. *Indicates bands also found in the MOCK-transfected lane that were not interrogated by MS. Data are representative of two independent experiments. Figure 2-1 presents the number of independent peptides and the complete list of proteins identified. The SCYL3 immune complex contained peptides ascribed to components of the COPI complex, such as COPA, COPB, COPB2, COPG1, COPG2, COPD, CASP, kinesin heavy chain, and heat-shock proteins. B, Validation of SCYL3 association with select putative protein partners. HEK293T cells were transfected with plasmids encoding FLAG-tagged versions of SCYL1, SCYL2, and SCYL3. Then, 36 h after transfection, protein lysates were prepared from SCYL1-, SCYL2-, SCYL3-, or MOCK-transfected cells and immunoprecipitated using the M2 affinity matrix. Cell lysates (L) and immune complexes (IP) were resolved by SDS-PAGE and analyzed by Western blot, using antibodies against CASP, COPG1/2, kinesin heavy chain (KinH), ezrin, and FLAG. C, Several subunits of the COPI complex associate with SCYL3. Indicated amounts of plasmid encoding the FLAG-tagged version of SCYL3 were transfected into HEK293T cells. Then, 36 h after transfection, cell lysates were prepared and immunoprecipitated using the anti-Flag M2 matrix. Immune complexes (IP) and cell lysates (L) were resolved by SDS-PAGE and analyzed by Western blot, using antibodies against COPA, COPB, COPB2, COPG (which recognizes both COPG1 and COPG2), and the FLAG epitope. D, Schematic representation of full-length and truncated versions of SCYL3 used to study SCYL3–COPG, SCYL3–CASP, and SCYL3–SCYL3 interactions. Numbering includes 8 aa of the FLAG epitope (purple). HEAT repeats are labeled 1 through 4. E, Distinct regions of SCYL3 bind to COPG1/2 and CASP. HEK293T cells were transfected with full-length or various FLAG-tagged truncated versions of SCYL3 illustrated in D. Then, 36 h after transfection, protein lysates were prepared from SCYL3- or MOCK-transfected cells and immunoprecipitated using the M2 affinity matrix. Cell lysates and immune complexes were resolved by SDS-PAGE and analyzed by Western blot, using antibodies against COPG1/2 and CASP. Ponceau staining was used to detect FLAG-SCYL3 from immune complexes. F, Homo-oligomerization of SCYL3. HEK293T cells were transfected with full-length or various FLAG-tagged truncated versions of SCYL3 (illustrated in D) together with an HA-tagged full-length version of SCYL3 as indicated. Then, 36 h after transfection, protein lysates from transfected cells were prepared and immunoprecipitated using the M2 affinity matrix (IP). Cell lysates (L) and immune complexes (IP) were resolved by SDS-PAGE and analyzed by Western blot, using antibodies against HA. Ponceau S staining was used to detect FLAG-SCYL3 from immune complexes. G, Schematic representation of SCYL3 domains required for homo-oligomerization, CASP and COPI interactions.
Peptides identified by LC-MS/MS. FLAG-SCYL3 was purified by immunoaffinity using M2 (anti-FLAG)-agarose beads. The immune complex was resolved on SDS-PAGE, and interacting partners were identified by MS (see Fig. 2). Abbreviations: LC-MS/MS, liquid chromatography - tandem mass spectrometry; SC, spectral count; TP, total peptides; GN, gene name; SV: Splice variant; OS; organism; PE, peptide. Download Figure 2-1, XLSX file (23KB, xlsx)
Selectivity of these interactions was determined by coimmunoprecipitation studies using FLAG-tagged versions of SCYL1, SCYL2, and SCYL3 expressed in Hek293T cells. Consistent with results of MS analyses, ectopically expressed SCYL3 interacted with both COPG1 and COPG2 as well as the Golgi-localized CASP (Fig. 2B). However, unlike previous studies (Sullivan et al., 2003), ezrin was not detected in the SCYL3 immune complex (Fig. 2B). Also, there was no strong interaction between SCYL3 and KINH (Fig. 2B). The interaction between SCYL3 and caprin 1 was nonspecific; all immune complexes, including the one from MOCK-transfected cells, contained caprin 1 (data not shown). Further, CASP as well as COPG1 and COPG2 associated specifically with SCYL3 (Fig. 2B), whereas only 1 COPG isoform (possibly COPG2; Hamlin et al., 2014) interacted with SCYL1. Neither COPG isoform nor CASP interacted with SCYL2 (Fig. 2B). COPA, COPB, and COPB2, which are components of the COPI complex, also interacted with SCYL3 (Fig. 2C), suggesting that SCYL3 associated with the whole protein complex.
SCYL3 domains involved in interactions with CASP and COPI were identified by coimmunoprecipitation studies, using FLAG-tagged full-length and truncated versions of SCYL3 expressed in Hek293T cells (Fig. 2D). COPG1 and COPG2 interacted with either the full-length or truncated version of SCYL3-containing residues 301–743 (including 8 residues of the FLAG epitope; Fig. 2E). No other truncated versions of SCYL3 interacted with COPG1 and COPG2 (Fig. 2E). Also, CASP interacted with the full-length version of SCYL3 as well as several truncated versions of SCYL3 but not with truncated versions containing residues 1–436 or 1–260 (Fig. 2E). These results indicate that distinct domains of SCYL3 are required for these interactions: the last 14 residues of SCYL3 are required for interaction with COPI, whereas a region immediately downstream of HEAT repeats is required for CASP binding.
A previous study showed that SCYL1 can form homo-oligomers and that oligomerization requires centrally located HEAT repeats (Hamlin et al., 2014). To test whether SCYL3 can also oligomerize and to identify the domains involved in this process, coimmunoprecipitation studies were performed using FLAG- and HA-tagged versions of SCYL3 expressed in Hek293T cells. SCYL3 formed oligomers, as indicated by presence of the HA-tagged version of SCYL3 in the FLAG–SCYL3 immune complex (Fig. 2F). Also, HA-tagged SCYL3 coimmunoprecipitated with several truncated versions of SCYL3, except for SCYL3-FLAG 301–743 and SCYL3-FLAG 199–552 (Fig. 2F). These results indicate that SCYL3 can form homo-oligomers and that SCYL3 oligomerization requires a region encompassing part of the pseudokinase domain, which intriguingly is also predicted to fold as a HEAT repeat (HEAT repeat number 1; Fig. 2G).
SCYL3 is dispensable for the core function of COPI and cellular migration
The biochemical and cell biological properties described here for SCYL3 suggested that, as proposed for SCYL1, SCYL3 may regulate COPI-mediated retrograde transport along the secretory pathway. To test this, mice deficient for Scyl3 were generated by conventional gene targeting in ES cells (Fig. 3A). Two independent ES cell clones were used to produce chimeras, and chimeras from both clones transmitted the null allele to their progeny. Scyl3+/− mice were viable and fertile and showed no overt abnormalities. Mating of Scyl3+/− mice yielded Scyl3+/+ (WT), Scyl3+/−, and Scyl3−/− mice in the expected 1:2:1 Mendelian ratio (Table 1). PCR genotyping (Fig. 3B), Western blot (Fig. 3C), and immunofluorescence staining in fibroblasts (Fig. 1E) confirmed the disruption of Scyl3. A 310-bp band corresponding to the null allele was detected by PCR amplification in Scyl3+/− and Scyl3−/− mice, and SCYL3 was detected in only WT but not in Scyl3−/− MEFs (Figs. 3C,E). Notably, despite its widespread expression in mouse tissues and potential role in regulating Golgi–ER and intra-Golgi trafficking, Scyl3−/− mice were viable, fertile, and exhibited no noticeable growth or morphologic abnormalities (Fig. 3D; Table 2) except for the presence of some angulated muscle fibers in rectus femoris (Table 2). Because SCYL3 is also strongly expressed in the thymus, lymphocyte development was examined in these mice. Scyl3−/− mice showed no overt differences in T- and B-cell development and homeostasis (data not shown). The lifespan of these mice was not determined, but they lived beyond 400 d without exhibiting overt abnormalities. These findings suggest that SCYL3 is dispensable for normal development of the mouse embryo and postnatal life.
Figure 3.
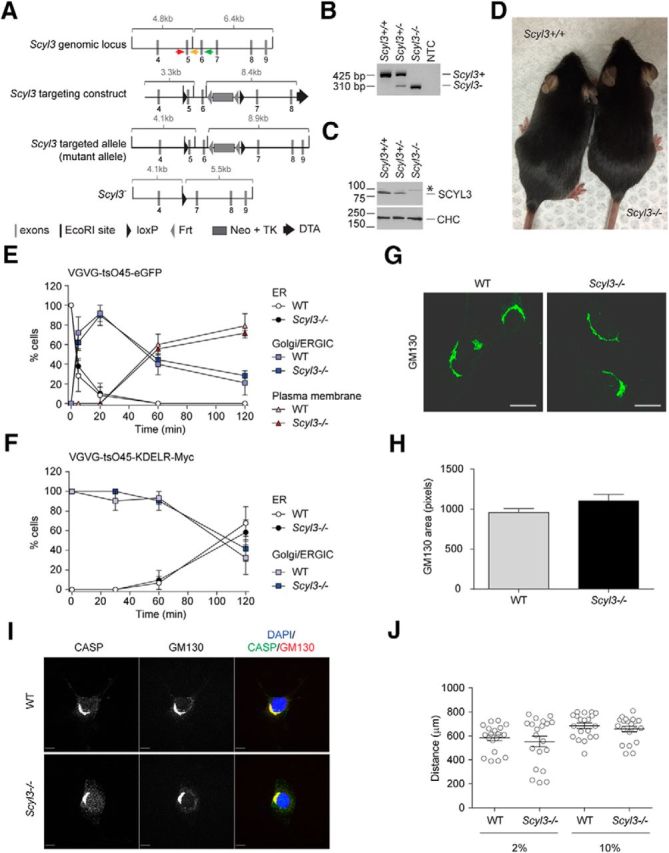
Scyl3 is dispensable for embryonic development and postnatal life, COPI function, Golgi size and morphology, CASP subcellular localization, and cell migration. A, Schematic representation of the Scyl3 locus (introns 3–9), targeting vector, and predicted Scyl3 mutant and null alleles. The Scyl3 gene is located on chromosome 1 and contains 14 exons. Only the region containing exons 4–9 is illustrated. Cre-mediated recombination of the Scyl3 locus to generate the null allele was performed in ES cells. The splicing of exon 4 in exons 7, 8, 9, or 10 causes frame shifts and premature STOP codons. Gray bars, Exons; black bars, EcoRI sites; black triangles, loxP; gray triangles, Frt sites; large gray box, neo-TK cassette; black arrow, diphtheria toxin cassette; red arrow, genotyping primer S3F01; orange arrow, genotyping primer S3R02; green arrow, genotyping primer S3R51. B, PCR-based genotyping of Scyl3+/+, Scyl3+/−, and Scyl3−/− mice. Genomic DNA from Scyl3+/+, Scyl3+/−, and Scyl3−/− mice was amplified by PCR, using primers S3F01, S3R51, and S3R02. Bands of 521 bp and 310 bp correspond to the WT (Scyl3+) and null allele (Scyl3−), respectively. NTC, no template control. C, SCYL3 expression in WT (Scyl3+/+), Scyl3+/−, and Scyl3−/− MEFs. Protein lysates were prepared from exponentially growing MEFs, resolved by SDS-PAGE, and analyzed by Western blot, using antibodies against SCYL3 or clathrin heavy chain (CHC) as loading control. D, Photograph of WT and Scyl−/− mice. Scyl3−/− mice were viable, fertile, and showed no overt abnormalities. E, VSVG-tsO45-eGFP forward movement in WT and Scyl3−/− MEFs. MEFs were transfected with VSVG-tsO45-eGFP. Then, 24 h after transfection, cells were incubated at 40°C for 12 h and then transferred to 32°C for various time points (0, 5, 20, 60, and 120 min), fixed and stained with GFP antibody, and imaged with a fluorescent microscope. The number of cells showing predominant ER, Golgi/ERGIC, or plasma membrane staining was determined and expressed as percentage of total number of cells analyzed for each independently derived cell line (WT, n = 20–25 cells per time point from 3 cell lines; Scyl3−/−, n = 20–25 cells per time point from 3 cell lines). F, VSVG-tsO45-KDELR-Myc retrograde movement in WT and Scyl3−/− MEFs. MEFs were transfected with VSVG-tsO45-KDELR-Myc and incubated at 32°C. Cells were then transferred to the nonpermissive temperature (40°C) for various time points (0, 30, 60, and 120 min) and fixed and stained with an anti-Myc antibody. The number of cells showing predominant ER or Golgi/ERGIC staining was determined and expressed as percentage of total number of cells analyzed for each independently derived cell line (WT, n = 20–25 cells per time point from 3 cell lines; Scyl3−/−, n = 20–25 cells per time point from 3 cell lines). G, H, Golgi size and morphology in WT and Scyl3-deficieint MEFs. G, Exponentially growing WT and Scyl3−/− MEFs were fixed and stained for CASP and GM130 and imaged by confocal microscopy. H, GM130-positive area (pixels) was determined in WT (n = 162 cells from 3 independently derived cell lines) and Scyl−/− MEFs (n = 142 cells from 3 independently derived cell lines). Data are expressed as the mean ± SEM. No significant differences in size or overall morphology were found between WT and Scyl3−/− MEFs. I, Absence of SCYL3 does not affect CASP localization to the Golgi apparatus. Confocal microscopy of exponentially growing WT and Scyl3−/− MEFs, using antibodies against CASP and GM130. Blue, DAPI staining; green, CASP; red, GM130. J, Migration of WT and Scyl3-deficient MEFs. The distance traveled by each cell line (in quintuplicate) over 24 h was measured. Data are expressed as the mean ± SEM of 15 measurements from three independently derived WT and Scyl3−/− MEFs cell lines. Scale bars, 10 μm
Table 1.
Mendelian distribution of genotypes obtained from the Scyl3+/− intercross*
| Scyl3+/+, N (%) | Scyl3+/+, N (%) | Scyl3−/−, N (%) | |
|---|---|---|---|
| Observed | 56 (23.9) | 127 (54.3) | 51 (21.8) |
| Expected | 58.5 (25) | 117 (50) | 58.5 (25) |
Mice were analyzed at weaning.
Table 2.
Histopathologic examination of Scyl3−/− mice*
| Organ | Histopathologic findings |
|---|---|
| Bone marrow | Normal |
| Cerebrum | Normal |
| Fat tissue | Normal |
| Heart | Normal |
| Kidney | Normal |
| Liver | Normal |
| Lung | Normal |
| Pancreas | Normal |
| Spinal cord | Normal |
| Spleen | Normal |
| Skeletal muscle | Sparse red angulated fibers |
| Testis | Normal |
| Thymus | Normal |
| Urinary bladder | Normal |
Three 8-week-old Scyl3−/− males were analyzed.
The absence of overt phenotypes in Scyl3-deficient mice was in stark contrast to studies showing that early embryonic lethality is associated with loss of COPI function in several model organisms, including mice (Faulstich et al., 1996; Coutinho et al., 2004; Hamamichi et al., 2008; Jayaram et al., 2008). This observation suggested that although SCYL3 can interact with components of the COPI complex, the pseudokinase may be dispensable for function of the coatomer. To test whether the absence of SCYL3 affected COPI-mediated function along the secretory pathway, retrograde transport of the VSVG-tsO45-KDELR-Myc chimera from the Golgi/ER-to-Golgi intermediate compartment (ERGIC) back to the ER was examined in WT and Scyl−/− MEFs. Because COPI also functions in forward transport within the Golgi apparatus (Szul and Sztul, 2011), forward movement of another chimeric protein VSVG-tsO45-eGFP from the ER to plasma membrane was also studied. Consistent with the lack of overt phenotypic changes in the absence of SCYL3 in vivo, both forward and reverse transport of chimeric proteins proceeded normally in Scyl3−/− MEFs (Fig. 3E,F).
Because COPI also regulates size and morphology of the Golgi apparatus (Guo et al., 1996, 2008; Presley et al., 2002), whether the absence of SCYL3 affected the size and/or morphology of the Golgi apparatus was tested by staining WT and Scyl3−/− MEFs for the Golgi marker GM130. Size and overall morphology of the Golgi apparatus were similar between WT and Scyl3−/− MEFs (Fig. 3G,H).
By virtue of its interaction with ezrin, which mediates interactions between the cytoplasmic tail of transmembrane proteins and the actin cytoskeleton (Neisch and Fehon, 2011), it was originally proposed that SCYL3 regulates cellular adhesion and/or migration (Sullivan et al., 2003). To determine whether SCYL3 regulates cellular migration in vitro, in vitro migration assays were performed using WT and Scyl3-deficient MEFs in the presence of low or high serum concentrations. Both WT and Scyl3-deficient MEFs traveled similar distances in the presence of low or high serum concentrations (Fig. 3J). These results indicate that SCYL3 is dispensable for cellular migration in vitro, which is consistent with the absence of an obvious phenotype in Scyl3-deficient mice.
Altogether, our findings indicate that although SCYL3 interacts with components of the COPI complex and CASP, it is dispensable for core functions of COPI and for localization of CASP along the secretory pathway. Also consistent with the absence of an essential role for SCYL3 in regulating COPI function, we found that unlike inhibition of COPI function in cultured cells which cause growth inhibition (Guo et al., 1996), Scyl3-deficient fibroblasts proliferated normally (data not shown). Moreover, we found no evidence that SCYL3 regulates cellular migration both in vitro and in vivo.
COPI and CASP interactions contribute to proper localization of SCYL3 along the secretory pathway
Because SCYL3 did not regulate COPI function and CASP localization, we tested whether COPI and/or CASP regulated the localization of SCYL3 along the secretory pathway. To determine whether CASP regulated the localization of SCYL3 along the secretory pathway, SV40-transformed MEFs lacking CASP were generated by clustered regularly interspaced short palindromic repeat (CRISPR) and CRISPR-associated protein 9 (Cas9) technology. CASP is encoded by the Cut-like homeobox 1 (Cux1) gene, which also encodes the CCAAT-displacement protein (CDP; also known as Cux1). The mRNA transcripts of CASP and CDP share the first 14 exons of the gene but then diverge to encode CDP (Cux1) and CASP (Fig. 4A). To inactivate CASP, 2 sgRNAs targeting introns 14 and 17 of CASP (Fig. 4A) were used to generate cells in which CASP exons 15–17 were excised. PCR amplification (Fig. 4B) and sequencing of amplicons (data not shown) confirmed accurate deletion of CASP exons 15–17 in selected SV40-transformed MEFs clones. To confirm that deletion of exons 15–17 resulted in gene inactivation, Western blot analyses and immunofluorescence staining using antibodies against CASP and GM130 were performed. Deletion of exons 15–17 of CASP eliminated CASP expression (Fig. 4C,D). Also, deletion of CASP did not affect the distribution of GM130 (Fig. 4D). To test whether CASP regulated the subcellular localization of SCYL3, immunofluorescence staining in both WT and CASP-KO cell lines was performed using antibodies against SCYL3 and GM130. SCYL3 localized to the Golgi apparatus in both WT and CASP-KO cell lines, suggesting that CASP is dispensable for the normal distribution of SCYL3 in cultured cells (Fig. 4E).
To test the biological significance of the SCYL3–COPI interaction, subcellular distribution of SCYL3 was compared with that of a mutant form of SCYL3 that could not interact with the COPI complex (SCYL3-1-721). Because SCYL3 can homo-oligomerize (Fig. 2F,G), these experiments were performed in Scyl3-deficient MEFs to avoid interaction between exogenous and endogenous SCYL3. Both WT and mutant forms of SCYL3 localized to the Golgi apparatus (Fig. 4F), indicating that the SCYL3–COPI interaction was dispensable for proper localization of SCYL3 to the Golgi apparatus. Interestingly, however, a mutant form of SCYL3 that could not interact with CASP and COPI (SCYL-1-428) did not localize to the Golgi apparatus in Scyl3-deficient MEFs (Fig. 4F). These observations support that both CASP and COPI interactions are required for proper localization of SCYL3 along the secretory pathway.
SCYL1 and SCYL3 are dispensable for COPI function but play overlapping role in maintaining motor neuron viability
Previous studies as well as our own unpublished data and results presented in Figure 2 indicated shared properties between SCYL1 and SCYL3 (Di et al., 2003; Burman et al., 2008, 2010; Hamlin et al., 2014). Like SCYL3, SCYL1 (1) interacts with components of the COPI complex, (2) interacts with the Golgi-associated golgin GORAB, and (3) localizes with COPI-positive vesicles at the ERGIC and the Golgi apparatus. However, although knockdown and overexpression experiments in HeLa cells supported a role for SCYL1 in regulating Golgi morphology, size, and function by regulating COPI-mediated retrograde transport (Burman et al., 2008, 2010; Hamlin et al., 2014), we found no evidence supporting a role for SCYL1 in regulating these functions in MEFs (Pelletier et al., 2012). Moreover, unlike targeted deletion of various components of the COPI complex in mice and other model organisms, which causes early embryonic lethality (Faulstich et al., 1996; Coutinho et al., 2004; Hamamichi et al., 2008; Jayaram et al., 2008), loss of SCYL1 function in mice does not result in embryonic lethality (Blot et al., 1995; Schmidt et al., 2007; Pelletier et al., 2012) suggesting that SCYL1 is also dispensable for the core function of COPI in vivo. At that time, we speculated that another member of the SCY1-like family of proteins may compensate for the absence of SCYL1 (Pelletier et al., 2012). The biochemical and cell biological properties of SCYL3, unlike those of SCYL2 (Gingras et al., 2015; Pelletier, 2016), strongly suggested that SCYL3 plays this role. To test whether SCYL1 and SCYL3 played overlapping or redundant function(s) in vivo, mice deficient for both Scyl1 and Scyl3 were generated. Targeted disruption of both genes in mice did not cause embryonic lethality, and Scyl1−/−;Scyl3−/− mice were born at normal Mendelian ratios at weaning (Table 3), suggesting that SCYL1 and SCYL3 paly at most an accessory role, if any, in regulating COPI function in vivo.
Table 3.
Genotypes of mice (n = 187) from Scyl1+/−;Scyl3+/− intercross*
| Scyl3+/+ | Scyl3+/− | Scyl3−/− | |
|---|---|---|---|
| Scyl1+/+ | 17 | 33 | 9 |
| Scyl1+/− | 21 | 44 | 17 |
| Scyl1−/− | 12 | 21 | 13 |
Mouse genotypes were analyzed at weaning.
Importantly, however, we found that loss of SCYL3 accelerated the onset of the motor phenotype and growth defects associated with the absence of SCYL1 (Fig. 5; Movies 1–4). At birth, Scyl1−/−;Scyl3−/− mice were indistinguishable from WT, Scyl1−/−, and Scyl3−/− littermates. Similar to Scyl1−/− mice, Scyl1−/−;Scyl3−/− mice exhibited growth retardation that was visible by 3 weeks of age and continued throughout their lifetime. Four-week-old Scyl1−/−;Scyl3−/− male mice were 42% smaller than their WT or Scyl3−/− male littermates and 18% smaller than Scyl1−/− male mice (Fig. 5A,B). At 8 weeks of age, weights of Scyl1−/−;Scyl3−/− male mice were 24% lower than those for Scyl1−/− littermates.
Figure 5.

Scyl3 deficiency exacerbates growth and motor phenotypes associated with the absence of Scyl1. A, Representative photograph of 8-week-old (wo) Scyl1−/− and Scyl1−/−;Scyl3−/− male littermates showing size differences between these genotypes. B, Body weight of 4- and 8-week-old WT (4-week-old mice, n = 22; 8-week-old mice, n = 13), Scyl1−/− (4-week-old mice, n = 22; 8-week-old mice, n = 15), Scyl3−/− (4-week-old mice, n = 12; 8-week-old mice, n = 8), and Scyl1−/−;Scyl3−/− (4-week-old mice, n = 8; 8-week-old mice, n = 3) males. Data are expressed as mean ± SEM. P values, determined by one-tailed Student's t test, are indicated on the graph. C, Disease progression in 4- and 8-week-old WT (4-week-old mice, n = 11; 8-week-old mice, n = 11), Scyl1−/− (4-week-old mice, n = 17; 8-week-old mice, n = 14), Scyl3−/− (4-week-old mice, n = 12; 8-week-old mice, n = 4), and Scyl1−/−;Scyl3−/− (4-week-old mice, n = 13; 8-week-old mice, n = 3) mice. Disease progression in mice was assessed by using an objective grading system as described in Materials and Methods. Data are expressed as mean ± SEM. P values, determined by the one-tailed Student's t test, are indicated on the graph. D, Motor defects in 4- and 8-week-old WT (4-week-old mice, n = 10; 8-week-old mice, n = 12), Scyl1−/− (4-week-old mice, n = 29; 8-week-old mice, n = 12), Scyl3−/− (4-week-old mice, n = 16; 8-week-old mice, n = 12) and Scyl1−/−;Scyl3−/− mice (4-week-old mice, n = 9; 8-week-old mice, n = 3). The inverted grid test was performed on 4- and 8-week-old mice as described in Materials and Methods. Data are expressed as mean ± SEM. P values, determined by the one-tailed Student's t test, are indicated on the graph.
Movie 1.
The movie shows the normal gait of a 6-week-old WT mouse.
Movie 2.
The movie shows the abnormal gait of a 6-week-old Scyl1−/− mouse.
Movie 3.
The movie shows a 6-week-old Scyl1−/−;Scyl3−/− mouse moving by pulling its body forward using its forelimbs. Note the flattening of the pelvis and stiffness and complete paralysis of the hindlimbs.
Movie 4.
The movie shows a 13-week-old Scyl1−/− mouse moving by pulling its body forward using its forelimbs. Note the flattening of the pelvis and stiffness and complete paralysis of the hindlimbs.
Scyl1−/− and Scyl1−/−;Scyl3−/− mice also developed progressive motor dysfunction leading to a paralytic state (Movies 1–4). Scyl1−/−;Scyl3−/− mice consistently looked worse than Scyl1−/− mice at a same age. Eventually, Scyl1-deficient mice catch up with Scyl1−/−;Scyl3−/− mice and become paralyzed (Movie 4). Using a previously established scoring system (see Materials and Methods), disease progression in Scyl1−/− and Scyl1−/−;Scyl3−/− mice was followed in a blinded manner. By 4 weeks of age, there were significant differences (p = 0.0321) between scoring averages of Scyl1−/− and Scyl1−/−;Scyl3−/− mice (Fig. 5C). Similarly, in 8-week-old animals, there was a significant (p = 0.0399) but smaller difference between both groups (Fig. 5C). In the mesh grip test, to assess motor function and muscle strength, Scyl1−/−;Scyl3−/− mice remained suspended on the inverted cage for a significantly shorter time period than did Scyl1−/− littermates (p = 0.0302) at 4 but not 8 weeks of age (Fig. 5D). That is, 4-week-old control and Scyl3−/− mice remained suspended for a mean time of >120 s, whereas Scyl1−/− and Scyl1−/−;Scyl3−/− mice remained suspended for a mean ± SEM of only 20.2 ± 5.3 and 0.4 ± 0.2 s, respectively. At 8 weeks of age, WT and Scyl3−/− mice still remained suspended for >120 s, whereas Scyl1−/− and Scyl1−/−;Scyl3−/− mice could remain suspended only for a fraction of a second (Fig. 5D). Together, these results indicate that absence of SCYL3 accelerated the onset of the motor phenotype associated with Scyl1 deletion. Onset of disease in Scyl1−/−;Scyl3−/− mice coincided with the growth phenotype.
As shown previously (Pelletier et al., 2012) and illustrated here, Scyl1 deletion causes multifocal neurogenic atrophy of some muscle groups, with the rectus femoris and bicep brachii being the most severely affected (Fig. 6). Because Scyl3 deletion worsened the motoric phenotype of Scyl1−/− mice (Fig. 6; Movies 1–3), we studied whether changes were associated with increased deterioration of muscle fibers in both rectus femoris (Fig. 6A–E) and bicep brachii (Fig. 6F–J), peripheral nerves (sciatic nerves; Fig. 7), and spinal motor neurons (Fig. 8). Although the total number of muscle fibers in rectus femoris and bicep brachii did not significantly differ between genotypes and age groups, changes in the size and shape of muscles and muscle fibers were observed in Scyl1−/− and Scyl1−/−;Scyl3−/− mice (Fig. 6). Mean CSA of rectus femoris and bicep brachii was reduced by 50–70% in 4- and 8-week-old animals Scyl1−/− and Scyl1−/−;Scyl3−/− compared with WT mice (Fig. 6B,G). The reduction in muscle CSA was accompanied by a 40–75% reduction in mean fiber CSA of both muscles in 4- and 8-week-old Scyl1−/− and Scyl1−/−;Scyl3−/− compared with WT mice. Significant reduction in the mean fiber CSA was also observed in both rectus femoris and bicep brachii of 4-week-old Scyl1−/−;Scyl3−/− mice compared with 4-week-old Scyl1−/− mice; a change that correlated with an increase number of angulated/atrophied muscle fiber, a characteristic feature of muscles that lose their innervations (Fig. 6A,F,E,J). Together our results indicate that absence Scyl1 in mice causes muscle pathology reminiscent of denervation atrophy and loss of Scyl3 accelerated the onset of the muscle phenotype.
Figure 6.

Myopathologic abnormalities in Scyl1−/− and Scyl1−/−;Scyl3−/− mice. A, Representative micrographs of H&E-stained sections of rectus femoris from 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice. B, Muscle cross-sectional area of rectus femoris of 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice (n = 3 for each genotype and age group). C, Total number of muscle fibers in rectus femoris of 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice (n = 3 for each genotype and age group). D, Muscle fiber cross-sectional area of rectus femoris of 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice (n = 3 for each genotype and age group). E, Percentage of angulated/atrophied fibers in rectus femoris of 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice (n = 3 for each genotype and age group). F, Representative micrographs of H&E-stained sections of bicep brachii from 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice. G, Muscle cross-sectional area of bicep brachii of 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice (n = 3 for each genotype and age group). H, Total number of muscle fibers in bicep brachii of 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice (n = 3 for each genotype and age group). I, Muscle fiber cross-sectional area of bicep brachii of 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice (n = 3 for each genotype and age group). J, Percentage of angulated/atrophied fibers in bicep brachii of 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice (n = 3 for each genotype and age group). Data are expressed as mean ± SEM. P values determined by two-tailed Student's t test. a, WT versus Scyl1−/−, p < 0.05; b, WT versus Scyl1−/−; Scyl3−/−, p < 0.05; c, Scyl1−/− versus Scyl1−/−;Scyl3−/−, p < 0.05; d, WT versus Scyl3−/−, p < 0.05.
Figure 7.

Loss of large-caliber axons in Scyl1−/− and Scyl1−/−;Scyl3−/− mice. A, Representative micrograph of toluidine blue-stained semithin sections of sciatic nerve from 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice. B, Myelinated axon counts in sciatic nerve of 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice (n = 3 for each age group and genotype). Data are expressed as mean ± SEM. P values, determined by the one-tailed Student's t test, are indicated on the graph. C, Histogram of the frequency of myelinated axons in sciatic nerves of 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice (n = 3 for each age group and genotype) as a function of axon diameter. Data are expressed as mean ± SEM. P values determined by two-tailed Student's t test. a, WT versus Scyl1−/−, p < 0.05; b, WT versus Scyl1−/−; Scyl3−/−, p < 0.05; c, Scyl1−/− versus Scyl1−/−;Scyl3−/−, p < 0.05.
Figure 8.

Loss of large motor neurons in the ventral horn of Scyl1−/− and Scyl1−/−;Scyl3−/− mice. A, Representative micrographs of H&E-stained lumbar ventral horn motor neurons in 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice. Arrows indicate healthy motor neurons. White triangles indicate motor neurons exhibiting rarefaction of cytosolic organelle. Black circles indicate motor neurons exhibiting central chromatolysis. B, Higher-magnification micrograph of healthy motor neurons (top left) and motor neurons exhibiting central chromatolysis (bottom left) or rarefaction of cytosolic organelles (right). Glial cells are in close contact with two degenerative motor neurons (arrowheads). Arrows indicate healthy motor neurons. White triangles indicate motor neurons exhibiting rarefaction of cytosolic organelle. Black circles indicate motor neurons exhibiting central chromatolysis. Black arrowheads show microglial cells surrounding and ingesting degenerated neurons (neuronophagia). C, Numbers of ventral horn motor neuron in 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice (n = 3 for each age group and genotype). The number of healthy motor neurons was determined as described in Materials and Methods. Data are expressed as mean ± SEM. P values, determined by one-tailed Student's t test, are indicated on the graph. D, Quantification of lumbar ventral horn motor neurons showing central chromatolysis in WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice (n = 3 for each age group and genotype). Data are expressed as mean ± SEM. P values, determined by one-tailed Student's t test, are indicated on the graph. E, Quantification of motor neurons with rarefaction of cytosolic organelles in the ventral horn of 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice (n = 3 for each age group and genotype). Data are expressed as mean ± SEM. P values, determined by one-tailed Student's t test, are indicated on the graph. F, G, Neuroinflammation in the spinal ventral horn of Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice. Immunohistochemistry using antibodies against GFAP (F) and Iba1 (G) on spinal ventral horn sections of 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice. Data are expressed as mean ± SEM. P values determined by two-tailed Student's t test. a, WT versus Scyl1−/−, p < 0.05; b, WT versus Scyl1−/−; Scyl3−/−, p < 0.05; c, Scyl1−/− versus Scyl1−/−;Scyl3−/−, p < 0.05; d, WT versus Scyl3−/−, p < 0.05.
Because muscles of Scyl1−/−,and Scyl1−/−;Scyl3−/− mice presented features characteristic of denervation atrophy, we assessed whether combined deficiencies also accelerated the onset of peripheral neuropathy associated with the absence of Scyl1 (Pelletier et al., 2012). Sciatic nerves of 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice were examined by light microscopy. As shown in Figure 7, the number and size distribution of myelinated axons were similar between 4-week-old WT, Scyl3−/− and Scyl1−/− mice (Fig. 7A–C). In contrast, the number of large caliber axons (4–5 μm or >5 μm) and total number of myelinated fibers were already significantly reduced in Scyl1−/−;Scyl3−/− mice compared with WT, Scyl1−/− and Scyl3−/− mice. The number and size distribution of myelinated axons in 8-week-old Scyl1−/− mice were significantly reduced compared with 8-week-old WT mice but no significant differences were found between 8-week-old Scyl1−/− and Scyl1−/−;Scyl3−/− mice.
Next, we assessed whether changes observed in peripheral nerves also translated to the ventral horn of the lumbar spinal cord. Spinal cord sections from 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice were stained with H&E and examined by light microscopy. In 4-week-old mice, the number of large motor neurons in the ventral horn of the spinal cord was 62% lower in Scyl1−/−;Scyl3−/− mice than in Scyl1−/− mice (Fig. 8A,C). By 8 weeks of age, the number of large motor neurons in the spinal ventral horn was similar in Scyl1−/− and Scyl1−/−; Scyl3−/− mice but lower than in WT and Scyl3−/− mice (Fig. 8A,C). There was also an increase in the number of cells exhibiting central chromatolysis or rarefaction of cytosolic organelles (Fig. 8B,D,E). At 4 weeks of age, numbers of chromatolytic cells (Fig. 8D) and cells with rarefaction of cytosolic organelles were higher in Scyl1−/−;Scyl3−/− mice compared with WT or Scyl1−/− mice (Fig. 8E). At 8 weeks of age, number of chromatolytic neurons and cells with rarefaction of cytosolic organelles in Scyl1−/− and Scyl1−/−;Scyl3−/− mice were similar (Fig. 8E). We also observed neuronophagia in the ventral horn of 8-week-old Scyl1−/− and Scyl1−/−;Scyl3−/− mice (Fig. 8B, arrowheads). Together, our results indicate that absence Scyl1 in mice causes spinal motor neuron degeneration and loss of large caliber axons in peripheral nerves and that combined deficiencies accelerated disease onset.
We have previously shown that loss of large motor neurons in Scyl1-deficient mice is accompanied by inflammation (Pelletier et al., 2012). In the spinal cord of Scyl1−/− and Scyl1−/−;Scyl3−/− mice, immunoreactivity for both GFAP and IBA1 was conspicuous (Fig. 8F,G).The number of GFAP-positive and Iba1-positive cells in the spinal ventral horn of Scyl1−/− was increased 5.0- and 4.1-fold respectively. In Scyl1−/−;Scyl3−/− mice, the number of GFAP+ and IbaI+ cells were increased 7.2- and 4.5-fold, respectively. Interestingly, GFAP immunoreactivity in the ventral horn of Scyl3−/− mice was slightly greater than that in WT mice, suggesting that Scyl3−/− mice might be more prone to inflammatory responses.
Because SCYL1 and SCYL3 are expressed in the nervous system, we also examined the overall architecture of the brain in these mice. Aside from a small reduction in the size of the cerebrum and cerebellum, which are likely due to the growth defect associated with the loss of Scyl1 and Scyl3, there was no brain region-specific atrophy. Similarly, aside from the selective loss of large caliber axons and ventral horn motor neurons, there were no obvious defects in regions other than the ventral horn of the spinal cord, including dorsal root ganglia (data not shown). Together, these findings indicate that absence of SCYL1 or combined deficiencies causes a motor neuron selective degenerative disorder in mice.
TDP-43 pathology in spinal motor neurons of Scyl1−/− and Scyl1−/−;Scyl3−/− mice
Histopathological examination of Scyl1−/− and Scyl1−/−;Scyl3−/− mice indicated that both SCYL1 and SCYL3 contribute to maintaining motor neuron viability. We previously showed that loss of SCYL1 function in mice caused an early onset motor neuron disorder characterized by cytoplasmic accumulation of TDP-43 in large motor neurons of the ventral horn. We also showed that SCYL1 is predominantly expressed in large motor neurons of the spinal cord and acts in a neural-cell autonomous manner to maintain motor neuron viability (Pelletier et al., 2012). Neuron-specific deletion of SCYL1 in mice recapitulated the phenotypic changes found in Scyl1-deficient mice (Pelletier et al., 2012). Although SCYL1 is expressed to some level in muscles, muscle-specific deletion of Scyl1 did not produce any obvious phenotype (Pelletier et al., 2012). To gain insights into the disease associated with the loss of Scyl1 and Scyl3, we examined the distribution of SCYL1 and SCYL3 in the brain, spinal cord, and skeletal muscles of WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice (Fig. 9A). We found that whereas SCYL1 is expressed in the brain, spinal cord, and to a lesser extent in skeletal muscles, SCYL3 is uniquely expressed in the brain and spinal cord (Fig. 9B). No detectable expression of SCYL3 was found in skeletal muscles. Immunohistochemistry further revealed that like SCYL1, which is predominantly expressed in large lower motor neurons (Fig. 9B), SCYL3 is also expressed in spinal motor neurons, a pattern that was absent in spinal cord sections from Scyl3-deficient mice (Fig. 9B). The overlapping expression pattern of SCYL1 and SCYL3 in large motor neuron suggest that both proteins may contribute to spinal motor neuron viability in cell autonomous manner.
Figure 9.

Nuclear-to-cytoplasmic redistribution of TDP-43 in spinal motor neurons of Scyl1−/− and Scyl1−/−;Scyl3−/− mice. A, Expression of SCYL1 and SCYL3 in brain and spinal cord. Tissue distribution of SCYL proteins in WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice. Protein extracts prepared from various mouse tissues were resolved by SDS-PAGE and analyzed by Western blot, using antibodies against SCYL1, SCYL3, β-actin. Ponceau staining of the membrane is also shown as loading control. *Indicates nonspecific band. B, Overlapping expression of SCYL1 and SCYL1 in spinal motor neurons. Immunohistochemical staining of spinal cord cross-sections from WT, Scyl1−/− and Scyl3−/− mice, using antibodies against SCYL1 or SCYL3. Arrows indicate expression of SCYL1 or SCYL3 in spinal motor neurons. C, Immunohistochemistry using antibodies against TDP-43 on lumbar spinal ventral horn sections from 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice. Arrows indicate spinal motor neurons with relocalized TDP-43. D, Quantification of cells exhibiting TDP-43 pathology in ventral horn of 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice (n = 3 for each age group and genotype). Data are expressed as mean ± SEM. P values, determined by the one-tailed Student's t test, are indicated on the graph. a, WT versus Scyl1−/−, p < 0.05; b, WT versus Scyl1−/−; Scyl3−/−, p < 0.05; c, Scyl1−/− versus Scyl1−/−;Scyl3−/−, p < 0.05.
To gain mechanistic insights into the degenerative process initiated by the loss of SCYL1 and SCYL3, we examined spinal ventral horn motor neurons for changes in the distribution of motor neuron diseases-associated RNA binding proteins. We have previously shown that loss of SCYL1 function results in aberrant accumulation of the TDP-43 into cytosolic aggregates within spinal motor neurons (Pelletier et al., 2012). To test whether loss of SCYL3 function also influenced TDP-43 proteostasis and whether combined deficiencies resulted in an increased number of motor neurons with TDP-43 pathology, immunohistochemical analyses were performed on the spinal ventral horn from 4- and 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice. As shown previously (Pelletier et al., 2012), disruption of Scyl1 caused redistribution of TDP-43 from the nucleus to cytosolic granules in 4- and 8-week-old mice (Fig. 9C,D). Interestingly, disruption Scyl3 exacerbated this phenotype in 4- but not 8-week-old mice which correlated with earlier disease onset in Scyl1−/−;Scyl3−/− mice (Fig. 9C,D). In addition to TDP-43, other RBPs (e.g., FUS/TLS and hnRNPA2/B1) have been associated with motor neuron diseases (Corrado et al., 2009; Kim et al., 2013). To test whether disease onset in Scyl1−/−;Scyl3−/− mice was associated solely with changes in TDP-43 proteostasis or due to a combination of proteostatic defects in additional ALS-associated RBPs, immunohistochemical staining using antibodies against FUS/TLS and hnRNPA2/B1 was performed in the spinal ventral horn of 8-week-old WT, Scyl1−/−, Scyl3−/−, and Scyl1−/−;Scyl3−/− mice. The localization of FUS/TLS or hnRNPA2/B1 in Scyl1−/− or Scyl1−/−;Scyl3−/− did not change (data not shown). Together, these findings demonstrate that disease onset in Scyl1−/−;Scyl3−/− mice is uniquely associated with changes in TDP-43 proteostasis. Our findings also suggest a link between SCYL proteins and the regulation of TDP-43 proteostasis in vivo.
Discussion
Using a combination of cell biological, biochemical, and genetic approaches, we show that SCYL1 and SCYL3 share biochemical and cell biological properties and play overlapping function in vivo. The overlapping role of SCYL1 and SCYL3 in maintaining motor neuron viability in vivo is supported by their similar localization along the secretory pathway, their similar molecular interactions, their overlapping expression in spinal motor neurons, and the worsening, in Scyl1−/−; Scyl3−/− mice, of the motor phenotype caused by the loss of Scyl1. Although loss of SCYL3 function in mice did not produce any overt abnormalities, loss of SCYL3 accelerated the onset of the motor phenotype caused by Scyl1 disruption. Onset and progression of motor dysfunction leading to a paralytic state in Scyl1−/− and Scyl1−/−;Scyl3−/− mice correlated with the loss of muscle mass, muscle fiber atrophy, loss of large caliber axons in peripheral nerves, and loss of spinal motor neurons and occurred at a younger age in Scyl1−/−;Scyl3−/− compared with Scyl1−/− mice.
The phenotypic changes described here are consistent with myopathies of neurogenic origin similar to those described in mouse models of spinal muscular atrophy (SMA; Cifuentes-Diaz et al., 2002; Monani et al., 2003). In one of these models, which involves the A2G missense mutation in the Smn gene, a ∼30% reduction of spinal motor neurons was associated with muscle atrophy, muscle weakness, and reduced activity (Monani et al., 2003). In a second SMA model, carrying a homozygous deletion of Smn exon 7 directed specifically to neurons, a ∼30% reduction in the number of spinal motor neurons was also associated with muscle denervation and progressive loss of motor function leading to a paralytic state (Cifuentes-Diaz et al., 2002). Here we show that an ∼80% reduction in the number of spinal motor neurons resulting from the loss of Scyl1 or the combined losses of Scyl1 and Scyl3 correlated with the loss of muscle mass, muscle fiber atrophy, and paralysis. Interestingly, both disorders result from the loss of ubiquitously expressed proteins thought to regulate essential cellular functions (Monani, 2005).
The mechanisms underlying motor neuron death in Scyl-deficient mice is unclear. As shown here, SCYL1 and SCYL3 are widely expressed in mammalian tissues and both proteins are proposed to play essential “housekeeping” cellular functions. However, whether these functions are truly regulated by SCYL1 and/or SCYL3 and whether they contribute to maintaining motor neuron viability in vivo remain to be determined. Because of its localization along the secretory pathway, in particular at the Golgi and ERGIC, and its interaction with components of the COPI complex, COPI-associated factors, and Golgi-associated proteins, SCYL1 is proposed to regulate the size and morphology of the Golgi apparatus via the regulation of Golgi- and ERGIC-to-ER retrograde transport (Burman et al., 2008, 2010; Hamlin et al., 2014; Pelletier, 2016). Another essential cellular function thought to be regulated by SCYL1 is the nucleocytoplasmic shuttling of tRNA molecules. SCYL1 was shown to interact with components of the nuclear pore complex including Nup98 and Nup107; the nuclear tRNA export receptors XPOT and XPO5; as well as the small GTPase Ran; and facilitate tRNA export from the nucleus (McGuire and Mangroo, 2007; Chafe and Mangroo, 2010). In addition to regulating intracellular trafficking, SCYL1 was shown to regulate turnover of the RE-1 silencing transcription factor; a transcriptional repressor whose degradation is essential for expression of genes that are critical to the development nervous system (Karlin et al., 2014). In the same vein, SCYL3 was originally identified in a yeast 2-hybrid screen for proteins interacting with the C-terminus of ezrin and this interaction suggested a role for SCYL3 in regulating cell adhesion complexes and migration (Sullivan et al., 2003). However, we found no evidence of the SCYL3–ezrin interaction or that SCYL3 regulates cell migration in vivo or in vitro. Cell migration defects in mice lead to severe embryonic malformations and result in early embryonic lethality or birth defects (Kurosaka and Kashina, 2008). Ezrin-deficient mice did not survive for long (Saotome et al., 2004), whereas Scyl3-deficient mice lived beyond 400 d without showing overt abnormalities. Knockdown studies did not show an essential, nonredundant role for SCYL1 in tRNA shuttling (McGuire and Mangroo, 2007; Chafe and Mangroo, 2010) and another independent study could also not validate these findings (Enkler, 2014). Moreover, findings in our laboratory revealed that SCYL1 does not regulate REST turnover (Gingras et al., 2017) and preliminary studies using genetically modified MEFs revealed that SCYL1 and SCYL3 are dispensable for REST turnover, indicating that neurodegenerative processes engaged by SCYL1 and SCYL3 loss do not involve deregulation of REST turnover. Finally, although the interaction of SCYL1 with components of the COPI complex is established, its role in regulating size and morphology of the Golgi apparatus through COPI-mediated retrograde transport is disputed (Pelletier et al., 2012; Pelletier, 2016). SCYL1 did not regulate retrograde transport of VSVG-KDELR-c-Myc, Golgi size or morphology in MEFs (Pelletier et al., 2012). Although we provide evidence that like SCYL1, SCYL3 self-assembles and interacts with the golgin CASP as well as components of the COPI complex, we found that SCYL3 does not regulate COPI function in vitro and in vivo. Instead, we found that these interactions are required for the proper localization of SCYL3 along the secretory pathway. We also ruled out the possibility of redundant functions between SCYL1 and SCYL3 in regulating COPI by generating Scyl1−/−;Scyl3−/− mice and found that combined deficiencies did not recapitulate early embryonic lethality associated with loss of COPI function in vivo (Faulstich et al., 1996; Coutinho et al., 2004; Hamamichi et al., 2008; Jayaram et al., 2008). Thus, SCYL1 and SCYL3 are dispensable for core functions of COPI in vivo and both proteins likely play a distinct role along the secretory pathway that supports motor neuron health.
One such function may be the regulation of TDP-43 proteostasis. TDP-43 is a ubiquitously expressed DNA/RNA binding protein that was identified several years ago as the major constituent of cytoplasmic inclusions in motor neurons of ALS and FTLD_TDP patients (Neumann et al., 2006). Mounting evidence suggest that accumulation of TDP-43 within cytoplasmic aggregates in spinal motor neurons negatively affects its normal function and drives neurodegeneration (Lee et al., 2011). Here, we show that spinal motor neurons of Scyl1−/− and Scyl1−/−;Scyl3−/− mice display TDP-43 pathology (Fig. 9C,D) and relocalization of TDP-43 in Scyl1−/−;Scyl3−/− mice occurred at a younger age compared with Scyl1-deficient mice and correlated with disease onset. The molecular pathways linking SCYL proteins and TDP-43 proteostasis however remains to be determined.
Interestingly, however, recent studies have linked COPI to mRNA transport along axons of motor neurons and suggested a role for survival motor neuron (SMN) in regulating this process (Custer et al., 2013; Todd et al., 2013; Li et al., 2015). Specifically, they revealed that COPI complexes interact with specific sets of RNAs enriched in G-quadruplex motifs, associated with fragile X mental retardation protein, or encoding plasma membrane and cytoplasmic proteins involved in signal transduction (Todd et al., 2013). Interestingly, SMN, whose expression is altered spinal muscular atrophy, may function as a scaffolding protein linking specific RNA molecules to the COPI complex (Todd et al., 2013; Li et al., 2015). Importantly, the RNA-trafficking function of COPI is likely independent of its Golgi-to-ER and/or intra-Golgi transport function, as cells expressing mutant COPA that abrogates SMN protein interaction but not COPI function in retrograde transport do not support neurite outgrowth in vitro and in vivo as does SMN depletion (Li et al., 2015). SCYL1 and SCYL3 might function similarly by linking TDP-43 mRNA granules to COPI for axonal transport. Elucidating the mechanisms underlying motor neuron death in Scyl1−/− and Scyl1−/−;Scyl3−/− mice will be the main focus of future work.
Footnotes
This work was supported by ALSAC. We thank Dr. Vani Shanker for editing the paper, and members of the Veterinary Pathology Core Laboratory, the Histology Laboratory, the Hartwell Center for Bioinformatics and Biotechnology, and the Transgenic/Gene Knockout Shared Resource, particularly Jack Sublett, Ling Li, and Taylor Walker, for their help in generating and characterizing mouse lines.
The authors declare no competing financial interests.
References
- Blot S, Poirier C, Dreyfus PA (1995) The mouse mutation muscle deficient (mdf) is characterized by a progressive motoneuron disease. J Neuropathol Exp Neurol 54:812–825. [PubMed] [Google Scholar]
- Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR (2006) Emerging roles of pseudokinases. Trends Cell Biol 16:443–452. 10.1016/j.tcb.2006.07.003 [DOI] [PubMed] [Google Scholar]
- Burman JL, Bourbonniere L, Philie J, Stroh T, Dejgaard SY, Presley JF, McPherson PS (2008) Scyl1, mutated in a recessive form of spinocerebellar neurodegeneration, regulates COPI-mediated retrograde traffic. J Biol Chem 283:22774–22786. 10.1074/jbc.M801869200 [DOI] [PubMed] [Google Scholar]
- Burman JL, Hamlin JN, McPherson PS (2010) Scyl1 regulates Golgi morphology. PloS One 5:e9537. 10.1371/journal.pone.0009537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chafe SC, Mangroo D (2010) Scyl1 facilitates nuclear tRNA export in mammalian cells by acting at the nuclear pore complex. Mol Biol Cell 21:2483–2499. 10.1091/mbc.E10-03-0176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Diaz C, Nicole S, Velasco ME, Borra-Cebrian C, Panozzo C, Frugier T, Millet G, Roblot N, Joshi V, Melki J (2002) Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum Mol Genet 11:1439–1447. 10.1093/hmg/11.12.1439 [DOI] [PubMed] [Google Scholar]
- Corrado L, Del Bo R, Castellotti B, Ratti A, Cereda C, Penco S, Sorarù G, Carlomagno Y, Ghezzi S, Pensato V, Colombrita C, Gagliardi S, Cozzi L, Orsetti V, Mancuso M, Siciliano G, Mazzini L, Comi GP, Gellera C, et al. (2009) Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J Med Genet 47:190–194. [DOI] [PubMed] [Google Scholar]
- Coutinho P, Parsons MJ, Thomas KA, Hirst EM, Saúde L, Campos I, Williams PH, Stemple DL (2004) Differential requirements for COPI transport during vertebrate early development. Dev Cell 7:547–558. 10.1016/j.devcel.2004.07.020 [DOI] [PubMed] [Google Scholar]
- Custer SK, Todd AG, Singh NN, Androphy EJ (2013) Dilysine motifs in exon 2b of SMN protein mediate binding to the COPI vesicle protein alpha-COP and neurite outgrowth in a cell culture model of spinal muscular atrophy. Hum Mol Genet 22:4043–4052. 10.1093/hmg/ddt254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Y, Li J, Fang J, Xu Z, He X, Zhang F, Ling J, Li X, Xu D, Li L, Li YY, Huo K (2003) Cloning and characterization of a novel gene which encodes a protein interacting with the mitosis-associated kinase-like protein NTKL. J Hum Genet 48:315–321. 10.1007/s10038-003-0031-5 [DOI] [PubMed] [Google Scholar]
- Enkler L. (2014) Le complexe multisynthetasique AME de la levure: dynamique de l'edifice et roles noncannoniques de ses composants. Universite de Strasbourg Thesis. [Google Scholar]
- Faulstich D, Auerbach S, Orci L, Ravazzola M, Wegchingel S, Lottspeich F, Stenbeck G, Harter C, Wieland FT, Tschochner H (1996) Architecture of coatomer: molecular characterization of delta-COP and protein interactions within the complex. J Cell Biol 135:53–61. 10.1083/jcb.135.1.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingham AK, Pfeifer AC, Munro S (2002) CASP, the alternatively spliced product of the gene encoding the CCAAT-displacement protein transcription factor, is a golgi membrane protein related to giantin. Mol Biol Cell 13:3761–3774. 10.1091/mbc.E02-06-0349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras S, Earls LR, Howell S, Smeyne RJ, Zakharenko SS, Pelletier S (2015) SCYL2 protects CA3 pyramidal neurons from excitotoxicity during functional maturation of the mouse hippocampus. J Neurosci 35:10510–10522. 10.1523/JNEUROSCI.2056-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras S, Kuliyev E, Pelletier S (2017) SCYL1 does not regulate REST expression and turnover. PloS One 12:e0178680. 10.1371/journal.pone.0178680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Penman M, Trigatti BL, Krieger M (1996) A single point mutation in epsilon-COP results in temperature-sensitive, lethal defects in membrane transport in a chinese hamster ovary cell mutant. J Biol Chem 271:11191–11196. 10.1074/jbc.271.19.11191 [DOI] [PubMed] [Google Scholar]
- Guo Y, Punj V, Sengupta D, Linstedt AD (2008) Coat-tether interaction in Golgi organization. Mol Biol cell 19:2830–2843. 10.1091/mbc.E07-12-1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamichi S, Rivas RN, Knight AL, Cao S, Caldwell KA, Caldwell GA (2008) Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson's disease model. Proc Natl Acad Sci U S A 105:728–733. 10.1073/pnas.0711018105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlin JN, Schroeder LK, Fotouhi M, Dokainish H, Ioannou MS, Girard M, Summerfeldt N, Melançon P, McPherson PS (2014) Scyl1 scaffolds class II arfs to specific subcomplexes of coatomer through the gamma-COP appendage domain. J Cell Sci 127:1454–1463. 10.1242/jcs.136481 [DOI] [PubMed] [Google Scholar]
- Jayaram SA, Senti KA, Tiklová K, Tsarouhas V, Hemphälä J, Samakovlis C (2008) COPI vesicle transport is a common requirement for tube expansion in Drosophila. PloS One 3:e1964. 10.1371/journal.pone.0001964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin KL, Mondal G, Hartman JK, Tyagi S, Kurley SJ, Bland CS, Hsu TY, Renwick A, Fang JE, Migliaccio I, Callaway C, Nair A, Dominguez-Vidana R, Nguyen DX, Osborne CK, Schiff R, Yu-Lee LY, Jung SY, Edwards DP, Hilsenbeck SG, et al. (2014) The oncogenic STP axis promotes triple-negative breast cancer via degradation of the REST tumor suppressor. Cell Rep 9:1318–1332. 10.1016/j.celrep.2014.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, et al. (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467–473. 10.1038/nature11922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaka S, Kashina A (2008) Cell biology of embryonic migration. Birth Defects Res C Embryo Today 84:102–122. 10.1002/bdrc.20125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Lee VM, Trojanowski JQ (2011) Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci 13:38–50. 10.1038/nrn3121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Custer SK, Gilson T, Hao le T, Beattie CE, Androphy EJ (2015) α-COP binding to the survival motor neuron protein SMN is required for neuronal process outgrowth. Hum Mol Genet 24:7295–7307. 10.1093/hmg/ddv428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG (2003) A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 13:476–484. 10.1101/gr.749203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire AT, Mangroo D (2007) Cex1p is a novel cytoplasmic component of the saccharomyces cerevisiae nuclear tRNA export machinery. EMBO J 26:288–300. 10.1038/sj.emboj.7601493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monani UR. (2005) Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron 48:885–896. 10.1016/j.neuron.2005.12.001 [DOI] [PubMed] [Google Scholar]
- Monani UR, Pastore MT, Gavrilina TO, Jablonka S, Le TT, Andreassi C, DiCocco JM, Lorson C, Androphy EJ, Sendtner M, Podell M, Burghes AH (2003) A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J Cell Biol 160:41–52. 10.1083/jcb.200208079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neisch AL, Fehon RG (2011) Ezrin, Radixin and Moesin: key regulators of membrane-cortex interactions and signaling. Curr Opin Cell Biol 23:377–382. 10.1016/j.ceb.2011.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- Pelletier S. (2016) SCYL pseudokinases in neuronal function and survival. Neural Regen Res 11:42–44. 10.4103/1673-5374.175040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier S, Gingras S, Funakoshi-Tago M, Howell S, Ihle JN (2006) Two domains of the erythropoietin receptor are sufficient for Jak2 binding/activation and function. Mol Cell Biol 26:8527–8538. 10.1128/MCB.01035-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier S, Gingras S, Howell S, Vogel P, Ihle JN (2012) An early onset progressive motor neuron disorder in Scyl1-deficient mice is associated with mislocalization of TDP-43. J Neurosci 32:16560–16573. 10.1523/JNEUROSCI.1787-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier S, Gingras S, Green DR (2015) Mouse genome engineering via CRISPR-Cas9 for study of immune function. Immunity 42:18–27. 10.1016/j.immuni.2015.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J (1997) ER-to-Golgi transport visualized in living cells. Nature 389:81–85. 10.1038/38001 [DOI] [PubMed] [Google Scholar]
- Presley JF, Ward TH, Pfeifer AC, Siggia ED, Phair RD, Lippincott-Schwartz J (2002) Dissection of COPI and Arf1 dynamics in vivo and role in Golgi membrane transport. Nature 417:187–193. 10.1038/417187a [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin SL, Rahimtula M, Mearow KM (2006) A method to assess multiple aspects of the motile behaviour of adherent PC12 cells on applied biological substrates. J Neurosci Methods 156:55–63. 10.1016/j.jneumeth.2006.02.011 [DOI] [PubMed] [Google Scholar]
- Saotome I, Curto M, McClatchey AI (2004) Ezrin is essential for epithelial organization and villus morphogenesis in the developing intestine. Dev Cell 6:855–864. 10.1016/j.devcel.2004.05.007 [DOI] [PubMed] [Google Scholar]
- Schmidt WM, Kraus C, Höger H, Hochmeister S, Oberndorfer F, Branka M, Bingemann S, Lassmann H, Müller M, Macedo-Souza LI, Vainzof M, Zatz M, Reis A, Bittner RE (2007) Mutation in the Scyl1 gene encoding amino-terminal kinase-like protein causes a recessive form of spinocerebellar neurodegeneration. EMBO Rep 8:691–697. 10.1038/sj.embor.7401001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt WM, Rutledge SL, Schüle R, Mayerhofer B, Züchner S, Boltshauser E, Bittner RE (2015) Disruptive SCYL1 mutations underlie a syndrome characterized by recurrent episodes of liver failure, peripheral neuropathy, cerebellar atrophy, and ataxia. Am J Hum Genet 97:855–861. 10.1016/j.ajhg.2015.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan A, Uff CR, Isacke CM, Thorne RF (2003) PACE-1, a novel protein that interacts with the C-terminal domain of ezrin. Exp Cell Res 284:224–238. 10.1016/S0014-4827(02)00054-X [DOI] [PubMed] [Google Scholar]
- Szul T, Sztul E (2011) COPII and COPI traffic at the ER–Golgi interface. Physiology 26:348–364. 10.1152/physiol.00017.2011 [DOI] [PubMed] [Google Scholar]
- Todd AG, Lin H, Ebert AD, Liu Y, Androphy EJ (2013) COPI transport complexes bind to specific RNAs in neuronal cells. Hum Mol Genet 22:729–736. 10.1093/hmg/dds480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey KJ, Hammond JW (2009) Traffic control: regulation of kinesin motors. Nat Rev Mol Cell Biol 10:765–777. 10.1038/nrm2782 [DOI] [PubMed] [Google Scholar]
- Yang JS, Zhang L, Lee SY, Gad H, Luini A, Hsu VW (2006) Key components of the fission machinery are interchangeable. Nat Cell Biol 8:1376–1382. 10.1038/ncb1503 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Peptides identified by LC-MS/MS. FLAG-SCYL3 was purified by immunoaffinity using M2 (anti-FLAG)-agarose beads. The immune complex was resolved on SDS-PAGE, and interacting partners were identified by MS (see Fig. 2). Abbreviations: LC-MS/MS, liquid chromatography - tandem mass spectrometry; SC, spectral count; TP, total peptides; GN, gene name; SV: Splice variant; OS; organism; PE, peptide. Download Figure 2-1, XLSX file (23KB, xlsx)