

Abstract
μ-Opioid receptor agonists are widely used within the contemporary treatment of pain, but abrupt opioid suspension, even after short-term infusion, can paradoxically increase the sensitivity to noxious stimuli, a phenomenon that has been, for example, reported after application of the fast-acting μ-opioid receptor agonist remifentanil. To investigate the mechanisms underlying the effects of discontinuation of remifentanil application on pain processing in the human CNS, we analyzed neuronal responses to thermal stimuli before and after a short-term infusion of remifentanil (30 min 0.1 μg/kg body weight/min) compared with control in the brain, brainstem, and spinal cord in drug-naive male volunteers using fMRI. Subsequent to remifentanil suspension, we observed reduced heat pain thresholds and increased neuronal responses in pain-encoding as well as in key regions of the descending pain-modulatory system, such as the periaqueductal gray matter, the nucleus cuneiformis, and the rostral ventromedial medulla. Moreover, the spinal pain-related multivoxel activity pattern showed an opioid-specific change after drug suspension. Importantly, remifentanil suspension increased the functional coupling between the nucleus cuneiformis and the rostral anterior cingulate cortex, and the coupling strength between the rostral anterior cingulate cortex and the nucleus cuneiformis correlated negatively with the individual pain threshold after opioid suspension. These findings demonstrate that, already subsequent to a short-term infusion of the μ-opioid receptor agonist remifentanil, signaling in the descending pain-modulatory system is fundamentally altered and that these changes are directly related to the behavioral sensitivity to pain.
SIGNIFICANCE STATEMENT Opioids are widely used in modern medicine, but, in addition to their known side effects, it is increasingly recognized that opioids can also increase sensitivity to pain subsequent to their use. Using the fast-acting μ-opioid receptor agonist remifentanil and fMRI in healthy male volunteers, this study demonstrates how signaling changes occur along the entire descending pain-modulatory pathway after opioid discontinuation and how these alterations are closely linked to increased behavioral pain sensitivity. Particularly by revealing modified responses in pain-modulatory brainstem regions that have been previously demonstrated to be causally involved in acute opioid withdrawal effects in rodents, the data provide a plausible neuronal mechanism by which the increased sensitivity to pain after opioid suspension is mediated in humans.
Keywords: fMRI, opioids, pain, remifentanil
Introduction
Therapeutically applied opioids exert their pharmacodynamic properties through an interaction with the complex endogenous opioid system shifting the balance between pronociception and antinociception and further homeostatic functions on a cellular, systems, and behavioral level toward a “protective” hypoalgesic state (Fields, 2004; Pasternak and Pan, 2013). However, abrupt discontinuation of opioid application, even after short-term application, has been shown to potentially bring an opposite state about, which is characterized by reduced pain thresholds and increased sensitivity to noxious stimulation (Rossbach, 1880; Mao, 2002; Angst et al., 2003; Koppert et al., 2003a,b; Angst and Clark, 2006; M. Lee et al., 2011), indicating the intricacy of the interaction between exogenously applied opioids and endogenous systems (Célèrier et al., 2001).
While the degree of manifestation and its relevance under clinical conditions are still the subject of investigation (Angst and Clark, 2006; Fishbain et al., 2009; Fletcher and Martinez, 2014; Volkow and McLellan, 2016; Yu et al., 2016), basic research studies consistently showed the existence of this class of phenomena under controlled laboratory conditions (Vanderah et al., 2000; Célèrier et al., 2001; Mao, 2002; Angst and Clark, 2006). With regard to the underlying mechanisms that can explain increased sensitivity to pain after opioid suspension, several explanations have been suggested (Vanderah et al., 2000; Gardell et al., 2002; Brown and Russell, 2004; Wang et al., 2005; Simonin et al., 2006; Drdla et al., 2009; Heinl et al., 2011; Chu et al., 2015; Roeckel et al., 2016). One prominent hypothesis is based on an altered equilibrium between ascending nociceptive signals and descending pain-modulatory systems (Kaplan and Fields, 1991; Heinricher et al., 2001; Vanderah et al., 2001a,b; Bie et al., 2003; Ossipov et al., 2003; Drdla et al., 2009; Wanigasekera et al., 2011). While several studies examined direct effects of opioid application in the human brain (Petrovic et al., 2002; Wise et al., 2002, 2004; Leppä et al., 2006; Atlas et al., 2012), demonstrating an involvement of descending pain-modulatory systems in opioid analgesia in humans, potential changes in the functionality of this system subsequent to opioid application have not been investigated in detail in humans so far. Hence, the current study aimed at addressing this hypothesis.
Investigating this hypothesis requires an experimental approach that is sensitive to neuronal responses in pain-encoding and pain-modulatory systems and to their dependence on neurochemical processes. To this end, we realized two pharmacological fMRI experiments using thermal pain stimuli and a short-term infusion of the μ-opioid receptor agonist remifentanil (0.1 μg/kg body weight/min for 30 min). In the first fMRI experiment (n = 32), BOLD responses were assessed in the brain and brainstem. In the second experiment (n = 30), responses were measured in the spinal cord. Both experiments compared BOLD responses in a between-group design between a baseline condition before opioid application and the state after remifentanil suspension with a precisely matched saline control condition to account for confounding effects, such as time (Fig. 1A). We implemented an injury-free stimulation protocol using slowly ascending thermal ramps (applied to the left forearm) starting from a physical intensity causing a slight warm sensation up to a painful level (Fig. 1B). This approach allowed us to disentangle BOLD signal changes related to the warm and painful period of thermal stimulation, to assess heat pain thresholds (HPTs), and to depict the temporal dynamics of BOLD signal changes along the entire neuraxis during the transition from nonpainful to painful perception and to finally analyze the effects of remifentanil suspension on these parameters. The results reveal opioid discontinuation-related effects at all levels of the CNS and elucidate how BOLD signal changes in the involved brain regions and altered functional coupling in the descending pain-modulatory system relate to the behavioral increase in pain sensitivity after discontinuation of remifentanil.
Figure 1.

Experimental procedure. A, Both fMRI experiments used the same experimental protocol. Volunteers were randomly assigned to either a control or a remifentanil group. After measuring BOLD responses to thermal stimuli during a baseline fMRI session, volunteers in the remifentanil group received an intravenous infusion of 0.1 μg remifentanil per kilogram body weight per minute for 30 min. Volunteers in the control group received the same amount of physiological saline solution for the same period. After a waiting period of 1 h, a second identical fMRI session followed. B, During each session, we applied 13 ascending thermal stimuli to the left radial forearm (dermatome C6), starting at a temperature of 32°C and rising over 30 s with a constant slope of 0.5°C per second up to 47°C. This stimulation caused a nonpainful warm sensation during the first seconds of stimulation and a painful sensation during the latter phase. During the first, seventh, and 13th trials, participants indicated by pressing a button (green triangle) when their HPT was reached. Subsequently to the thermal stimulus, after a short pause, participants rated the level of pain present during that trial on a VAS: 0–100, “no pain” to “unbearable pain.” Finally, a variable intertrial interval (ITI) followed.
Materials and Methods
Experiment 1: brain fMRI
Participants.
A total of 38 healthy male volunteers (26.5 ± 1.2 years of age, mean ± SEM) were acquired and found to be eligible to participate in the first fMRI experiment investigating opioid suspension-related effects in the brain.
To avoid possible gender differences (Bodnar and Kest, 2010; Niesters et al., 2010; C. W. Lee and Ho, 2013), we only studied male volunteers. Exclusion criteria were abnormal HPT at the site of stimulus application, any history of neurological disease, including any history of pain syndrome, any other serious acute or chronic illness, current medication, intolerance to remifentanil, and any pain event within the last week. All participants underwent a medical examination and gave written consent before the experiment. The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the Medical Council of Hamburg and followed the ethical guidelines of the International Association for the Study of Pain. Participants were randomly assigned to either remifentanil or a saline group. Data of 6 participants had to be excluded (due to discomfort within the scanner and premature termination of the experiment, 2 participants; incomplete ratings, 2 participants; technical malfunction, 2 participants), leaving 32 complete datasets (remifentanil group: n = 16). Groups did not differ significantly regarding age, body weight, initial HPT, depression score, and pain vigilance and awareness score (Table 1). Aftereffects of remifentanil and remifentanil effects on participant's mood can be obtained from Tables 2–5.
Table 1.
Participant characteristicsa
| Saline control group | Remifentanil group | |
|---|---|---|
| Experiment 1 | ||
| Age | 27.2 ± 1.1 years | 25.7 ± 1.1 years |
| Body weight | 77.6 ± 2.0 kg | 79.8 ± 2.6 kg |
| Initial HPT | 43.7 ± 0.8°C | 44.6 ± 0.6°C |
| Depression score | 9.7 ± 1.0 | 8.6 ± 0.7 |
| Pain awareness score | 52.7 ± 3.8 | 52.6 ± 4.3 |
| Experiment 2 | ||
| Age | 27.4 ± 1.1 years | 27.5 ± 1.1 years |
| Body weight | 76.7 ± 2.5 kg | 76.4 ± 1.1 kg |
| Initial HPT | 41.3 ± 0.6°C | 42.4 ± 0.6°C |
| Depression score | 8.2 ± 0.5 | 8.5 ± 0.5 |
| Pain awareness score | 51.5 ± 3.5 | 56.5 ± 3.7 |
aData are the average group values (mean ± SEM).
Table 2.
Effects of remifentanil suspension on mood: Experiment 1a
| SC T1 |
OS T1 |
SC T2 |
OS T2 |
|||||
|---|---|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | |
| Session 1 | ||||||||
| Drowsy/alert | 2.31 | 0.45 | 2.11 | 0.41 | 3.52 | 0.59 | 5.20 | 0.61 |
| Excited/calm | 2.11 | 0.33 | 1.91 | 0.32 | 1.58 | 0.43 | 1.48 | 0.37 |
| Feeble/strong | 2.45 | 0.33 | 2.13 | 0.36 | 3.06 | 0.49 | 4.34 | 0.49 |
| Clear-headed/muzzy | 7.95 | 0.57 | 8.77 | 0.33 | 6.70 | 0.72 | 5.08 | 0.57 |
| Clumsy/well-coordinated | 3.16 | 0.44 | 2.45 | 0.53 | 3.31 | 0.56 | 4.47 | 0.63 |
| Energetic/lethargic | 5.83 | 0.45 | 6.72 | 0.58 | 6.53 | 0.51 | 5.48 | 0.66 |
| Discontented/contended | 2.08 | 0.48 | 1.66 | 0.47 | 1.30 | 0.24 | 2.73 | 0.63 |
| Tranquil/troubled | 7.78 | 0.59 | 8.97 | 0.27 | 8.28 | 0.40 | 7.83 | 0.42 |
| Quick-witted/mentally slow | 7.97 | 0.43 | 8.59 | 0.29 | 7.16 | 0.62 | 5.97 | 0.47 |
| Relaxed/tense | 7.95 | 0.33 | 8.20 | 0.47 | 8.42 | 0.34 | 8.11 | 0.35 |
| Dreamy/attentive | 3.02 | 0.50 | 1.70 | 0.35 | 3.47 | 0.61 | 4.39 | 0.57 |
| Proficient/incompetent | 8.05 | 0.36 | 8.81 | 0.24 | 7.61 | 0.41 | 6.81 | 0.48 |
| Sad/happy | 2.33 | 0.42 | 1.11 | 0.22 | 1.88 | 0.42 | 1.72 | 0.40 |
| Amicable/antagonistic | 8.23 | 0.33 | 9.08 | 0.22 | 8.47 | 0.25 | 8.27 | 0.30 |
| Bored/interested | 1.73 | 0.25 | 1.42 | 0.25 | 2.17 | 0.40 | 2.48 | 0.50 |
| Gregarious/withdrawn | 5.50 | 0.47 | 6.56 | 0.52 | 5.53 | 0.57 | 4.94 | 0.63 |
| Insecure/secure | 2.20 | 0.24 | 1.33 | 0.20 | 1.97 | 0.35 | 2.41 | 0.39 |
| SC T3 |
OS T3 |
SC T4 |
OS T4 |
|||||
|---|---|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | |
| Session 2 | ||||||||
| Drowsy/alert | 1.66 | 0.32 | 2.23 | 0.33 | 3.94 | 0.61 | 3.61 | 0.60 |
| Excited/calm | 1.50 | 0.31 | 1.81 | 0.40 | 1.69 | 0.39 | 1.69 | 0.30 |
| Feeble/strong | 1.92 | 0.43 | 2.61 | 0.46 | 3.02 | 0.44 | 3.42 | 0.46 |
| Clear-headed/muzzy | 8.13 | 0.45 | 7.45 | 0.42 | 6.28 | 0.60 | 7.19 | 0.47 |
| Clumsy/well-coordinated | 2.77 | 0.62 | 2.84 | 0.48 | 4.20 | 0.58 | 2.91 | 0.45 |
| Energetic/lethargic | 7.02 | 0.59 | 7.41 | 0.41 | 5.89 | 0.59 | 6.70 | 0.48 |
| Discontented/contended | 1.20 | 0.28 | 1.64 | 0.39 | 1.73 | 0.27 | 1.55 | 0.31 |
| Tranquil/troubled | 8.06 | 0.44 | 7.63 | 0.55 | 8.38 | 0.31 | 8.08 | 0.30 |
| Quick-witted/mentally slow | 7.58 | 0.53 | 7.47 | 0.49 | 6.80 | 0.60 | 6.86 | 0.57 |
| Relaxed/tense | 8.31 | 0.27 | 8.23 | 0.30 | 8.03 | 0.35 | 8.55 | 0.32 |
| Dreamy/attentive | 2.55 | 0.51 | 2.73 | 0.41 | 3.63 | 0.57 | 3.38 | 0.59 |
| Proficient/incompetent | 8.23 | 0.33 | 7.91 | 0.33 | 7.69 | 0.40 | 7.00 | 0.50 |
| Sad/happy | 2.19 | 0.48 | 1.53 | 0.32 | 1.95 | 0.38 | 1.75 | 0.38 |
| Amicable/antagonistic | 8.38 | 0.34 | 8.53 | 0.28 | 8.19 | 0.29 | 8.55 | 0.34 |
| Bored/interested | 2.23 | 0.36 | 2.08 | 0.37 | 2.61 | 0.42 | 2.86 | 0.44 |
| Gregarious/withdrawn | 6.19 | 0.65 | 6.13 | 0.57 | 5.75 | 0.64 | 5.22 | 0.54 |
| Insecure/secure | 1.83 | 0.32 | 1.61 | 0.24 | 1.94 | 0.35 | 1.67 | 0.26 |
aBefore and after each experimental session, subjects had to rate their current mood on a 17 item questionnaire. Each item consisted of a VAS with opposing verbal descriptors at each end. Ratings were scored from 0 to 10. Data are the average group scores (mean ± SEM). SC, Saline control group; OS, opioid suspension group; T1, before Session 1; T2, after Session 1; T3, before Session 2; T4, after Session 2.
Table 5.
Adverse effects of remifentanil: Experiment 2a
| SC T1 |
OS T1 |
SC T2 |
OS T2 |
|||||
|---|---|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | |
| Headache | 1.27 | 0.15 | 1.27 | 0.12 | 1.27 | 0.12 | 1.27 | 0.12 |
| Dizziness | 1.00 | 0 | 1.00 | 0 | 1.00 | 0 | 1.00 | 0 |
| Nausea | 1.00 | 0 | 1.00 | 0 | 1.13 | 0.09 | 1.20 | 0.11 |
| Sedation | 2.07 | 0.32 | 1.33 | 0.16 | 1.60 | 0.21 | 1.93 | 0.32 |
| Blurred vision | 1.00 | 0 | 1.00 | 0 | 1.00 | 0 | 1.00 | 0 |
| Dry skin | 1.00 | 0 | 1.00 | 0 | 1.07 | 0.07 | 1.07 | 0.07 |
| Dry mouth | 1.00 | 0 | 1.00 | 0 | 1.00 | 0 | 1.00 | 0 |
| SC T3 |
OS T3 |
SC T4 |
OS T4 |
|||||
|---|---|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | |
| Headache | 1.33 | 0.16 | 1.20 | 0.14 | 1.40 | 0.19 | 1.53 | 0.19 |
| Dizziness | 1.07 | 0.07 | 1.13 | 0.09 | 1.00 | 0 | 1.00 | 0 |
| Nausea | 1.33 | 0.13 | 1.13 | 0.09 | 1.33 | 0.16 | 1.20 | 0.11 |
| Sedation | 1.47 | 0.24 | 1.33 | 0.16 | 1.73 | 0.30 | 1.60 | 0.25 |
| Blurred vision | 1.13 | 0.09 | 1.20 | 0.11 | 1.07 | 0.07 | 1.07 | 0.07 |
| Dry skin | 1.07 | 0.07 | 1.20 | 0.11 | 1.13 | 0.09 | 1.20 | 0.11 |
| Dry mouth | 1.00 | 0 | 1.00 | 0 | 1.07 | 0.07 | 1.20 | 0.14 |
aBefore and after each session, subjects also answered a 7 item questionnaire regarding adverse effects. Each adverse effect could be rated as “inexistent,” “very weak,” “weak,” “moderate,” “strong,” “very strong,” and “extremely strong” (scores 1–7). SC, Saline control group; OS, opioid suspension group; T1, before Session 1; T2, after Session 1; T3, before Session 2; T4, after Session 2.
Table 3.
Effects of remifentanil suspension on mood: Experiment 2a
| SC T1 |
OS T1 |
SC T2 |
OS T2 |
|||||
|---|---|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | |
| Session 1 | ||||||||
| Drowsy/alert | 2.93 | 0.43 | 2.53 | 0.49 | 5.23 | 0.51 | 3.62 | 0.49 |
| Excited/calm | 2.33 | 0.41 | 1.83 | 0.30 | 1.33 | 0.34 | 1.43 | 0.39 |
| Feeble/strong | 2.85 | 0.47 | 2.28 | 0.35 | 3.77 | 0.55 | 2.72 | 0.51 |
| Clear-headed/muzzy | 8.22 | 0.47 | 8.48 | 0.47 | 6.00 | 0.69 | 7.10 | 0.67 |
| Clumsy/well-coordinated | 2.87 | 0.43 | 2.58 | 0.40 | 4.33 | 0.57 | 3.60 | 0.51 |
| Energetic/lethargic | 6.52 | 0.51 | 6.90 | 0.40 | 4.92 | 0.55 | 9.45 | 3.28 |
| Discontented/contended | 2.50 | 0.59 | 1.55 | 0.22 | 1.97 | 0.38 | 1.82 | 0.36 |
| Tranquil/troubled | 7.97 | 0.52 | 7.48 | 0.64 | 7.92 | 0.40 | 8.00 | 0.37 |
| Quick-witted/mentally slow | 7.78 | 0.52 | 8.42 | 0.48 | 6.67 | 0.55 | 7.27 | 0.73 |
| Relaxed/tense | 7.23 | 0.48 | 8.03 | 0.38 | 7.53 | 0.57 | 8.47 | 0.36 |
| Dreamy/attentive | 3.17 | 0.60 | 2.33 | 0.55 | 4.23 | 0.59 | 3.70 | 0.71 |
| Proficient/incompetent | 7.90 | 0.50 | 8.45 | 0.43 | 6.88 | 0.62 | 8.22 | 0.31 |
| Sad/happy | 2.45 | 0.54 | 1.57 | 0.31 | 3.35 | 0.78 | 1.77 | 0.44 |
| Amicable/antagonistic | 8.45 | 0.35 | 9.00 | 0.27 | 7.73 | 0.46 | 8.82 | 0.32 |
| Bored/interested | 2.27 | 0.36 | 1.77 | 0.41 | 3.00 | 0.68 | 1.85 | 0.47 |
| Gregarious/withdrawn | 5.78 | 0.53 | 5.72 | 0.29 | 5.33 | 0.55 | 5.38 | 0.43 |
| Insecure/secure | 2.48 | 0.55 | 1.95 | 0.34 | 2.28 | 0.51 | 1.80 | 0.40 |
| SC T3 |
OS T3 |
SC T4 |
OS T4 |
|||||
|---|---|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | |
| Session 2 | ||||||||
| Drowsy/alert | 3.20 | 0.53 | 2.92 | 0.40 | 4.53 | 0.54 | 3.72 | 0.57 |
| Excited/calm | 1.65 | 0.28 | 1.38 | 0.32 | 2.18 | 0.44 | 1.22 | 0.24 |
| Feeble/strong | 2.90 | 0.50 | 3.45 | 0.78 | 3.68 | 0.46 | 3.32 | 0.53 |
| Clear-headed/muzzy | 7.35 | 0.60 | 8.17 | 0.54 | 6.00 | 0.65 | 7.82 | 0.43 |
| Clumsy/well-coordinated | 3.02 | 0.45 | 2.62 | 0.61 | 4.73 | 0.50 | 3.63 | 0.43 |
| Energetic/lethargic | 6.38 | 0.56 | 6.85 | 0.58 | 5.38 | 0.55 | 6.32 | 0.50 |
| Discontented/contended | 1.65 | 0.48 | 1.98 | 0.62 | 1.83 | 0.40 | 1.70 | 0.56 |
| Tranquil/troubled | 7.45 | 0.59 | 8.35 | 0.31 | 7.85 | 0.32 | 8.40 | 0.32 |
| Quick-witted/mentally slow | 8.10 | 0.37 | 8.58 | 0.39 | 7.50 | 0.57 | 7.50 | 0.59 |
| Relaxed/tense | 7.73 | 0.57 | 8.48 | 0.35 | 7.33 | 0.62 | 8.25 | 0.37 |
| Dreamy/attentive | 2.65 | 0.36 | 1.88 | 0.39 | 3.78 | 0.65 | 2.58 | 0.51 |
| Proficient/incompetent | 7.60 | 0.46 | 8.68 | 0.35 | 7.17 | 0.55 | 8.35 | 0.36 |
| Sad/happy | 2.05 | 0.51 | 1.17 | 0.22 | 2.82 | 0.58 | 1.58 | 0.33 |
| Amicable/antagonistic | 8.15 | 0.47 | 8.87 | 0.29 | 7.55 | 0.63 | 9.02 | 0.29 |
| Bored/interested | 2.85 | 0.63 | 1.60 | 0.32 | 3.62 | 0.73 | 1.93 | 0.46 |
| Gregarious/withdrawn | 5.55 | 0.58 | 5.90 | 0.44 | 5.58 | 0.43 | 5.65 | 0.39 |
| Insecure/secure | 1.98 | 0.46 | 1.42 | 0.24 | 2.42 | 0.50 | 1.30 | 0.28 |
aBefore and after each experimental session, subjects had to rate their current mood on a 17 item questionnaire. Each item consisted of a VAS with opposing verbal descriptors at each end. Ratings were scored from 0 to 10. Data are the average group scores (mean ± SEM). SC, Saline control group; OS, opioid suspension group; T1, before Session 1; T2, after Session 1; T3, before Session 2; T4, after Session 2.
Table 4.
Adverse effects of remifentanil: Experiment 1a
| SC T1 |
OS T1 |
SC T2 |
OS T2 |
|||||
|---|---|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | |
| Headache | 1.38 | 0.13 | 1.06 | 0.06 | 1.19 | 0.10 | 1.06 | 0.06 |
| Dizziness | 1.00 | 0 | 1.00 | 0 | 1.00 | 0 | 1.00 | 0 |
| Nausea | 1.00 | 0 | 1.00 | 0 | 1.06 | 0.06 | 1.06 | 0.06 |
| Sedation | 1.44 | 0.13 | 1.13 | 0.09 | 1.44 | 0.24 | 1.38 | 0.18 |
| Blurred vision | 1.00 | 0 | 1.00 | 0 | 1.00 | 0 | 1.00 | 0 |
| Dry skin | 1.00 | 0 | 1.00 | 0 | 1.13 | 0.09 | 1.00 | 0 |
| Dry mouth | 1.00 | 0 | 1.00 | 0 | 1.25 | 0.11 | 1.00 | 0 |
| SC T3 |
OS T3 |
SC T4 |
OS T4 |
|||||
|---|---|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | |
| Headache | 1.06 | 0.06 | 1.06 | 0.06 | 1.06 | 0.06 | 1.06 | 0.06 |
| Dizziness | 1.06 | 0.06 | 1.00 | 0 | 1.13 | 0.09 | 1.13 | 0.09 |
| Nausea | 1.00 | 0 | 1.00 | 0 | 1.13 | 0.09 | 1.06 | 0.06 |
| Sedation | 1.75 | 0.31 | 1.50 | 0.20 | 1.75 | 0.21 | 2.00 | 0.39 |
| Blurred vision | 1.19 | 0.10 | 1.13 | 0.09 | 1.00 | 0 | 1.00 | 0 |
| Dry skin | 1.06 | 0.06 | 1.00 | 0 | 1.25 | 0.11 | 1.19 | 0.10 |
| Dry mouth | 1.00 | 0 | 1.00 | 0 | 1.13 | 0.09 | 2.56 | 1.30 |
aBefore and after each session, subjects also answered a 7 item questionnaire regarding adverse effects. Each adverse effect could be rated as “inexistent,” “very weak,” “weak,” “moderate,” “strong,” “very strong,” and “extremely strong” (scores 1–7). SC, Saline control group; OS, opioid suspension group; T1, before Session 1; T2, after Session 1; T3, before Session 2; T4, after Session 2.
Experimental procedures.
To study the effects of suspension from acute remifentanil application in the CNS, we used two pharmacological groups (remifentanil and saline, randomized single-blind protocol) and two experimental sessions (before and after application of the test substance; Fig. 1). We implemented a new thermal stimulation protocol that allowed us to obtain information regarding the intensity component of the pain perception, the HPT, and the dynamic development of the BOLD signal over time. To this end, we applied during each session 13 ascending thermal ramps (Fig. 1) with a Peltier-Thermode (TSA-II, Medoc) to the left radial forearm (dermatome C6), starting at baseline temperature of 32°C and rising over 30 s with a constant slope of 0.5°C per second up to 47°C. Pilot experiments had shown that this slope and peak temperature are well tolerable for almost all healthy young adults. At the beginning of each thermal ramp (i.e., shortly before the thermal stimulus becomes noticeable), a white crosshair that was displayed in the center of the screen during the entire intertrial interval changed its color into red to indicate the onset of the new trial to the participant and to avoid surprise. This crosshair stayed red until a visual analog scale (VAS) was presented (see below).
During the first, seventh, and 13th trial, participants had to indicate by pressing a button when their HPT was reached following the instructions of the quantitative sensory testing protocol of the German Research Network on Neuropathic Pain (Rolke et al., 2006). However, in contrast to a typical method-of-limits assessment of the HPT, the temperature did not return to baseline after the participant indicated the HPT, but further increased until the end of the thermal ramp was reached, hence causing a painful period for some seconds. We only assessed the HPT during three trials, which allowed us to estimate the threshold for the intermediate trails (see Behavioral data analysis) and to avoid button-press-related motor activity in the CNS during the remaining trials. Subsequent to each thermal ramp, after a short pause of 6 s, participants had to rate the level of pain present during the painful phase of that trial using a VAS (endpoints labeled with 0 = “no pain” and 100 = “unbearable pain”) within a period of 12 s. Finally, a variable intertrial interval (60 ± 5 s) followed during which a white crosshair was presented. Presentation software (Neurobehavioral Systems) was used for stimulus control, recording of scanner pulses, and behavioral responses.
Remifentanil application.
Access to a cubital vein was established before the first scanning session. After the first fMRI session, participants received an intravenous infusion of remifentanil (0.1 μg/kg body weight/min; Ultiva, GlaxoSmithKline) or the same amount of 0.9% saline solution for 30 min outside the scanner. This remifentanil dose is a clinically relevant and falls within the dose range that is required for the relief of acute severe clinical pain (Bowdle et al., 1996; Volmanen et al., 2002). While experimental evidence indicates that higher remifentanil doses elicit stronger effects on pain after discontinuation (Guignard et al., 2000; Koppert et al., 2003a), the chosen dose still exhibits a low risk of unwanted drug effects and has been shown to elicit reliable effects on pain after discontinuation (Koppert et al., 2003a,b).
Remifentanil has a fast onset and likewise a rapid offset/washout due to hydrolysis by nonspecific plasma esterases with a half-life time of a few minutes, and the plasma concentration 30 min after infusion can be estimated as almost zero (Wise et al., 2002; Angst et al., 2003). During the infusion period, all participants received 3 L oxygen per minute via a respiratory mask, and ECG, SpO2, and noninvasive blood pressure were monitored. The infusion with the test substance was followed by a 1 h break before the second scanning session started. Pilot experiments had suggested that behavioral effects might increase after this period. In accordance with the very short half-life period of remifentanil, shortly after suspension of the infusion, physiological parameters of all participants were within normal range.
MRI data acquisition.
fMRI data were acquired on a 3 tesla system (Magnetom Tim Trio, Siemens) equipped with a 32-channel head coil. Forty transversal slices (slice thickness, 2 mm; gap, 1 mm) were acquired in each volume (repetition time: 2.4 s, echo time: 25 ms, flip angle: 80°, FOV: 216 × 216 mm2, matrix: 108 × 108; GRAPPA with acceleration factor 2, and 48 reference lines) using a T2*-weighted gradient-echo EPI sequence. Slice orientation was tilted by ∼°30. We also acquired high-resolution (1 × 1 × 1 mm3 voxel size) T1-weighted images for each subject using a 3D MPRAGE sequence (repetition time 2.3 s, echo time 2.98 ms, readout flip angle 9°, inversion time 1.1 s, FOV 192 × 240 × 256 mm3).
fMRI data preprocessing.
fMRI data preprocessing and statistical analyses were performed using SPM12 (Wellcome Trust Centre for Neuroimaging, London). The first five EPI volumes of each session were discarded to exclude T1-saturation effects. Functional images were corrected for time differences in slice acquisition time and motion (6 df). Structural T1-weighted images were coregistered to the functional images and subsequently SPM12-segmented. The obtained normalization parameters were applied to the functional images, which were resampled with a resolution of 2 × 2 × 2 mm3. Finally, functional images were smoothed with an 8 mm FWHM isotropic Gaussian kernel.
Because we specifically hypothesized brainstem structures to be involved in the mechanisms underlying opioid suspension-related effects, we used a second preprocessing strategy for the preprocessing of the brainstem that is specifically optimized for this region. To minimize the effect of movements of cortical regions on the motion correction procedure of the brainstem, the cost function of the realignment algorithm was weighted by an automatically created cylindrical binary mask that only included the brainstem and directly surrounding tissues (Napadow et al., 2006). Within this mask, highly variant regions (i.e., CSF) were excluded, to minimize the influence of strong-intensity fluctuations on the motion-correction procedure and to base the image registration only on brainstem movements. For the brainstem, EPI images were resampled after normalization with a resolution of 1 × 1 × 1 mm3 and smoothed with a 4 mm FWHM isotropic Gaussian kernel (Beissner, 2015).
Statistical analysis: behavior.
All behavioral data were analyzed in MATLAB version 8.4 (The MathWorks). We calculated the mean and SEM of pain ratings and HPTs for each session and group. To test for effects of remifentanil on behavioral pain measures, we compared differences of the mean pain ratings (based on all 13 trials) and thresholds (based on the first, seventh, and 13th trial) of the first and the second session between groups using a two-sample t test. Results were considered significant at p ≤ 0.05 (one-tailed based on the a priori hypothesis). To assign painful and warm periods in trials where the HPT was not assessed, we estimated the time point of the HPT based on the arithmetic mean of the HPT of the seventh and the 13th trials. This approach is based on a pilot experiment (n = 10) where participants had to indicate their HPTs during all 13 trials. The chosen approach yielded the least sum of error squares between estimated and recorded thresholds compared with other interpolations techniques.
Statistical analysis: fMRI.
The statistical fMRI data analysis was performed using a GLM approach. Analyses were performed separately for the imaging data preprocessed with the brain and the brainstem optimized preprocessing approach. For structures below the diencephalon, we report results from analyses that used the brainstem-adjusted preprocessing procedure. To identify brain regions responding to the warm and painful phase of the ascending thermal stimulation, the analysis of the imaging data commenced by specifying a basis model at the single-subject level implementing the warm and the painful phase of the ascending thermal ramps as separate box-car regressors, separately for trails during which subjects indicated their HPT via button press (first, seventh, and 13th trials) or not (second to the sixth and the eighth to the 12th trials). Only β estimations of the latter regressors were used for subsequent analyses to ensure that these analyses do not depict any button-press-related motor activity. The model also specified a box-car regressor for the VAS rating, a linear increasing regressor for the duration of each thermal ramp, and finally a session constant for each session, thus comprising altogether 12 regressors. Box-car functions were subsequently convolved with the canonical hemodynamic response function as implemented in SPM12, and data were subjected to temporal high-pass filtering with a cutoff period of 128 s. After model estimations, β images corresponding to the warm and the painful phase of trials without button presses were raised to a second-level random-effects analysis.
To capture the temporal dynamics of signal changes in response to the thermal stimulation protocol, we also set up a first-level analysis using a finite impulse response set as the basis function using a window length of 36 s (locked to the onset of each thermal ramp) and 15 bins (1 bin = 2.4 s). Trials during which subjects performed button presses and trials without button presses were again modeled separately. Each first-level design matrix of this model thus comprised 62 regressors (2 sessions × 2 conditions × 15 bins plus 2 sessions constants). Data were likewise subjected to temporal high-pass filtering with a cutoff period of 128 s. After model estimations, we defined contrasts for the time periods before and after exceeding the HPT. As the HPT naturally varies across participants, our linear contrast vector was defined as the fixed time window during which all participants were below or above the HPT to ensure that the group analysis of the nonpainful phase is not confounded by pain responses and vice versa. Contrast images corresponding to the warm and the painful period were then raised to a second-level random-effects analysis using a SPM-flexible factorial design considering pharmacological condition as a group factor. To illustrate the peristimulus time course in brain region, we also specified a similar second-level analysis incorporating the β weights for all bins.
To identify brain regions that are related to intraindividual variations of pain intensity perception and potentially modulated by opioid suspension, we also specified a first-level analysis model that included the trialwise individual pain ratings as a parametric modulator of the regressor representing the nonpainful and painful phase of stimulation respectively. The model was otherwise identical to our basis model (see above).
To further test for a modified signaling in the descending pain-modulatory network caused by opioid suspension, we used the nucleus cuneiformis (NCF) and the periaqueductal gray matter (PAG) (which were revealed by the opioid suspension specific interaction contrast) as seed voxels for functional connectivity analyses. We set a sphere of 6 mm diameter around the respective coordinate and extracted the mean time series for each subject for each session. These time series were then used as regressors in subject-specific design matrices. To remove stimulus-related linear trends from the data, we also included a linear increasing regressor for the duration of each thermal ramp in the design matrices. The resulting β images were subsequently raised to the second-level analysis, where we observed a significant pharmacological interaction effect for the analyses using the left and the right NCF as the seed voxel in the rostral anterior cingulate cortex (rACC). To better account for symmetry in our analysis, we subsequently also extracted the time series around the observed rACC coordinates and correlated the two time series. The resulting correlation coefficients for each session for each subject were Fisher z-transformed. Differences in the correlation strength between sessions were compared between groups using a two-sample t test. To test for the functional relevance of the findings, we correlated on the individual level the coupling strength with the HPT. As both vectors contained extreme values, we report Spearman's rank correlation coefficient. To test for group differences, we Fisher z-transformed the correlation coefficients (Myers and Sirois, 2006) and calculated z statistics. Finally, we used a similar analysis to test for a correlation between the individual nucleus cuneiformis–rostral ventromedial medulla (RVM) coupling strength and the HPT.
Results were considered significant at p < 0.05, corrected for multiple comparisons using the FWE rate based on the Gaussian random field theory. Based on our hypothesis of an altered signaling in the descending pain-modulatory system after opioid suspension, we defined the rACC, the PAG, the NCF, and the RVM as a priori regions of interest (ROIs). Correction was based on peak coordinates obtained from previous studies on pain processing and pain modulation. The rACC was corrected using a sphere of 12 mm radius; brainstem regions were corrected using a sphere of 6 mm radius. Outside a priori ROIs, FWE correction was performed for all voxels. We also report uncorrected p values with a threshold of p < 0.001. Findings in the brain are reported in square brackets stating x, y, z coordinates in the MNI space. For illustration purposes, statistical maps of the brain are thresholded at p < 0.005 uncorrected for multiple comparisons.
Experiment 2: high-resolution fMRI of the cervical spinal cord
Participants.
A total of 34 healthy male volunteers (26.5 ± 1.2 years of age) participated in the second experiment investigating opioid suspensions effects at the spinal level. Inclusion and exclusion criteria and ethical guidelines were the same as in Experiment 1 (see above). Data of 4 participants had to be excluded due to premature termination of the experiment (2 participants) and image artifacts (2 participants), leaving 30 complete datasets (remifentanil group: n = 15). Both groups did not differ significantly regarding age, body weight, initial HPT, depression score, and pain vigilance and awareness score (Table 1). Aftereffects of remifentanil and remifentanil effects on participant's mood can be again obtained from Tables 2–5. The experimental procedures and the drug application were identical to the first experiment.
MRI data acquisition.
fMRI data were acquired on the same system as Experiment 1, equipped with a 12-channel head coil and an additional 4-channel neck coil with the target region of the spinal cord being centered in the magnet's isocenter (EPI parameter: repetition time 1040 ms, echo time: 34 ms, flip angle: 60°, FOV: 132 × 130 mm2, matrix: 110 × 108; GRAPPA acceleration factor of 2, and 24 reference lines).
In short, we acquired 12 slices positioned approximately perpendicular to the spinal cord, using a slice thickness of 3.5 mm and an in-plane resolution of 1.2 × 1.2 mm2. Saturation pulses were applied anterior and posterior to the target region to avoid ghosting. To minimize signal intensity variations along the z-axis caused by magnetic susceptibility differences in the spinal canal, a slice-specific z-shim gradient momentum was used (Finsterbusch et al., 2012; Sprenger et al., 2012). We also acquired T2*-weighted anatomical images of the spinal target region using a Multiple Echo Data Image Combination sequence (12 transversal slices, slice thickness 3.5 mm, 0.57 × 0.57 mm2 in-plane resolution, repetition time 473 ms, echo time 22 ms, flip angle 20°, FOV 256 × 184 mm2, GRAPPA, acceleration factor 2, and 24 reference lines). Finally, we acquired high-resolution (1 × 1 × 1 mm3) T1-weighted anatomical images using a 3D-MPRAGE sequence (repetition time 2.3 s, echo time 3.43 ms, readout flip angle 9°, inversion time 1.1 s, FOV 192 × 240 × 256 mm3).
Physiological data acquisition.
To allow for retrospective physiological noise correction, the pulsoximetric pulse-curve (left index finger) and respiratory chest movements (using a respiratory belt) were measured using a clinical monitoring unit (Expression, In vivo). Data were recorded together with the scanner pulses using Spike2 software (Cambridge Electronic Design) to ensure timing accuracy.
fMRI data preprocessing.
After the first 5 EPI volumes were discarded, we corrected spinal EPIs for motion by using the same motion correction procedure that we used for the brainstem (see above).
We then spatially normalized the T1-weighted structural images following an approach similar to the one described by Geuter and Büchel (2013). After the subject-specific first-level analysis (see below), we applied the previously determined normalization parameters to the β images and resampled images to a resolution of 0.75 × 0.75 × 0.75 mm3. Finally, β images were smoothed using a 3D isotropic Gaussian kernel with an FWHM of 2 mm.
Statistical analysis: behavior.
The behavioral data analysis was identical to Experiment 1.
Statistical analysis: fMRI.
For the statistical analysis of the spinal data, we followed the same basic analysis approach on the single-subject and group level that we used for the analysis of the brain data (see above). Given that the influence of physiological noise is much stronger at the spinal level compared with the brain (Kong et al., 2012; Fratini et al., 2014; Eippert et al., 2017), we additionally corrected for this potential confound. Similar to previous studies (Eippert et al., 2009b; Sprenger et al., 2012, 2015), we used the selective averaging approach described by Deckers et al. (2006) to generate regressors representing cardiac and respiratory effects (using 10 bins for cardiac and respiratory effects, respectively) that were subsequently included in the subject-specific first-level design matrices. We also corrected for CSF-related signal fluctuations in the spinal EPI time series (Brooks et al., 2008). Therefore, we extracted the time courses of voxels from the spinal canal whose variance lay in the top 10th percentile and generated 1–3 regressors explaining 95% of the variance of these time series by using a principal component analysis. These regressors were likewise included in the first-level design matrices.
Results were considered significant at p < 0.05, corrected for multiple comparisons using the FWE rate. Approaches adjusting p values to control FWE rates based on the Gaussian random field theory are not suitable for very small cylindrical structures, such as the spinal cord (Worsley et al., 1992; Brooks, 2014). Similar to a previous study (Sprenger et al., 2015), we therefore used nonparametric permutation tests (Nichols and Holmes, 2002) using the Statistical Non-Parametric Mapping toolbox (SnPM13; http://warwick.ac.uk/snpm) to report FWE-corrected p values in the spinal cord. Estimations were based on 5000 permutations (Nichols and Holmes, 2002).
Based on observations from previous spinal fMRI studies (Eippert et al., 2009b; Sprenger et al., 2012, 2015), which consistently revealed BOLD responses caused by painful stimulation in the dermatome C6 in the ipsilateral dorsal horn of the spinal segment C6, we defined this region as a priori ROI. For this region, the multiple-comparison correction was restricted to a sphere of a 5 mm radius centered on the average coordinate from the previous studies. Outside this region, FWE correction was performed for all voxels.
In addition to the GLM analysis, we also performed a multivoxel pattern analysis (MVPA) using a linear support vector machine to decode the saline and remifentanil condition based on the corresponding spinal activity patterns as determined by the individual GLM contrast images. The MVPA was restricted to a region that showed in accordance with the functional neuroanatomy of the nociceptive system a significant BOLD response to the painful phase of thermal stimulation in the ipsilateral dorsal horn of the spinal segment C6 (thresholded at p < 0.05 uncorrected). We used a leave-one-subject-out cross-validation and compared the average accuracy across subjects to chance (50%) using a nonparametric permutation test. Finally, we conducted a multivoxel pattern searchlight analysis over the entire measured volume with a radius of 4 mm that was otherwise identical to the initial MVPA.
Results
Effects subsequent to remifentanil short-term infusion on behavior and BOLD responses to ascending thermal ramps in the brain
We first tested whether our experimental protocol resulted in a behaviorally measurable aftereffect of remifentanil suspension on pain perception. Both experiments yielded very comparable behavioral results (Fig. 2). The latter phase of thermal stimulation provoked a clearly painful sensation across all participants that remained approximately stable over trials and did not significantly differ between groups before application of the test substance (two-sample t test: Experiment 1, t(30) = 0.16, p = 0.88; Experiment 2, t(28) = −0.48, p = 0.63). We observed no significant effect of remifentanil suspension on pain intensity ratings (two-sample t test, Session 2 − Session 1: Experiment 1, t(30) = −0.93, p = 0.18; Experiment 2, t(28) = −0.02, p = 0.49). However, in both independent experiments, remifentanil suspension caused a clear effect on HPTs. While thresholds did not differ between both groups during the first experimental session (two-sample t test: Experiment 1, t(30) = −0.5, p = 0.63; Experiment 2, t(28) = −0.1, p = 0.93), we observed a decreased HPT in the second session after remifentanil application (Experiment 1, −0.37 ± 0.3°C; Experiment 2, −0.32 ± 0.4°C) and an increased HPT following saline application (Experiment 1, 0.25 ± 0.1°C; Experiment 2, 0.56 ± 0.2°C), leading to a significant pharmacological interaction effect in both experiments (two-sample t test: Experiment 1, t(30) = 2.2, p = 0.02; Experiment 2, t(28) = 1.9, p = 0.04).
Figure 2.
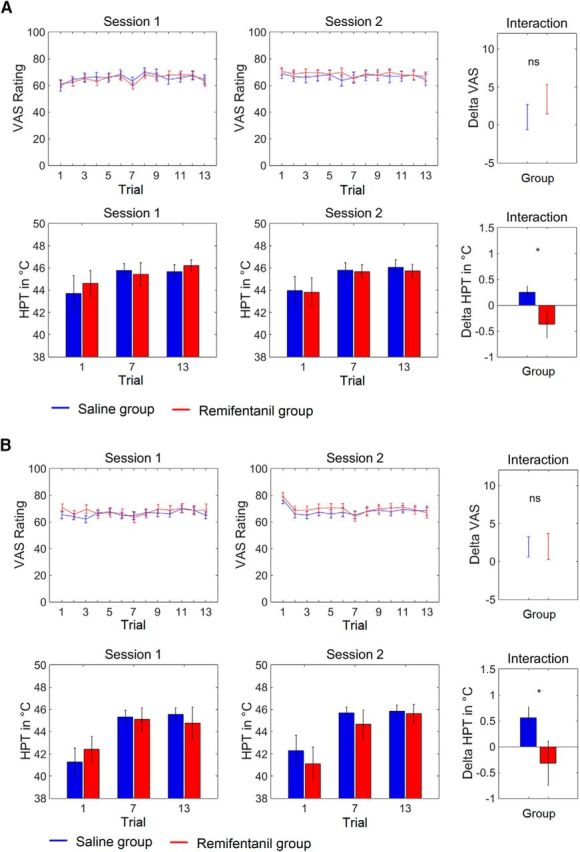
Behavioral effects of remifentanil suspension. A, Experiment 1. B, Experiment 2. VAS ratings of both experiments show that the latter phase of thermal stimulation caused a painful sensation that remained approximately stable over time and did not significantly differ between pharmacological groups before and after application of the test substance. In contrast, remifentanil suspension reduces the HPT compared with saline application. The interaction effects (rightmost panels) show the differences of the average pain ratings and HPTs, respectively, between the first and the second session for both groups separately. Error bars indicate SEM. *p < 0.05.
Next, we analyzed BOLD responses to the thermal stimuli in the brain separately for the nonpainful and painful period of stimulation and whether these responses would be modulated after remifentanil infusion. We identified a number of brain regions in response to thermal stimulation that have been previously shown to typically exhibit increased levels of activity during the processing of warm and painful stimuli by a large body of evidence (Apkarian et al., 2005; Duerden and Albanese, 2013). For the painful phase of stimulation this comprises regions, such as the bilateral insula, the anterior cingulate cortex, the primary and secondary somatosensory cortex (Fig. 3; Table 6).
Figure 3.
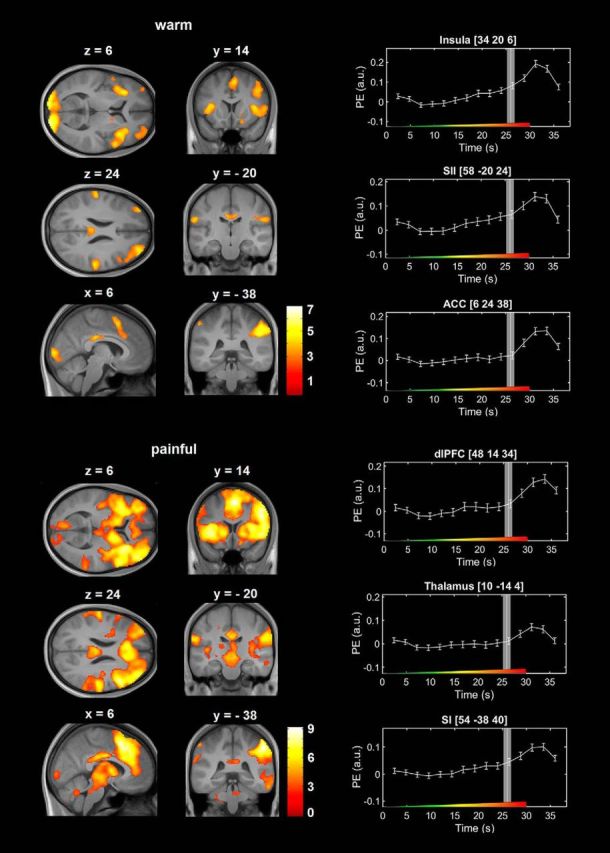
BOLD responses to the nonpainful and painful phase of ascending thermal stimulation in the brain during the baseline session across all participants. The marked activation of the visual cortex during the nonpainful phase of stimulation is most likely explained by the change of the color of the visually presented crosshair at the beginning of each trial. Right, Peristimulus time courses from selected brain regions. Vertical white lines indicate the HPT in seconds after stimulus onset. Gray bars represent the SEM of the HPT. Color bars represent t values. Error bars indicate SEM. Colored ramp at the bottom of each box represents the ascending thermal stimulus. PE (a.u.), Parameter estimates (arbitrary units).
Table 6.
BOLD responses to the nonpainful and painful phase of ascending thermal stimulation in the brain during the baseline sessiona
| Region | x | y | z | t | p |
|---|---|---|---|---|---|
| Nonpainful phase | |||||
| Anterior cingulate cortex | 8 | 24 | 30 | 3.31 | <0.001 |
| Calcarine gyrus | −12 | −100 | 0 | 6.95 | <0.001 |
| Cerebellum | 28 | −72 | −22 | 4.43 | <0.001 |
| 42 | −54 | −32 | 3.68 | <0.001 | |
| −36 | −66 | −52 | 3.39 | <0.001 | |
| Inferior frontal gyrus | 38 | 28 | −2 | 5.12 | <0.001 |
| −54 | 6 | 10 | 3.51 | <0.001 | |
| Inferior parietal lobule | −36 | −48 | 40 | 5.57 | <0.001 |
| −50 | −50 | 52 | 4.68 | <0.001 | |
| Left insula | −32 | 26 | 8 | 4.90 | <0.001 |
| Middle frontal gyrus | 38 | 52 | 20 | 4.80 | <0.001 |
| −36 | 48 | 20 | 4.44 | <0.001 | |
| Middle orbital gyrus | 40 | 48 | 4 | 3.37 | <0.001 |
| Postcentral gyrus | −62 | −22 | 24 | 4.20 | <0.001 |
| Posterior cingulate cortex | −4 | −26 | 28 | 4.83 | <0.001 |
| 6 | −14 | 30 | 3.17 | <0.001 | |
| Posterior-medial frontal | 4 | 12 | 52 | 4.24 | <0.001 |
| Precuneus | −14 | −64 | 36 | 3.77 | <0.001 |
| 12 | −66 | 36 | 3.37 | <0.001 | |
| Rolandic operculum | 56 | −24 | 24 | 4.54 | <0.001 |
| 46 | −24 | 28 | 3.17 | <0.001 | |
| Superior orbital gyrus | 18 | 16 | −16 | 3.62 | <0.001 |
| Supramarginal gyrus | 52 | −42 | 34 | 6.17 | <0.001 |
| Painful phase | |||||
| Calcarine gyrus | −8 | −98 | −6 | 5.06 | <0.001 |
| Cerebellum | −32 | −56 | −34 | 7.82 | <0.001 |
| −30 | −66 | −30 | 7.79 | <0.001 | |
| −10 | −74 | −30 | 7.41 | <0.001 | |
| 34 | −56 | −32 | 5.13 | <0.001 | |
| 26 | −46 | −30 | 3.47 | <0.001 | |
| 38 | −66 | −28 | 3.31 | <0.001 | |
| 18 | −70 | −52 | 3.66 | <0.001 | |
| 20 | −64 | −58 | 3.54 | <0.001 | |
| 14 | −76 | −48 | 3.52 | <0.001 | |
| 2 | −56 | −32 | 3.65 | <0.001 | |
| 14 | −58 | −32 | 3.19 | <0.001 | |
| 38 | −56 | −52 | 3.26 | <0.001 | |
| −10 | −58 | −54 | 3.19 | <0.001 | |
| 20 | −50 | −24 | 3.17 | <0.001 | |
| Inferior parietal lobule | −36 | −48 | 40 | 5.66 | <0.001 |
| −54 | −50 | 50 | 5.39 | <0.001 | |
| −56 | −38 | 52 | 5.08 | <0.001 | |
| Inferior temporal gyrus | 60 | −30 | −18 | 4.92 | <0.001 |
| Insula | −34 | 18 | −2 | 8.97 | <0.001 |
| Linual gyrus | −10 | −86 | 0 | 4.98 | <0.001 |
| 14 | −90 | 2 | 4.21 | <0.001 | |
| Middle frontal gyrus | −52 | 10 | 44 | 4.21 | <0.001 |
| Middle temporal gyrus | 62 | −32 | −8 | 5.33 | <0.001 |
| 62 | −46 | 6 | 4.11 | <0.001 | |
| Precentral gyrus | −42 | −2 | 42 | 4.80 | <0.001 |
| −44 | −2 | 58 | 4.03 | <0.001 | |
| Precuneus | 14 | −62 | 38 | 4.16 | <0.001 |
| −12 | −64 | 36 | 3.61 | <0.001 | |
| Superior medial gyrus | 0 | 20 | 44 | 9.14 | <0.001 |
| 48 | −48 | 8 | 3.36 | <0.001 | |
| Supramarginal gyrus | 52 | −36 | 46 | 8.69 | <0.001 |
aCoordinates are denoted by x, y, z in mm (MNI space), and strength of activation is expressed in t scores (df = 120).
We subsequently tested whether BOLD responses in the brain were modulated subsequent to opioid application using t tests. After remifentanil suspension, we observed increased responses to the painful phase of stimulation (Session 2 − Session 1) that were stronger compared with the control group in the posterior insula (−40, −20, 12, t(120) = 3.47, punc.< 0.001), the contralateral thalamus (8, −22, 10, t(120) = 3.88, punc.< 0.001) and the amygdala (26, −12, −18, t(120) = 3.35, punc.< 0.001; Fig. 4). The reverse contrast did not reveal any significant results. Likewise, our analysis revealed no significant differences between groups regarding the nonpainful phase.
Figure 4.
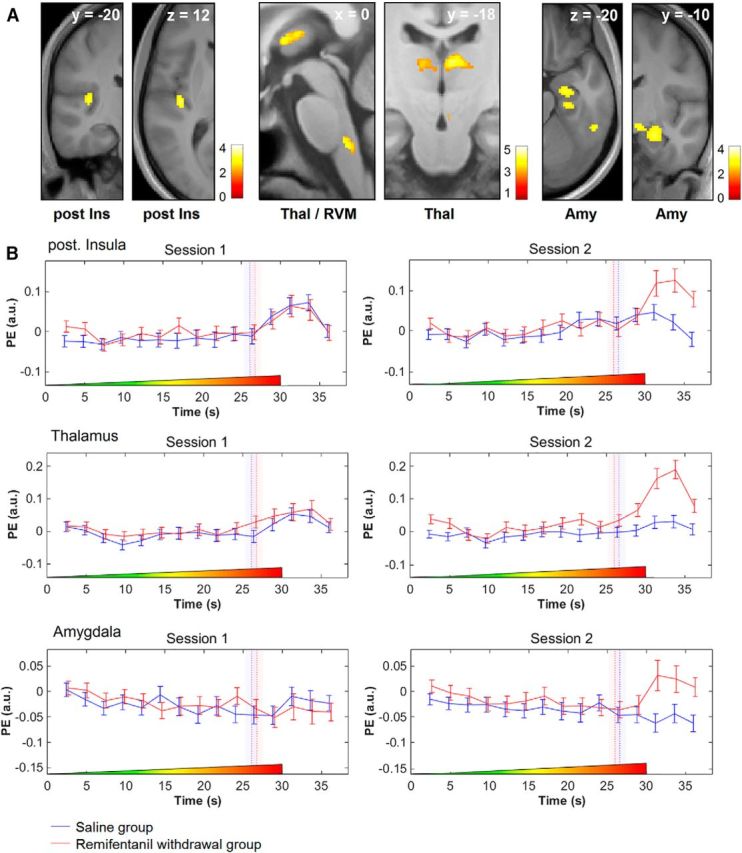
Increased BOLD responses after remifentanil discontinuation during the painful period of stimulation compared with control. A, Activation maps show that BOLD responses in the posterior insula (post Ins), the thalamus (Thal), the amygdala (Amy), and the rostral ventro-medial medulla (RVM) are stronger after discontinuation of remifentanil compared with the saline control group during the painful period of stimulation. The visualization threshold is set to p < 0.005 uncorrected. Color bars represent t values. B, The peristimulus time course in the posterior insula, thalamus, and the amygdala shows a very similar progression during the first session for both groups. After discontinuation of remifentanil, the peristimulus time course displays a selective increase after exceeding the HPT in these regions. The time course of the control group is slightly shifted to the left to avoid overlapping of error bars. Dashed vertical lines indicate the HPT in seconds. Bars in transparent color represent the SEM of the HPT. Notice the shift of HPTs from the first to the second experimental session. Colored ramps at the bottom of each box represent the temporal profile of ascending thermal stimulus. Error bars indicate SEM. PE (a.u.), Parameter estimates (arbitrary units).
Remifentanil suspension-related effects on descending pain-modulatory circuits
Based on the hypothesis that, subsequent to remifentanil application, the processing of pain-related signals would be altered in brainstem structures well known to play a pivotal role in various aspects of pain modulation, including opioid action (Basbaum and Fields, 1984; Millan, 2002; Mason, 2012) and amplification of nociceptive signals (Ji et al., 2003; Ren and Dubner, 2007; M. C. Lee et al., 2008), we next analyzed this region using a brainstem-optimized image preprocessing approach. Following remifentanil suspension, we observed increased BOLD responses to the painful phase of stimulation that were significantly stronger elevated compared with the control group in a region that can be most likely attributed to the RVM (presumably the nucleus raphe magnus and the adjacent nucleus reticularis gigantocellularis; 0, −36, −43, t(120) = 4.01, p = 0.001; Fig. 4A). The reverse contrast did not reveal any significant differences.
More importantly, with regard to mechanisms underlying the observed effect of remifentanil suspension on the pain threshold, we subsequently analyzed the period before reaching the HPT. This analysis showed increased BOLD responses after remifentanil suspension compared with control bilaterally in the midbrain reticular formation's NCF (right: 9, −26, −13, t(120) = 3.36, p = 0.04; left: −12, −27, −12, t(120) = 3.75, p = 0.01; Fig. 5) and in two portions of the PAG matter (ventrolateral PAG, −2, −29, −11, t(120) = 3.92, p = 0.01; caudal portion of the ventromedial PAG, 1, −36, −24, t(120) = 3.79, punc.< 0.001).
Figure 5.
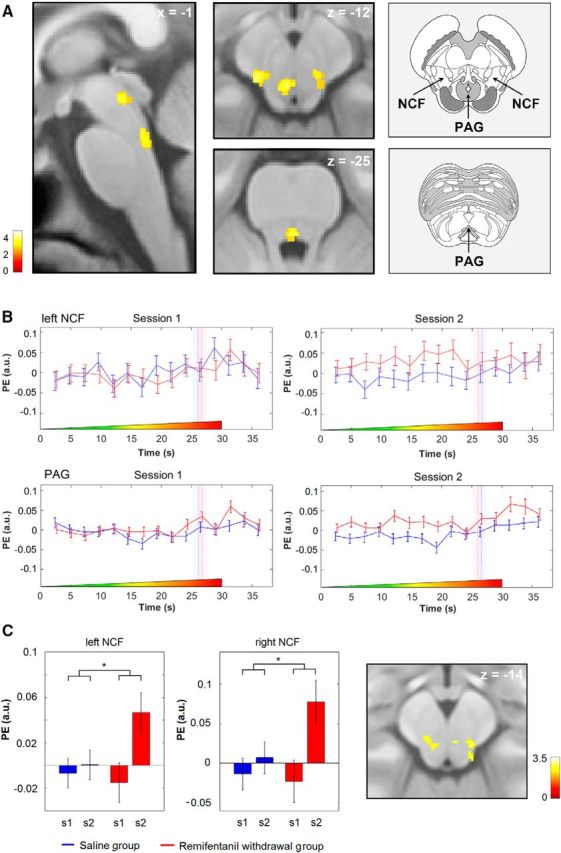
Increased BOLD responses after discontinuation of remifentanil during the nonpainful period of stimulation compared with control and parametric modulation. A, Activation maps show that BOLD responses in the bilateral NCF and in two portions of the PAG are stronger after discontinuation of remifentanil compared with the saline control group during the nonpainful phase of stimulation. Right, Anatomical images represent the localization of these structures in the brainstem. Modified with permission from Naidich (2009). B, The peristimulus time course in the left NCF and the PAG shows a BOLD signal increase during the nonpainful phase of stimulation after remifentanil suspension compared with control. The time course of the control group was slightly shifted to the left to avoid overlapping of error bars. Dashed vertical lines indicate the HPTs in seconds after stimulus onset. Bars in transparent color represent SEM of the HPT. Colored ramps at the bottom of each box represent the ascending thermal stimulus. C, The peak voxel parameter estimates of an analysis using the trial-by-trial VAS ratings as parametric modulator show a positive correlation between the heat pain ratings and the trialwise responses in the NCF during the nonpainful phase of stimulation exclusively after opioid suspension. Right, Statistical map represents the localization of this finding in the brainstem. The visualization threshold is set to p < 0.005 uncorrected. Error bars indicate SEM. Color bars represent t values. PE (a.u.), Parameter estimates (arbitrary units). *p < 0.05.
A subsequent analysis revealed that, after remifentanil application, parametric trial-by-trial responses during the nonpainful period correlated positively with the behavioral pain ratings in the previously identified NCF, but not in any of the other experimental sessions (right: 10, −28, −14, t(60) = 2.88, p < 0.05; left: −7, −26, −13, t(60) = 2.63, p < 0.05; Fig. 5C). At both peaks, parametric responses were significantly stronger after remifentanil suspension compared with the saline control condition (right: t(60) = 2.1 p < 0.05; left: t(60) = 2.13, p < 0.05).
Altered functional connectivity in the descending pain-modulatory system predicts remifentanil offset-related increase in pain sensitivity
As μ-opioid receptor agonists unfold part of their analgesic properties by acting on the pain-modulatory system (Barton et al., 1980; Fields, 2004; Haghparast et al., 2008), changing the functional connectivity within this system during exogenous opioid application (Petrovic et al., 2002) and opioid-dependent endogenous analgesia (Petrovic et al., 2002; Eippert et al., 2009a; Sprenger et al., 2012), and previous studies suggesting altered functionality of this system to be crucially linked to opioid related increase in pain sensitivity (Kaplan and Fields, 1991; Heinricher et al., 2001; Vanderah et al., 2001a,b; Ossipov et al., 2003), we next directly investigated whether such connectivity changes can be observed in humans subsequent to remifentanil short-term application.
Our functional connectivity analysis revealed significant functional coupling between the right NCF and the rACC (−2, 42, −2) that exhibited an opioid-suspension-related pharmacological interaction effect. During the baseline session, we observed a similar functional coupling between the right NCF and the rACC in both groups (saline group, r' = 0.16 ± 0.03; remifentanil group, r' = 0.13 ± 0.03). However, while NCF-rACC coupling showed a reduction during the second session in the saline group (r' = 0.06 ± 0.04), NCF-rACC coupling increased after remifentanil application (r' = 0.18 ± 0.03), leading to a significant pharmacological interaction effect (t(30) = 2.24, p = 0.02; Fig. 6A–C).
Figure 6.

Functional connectivity of the right NCF and the rACC. A, Approximate location of the NCF and the rACC depicted on the mediosagittal plane of the average structural image. To test for NCF-rACC connectivity, we first determined the seed voxel by the contrast testing for increased responses after remifentanil discontinuation during the nonpainful phase of stimulation (see Fig. 5). B, Subsequently, we used the time series from the NCF as predictor in a new GLM analysis to test for remifentanil discontinuation-related changes in connectivity. We identified the rACC in a second-level group analysis. C, We finally extracted also the time series from the rACC and correlated the rACC time series with those from the NCF. While the intraindividual strength of coupling (as represented by the average correlation coefficient) showed a marked decrease from Session 1 (s1) to Session 2 (s2) in the control group, the coupling strength increased during the second session after remifentanil suspension, leading to a significant interaction effect between groups. Error bars indicate SEM. *p < 0.05. D, The individual NCF-rACC coupling strength shows a negative correlation with the HPT after discontinuation of remifentanil (right), but not after saline application (left). Right, The fitted line was determined by a robust regression.
With regard to the functional relevance of this finding, we observed that, after remifentanil short-term infusion, the individual strength of this coupling showed a strong negative correlation with the HPTs (r = −0.7, p = 0.004; Fig. 6D); that is, the stronger the individual opioid-offset related NCF-rACC coupling, the lower the HPT. This correlation was significantly more negative compared with the saline group (z = 2.6, p = 0.01).
Similarly to the right NCF, we observed functional coupling between the left NCF and the rACC (−16, 46, 2) that likewise exhibited a pharmacological interaction effect (t(30) = 3.5, p < 0.001).
If rACC-NCF coupling is involved in a pain facilitating mechanism after opioid application, it can be expected that NCF activity subsequently also predicts activity in the RVM, which receives direct fiber input from the NCF (Edwards, 1975; Zemlan and Behbehani, 1988) and takes a key position in descending pain modulation (Millan, 2002; Heinricher et al., 2009). In line with this hypothesis, we also observed functional coupling between the NCF and the RVM that increased after remifentanil suspension (remifentanil group session 1, r' = −0.08 ± 0.02, remifentanil group session 2, r' = 0.13 ± 0.03), but the strength of coupling displayed no pharmacological interaction effect (t(30) = 0.03, p > 0.05). However, similar to the rACC-NCF coupling, the individual strength of the right NCF-RVM coupling correlated negatively with the HPT after remifentanil application (r = −0.56, p = 0.03), and this correlation was significantly stronger compared with the saline control group (z = 2.1, p = 0.02).
We also observed functional coupling between the PAG and the rACC that was increased after remifentanil suspension. However, the pharmacological interaction failed significance (t(30) = 1.4, p = 0.09).
BOLD responses subsequent to remifentanil short-term application at the spinal level
We finally investigated neuronal responses at the spinal level using high-resolution fMRI of the human cervical spinal cord. To localize the core region of nociceptive processing in the spinal cord in response to signals originating from painful stimulation to the left radial forearm (dermatome C6), we first determined BOLD responses to the painful phase of stimulation before application of the test substance across all participants. In accordance with the functional neuroanatomy of the nociceptive system, we observed a marked BOLD response in the dorsal horn of the spinal segment C6 ipsilateral to the side of stimulation (t(112) = 8.48, p < 0.001; Fig. 7A–C). No further significant clusters and no negative responses were detected within the spinal cord. We subsequently analyzed the effect of opioid suspension on BOLD responses at this site. We observed no significant pharmacological interaction effect. Also, extending our analysis to the entire measured volume did not reveal a significant modulation of BOLD responses by remifentanil suspension.
Figure 7.

Spinal results. A, Spinal BOLD response to the painful phase of stimulation during the baseline session across all participants. The activation can be most likely attributed to the spinal segment C6. Green line indicates the transverse section (C). B, Enlarged detail of the sagittal depiction (A). C, BOLD responses overlaid on a transversal section of a structural T2*-weighted image. The figure shows that the peak of the BOLD response is located in deeper sections the dorsal horn, ipsilateral to the side of stimulation (left). Color bar represents t values. The visualization threshold is set to p < 0.005 uncorrected. D, The parameter estimates at the peak voxel of the BOLD response to painful stimulation show a comparable BOLD response to the painful phase of stimulation (p) in both groups during the baseline session (s1), but no detectable response for the nonpainful period of thermal stimulation (w). BOLD responses to painful stimulation are strongly reduced during the second experimental session (s2) hindering the uncovering of opioid suspension-related group differences. Error bars indicate SEM. E, However, an MVPA was able to successfully decode the pharmacological condition based on the activity patterns during the second experimental session in a previously identified region in the spinal cord that showed a strong positive BOLD response to painful stimulation (see Results). To investigate the specificity of our findings regarding the involved neuronal pathway, we subsequently conducted a searchlight MVPA across the entire measured volume that likewise compared the spinal activity patterns between the remifentanil and the saline group during the second session. The figure shows spatial coincidence of high predictive values in the accuracy map (red, threshold set to average accuracy > 75%) with the region in the spinal cord that showed a significant BOLD response to painful stimulation (green).
To further pursue the assumption of an opioid evoked modulation of neuronal activity in the spinal cord, we subsequently analyzed the ROI in the spinal cord that showed a BOLD response to painful stimulation in our previous analysis with an MVPA. For this analysis, we used a linear support vector machine classifier and a leave-one-subject-out cross-validation to decode the pharmacological condition based on the spinal activity patterns in response to the painful phase of stimulation. While the MVPA was not able to decode the pharmacological condition during the baseline session (average accuracy 56.7%, not significant), it was successfully able to decode the remifentanil suspension and the saline condition better than chance after application of the test substance (average accuracy 83.3%, p = 0.005). An MVPA based on the activity patterns of the nonpainful phase of stimulation was not able to decode the pharmacological condition.
To demonstrate the specificity of our ROI-based finding regarding the involved neuronal pathway, we subsequently conducted a searchlight MVPA across the entire measured volume, which quantitatively measures the degree to which spatial activation patterns predict the pharmacological condition. This analysis showed the highest values at the initially determined site of the BOLD response to painful stimulation in the dorsal horn of the spinal segment C6 (Fig. 7D).
Discussion
Performing two fMRI experiments with optimized protocols for the respective CNS regions allowed us to study remifentanil-induced neuronal changes along the entire central pain pathway. Remifentanil discontinuation lowered HPTs and increased BOLD responses to painful stimulation in brain regions that exhibit high μ-opioid receptor density and that are crucially involved in nociceptive processing, such as the thalamus, posterior insula, amygdala, and RVM. Already before reaching a painful level of stimulation, remifentanil suspension increased neuronal responses in the PAG and NCF. Opioid discontinuation increased functional connectivity between NCF and rACC, and both the strength of rACC-NCF and NCF-RVM connectivity correlated negatively with the individual HPT after application of remifentanil. Finally, we observed an opioid-suspension-specific modulation of the spinal multivoxel activity pattern. Our results thus delineate specific opioid-dependent alterations in pain-encoding and pain-modulatory systems in humans after short-term application of remifentanil that are directly related to the behavioral sensitivity to pain.
Experiments in both animal and human volunteers have demonstrated increased sensitivity to pain after exposure to opioids across many different nociceptive setups and routes of administration. Whereas most human studies investigated the effect of opioid suspension in models of preexisting hyperalgesia (Petersen et al., 2001; Angst et al., 2003; Hood et al., 2003), the current study extends previous observations by showing that short-term infusion of remifentanil affects HPTs and central pain processing after opioid discontinuation in an injury-free experimental model without presensitizing the skin. Under these experimental conditions, remifentanil caused a reliable effect on HPTs, whereas heat pain ratings were not altered. This observation is interesting as it highlights a partial independence of the underlying CNS mechanisms and is in line with human studies showing no effect (Angst et al., 2003; Hood et al., 2003) of opioid suspension on thermal pain ratings but on other experimental parameters.
Altered signaling after opioid suspension in the brain and brainstem
In the brain, we observed increased BOLD responses during painful stimulation subsequent to remifentanil discontinuation in the thalamus, amygdala, and posterior insula. All three regions are involved in nociceptive processing, belong to the regions exhibiting a high opioid receptor density (Henriksen and Willoch, 2008), and are directly modulated during μ-opioid receptor agonist application (Wise et al., 2002; Leppä et al., 2006; Atlas et al., 2012). A series of animal studies indicated the paraventricular thalamus and the amygdala to be related to the highly aversive withdrawal symptoms after prolonged opioid consumption (Gracy et al., 2001; Watanabe et al., 2002; Zhu et al., 2016). However, given that short-term opioid exposure generated only weak effects on mood and parameters of well-being after discontinuation, it appears unlikely that the observed BOLD changes in the amygdala reflect fear, aversiveness, or stress effects related to pain.
In the brainstem, we observed BOLD responses in the RVM that were increased after opioid application during painful stimulation. The RVM is well established to be an essential source of bulbospinal inhibitory and facilitatory modulation of nociceptive inputs receiving numerous inputs from other brainstem regions, such as the PAG and the NCF (Millan, 2002). Importantly, our finding is in agreement with a series of conclusive rodent studies showing a crucial contribution of this region to the occurrence of opioid-induced increased pain sensitivity (Bederson et al., 1990; Kaplan and Fields, 1991; Bie et al., 2003; Neubert et al., 2004). These studies demonstrated a strong increase in on-cell activity and silencing of off-cell activity after naloxone-precipitated opioid withdrawal, thus displaying an inverted picture of acute opioid effects, subsequently leading to a descending facilitation of pain (Vanderah et al., 2001b).
Because we did not record BOLD responses during remifentanil application, we cannot know which direct effects remifentanil exerted on the neuronal circuits showing altered signaling after discontinuation. However, previous studies have shown that the used and even lower doses of remifentanil dose cause marked behavioral analgesia and both reduced neuronal responses to noxious stimuli in pain-related brain regions and altered functional coupling with the brainstem (Petrovic et al., 2002; Wise et al., 2002, 2004; Koppert et al., 2003a,b).
To capture neuronal mechanisms underlying the decreased HPTs, we also analyzed the time period before the threshold. After opioid discontinuation, we observed BOLD responses in the PAG and the bilateral NCF that were exclusively elevated before the HPT. The NCF is an important brainstem site for pain-modulatory processes (Gray and Dostrovsky, 1983; Richter and Behbehani, 1991), involved in the mediation of acute opioid effects (Haghparast et al., 2007, 2008; Ronaghi et al., 2011), negative and positive expectation effects (Keltner et al., 2006; Wager et al., 2007), and most notably in central sensitization processes (Zambreanu et al., 2005; M. C. Lee et al., 2008; Wanigasekera et al., 2016), including opioid withdrawal (Wanigasekera et al., 2011). The NCF receives ample input from spinal lamina I neurons (McMahon and Wall, 1985; Hylden et al., 1986) but also strong input from the PAG, which in turn receives inputs from the rACC, hypothalamus, and amygdala (Mantyh, 1982; An et al., 1998; Floyd et al., 2000). On the other hand, the NCF mainly projects to the RVM, which exerts direct control over dorsal horn neurons (Fields et al., 1995; Zhuo and Gebhart, 1997). Interestingly, we observed the BOLD increase after opioid withdrawal in the NCF directly before the HPT, which is reminiscent of the fact that on-cells fire shortly before the occurrence of nociceptive withdrawal reflexes (Heinricher et al., 1987; Haws et al., 1989). Similar to our study, Wanigasekera et al. (2011) observed heightened BOLD responses in the NCF in a subset of individuals that showed behavioral pain increase after short-term remifentanil exposure. The authors additionally reported a negative correlation between withdrawal-related rise in NCF activity and changes in pain scores pointing at a possible inhibitory role of the NCF in responders. In contrast, our data indicate a predominantly pain-facilitatory role. Such a facilitatory influence of the NCF after opioid withdrawal appears plausible in view of the response characteristics of on-cells (Haws et al., 1989; Pan et al., 1990; Neubert et al., 2004), electrophysiological features (Haghparast et al., 2010), and a mainly excitatory effect of the NCF on the RVM (Richter and Behbehani, 1991).
Opioid suspension and spinal cord processing of thermal stimuli
In accordance with previous studies, we were able to detect robust pain-related BOLD responses in the ipsilateral spinal cord with the strongest response centered in deeper portions of the dorsal horn. The activation of heat-sensitive nociceptors by a noxious thermal stimulus induces a considerably stronger response in the spinal cord compared with a non-noxious warm stimulus (Coghill et al., 1991, 1993), potentially even triggering NMDA receptor unblocking from Mg2+ after repetitive stimulation (Dickenson and Sullivan, 1990; Sandkühler, 2009). In line with the present results, previous spinal fMRI studies reported only weak or absent BOLD responses after non-noxious thermal stimulation (Brooks et al., 2002; Summers et al., 2010; Weber et al., 2016), but frequently responses to noxious stimulation (Kolesar et al., 2015). Pain-related BOLD responses in the spinal cord showed a marked decrease during the second experimental session. Reduced responses to repeated thermal stimuli have been previously reported in multisession experiments (Letzen et al., 2014; Quiton et al., 2014), and similar phenomena are generally recognized in context of stimulus repetition and are presumably related to firing-rate adaptation and synaptic depression (Grill-Spector et al., 2006; Nickel et al., 2014). In the present study, this assumption is supported by the observation that, during the second session, also BOLD responses in the brain were weaker in the saline group. Given the considerably higher level of physiological noise in spinal fMRI (Fratini et al., 2014), the detection of responses to thermal stimuli during the second session and consequently of opioid-induced neuronal changes was therefore hampered at this level. However, using a more sensitive multivariate approach allowed us to successfully decode the pharmacological condition based on the spinal activity patterns after application of the test substance. This was not possible for the baseline session, hence indicating an opioid-suspension-induced modulation of neuronal activity.
Effects of remifentanil suspension on functional connectivity
We finally attempted to functionally link structures within the descending pain-modulatory system by using functional connectivity analyses. Our analysis revealed significantly enhanced functional connectivity between NCF and rACC after remifentanil discontinuation. Moreover, we observed enhanced functional connectivity between rACC and PAG, but without significant pharmacological interaction. Given that the NCF receives input from the rACC mainly indirectly via connections with the PAG that in turn receives direct input from the rACC, it appears plausible that the functional connectivity between rACC and NCF is structurally mediated via the PAG. There is a large body of evidence showing rACC–brainstem connectivity to be an important functional pathway mediating influences of opioid-receptor-rich forebrain regions to phylogenetically old brainstem sites that exert direct control over the spinal cord in different types of endogenous analgesia (Petrovic et al., 2002; Valet et al., 2004; Eippert et al., 2009a). Although it is known that this pathway can be recruited by both exogenous opioid application and cognitive/motivational factors, our study now shows that abrupt opioid suspension most likely sets the functionality of this system into a pronociceptive mode. In support of this view, we could show that the opioid-withdrawal-enhanced functional connectivity within the descending pain-modulatory system is of functional relevance, as both the individual strength of the rACC-NCF and of the NCF-RVM coupling correlated negatively with the HPT, thus linking the signaling within the most rostral portions of the descending pain-modulatory system with the RVM, which directly controls spinal nociceptive transmission.
In conclusion, our study indicates remifentanil-suspension-related changes along the entire central pain pathway, particularly in key structures of the descending pain-modulatory system. Both neuronal signaling within and functional connectivity between these structures corresponded with the behavioral pain sensitivity. Our findings thus demonstrate a crucial involvement of this system in increased sensitivity to pain subsequent to opioid application in humans.
Footnotes
C.S. was supported by German Research Foundation SP-1668. C.B. was supported by German Research Foundation SFB 936 Project A06 and SFB TRR 134. C.B. and C.S. were supported by European Research Council ERC-2010-AdG_20100407.
The authors declare no competing financial interests.
References
- An X, Bandler R, Ongür D, Price JL (1998) Prefrontal cortical projections to longitudinal columns in the midbrain periaqueductal gray in macaque monkeys. J Comp Neurol 401:455–479. [DOI] [PubMed] [Google Scholar]
- Angst MS, Clark JD (2006) Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology 104:570–587. 10.1097/00000542-200603000-00025 [DOI] [PubMed] [Google Scholar]
- Angst MS, Koppert W, Pahl I, Clark DJ, Schmelz M (2003) Short-term infusion of the mu-opioid agonist remifentanil in humans causes hyperalgesia during withdrawal. Pain 106:49–57. 10.1016/S0304-3959(03)00276-8 [DOI] [PubMed] [Google Scholar]
- Apkarian AV, Bushnell MC, Treede RD, Zubieta JK (2005) Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain 9:463–484. 10.1016/j.ejpain.2004.11.001 [DOI] [PubMed] [Google Scholar]
- Atlas LY, Whittington RA, Lindquist MA, Wielgosz J, Sonty N, Wager TD (2012) Dissociable influences of opiates and expectations on pain. J Neurosci 32:8053–8064. 10.1523/JNEUROSCI.0383-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton C, Basbaum AI, Fields HL (1980) Dissociation of supraspinal and spinal actions of morphine: a quantitative evaluation. Brain Res 188:487–498. 10.1016/0006-8993(80)90047-5 [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Fields HL (1984) Endogenous pain control systems: brainstem spinal pathways and endorphin circuitry. Annu Rev Neurosci 7:309–338. 10.1146/annurev.ne.07.030184.001521 [DOI] [PubMed] [Google Scholar]
- Bederson JB, Fields HL, Barbaro NM (1990) Hyperalgesia during naloxone-precipitated withdrawal from morphine is associated with increased on-cell activity in the rostral ventromedial medulla. Somatosens Mot Res 7:185–203. 10.3109/08990229009144706 [DOI] [PubMed] [Google Scholar]
- Beissner F. (2015) Functional MRI of the brainstem: common problems and their solutions. Clin Neuroradiol 25 [Suppl. 2]:251–257. [DOI] [PubMed] [Google Scholar]
- Bie B, Fields HL, Williams JT, Pan ZZ (2003) Roles of alpha1- and alpha2-adrenoceptors in the nucleus raphe magnus in opioid analgesia and opioid abstinence-induced hyperalgesia. J Neurosci 23:7950–7957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar RJ, Kest B (2010) Sex differences in opioid analgesia, hyperalgesia, tolerance and withdrawal: central mechanisms of action and roles of gonadal hormones. Horm Behav 58:72–81. 10.1016/j.yhbeh.2009.09.012 [DOI] [PubMed] [Google Scholar]
- Bowdle TA, Camporesi EM, Maysick L, Hogue CW Jr, Miguel RV, Pitts M, Streisand JB (1996) A multicenter evaluation of remifentanil for early postoperative analgesia. Anesth Analg 83:1292–1297. 10.1213/00000539-199612000-00028 [DOI] [PubMed] [Google Scholar]
- Brooks JC. (2014) Physiological noise modeling and analysis for spinal cord fMRI. In: Quantitative MRI of the spinal cord (Wheeler-Kingshott C, ed), pp 240–257. Burlington, MA: Elsevier Science. [Google Scholar]
- Brooks JC, Nurmikko TJ, Bimson WE, Singh KD, Roberts N (2002) fMRI of thermal pain: effects of stimulus laterality and attention. Neuroimage 15:293–301. 10.1006/nimg.2001.0974 [DOI] [PubMed] [Google Scholar]
- Brooks JC, Beckmann CF, Miller KL, Wise RG, Porro CA, Tracey I, Jenkinson M (2008) Physiological noise modelling for spinal functional magnetic resonance imaging studies. Neuroimage 39:680–692. 10.1016/j.neuroimage.2007.09.018 [DOI] [PubMed] [Google Scholar]
- Brown CH, Russell JA (2004) Cellular mechanisms underlying neuronal excitability during morphine withdrawal in physical dependence: lessons from the magnocellular oxytocin system. Stress 7:97–107. 10.1080/10253890410001727776 [DOI] [PubMed] [Google Scholar]
- Célèrier E, Laulin JP, Corcuff JB, Le Moal M, Simonnet G (2001) Progressive enhancement of delayed hyperalgesia induced by repeated heroin administration: a sensitization process. J Neurosci 21:4074–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu LF, Lin JC, Clemenson A, Encisco E, Sun J, Hoang D, Alva H, Erlendson M, Clark JD, Younger JW (2015) Acute opioid withdrawal is associated with increased neural activity in reward-processing centers in healthy men: a functional magnetic resonance imaging study. Drug Alcohol Depend 153:314–322. 10.1016/j.drugalcdep.2015.04.019 [DOI] [PubMed] [Google Scholar]
- Coghill RC, Price DD, Hayes RL, Mayer DJ (1991) Spatial distribution of nociceptive processing in the rat spinal cord. J Neurophysiol 65:133–140. 10.1152/jn.1991.65.1.133 [DOI] [PubMed] [Google Scholar]
- Coghill RC, Mayer DJ, Price DD (1993) The roles of spatial recruitment and discharge frequency in spinal cord coding of pain: a combined electrophysiological and imaging investigation. Pain 53:295–309. 10.1016/0304-3959(93)90226-F [DOI] [PubMed] [Google Scholar]
- Deckers RH, van Gelderen P, Ries M, Barret O, Duyn JH, Ikonomidou VN, Fukunaga M, Glover GH, de Zwart JA (2006) An adaptive filter for suppression of cardiac and respiratory noise in MRI time series data. Neuroimage 33:1072–1081. 10.1016/j.neuroimage.2006.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickenson AH, Sullivan AF (1990) Differential effects of excitatory amino acid antagonists on dorsal horn nociceptive neurones in the rat. Brain Res 506:31–39. 10.1016/0006-8993(90)91195-M [DOI] [PubMed] [Google Scholar]
- Drdla R, Gassner M, Gingl E, Sandkühler J (2009) Induction of synaptic long-term potentiation after opioid withdrawal. Science 325:207–210. 10.1126/science.1171759 [DOI] [PubMed] [Google Scholar]
- Duerden EG, Albanese MC (2013) Localization of pain-related brain activation: a meta-analysis of neuroimaging data. Hum Brain Mapp 34:109–149. 10.1002/hbm.21416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SB. (1975) Autoradiographic studies of the projections of the midbrain reticular formation: descending projections of nucleus cuneiformis. J Comp Neurol 161:341–358. 10.1002/cne.901610306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eippert F, Bingel U, Schoell ED, Yacubian J, Klinger R, Lorenz J, Büchel C (2009a) Activation of the opioidergic descending pain control system underlies placebo analgesia. Neuron 63:533–543. 10.1016/j.neuron.2009.07.014 [DOI] [PubMed] [Google Scholar]
- Eippert F, Finsterbusch J, Bingel U, Büchel C (2009b) Direct evidence for spinal cord involvement in placebo analgesia. Science 326:404. 10.1126/science.1180142 [DOI] [PubMed] [Google Scholar]
- Eippert F, Kong Y, Jenkinson M, Tracey I, Brooks JC (2017) Denoising spinal cord fMRI data: approaches to acquisition and analysis. Neuroimage 154:255–266. 10.1016/j.neuroimage.2016.09.065 [DOI] [PubMed] [Google Scholar]
- Fields H. (2004) State-dependent opioid control of pain. Nat Rev Neurosci 5:565–575. 10.1038/nrn1431 [DOI] [PubMed] [Google Scholar]
- Fields HL, Malick A, Burstein R (1995) Dorsal horn projection targets of ON and OFF cells in the rostral ventromedial medulla. J Neurophysiol 74:1742–1759. 10.1152/jn.1995.74.4.1742 [DOI] [PubMed] [Google Scholar]
- Finsterbusch J, Eippert F, Büchel C (2012) Single, slice-specific z-shim gradient pulses improve T2*-weighted imaging of the spinal cord. Neuroimage 59:2307–2315. 10.1016/j.neuroimage.2011.09.038 [DOI] [PubMed] [Google Scholar]
- Fishbain DA, Cole B, Lewis JE, Gao J, Rosomoff RS (2009) Do opioids induce hyperalgesia in humans? An evidence-based structured review. Pain Med 10:829–839. 10.1111/j.1526-4637.2009.00653.x [DOI] [PubMed] [Google Scholar]
- Fletcher D, Martinez V (2014) Opioid-induced hyperalgesia in patients after surgery: a systematic review and a meta-analysis. Br J Anaesth 112:991–1004. 10.1093/bja/aeu137 [DOI] [PubMed] [Google Scholar]
- Floyd NS, Price JL, Ferry AT, Keay KA, Bandler R (2000) Orbitomedial prefrontal cortical projections to distinct longitudinal columns of the periaqueductal gray in the rat. J Comp Neurol 422:556–578. [DOI] [PubMed] [Google Scholar]
- Fratini M, Moraschi M, Maraviglia B, Giove F (2014) On the impact of physiological noise in spinal cord functional MRI. J Magn Reson Imaging 40:770–777. 10.1002/jmri.24467 [DOI] [PubMed] [Google Scholar]
- Gardell LR, Wang R, Burgess SE, Ossipov MH, Vanderah TW, Malan TP Jr, Lai J, Porreca F (2002) Sustained morphine exposure induces a spinal dynorphin-dependent enhancement of excitatory transmitter release from primary afferent fibers. J Neurosci 22:6747–6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuter S, Büchel C (2013) Facilitation of pain in the human spinal cord by nocebo treatment. J Neurosci 33:13784–13790. 10.1523/JNEUROSCI.2191-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracy KN, Dankiewicz LA, Koob GF (2001) Opiate withdrawal-induced fos immunoreactivity in the rat extended amygdala parallels the development of conditioned place aversion. Neuropsychopharmacology 24:152–160. 10.1016/S0893-133X(00)00186-X [DOI] [PubMed] [Google Scholar]
- Gray BG, Dostrovsky JO (1983) Descending inhibitory influences from periaqueductal gray, nucleus raphe magnus, and adjacent reticular formation: I. Effects on lumbar spinal cord nociceptive and nonnociceptive neurons. J Neurophysiol 49:932–947. 10.1152/jn.1983.49.4.932 [DOI] [PubMed] [Google Scholar]
- Grill-Spector K, Henson R, Martin A (2006) Repetition and the brain: neural models of stimulus-specific effects. Trends Cogn Sci 10:14–23. 10.1016/j.tics.2005.11.006 [DOI] [PubMed] [Google Scholar]
- Guignard B, Bossard AE, Coste C, Sessler DI, Lebrault C, Alfonsi P, Fletcher D, Chauvin M (2000) Acute opioid tolerance: intraoperative remifentanil increases postoperative pain and morphine requirement. Anesthesiology 93:409–417. 10.1097/00000542-200008000-00019 [DOI] [PubMed] [Google Scholar]
- Haghparast A, Gheitasi IP, Lashgari R (2007) Involvement of glutamatergic receptors in the nucleus cuneiformis in modulating morphine-induced antinociception in rats. Eur J Pain 11:855–862. 10.1016/j.ejpain.2006.12.010 [DOI] [PubMed] [Google Scholar]
- Haghparast A, Ordikhani-Seyedlar M, Ziaei M (2008) Electrolytic lesion of the nucleus raphe magnus reduced the antinociceptive effects of bilateral morphine microinjected into the nucleus cuneiformis in rats. Neurosci Lett 438:351–355. 10.1016/j.neulet.2008.04.072 [DOI] [PubMed] [Google Scholar]
- Haghparast A, Naderi N, Khani A, Lashgari R, Motamedi F (2010) Formalin-induced differential activation of nucleus cuneiformis neurons in the rat: an electrophysiological study. J Pain 11:32–43. 10.1016/j.jpain.2009.05.005 [DOI] [PubMed] [Google Scholar]
- Haws CM, Williamson AM, Fields HL (1989) Putative nociceptive modulatory neurons in the dorsolateral pontomesencephalic reticular formation. Brain Res 483:272–282. 10.1016/0006-8993(89)90171-6 [DOI] [PubMed] [Google Scholar]
- Heinl C, Drdla-Schutting R, Xanthos DN, Sandkühler J (2011) Distinct mechanisms underlying pronociceptive effects of opioids. J Neurosci 31:16748–16756. 10.1523/JNEUROSCI.3491-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinricher MM, Cheng ZF, Fields HL (1987) Evidence for two classes of nociceptive modulating neurons in the periaqueductal gray. J Neurosci 7:271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinricher MM, McGaraughty S, Tortorici V (2001) Circuitry underlying antiopioid actions of cholecystokinin within the rostral ventromedial medulla. J Neurophysiol 85:280–286. 10.1152/jn.2001.85.1.280 [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Tavares I, Leith JL, Lumb BM (2009) Descending control of nociception: specificity, recruitment and plasticity. Brain Res Rev 60:214–225. 10.1016/j.brainresrev.2008.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen G, Willoch F (2008) Imaging of opioid receptors in the central nervous system. Brain 131:1171–1196. 10.1093/brain/awm255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood DD, Curry R, Eisenach JC (2003) Intravenous remifentanil produces withdrawal hyperalgesia in volunteers with capsaicin-induced hyperalgesia. Anesth Analg 97:810–815. [DOI] [PubMed] [Google Scholar]
- Hylden JL, Hayashi H, Dubner R, Bennett GJ (1986) Physiology and morphology of the lamina I spinomesencephalic projection. J Comp Neurol 247:505–515. 10.1002/cne.902470410 [DOI] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ (2003) Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci 26:696–705. 10.1016/j.tins.2003.09.017 [DOI] [PubMed] [Google Scholar]
- Kaplan H, Fields HL (1991) Hyperalgesia during acute opioid abstinence: evidence for a nociceptive facilitating function of the rostral ventromedial medulla. J Neurosci 11:1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keltner JR, Furst A, Fan C, Redfern R, Inglis B, Fields HL (2006) Isolating the modulatory effect of expectation on pain transmission: a functional magnetic resonance imaging study. J Neurosci 26:4437–4443. 10.1523/JNEUROSCI.4463-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesar TA, Fiest KM, Smith SD, Kornelsen J (2015) Assessing nociception by fMRI of the human spinal cord: a systematic review. Magn Reson Insights 8:31–39. 10.4137/MRI.S23556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y, Jenkinson M, Andersson J, Tracey I, Brooks JC (2012) Assessment of physiological noise modelling methods for functional imaging of the spinal cord. Neuroimage 60:1538–1549. 10.1016/j.neuroimage.2011.11.077 [DOI] [PubMed] [Google Scholar]
- Koppert W, Angst M, Alsheimer M, Sittl R, Albrecht S, Schüttler J, Schmelz M (2003a) Naloxone provokes similar pain facilitation as observed after short-term infusion of remifentanil in humans. Pain 106:91–99. 10.1016/S0304-3959(03)00294-X [DOI] [PubMed] [Google Scholar]
- Koppert W, Sittl R, Scheuber K, Alsheimer M, Schmelz M, Schüttler J (2003b) Differential modulation of remifentanil-induced analgesia and postinfusion hyperalgesia by S-ketamine and clonidine in humans. Anesthesiology 99:152–159. 10.1097/00000542-200307000-00025 [DOI] [PubMed] [Google Scholar]
- Lee CW, Ho IK (2013) Sex differences in opioid analgesia and addiction: interactions among opioid receptors and estrogen receptors. Mol Pain 9:45. 10.1186/1744-8069-9-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Silverman SM, Hansen H, Patel VB, Manchikanti L (2011) A comprehensive review of opioid-induced hyperalgesia. Pain Physician 14:145–161. [PubMed] [Google Scholar]
- Lee MC, Zambreanu L, Menon DK, Tracey I (2008) Identifying brain activity specifically related to the maintenance and perceptual consequence of central sensitization in humans. J Neurosci 28:11642–11649. 10.1523/JNEUROSCI.2638-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppä M, Korvenoja A, Carlson S, Timonen P, Martinkauppi S, Ahonen J, Rosenberg PH, Aronen HJ, Kalso E (2006) Acute opioid effects on human brain as revealed by functional magnetic resonance imaging. Neuroimage 31:661–669. 10.1016/j.neuroimage.2005.12.019 [DOI] [PubMed] [Google Scholar]
- Letzen JE, Sevel LS, Gay CW, O'Shea AM, Craggs JG, Price DD, Robinson ME (2014) Test-retest reliability of pain-related brain activity in healthy controls undergoing experimental thermal pain. J Pain 15:1008–1014. 10.1016/j.jpain.2014.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantyh PW. (1982) Forebrain projections to the periaqueductral gray in the monkey, with observations in the cat and rat. J Comp Neurol 206:146–158. 10.1002/cne.902060205 [DOI] [PubMed] [Google Scholar]
- Mao J. (2002) Opioid-induced abnormal pain sensitivity: implications in clinical opioid therapy. Pain 100:213–217. 10.1016/S0304-3959(02)00422-0 [DOI] [PubMed] [Google Scholar]
- Mason P. (2012) Medullary circuits for nociceptive modulation. Curr Opin Neurobiol 22:640–645. 10.1016/j.conb.2012.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon SB, Wall PD (1985) Electrophysiological mapping of brainstem projections of spinal cord lamina I cells in the rat. Brain Res 333:19–26. 10.1016/0006-8993(85)90119-2 [DOI] [PubMed] [Google Scholar]
- Millan MJ. (2002) Descending control of pain. Prog Neurobiol 66:355–474. 10.1016/S0301-0082(02)00009-6 [DOI] [PubMed] [Google Scholar]
- Myers L, Sirois MJ (2006) Spearman correlation coefficients, differences between. New York, NY: Wiley StatsRef. [Google Scholar]
- Naidich TP. (2009) Duvernoy's atlas of the human brain stem and cerebellum. Springer 2009 10.1007/978-3-211-73971-6 [DOI] [Google Scholar]
- Napadow V, Dhond R, Kennedy D, Hui KK, Makris N (2006) Automated brainstem co-registration (ABC) for MRI. Neuroimage 32:1113–1119. 10.1016/j.neuroimage.2006.05.050 [DOI] [PubMed] [Google Scholar]
- Neubert MJ, Kincaid W, Heinricher MM (2004) Nociceptive facilitating neurons in the rostral ventromedial medulla. Pain 110:158–165. 10.1016/j.pain.2004.03.017 [DOI] [PubMed] [Google Scholar]
- Nichols TE, Holmes AP (2002) Nonparametric permutation tests for functional neuroimaging: a primer with examples. Hum Brain Mapp 15:1–25. 10.1002/hbm.1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel FT, Ott S, Möhringer S, Saake M, Dörfler A, Seifert F, Maihöfner C (2014) Brain correlates of short-term habituation to repetitive electrical noxious stimulation. Eur J Pain 18:56–66. 10.1002/j.1532-2149.2013.00339.x [DOI] [PubMed] [Google Scholar]
- Niesters M, Dahan A, Kest B, Zacny J, Stijnen T, Aarts L, Sarton E (2010) Do sex differences exist in opioid analgesia? A systematic review and meta-analysis of human experimental and clinical studies. Pain 151:61–68. 10.1016/j.pain.2010.06.012 [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, Vanderah TW, Porreca F (2003) Induction of pain facilitation by sustained opioid exposure: relationship to opioid antinociceptive tolerance. Life Sci 73:783–800. 10.1016/S0024-3205(03)00410-7 [DOI] [PubMed] [Google Scholar]
- Pan ZZ, Williams JT, Osborne PB (1990) Opioid actions on single nucleus raphe magnus neurons from rat and guinea-pig in vitro. J Physiol 427:519–532. 10.1113/jphysiol.1990.sp018185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak GW, Pan YX (2013) Mu opioids and their receptors: evolution of a concept. Pharmacol Rev 65:1257–1317. 10.1124/pr.112.007138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KL, Jones B, Segredo V, Dahl JB, Rowbotham MC (2001) Effect of remifentanil on pain and secondary hyperalgesia associated with the heat-capsaicin sensitization model in healthy volunteers. Anesthesiology 94:15–20. 10.1097/00000542-200101000-00008 [DOI] [PubMed] [Google Scholar]
- Petrovic P, Kalso E, Petersson KM, Ingvar M (2002) Placebo and opioid analgesia: imaging a shared neuronal network. Science 295:1737–1740. 10.1126/science.1067176 [DOI] [PubMed] [Google Scholar]
- Quiton RL, Keaser ML, Zhuo J, Gullapalli RP, Greenspan JD (2014) Intersession reliability of fMRI activation for heat pain and motor tasks. Neuroimage Clin 5:309–321. 10.1016/j.nicl.2014.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K, Dubner R (2007) Pain facilitation and activity-dependent plasticity in pain-modulatory circuitry: role of BDNF-TrkB signaling and NMDA receptors. Mol Neurobiol 35:224–235. 10.1007/s12035-007-0028-8 [DOI] [PubMed] [Google Scholar]
- Richter RC, Behbehani MM (1991) Evidence for glutamic acid as a possible neurotransmitter between the mesencephalic nucleus cuneiformis and the medullary nucleus raphe magnus in the lightly anesthetized rat. Brain Res 544:279–286. 10.1016/0006-8993(91)90065-4 [DOI] [PubMed] [Google Scholar]
- Roeckel LA, Le Coz GM, Gavériaux-Ruff C, Simonin F (2016) Opioid-induced hyperalgesia: cellular and molecular mechanisms. Neuroscience 338:160–182. 10.1016/j.neuroscience.2016.06.029 [DOI] [PubMed] [Google Scholar]
- Rolke R, Baron R, Maier C, Tölle TR, Treede RD, Beyer A, Binder A, Birbaumer N, Birklein F, Bötefür IC, Braune S, Flor H, Huge V, Klug R, Landwehrmeyer GB, Magerl W, Maihöfner C, Rolko C, Schaub C, Scherens A, et al. (2006) Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): standardized protocol and reference values. Pain 123:231–243. 10.1016/j.pain.2006.01.041 [DOI] [PubMed] [Google Scholar]
- Ronaghi A, Ebrahimzadeh M, Haghparast A (2011) Contribution of the nucleus cuneiformis to the antinociceptive effects of systemic morphine on inflammatory pain in rats. Basic Clin Neurosci 3:35–43. [Google Scholar]
- Rossbach MJ. 1880. Ueber die Gewoehnung an Gifte. Pflügers Arch Eur J Physiol 21:213–225. [Google Scholar]
- Sandkühler J. (2009) Models and mechanisms of hyperalgesia and allodynia. Physiol Rev 89:707–758. 10.1152/physrev.00025.2008 [DOI] [PubMed] [Google Scholar]
- Simonin F, Schmitt M, Laulin JP, Laboureyras E, Jhamandas JH, MacTavish D, Matifas A, Mollereau C, Laurent P, Parmentier M, Kieffer BL, Bourguignon JJ, Simonnet G (2006) RF9, a potent and selective neuropeptide FF receptor antagonist, prevents opioid-induced tolerance associated with hyperalgesia. Proc Natl Acad Sci U S A 103:466–471. 10.1073/pnas.0502090103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenger C, Eippert F, Finsterbusch J, Bingel U, Rose M, Büchel C (2012) Attention modulates spinal cord responses to pain. Curr Biol 22:1019–1022. 10.1016/j.cub.2012.04.006 [DOI] [PubMed] [Google Scholar]
- Sprenger C, Finsterbusch J, Büchel C (2015) Spinal cord-midbrain functional connectivity is related to perceived pain intensity: a combined spino-cortical FMRI study. J Neurosci 35:4248–4257. 10.1523/JNEUROSCI.4897-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers PE, Ferraro D, Duzzi D, Lui F, Iannetti GD, Porro CA (2010) A quantitative comparison of BOLD fMRI responses to noxious and innocuous stimuli in the human spinal cord. Neuroimage 50:1408–1415. 10.1016/j.neuroimage.2010.01.043 [DOI] [PubMed] [Google Scholar]
- Valet M, Sprenger T, Boecker H, Willoch F, Rummeny E, Conrad B, Erhard P, Tolle TR (2004) Distraction modulates connectivity of the cingulo-frontal cortex and the midbrain during pain: an fMRI analysis. Pain 109:399–408. 10.1016/j.pain.2004.02.033 [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Gardell LR, Burgess SE, Ibrahim M, Dogrul A, Zhong CM, Zhang ET, Malan TP Jr, Ossipov MH, Lai J, Porreca F (2000) Dynorphin promotes abnormal pain and spinal opioid antinociceptive tolerance. J Neurosci 20:7074–7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderah TW, Ossipov MH, Lai J, Malan TP Jr, Porreca F (2001a) Mechanisms of opioid-induced pain and antinociceptive tolerance: descending facilitation and spinal dynorphin. Pain 92:5–9. 10.1016/S0304-3959(01)00311-6 [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP Jr, Lai J, Porreca F (2001b) Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J Neurosci 21:279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, McLellan AT (2016) Opioid abuse in chronic pain: misconceptions and mitigation strategies. N Engl J Med 374:1253–1263. 10.1056/NEJMra1507771 [DOI] [PubMed] [Google Scholar]
- Volmanen P, Akural EI, Raudaskoski T, Alahuhta S (2002) Remifentanil in obstetric analgesia: a dose-finding study. Anesth Analg 94:913–917. 10.1097/00000539-200204000-00026 [DOI] [PubMed] [Google Scholar]
- Wager TD, Scott DJ, Zubieta JK (2007) Placebo effects on human mu-opioid activity during pain. Proc Natl Acad Sci U S A 104:11056–11061. 10.1073/pnas.0702413104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Friedman E, Olmstead MC, Burns LH (2005) Ultra-low-dose naloxone suppresses opioid tolerance, dependence and associated changes in mu opioid receptor-G protein coupling and gbetagamma signaling. Neuroscience 135:247–261. 10.1016/j.neuroscience.2005.06.003 [DOI] [PubMed] [Google Scholar]
- Wanigasekera V, Lee MC, Rogers R, Hu P, Tracey I (2011) Neural correlates of an injury-free model of central sensitization induced by opioid withdrawal in humans. J Neurosci 31:2835–2842. 10.1523/JNEUROSCI.5412-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanigasekera V, Mezue M, Andersson J, Kong Y, Tracey I (2016) Disambiguating pharmacodynamic efficacy from behavior with neuroimaging: implications for analgesic drug development. Anesthesiology 124:159–168. 10.1097/ALN.0000000000000924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Nakagawa T, Yamamoto R, Maeda A, Minami M, Satoh M (2002) Involvement of glutamate receptors within the central nucleus of the amygdala in naloxone-precipitated morphine withdrawal-induced conditioned place aversion in rats. Jpn J Pharmacol 88:399–406. 10.1254/jjp.88.399 [DOI] [PubMed] [Google Scholar]
- Weber KA 2nd, Chen Y, Wang X, Kahnt T, Parrish TB (2016) Functional magnetic resonance imaging of the cervical spinal cord during thermal stimulation across consecutive runs. Neuroimage 143:267–279. 10.1016/j.neuroimage.2016.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RG, Rogers R, Painter D, Bantick S, Ploghaus A, Williams P, Rapeport G, Tracey I (2002) Combining fMRI with a pharmacokinetic model to determine which brain areas activated by painful stimulation are specifically modulated by remifentanil. Neuroimage 16:999–1014. 10.1006/nimg.2002.1146 [DOI] [PubMed] [Google Scholar]
- Wise RG, Williams P, Tracey I (2004) Using fMRI to quantify the time dependence of remifentanil analgesia in the human brain. Neuropsychopharmacology 29:626–635. 10.1038/sj.npp.1300364 [DOI] [PubMed] [Google Scholar]
- Worsley KJ, Evans AC, Marrett S, Neelin P, others (1992) A three-dimensional statistical analysis for CBF activation studies in human brain. J Cereb Blood Flow Metab 12:900–918. 10.1038/jcbfm.1992.127 [DOI] [PubMed] [Google Scholar]
- Yu EH, Tran DH, Lam SW, Irwin MG (2016) Remifentanil tolerance and hyperalgesia: short-term gain, long-term pain? Anaesthesia 71:1347–1362. 10.1111/anae.13602 [DOI] [PubMed] [Google Scholar]
- Zambreanu L, Wise RG, Brooks JC, Iannetti GD, Tracey I (2005) A role for the brainstem in central sensitisation in humans: evidence from functional magnetic resonance imaging. Pain 114:397–407. 10.1016/j.pain.2005.01.005 [DOI] [PubMed] [Google Scholar]
- Zemlan FP, Behbehani MM (1988) Nucleus cuneiformis and pain modulation: anatomy and behavioral pharmacology. Brain Res 453:89–102. 10.1016/0006-8993(88)90146-1 [DOI] [PubMed] [Google Scholar]
- Zhu Y, Wienecke CF, Nachtrab G, Chen X (2016) A thalamic input to the nucleus accumbens mediates opiate dependence. Nature 530:219–222. 10.1038/nature16954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M, Gebhart GF (1997) Biphasic modulation of spinal nociceptive transmission from the medullary raphe nuclei in the rat. J Neurophysiol 78:746–758. 10.1152/jn.1997.78.2.746 [DOI] [PubMed] [Google Scholar]