

Abstract
In toxin-based models of Parkinson's disease (PD), striatal projection neurons (SPNs) exhibit dendritic atrophy and spine loss concurrent with an increase in excitability. Chronic l-DOPA treatment that induces dyskinesia selectively restores spine density and excitability in indirect pathway SPNs (iSPNs), whereas spine loss and hyperexcitability persist in direct pathway SPNs (dSPNs). These alterations have only been characterized in toxin-based models of PD, raising the possibility that they are an artifact of exposure to the toxin, which may engage compensatory mechanisms independent of the PD-like pathology or due to the loss of dopaminergic afferents. To test all these, we studied the synaptic remodeling in Pitx3−/− or aphakia mice, a genetic model of PD, in which most of the dopamine neurons in the substantia nigra fail to fully differentiate and to innervate the striatum. We made 3D reconstructions of the dendritic arbor and measured excitability in identified SPNs located in dorsal striatum of BAC-Pitx3−/− mice treated with saline or l-DOPA. Both dSPNs and iSPNs from BAC-Pitx3−/− mice had shorter dendritic trees, lower spine density, and more action potentials than their counterparts from WT mice. Chronic l-DOPA treatment restored spine density and firing rate in iSPNs. By contrast, in dSPNs, spine loss and hyperexcitability persisted following l-DOPA treatment, which is similar to what happens in 6-OHDA WT mice. This indicates that dopamine-mediated synaptic remodeling and plasticity is independent of dopamine innervation during SPN development and that Pitx3−/− mice are a good model because they develop the same pathology described in the toxins-based models and in human postmortem studies of advanced PD.
SIGNIFICANCE STATEMENT As the only genetic model of Parkinson's disease (PD) that develops dyskinesia, Pitx3−/− mice reproduce the behavioral effects seen in humans and are a good system for studying dopamine-induced synaptic remodeling. The studies we present here establish that the structural and functional synaptic plasticity that occur in striatal projection neurons in PD and in l-DOPA-induced dyskinesia are specifically due to modulation of the neurotransmitter dopamine and are not artifacts of the use of chemical toxins in PD models. In addition, our findings provide evidence that synaptic plasticity in the Pitx3−/− mouse is similar to that seen in toxin models despite its lack of dopaminergic innervation of the striatum during development. Pitx3−/− mice reproduced the alterations described in patients with advanced PD and in well accepted toxin-based models of PD and dyskinesia. These results further consolidate the fidelity of the Pitx3−/− mouse as a PD model in which to study the morphological and physiological remodeling of striatal projection neurons by administration of l-DOPA and other drugs.
Keywords: aphakia, L-DOPA, dyskinesia, Parkinson's disease, striatum
Introduction
The striatum, a major component of the basal ganglia motor circuit, is mainly composed of striatal projection neurons (SPNs), which are divided into two populations based on their divergent gene expression and circuit function: the direct SPNs (dSPNs), which contain D1 receptors and facilitate movement; and the indirect SPNs (iSPNs), which contain D2 receptors and inhibit movement (Albin et al., 1989). Because of this dichotomous dopamine receptor expression, dopamine plays a fundamental role in movement (Gerfen and Surmeier, 2011; Silberberg and Bolam, 2015). In addition, the SPNs are the only striatal neurons with dendritic spines, highly specialized dendritic structures that regulate synaptic strength (Kawaguchi, 1997). While SPNs receive glutamatergic and dopaminergic inputs on the same spines (Silberberg and Bolam, 2015), the localization of the dopaminergic inputs on the neck of the spines gives dopamine a central role in regulating spine function (Bolam et al., 2000; Deutch et al., 2007). Dopamine facilitates spine formation on SPNs via D1 and D2 receptors (Fasano et al., 2013), and the loss of striatal dopamine in Parkinson's disease (PD) reduces the dendritic length and spine density to a similar extent in both types of SPNs (Solís et al., 2007; Fieblinger et al., 2014; Suarez et al., 2014, 2016; Gagnon et al., 2017). Curiously, this spine loss produces opposite changes in synaptic transmission, facilitating synaptic strength in dSPNs and reducing it in iSPNs (Suarez et al., 2016). Dopamine loss also increases the intrinsic excitability of both SPN types, but by opposite mechanisms, as follows: via synaptic facilitation in dSPNs and by reduced activation of D2 receptors in iSPNs (Suarez et al., 2016).
Replacing striatal dopamine with its precursor, l-3,4-dihydroxyphenylalanine (l-DOPA), remains the most effective treatment for PD, but also induces dyskinesias with prolonged treatment. In animal models of PD, chronic l-DOPA treatment produces dyskinesia and induces opposite patterns of synaptic remodeling in iSPNs and dSPNs. In iSPNs, spine density and intrinsic excitability are recovered in dyskinetic mice, but synaptic transmission remains reduced. However, in dSPNs in the same dyskinetic mice, spine loss and hyperexcitability persist and synaptic transmission is facilitated because the spines are enlarged, with bigger heads, necks, and postsynaptic densities (Suarez et al., 2016), possibly due to D1 receptor hypersensitization (Santini et al., 2007; Darmopil et al., 2009; Murer and Moratalla, 2011; Ruiz-DeDiego et al., 2015b; Solís et al., 2017).
Thus far, these structural and functional changes have been studied after dopamine denervation using chemical toxin models of PD, making it impossible to differentiate between changes caused by exposure to the toxins and those caused by the lack of dopamine per se or whether the effects of l-DOPA are influenced by the loss of pre-existing dopaminergic fibers or not. Moreover, toxin models are unilateral models of PD, whereas in humans the disease affects both hemispheres.
To test whether these changes in SPNs are specifically due to the lack of dopamine, we used the following genetic model of PD: the Aphakia or Pitx3−/− mice, which exhibit selective bilateral dopamine depletion in the striatum (Nunes et al., 2003; van den Munckhof et al., 2003; Smidt et al., 2004) because most of their substantia nigra (SN) dopaminergic neurons fail to fully differentiate and innervate the SPNs. This lack of dopamine produces PD-like motor impairment (Hwang et al., 2005; Ardayfio et al., 2008; Solís et al., 2015; Filali and Lalonde, 2016), which is reversed with acute l-DOPA treatment; however, after chronic treatment the mice develop dyskinesia (Ding et al., 2007; Iderberg et al., 2012; Solís et al., 2015). As SPNs of Pitx3−/− mice have never been innervated by dopamine, the Pitx3−/− mouse represents a very useful model in which to investigate whether the synaptic remodeling of SPNs in the absence of nigrostriatal circuit is similar to that described in toxin-based PD models and whether l-DOPA exposure recovers these changes.
Materials and Methods
Animals and treatment
This study was performed in 3- to 4-month-old male hemizygous BAC-transgenic mice (D1R-tomato and D2R-eGFP, C57BL/6) crossed with WT or with homozygous Pitx3−/− (C57BL/6) mice. Animals were housed and maintained following the guidelines of European Union Council Directive (86/609/European Economic Community). WT, Pitx3−/−, and BAC-Pitx3−/− mice received a daily intraperitoneal injection of 10 mg/kg benserazide hydrochloride (Sigma-Aldrich) followed 20 min later by an intraperitoneal injection of 25 mg/kg l-DOPA (Sigma-Aldrich) or two saline injections 20 min apart.
Behavior
Rotarod test.
To evaluate motor coordination and balance in WT and Pitx3−/− mice, we used the rotarod test (UgoBasile) as previously described (Chen et al., 2003; González-Aparicio and Moratalla, 2014). Mice were habituated to the rod before the test sessions with a 1 min training session, during which the rod was set at a constant speed of 10 rpm, and mice were placed back on the rod if they fell. During the subsequent training phase, mice were placed on the rod for 5 min per session for six sessions at increasing velocity (4–10 rpm), and the latency to fall from the rod was measured.
Challenging beam traversal test.
Motor performance was measured with a beam test described by Hwang et al. (2005). Briefly, mice received 2 d of training on the beam before testing. On the test day, a mesh grid (0.8 cm2 holes) was placed over the beam, leaving 1 cm of space between the grid and the beam surface. Mice were then videotaped while crossing the grid-covered (challenging) beam during five trials, and the total time and number of steps to traverse the beam were scored for each trial.
Dyskinesia.
l-DOPA-induced dyskinesia consisted primarily of abnormal stereotypic paw movements. The quantification of the dyskinetic events was performed manually using the protocol previously described (Ding et al., 2007; Solís et al., 2015). Briefly, 60 min after l-DOPA or saline injection, mice were individually placed in glass cylinders, and the time spent in three to four paw dyskinesia was scored over 5 min.
Immunohistochemistry
On day 15 of l-DOPA or saline treatment (1 h after injection), mice were anesthetized and transcardially perfused with 4% paraformaldehyde, pH 7.4. Brains were postfixed for 24 h in the same solution. Coronal sections were obtained on a vibratome (Leica; RRID:SCR_008960). The cutting sequence was 200-, 30-, 30-, 30-μm-thick sections throughout the entire striatum, following the protocol previously described by Suárez et al. (2014). The 30 μm sections were used for TH (1:1000; catalog #AB1542, Millipore; RRID:AB_11213126) and FosB (H-237; 1:15,000; catalog #sc-28213, Santa Cruz Biotechnology; RRID:AB_2106911) immunostaining as previously described (Darmopil et al., 2009; Ares-Santos et al., 2014; Ruiz-DeDiego et al., 2015a).
Single-cell microinjection and 3D spine analysis
The 200 μm sections were used for Lucifer yellow injections and morphological reconstruction of SPNs. dSPNs (red fluorescence) and iSPNs (green fluorescence) were impaled with a micropipette containing 8% Lucifer yellow (Sigma-Aldrich), injected with 10–20 nA of hyperpolarizing current, and posterior sections were processed for immunocytochemistry with the anti-Lucifer yellow antibody (1:100,000; manufactured) as described previously (Suárez et al., 2014, 2016). Neurolucida version 8 (MicroBrightField; RRID:SCR_001775) was used to trace 3D dendritic arbors of SPNs and to mark spines. For spine subtype quantification, fluorescence high-magnification (63× oil) confocal images were deconvolved using Leica LAS AF Image Acquisition Software version 2.5 (RRID:SCR_013673), and spine analysis was performed using the semiautomated software NeuronStudio (http://research.mssm.edu/cnic/tools-ns.html; RRID:SCR_013798) as previously described (Rodriguez et al., 2008; Dumitriu et al., 2012; Suarez et al., 2016). Only neurons that were completely filled and located in totally denervated areas were included for analysis. Quantifications were performed by a researcher blind to the experimental conditions, and six to eight neurons per animal were analyzed (Table 1).
Table 1.
Numbers of mice and SPNs for morphological analysis
| Treatment (n) | D1R-tmt mice | dSPNs analyzed | D2R-GFP mice | iSPNs analyzed | |
|---|---|---|---|---|---|
| WT | Saline (14) | 8 | 52 | 6 | 37 |
| l-DOPA (8) | 4 | 18 | 4 | 23 | |
| BAC-Pitx3−/− | Saline (10) | 5 | 26 | 5 | 33 |
| l-DOPA (10) | 5 | 32 | 5 | 30 |
D1R, D1 receptor; D2R, D2 receptor; tmt, tomato protein.
Electrophysiology
After 15 d of l-DOPA or saline treatment, mice were decapitated, and their brains were removed and submerged in ice-cold Krebs-Ringer-bicarbonate (KRB) solution containing the following (in mm): 119 NaCl, 26.2 NaHCO3, 2.5 KCl, 1 KH2PO4, 1.3 MgSO4 2.5 CaCl2, and 11 glucose, gassed with 95% O2 and 5% CO2. Transverse corticostriatal slices (350 μm thick) were cut and recovered for >1 h at room temperature. Recordings were performed in a submersion-type chamber continuously perfused (2 ml/min) with standard KRB solution at 31–32°C in the presence 100 μm picrotoxin (Sigma-Aldrich) to block GABAA receptors. SPNs located in dorsal striatum were identified using infrared differential interference contrast on an upright DM6000FS Leica microscope, controlled by LASAF software. Red and green fluorescence, respectively, were used to identify dSPNs and iSPNs. Sharp electrodes for intracellular recording were filled with 2 m KCl (50–80 MΩ). Signals were recorded with an Axoclamp-2B Amplifier (Molecular Devices) used in bridge mode, and stored and analyzed using a digital system (pClamp version 10.0, Molecular Devices; RRID:SCR_011323). Only cells (one to two neurons per slice) with a stable resting membrane potential (RMP) more hyperpolarized than −70 mV and with an apparent input resistance (Rin) of >30 MΩ were considered in this study (Table 2). The firing rate was determined by injecting depolarizing current steps (200 ms) through the patch electrode and measuring the resulting spikes. Action potentials (APs) were elicited by 200 ms, rectangular, 0.3–0.8 nA depolarizing pulses.
Table 2.
Numbers of mice and SPNs for electrophysiological analysis
| Treatment (n) | D1R-tmt mice | dSPNs analyzed | D2R-GFP mice | iSPNs analyzed | |
|---|---|---|---|---|---|
| WT | Saline (15) | 7 | 7 | 8 | 9 |
| BAC-Pitx3−/− | Saline (13) | 7 | 8 | 6 | 7 |
| l-DOPA (18) | 8 | 9 | 10 | 11 |
D1R, D1 receptor; D2R, D2 receptor; tmt, tomato protein.
Statistical analysis
Data are expressed as mean ± SEM. Statistical evaluations and graphs were generated using SigmaPlot version 12.0 (Systat Software; RRID:SCR_003210). Statistical differences were assessed by t test and one-way or two-way ANOVA followed by Bonferroni's t test or Kruskal–Wallis analysis followed by Dunn′s test for nonparametric data. A value of p < 0.05 was considered statistically significant.
Results
BAC transgene expression in SPNs does not alter the motor impairment in Pitx3−/− mice
To identify dSPNs and iSPNs in the Aphakia or Pitx3−/− mice, we mated them with D1R-tomato and D2R-eGFP BAC transgenic mice. We first tested that this cross does not alter motor impairments described in Pitx3−/− mice (Hwang et al., 2005; Beeler et al., 2010; Le et al., 2015; Solís et al., 2015); thus, we evaluated motor coordination tasks. We found that Pitx3−/− and BAC-Pitx3−/− mice remained on the rotarod for less time than WT mice (Fig. 1A). In the challenging beam test, they also needed much more time and more steps to finish the task (126.4 ± 10.62 and 120.1 ± 4.2 steps in Pitx3−/− and BAC-Pitx3−/−, respectively) compared with WT mice (70.6 ± 2.34 steps; one-way ANOVA: F(2,12) = 12.74, p = 0.02; Fig. 1B). We observed no difference in motor skills between Pitx3−/− and BAC-Pitx3−/− mice (Fig. 1A,B), indicating that transgene expression does not interact with the Pitx3−/− motor PD-like phenotype.
Figure 1.

Motor alterations and dyskinesia in BAC-Pitx3−/− mice. A, Latency to fall in the rotarod test. B, Time and number of steps to cross the challenging beam traversal test. C, The three to four paw dyskinesia score. *p < 0.01 vs WT mice; Kruskal–Wallis following Dunn's test for A; one-way (for B) or two-way ANOVA (for C) following Bonferroni's post-test. Sal, saline.
The Pitx3−/− mouse is the only genetic model of PD that develops dyskinesia following l-DOPA administration (Ding et al., 2007; Solís et al., 2015) and reproduces the biochemical changes induced by l-DOPA treatment in the denervated striatum (Ding et al., 2007, Espadas et al., 2012; Li and Zhou, 2013; Solís et al., 2015). We next confirmed that the expression of the BAC transgene did not alter dyskinesia or the expression pattern of FosB in these mice. Indeed, l-DOPA treatment (25 mg/kg) produced three to four paw dyskinesia in the BAC-Pitx3−/− mice that was indistinguishable from that in the Pitx3−/− mice (Fig. 1C). In addition, FosB expression was also similar in both groups of mice (data not shown).
Shrinking of SPNs dendritic arbors in BAC-Pitx3−/− mice
Postmortem studies in patients with PD have revealed marked atrophy of SPN dendritic arbors in the denervated putamen (McNeill et al., 1988), which has not been reproduced in all PD animal models (Moratalla et al., 2016). We next examined whether the BAC-Pitx3−/− mice exhibit the same pathology as human patients. Because only the SPNs located in the totally denervated areas (with <10% of control TH staining and >1500 FosB-positive cells/mm2) are affected by the lack of dopamine (Suárez et al., 2014), we morphologically reconstructed the SPNs located in this zone. To this issue, identified dSPNs (red fluorescent) and iSPNs (green fluorescent) were filled with Lucifer yellow dye in coronal sections, and adjacent sections were stained with TH and/or FosB to delineate the striatal areas without nigral afferents (Fig. 2).
Figure 2.

Morphological reconstruction of striatal SPNs located in dorsal striatum without dopaminergic fibers. Serial sections showing FosB, TH, and LY immunostaining in striatum of mice. The right-most images show high-magnification microphotographs of the SPNs depicted in the LY pictures. The outline shows the boundaries of striatal areas without dopaminergic fibers. Note the FosB expression in the totally denervated striatal areas. LY, Lucifer Yellow; TH, tyrosine hydroxilase.
In the BAC-Pitx3−/− mice, the complexity of the dendritic tree was reduced in both types of SPNs. The total length of the dendrites was homogeneously reduced along the length of the dendritic tree as shown by the Sholl analysis. Moreover, the maximal dendritic length in the BAC-Pitx3−/− mice was 188 ± 9 μm, whereas in the WT mice it was 213 ± 6 μm (t test, p < 0.05; Fig. 3). In addition, the number of the branches or intersection points was reduced homogeneously along the dendritic arbor. This reduction was accompanied by a decrease in the maximum branch order of the dendrites, but not by a decreased of the number of primary dendrites. l-DOPA treatment, which did not alter the dendritic arbor in WT mice, did not significantly reverse the reduction of the dendritic length in BAC-Pitx3−/− mice (Fig. 3).
Figure 3.
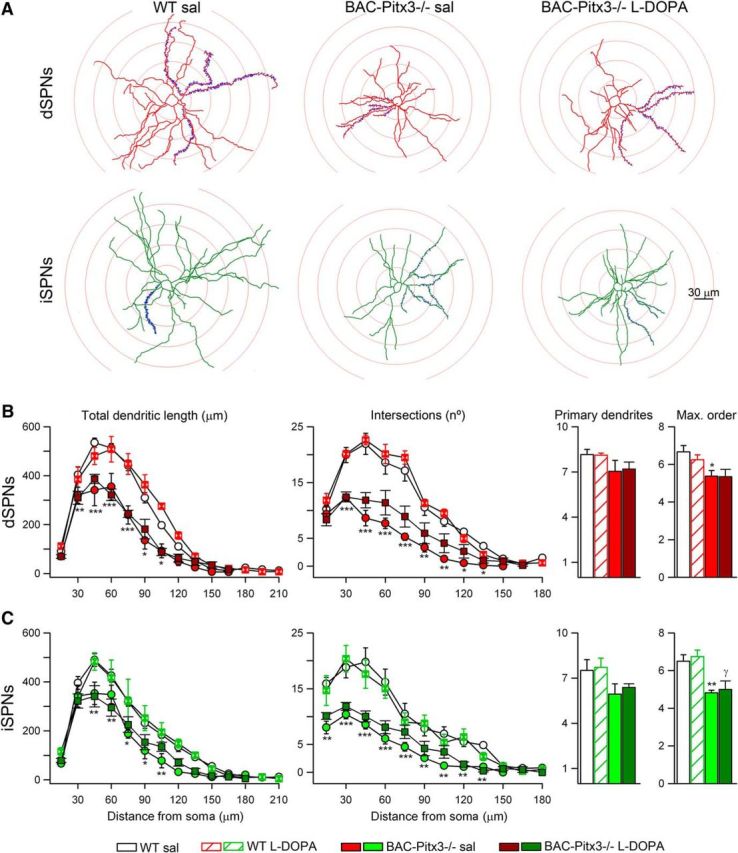
Dendritic length is reduced in both SPNs in BAC-Pitx3−/− mice. A, Illustrations of the Sholl analysis of SPNs. B, Sholl analysis of the total dendritic length and the number of intersections of dSPNs. Number of primary dendrites and maximum dendritic order in dSPNs. C, Quantification of dendritic tree in iSPNs. *p < 0.05; **p < 0.01; ***p < 0.001 vs WT; one-way or two-way (for Sholl analysis) ANOVA following Bonferroni's post-test. Max., Maximum; sal, saline.
Dopamine modulates spine density of SPN in BAC-Pitx3−/− mice
The reduction of striatal dendritic spines found in PD patients (Stephens et al., 2005; Zaja-Milatovic et al., 2005) has been reproduced in rodent models of PD using MPTP or 6-OHDA lesions (Suárez et al., 2014; Fieblinger and Cenci, 2015; Moratalla et al., 2016; Nishijima et al., 2017; Villalba and Smith, 2018). In these PD models, chronic l-DOPA treatment selectively restores the spine density in iSPNs (Fieblinger et al., 2014; Suárez et al., 2014, 2016). Therefore, we next studied whether spine loss is also observed in BAC-Pitx3−/− mice in which dorsal striatal neurons were never innervated by dopamine and whether l-DOPA can modulate these spines. In BAC-Pitx3−/−, the spine density is reduced in both types of SPNs along the dendritic tree (Fig. 4). After l-DOPA treatment, spine loss persisted in dSPNs (Fig. 4A,B), but it recovered in iSPNs to WT values in both proximal and distal parts of the dendrite (Fig. 4B). l-DOPA treatment did not affect spine density in WT mice.
Figure 4.
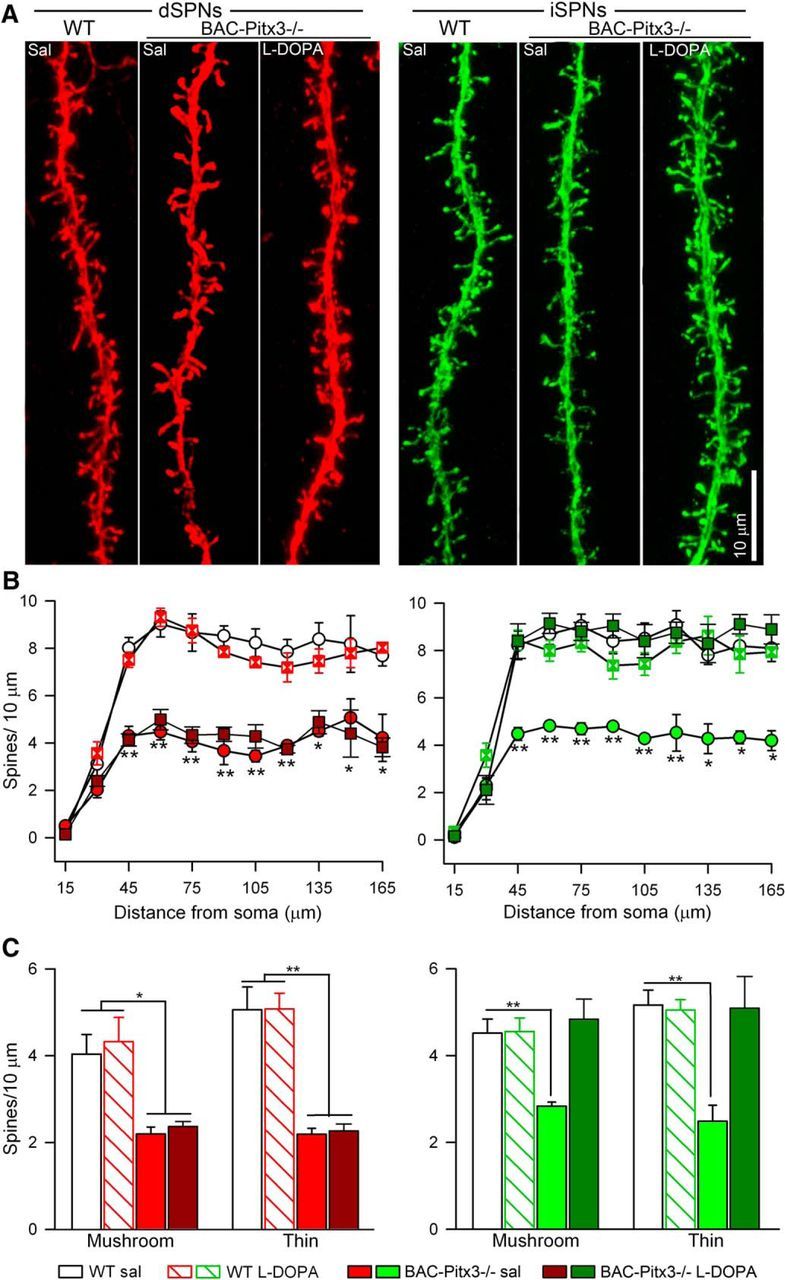
Spine density is reduced in both SPNs in BAC-Pitx3−/− mice. A, Confocal images of dendrites of SPNs. B, Sholl analysis of spine density. C, Spine density of mushroom and thin spines in SPNs. *p < 0.05, **p < 0.01 vs WT; one-way or two-way (for Sholl analysis) ANOVA following Bonferroni's post-test. Sal, saline.
Our previous study has demonstrated that dopamine depletion in 6-OHDA-exposed mice reduced the numbers of the main morphologically defined types of striatal spines (mushroom and thin) to a similar extent (Suarez et al., 2016). To determine whether this also occurs in BAC-Pitx3−/− mice, we quantified each of the subtypes of spines in dSPNs and iSPNs using the semiautomated software NeuronStudio. As in the 6-OHDA-lesioned mice, mushroom and thin spines were reduced in both types of SPNs in BAC-Pitx3−/− mice. l-DOPA treatment did not change the density or the proportion of the spines in WT mice in either dSPNs or iSPNs. However, in dyskinetic animals, l-DOPA produced in iSPNs a recovery in the density of both mushroom spines (4.51 ± 0.33 vs 4.84 ± 0.46 spines in 10 μm in WT and BAC-Pitx3−/−, respectively) and thin spines (5.16 ± 0.34 vs 5.09 ± 0.73 spines in 10 μm in WT and BAC-Pitx3−/− mice, respectively), whereas it did not alter the density of spines in dSNPs of mushroom spines (4.51 ± 0.33 vs 2.37 ± 0.11 spines in 10 μm in WT and BAC-Pitx3−/− mice, respectively) and thin spines (5.16 ± 0.34 vs 2.27 ± 0.06 spines in 10 μm in WT and BAC-Pitx3−/− mice, respectively; Fig. 4C).
Increased excitability of both types of SPNs in BAC-Pitx3−/− mice
Because the loss of dopamine increases intrinsic excitability in rodent models of PD (Calabresi et al., 1993a; Azdad et al., 2009; Suárez et al., 2014, 2016), we next assessed the spiking in SPNs in BAC-Pitx3−/− mice. We injected depolarizing steps of current through a recording electrode positioned in the soma of identified dSPNs and iSPNs located in striatal zones without dopaminergic fibers (Fig. 5A).
Figure 5.

Intrinsic excitability is increased in both SPNs in BAC-Pitx3−/− mice. A, Scheme of a coronal slice depicting the striatal area without dopaminergic inputs in gray (left) and photographs (40×) of representative registered neurons in the gray area from a BAC-Pitx3−/− D1R-tomato mouse. B, Representative current-clamp recordings showing dSPNs and iSPNs recorded at 0.3 nA. C, Frequency of action potential evoked with depolarizing current in dSPNs and iSPNs. *p < 0.05; **p < 0.001 saline or l-DOPA BAC-Pitx3−/− mice vs WT mice; two-way ANOVA following Bonferroni's post-test. D, Histograms show the mean firing rate at 0.3nA to facilitate the comparison between dSPNs with iSPNs. *p < 0.05 dSPNs vs iSPNs t-test. Sal/sal, Saline; Str, striatum; Cx cerebral cortex.
In WT mice, dSPNs were less excitable than iSPNs since they had less evoked action potential for all tested intensities (F(1, 78) = 6.60, p = 0.013; Two-way ANOVA; Figure 5B–D), in agreement with previous results (Kreitzer and Malenka, 2007; Ade et al., 2008; Cepeda et al., 2008; Gertler et al., 2008; Planert et al., 2013; Maurice et al., 2015; Ketzef et al, 2017). The evoked action potential properties between dSPNs and iSPNs were not significant different in threshold, amplitude, duration or amplitude after hyperpolarization (AHP) (Table 3). However, the rheobase was slightly higher in dSPNs from (0. ± 0.02) to (0.31 ± 0.02) compared with iSPNs (0.21 ± 0.03), p = 0.07 t-test, in agreement with previous results (Planert et al., 2013; Maurice et al., 2015).
Table 3.
Intrinsic membrane properties of SPNs
| dSPNs |
iSPNs |
|||||
|---|---|---|---|---|---|---|
| WT Saline (7) | BAC-Pitx3−/− |
WT Saline (9) | BAC-Pitx3−/− |
|||
| Saline (8) | l-DOPA (9) | Saline (7) | l-DOPA (11) | |||
| Passive membrane properties | ||||||
| RMP (mV) | −88.9 ± 4.0 | −89.6 ± 3.0 | −91.7 ± 3.4 | −89.4 ± 1.9 | −90.2 ± 4.5 | −92.9 ± 1.4 |
| Rin (MΩ) | 46.31 ± 5.2 | 44.7 ± 5.70 | 33.2 ± 1.5 | 41.3 ± 3.2 | 39.9 ± 5.4 | 37.7 ± 3.3 |
| Active membrane properties | ||||||
| AP threshold (mV) | −45.5 ± 1.6 | −51.7 ± 1.7* | −48.6 ± 2.2 | −45.9 ± 2.2 | −53.7 ± 1.1* | −48.4 ± 1.1 |
| AP amplitude (mV) | 86.9 ± 6.9 | 87.3 ± 8.1 | 76.3 ± 11.6 | 87.4 ± 5.1 | 78.7 ± 6.4 | 83.8 ± 5.3 |
| AP width (ms) | 0.71 ± 0.05 | 0.74 ± 0.11 | 0.54 ± 0.11 | 0.72 ± 0.06 | 0.77 ± 0.12 | 0.57 ± 0.04 |
| Rheobase (nA) | 0.31 ± 0.02 | 0.22 ± 0.02* | 0.21 ± 0.01** | 0.20 ± 0.04 | 0.21 ± 0.03 | 0.29 ± 0.02 |
| AHP amplitude (mV) | 4.96 ± 0.98 | 5.50 ± 0.94 | 5.35 ± 2.38 | 4.96 ± 0.98 | 5.83 ± 1.12 | 7.27 ± 1.33 |
Data are presented as the mean ± SEM. The number of SPNs recorded in each condition is expressed in parentheses following the condition.
*p < 0.05;
**p < 0.01 vs WT mice; one-way ANOVA following Bonferroni's post-test.
We observed that the same depolarizing current evoked significantly more action potentials in both dSPNs and iSPNs of BAC-Pitx3−/− mice than WT mice at all tested intensities (two-way ANOVA, p < 0.001; Fig. 5B,C), indicating increased excitability in BAC-Pitx3−/− mice. While the RMP and Rin values were unchanged in BAC-Pitx3−/− mice (Table 3), the action potential threshold was lower compared with WT animals (BAC-Pitx3−/− vs WT respectively: −51.74 ± 1.75 vs −45.48 ± 1.61 mV in dSPNs and −53.72 ± 1.12 vs −45.93 ± 2.18 in iSPNs; two-way ANOVA, F(2,41) = 10.1 p < 0.001; Table 3).
In addition, in the BAC-Pitx3−/− mice no differences in the excitability between dSPNs and iSPNs (F(1,36) = 3.27, p = 0.076; Two-way ANOVA; Figure 5D) were found, in line with previous results in 6-OHDA toxin models of PD (Ketzef et al., 2017).
After chronic l-DOPA treatment, the firing rate remained increased in dSPNs, but was indistinguishable from that of WT mice in iSPNs (Fig. 5) coinciding with the recovered of the action potential threshold (Table 3).
This recovery resulted in a lower excitability of iSPN compared with dSPNs in L-DOPA-treated BAC-Pitx3−/− mice (F(1,86) = 8.12, p = 0.006; Two-way ANOVA), in agreement with previous results using 6-OHDA toxin models of PD (Fieblinger et al, 2014; Suarez et al., 2016).
Enhanced D1 receptor sensitivity in BAC-Pitx3−/− mice contributes to dyskinesia
In Pitx3−/− mice, the dorsal SPNs have never been innervated by dopamine; thus, we next evaluated whether dopamine receptor sensitization is changed in these mice. Because dopamine receptors modulate the intrinsic excitability of SPNs (D1 receptor increases spiking in dSPNs, whereas D2 receptor decreases it in iSPNs; Gerfen and Surmeier, 2011; Planert et al., 2013), we studied the changes in spiking in the presence of specific receptor ligands.
Dyskinesia is associated with overactivation of D1 receptors (Santini et al., 2007; Darmopil et al., 2009; Murer and Moratalla, 2011; Ruiz-DeDiego et al., 2015b; Solís et al., 2017); thus, we chose a low dose of D1 receptor agonist (SKF38393, 3 μm) that did not affect the firing rate in WT mice (Fig. 6A) to avoid saturation of the responses in dyskinetic animals. In dSPNs from BAC-Pitx3−/− mice, bath application of the D1 agonist did not alter the already increased firing rate in saline-treated mice (Fig. 6A). However, in dyskinetic mice the bath application of D1 agonist increased spiking at all injected currents (Fig. 6A; two-way ANOVA: F(2,80) = 41.22, p < 0.001). Moreover, this potentiation was completely blocked by SCH23390 (10 μm), a D1 receptor antagonist (Fig. 6A), confirming that this effect was mediated by D1 receptor activation. These data indicate that D1 receptor signaling is hypersensitive in BAC-Pitx3−/− mice after chronic l-DOPA treatment, similar to previous findings in the 6-OHDA-lesioned mouse model (Suárez et al., 2014).
Figure 6.

Dopamine D1 receptors are hypersensitized and D2 receptors desensitized in L-DOPA-treated BAC-Pitx3−/− dyskinetic mice. A, Firing rate of dSPNs before (baseline) and during D1 agonist bath perfusion in WT and BAC-Pitx3−/− mice treated with saline or l-DOPA, respectively. D1 antagonist (blue) blocks the increase in firing rate produced by D1 agonist. The gray trace represents the WT curve to facilitate comparison. Top, Representative current-clamp recordings at 0.4 nA. B, Firing rate of iSPNs before (baseline) and during D2 agonist and D2 antagonist bath perfusion in WT and BAC-Pitx3−/− mice treated with saline or l-DOPA, respectively. Note that in dyskinetic BAC-Pitx3−/− mice, only high doses of D2 agonist decreased the firing rate. Top, Representative current-clamp recording at 0.4 nA. Two-way ANOVA following Bonferroni's post-test: *p < 0.05; **p < 0.001; ***p < 0.001 baseline vs dopamine receptor agonist.
In iSPNs, the application of D2 receptor agonist (quinpirole 3 μm) decreased the spiking rate in BAC-Pitx3−/− mice treated with saline and WT mice to a similar extent (Fig. 6B). This effect was completely reversed during bath application of sulpiride (10 μm), a D2 receptor antagonist (Fig. 6B). By contrast, in dyskinetic mice, the same dose of quinpirole had no effect on the firing rate at any level of injected current tested (Fig. 6B), suggesting that D2 receptors are desensitized in dyskinesia.
We then, analyzed the excitability of SPNs and found that in WT mice, dSPNs are less excitable than iSPNs at all tested intensities when the area under the curve was analyzed (F(1,69) = 13.25, p < 0.01; Two-way ANOVA), although point to point comparison did not reached statistical significance (Figure 7A). This difference was loss in BAC-Pitx3−/− mice (F(1,60) = 0.65, p = 0.43; Two-way ANOVA). Noticeably, the firing rate of dSPNs did not change during bath application of low doses of SKF38383, a D1 receptor agonist (Figure 7B), while it was reduced in iSPNs after low doses of quinpirole, a D2 receptor agonist (Figure 7C). L-DOPA treatment produced a slight reduction in the excitability of dSPNs (Figure 7A) and a robust reduction in iSPNs at all tested intensities (F(1,69) = 4.67, p = 0.035; Two-way ANOVA). Moreover, the firing rate was increased in dSPNs with low doses of SKF38393 (Figure 7B) but it was not altered after quinpirole in iSPNs (Figure 7C). These results suggest that D1 receptors are hypersensitized while D2 receptors are desensitized in dyskinetic BAC-Pitx3−/− mice treated with L-DOPA.
Figure 7.

Excitability of dSPNs and iSPNs after D1 or D2 agonist. A, Frequency of evoked action potential at 0.4nA in WT and in BAC-Pitx3−/− mice. B, Firing rate in dSPNs and iSPNs during bath application of 3 μM SKF38393 (D1 agonist) or 1 μM quinpirole (D2 agonist). *p < 0.05; **p < 0.005; ***p < 0.001, Student-t-test.
Discussion
Here we demonstrated that Pitx3−/− mice, in which SPNs have never been innervated by dopamine, exhibit the same structural and electrophysiological alterations described in the 6-OHDA lesion model of PD and dyskinesia, specifically as follows: (1) diminished dendritic arborization and decreased density spines in both dSPNs and iSPNs; (2) selective increase by l-DOPA treatment of dendritic spines in iSPNs, but not in dSPNs; (3) increased intrinsic excitability of both types of SPNs; (4) selective decrease by l-DOPA treatment of excitability in iSPNs, but not in dSPNs; and (5) l-DOPA-induced hypersensitized D1 receptor responses and desensitized D2 receptor responses.
Only ∼10–15% of PD cases have a genetic etiology (Klein and Westenberger, 2012). Nevertheless, genetic models of PD provide a very helpful tool to study the synaptic plasticity mechanisms underlying PD and to develop novel therapies. Several genetic models of PD have been developed that recapitulate PD motor impairments and reversal by acute l-DOPA treatment, but only the Pitx3−/− mouse mimics the development of chronic l-DOPA-induced dyskinesia that is seen clinically (Hwang et al., 2005; Ding et al., 2007; Espadas et al., 2012; Li and Zhou, 2013; Solís et al., 2015).
BAC-Pitx3−/− mice exhibit the same motor impairment and dyskinetic behavior that those described in Pitx3−/− mice. Although the BAC-Pitxt3−/− mice are blind, because the Pitx3 factor is involved in eye lens development (Semina et al., 1997), previous results demonstrate that their motor deficits are specifically due to the lack of striatal dopamine but not to their blindness (Ardayfio et al., 2008).
In human PD, the involvement of PITX3 is controversial: several studies found an association between the PITX3 gene polymorphism and PD (Fuchs et al., 2009; Bergman et al., 2010; Le et al., 2011; Tang et al., 2012; Qiu et al., 2014), but others did not (Cai et al., 2011; Jiménez-Jiménez et al., 2014).
The main aim of this study was to determine whether the structural and synaptic plasticity previously described in validated models of dyskinesia are also observed in the Pitx3−/− mouse model of PD. The synaptic plasticity alterations observed in toxin models of PD could be a consequence of the use of the toxin, of compensatory mechanisms originating from the contralateral nonlesioned hemisphere, or of the interactions among multiple factors, so it was important to consolidate these results using a genetic and bilateral model of PD to circumvent all of these caveats.
We demonstrated that both types of SPNs in the BAC-Pitx3−/− mice have shorter dendritic lengths than SPNs in WT animals. Although the number of the primary dendrites did not change, the maximal branch order of the dendrites was lower than WT animals. This is similar to what is observed in PD patients (McNeill et al., 1988) and in animal models of PD after dopamine degeneration (Solís et al., 2007; Fieblinger et al., 2014). In the Pitx3−/− mice, the SPNs develop and mature without dopamine afferents; these results indicate that dopamine is essential for proper SPN dendritic tree complexity. In addition, l-DOPA treatment does not restore SPN dendritic tree complexity in BAC-Pitx3−/− mice, which is in agreement with the findings of the study by McNeill et al. (1988), in which all the patients received l-DOPA treatment chronically, or with the findings of the study by Fieblinger et al. (2014) using the 6-OHDA animal model, presumably because l-DOPA does not produce dopaminergic reinnervation.
Our results demonstrate that the absence of dopamine during SPN development reduces the spine density in both types of SPNs in BAC-Pitx3−/− mice, indicating a central role for dopamine in regulating striatal morphology. In fact, in LRKK2 mutant mice, another genetic model of PD that maintains striatal dopamine levels, spine density does not change in SPNs (Matikainen-Ankney et al., 2016). Our experimental model does not allow us to determine whether the requirement for dopamine for normal spine density occurs during development, in adulthood, or both. However, previous studies using organotypic corticostriatal cocultures have shown that dopamine is essential both for spine formation during development and for spine maintenance in mature SPNs (Deutch, 2006; Deutch et al., 2007; Neely et al., 2007; Garcia et al., 2010; Tian et al., 2010), and that both are D1 and D2 receptor dependent (Fasano et al., 2013). In this line, our results show that the lack of dopamine reduces spine density similarly in dSPNs and iSPNs, demonstrating that both D1 and D2 receptors are necessary for spine formation and maintenance. These results are in agreement with our previous results showing that the lack of dopamine in PD reduces spine density in both types of SPNs (Suárez et al., 2014, 2016; Gagnon et al., 2017). In addition, spine loss occurred mainly in the predominant types of spines, mushroom and thin, which is in agreement with our findings in the 6-OHDA mice model (Suarez et al., 2016). Because mushroom spines control the synaptic connectivity of the neurons (Bourne and Harris, 2008), it is possible that corticostriatal synapses decrease in Pitx3−/− mice, as happens in the 6-OHDA rodent models (Zhang et al., 2013).
Along with lower spine density, both SPNs exhibit increased intrinsic excitability in BAC-Pitx3−/− mice due to a decreased AP threshold, as in 6-OHDA-lesioned mice (Suarez et al., 2016). The different excitability between dSPNs and iSPNs in naïve animals is loss after dopamine depletion (Fieblinger et al., 2014; Maurice et al., 2015; Ketzef et al., 2017). We now show that this loss is due to the lack of dopamine but not to compensatory processes after lesion because both SPNs exhibit similar excitability in Pixt3−/− mice. While we have not determined the conductance changes that underlie this phenomenon in the BAC-Pitx3−/− mice, previous studies indicate that in 6-OHDA models of PD, increases SPN excitability are mediated by both potassium IA and calcium Cav1.3 channels (Day et al., 2008; Azdad et al., 2009). Small-conductance Ca2+-activated K+ channels could also be involved since the activation of these channels limits the spiking frequency and their expression is increased in 6-OHDA-lesioned mice (Trusel et al., 2015).
Another possibility is that hyperexcitability is the result of changes in striatal microcircuits because the cholinergic (Plata et al., 2013; Pérez-Ortega et al., 2016) and fast-spiking (Szydlowski et al., 2013) interneurons that modulate the excitability of SPNs are altered in PD models. This hyperexcitability could be a compensatory mechanism, similar to hippocampal synaptic scaling (Turrigiano, 2008), to maintain global striatal activity despite lower depolarized input due to dendritic spine loss (Azdad et al., 2009; Suárez et al., 2014). Consistent with this, in 6-OHDA-lesioned mice, dSPNs and iSPNs exhibit attenuated cortical synaptic transmission (Escande et al., 2016; Suarez et al., 2016).
Another important result of our study is that l-DOPA treatment increases the number of spines in iSPNs, even though these SPNs have never been innervated by dopamine, which is in line with those found in denervated neurons treated with 6-OHDA (Fieblinger et al., 2014; Suárez et al., 2014, 2016). The spine remodeling of iSPNs coincided with the decrease of the firing rate to the values of WT mice due to an increase in the threshold of the AP. Thus, these results suggested that the increase in spine density compensates for changes in the excitability, decreasing the firing rate to normal values in SPNs. Although the resolution of hyperexcitability could be due to the inhibitory effect of D2 receptor activation on firing rate (Gerfen and Surmeier, 2011), this seems unlikely since our results show that the D2 receptor is desensitized in BAC-Pitx3−/− dyskinetic mice.
By contrast, in dSPNs, the lower spine number and the hyperexcitability persist after chronic l-DOPA treatment. These data are in agreement with previous results using l-DOPA in 6-OHDA-lesioned mice (Fieblinger et al., 2014; Suárez et al., 2014, 2016). Our results show that l-DOPA-induced dyskinesia is accompanied by a marked sensitization of the D1 receptor stimulation in dSNPs, which is in line with previous results using dyskinetic mice (Pavón et al., 2006; Santini et al., 2007; Darmopil et al., 2009; Ruiz-DeDiego et al., 2015a,b; F Hernández et al., 2017). Because D1 receptor activation increases the dSPN firing rate (Gerfen and Surmeier, 2011; Planert et al., 2013), it is possible that this overactivation of D1 receptors maintains the hyperexcitability in BAC-Pitx3−/− mice. In slices from sham-operated WT mice, the doses of l-DOPA used to activate D1 receptor did not affect SPN excitability (Calabresi et al., 1993b; Suárez et al., 2014), suggesting that the hyperexcitability we observe in the Pitx3−/− mice is an abnormal response of SPNs that have never received dopaminergic afferents, which is further enhanced in dyskinesia. This striatal hypersensitivity to D1 receptor stimulation may mediate the widespread molecular and electrophysiological changes observed in dyskinetic animals and patients (Boraud et al., 2001; Guigoni et al., 2005; Halje et al., 2012; Trusel et al., 2015; Araque et al., 2017). Together, these results indicate that in BAC-Pitx3−/− dyskinetic mice there is a functional imbalance in the striatal projection neurons, as follows: the dSPNs have an increased firing rate and the D1 receptor is sensitized, whereas the D2 receptor is desensitized in iSPNs.
In summary, we found that BAC-Pitx3−/− mice reproduced the alterations described in patients with advanced PD and in well accepted toxin models of PD and l-DOPA-induced dyskinesia. These results demonstrate that dopamine modulates spine and synaptic plasticity in dSPNs and iSPNs, independent of homeostatic changes induced by the use of chemical toxins. In addition, our results further establish Pitx3−/− mice as a robust model for the investigation of striatal circuity function in PD and dyskinesia.
Footnotes
This work was supported by grants from the Spanish Ministries of Economía, Industria y Competitividad (SAF2016-78207-R and PCIN-2015-098) and of Sanidad Servicios Sociales e Igualdad, Instituto de Salud Carlos III (ISCIII), Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas [CIBERNED] (CB06/05/0055, PNSD2016I033), the Ramón Areces Foundation (172275), and from Secretaría de Ciencia, Tecnología e Innovacíon [SECITI] from Ciudad de Mexico (037/2016) to R.M., J.R.G.-M. received scholarships from Consejo Nacional de Ciencia y Tecnología and SECITI of Ciudad de México. L.M.S., S.A., and R.M. were awarded the First Prize and Special Prize at the XV Archimedes University Competition from the Spanish Ministry of Educación Cultura y Deporte and ONCE Foundation. We thank Beatriz Pro and Emilia Rubio for technical assistance and Dr. J. DeFelipe for providing the Lucifer yellow antibody.
The authors declare no competing financial interests.
References
- Ade KK, Janssen MJ, Ortinski PI, Vicini S (2008) Differential tonic GABA conductances in striatal medium spiny neurons. J Neurosci 28:1185–1197. 10.1523/JNEUROSCI.3908-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB (1989) The functional anatomy of basal ganglia disorders. Trends Neurosci 12:366–375. 10.1016/0166-2236(89)90074-X [DOI] [PubMed] [Google Scholar]
- Araque A, Castillo PE, Manzoni OJ, Tonini R (2017) Synaptic functions of endocannabinoid signaling in health and disease. Neuropharmacology 124:13–24. 10.1016/j.neuropharm.2017.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardayfio P, Moon J, Leung KK, Youn-Hwang D, Kim KS (2008) Impaired learning and memory in Pitx3 deficient aphakia mice: a genetic model for striatum-dependent cognitive symptoms in Parkinson's disease. Neurobiol Dis 31:406–412. 10.1016/j.nbd.2008.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ares-Santos S, Granado N, Espadas I, Martinez-Murillo R, Moratalla R (2014) Methamphetamine causes degeneration of dopamine cell bodies and terminals of the nigrostriatal pathway evidenced by silver staining. Neuropsychopharmacol 39:1066–1080. 10.1038/npp.2013.307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azdad K, Chàvez M, Don Bischop P, Wetzelaer P, Marescau B, De Deyn PP, Gall D, Schiffmann SN (2009) Homeostatic plasticity of striatal neurons intrinsic excitability following dopamine depletion. PLoS One 4:e6908. 10.1371/journal.pone.0006908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler JA, Cao ZF, Kheirbek MA, Ding Y, Koranda J, Murakami M, Kang UJ, Zhuang X (2010) Dopamine-dependent motor learning: insight into levodopa's long-duration response. Ann Neurol 67:639–647. 10.1002/ana.21947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman O, Håkansson A, Westberg L, Nordenström K, Carmine Belin A, Sydow O, Olson L, Holmberg B, Eriksson E, Nissbrandt H (2010) PITX3 polymorphism is associated with early onset Parkinson's disease. Neurobiol Aging 31:114–117. 10.1016/j.neurobiolaging.2008.03.008 [DOI] [PubMed] [Google Scholar]
- Bolam JP, Hanley JJ, Booth PA, Bevan MD (2000) Synaptic organisation of the basal ganglia. J Anat 196:527–542. 10.1046/j.1469-7580.2000.19640527.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boraud T, Bezard E, Bioulac B, Gross CE (2001) Dopamine agonist-induced dyskinesias are correlated to both firing pattern and frequency alterations of pallidal neurones in the MPTP-treated monkey. Brain 124:546–557. 10.1093/brain/124.3.546 [DOI] [PubMed] [Google Scholar]
- Bourne JN, Harris KM (2008) Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci 31:47–67. 10.1146/annurev.neuro.31.060407.125646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Ding H, Gu Z, Baskys A, Ma J, Chan P (2011) PITX3 polymorphism is not associated with Parkinson's disease in a Chinese population. Neurosci Lett 505:260–e262. 10.1016/j.neulet.2011.10.034 [DOI] [PubMed] [Google Scholar]
- Calabresi P, Mercuri NB, Sancesario G, Bernardi G (1993a) Electrophysiology of dopamine-denervated striatal neurons. Implications for Parkinson's disease. Brain 116:433–452. 10.1093/brain/116.2.433 [DOI] [PubMed] [Google Scholar]
- Calabresi P, Pisani A, Mercuri NB, Bernardi G (1993b) Heterogeneity of metabotropic glutamate receptors in the striatum: electrophysiological evidence. Eur J Neurosci 5:1370–1377. 10.1111/j.1460-9568.1993.tb00923.x [DOI] [PubMed] [Google Scholar]
- Cepeda C, André VM, Yamazaki I, Wu N, Kleiman-Weiner M, Levine MS (2008) Differential electrophysiological properties of dopamine D1 and D2 receptors-containing striatal medium-sized spiny neurons. Eur J Neurosci 27:671–682. 10.1111/j.1460-9568.2008.06038.x [DOI] [PubMed] [Google Scholar]
- Chen JF, Moratalla R, Yu L, Martín AB, Xu K, Bastia E, Hackett E, Alberti I, Schwarzschild MA (2003) Inactivation of adenosine A2A receptors selectively attenuates amphetamine-induced behavioral sensitization. Neuropsychopharmacology 28:1086–1095. [DOI] [PubMed] [Google Scholar]
- Darmopil S, Martín AB, De Diego IR, Ares S, Moratalla R (2009) Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol Psychiatry 66:603–613. 10.1016/j.biopsych.2009.04.025 [DOI] [PubMed] [Google Scholar]
- Day M, Wokosin D, Plotkin JL, Tian X, Surmeier DJ (2008) Differential excitability and modulation of striatal medium spiny neuron dendrites. J Neurosci 28:11603–11614. 10.1523/JNEUROSCI.1840-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutch AY. (2006) Striatal plasticity in parkinsonism: dystrophic changes in medium spiny neurons and progression in Parkinson's disease. J Neural Transm Suppl (70):67–70. [DOI] [PubMed] [Google Scholar]
- Deutch AY, Colbran RJ, Winder DJ (2007) Striatal plasticity and medium spiny neuron dendritic remodeling in parkinsonism. Parkinsonism Relat Disord 13 [Suppl 3]:S251–S258. 10.1016/S1353-8020(08)70012-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Restrepo J, Won L, Hwang DY, Kim KS, Kang UJ (2007) Chronic 3,4-dihydroxyphenylalanine treatment induces dyskinesia in aphakia mice, a novel genetic model of Parkinson's disease. Neurobiol Dis 27:11–23. 10.1016/j.nbd.2007.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitriu D, Laplant Q, Grossman YS, Dias C, Janssen WG, Russo SJ, Morrison JH, Nestler EJ (2012) Subregional, dendritic compartment, and spine subtype specificity in cocaine regulation of dendritic spines in the nucleus accumbens. J Neurosci 32:6957–6966. 10.1523/JNEUROSCI.5718-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escande MV, Taravini IR, Zold CL, Belforte JE, Murer MG (2016) Loss of homeostasis in the direct pathway in a mouse model of asymptomatic Parkinson's disease. J Neurosci 36:5686–5698. 10.1523/JNEUROSCI.0492-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espadas I, Darmopil S, Vergaño-Vera E, Ortiz O, Oliva I, Vicario-Abejón C, Martín ED, Moratalla R (2012) L-DOPA-induced increase in TH-immunoreactive striatal neurons in parkinsonian mice: insights into regulation and function. Neurobiol Dis 48:271–281. 10.1016/j.nbd.2012.07.012 [DOI] [PubMed] [Google Scholar]
- Fasano C, Bourque MJ, Lapointe G, Leo D, Thibault D, Haber M, Kortleven C, Desgroseillers L, Murai KK, Trudeau LÉ (2013) Dopamine facilitates dendritic spine formation by cultured striatal medium spiny neurons through both D1 and D2 dopamine receptors. Neuropharmacology 67:432–443. 10.1016/j.neuropharm.2012.11.030 [DOI] [PubMed] [Google Scholar]
- F Hernández L, Castela I, Ruiz-DeDiego I, Obeso JA, Moratalla R (2017) Striatal activation by optogenetics induces dyskinesias in the 6-hydroxydopamine rat model of parkinson disease. Mov Disord 32:530–537. 10.1002/mds.26947 [DOI] [PubMed] [Google Scholar]
- Fieblinger T, Cenci MA (2015) Zooming in on the small: the plasticity of striatal dendritic spines in L-DOPA-induced dyskinesia. Mov Disord 30:484–493. 10.1002/mds.26139 [DOI] [PubMed] [Google Scholar]
- Fieblinger T, Graves SM, Sebel LE, Alcacer C, Plotkin JL, Gertler TS, Chan SC, Heiman M, Greengard P, Cenci MA, Surmeier DJ (2014) Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nat Comm 5:5316. 10.1038/ncomms6316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filali M, Lalonde R (2016) Neurobehavioral anomalies in the Pitx3/ak murine model of Parkinson's disease and MPTP. Behav Genet 46:228–241. 10.1007/s10519-015-9753-3 [DOI] [PubMed] [Google Scholar]
- Fuchs J, Mueller JC, Lichtner P, Schulte C, Munz M, Berg D, Wüllner U, Illig T, Sharma M, Gasser T (2009) The transcription factor PITX3 is associated with sporadic Parkinson's disease. Neurobiol Aging 30:731–738. 10.1016/j.neurobiolaging.2007.08.014 [DOI] [PubMed] [Google Scholar]
- Gagnon D, Petryszyn S, Sanchez MG, Bories C, Beaulieu JM, De Koninck Y, Parent A, Parent M (2017) Striatal neurons expressing D1 and D2 receptors are morphologically distinct and differently affected by dopamine denervation in mice. Sci Rep 7:41432. 10.1038/srep41432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia BG, Neely MD, Deutch AY (2010) Cortical regulation of striatal medium spiny neuron dendritic remodeling in parkinsonism: modulation of glutamate release reverses dopamine-depletion-induced dendritic spine loss. Cereb Cortex 20:2423–2432. 10.1093/cercor/bhp317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ (2011) Modulation of striatal projection systems by dopamine. Annu Rev Neurosci 34:441–466. 10.1146/annurev-neuro-061010-113641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler TS, Chan CS, Surmeier DJ (2008) Dichotomous anatomical properties of adult striatal medium spiny neurons. J Neurosci 28:10814–10824. 10.1523/JNEUROSCI.2660-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Aparicio R, Moratalla R (2014) Oleoylethanolamide reduces L-DOPA-induced dyskinesia via TRPV1 receptor in a mouse model of Parkinson's disease. Neurobiol Dis 62:416–425. 10.1016/j.nbd.2013.10.008 [DOI] [PubMed] [Google Scholar]
- Guigoni C, Dovero S, Aubert I, Li Q, Bioulac BH, Bloch B, Gurevich EV, Gross CE, Bezard E (2005) Levodopa-induced dyskinesia in MPTP-treated macaques is not dependent on the extent and pattern of nigrostrial lesioning. Eur J Neurosci 22:283–287. 10.1111/j.1460-9568.2005.04196.x [DOI] [PubMed] [Google Scholar]
- Halje P, Tamtè M, Richter U, Mohammed M, Cenci MA, Petersson P (2012) Levodopa-induced dyskinesia is strongly associated with resonant cortical oscillations. J Neurosci 32:16541–16551. 10.1523/JNEUROSCI.3047-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang DY, Fleming SM, Ardayfio P, Moran-Gates T, Kim H, Tarazi FI, Chesselet MF, Kim KS (2005) 3,4-Dihydroxyphenylalanine reverses the motor deficits in Pitx3-deficient aphakia mice: behavioral characterization of a novel genetic model of Parkinson's disease. J Neurosci 25:2132–2137. 10.1523/JNEUROSCI.3718-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iderberg H, Francardo V, Pioli EY (2012) Animal models of L-DOPA-induced dyskinesia: an update on the current options. Neuroscience 211:13–27. 10.1016/j.neuroscience.2012.03.023 [DOI] [PubMed] [Google Scholar]
- Jiménez-Jiménez FJ, García-Martín E, Alonso-Navarro H, Agúndez JA (2014) PITX3 and risk for Parkinson's disease: a systematic review and meta-analysis. Eur Neurol 71:49–56. 10.1159/000353981 [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y. (1997) Neostriatal cell subtypes and their functional roles. Neurosci Res 27:1–8. 10.1016/S0168-0102(96)01134-0 [DOI] [PubMed] [Google Scholar]
- Ketzef M, Spigolon G, Johansson Y, Bonito-Oliva A, Fisone G, Silberberg G (2017) Dopamine depletion impairs bilateral sensory processing in the striatum in a pathway-dependent manner. Neuron 94:855–865.e5. 10.1016/j.neuron.2017.05.004 [DOI] [PubMed] [Google Scholar]
- Klein C, Westenberger A (2012) Genetics of Parkinson's disease. Cold Spring Harb Perspect Med 2:a008888. 10.1101/cshperspect.a008888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC (2007) Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature 445:643–647. 10.1038/nature05506 [DOI] [PubMed] [Google Scholar]
- Le W, Nguyen D, Lin XW, Rawal P, Huang M, Ding Y, Xie W, Deng H, Jankovic J (2011) Transcription factor PITX3 gene in Parkinson's disease. Neurobiol Aging 32:750–753. 10.1016/j.neurobiolaging.2009.03.015 [DOI] [PubMed] [Google Scholar]
- Le W, Zhang L, Xie W, Li S, Dani JA (2015) Pitx3 deficiency produces decreased dopamine signaling and induces motor deficits in Pitx3(−/−) mice. Neurobiol Aging 36:3314–3320. 10.1016/j.neurobiolaging.2015.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhou FM (2013) Parallel dopamine D1 receptor activity dependence of L-DOPA induced normal movement and dyskinesia in mice. Neuroscience 236:66–76. 10.1016/j.neuroscience.2012.12.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matikainen-Ankney BA, Kezunovic N, Mesias RE, Tian Y, Williams FM, Huntley GW, Benson DL (2016) Altered development of synapse structure and function in striatum caused by Parkinson's disease-linked LRRK2–G2019S mutation. J Neurosci 36:7128–7141. 10.1523/JNEUROSCI.3314-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Liberge M, Jaouen F, Ztaou S, Hanini M, Camon J, Deisseroth K, Amalric M, Kerkerian-Le Goff L, Beurrier C (2015) Striatal cholinergic interneurons control motor behavior and basal ganglia function in experimental parkinsonism. Cell Rep 13:657–666. 10.1016/j.celrep.2015.09.034 [DOI] [PubMed] [Google Scholar]
- McNeill TH, Brown SA, Rafols JA, Shoulson I (1988) Atrophy of medium spiny I striatal dendrites in advanced Parkinson's disease. Brain Res 455:148–152. 10.1016/0006-8993(88)90124-2 [DOI] [PubMed] [Google Scholar]
- Moratalla R, Solis O, Suarez LM (2016) Morphological plasticity in the striatum associated with dopamine dysfunction. In: Handbook of basal ganglia structure and function, Ed 2 (Steiner H, Tseng KY, eds), pp 755–70. Amsterdam, the Netherlands: Elsevier. [Google Scholar]
- Murer MG, Moratalla R (2011) Striatal signaling in L-DOPA-induced dyskinesia: common mechanisms with drug abuse and long term memory involving D1 dopamine receptor stimulation. Front Neuroanat 5:51. 10.3389/fnana.2011.00051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely MD, Schmidt DE, Deutch AY (2007) Cortical regulation of dopamine depletion-induced dendritic spine loss in striatal medium spiny neurons. Neuroscience 149:457–464. 10.1016/j.neuroscience.2007.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijima H, Ueno T, Funamizu Y, Ueno S, Tomiyama M (2017) Levodopa treatment and dendritic spine pathology. Mov Disord. Advance online publication. Retrieved February 26, 2018. doi: 10.1002/mds.27172. 10.1002/mds.27172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes I, Tovmasian LT, Silva RM, Burke RE, Goff SP (2003) Pitx3 is required for development of substantia nigra dopaminergic neurons. Proc Natl Acad Sci U S A 100:4245–4250. 10.1073/pnas.0230529100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavón N, Martín AB, Mendialdua A, Moratalla R (2006) ERK phosphorylation and FosB expression are associated with L-DOPA-induced dyskinesia in hemiparkinsonian mice. Biol Psychiatry 59:64–74. 10.1016/j.biopsych.2005.05.044 [DOI] [PubMed] [Google Scholar]
- Pérez-Ortega J, Duhne M, Lara-González E, Plata V, Gasca D, Galarraga E, Hernández-Cruz A, Bargas J (2016) Pathophysiological signatures of functional connectomics in parkinsonian and dyskinetic striatal microcircuits. Neurobiol Dis 91:347–361. 10.1016/j.nbd.2016.02.023 [DOI] [PubMed] [Google Scholar]
- Planert H, Berger TK, Silberberg G (2013) Membrane properties of striatal direct and indirect pathway neurons in mouse and rat slices and their modulation by dopamine. PLoS One 8:e57054. 10.1371/journal.pone.0057054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plata V, Duhne M, Pérez-Ortega J, Hernández-Martinez R, Rueda-Orozco P, Galarraga E, Drucker-Colín R, Bargas J (2013) Global actions of nicotine on the striatal microcircuit. Front Syst Neurosci 7:78. 10.3389/fnsys.2013.00078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu G, Fu C, Liang GH (2014) Association between PITX3 promoter polymorphism and risk of Parkinson's disease: the impact of ethnicity and onset age. Neurosci Lett 561:128–133. 10.1016/j.neulet.2013.12.054 [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL (2008) Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One 3:e1997. 10.1371/journal.pone.0001997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-DeDiego I, Mellstrom B, Vallejo M, Naranjo JR, Moratalla R (2015a) Activation of DREAM (downstream regulatory element antagonistic modulator), a calcium-binding protein, reduces L-DOPA-induced dyskinesias in mice. Biol Psychiatry 77:95–105. 10.1016/j.biopsych.2014.03.023 [DOI] [PubMed] [Google Scholar]
- Ruiz-DeDiego I, Naranjo JR, Hervé D, Moratalla R (2015b) Dopaminergic regulation of olfactory type G-protein α subunit expression in the striatum. Mov Disord 30:1039–1049. 10.1002/mds.26197 [DOI] [PubMed] [Google Scholar]
- Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault JA, Hervé D, Greengard P, Fisone G (2007) Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in l-DOPA-induced dyskinesia. J Neurosci 27:6995–7005. 10.1523/JNEUROSCI.0852-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semina EV, Reiter R S, Murray J C (1997) Isolation of a new homeobox gene belonging to the Pitx/Rieg family: expression during lens development and mapping to the aphakia region on mouse chromosome 19. Hum Mol Genet 6:2109–2116. [DOI] [PubMed] [Google Scholar]
- Silberberg G, Bolam JP (2015) Local and afferent synaptic pathways in the striatal microcircuitry. Curr Opin Neurobiol 33:182–187. 10.1016/j.conb.2015.05.002 [DOI] [PubMed] [Google Scholar]
- Smidt MP, Smits SM, Bouwmeester H, Hamers FP, van der Linden AJ, Hellemons AJ, Graw J, Burbach JP (2004) Early developmental failure of substantia nigra dopamine neurons in mice lacking the homeodomain gene Pitx3. Development 131:1145–1155. 10.1242/dev.01022 [DOI] [PubMed] [Google Scholar]
- Solís O, Limón DI, Flores-Hernández J, Flores G (2007) Alterations in dendritic morphology of the prefrontal cortical and striatum neurons in the unilateral 6-OHDA-rat model of Parkinson's disease. Synapse 61:450–458. 10.1002/syn.20381 [DOI] [PubMed] [Google Scholar]
- Solís O, Espadas I, Del-Bel EA, Moratalla R (2015) Nitric oxide synthase inhibition decreases l-DOPA-induced dyskinesia and the expression of striatal molecular markers in Pitx3(−/−) aphakia mice. Neurobiol Dis 73:49–59. 10.1016/j.nbd.2014.09.010 [DOI] [PubMed] [Google Scholar]
- Solís O, Garcia-Montes JR, González-Granillo A, Xu M, Moratalla R (2017) Dopamine D3 receptor modulates L-DOPA-induced dyskinesia by targeting D1 receptor-mediated striatal signaling. Cereb Cortex 27:435–446. 10.1093/cercor/bhv231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens B, Mueller AJ, Shering AF, Hood SH, Taggart P, Arbuthnott GW, Bell JE, Kilford L, Kingsbury AE, Daniel SE, Ingham CA (2005) Evidence of a breakdown of corticostriatal connections in Parkinson's disease. Neuroscience 132:741–754. 10.1016/j.neuroscience.2005.01.007 [DOI] [PubMed] [Google Scholar]
- Suarez LM, Solís O, Caramés JM, Taravini IR, Solís JM, Murer MG, Moratalla R (2014) L-DOPA treatment selectively restores spine density in dopamine receptor D2-expressing projection neurons in dyskinetic mice. Biol Psychiatry 75:711–722. 10.1016/j.biopsych.2013.05.006 [DOI] [PubMed] [Google Scholar]
- Suarez LM, Solis O, Aguado C, Lujan R, Moratalla R (2016) L-DOPA-induced dyskinesia is associated with opposite synaptic changes in D1 and D2 striatal projection neurons. Cereb Cortex 26:4253–4264. 10.1093/cercor/bhw263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szydlowski SN, Pollak Dorocic I, Planert H, Carlén M, Meletis K, Silberberg G (2013) Target selectivity of feedforward inhibition by striatal fast-spiking interneurons. J Neurosci 33:1678–1683. 10.1523/JNEUROSCI.3572-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Zhao S, Wang M, Sheth A, Zhao Z, Chen L, Fan X, Chen L (2012) Meta-analysis of association between PITX3 gene polymorphism and Parkinson's disease. J Neurol Sci 317:80–86. 10.1016/j.jns.2012.02.025 [DOI] [PubMed] [Google Scholar]
- Tian X, Kai L, Hockberger PE, Wokosin DL, Surmeier DJ (2010) MEF-2 regulates activity-dependent spine loss in striatopallidal medium spiny neurons. Mol Cell Neurosci 44:94–108. 10.1016/j.mcn.2010.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trusel M, Cavaccini A, Gritti M, Greco B, Saintot PP, Nazzaro C, Cerovic M, Morella I, Brambilla R, Tonini R (2015) Coordinated regulation of synaptic plasticity at striatopallidal and striatonigral neurons orchestrates motor control. Cell Rep 13:1353–1365. 10.1016/j.celrep.2015.10.009 [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. (2008) The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135:422–435. 10.1016/j.cell.2008.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Munckhof P, Luk KC, Ste-Marie L, Montgomery J, Blanchet PJ, Sadikot AF, Drouin J (2003) Pitx3 is required for motor activity and for survival of a subset of midbrain dopaminergic neurons. Development 130:2535–2542. 10.1242/dev.00464 [DOI] [PubMed] [Google Scholar]
- Villalba RM, Smith Y (2018) Loss and remodeling of striatal dendritic spines in Parkinson's disease: from homeostasis to maladaptive plasticity? J Neural Transm (Vienna) 125:431–447 10.1007/s00702-017-1735-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaja-Milatovic S, Milatovic D, Schantz AM, Zhang J, Montine KS, Samii A, Deutch AY, Montine TJ (2005) Dendritic degeneration in neostriatal medium spiny neurons in parkinson disease. Neurology 64:545–547. 10.1212/01.WNL.0000150591.33787.A4 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Meredith GE, Mendoza-Elias N, Rademacher DJ, Tseng KY, Steece-Collier K (2013) Aberrant restoration of spines and their synapses in l-DOPA-induced dyskinesia: involvement of corticostriatal but not thalamostriatal synapses. J Neurosci 33:11655–11667. 10.1523/JNEUROSCI.0288-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]