

Abstract
There is substantial interest in memory reconsolidation as a target for the treatment of anxiety disorders, such as post-traumatic stress disorder. However, its applicability is restricted by reconsolidation-resistant boundary conditions that constrain the initial memory destabilization. In this study, we investigated whether the induction of synaptic protein degradation through autophagy modulation, a major protein degradation pathway, can enhance memory destabilization upon retrieval and whether it can be used to overcome these conditions. Here, using male mice in an auditory fear reconsolidation model, we showed that autophagy contributes to memory destabilization and its induction can be used to enhance erasure of a reconsolidation-resistant auditory fear memory that depended on AMPAR endocytosis. Using male mice in a contextual fear reconsolidation model, autophagy induction in the amygdala or in the hippocampus enhanced fear or contextual memory destabilization, respectively. The latter correlated with AMPAR degradation in the spines of the contextual memory-ensemble cells. Using male rats in an in vivo LTP reconsolidation model, autophagy induction enhanced synaptic destabilization in an NMDAR-dependent manner. These data indicate that induction of synaptic protein degradation can enhance both synaptic and memory destabilization upon reactivation and that autophagy inducers have the potential to be used as a therapeutic tool in the treatment of anxiety disorders.
SIGNIFICANCE STATEMENT It has been reported that inhibiting synaptic protein degradation prevents memory destabilization. However, whether the reverse relation is true and whether it can be used to enhance memory destabilization are still unknown. Here we addressed this question on the behavioral, molecular, and synaptic levels, and showed that induction of autophagy, a major protein degradation pathway, can enhance memory and synaptic destabilization upon reactivation. We also show that autophagy induction can be used to overcome a reconsolidation-resistant memory, suggesting autophagy inducers as a potential therapeutic tool in the treatment of anxiety disorders.
Keywords: AMPA receptors, long-term potentiation, NMDA receptors, post-traumatic stress disorder, protein degradation, reconsolidation
Introduction
Retrieval of long-term memories can induce a destabilization process that returns them into a labile state, which is followed by a protein synthesis-dependent reconsolidation process that serves to strengthen or update the original memories (Nader et al., 2000; Besnard et al., 2012; Finnie and Nader, 2012; Inaba et al., 2015; Lee et al., 2017). Blocking reconsolidation has been suggested as a tool to weaken traumatic memories in anxiety disorders, such as post-traumatic stress disorder (PTSD). However, the initial destabilization step is challenging when memories are formed under extremely stressful conditions, and it might require pharmacological assistance (Tronson and Taylor, 2007; Pitman, 2011; Besnard et al., 2012; Sevenster et al., 2012; Kindt and van Emmerik, 2016). It has been reported that inhibition of synaptic protein degradation, through blocking the ubiquitin-proteasome system, prevents memory destabilization (Lee et al., 2008). However, whether induction of synaptic protein degradation can be used to enhance memory destabilization is yet to be tested.
Macro-autophagy, hereafter referred to as autophagy, is a major protein degradation pathway where a newly synthesized isolation membrane sequesters a small portion of the cytoplasm to form a multilamellar vesicle called an autophagosome. To degrade the entrapped contents, autophagosomes fuse into the endosome-lysosome system (Mizushima and Komatsu, 2011; Yamamoto and Yue, 2014). The process of autophagosome synthesis is orchestrated by molecular machinery consisting of the autophagy-related genes (Atg) found in yeast, and their mammalian homologs (Mizushima et al., 2011; Ohsumi, 2014). In the brain, autophagy plays an important role in neurodegenerative diseases (Yamamoto and Yue, 2014) and is essential for the development of a healthy brain (Hara et al., 2006; Komatsu et al., 2006; Liang et al., 2010). It has been suggested that neurons may have adapted autophagy to suit their complex needs, including contribution to synaptic function (Bingol and Sheng, 2011; Mizushima and Komatsu, 2011; Shehata and Inokuchi, 2014; Yamamoto and Yue, 2014). In line with this idea, autophagosomes are found not only in the neuron's soma and axons but also in the dendrites (Hollenbeck, 1993; Shehata et al., 2012), Also, autophagy contributes to the degradation of the endocytosed GABAR in Caenorhabditis elegans and of the AMPARs upon chemical LTD in cultured neurons (Rowland et al., 2006; Shehata et al., 2012). Both GABAR and AMPAR play pivotal roles in the synaptic plasticity models of LTD and LTP, which are causally correlated with memory (Kessels and Malinow, 2009; Squire and Kandel, 2009; Nabavi et al., 2014). Moreover, the regulation of autophagy intersects with protein synthesis regulation at the mammalian target of rapamycin (mTOR) and the phosphatidylinositol-3-monophosphate kinase (PI3K) and by careful consideration of the discrepancy in the effects of the mTOR and PI3K modulators on memory processes, autophagy is suggested to play a role in memory reconsolidation (Chen et al., 2005; Gafford et al., 2011; Shehata and Inokuchi, 2014). In the present study, we tested the hypothesis that autophagy could play a role in synaptic and memory destabilization and therefore, the induction of autophagic protein degradation can be used to enhance erasure of reconsolidation-resistant fear memories.
Materials and Methods
Drugs and peptides
Anisomycin (Sigma-Aldrich) was dissolved in a minimum quantity of HCl, diluted with PBS, and adjusted to pH 7.4 with NaOH. Ifenprodil tartrate (Sigma-Aldrich) and trifluoperazine dihydrochloride (Sigma-Aldrich) were dissolved in PBS. Spautin-1 (Sigma-Aldrich) was dissolved in DMSO and diluted with equal volume of saline. The retro-inverso Tat-beclin 1 peptide D-amino acid sequence (RRRQRRKKRGYGGTGFEGDHWIEFTANFVNT; synthesized by GenScript through Funakoshi) was dissolved in either PBS (tBC) or anisomycin solution (Ani+tBC). The control d-Tat peptide d-amino acid sequence (YGRKKRRQRRR; EMC Microcollections) was dissolved in PBS (d-Tat). The Tat-GluA23Y peptide l-amino acid sequence (YGRKKRRQRRRYKEGYNVYG, AnaSpec) and its control Tat-GluA23A peptide l-amino acid sequence (YGRKKRRQRRRAKEGANVAG; AnaSpec) were both dissolved in PBS (GluA23Y or GluA23A, respectively). All peptides were aliquoted into single experiment volumes and stored at −80°C.
Experiments on mice
All procedures involving the use of animals were conducted in compliance with the guidelines of the National Institutes of Health and were approved by the Animal Care and Use Committee of the University of Toyama, Japan. Eight-week-old male C57BL/6J mice (IMSR catalog #JAX:000664, RRID:IMSR_JAX:000664) were purchased from Sankyo Labo Service, and the c-fos-tTA mice were purchased from the Mutant Mouse Regional Resource Center (MMRRC catalog #031756-MU, RRID:MMRRC_031756-MU). The progeny for the c-fos-tTA line was generated using in vitro fertilization of eggs from C57BL/6J mice, as described previously (Ohkawa et al., 2015). Mice were maintained on a 12 h light-dark cycle at 24 ± 3°C and 55 ± 5% humidity and were given food and water ad libitum and housed with littermates until surgery.
Stereotactic surgery and drug infusion in mice.
Mice were 8–10 weeks old at the time of surgery. Mice were anesthetized with isoflurane, given an intraperitoneal injection of pentobarbital solution (80 mg/kg of body weight), and then placed in a stereotactic apparatus (Narishige). Mice were then bilaterally implanted with a stainless guide cannula (PlasticsOne). For targeting the CA1, the guide cannula was positioned 1.8 mm posterior, 1.55 mm lateral, and 1.5 (C57BL/6J) or 1.0 mm (c-fos-tTA mice) ventral to the bregma. For targeting the BLA, the guide cannula was positioned 1.5 mm posterior, 3.3 mm lateral, and 3.4 mm ventral to the bregma. For targeting the lateral amygdala (LA), the guide cannula was positioned 1.7 mm posterior, 3.4 mm lateral, and 2.6 mm ventral to the bregma. After surgery, a cap or dummy cannula (PlasticsOne) was inserted into the guide cannula, and mice were allowed to recover for at least 7 d in individual home cages before the experiment. Mice were excluded from the experiment if showing an abnormal motility behavior after the recovery period or if the cannula was misplaced in position (∼10%). Mice in the NoFS condition were not cannulated.
All drug infusions were done under isoflurane anesthesia, using an injection cannula with a 0.25 mm internal diameter (PlasticsOne), and extending beyond the end of the guide cannula by 0.5 mm for the CA1, or by 1.5 mm for the BLA and LA. The drug infusion rate was 0.2 μl/min for the CA1 in C57BL/6J mice, or 0.1 μl/min for the CA1 in c-fos-tTA mice, and the BLA and LA. Following drug infusion, the injection cannula was left in place for 2 min to allow for drug diffusion. For the reconsolidation experiments, immediately after retrieval 1 μl of drug solution was injected into the CA1 in C57BL/6J mice, or 0.5 μl was injected into the CA1 in c-fos-tTA mice, the BLA and LA. In all of these reconsolidation experiments, 1 μl of drug solution contained either PBS, 125 μg of anisomycin, 20 μg of tBC, or 125 μg anisomycin + 20 μg tBC. For autophagy inhibition, 0.5 μl of a solution containing 8.3 μg spautin-1 or vehicle was injected into LA. For blocking AMPAR endocytosis, 0.5 μl of solution containing 20 ng of GluA23Y or GluA23A was injected into the LA.
Lysate preparation and immunoblot analysis.
Drugs were infused into the CA1 or amygdala of one hemisphere of the C57BL/6J mice, as described above. Four hours later, their brains were removed and cut into 1 mm slices, placed on ice, and the hippocampus or amygdala from each hemisphere was dissected under a binocular microscope, rapidly frozen on dry ice, and stored at −80°C. Samples were then sonicated in RIPA buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 2 mm EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mm NaF) containing a protease inhibitor mixture (cOmplete ULTRA tablets, Roche Diagnostics) and a phosphatase inhibitor mixture (PhosSTOP tablets, Roche Diagnostics). Samples were then centrifuged at 14,000 rpm for 15 min at 4°C, and supernatants were stored at −30°C until use. Measurement of protein concentration, immunoblotting for LC3 detection (Abcam catalog #ab48394, RRID:AB_881433), visualization and quantitation were performed as previously described (Shehata et al., 2012).
Contextual fear conditioning.
All behavioral sessions were conducted during the light cycle, in a dedicated soundproof behavioral room (Yamaha), described here as Room A. The conditioning specific context was a square chamber (Chamber A) with a Plexiglas front, off-white side- and back-walls (length 175 × width 165 × height 300 mm) and a floor consisting of stainless-steel rods connected to an electric shock generator. The distinct context was a circular chamber (Chamber B) with opaque reddish walls (diameter 235 mm × height 225 mm) and a smooth gray floor. One day before the experiment, mice were left undisturbed on a waiting rack for 2 h for habituation purposes. On the day of the experiment, mice were left undisturbed on the waiting rack for at least 30 min before and after each session, and during the experiment. In each session, one mouse in its home cage was moved into Room A. During the conditioning or reconditioning sessions, mice were placed in Chamber A and allowed to explore for 148 s, before receiving one footshock (2 s, 0.4 mA). They then remained for 30 s, before being moved back to their home cages and returned to the waiting rack. During the retrieval session (T1), mice were placed back into Chamber A for 3 min, then immediately subjected to isoflurane anesthesia and drug infusion. Mice in the NoFS condition were manipulated identically, with the exceptions that the shock generator was turned off. During the test sessions, mice were placed back into Chamber A (T2 and T4) for 5 min, and 1 h later into Chamber B (T3) for 5 min. Mice remained on the waiting rack during the 1 h interval. In all behavioral sessions, chambers were cleaned with 70% ethanol and water between each mouse, and kept odorless to the experimenter.
Auditory fear conditioning.
Different chambers were used for each auditory fear conditioning session. Context exploration and conditioning were performed in Chamber A. Retrieval sessions were performed in a circular chamber (Chamber C) with opaque black walls (diameter 215 mm × height 340 mm) and a smooth gray floor. Test sessions were performed in a circular chamber (Chamber D) with opaque reddish walls (diameter 235 mm × height 310 mm) and a smooth gray floor. After recovery from surgery, a maximum of 6 mice were moved with their home cages on racks in the maintenance room to a soundproof (Yamaha) waiting room (Room B). Mice were left undisturbed for at least 15 min before and after each session and during the experiment. In each session, one mouse in its home cage was moved into Room A. During the context exploration sessions, mice were placed in Chamber A and allowed to explore for 5 min per day for 2 d. During the conditioning sessions, mice were placed in Chamber A for 2 min and then received one or three tones (30 s, 65 dB, 7 kHz), coterminating with a shock (2 s, 0.4 mA), with an interval of 30 s. After the last shock, mice remained for 30 s and were then returned to their home cages and to Room B. During the retrieval sessions, mice were placed into Chamber C for 2 min before receiving a tone (30 s, 65 dB, 7 kHz); then 30 s later, mice were subjected to isoflurane anesthesia and drug infusion before being returned to Room B. For autophagy inhibition or blocking of AMPAR endocytosis, mice were subjected to isoflurane anesthesia and drug infusion 75 min before the retrieval sessions. During test sessions, mice were placed in Chamber D for 2 min and then received a tone (30 s, 65 dB, 7 kHz).
Behavioral analysis.
All experiments were conducted using a video tracking system (Muromachi Kikai) to measure the freezing behavior of the animals. Freezing was defined as a complete absence of movement, except for respiration. We started scoring the duration of the freezing response after 1 s of sustained freezing behavior. All behavioral sessions were digitally recorded using Bandicam software (Bandisoft). Occupancy plots representing the maximum occupancy of the mouse center in the defined context space during each session were generated by analyzing the screen recorded movies using ANY-maze software (RRID:SCR_014289, Stoelting). Mice were assessed as completely amnesic when they: (1) showed at least a 50% decrease in freezing level after drug infusion compared with the level before treatment, and (2) showed a freezing level in the conditioning or the distinct contexts within the 95% CI of the freezing level of the NoFS condition (used as a reference for normal mouse behavior).
Plasmid construction, lentivirus preparation, and infection.
For plasmid construction, mCherry (Clontech) was amplified by PCR using the following primers, sense: gggggatccgccaccatggtgagcaagggcgaggagg; antisense: ggggtcgaccccgggctacttgtacagctcgtcc. The resulting fragment was then used to replace the EYFP fragment at the BamHI-Sall sites in pBS-TRE3G-EYFP to produce the pBS-TRE3G-mCherry plasmid. The pBS-TRE3G-EYFP plasmid is a pBluescript II SK+ plasmid (Stratagene) containing the third-generation tTA-responsive TRE3G promoter sequence, derived from pTRE3G-IRES (Clontech, 631161) fused to EYFP. Finally, the TRE3G-mCherry fragment was subcloned into the STB plasmid using the SpeI/XbaI-XmaI sites to produce the pLenti-TRE3G-mCherry plasmid, which was used for the lentivirus preparation as previously described (Ohkawa et al., 2015). The viral titer was ∼5 × 109 IU/ml. Virus infection into CA1 of the c-fos-tTA mice (18–20 weeks old) was performed during the surgery for drug cannula fixation. Lentivirus (0.5 μl/site) was introduced through an injection cannula inserted into the guide cannula and left protruding by 0.5 mm. The injection rate was 0.1 μl/min, and the cannula was left in place for 20 min after the end of the injection, before being slowly withdrawn.
Labeling of the memory-ensemble cells.
Labeling of the memory-ensemble cells associated with contextual fear was performed in a similar manner to the experiments on contextual fear conditioning. The experiment was performed on lentivirus-injected c-fos-tTA mice, maintained since weaning on food containing 40 mg/kg doxycycline. Two weeks after lentivirus infection, mice were subjected to the waiting rack for 2 h for habituation purposes. One day later, doxycycline was removed and mice were maintained on normal food. Two days after doxycycline removal, mice were subjected to a contextual fear conditioning session as mentioned above. Six hours later, the feed for the mice was changed to food containing 1000 mg/kg doxycycline. The retrieval session and drug infusion were performed as mentioned above. One day after drug infusion, the mice were deeply anesthetized with an overdose of pentobarbital solution, and perfused transcardially with PBS, pH 7.4, followed by 4% PFA in PBS. The brains were removed, further postfixed by immersion in PFA for 16–24 h at 4°C, equilibrated in 30% sucrose in PBS for 36–48 h at 4°C, and then stored at −80°C.
Immunohistochemistry.
Double labeling primary antibodies from the same host species (rabbit) were used for GluA1 and mCherry staining. Several incisions were made to label the right side of the brains, and they were then cut into 50 μm coronal sections using a cryostat and transferred to 12-well cell culture plates (Corning) containing PBS. After washing with PBS, the floating sections were treated with blocking buffer (5% normal donkey serum; S30, Bioscience Research Reagents by EMD Millipore) in 0.3% Triton X-100-PBS (TPBS) at room temperature (RT) for 1 h. They were then treated with anti-GluA1 antibody (1:500; Millipore catalog #AB1504, RRID:AB_2113602) in blocking buffer at 4°C for 36–40 h. After three 10 min washes with 0.1% PBST (the procedure for further mentions of washing in this paragraph), sections were incubated with donkey anti-rabbit IgG-AlexaFluor-488 secondary antibody in blocking buffer (Invitrogen catalog #A-21206, RRID:AB_141708) at RT for 4 h. After washing, sections were incubated with 5% normal rabbit serum (Jackson ImmunoResearch Laboratories) in 0.3% TPBS at RT for 1 h. Following washing, sections were incubated with 4% Fab Fragment Donkey Anti-rabbit IgG (Jackson ImmunoResearch Laboratories) in 0.3% TPBS at RT for 2 h. Sections were then washed and treated with anti-mCherry antibody (1:500; Clontech Laboratories catalog #632496, RRID:AB_10013483) in blocking buffer at 4°C for 36–40 h. After washing, sections were incubated with donkey anti-rabbit IgG-AlexaFluor546 secondary antibody in blocking buffer (Thermo Fisher Scientific catalog #A10040, RRID:AB_2534016) at RT for 4 h. Sections were then washed and treated with DAPI (1 μg/ml, Roche Diagnostics, 10236276001), then washed three times with PBS. Sections were then mounted on glass slides with ProLong Gold antifade reagent (Invitrogen).
Confocal microscopy and analysis of puncta.
Images were acquired using an LSM 780 confocal microscope (Carl Zeiss). First, a Plan-Apochromat 5× objective lens was used to check for the treatment side, and then low-magnification images of the CA1 radiatum were acquired for each selected hemisphere using a Plan-Apochromat 20× objective lens. High-magnification images for dendrites and spines were acquired using a Plan-Apochromat 63×/1.4 oil DIC objective lens. All acquisition parameters were kept constant within each magnification. To detect GluA1 puncta and the mCherry-labeled dendrites and spines, high resolution (4096 × 4096) images were acquired by collecting z stacks (5 slices at 0.6 μm thickness, and 0.3 μm interval). After performing a digital zoom (7×), maximum intensity projection images were created with ZEN 2.1 Black (Carl Zeiss) and further processed with Gamma correction at γ = 1.5 (for mCherry-labeled spines and Total GluA1 puncta) or γ = 5 (for GluA1 strong signal). ImageJ software (RRID:SCR_003070; National Institutes of Health) was used to apply a constant threshold to the green channel to create binary images for both Total GluA1 puncta (GluA(Tot)+) and GluA1 strong signal (GluA1(Str)+). Both puncta were automatically counted using the Analyze particles function with a particle size of > 50 pixel2 for GluA(Tot)+ or > 100 pixel2 for GluA1(Str)+, and a circularity of 0.2–1.0. Any fused puncta were manually separated before automatic counting. Overlaps between the GluA1(Str)+ puncta and mCherry+ spines were manually counted, guided by the green and red thresholding in ImageJ. The mCherry+ only spines did not overlap with any GluA1(Tot)+ puncta. Three hemispheres were analyzed for each of the PBS or Ani+tBC treatments from 4 mice. For each hemisphere, data from four analyzed maximum intensity projection images were averaged.
Experiments on rats
All procedures involving the use of animals were conducted in compliance with the guidelines of the National Institutes of Health and were approved by the Animal Care and Use Committee of the University of Toyama, Japan. Eight-week-old male Wistar ST rats were purchased from Sankyo Labo Service and maintained on a 12 h light-dark cycle at 24 ± 3°C and 55 ± 5% humidity. They were given food and water ad libitum and housed with littermates until surgery.
Stereotactic surgery and drug infusion in rats.
Previously described surgical procedures were used with some modifications (Okubo-Suzuki et al., 2016). Rats were 8–10 weeks old at the time of surgery. Rats were anesthetized with isoflurane, given an intraperitoneal injection of pentobarbital solution (55 mg/kg of body weight), and then placed in a stereotactic apparatus (Narishige). In brief, a bipolar stimulating electrode and a monopolar recording electrode, both made of tungsten wire, were stereotaxically positioned to stimulate the perforant pathway (angular bundle) while recording in the dentate gyrus. The stimulating electrode was positioned 7.5 ± 0.3 mm posterior, 4.4 ± 0.3 mm lateral, and 4.7 ± 0.3 mm ventral to the bregma. The recording electrode was positioned ipsilaterally 4.0 ± 0.3 mm posterior, 2.5 ± 0.3 mm lateral, and 3.8 ± 0.3 mm ventral to the bregma.
For intracerebroventricular infusion, a stainless-steel guide cannula (Eicom) was positioned ipsilaterally 0.7 ± 0.3 mm posterior, 1.6 ± 0.3 mm lateral, and 4.0 mm ventral to the bregma. After surgery, a dummy cannula (Eicom), which extended 1.0 mm beyond the end of the guide cannula, was inserted into the guide cannula. After surgery, rats were given subcutaneous injection of lidocaine as analgesic and were allowed to recover for at least 10 d in individual home cages before the experiment. Intracerebroventricular drug infusion was performed on unanesthetized freely moving rats, using an injection cannula (Eicom) that extended 0.5 mm beyond the end of the guide cannula, with an infusion rate of 1 μl/min. Following drug infusion, the injection cannula was left in place for 5 min to allow for drug diffusion.
In vivo electrophysiology on freely moving rats.
The LTP experiments were modified from those previously described (Okubo-Suzuki et al., 2016). After recovery from surgery, the input–output curves were determined as a function of current intensity (0.1–1.0 mA), and the intensity of the stimulus current required to elicit the maximum fEPSP slope (MAX) was determined for each animal. The stimulus current intensity was set to elicit 50% of MAX. Three days later, 400 Hz was performed. The 400 Hz stimulation used for LTP induction consisted of 10 trains with 1 min intertrain intervals, with each train consisting of five bursts of 10 pulses at 400 Hz, delivered at 1 s interburst intervals, giving a total of 500 pulses. The 8 Hz stimulation, which was performed as a reactivation stimulation, consisted of 128 pulses at 8 Hz. The fEPSP slope was monitored by delivering test pulses at 0.05 Hz for 15 min before (PreStim), and 5 min after (PostStim) stimulation. For testing the dependency of the stimulation on protein synthesis, 5 μl PBS or 5 μl of a solution containing 400 μg of anisomycin, was infused directly after the PostStim recording. For LTP reconsolidation experiments, LTP (400 Hz stimulation) was performed 3 d after MAX, and 1 d later, the 8 Hz reactivation stimulation was performed. As mentioned above, the fEPSP slope was monitored both before (PreLTP) and 5 min after LTP induction (PostLTP), and before (PreReact) and 5 min after (PostReact) the 8 Hz reactivation stimulation. Immediately after PostReact recording, rats received a 5 μl intracerebroventricular drug infusion, containing PBS, 400 μg of anisomycin, 100 μg of tBC, or 400 μg of anisomycin + 100 μg of tBC, as described above. For inhibition of NMDAR-2B, 5 μl of a solution containing 5 μg of ifenprodil tartrate was intracerebroventricularly infused immediately before the PreReact recording. The fEPSP slope was monitored over the following 3 d. Rats were excluded when showing abnormal behavior after surgery, LTP was not induced from the first trial, or the cannula or the electrodes were misplaced in position.
Experimental design and statistical analysis
In figure legends, n indicates the number of animals per treatment condition unless otherwise indicated. All experiments were performed at least three times with lots of 3–6 animals each. Treatments were counterbalanced for each lot. Animals were blindly and randomly allocated for each treatment condition. Statistical analysis was performed using Prism version 6.01 or InStat version 3.1 (GraphPad Software). Data from two conditions were compared using two-tailed unpaired Student's t tests. Multiple-condition comparisons were assessed using ANOVA with post hoc tests as described in Results. p values were considered significant if <0.05. Quantitative data are presented as mean ± SEM.
Results
Autophagy contributes to fear memory destabilization
To modulate autophagy activity within the time window of reconsolidation, we pharmacologically targeted the Beclin1 protein, which is part of the Beclin1-Atg14L-Vps34 lipid kinase complex that is involved in the autophagosome synthesis. This will specifically modulate autophagy activity without affecting endocytosis, mTOR, or PI3K activity (Vanhaesebroeck et al., 2010; Liu et al., 2011; Shoji-Kawata et al., 2013; Marsh and Debnath, 2015; De Leo et al., 2016). For autophagy induction, we used the cell-permeable tat-beclin1 peptide (tBC), which is composed of the human immunodeficiency virus-1 transduction domain attached to the necessary and sufficient peptide sequence of the beclin1 protein (Shoji-Kawata et al., 2013). The tBC peptide induces autophagy in the brains of mice neonates when systemically injected (Shoji-Kawata et al., 2013), and induces autophagy in the amygdala of adult mice when directly infused as monitored through the conversion of the light chain protein 3 (LC3), an autophagosome-specific marker, from its inactive form (LC3-I) to the lipidated active form (LC3-II). For autophagy inhibition, we used spautin-1, which promotes the degradation of the Beclin1-Atg14L-Vps34 complex through inhibiting the ubiquitin-specific peptidases that target the beclin1 subunit of the complex (Liu et al., 2011). Infusion of spautin-1 into the amygdala inhibited both the basal and the tBC-induced autophagic activity (LC3-II/LC3-I: one-way ANOVA, F(3,12) = 6.597, p = 0.007, Tukey's post hoc test; and total LC3: one-way ANOVA, F(3,12) = 0.8254, p = 0.5048; Fig. 1A,B).
Figure 1.

Autophagy contributes to fear memory destabilization. A, Representative LC3 immunoblot from mouse amygdala lysates showing autophagy induction by tBC and inhibition by spautin-1 (Spautin). LC3-II is an autophagosome-specific marker. B, Quantitation of the immunoblot signal intensity represented as percentage relative to a Veh/PBS sample (n = 4 mice/condition). C, Design for the 1FS-AFC experiments. D, Average percentage freezing during tone at T1 and T2 showing that blocking autophagy significantly decreased the amnesic effect of anisomycin (n = 10 or 11 mice/condition). E, Average percentage freezing during tone at T1 and T2 showing that, when the amnesic effect of anisomycin (Ani) was complete, autophagy induction did not have any further effect. No reactivation (NR) control showed no amnesic effect. No injections were done before T1 (n = 7–9 mice/ condition). Error bars indicate mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001. Veh, Vehicle.
To examine the effect of autophagy modulation on memory destabilization, we used a reconsolidation model of fear conditioning. Fear conditioning is an associative learning procedure, in which animals learn to associate a specific auditory cue (auditory fear conditioning) or context (contextual fear conditioning), which is a conditioned stimulus (CS), with a foot shock, an unconditioned stimulus (US). When animals are subjected to the CS, they recall the fear memory, resulting in a freezing response.
When a one tone-footshock pair was used for auditory fear conditioning (1FS-AFC), anisomycin infusion into the LA after tone retrieval led to a significant decrease in the tone-elicited freezing response compared with the vehicle-infused condition (Fig. 1C–E), in agreement with previous reports (Nader et al., 2000; Suzuki et al., 2004; Mamiya et al., 2009). Anisomycin produced a retrieval-specific retrograde amnesia as anisomycin administration without the retrieval session had no effect on tone fear memory (Fig. 1E). Inhibiting autophagy through spautin-1 infusion into the LA before retrieval partially blocked the anisomycin amnesic effect, indicating that autophagy contributes to the memory destabilization process (two-way repeated-measures ANOVA, interaction F(3,37) = 4.224, p = 0.0115; Bonferroni's post hoc test for within-condition comparison and Newman–Keuls test for between-conditions comparison; Fig. 1C,D). Anisomycin administration alone resulted in almost complete fear memory amnesia of the weak auditory fear conditioning (1FS-AFC), leaving no space for a further decrease in the tone-elicited freezing response. Therefore, autophagy induction combined with anisomycin (Ani+tBC) did not show any additional amnesic effect over anisomycin administration alone (two-way repeated-measures ANOVA, interaction F(2,22) = 1.594, p = 0.2257; Holm–Sidak's post hoc test; Fig. 1C,E).
Autophagy overcomes a reconsolidation-resistant boundary condition that is AMPAR endocytosis-dependent
Next, we examined the effect of autophagy induction on stronger auditory fear conditioning by increasing memory strength using three tone-FS pairs (3FS-AFC), generating a reconsolidation-resistant boundary condition. In the 3FS-AFC, anisomycin infusion into the LA after retrieval did not show any significant effect on the tone-elicited freezing response in comparison with the vehicle-infused condition. By contrast, Ani+tBC infusion after retrieval significantly reduced the tone-elicited freezing response levels, indicating that autophagy induction enhances memory destabilization beyond the fear memory reconsolidation-resistant boundary condition. Without the retrieval session, Ani+tBC administration in the 3FS-AFC had no effect on auditory fear memory, indicating that a retrieval-specific process is necessary for the autophagy-enhancing effect on memory destabilization (two-way repeated-measures ANOVA, interaction F(3,30) = 3.476, p = 0.0281; Bonferroni's post hoc test for within-condition comparison and Kramer test for between-conditions comparison; Fig. 2A,B). Collectively, the results obtained from both the inhibition and the induction of autophagy indicate a causal relationship between autophagy activity and memory destabilization.
Figure 2.

Autophagy overcomes a reconsolidation-resistant boundary condition in an AMPAR endocytosis-dependent manner. A, Design for the 3FS-AFC experiments. The experiment was performed either with no injection before T1 or with injection of Tat-GluA2 peptides: GluA23Y, for blocking AMPA receptor endocytosis; or GluA23A, as a negative control. B, Average percentage freezing during tone at T1 and T2 showing that Ani+tBC showed significant retrograde amnesia, whereas the anisomycin alone condition showed no amnesic effect (n = 7–10 mice/condition). C, Average percentage freezing during tone at T1 and T2 showing that blocking AMPAR endocytosis abolished the amnesic effect of autophagy induction (n = 8–11 mice/condition). Error bars indicate mean ± SEM. *p < 0.05, **p < 0.01. NR, No reactivation (no T1).
We attempted to elucidate how autophagy modulates memory destabilization. As AMPARs are endocytosed after memory retrieval, we hypothesized that the autophagosome may fuse with endosomes carrying AMPAR and dictate their fate to lysosomal degradation (Rao-Ruiz et al., 2011; Shehata et al., 2012; Shehata and Inokuchi, 2014). Therefore, blocking endocytosis would block the autophagy effect on memory destabilization. The neural activity-dependent endocytosis of AMPAR relies on the carboxy-tail of GluA2, and the use of the synthetic peptide Tat-GluA23Y is well established in attenuating activity-induced, but not constitutive, GluA2-dependent synaptic removal of AMPARs (Kim et al., 2001; Lee et al., 2002; Ahmadian et al., 2004; Scholz et al., 2010). In the 3FS-AFC, Tat-GluA23Y peptide infusion into the LA before retrieval completely blocked the Ani+tBC amnesic effect, whereas the control mutant peptide Tat-GluA23A had no effect (two-way repeated-measures ANOVA, interaction F(3,35) = 4.787, p = 0.0067; Bonferroni's post hoc test for within-condition comparison and Tukey's post hoc test for between-conditions comparison; Fig. 2A,C).
Autophagy enhances retrograde amnesia of fear memory in contextual fear conditioning when targeted to the amygdala
We further investigated the autophagy induction effect on reconsolidation of contextual fear conditioning. In contextual fear conditioning, the CS is a specific context, and the memory of the details of that context triggers a freezing response that is greater than that triggered by any other distinct context (Fanselow, 2000). Typically, inhibition of protein synthesis after the CS retrieval leads to a certain degree of retrograde amnesia (Besnard et al., 2012; Finnie and Nader, 2012). To assess the degree of the retrograde amnesia, we compared it with that of a reference condition exposed to the same contexts without receiving any shock (NoFS). After contextual fear conditioning, an anisomycin infusion into the BLA after memory retrieval led to a decrease in the freezing response in comparison with the vehicle-infused condition (Fig. 3A,B) (Suzuki et al., 2004; Mamiya et al., 2009). Nevertheless, the freezing response after anisomycin administration was significantly higher than that in the NoFS condition, in both the specific and distinct contexts, implying that the resultant retrograde amnesia was only partial. After Ani+tBC administration, the average freezing response dramatically reduced, reaching no statistical significant difference from the NoFS condition in both contexts (two-way repeated-measures ANOVA, interaction F(8,102) = 10.19, p = 0.0001; Bonferroni's post hoc test for within-condition comparison and Tukey's post hoc test for between-conditions comparison; T2: Ani vs NoFS p = 0.0013, Ani+tBC vs NoFS p = 0.2039; T3: Ani vs NoFS p = 0.0222, Ani+tBC vs NoFS p = 0.9919; Fig. 3B). Further, we assessed the level of amnesia for each mouse where those showing a freezing response within the CI of the NoFS condition were classified as complete amnesic mice (see Materials and Materials and Methods; Fig. 3C). In the Ani+tBC administered condition, 5 of 12 mice were regarded as complete amnesic; in contrast, only 1 of 12 mice in the anisomycin administered condition (Fig. 3D). Within the Ani+tBC condition, the average freezing response of the complete amnesic mice was as high as the remaining mice and dropped to be as low as the NoFS mice after Ani+tBC administration (two-way ANOVA, interaction F(2,19) = 27.15, p = 0.0001, Tukey's post hoc test; T1: Ani+tBC(complete) vs Ani+tBC(incomplete) p = 0.9616, T2: Ani+tBC(complete) vs NoFS p = 0.9591; Fig. 3E). When the complete amnesic mice were subjected to a reconditioning session, they regained the freezing response to levels matching the preamnesic freezing levels, indicating an intact capacity for fear expression (one-way ANOVA, F(2,11) = 11.316, p = 0.0021, Tukey's post hoc test; Fig. 3F). Together, these behavioral data indicate that induction of autophagy enhanced the amnesic effect of protein synthesis inhibition after retrieval and resulted in an enhanced level of retrograde amnesia.
Figure 3.

Autophagy enhances fear memory destabilization in contextual fear conditioning when targeted to amygdala. A, Design for contextual fear conditioning reconsolidation and reconditioning experiments. B, Average percentage freezing during retrieval (T1), and after the drugs were infused into the BLA when tested in the conditional stimulus context (T2) and in a distinct context (T3). Freezing levels at both T2 and T3 is showing a significant enhancement of anisomycin (Ani) amnesic effect on fear memory when combined with autophagy induction (n = 10–12 mice/condition). C, Plot of individual mice freezing level at T2 against their freezing level at T3, for the assessment of complete fear amnesia after contextual fear conditioning. Red cross and yellow dashed lines indicate a hypothetical point calculated from double the SD for the freezing of the no foot shock (NoFS) condition at T2 and T3, where most of mice received Ani+tBC treatment behaved as the NoFS condition. D, Individual data for the complete and incomplete amnesic mice of the Ani+tBC condition compared with the anisomycin alone condition. Dashed red line indicates a complete amnesic mouse in the anisomycin only condition. E, Average percentage freezing for the complete and incomplete amnesic mice of the Ani+tBC condition compared with the NoFS condition (n = 5–10 mice/condition). F, The complete amnesic mice showed a normal freezing response 1 d after a reconditioning session (T4). Error bars indicate mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Autophagy enhances retrograde amnesia of contextual memory when targeted to the hippocampus and AMPAR degradation in the spines of the memory-ensemble cells
We next examined the generality of the autophagy induction effect on other brain areas by targeting the CA1 region of the hippocampus. The tBC peptide induced autophagy in the hippocampus of adult mice when directly infused (LC3-II/LC3-I: one-way ANOVA, F(2,10) = 5.429, p = 0.0253, Tukey's post hoc test and total LC3: one-way ANOVA, F(2,10) = 0.1282, p = 0.8811; Fig. 4A,B). As with the results from the BLA, anisomycin infusion into the CA1 after memory retrieval led to a decrease in the freezing response (two-way repeated-measures ANOVA, interaction F(8,106) = 9.027, p = 0.0001; Bonferroni's post hoc test for within-condition comparison and Tukey's post hoc test for between-conditions comparison; Fig. 4C,D) (Mamiya et al., 2009). Ani+tBC infusion into CA1 significantly decreased the discrimination between the conditioning specific and the distinct contexts (two-way repeated-measures ANOVA, interaction F(3,44) = 7.211, p = 0.0005; Holm–Sidak's post hoc test for within-condition comparison and Newman–Keuls test for between-conditions comparison; Fig. 4E,F) without affecting the fear memory itself in the conditioning specific context (Fig. 4D,E). The same result was obtained when another autophagy inducer, trifluoperazine, was combined with anisomycin (two-way repeated-measures ANOVA, interaction F(3,18) = 5.328, p = 0.0084; Bonferroni's post hoc test for within-condition comparison and Tukey's post hoc test for between-conditions comparison; Fig. 4G). These results indicate that context information was erased from CA1, whereas fear information was intact, leading to fear memory generalization. Together, these findings indicate that the enhanced memory destabilization resulting from induction of autophagy is not restricted to one brain area, and that the behavioral outcome of autophagy induction differs in accordance with the main information encoded in the target brain area.
Figure 4.

Autophagy enhances context memory destabilization in contextual fear conditioning when targeted to hippocampus. A, Representative immunoblot from hippocampal lysates collected 4 h after unilateral drug infusion into CA1. B, Quantitation of the signal intensity represented as percentage relative to a control PBS sample (n = 4 or 5 per condition). C, Design for the contextual fear conditioning reconsolidation experiment. D, Average percentage freezing during retrieval (T1), and after the drugs were infused into the hippocampal CA1 region when tested in the conditional stimulus context (T2) and in a distinct context (T3). Combined Ani+tBC induction showed higher freezing levels at T3 compared with anisomycin alone condition (n = 10–14 mice/condition). E, Data from D represented as the freezing level at T2 or T3 relative to T1, showing the loss of context discrimination after Ani+tBC combined treatment. F, A representative occupancy plot for a mouse per condition at T3 from D. G, Freezing level at T2 or T3 relative to T1 after autophagy induction by infusion of AMPAR into the hippocampal CA1 region alone or combined with anisomycin. As with tBC, AMPAR combined with anisomycin resulted in loss of context discrimination (n = 5 or 6 mice/condition). Error bars indicate mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We tested the involvement of autophagy in the degradation of the endocytosed AMPAR after retrieval using contextual fear conditioning and benefiting from the dendrite orientation in the CA1 radiatum. We quantified the level of AMPAR colocalizing with the spines of the neurons holding the memory trace after the amnesic treatments. To label the memory-ensemble cells in the CA1 region, lentivirus expressing mCherry under the control of the tetracycline response element was injected into c-fos-tTA transgenic mice, which had been maintained on a diet containing doxycycline, except for the period spanning 2 d before and 6 h after the contextual fear conditioning session (Fig. 5A,B) (Reijmers et al., 2007; Ohkawa et al., 2015). Following retrieval, vehicle or Ani+tBC was unilaterally infused into the CA1, and changes in the GluA1, an AMPAR subunit, staining and its overlap with the mCherry spines (representing the spines of the memory-ensemble cells) were checked 1 d later, reflecting their status at the Test 2 session (Fig. 5B,C). The GluA1 puncta were classified into strong or weak signals according to their fluorescent intensity and reflecting the level of AMPAR enrichment. The total GluA1 puncta, the ratio of strong signals to the total GluA1 puncta, and the number of mCherry-only spines did not significantly differ between the two conditions (unpaired Student's t tests; Fig. 5D,E,I–K). Only the overlap between the GluA1 strong signals and mCherry spines was significantly lower in the Ani+tBC condition compared with the vehicle condition and not significantly different from the chance level (two-way repeated-measures ANOVA, interaction F(1,4) = 12.4, p = 0.0244; Bonferroni's post hoc test; Fig. 5F–H). This decrease in AMPAR enrichment in the spines of ensemble-cells to the chance level is in accordance with the behavioral data showing a decrease in contextual memory and hence loss of context discrimination (Fig. 4E).
Figure 5.
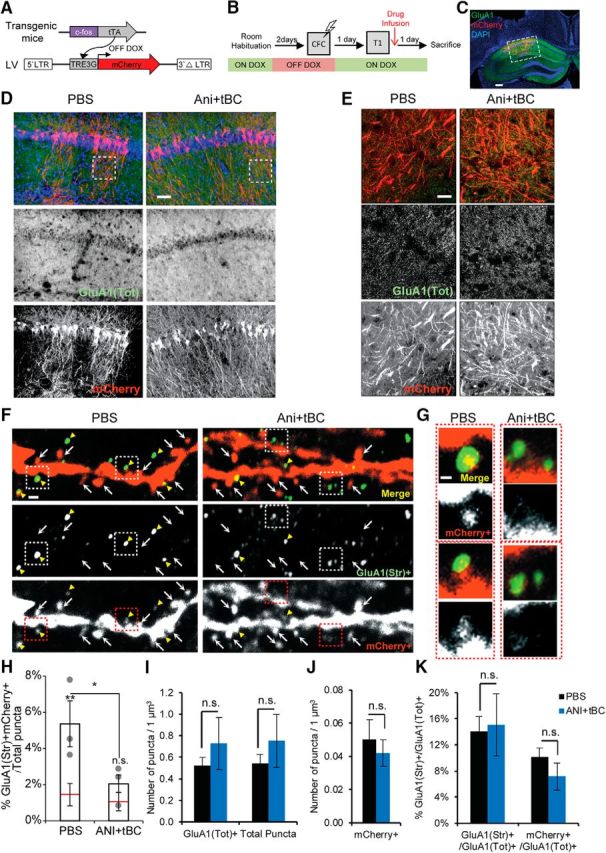
Autophagy enhances AMPAR degradation in the spines of memory-ensemble cells. A, Lentivirus (LV)-mediated labeling of the spines of the memory-ensemble cells with mCherry in the c-fos-tTA transgenic mice. B, Experimental design for checking the effect of autophagy induction on AMPAR expression and distribution in memory-ensemble cells. C, Images showing immunohistochemical staining for mCherry (red), and endogenous GluA1 (green) in an Ani+tBC treated hemisphere. Scale bar, 400 μm. D, E, Low-magnification images (D; scale bar, 50 μm) and higher-magnification maximum intensity projection images (E; scale bar, 20 μm) showing that Ani+tBC treatment did not affect the overall AMPAR signals compared with PBS control. F, G, Representative dendrites for each treatment condition showing less colocalization of the mCherry-stained spines (mCherry+) with the GluA1-strongly stained puncta (GluA1(Str)+) in the Ani+tBC condition than in the PBS condition. Scale bar, 500 nm. Insets are shown in G. Yellow arrowheads indicate colocalization. Arrows indicate mCherry+-only spines. G, Higher-magnification images for two spines per condition. Scale bar, 200 nm. H, Quantitation for the colocalization of mCherry+ spines with the GluA1(Str)+ puncta per total puncta counted and the chance level (red line). The overlap between mCherry+ spines and the GluA1(Str)+ decreased to chance level after Ani+tBC treatment (n = 3 hemispheres/condition; four images/hemisphere). I–K, No significant difference between PBS- or Ani+tBC-injected hemispheres. I, The total GluA1 puncta (GluA1(Tot)+) or total counted puncta. J, The mCherry-labeled spines (mCherry+). K, The ratio of the GluA1 puncta with a strong signal (GluA1(Str)+) or in the mCherry+ spines with GluA1(Tot) + puncta (n = 3 hemispheres/condition; four images/hemisphere). Error bars indicate mean ± SEM. *p < 0.05, **p < 0.01. DOX, Doxycycline.
Autophagy destabilizes synaptic plasticity in an LTP reconsolidation model
Finally, we tested the effect of autophagy induction on synaptic destabilization using an in vivo LTP system in rats, in which a protein synthesis-dependent long-lasting LTP was induced in the dentate gyrus by 400 Hz high-frequency stimulation of the perforant path (unpaired Student's t test, 400 Hz: p = 0.0371; Fig. 6A,B) (Fukazawa et al., 2003). To model synaptic reconsolidation, the perforant path was reactivated by a protein synthesis-dependent 8 Hz stimulation (unpaired Student's t test, 8 Hz: p = 0.0086; Fig. 6B) 1 d after LTP induction; this to resensitize the LTP to the protein synthesis inhibitor anisomycin, thereby mimicking behavioral reconsolidation (Fig. 6C) (Okubo-Suzuki et al., 2016). Anisomycin treatment significantly decreased the fEPSP slope 1 d after 8 Hz reactivation compared with the vehicle condition. However, this effect was only partial, as the fEPSP slope was still higher than the baseline level before LTP induction. Following Ani+tBC treatment, LTP destabilization was almost complete and the fEPSP slope was not significantly higher than the baseline level. This enhancement of synaptic destabilization was not observed when anisomycin administration was combined with the unfused Tat peptide (d-Tat) (two-way repeated-measures ANOVA, interaction F(20,235) = 4.279, p = 0.0001; Holm–Sidak's post hoc test; Fig. 6D,E). These data indicate that induction of autophagy enhanced the synaptic destabilization triggered by the 8 Hz reactivation. Furthermore, behavioral reconsolidation is dependent on NMDARs, and GluN2B-containing NMDAR (NMDAR-2B) is required for memory destabilization after recall (Ben Mamou et al., 2006; Milton et al., 2013). In our synaptic reconsolidation model, ifenprodil, a selective NMDAR-2B antagonist, blocked the LTP-destabilizing effect of anisomycin administration, mimicking behavioral reconsolidation. Also, ifenprodil completely blocked the LTP-destabilizing effect of Ani+tBC administration (two-way repeated-measures ANOVA, interaction F(10,105) = 0.5138, p = 0.8771; Holm–Sidak's post hoc test; Fig. 6F,G). These data indicate that NMDAR-2B was involved in physiological destabilization in our in vivo synaptic reconsolidation model, and demonstrate that the effect of enhanced autophagy on synapse destabilization is downstream of NMDAR-2B.
Figure 6.

Autophagy induction enhances synaptic destabilization of LTP in freely moving rats. A, Diagrams and images of hematoxylin-stained slices for the stimulation electrode, the recording electrode, and the drug injection cannula. Arrows indicate corresponding scars. B, In the in vivo LTP, both 400 and 8 Hz stimulations were protein synthesis-dependent. Anisomycin (Ani) or PBS was infused 5 min after the 400 or 8 Hz stimulation and percentage fEPSP slope was calculated on day 2 relative to the prestimulation level on day 1 (n = 5–7 rats/condition). C, Design for the LTP reconsolidation experiment; fEPSP was recorded immediately before (pre) and after (post) LTP induction and reactivation, and for 3 consecutive days after intracerebroventricular drug infusion. D, Percentage fEPSP slope relative to the preLTP level showing that autophagy induction using tBC, but not the unfused control peptide d-Tat, significantly enhanced synaptic destabilization compared with anisomycin (Ani) only treatment. No injections were given before reactivation (n = 10 or 11 rats/condition). E, Representative waveform traces and enlarged portion of slope (inset) for each treatment from D. F, Percentage fEPSP slope relative to the preLTP level when ifenprodil (IFN), an NMDA receptor blocker, was injected before 8 Hz reactivation. IFN completely blocked the synaptic destabilization effect of the anisomycin only and the Ani+tBC treatments (n = 8 rats/condition). G, Representative waveform traces and enlarged portion of slope (inset) for each treatment from F. Error bars indicate mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Discussion
Our results indicate that autophagy contributes to memory destabilization and that its induction enhances memory destabilization, including a reconsolidation-resistant one, and the degradation of the endocytosed AMPAR in the spines of memory ensemble-neurons. Also, autophagy induction enhances synaptic destabilization in an NMDAR-dependent manner (Fig. 7A,B). A consistent finding through our study is that autophagy induction alone, through tBC administration after reactivation, did not show any significant amnesic effect in the reconsolidation-resistant boundary condition auditory fear conditioning, contextual fear conditioning, and synaptic reconsolidation models. This indicates that, regardless of the degree of destabilization, if the protein synthesis is not compromised within a certain time window following reactivation, the synthesized proteins have the capacity to regain synaptic plasticity and memory (Fig. 7B). This demonstrates the capacity of protein synthesis in the restabilization of synapses and the reinstatement of specific memories.
Figure 7.
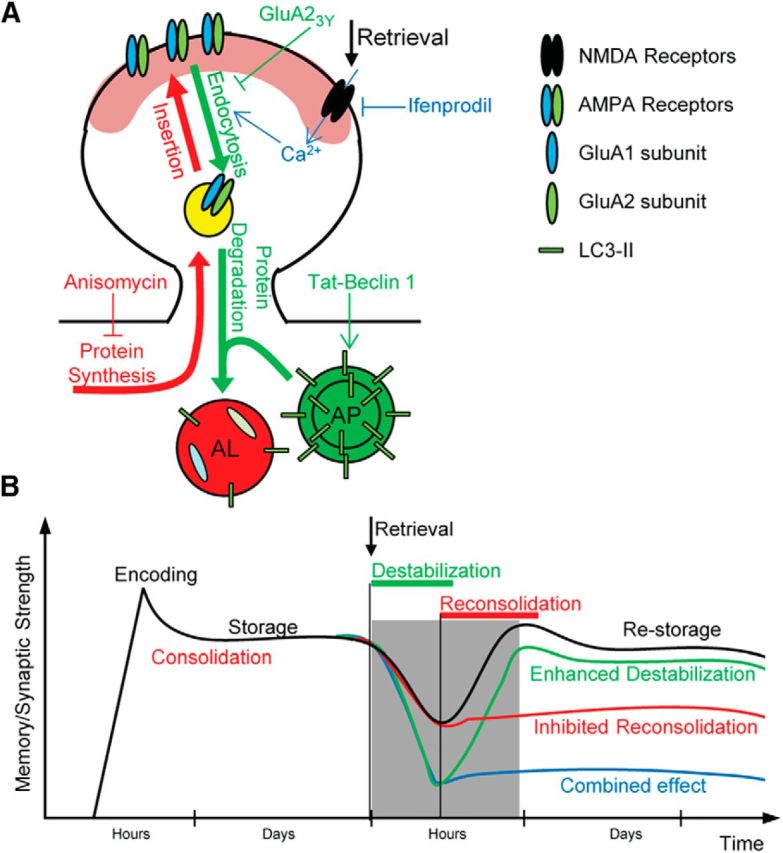
Models for the effect of autophagy on synaptic and memory destabilization. A, Hypothesized molecular mechanism for the effect of autophagy induction on synaptic and memory destabilization. Memory retrieval leads to NMDAR activation, which stimulates both autophagy and the endocytosis of AMPAR in the activated neurons. Autophagosomes (AP) fuse with the endosomes carrying internalized AMPARs, forming autolysosomes (AL), and dictate their fate to lysosomal degradation (green arrows). During the reconsolidation process, newly synthesized proteins, including AMPARs, are delivered to the synaptic surface replacing the degraded ones (red arrows). In the present study, ifenprodil (IFN) was used to block NMDA receptor activation, GluA23Y peptide to block endocytosis of AMPARs, tBC peptide to induce autophagy, and anisomycin (Ani) to inhibit protein synthesis. B, Hypothesized model explaining the time line of synaptic and memory strength changes by autophagy. After recall, consolidated synaptic plasticity and memory strength are usually physiologically destabilized and return to a labile state, after which a protein synthesis-dependent reconsolidation process is required for restabilization (black line). The labile state (destabilization) is inferred from the decreased synaptic strength and the retrograde amnesia produced by the protein synthesis inhibition (red line). Autophagy induction when combined with protein synthesis inhibition leads to a greater decrease in synaptic strength and enhanced retrograde amnesia, indicating enhanced destabilization (blue line). Autophagy induction alone does not affect synaptic or memory strength (green line).
AMPARs are heterotetrameric complexes composed of various combinations of four subunits (GluA1–4), with the GluA1/2 and GluA2/3 tetramers being the two major subtypes (Wenthold et al., 1996). The amount of synaptic GluA2-containing AMPARs correlates with long-term memory maintenance and strength (Yao et al., 2008; Migues et al., 2010, 2014; Dong et al., 2015). Blocking the endocytosis of GluA2-containing AMPARs inhibits the induction of LTD, but not LTP, without affecting basal synaptic transmission (Ahmadian et al., 2004; Brebner et al., 2005; Dalton et al., 2008; Scholz et al., 2010). More relevant is its involvement in memory destabilization, where it does not affect the acquisition or the retrieval of conditioned fear memory (Rao-Ruiz et al., 2011; Hong et al., 2013). These reports are in agreement with our hypothesis that autophagy works through enhancing lysosomal degradation of the endosomes carrying AMPAR, as evidenced by our demonstration that GluA2-dependent AMPAR endocytosis is a prerequisite for autophagy to affect memory destabilization. Additionally, GluA2-dependent AMPAR endocytosis correlates with the decay of LTP and the natural active forgetting of long-term memories (Hardt et al., 2014; Dong et al., 2015; Migues et al., 2016), which suggests that autophagy may play a role in the forgetting of consolidated memories through the gradual synaptic loss of AMPAR overtime and, hence, memory loss.
The GluA1 subunit acts dominantly over other subunits to determine the direction of AMPAR to the surface, and is correlated with synaptic potentiation, LTP, and fear memory (Ehlers, 2000; Shi et al., 2001; Malinow and Malenka, 2002; Lee et al., 2004; Rumpel et al., 2005). A mouse model lacking GluA1 subunit expression exhibits impaired hippocampus-dependent spatial memory (Reisel et al., 2002; Sanderson et al., 2007). Contextual fear conditioning recruits newly synthesized GluA1-containing AMPAR into the spines of the hippocampal memory-ensemble cells in a learning-specific manner (Matsuo et al., 2008). Inhibitory avoidance, a hippocampus-dependent contextual fear-learning task, delivers GluA1-containing AMPARs into the CA1 synapses in the dorsal hippocampus, where they are required for encoding contextual fear memories (Mitsushima et al., 2011). Inhibition of the cAMP response element-binding protein, a key transcription factor implicated in synaptic plasticity and memory, is associated with a specific reduction in the AMPAR subunit of GluA1 within the postsynaptic densities, and impaired contextual fear conditioning (Middei et al., 2013). These reports are in agreement with our use of spine enrichment with GluA1-containing AMPAR as a molecular reflection of synaptic and contextual memory strength in the CA1 region of the hippocampus.
d-Cycloserine, an NMDAR agonist, prepares resistant memories for destabilization (Bustos et al., 2010). Noteworthy, d-cycloserine also enhances memory update (fear extinction) by increasing GluA2-containing AMPAR endocytosis, and augments NMDAR-2B-dependent hippocampal LTD (Duffy et al., 2008; Bai et al., 2014). Therefore, autophagy might be a potential downstream mechanism by which d-cycloserine facilitates destabilization.
The ubiquitin-proteasome system has been implicated in synaptic plasticity, learning, and memory through remodeling the protein decomposition of the postsynaptic density (Hegde et al., 1993, 1997; Bingol et al., 2010; Bingol and Sheng, 2011; Lip et al., 2017). Regarding memory destabilization, retrieval of conditioned fear memory in mice was found to increase the polyubiquitination and degradation of the Shank and guanylate kinase-associated protein proteins in the mouse hippocampus (Lee et al., 2008). Also, the retrieval of cocaine-induced conditioned place preference was found to increase the polyubiquitination and degradation of the N-ethylmaleimide-sensitive fusion protein in the nucleus accumbens core (Ren et al., 2013). In both studies, the protreosome inhibitor lactacystin rescued the memory impairment induced by anisomycin and prevented the extinction of the fear or the reward memory. Therefore, the ubiquitin-proteasome system mainly contributes to memory destabilization through affecting proteins, within the postsynaptic density, with altered polyubiquitination dynamics. On the other hand, autophagy contributes to memory destabilization through affecting the endocytosed proteins from the synaptic surface, such as AMPAR. This implies that both protein degradation systems might not have redundant functions in memory destabilization.
The hippocampal CA1 region encodes mainly spatial and contextual (CS) information, while the fear (FS or US) memory itself is encoded by the amygdala (LeDoux, 2000; Maren et al., 2013), which agrees with the results from the present study using contextual fear conditioning, where manipulation of BLA neurons led to complete retrograde amnesia, whereas manipulation of CA1 neurons led to a generalization of fear. Therefore, targeting of the proper brain region is necessary to achieve the desired behavioral response. This highlights the importance of targeting the fear memory to successfully alleviate PTSD symptoms, rather than any other associated memory within the entire network.
Human studies that used reconsolidation intervention, indirectly through the noradrenergic β-blocker propranolol, to treat PTSD patients showed a variability of the treatment success, which might be attributed to internal factors, such as personality characteristics and genetic background, or to external factors, such as the strength of the initial fear experience or substance use (Kindt et al., 2009; Soeter and Kindt, 2010, 2011; Bos et al., 2014; Wood et al., 2015; Kindt and van Emmerik, 2016). The success of the intervention depends on whether the memory retrieval session can trigger memory destabilization where only retrieval experiences that involve novel or unexpected information can show a memory destabilizing effect (Sevenster et al., 2012, 2013, 2014). In our study, we also have observed variation in the levels of amnesia among individual animals having the same genetic background and receiving the same conditioning, retrieval, and treatment conditions (Fig. 3D). Thus, the above factors could not explain such variability as animal experiments are done under controlled conditions compared with human experiments. Nevertheless, the Ani+tBC resulted in a higher rate of complete amnesia on the individual level, which might indicate that the factors affecting the initial destabilization are still the main player to explain such variability.
We showed here that autophagy destabilizes resistant memories formed under stressful conditions, suggesting autophagy as a potential target for clinical applications. Because of the growing interest in finding autophagy inducers for several applications, many FDA-approved autophagy inducers already exist, including known antipsychotic and antidepressant drugs, and more specific ones are on their way (Levine et al., 2015; Morel et al., 2017). This increases the feasibility of using autophagy inducers for future therapeutic applications, including PTSD treatment.
Footnotes
This work was supported by Ministry of Education, Culture, Sports, Science, and Technology Grant-in-Aid for Scientific Research on Innovative Areas “Memory dynamism” JP25115002, Japan Society for the Promotion of Science KAKENHI Grant JP23220009, Japan Science and Technology Agency Core Research for Evolutional Science and Technology Program JPMJCR13W1, the Mitsubishi Foundation, and the Uehara Memorial Foundation. K.I. was supported by the Takeda Science Foundation. M.S. was supported by Japan Society for the Promotion of Science KAKENHI Grant-in-Aid for Young Scientists JP25830007. K.A. was supported by the Otsuka Toshimi Scholarship Foundation. We thank Noriaki Ohkawa for assistance with c-fos-tTA mice; Reiko Okubo-Suzuki and Yoshito Saitoh for assistance with electrophysiology; Sakurako Kosugi for lentivirus preparation; and Shuhei Tsujimura for maintenance of mice.
The authors declare no competing financial interests.
References
- Ahmadian G, Ju W, Liu L, Wyszynski M, Lee SH, Dunah AW, Taghibiglou C, Wang Y, Lu J, Wong TP, Sheng M, Wang YT (2004) Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J 23:1040–1050. 10.1038/sj.emboj.7600126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Zhou L, Wu X, Dong Z (2014) D-serine enhances fear extinction by increasing GluA2-containing AMPA receptor endocytosis. Behav Brain Res 270:223–227. 10.1016/j.bbr.2014.05.025 [DOI] [PubMed] [Google Scholar]
- Ben Mamou C, Gamache K, Nader K (2006) NMDA receptors are critical for unleashing consolidated auditory fear memories. Nat Neurosci 9:1237–1239. 10.1038/nn1778 [DOI] [PubMed] [Google Scholar]
- Besnard A, Caboche J, Laroche S (2012) Reconsolidation of memory: a decade of debate. Prog Neurobiol 99:61–80. 10.1016/j.pneurobio.2012.07.002 [DOI] [PubMed] [Google Scholar]
- Bingol B, Sheng M (2011) Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron 69:22–32. 10.1016/j.neuron.2010.11.006 [DOI] [PubMed] [Google Scholar]
- Bingol B, Wang CF, Arnott D, Cheng D, Peng J, Sheng M (2010) Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell 140:567–578. 10.1016/j.cell.2010.01.024 [DOI] [PubMed] [Google Scholar]
- Bos MG, Beckers T, Kindt M (2014) Noradrenergic blockade of memory reconsolidation: a failure to reduce conditioned fear responding. Front Behav Neurosci 8:412. 10.3389/fnbeh.2014.00412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S, Phelps L, Phillips AG, Wang YT (2005) Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science 310:1340–1343. 10.1126/science.1116894 [DOI] [PubMed] [Google Scholar]
- Bustos SG, Giachero M, Maldonado H, Molina VA (2010) Previous stress attenuates the susceptibility to Midazolam's disruptive effect on fear memory reconsolidation: influence of pre-reactivation d-cycloserine administration. Neuropsychopharmacology 35:1097–1108. 10.1038/npp.2009.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Garelick MG, Wang H, Lil V, Athos J, Storm DR (2005) PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat Neurosci 8:925–931. 10.1038/nn1482 [DOI] [PubMed] [Google Scholar]
- Dalton GL, Wang YT, Floresco SB, Phillips AG (2008) Disruption of AMPA receptor endocytosis impairs the extinction, but not acquisition of learned fear. Neuropsychopharmacology 33:2416–2426. 10.1038/sj.npp.1301642 [DOI] [PubMed] [Google Scholar]
- De Leo MG, Staiano L, Vicinanza M, Luciani A, Carissimo A, Mutarelli M, Di Campli A, Polishchuk E, Di Tullio G, Morra V, Levtchenko E, Oltrabella F, Starborg T, Santoro M, Di Bernardo D, Devuyst O, Lowe M, Medina DL, Ballabio A, De Matteis MA (2016) Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat Cell Biol 18:839–850. 10.1038/ncb3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Han H, Li H, Bai Y, Wang W, Tu M, Peng Y, Zhou L, He W, Wu X, Tan T, Liu M, Wu X, Zhou W, Jin W, Zhang S, Sacktor TC, Li T, Song W, Wang YT (2015) Long-term potentiation decay and memory loss are mediated by AMPAR endocytosis. J Clin Invest 125:234–247. 10.1172/JCI77888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S, Labrie V, Roder JC (2008) D-serine augments NMDA-NR2B receptor-dependent hippocampal long-term depression and spatial reversal learning. Neuropsychopharmacology 33:1004–1018. 10.1038/sj.npp.1301486 [DOI] [PubMed] [Google Scholar]
- Ehlers MD. (2000) Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28:511–525. 10.1016/S0896-6273(00)00129-X [DOI] [PubMed] [Google Scholar]
- Fanselow MS. (2000) Contextual fear, gestalt memories, and the hippocampus. Behav Brain Res 110:73–81. 10.1016/S0166-4328(99)00186-2 [DOI] [PubMed] [Google Scholar]
- Finnie PS, Nader K (2012) The role of metaplasticity mechanisms in regulating memory destabilization and reconsolidation. Neurosci Biobehav Rev 36:1667–1707. 10.1016/j.neubiorev.2012.03.008 [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K (2003) Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron 38:447–460. 10.1016/S0896-6273(03)00206-X [DOI] [PubMed] [Google Scholar]
- Gafford GM, Parsons RG, Helmstetter FJ (2011) Consolidation and reconsolidation of contextual fear memory requires mammalian target of rapamycin-dependent translation in the dorsal hippocampus. Neuroscience 182:98–104. 10.1016/j.neuroscience.2011.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889. 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- Hardt O, Nader K, Wang YT (2014) GluA2-dependent AMPA receptor endocytosis and the decay of early and late long-term potentiation: possible mechanisms for forgetting of short- and long-term memories. Philos Trans R Soc Lond B Biol Sci 369:20130141. 10.1098/rstb.2013.0141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN, Goldberg AL, Schwartz JH (1993) Regulatory subunits of cAMP-dependent protein kinases are degraded after conjugation to ubiquitin: a molecular mechanism underlying long-term synaptic plasticity. Proc Natl Acad Sci U S A 90:7436–7440. 10.1073/pnas.90.16.7436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN, Inokuchi K, Pei W, Casadio A, Ghirardi M, Chain DG, Martin KC, Kandel ER, Schwartz JH (1997) Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell 89:115–126. 10.1016/S0092-8674(00)80188-9 [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ. (1993) Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol 121:305–315. 10.1083/jcb.121.2.305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong I, Kim J, Kim J, Lee S, Ko HG, Nader K, Kaang BK, Tsien RW, Choi S (2013) AMPA receptor exchange underlies transient memory destabilization on retrieval. Proc Natl Acad Sci U S A 110:8218–8223. 10.1073/pnas.1305235110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba H, Tsukagoshi A, Kida S (2015) PARP-1 activity is required for the reconsolidation and extinction of contextual fear memory. Mol Brain 8:63. 10.1186/s13041-015-0153-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Malinow R (2009) Synaptic AMPA receptor plasticity and behavior. Neuron 61:340–350. 10.1016/j.neuron.2009.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Chung HJ, Lee HK, Huganir RL (2001) Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc Natl Acad Sci U S A 98:11725–11730. 10.1073/pnas.211132798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindt M, van Emmerik A (2016) New avenues for treating emotional memory disorders: towards a reconsolidation intervention for posttraumatic stress disorder. Ther Adv Psychopharmacol 6:283–295. 10.1177/2045125316644541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindt M, Soeter M, Vervliet B (2009) Beyond extinction: erasing human fear responses and preventing the return of fear. Nat Neurosci 12:256–258. 10.1038/nn.2271 [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884. 10.1038/nature04723 [DOI] [PubMed] [Google Scholar]
- LeDoux JE. (2000) Emotion circuits in the brain. Annu Rev Neurosci 23:155–184. 10.1146/annurev.neuro.23.1.155 [DOI] [PubMed] [Google Scholar]
- Lee JL, Nader K, Schiller D (2017) An update on memory reconsolidation updating. Trends Cogn Sci 21:531–545. 10.1016/j.tics.2017.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Choi JH, Lee N, Lee HR, Kim JI, Yu NK, Choi SL, Lee SH, Kim H, Kaang BK (2008) Synaptic protein degradation underlies destabilization of retrieved fear memory. Science 319:1253–1256. 10.1126/science.1150541 [DOI] [PubMed] [Google Scholar]
- Lee SH, Liu L, Wang YT, Sheng M (2002) Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron 36:661–674. 10.1016/S0896-6273(02)01024-3 [DOI] [PubMed] [Google Scholar]
- Lee SH, Simonetta A, Sheng M (2004) Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron 43:221–236. 10.1016/j.neuron.2004.06.015 [DOI] [PubMed] [Google Scholar]
- Levine B, Packer M, Codogno P (2015) Development of autophagy inducers in clinical medicine. J Clin Invest 125:14–24. 10.1172/JCI73938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CC, Wang C, Peng X, Gan B, Guan JL (2010) Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem 285:3499–3509. 10.1074/jbc.M109.072389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lip PZ, Demasi M, Bonatto D (2017) The role of the ubiquitin proteasome system in the memory process. Neurochem Int 102:57–65. 10.1016/j.neuint.2016.11.013 [DOI] [PubMed] [Google Scholar]
- Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, Furuya T, Jin M, Zhu Z, Wang H, Yu J, Li Y, Hao Y, Choi A, Ke H, Ma D, Yuan J (2011) Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147:223–234. 10.1016/j.cell.2011.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25:103–126. 10.1146/annurev.neuro.25.112701.142758 [DOI] [PubMed] [Google Scholar]
- Mamiya N, Fukushima H, Suzuki A, Matsuyama Z, Homma S, Frankland PW, Kida S (2009) Brain region-specific gene expression activation required for reconsolidation and extinction of contextual fear memory. J Neurosci 29:402–413. 10.1523/JNEUROSCI.4639-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Phan KL, Liberzon I (2013) The contextual brain: implications for fear conditioning, extinction and psychopathology. Nat Rev Neurosci 14:417–428. 10.1038/nrn3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh T, Debnath J (2015) Ironing out VPS34 inhibition. Nat Cell Biol 17:1–3. [DOI] [PubMed] [Google Scholar]
- Matsuo N, Reijmers L, Mayford M (2008) Spine-type-specific recruitment of newly synthesized AMPA receptors with learning. Science 319:1104–1107. 10.1126/science.1149967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middei S, Houeland G, Cavallucci V, Ammassari-Teule M, D'Amelio M, Marie H (2013) CREB is necessary for synaptic maintenance and learning-induced changes of the AMPA receptor GluA1 subunit. Hippocampus 23:488–499. 10.1002/hipo.22108 [DOI] [PubMed] [Google Scholar]
- Migues PV, Hardt O, Wu DC, Gamache K, Sacktor TC, Wang YT, Nader K (2010) PKMzeta maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat Neurosci 13:630–634. 10.1038/nn.2531 [DOI] [PubMed] [Google Scholar]
- Migues PV, Hardt O, Finnie P, Wang YW, Nader K (2014) The maintenance of long-term memory in the hippocampus depends on the interaction between N-ethylmaleimide-sensitive factor and GluA2. Hippocampus 24:1112–1119. 10.1002/hipo.22295 [DOI] [PubMed] [Google Scholar]
- Migues PV, Liu L, Archbold GE, Einarsson EÖ, Wong J, Bonasia K, Ko SH, Wang YT, Hardt O (2016) Blocking synaptic removal of GluA2-containing AMPA receptors prevents the natural forgetting of long-term memories. J Neurosci 36:3481–3494. 10.1523/JNEUROSCI.3333-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton AL, Merlo E, Ratano P, Gregory BL, Dumbreck JK, Everitt BJ (2013) Double dissociation of the requirement for GluN2B- and GluN2A-containing NMDA receptors in the destabilization and restabilization of a reconsolidating memory. J Neurosci 33:1109–1115. 10.1523/JNEUROSCI.3273-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsushima D, Ishihara K, Sano A, Kessels HW, Takahashi T (2011) Contextual learning requires synaptic AMPA receptor delivery in the hippocampus. Proc Natl Acad Sci U S A 108:12503–12508. 10.1073/pnas.1104558108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147:728–741. 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27:107–132. 10.1146/annurev-cellbio-092910-154005 [DOI] [PubMed] [Google Scholar]
- Morel E, Mehrpour M, Botti J, Dupont N, Hamaï A, Nascimbeni AC, Codogno P (2017) Autophagy: a druggable process. Annu Rev Pharmacol Toxicol 57:375–398. 10.1146/annurev-pharmtox-010716-104936 [DOI] [PubMed] [Google Scholar]
- Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R (2014) Engineering a memory with LTD and LTP. Nature 511:348–352. 10.1038/nature13294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE (2000) Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406:722–726. 10.1038/35021052 [DOI] [PubMed] [Google Scholar]
- Ohkawa N, Saitoh Y, Suzuki A, Tsujimura S, Murayama E, Kosugi S, Nishizono H, Matsuo M, Takahashi Y, Nagase M, Sugimura YK, Watabe AM, Kato F, Inokuchi K (2015) Artificial association of pre-stored information to generate a qualitatively new memory. Cell Rep 11:261–269. 10.1016/j.celrep.2015.03.017 [DOI] [PubMed] [Google Scholar]
- Ohsumi Y. (2014) Historical landmarks of autophagy research. Cell Res 24:9–23. 10.1038/cr.2013.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okubo-Suzuki R, Saitoh Y, Shehata M, Zhao Q, Enomoto H, Inokuchi K (2016) Frequency-specific stimulations induce reconsolidation of long-term potentiation in freely moving rats. Mol Brain 9:36. 10.1186/s13041-016-0216-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman RK. (2011) Will reconsolidation blockade offer a novel treatment for posttraumatic stress disorder? Front Behav Neurosci 5:11. 10.3389/fnbeh.2011.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao-Ruiz P, Rotaru DC, van der Loo RJ, Mansvelder HD, Stiedl O, Smit AB, Spijker S (2011) Retrieval-specific endocytosis of GluA2-AMPARs underlies adaptive reconsolidation of contextual fear. Nat Neurosci 14:1302–1308. 10.1038/nn.2907 [DOI] [PubMed] [Google Scholar]
- Reijmers LG, Perkins BL, Matsuo N, Mayford M (2007) Localization of a stable neural correlate of associative memory. Science 317:1230–1233. 10.1126/science.1143839 [DOI] [PubMed] [Google Scholar]
- Reisel D, Bannerman DM, Schmitt WB, Deacon RM, Flint J, Borchardt T, Seeburg PH, Rawlins JN (2002) Spatial memory dissociations in mice lacking GluR1. Nat Neurosci 5:868–873. 10.1038/nn910 [DOI] [PubMed] [Google Scholar]
- Ren ZY, Liu MM, Xue YX, Ding ZB, Xue LF, Zhai SD, Lu L (2013) A critical role for protein degradation in the nucleus accumbens core in cocaine reward memory. Neuropsychopharmacology 38:778–790. 10.1038/npp.2012.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland AM, Richmond JE, Olsen JG, Hall DH, Bamber BA (2006) Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J Neurosci 26:1711–1720. 10.1523/JNEUROSCI.2279-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R (2005) Postsynaptic receptor trafficking underlying a form of associative learning. Science 308:83–88. 10.1126/science.1103944 [DOI] [PubMed] [Google Scholar]
- Sanderson DJ, Gray A, Simon A, Taylor AM, Deacon RM, Seeburg PH, Sprengel R, Good MA, Rawlins JN, Bannerman DM (2007) Deletion of glutamate receptor-A (GluR-A) AMPA receptor subunits impairs one-trial spatial memory. Behav Neurosci 121:559–569. 10.1037/0735-7044.121.3.559 [DOI] [PubMed] [Google Scholar]
- Scholz R, Berberich S, Rathgeber L, Kolleker A, Köhr G, Kornau HC (2010) AMPA receptor signaling through BRAG2 and Arf6 critical for long-term synaptic depression. Neuron 66:768–780. 10.1016/j.neuron.2010.05.003 [DOI] [PubMed] [Google Scholar]
- Sevenster D, Beckers T, Kindt M (2012) Retrieval per se is not sufficient to trigger reconsolidation of human fear memory. Neurobiol Learn Mem 97:338–345. 10.1016/j.nlm.2012.01.009 [DOI] [PubMed] [Google Scholar]
- Sevenster D, Beckers T, Kindt M (2013) Prediction error governs pharmacologically induced amnesia for learned fear. Science 339:830–833. 10.1126/science.1231357 [DOI] [PubMed] [Google Scholar]
- Sevenster D, Beckers T, Kindt M (2014) Prediction error demarcates the transition from retrieval, to reconsolidation, to new learning. Learn Mem 21:580–584. 10.1101/lm.035493.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehata M, Inokuchi K (2014) Does autophagy work in synaptic plasticity and memory? Rev Neurosci 25:543–557. 10.1515/revneuro-2014-0002 [DOI] [PubMed] [Google Scholar]
- Shehata M, Matsumura H, Okubo-Suzuki R, Ohkawa N, Inokuchi K (2012) Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J Neurosci 32:10413–10422. 10.1523/JNEUROSCI.4533-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R (2001) Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105:331–343. 10.1016/S0092-8674(01)00321-X [DOI] [PubMed] [Google Scholar]
- Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, Huerta C, Virgin HW, Helms JB, Eerland R, Tooze SA, Xavier R, Lenschow DJ, Yamamoto A, King D, Lichtarge O, et al. (2013) Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494:201–206. 10.1038/nature11866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeter M, Kindt M (2010) Dissociating response systems: erasing fear from memory. Neurobiol Learn Mem 94:30–41. 10.1016/j.nlm.2010.03.004 [DOI] [PubMed] [Google Scholar]
- Soeter M, Kindt M (2011) Disrupting reconsolidation: pharmacological and behavioral manipulations. Learn Mem 18:357–366. 10.1101/lm.2148511 [DOI] [PubMed] [Google Scholar]
- Squire LR, Kandel ER (2009) Memory: from mind to molecules, Ed 2 Greenwood Village, CO: Roberts. [Google Scholar]
- Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S (2004) Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci 24:4787–4795. 10.1523/JNEUROSCI.5491-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronson NC, Taylor JR (2007) Molecular mechanisms of memory reconsolidation. Nat Rev Neurosci 8:262–275. 10.1038/nrn2090 [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B (2010) The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol 11:329–341. 10.1038/nrm2882 [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J II, Niedzielski AS (1996) Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci 16:1982–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood NE, Rosasco ML, Suris AM, Spring JD, Marin MF, Lasko NB, Goetz JM, Fischer AM, Orr SP, Pitman RK (2015) Pharmacological blockade of memory reconsolidation in posttraumatic stress disorder: three negative psychophysiological studies. Psychiatry Res 225:31–39. 10.1016/j.psychres.2014.09.005 [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Yue Z (2014) Autophagy and its normal and pathogenic states in the brain. Annu Rev Neurosci 37:55–78. 10.1146/annurev-neuro-071013-014149 [DOI] [PubMed] [Google Scholar]
- Yao Y, Kelly MT, Sajikumar S, Serrano P, Tian D, Bergold PJ, Frey JU, Sacktor TC (2008) PKM zeta maintains late LTP by N-ethylmaleimide-sensitive factor/GluR2-dependent trafficking of postsynaptic AMPA receptors. J Neurosci 28:7820–7827. 10.1523/JNEUROSCI.0223-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]