

Abstract
Disturbances in prefrontal cortical (PFC) dopamine (DA) transmission are well established features of psychiatric disorders involving pathological memory processing, such as post-traumatic stress disorder and opioid addiction. Transmission through PFC DA D4 receptors (D4Rs) has been shown to potentiate the emotional salience of normally nonsalient emotional memories, whereas transmission through PFC DA D1 receptors (D1Rs) has been demonstrated to selectively block recall of reward- or aversion-related associative memories. In the present study, using a combination of fear conditioning and opiate reward conditioning in male rats, we examined the role of PFC D4/D1R signaling during the processing of fear-related memory acquisition and recall and subsequent sensitivity to opiate reward memory formation. We report that PFC D4R activation potentiates the salience of normally subthreshold fear conditioning memory cues and simultaneously potentiates the rewarding effects of systemic or intra-ventral tegmental area (VTA) morphine conditioning cues. In contrast, blocking the recall of salient fear memories with intra-PFC D1R activation, blocks the ability of fear memory recall to potentiate systemic or intra-VTA morphine place preference. These effects were dependent upon dissociable PFC phosphorylation states involving calcium-calmodulin-kinase II or extracellular signal-related kinase 1–2, following intra-PFC D4 or D1R activation, respectively. Together, these findings reveal new insights into how aberrant PFC DAergic transmission and associated downstream molecular signaling pathways may modulate fear-related emotional memory processing and concomitantly increase opioid addiction vulnerability.
SIGNIFICANCE STATEMENT Post-traumatic stress disorder is highly comorbid with addiction. In this study, we use a translational model of fear memory conditioning to examine how transmission through dopamine D1 or D4 receptors, in the prefrontal cortex (PFC), may differentially control acquisition or recall of fear memories and how these mechanisms might regulate sensitivity to the rewarding effects of opioids. We demonstrate that PFC D4 activation not only controls the salience of fear memory acquisition, but potentiates the rewarding effects of opioids. In contrast, PFC D1 receptor activation blocks recall of fear memories and prevents potentiation of opioid reward effects. Together, these findings demonstrate novel PFC mechanisms that may account for how emotional memory disturbances might increase the addictive liability of opioid-class drugs.
Keywords: dopamine, fear memory, opioids, prefrontal cortex, PTSD, ventral tegmental area
Introduction
Disturbances in emotional memory processing and salience misattribution deficits are cardinal features of post-traumatic stress disorder (PTSD) and addiction, both of which involve the pathological recall of memories associated with trauma or drug-related conditioned memories (Sripada et al., 2012; Bowers and Ressler, 2015). In addition to these common psychopathological features, 50–60% of PTSD patients experience some form of cooccurring drug dependence (DeHaas et al., 2002; Conrod and Stewart, 2003). Opioid addiction represents one of the most intractable forms of drug dependence and occurs frequently in PTSD, where it is associated with poorer prognoses and treatment outcomes (Mills et al., 2005, 2007; Tull et al., 2010; Dell'Osso et al., 2014; Banerjee et al., 2016). Nevertheless, the neurobiological mechanisms linking the recall of traumatic memories to increased vulnerability to drugs of abuse remain poorly understood.
Neurons in the prefrontal cortex (PFC) are involved in the encoding of both fear- and opiate-related associative memories (Laviolette et al., 2005; Laviolette and Grace, 2006; Sun et al., 2011, Sun and Laviolette, 2012). Importantly, these PFC-dependent memory processes are strongly modulated by dopamine (DA) receptor transmission (Laviolette et al., 2005; Lauzon et al., 2009, 2013). We have previously identified dissociable roles for intra-PFC transmission through DA D1 receptor (D1R) versus D4 receptor (D4R) subtypes during distinct phases of emotional memory processing. Thus, whereas PFC D4R activation is selectively involved in controlling the salience of fear-related associative memory formation during memory encoding, D1R transmission selectively controls the recall of phase of these memories (Laviolette et al., 2005; Lauzon et al., 2009, 2013). Specifically, PFC D4R activation dramatically potentiates the salience of normally subthreshold fear-related associative memory cues. In contrast, D1R activation selectively blocks the recall of both associative fear-related and reward-related associative memories, without influencing the salience of these memories during memory acquisition (Lauzon et al., 2009, 2013). In addition, these PFC DAergic memory mechanisms are functionally associated with downstream signaling through the extracellular signal-related kinase 1–2 (ERK 1/2) and calcium/calmodulin kinase-α II (CaMKII-α), directly in the PFC (Lauzon et al., 2013; Rosen et al., 2016), two signaling substrates that are involved in the processing of both fear- and reward-related associative memories via D1R and D4R transmission (Lauzon et al., 2013; Lyons et al., 2013).
Given the importance of PFC DAergic transmission in modulating emotional memory and its role in PTSD and addiction-related memory formation, we hypothesized that the ability of intra-PFC D4R and D1R transmission to modulate fear- and reward-related memory encoding or recall might involve dissociable mechanisms which regulate sensitivity to the rewarding properties of opioids. Using a combination of fear conditioning, morphine conditioned place preference (CPP) and localized protein expression analyses in rats, we examined how PFC D4R versus D1R-mediated modulation of fear memory encoding versus recall may modulate sensitivity to opiate-related reward memory and their associated downstream molecular memory signaling pathways, ERK 1/2 and CaMKII-α. We report that intra-PFC D4R or D1R activation differentially regulates the acquisition versus recall stages of fear memory processing and concomitantly controls the rewarding effects of both systemic or intra-ventral tegmental area (VTA) morphine conditioning cues. These shared phenomena were controlled via local PFC activation of distinct phosphorylation states involving D4R-CaMKII-α or D1R-ERK 1/2 signaling pathways. These findings identify a novel PFC DA-dependent mechanism linking the salience and recall of aversive memory formation with increased vulnerability to opioid-related reward cues.
Materials and Methods
Animals and surgery.
Experimental animals were adult (350–400 g) male Sprague-Dawley rats (Charles River Laboratories). A total of n = 149 rats were used in the present study. All procedures were conducted in accordance with Canadian Council of Animal Care guidelines and Western University Institutional Animal Care and Use Committees. Rats were anesthetized with a ketamine (80 mg/ml)-xylazine (6 mg/ml) mixture and placed into a stereotaxic device (Kopf Instruments). Stainless steel guide cannulae (22 gauge; Plastics One) were bilaterally implanted using the following stereotaxic coordinates (in mm). For the PFC (15° angle): from bregma, AP +2.9, L ±1.9, and from the dural surface, V −3.0. For the VTA (10° angle): from bregma, AP −5.0, L ±2.3, and from the dural surface, V −8.0.
Behavioral conditioning procedure: fear memory and morphine CPP conditioning.
A simplified schematic summary of experimental conditioning procedures showing the timing of intra-PFC DA D1R or D4R treatments is presented in Figure 1, A and B. To examine the effects of associative fear memory recall on subsequent sensitivity to morphine reward effects, we used a novel behavioral conditioning assay combining our previously described olfactory fear conditioning protocol (Lauzon et al., 2009) with our previously described unbiased morphine place conditioning procedure, using a sub-reward threshold conditioning dose of systemic or bilateral intra-VTA morphine microinfusions (0.05 mg/kg, i.p.; 250 ng/0.5 μl), which we have previously reported produce no significant rewarding CPP under normal conditions (Bishop et al., 2011; De Jaeger et al., 2013). Olfactory fear conditioning took place in one of two distinct environments, counterbalanced within groups: Environment A was a 30 × 30 inch Plexiglas box with black stripes on a white background, whereas Environment B had black dots on a white background with a grid shock floor. Olfactory cues were either an almond or peppermint scent, randomly assigned, as previously reported (Laviolette et al., 2005; Lauzon et al., 2009). Olfactory cues are delivered via an automated olfactometer controlled by solenoid valves and delivered into the ventilated conditioning chamber through which odors are cleared into a scavenging unit following the cue delivery. We used two conditioning levels of footshock: (1) A subthreshold footshock cue (0.4 mA), which we have previously demonstrated to produce no significant associative fear memory acquisition or (2) a suprathreshold footshock cue (0.8 mA) which we have previously reported produces a robust associative fear memory (Lauzon et al., 2009). On Days 1–2 of the conditioning procedure, rats (all experimental groups) were preconditioned to each of the fear conditioning chambers for 20 min and were then acclimatized for CPP experiments by being placed in a gray Plexiglas box corresponding to the dimensions of the conditioning environments for 30 min. On Day 3, rats were randomly assigned to one of the fear conditioning chambers where one of the odors (almond or peppermint) was presented to the animal for 19 s and a footshock was delivered (0.4 or 0.8 mA) for 1 s. For groups receiving challenge drug or VEH administration targeting the 'acquisition' phase of fear memory conditioning (Fig. 1B), intra-PFC microinfusions were administered immediately before the conditioning phase. During the training session, rats received 5 footshock-olfactory cue pairings once every 4 min for a total 20 min conditioning session. Following the fear conditioning training, rats were returned to their home cages for 24 h. On Day 4, rats were tested for fear memory recall in the alternate (safe) conditioning chamber, by presenting the olfactory cue (in the absence of footshock) previously paired with either sub or suprathreshold footshock the previous day. For groups receiving challenge drug or VEH administration targeting the “recall” phase of fear memory conditioning (Fig. 1A), intra-PFC microinfusions were administered immediately before the fear memory recall test. Freezing behavior (defined as the absence of all movement except respiration) was scored with video tracking software (AnyMaze) and analyzed off-line. Immediately after fear memory recall test, rats (in all experimental groups) were removed from the test chamber, received a sub-reward threshold intraperitoneal injection (0.05 mg/kg) or intra-VTA microinfusion (250 ng/0.5 μl) of morphine, and placed in a randomly assigned morphine conditioning environment for 30 min. Rats were then returned to their home cage for 24 h and on Day 5, received a subsequent control environment conditioning session wherein they received intraperitoneal or intra-VTA administration of saline vehicle, and placed in an alternate (control) place conditioning environment. Following this final CPP conditioning session, rats (in all experimental groups) were returned to home cages for 24 h. CPP conditioning environments were as previously described (Bishop et al., 2011). Briefly, one environment was white with a wire mesh floor, covered in wood chips. The other environment was black with a smooth Plexiglas floor, wiped down with 2% acetic acid solution. On Day 6, rats (in all experimental groups) were given a 10 min CPP recall test wherein they were given free choice in a CPP testing box to spend time in either saline or morphine-paired environments. Times spent in either environment were recorded with digital timers and analyzed off-line.
Figure 1.

Schematic summary of experimental timeline and behavioral conditioning procedures combining olfactory associative fear conditioning and morphine CPP assays. Intra-PFC D1R or D4R pharmacological activation was administered immediately before fear memory recall testing or acquisition phases, respectively.
Drug administration procedures.
For PFC microinfusions, stainless steel cannulae (22 gauge) were implanted bilaterally into the PFC and drugs were administered through a 28 gauge injector (Plastics One) via a Hamilton microsyringe. The selective CaMKII autophosphorylation inhibitor, autocamtide-2-related inhibitory peptide (AIP; Tocris Bioscience, 500 ng/0.5 μl) and morphine sulfate (Johnson-Matthey) were dissolved in physiological saline, with pH adjusted to 7.4. The selective D4R agonist, PD 168077 (5–50 ng/0.5 μl), the D1R agonist SKF81297 hydrobromide (10–100 ng/0.5 ml; Tocris Bioscience) and ERK 1/2 inhibitor, U0126 (1 μg/0.5 μl) were first dissolved in dimethyl-sulfoxide (DMSO) and then diluted with physiological saline to a final concentration of 5% DMSO in physiological saline. To examine the roles of intra-PFC DA D1R versus D4R activation in fear memory processing, we targeted two separate phases of fear memory processing: acquisition or recall. We have previously demonstrated dissociable roles for PFC D1R versus D4R activation in the control of fear-related memory processing such that PFC D1R activation selectively blocks recall of suprathreshold associative fear memories (but has no influence on fear memory acquisition; Lauzon et al., 2009, 2013). In contrast, D4R activation potentiates the salience of normally nonsalient fear memory cues, selectively during the acquisition phase (Laviolette et al., 2005; Lauzon et al., 2009, 2013). Accordingly, to block the recall of suprathreshold (0.8 mA) fear memories, rats received intra-PFC microinfusions of the D1R agonist (SKF 81297) immediately before the fear memory recall test (Fig. 1A). In contrast, to potentiate the salience of a normally nonsalient, subthreshold fear memory (0.4 mA), rats received intra-PFC D4R activation with the D4R agonist, PD168077, immediately before the fear memory acquisition training session (Fig. 1B). For studies involving sub-reward threshold intra-VTA morphine (250 μl/0.5 μl), microinfusions of morphine were performed immediately after fear memory recall testing.
Histological analyses.
At the conclusion of experiments, rats were anesthetized with sodium pentobarbital (Euthanyl) and transcardially perfused with isotonic saline followed by 10% formalin. Brain sections were mounted and stained with cresyl violet, and PFC or VTA cannulae placements were verified with light microscopy according to the anatomical boundaries defined by Paxinos and Watson (1986). Three rats were found to have cannulae placements outside the boundaries of the target regions and were excluded from analysis (indicated in associated results sections).
Western blot procedure.
To examine the local effects of intra-PFC D1R versus D4R activation on total or phosphorylated expression levels of ERK 1/2 or CaMKII-α total (tERK1/2, tCaMKII-α) or phosphorylated (pERK1/2, pERK1/2) expression levels, a subset of rats received intra-PFC SKF 81297 (100 ng; n = 6) or PD 168077 (50 ng; n = 6) immediately before being killed, following behavioral conditioning with either SKF 81297 (100 ng) or PD 168077 (50 ng), respectively. Brains were rapidly removed and flash frozen at −80°C. PFC sections were sliced on a freezing microtome and microdissections were performed around the injector sites, avoiding regions with reactive gliosis. PFC samples were then homogenized using a Dounce homogenizer and protein isolated using a Noindent P40/SDS lysis buffer (137 mm NaCl, 20 mm Tris, pH 8.0, 1% NP-40, 10% glycerol, and 0.1% SDS) containing protease and phosphatase inhibitors (Halt 100× inhibitor mixture, ThermoFisher). Following homogenization and centrifugation at 4°C to remove debris, samples were mixed with an equal volume of 2× Laemmli loading buffer and heated to 95°C for 5 min before storage at −80°C. 12.5 μg of protein from control (vehicle-treated; n = 6) or treatment [D1R (n = 6) or D4R (n = 6) agonist groups] samples were loaded onto 12% denaturing SDS-PAGE gels. Samples were then subjected to electrophoresis in a Bio-Rad Mini Protein 3 Western blotting apparatus with Tris/glycine/SDS buffer (Bio-Rad Cube Solutions) at 125 V for 1.5 h. Molecular weight standards (Froggabio BLUeye) were included on the gels to confirm the molecular weight of the proteins of interest. Following electrophoresis, protein was transferred from the gels onto a nitrocellulose membrane (Bio-Rad) using a Mini Trans-blot apparatus (Bio-Rad) with a tris/glycine/methanol solution (Bio-Rad Cube Solutions) at 75 V for 1 h. To identify changes in levels of phosphorylated or total proteins, blots were blocked with 5% nonfat dry milk in TBST for 1 h at room temperature with rocking. After blocking, membranes were incubated overnight in a solution containing 2.5% BSA in TBST along with antibodies directed against pERK1/2 (Thr202/Tyr204), tERK1/2, pCaMKII (Thr286), or tCaMKII along with α-tubulin (loading control) at 4°C with rocking. Following incubation with primary antibody, blots were washed in TBST ×3 for 15 min. Last, a solution containing 5% dried nonfat milk in TBST and secondary antibodies (LI-COR IRDye 680RD and IRDye 800CW-conjugated secondary antibodies; 1:10,000) was applied to the blots and left to incubate for 1 h at room temperature. Following three washes in TBST, the proteins of interest were imaged using a LI-COR Odyssey imaging system. Densitometry measurements were obtained using Kodak digital analysis software. Antibody source and dilutions were as follows: α-tubulin (1:1,000,000; Sigma-Aldrich), tERK1/2 (1:2000; Cell Signaling Technology), pERK1/2 (1:1000; Cell Signaling Technology), tCaMKIIα (rabbit, 1:1000; Cell Signaling Technology), and pCaMKII (rabbit, 1:1000; Cell Signaling Technology).
Data analyses.
Data were analyzed with one or two-way ANOVA where appropriate or Student's t tests. Post hoc analyses were performed with Fisher's least significant difference or Newman–Keuls tests where appropriate.
Results
Histological verification
Histological analyses revealed intra-PFC injector placements to be within the anatomical boundaries of the PFC as defined by Paxinos and Watson (1986). In Figure 2A, a representative microphotograph showing a typical intra-PFC cannulae injector placement is presented. In Figure 2B, a schematic representation of select experimental groups is presented. Symbols represent the following experimental groups: ● = PFC VEH controls receiving CS+ presentations during the fear recall test; □ = rats receiving SKF81297 100 ng/0.5 μl; ■ = rats receiving intra-PFC PD168077 50 ng/0.5 μl.
Figure 2.

Histological analysis of PFC injector placements. A, Representative microphotograph showing a typical intra-PFC injector placement. B, Schematic representation of bilateral intra-PFC injector placements. Figure symbols represent the following experimental groups: ● = PFC VEH controls receiving CS+ presentations during the fear recall test; □ = rats receiving SKF81297 100 ng/0.5 μl; ■ = rats receiving intra-PFC PD168077 50 ng/0.5 μl.
Fear memory recall potentiates morphine reward sensitivity
To first validate our behavioral assay examining the effects of fear memory recall on subsequent sensitivity to the rewarding conditioning properties of morphine, we first examined whether selectively recalling a previously acquired, suprathreshold (0.8 mA footshock) associative fear memory might potentiate the reward salience of a normally sub-reward conditioning dose of morphine (Bishop et al., 2011), in the period immediately following fear memory recall (see Materials and Methods; Fig. 1). Three groups of rats received bilateral intra-PFC cannulae implantations (n = 8 per group) and either (1) olfactory fear conditioning in the presence of the paired footshock stimulus followed by the presentation of the olfactory fear cue during the fear memory recall test: “shock with recall”; (2) olfactory conditioning in the absence of any footshock stimulus and the presentation of the unpaired olfactory cue during the fear memory recall test: “no shock control”; or (3) olfactory fear conditioning in the presence of the paired footshock stimulus followed by the fear memory recall test in the absence of the CS+: “shock/no recall”. All groups received intra-PFC VEH microinfusions before the fear conditioning assay. Comparing freezing behaviors during the fear memory recall test with one-way ANOVA revealed a significant main effect of group (F(2,23) = 170.39, p < 0.01). Post hoc analyses revealed that rats experiencing foot shock in the absence of the memory cue at testing displayed slightly increased freezing levels (22%) relative to the no shock control group (p < 0.01). However, rats receiving the CS+ cue presentation immediately before recall testing displayed dramatically higher freezing levels [relative to both the fear conditioning with no recall group, as well as rats that had experienced olfactory conditioning in the absence of foot shock conditioning (73%; p values <0.01); Fig. 3A]. Thus, consistent with previous reports (Lauzon et al., 2009, 2013; Laviolette et al., 2005), this level of footshock (0.8 mA) produces robust associative fear memories. Comparing morphine CPP scores between groups revealed a significant effect of group (F(2,47) = 24.9; p < 0.0001), treatment (F(2,47) = 181.42; p < 0.0001) and a group × treatment interaction on times spent in morphine versus saline-paired conditioning environments (F(2,47) = 106.74, p < 0.0001). Post hoc analyses revealed that whereas rats conditioned without footshock or rats conditioned with footshock but not receiving the CS+, displayed no significant differences in times spent in saline versus morphine-paired environments (p values >0.05), rats that had received the CS+ presentation during fear memory recall testing displayed significantly greater time in morphine versus saline-paired environments (p < 0.01) and greater times in morphine-paired environments relative to other experimental groups (p values <0.01; Fig. 3B). Thus, only rats that actively recalled the suprathreshold associative fear memory before sub-reward threshold morphine CPP conditioning, demonstrated potentiated morphine reward sensitivity.
Figure 3.

Effects of recalling a suprathreshold associative fear memory on subthreshold morphine reward CPP. A, Whereas rats tested after conditioning with the absence of footshock stimuli (n = 8) or with the presence of footshock and absence of a CS+ cue display (n = 8) low levels of associative freezing, rats conditioned with footshock and then presented with the CS+ fear memory cue (n = 8) immediately before testing displayed significantly greater freezing levels relative to other groups (p < 0.01). B, When tested for CPP after subthreshold (0.05 mg/kg) morphine CPP conditioning, only rats receiving footshock and fear memory recall cues during recall testing displayed a significant morphine reward CPP. *p < 0.05, **p < 0.01; and error bars represent mean ± SEM for this and all subsequent figures.
Intra-PFC DA D1R activation blocks the recall of salient fear memories and blocks morphine reward salience potentiation
Previous evidence has demonstrated that stimulation of PFC D1R transmission potently suppresses the recall of fear-related associative memories (Lauzon et al., 2013). Accordingly, we next sought to determine whether blocking the recall of a suprathreshold associative fear memory might similarly block the potentiation of morphine reward salience observed in our previous control study (Fig. 3). Thus, we challenged the recall of suprathreshold associative fear memory with bilateral microinfusions of the D1R agonist, SKF 81297, using a dose range (10–100 ng/0.5 μl) previously established to be behaviorally effective and pharmacologically selective, while having no impact on physiological measures of footshock sensitivity (Lauzon et al., 2013). Three separate groups of rats received either VEH (n = 6), 10 (n = 8), or 100 ng/0.5 μl (n = 8) of intra-PFC SKF 81297 immediately before the fear memory recall test. One-way ANOVA comparison of these groups with rats that had received intra-PFC VEH before the CS+ cue presentation revealed a significant effect of treatment on times spent freezing to CS+ presentations (F(2,23) = 109.9; p < 0.0001; Fig. 4A). Post hoc analyses revealed that intra-PFC D1R activation dose-dependently blocked recall of fear-related associative memory, relative to VEH controls (Fig. 4A), with the higher dose of 100 ng suppressing memory recall to a significantly greater extent relative to the lower dose of 10 ng and relative to VEH controls (p values < 0.01), as demonstrated by significant reductions in levels of freezing behavior following the CS+ presentations. Next, two-way ANOVA of CPP scores revealed a significant effect of group (F(2,43) = 7.03; p < 0.01), treatment (F(1,43) = 72.3; p < 0.0001), and a group × treatment interaction on times spent in morphine versus saline-paired conditioning environments (F(2,43) = 29.41; p < 0.0001). Post hoc analyses revealed that whereas rats receiving a lower dose of intra-PFC SKF 81297 (10 ng) showed a significant morphine reward CPP (p < 0.01; Fig. 4B), rats receiving the higher dose of 100 ng failed to demonstrate a significant morphine CPP (p > 0.05). Furthermore, both groups receiving intra-PFC D1R activation spent significantly less time in the morphine-paired environments relative to VEH controls (p values <0.01). Thus, blocking the recall of suprathreshold associative fear memory with intra-PFC D1R activation dose-dependently blocked the ability of fear memory recall to potentiate morphine reward CPP.
Figure 4.

Effects of PFC D1R activation on the recall of suprathreshold fear memory and morphine reward CPP behaviors. A, Relative to VEH (n = 6) controls, intra-PFC D1R activation with SKF81297 immediately before the recall test, dose-dependently blocks the recall of suprathreshold fear memories demonstrated by significantly lower levels of associative freezing in rats receiving 10 ng (n = 8) or 100 ng (n = 8) of intra-PFC SKF81297. B, Blockade of suprathreshold fear memory recall with SKF81297 similarly blocks the potentiation of subthreshold morphine reward CPP, demonstrated by attenuated morphine environment preferences in rats receiving 10 or 100 ng intra-PFC SKF81297. **p < 0.01.
Intra-PFC DA D4R activation potentiates the salience of subthreshold fear memory and morphine reward sensitivity
Previous evidence has demonstrated that stimulation of PFC D4R transmission is capable of strongly potentiating the emotional salience of normally subthreshold fear memory cues, both behaviorally, and in PFC neurons, selectively during the encoding of associative memories (Laviolette et al., 2005; Lauzon et al., 2009). Accordingly, we next examined whether amplifying the salience of a fear-related memory cue via PFC D4R stimulation might influence the subsequent processing of a sub-reward morphine CPP cue. First, we pharmacologically potentiated the salience of a normally subthreshold associative fear memory stimulus (0.4 mA) with bilateral intra-PFC D4R activation, using a previously characterized, effective and selective in vivo dose range that has been demonstrated to have no impact on physiological measures of footshock sensitivity (Laviolette et al., 2005; Lauzon et al., 2009; 5–50 ng/0.5 μl). Three groups of rats received either VEH (n = 9), 5 (n = 6), or 50 ng/0.5 μl (n = 7) of the selective DA D4 agonist, PD168077, immediately before the fear memory training (Fig. 1). One-way ANOVA comparison of these groups to rats that had received intra-PFC VEH before subthreshold fear conditioning training revealed a significant effect of treatment on percentage of time spent freezing to the CS+ presentation (F(2,23) = 29.8; p < 0.0001; Fig. 5A). Post hoc analyses revealed that intra-PFC D4R activation dose-dependently potentiated the salience of normally nonsalient fear conditioning memory cues relative to VEH controls, with rats receiving the higher dose of 50 ng displaying significantly more freezing behavior during CS+ presentations relative to either VEH or the 5 ng experimental groups (p values <0.01; Fig. 5A). Two-way ANOVA comparing morphine CPP scores between groups revealed a significant effect of group (F(2,47) = 6.18; p < 0.01), treatment (F(1,47) = 18.79; p < 0.01), and a group × treatment interaction on times spent in morphine versus saline-paired conditioning environments (F(2,47) = 5.5; p < 0.05). Post hoc analyses revealed that whereas rats receiving the higher dose of intra-PFC PD168077 (50 ng) showed a significant morphine reward CPP (p < 0.01), rats receiving the lower dose of 5 ng or VEH failed to demonstrate a significant morphine CPP (p values >0.05). Furthermore, both groups receiving intra-PFC D4R activation spent significantly more time in the morphine-paired environments relative to VEH controls (p values <0.05 and 0.01, respectively; Fig. 5B). Thus, potentiating the salience of a normally nonsalient fear conditioning cue during the memory acquisition phase concomitantly potentiated subsequent morphine reward salience. Although previous evidence has demonstrated that intra-PFC D1R versus D4R transmission modulate separate phases of associative memory formation (recall vs acquisition, respectively; Lauzon et al., 2009, 2013), we next sought to determine whether blocking the recall of a D4R-potentiated fear memory with PFC D1R stimulation immediately before recall, might similarly modulate the acquisition of sub-reward threshold morphine CPP. Thus, a separate experimental group (n = 6) received intra-PFC D1R activation (100 ng) immediately before the recall of a fear memory potentiated by PFC D4R activation during the acquisition phase. ANOVA comparing freezing times between VEH controls, rats receiving intra-PFC D4R activation before subthreshold fear conditioning and rats receiving intra-PFC D1R activation immediately before the fear recall test demonstrated a significant effect of treatment on percentage freezing times during the recall test (F(2,26) = 28.6; p < 0.0001) with post hoc analyses demonstrating that intra-PFC D1R activation immediately before recall significantly blocked the recall of the D4R-mediated potentiated fear memory (p < 0.01; Fig. 5A). Thus, intra-PFC D1R activation is not only capable of blocking the recall of suprathreshold associative fear memory (Fig. 4A), but also blocks the recall of a normally nonsalient fear memory potentiated by PFC D4R activation during memory acquisition. Two-way ANOVA comparing morphine CPP scores across these same groups revealed a significant group × treatment interaction (F(1,41) = 16.9; p < 0.0001), with post hoc analyses revealing that rats receiving intra-PFC D1R activation before D4R-modulated fear memory recall displayed no significant morphine CPP (p > 0.05) and spent significantly less time in morphine-paired environments relative to rats receiving intra-PFC D4R activation during the memory encoding phase (p < 0.01; Fig. 5B). Thus, blocking the recall of a D4R-mediated potentiated fear memory similarly blocked the potentiation of morphine reward CPP.
Figure 5.

Effects of PFC D4R activation on the acquisition of subthreshold fear memory and morphine reward CPP behaviors. A, Relative to VEH controls (n = 9), intra-PFC D4R activation with PD 168077 during fear memory acquisition, dose-dependently potentiates the formation of subthreshold fear memories demonstrated by significantly higher levels of associative freezing in rats receiving 50 ng intra-PFC PD 168077 (n = 7) but not a lower dose of 5 ng (n = 6). This effect is blocked by intra-PFC D1R activation with SKF 81297 (100 ng) immediately before recall testing (n = 6). B, Potentiation of normally subthreshold fear memory with PD 168077 similarly potentiated the reward salience of normally sub-reward threshold conditioning doses of morphine (0.05 mg/kg, i.p.) in these same groups. Again, this effect is also blocked when fear memory recall is prevented with intra-PFC SKF 81297 immediately before recall testing. *p < 0.05, **p < 0.01.
Intra-PFC DA D1R and D4R activation regulates morphine reward sensitivity in the VTA
The PFC and VTA share strong functional connections, with the VTA serving as a critical neural region for the primary rewarding effects of opioids, including morphine (Nader and van der Kooy, 1997; Lintas et al., 2011, 2012). In addition, the PFC is capable of potently regulating the reward salience of morphine directly in the VTA (Bishop et al., 2011). Accordingly, we next examined whether intra-PFC D1R/D4R modulation of systemic opiate reward salience may extend to the motivational effects of morphine directly in the VTA. Four groups of rats received combined bilateral PFC-VTA cannulations and were treated either with VEH versus intra-PFC SKF81297 (100 ng) immediately before suprathreshold fear memory recall testing or VEH versus PD168077 (50 ng) during fear conditioning training to a subthreshold footshock level (see Materials and Methods). For comparison with previous experiments using the systemic dose of sub-reward threshold morphine (Figs. 3–5), we used a dose of intra-VTA morphine that has previously been established as a sub-reward microinfusion dose (Nader and van der Kooy, 1997; Bishop et al., 2011; De Jaeger et al., 2013; i.e., fails to produce significant morphine CPP). In Figure 6A, a representative microphotograph of a typical intra-VTA injector location is presented. In Figure 6B, a schematic summary showing bilateral intra-VTA injector placements is presented. First, in rats receiving intra-PFC D1R activation with SKF81297 (n = 8) versus VEH (n = 7), comparison of freezing scores during the fear memory recall test for suprathreshold fear memory (0.8 mA) revealed a significant block of fear memory recall relative to VEH controls (t(14) = 8.45; p < 0.001; Fig. 6C). Comparing morphine CPP scores revealed a significant main effect of treatment (VTA saline vs morphine; F(1,29) = 10.74; p < 0.01) on times spent in intra-VTA morphine vs saline environments (Fig. 6D). Thus, consistent with effects observed in experiments using sub-reward threshold systemic conditioning doses of morphine, blockade of fear memory recall similarly blocked intra-VTA morphine reward CPP potentiation relative to VEH controls. Next, in rats receiving intra-PFC D4R activation with PD168077 (n = 7; one rat was removed due to cannula misplacement) versus VEH (n = 8), comparison of freezing scores during the fear memory recall test revealed a significant potentiation of normally subthreshold fear memory (0.4 mA), relative to VEH controls (t(14) = 7.48; p < 0.001; Fig. 6E). Comparing CPP scores revealed a significant effect of group (F(1,31) = 8.14; p < 0.01), treatment (F(1,31) = 10.72; p < 0.01) and a group × treatment interaction (F(1,31) = 28.3; p < 0.0001) on times spent in intra-VTA morphine vs saline environments (Fig. 6F). Post hoc analyses revealed that whereas VEH control rats failed to demonstrate a significant CPP for intra-VTA morphine-paired environments (p > 0.05), rats with potentiated fear memory acquisition displayed significant CPP for intra-VTA morphine environments, spending significantly more time in morphine versus saline environments and greater time in morphine-paired environments relative to VEH controls (p values <0.01; Fig. 6F). Thus, consistent with effects observed in experiments using sub-reward threshold systemic doses of morphine, potentiation of nonsalient fear memory similarly potentiates intra-VTA morphine reward CPP.
Figure 6.
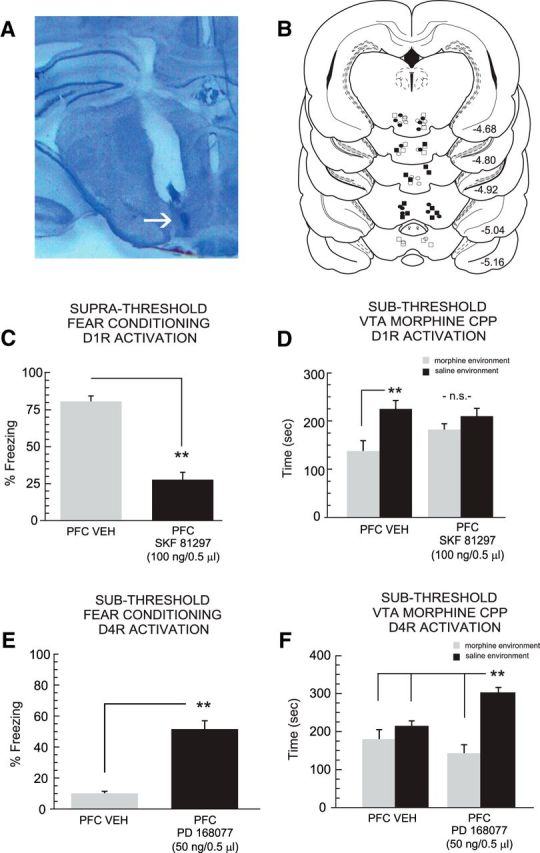
VTA histological analysis and effects of intra-PFC D1R or D4R activation on fear memory processing and intra-VTA morphine reward CPP behaviors. A, Microphotograph showing typical intra-VTA injector location. B, Schematic summary of bilateral intra-VTA cannulae injector locations; ○ = intra-PFC VEH; ● = intra-PFC PD168077 (50 ng/0.5 μl); ■ = intra-PFC SKF81297 (100 ng/0.5 μl). C, Intra-PFC SKF81297 administration immediately before fear memory recall significantly attenuates freezing behaviors. D, Relative to VEH controls (n = 7), rats receiving intra-PFC SKF81297 (n = 8) do not display morphine CPP. E, Intra-PFC PD168077 (n = 7) administration during fear memory acquisition significantly potentiates subthreshold associative fear memory and F, potentiates intra-VTA morphine reward CPP relative to VEH controls (n = 8). **p < 0.01.
Intra-PFC DA D1R activation blocks fear memory recall and morphine reward potentiation via selective upregulation of ERK 1/2 phosphorylation states
Signaling through the ERK 1/2 pathway is critically involved in the consolidation of morphine-reward memories directly in the PFC (Gholizadeh et al., 2013) and is functionally linked to transmission through the D1R system during morphine reward memory processing (Lyons et al., 2013; Rosen et al., 2016). In contrast, whereas CaMKII-α signaling is similarly involved in the processing of opiate-related reward memories, it has been functionally linked to transmission through the D2R system during the processing of opiate-related learning and memory behaviors (Lyons et al., 2013; Rosen et al., 2016). To examine the potential roles of either ERK 1/2 or CaMKII-α in PFC D1R-mediated blockade of fear memory recall and morphine reward salience, we next examined the local effects of intra-PFC D1R or D4R activation on PFC expression levels of total ERK 1/2 (tERK 1/2), phosphorylated ERK 1/2 (pERK 1/2), total CaMKII-α (tCaMKII-α), or phosphorylated CaMKII-α (pCaMKII-α; see Materials and Methods). First, comparing pERK-1 levels between groups revealed a significant effect of treatment on normalized densitometry levels (F(2,17) = 4.1; p < 0.05; Fig. 7A,B) with post hoc analyses revealing that rats receiving intra-PFC SKF 81297 (100 ng/0.5 μl; n = 6) had significantly elevated levels of pERK1 relative to both VEH controls (n = 6) or rats receiving the D4 agonist (PD 168077, 50 ng/0.5 μl; p values <0.05; n = 5). Similarly, comparing pERK-2 levels between groups revealed a significant effect of treatment on normalized densitometry levels (F(2,17) = 5.44; p < 0.05; Fig. 7A,C) with post hoc analyses revealing that rats receiving intra-PFC SKF 81297 had significantly elevated levels of pERK2 relative to both VEH controls (p < 0.01) or rats receiving the D4 agonist (p < 0.05). Next, comparing tERK-1 levels between groups revealed no significant effect of treatment on normalized densitometry levels (F(2,17) = 3.6; p > 0.05; Fig. 7D,E). In contrast, comparing tERK-2 levels between groups revealed a significant effect of treatment on normalized densitometry levels (F(2,17) = 6.01; p < 0.05; Fig. 7D,F) with post hoc analyses revealing that rats receiving either intra-PFC SKF 81297 or the D4 agonist showed significantly lower levels of tERK2 relative to VEH controls (p values < 0.05).
Figure 7.

Effects of PFC D1R activation on total and phosphorylated ERK 1/2 levels in the PFC and effects of intra-PFC ERK inhibition on PFC D1R modulation of fear and morphine reward behaviors. A, Sample Western blots showing PFC pERK 1/2 bands relative to loading controls in intra-PFC VEH (n = 6), SKF 81297 (100 ng; n = 6), or PD 168077 (50 ng; n = 5) -treated rats. B, C, Normalized densitometry values comparing average levels of pERK 1 or 2 following intra-PFC VEH, SKF 81297, or PD 168077 administration. D, Sample Western blots showing PFC tERK 1/2 bands relative to loading controls in intra-PFC VEH, SKF 81297 (100 ng), or PD 168077 (50 ng) -treated rats. E, F, Normalized densitometry values comparing average levels of tERK 1 or 2 following intra-PFC VEH, SKF 81297, or PD 168077 administration. G, Coadministration of SKF 81297 (100 ng) with the MEK 1/2 inhibitor U0126 (1 μg; n = 6) reverses the effects of intra-PFC D1R activation on blockade of fear memory recall, with U0126 having no effect on its own (n = 8), relative to SKF 81297 (100 ng) on its own (n = 7). H, Similarly, reversing the block of D1R-mediated memory recall with U0126 restores the potentiation of sub-reward threshold morphine reward CPP in these same groups. Rats receiving U0126 alone showed a normal potentiated morphine CPP. *p < 0.05, **p < 0.01.
Given our observation that PFC D1R activation selectively increased pERK 1/2 levels, we challenged the effects of PFC D1R activation on associative fear memory recall inhibition using the selective MEK 1/2 inhibitor, U0126. We selected a dose of U0126 (1 μg/0.5 μl) that we have previously reported to produce no nonselective deficits in associative learning (Gholizadeh et al., 2013; Lyons et al., 2013), for coadministration with our effective dose of intra-PFC SKF 81297 (100 ng/0.5 μl; n = 6; one rat was removed due to cannula misplacement) or U0126 alone (n = 7; one rat was removed due to cannula misplacement), immediately before the memory recall phase following suprathreshold fear conditioning. Comparing these groups with rats receiving intra-PFC SKF 81297 alone (n = 8), revealed a significant effect of group on freezing times (F(2,23) = 99.89; p < 0.0001; Fig. 7G). Post hoc analyses revealed that rats receiving coadministration of SKF81297 with U0126 displayed significantly greater freezing levels relative to rats receiving SKF81297 alone (p < 0.01), demonstrating that inhibition of PFC ERK 1/2 is sufficient to reverse the effects of PFC D1R activation on blockade of fear memory recall. Furthermore, rats receiving PFC U0126 alone displayed significantly greater freezing levels relative to rats receiving SKF81297 alone (p < 0.01), demonstrating that U0126 alone was not producing any nonspecific effects on the recall of a suprathreshold associative fear memory. Next, comparing morphine CPP scores across the same groups revealed a significant effect of treatment (F(1,37) = 19.85; p < 0.001) and a significant group × treatment interaction (F(2,37) = 8.59; p < 0.01) on times spent in morphine versus saline-paired environments recorded during the CPP test. Post hoc analyses revealed that rats receiving SKF81297 + U0126 administration as well as rats receiving U0126 alone showed significant CPP for sub-reward threshold morphine-paired environments (p values < 0.01; Fig. 7H) relative to rats receiving SKF81297 alone before the fear memory recall test. Thus, inhibition of PFC ERK 1/2 signaling is sufficient to reverse the D1R activation-induced blockade of fear memory recall and concomitantly restore the potentiation of morphine reward CPP behaviors.
Intra-PFC DA D4R activation potentiates fear memory acquisition and morphine reward sensitivity via selective upregulation of CaMKII-α phosphorylation states
Previous evidence has demonstrated that the ability of PFC D4R activation to modulate fear-related memory salience and associated neuronal activity states is functionally dependent upon CaMKII-α signaling and is functionally linked to D2R-mediated modulation of associative opiate reward memory processing (Gu et al., 2006; Yuen and Yan, 2011; Lauzon et al., 2013; Lyons et al., 2013). Therefore, we next examined the potential role of intra-PFC CaMKII-α expression and phosphorylation states in the mediation of D4R-activation induced modulation of fear and opiate-reward related associative memory processing. First, comparing pCaMKII-α levels between groups revealed a significant effect of treatment on normalized densitometry levels (F(2,14) = 6.56; p < 0.05; Fig. 8A) with post hoc analyses revealing that rats receiving intra-PFC PD 168077 (50 ng/0.5 μl) had significantly elevated levels of pCaMKII-α relative to VEH controls (n = 6). In contrast, receiving the D1 agonist (SKF 81297, 100 ng/0.5 μl; p values <0.05) showed no significant increase relative to VEH controls (p > 0.05). Comparing tCaMKII-α between groups revealed no significant effect of treatment on normalized densitometry levels (F(2,14) = 0.44; p > 0.05; Fig. 8B). Thus, intra-PFC D4R activation, but not D1R activation, selectively increases phosphorylation levels of CaMKII-α, without influencing total protein expression levels.
Figure 8.

Effects of PFC D4R activation on total and phosphorylated PFC CaMKII-α expression levels and effects of intra-PFC CaMKII-α phosphorylation inhibition on PFC D4R modulation of fear and morphine reward behaviors. A, Sample Western blots showing PFC pCaMKII-α bands relative to loading controls in intra-PFC VEH, SKF 81297 (100 ng), or PD 168077 (50 ng) -treated rats. B, Sample Western blots showing representative PFC tCaMKII-α bands relative to loading controls in intra-PFC VEH, SKF 81297 (100 ng), or PD 168077 (50 ng) -treated rats. C, Coadministration of PD 168077 (50 ng) with the CaMKII-α phosphorylation inhibitor AIP (1 μg; n = 8) reverses the effects of intra-PFC D4R activation on potentiation of subthreshold fear memory acquisition, with AIP having no effect on its own (n = 8), relative to PD 168077 (50 ng) on its own (n = 8). D, Similarly, reversing the potentiation of D4R-mediated fear memory recall with AIP blocks the potentiation of sub-reward threshold morphine reward CPP in these same groups. Rats receiving AIP alone show no potentiation of morphine reward CPP. **p < 0.01.
Given our observation of a selective increase in PFC pCaMKII-α levels following D4R activation, we next challenged the effects of PFC D4R activation on the potentiation of subthreshold fear memory formation by coadministering a selective inhibitor or CaMKII-α autophosphorylation, AIP with a dose (500 ng/0.5 μl) we have previously determined to produce no nonselective deficits in associative learning (Lyons et al., 2013). Experimental groups received intra-PFC PD168077 (50 ng/0.5 μl; n = 8) combined with AIP, AIP alone (n = 8), or PD168077 (50 ng/0.5 μl; n = 8) alone, immediately before fear conditioning with the subthreshold footshock conditioning cue (0.4 mA). Comparing freezing scores between groups revealed a significant effect of group on freezing times (F(2,23) = 32.28; p < 0.0001; Fig. 8C). Post hoc analyses revealed that rats receiving coadministration of PD168077 + AIP or AIP alone displayed significantly less freezing levels relative to rats receiving PD168077 alone (p < 0.01), demonstrating that inhibition of PFC CaMKII-α phosphorylation is sufficient to reverse the effects of PFC D4R activation on potentiation of normally nonsalient associative fear memory. Furthermore, rats receiving PFC AIP alone displayed no potentiation of subthreshold fear memory, demonstrating that AIP alone was not producing any nonspecific effects during the encoding phase of subthreshold associative fear memory. Next, comparing morphine CPP scores across the same groups revealed a significant effect of treatment (F(1,45) = 9.33; p < 0.01) and a significant group × treatment interaction (F(1,45) = 7.63; p < 0.01) on times spent in morphine versus saline-paired environments recorded during the CPP test. Post hoc analyses revealed that rats receiving PD168077 + AIP administration as well as rats receiving AIP alone showed no significant CPP for sub-reward threshold morphine-paired environments (p values >0.05; Fig. 8D) relative to rats receiving PD168077 during subthreshold fear memory training (p < 0.01). Thus, inhibition of PFC CaMKII-α phosphorylation is sufficient to reverse the D4R activation-induced potentiation of fear memory salience and concomitantly blocks the potentiation of morphine reward CPP.
Discussion
Abnormal processing of emotionally salient sensory information and associative memory is linked to PTSD and addiction-related PFC pathology (Bishop et al., 2011; Clausen et al., 2017; Malejko et al., 2017; O'Doherty et al., 2017). Beyond the role of learning and memory, the role of stress as a critical variable in drug abuse vulnerability is well established. Indeed, exposure to acute, unconditioned stressors, such as footshock, potently reinstates drug-seeking behaviors, including those linked to opioids and other drugs of abuse (Shaham and Stewart, 1994; Buczek et al., 1999; Shaham et al., 2000). However, the identification of common neural mechanisms responsible for processing traumatic memories as well as modulating vulnerability to drugs of abuse following their recall, remain elusive. The present study demonstrates that acute recall of a fear-related associative memory can potentiate sensitivity to normally non-rewarding systemic or intra-VTA morphine conditioning cues. Consistent with a generalized effect of footshock stressor exposure, fear conditioning involving exposure to suprathreshold footshock caused a slight increase in freezing behaviors when tested in the absence of the CS+ recall cue. However, only subjects that had been fear conditioned with suprathreshold footshock and exposed to the CS+ during the recall test displayed significant morphine reward CPP potentiation. This suggests that the recall alone of a salient, fear-related associative memory is sufficient to cause behavioral potentiation of normally non-rewarding, morphine-related conditioning cues. Given the dissociable roles of PFC D4R-D1R transmission in reward and aversion-related memory acquisition and recall, we examined the potential modulatory roles of these DA receptor subtypes in the acquisition or recall phases of fear memory and morphine reward learning and memory. We found that targeting distinct phases of fear memory processing (memory acquisition vs recall) via D1R versus D4R activation, concomitantly regulated both systemic or intra-VTA morphine CPP reward sensitivity via dissociable ERK 1/2 and CaMKII-α phosphorylation states in the PFC.
PFC D4R transmission modulates fear memory acquisition and morphine reward salience processing via CaMKII-α phosphorylation
Prefrontal cortical D4R transmission is strongly linked to the regulation of emotional salience attribution and related associative memory formation (Lauzon and Laviolette, 2010). Activation of PFC D4R transmission has previously been shown to strongly potentiate normally nonsalient emotional memory processing both behaviorally, and in terms of associative neuronal activity patterns in PFC neurons (Laviolette et al., 2005; Laviolette, 2007; Lauzon et al., 2009). In terms of D4R involvement in PTSD or addiction, genetic variations in the D4R have been reported to strongly influence the processing of psychological responses to traumatic stressors (Armbruster et al., 2009; Dragan and Oniszczenko, 2009; Bakermans-Kranenburg et al., 2011) and polymorphisms in the D4R are linked to opioid addiction vulnerability, including sensitivity to heroin-related associative cues (Kotler et al., 1997; Mel et al., 1998; Shao et al., 2006; McGeary, 2009). Anatomically, the PFC contains functional D4Rs localized on both inhibitory interneuron, and pyramidal output neuron populations (Zhong and Yan, 2016) capable of regulating cortical neuronal activity states (Ceci et al., 1999; Onn et al., 2006). For example, acute D4R activation has been shown to transiently increase and then decrease, PFC interneuron population activity (Zhong and Yan, 2016). Furthermore, PFC D4R transmission is involved in modulating activation of PFC neurons via inputs from emotional processing regions such as the basolateral amgydala (Floresco and Tse, 2007; Lauzon et al., 2009). Blockade of PFC D4R activation prevents associative increases in PFC pyramidal neuron populations (Laviolette et al., 2005), suggesting that D4R activation might potentiate the salience of normally nonsalient fear-cues by removing inhibitory input to PFC pyramidal output neurons. Given the ability of PFC neuronal activity states to modulate VTA DAergic neuronal activity levels and regulate sensitivity to fear-related conditioning cues (Draycott et al., 2014), one possibility is that PFC D4R activation may subsequently potentiate morphine-related reward cues, by amplifying subcortical DAergic activity levels and associated reward salience processing.
Functionally, PFC D4R transmission strongly regulates local PFC neuronal network dynamics and associative memory encoding by bidirectionally modulating CaMKII-α signaling. For example, D4R activation increases CaMKII-α phosphorylation during periods of low neuronal activity states (Gu et al., 2006) or during the encoding of normally nonsalient associative fear memories (Lauzon et al., 2012). In contrast, D4R activation inhibits CaMKII-α phosphorylation during states of high PFC neuronal activity or during the encoding of suprathreshold associative fear memory formation (Gu et al., 2006; Lauzon et al., 2012). In the present study, we found that acute PFC D4R activation strongly activated PFC CaMKII-α phosphorylation states and that blocking this effect, was sufficient to both prevent D4R-mediated potentiation of normally nonsalient fear memory, but to concomitantly block the potentiation of opiate-related reward CPP. Interestingly, there is considerable evidence linking PFC CaMKII-α signaling to both fear and opiate-reward related memory processing. For example, long-term opioid exposure modulates expression levels of total and phosphorylated CaMKII-α levels in the PFC and BLA, specifically during states of withdrawal (Lyons et al., 2013; Rosen et al., 2015). Functionally, blocking PFC CaMKII-α phosphorylation has been shown to prevent the formation of morphine CPP memories (Rosen et al., 2015, 2016) and blocks the acute consolidation of morphine CPP memories (Gholizadeh et al., 2013). This would suggest that acute opiate reward memory formation requires CaMKII-α signaling in the PFC and further, D4R activation of PFC CaMKII-α signaling may in turn amplify the formation of opiate-related associative memories, using a parallel mechanism to that observed with the potentiation of fear-related memory formation (Lauzon et al., 2012).
PFC D1R transmission modulates fear memory recall and morphine reward salience processing via ERK 1/2 phosphorylation
While there is currently little direct evidence linking disturbances in the D1R system to PTSD and addiction comorbidity, significant evidence has demonstrated a role for PFC D1R and associated ERK 1/2 signaling in the processing of both positive and aversive associative memories. Specifically, supranormal stimulation of PFC D1R transmission can independently block the recall of both fear and opiate-reward-related associative memories (Lauzon et al., 2009, 2013). In contrast, blockade of D1R transmission in noncortical regions, such as the BLA, can block the acquisition of associative morphine-related reward memories (Lyons et al., 2013). In the present study, PFC D1R activation blocked the recall of normally suprathreshold associative fear memories and concomitantly, prevented the potentiation of sub-reward threshold morphine CPP behaviors. This effect was correlated with increased phosphorylation of PFC ERK 1/2 and functionally, pharmacological blockade of ERK was sufficient to reverse the effects of PFC D1R stimulation on memory recall blockade. Furthermore, PFC D1R activation blocked recall of a D4R-mediated potentiated fear memory, which concomitantly blocked potentiation of sub-reward threshold morphine CPP, demonstrating that D1R activation is capable of overriding the effects of D4R modulation during the memory encoding phase and concomitantly blocking the potentiation of morphine CPP.
The ERK signaling pathway is functionally linked to PTSD and addictive behaviors both in terms of vulnerability and stress-related neuronal responses. For example, exposure to acute or prolonged stressors activates ERK phosphorylation in the amygdala and PFC, which has been linked to increased vulnerability to the reinforcing effects of alcohol and opioids (Bertotto et al., 2010, 2011; Wang et al., 2010; Xiao et al., 2011; Hauger et al., 2012). During opiate-related reward memory processing D1R transmission is linked to the ERK 1/2 signaling pathway during acquisition and consolidation phases (Gholizadeh et al., 2013; Lyons et al., 2013) and ERK expression levels are significantly elevated in the amygdala during states of opioid withdrawal (Lyons et al., 2013). Although PFC ERK 1/2 signaling has not previously been linked to suppression of either fear- or reward-related memory recall, blockade of ERK 1/2 in the central nucleus of the amygdala has been shown to prevent the recall of opiate-related reward memories (Li et al., 2008). Given the common roles of ERK in fear- and reward-related memory processing, the ability of PFC D1R activation to induce ERK 1/2 phosphorylation may underlie D1R-mediated common modulation of fear memory recall and opiate-reward sensitivity.
Nevertheless, important questions remain. For example, both D4R and D1R signaling are capable of mediating glutamatergic transmission through modulation of NMDA receptor activity states, via downstream CaMKII-α (Yuen et al., 2010) and ERK 1/2 signaling (Nagai et al., 2007), respectively. Given the critical role of PFC NMDA transmission in both the acquisition and recall of emotional associative memories (Bishop et al., 2011), future research should explore how these mechanisms might relate to NMDA-mediated modulation of memory acquisition and/or recall, in the context of fear- and reward-related memory processing. In contrast to our observed effects on ERK 1/2 phosphorylation states, both PFC D1R and D4R activation produced small, but significant decreases in total levels PFC ERK 2, whereas D4R activation caused a slight but significant decrease in total PFC ERK 1 expression levels. However, changes in total ERK 1/2 expression levels mediated by PFC D4R did not appear to modulate ERK 1/2 phosphorylation states, because we observed no concomitant changes in D4R-mediated ERK 1/2 phosphorylation. Importantly, because no pharmacological compound displays complete specificity, the possibility that other signaling pathways may be functionally involved in the present effects cannot be ruled out. Although beyond the scope of the present study, future experiments are required to fully explore the possible functional effects of D4R activation on total ERK 1/2 expression levels within the PFC and how these may impact the encoding of fear- or reward-related associative memory salience. In addition, future studies should explore how the effects of acute pharmacological activation of cortical D1R and D4R substrates may translate into underlying transmission or expression disturbances in these systems in the context of either PTSD or addiction.
In summary, the present findings reveal several novel mechanisms by which cortical DAergic transmission modulates the acquisition and recall of associative, fear-related memory while regulating sensitivity to opiate-related reward effects, directly in the mesolimbic pathway. Given the high comorbidity between PTSD and opioid dependence, these findings identify D4R-CaMKII-α and D1-ERK-1/2 signaling pathways in the PFC as potential biomarkers and mechanisms controlling how traumatic memory recall may serve to increase addiction liability.
Footnotes
This work was supported by the Canadian Institutes of Health Research (MOP 272999, 123378) and the Natural Science and Engineering Research Council of Canada.
The authors declare no competing financial interests.
References
- Armbruster D, Mueller A, Moser DA, Lesch KP, Brocke B, Kirschbaum C (2009) Interaction effect of D4 dopamine receptor gene and serotonin transporter promoter polymorphism on the cortisol stress response. Behav Neurosci 123:1288–1295. 10.1037/a0017615 [DOI] [PubMed] [Google Scholar]
- Bakermans-Kranenburg MJ, van Ijzendoorn MH, Caspers K, Philibert R (2011) DRD4 genotype moderates the impact of parental problems on unresolved loss or trauma. Attach Hum Dev 13:253–269. 10.1080/14616734.2011.562415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee G, Edelman EJ, Barry DT, Becker WC, Cerdá M, Crystal S, Gaither JR, Gordon AJ, Gordon KS, Kerns RD, Martins SS, Fiellin DA, Marshall BD (2016) Non-medical use of prescription opioids is associated with heroin initiation among US veterans: a prospective cohort study. Addiction 111:2021–2031. 10.1111/add.13491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertotto ME, Bussolino DF, Molina VA, Martijena ID (2010) Increased voluntary ethanol consumption and c-fos expression in selected brain areas induced by fear memory retrieval in ethanol withdrawn rats. Eur Neuropsychopharmacol 20:568–581. 10.1016/j.euroneuro.2010.02.014 [DOI] [PubMed] [Google Scholar]
- Bertotto ME, Maldonado NM, Bignante EA, Gorosito SV, Cambiasso MJ, Molina VA, Martijena ID (2011) ERK activation in the amygdala and hippocampus induced by fear conditioning in ethanol withdrawn rats: modulation by MK-801. Eur Neuropsychopharmacol 21:892–904. 10.1016/j.euroneuro.2011.01.001 [DOI] [PubMed] [Google Scholar]
- Bishop SF, Lauzon NM, Gholizadeh S, Bechard M, Laviolette SR (2011) NMDA receptor hypofunction in the prelimbic cortex increases sensitivity to the rewarding properties of opiates via dopaminergic and amygdalar substrates. Cereb Cortex 21:68–80. 10.1093/cercor/bhq060 [DOI] [PubMed] [Google Scholar]
- Bowers ME, Ressler KJ (2015) An overview of translationally informed treatments for PTSD: animal models of Pavlovian fear conditioning to human clinical trials. Biol Psychiatry 78:E15–E27. 10.1016/j.biopsych.2015.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczek Y, Lê AD, Wang A, Stewart J, Shaham Y (1999) Stress reinstates nicotine seeking but not sucrose solution seeking in rats. Psychopharmacology 144:183–188. 10.1007/s002130050992 [DOI] [PubMed] [Google Scholar]
- Ceci A, Brambilla A, Duranti P, Grauert M, Grippa N, Borsini F (1999) Effect of antipsychotic and selective dopaminergic antagonists on dopamine-induced facilitatory activity in prelimbic cortical pyramidal neurons: an in vitro study. Neuroscience 93:107–115. 10.1016/S0306-4522(99)00123-2 [DOI] [PubMed] [Google Scholar]
- Clausen AN, Francisco AJ, Thelen J, Bruce J, Martin LE, McDowd J, Simmons WK, Aupperle RL (2017) PTSD and cognitive symptoms relate to inhibition-related prefrontal activation and functional connectivity. Depress Anxiety 34:427–436. 10.1002/da.22613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrod PJ, Stewart SH (2003) Experimental studies exploring functional relations between post-traumatic stress disorder and substance use disorders. In: Trauma and substance use: causes, consequences, and treatment of comorbid disorders (Ouimette P, Browns P, eds.), pp 29–55. Washington, DC: American Psychological Association. [Google Scholar]
- DeHaas RAB, Calamari JE, Bair JP (2002) Anxiety sensitivity and the situational antecedents to drug and alcohol use: an evaluation of anxiety patients with substance use disorders. Cognit Ther Res 26:335–353. 10.1023/A:1016076911164 [DOI] [Google Scholar]
- De Jaeger X, Bishop SF, Ahmad T, Lyons D, Ng GA, Laviolette SR (2013) The effects of AMPA receptor blockade in the prelimbic cortex on systemic and ventral tegmental area opiate reward sensitivity. Psychopharmacology 225:687–695. 10.1007/s00213-012-2852-4 [DOI] [PubMed] [Google Scholar]
- Dell'Osso L, Rugani F, Maremmani AG, Bertoni S, Pani PP, Maremmani I (2014) Towards a unitary perspective between post-traumatic stress disorder and substance use disorder: heroin use disorder as case study. Compr Psychiatry 55:1244–1251. 10.1016/j.comppsych.2014.03.012 [DOI] [PubMed] [Google Scholar]
- Dragan WŁ, Oniszczenko W (2009) The association between dopamine D4 receptor exon III polymorphism and intensity of PTSD symptoms among flood survivors. Anxiety Stress Coping 22:483–495. 10.1080/10615800802419407 [DOI] [PubMed] [Google Scholar]
- Draycott B, Loureiro M, Ahmad T, Tan H, Zunder J, Laviolette SR (2014) Cannabinoid transmission in the prefrontal cortex bi-phasically controls emotional memory formation via functional interactions with the ventral tegmental area. J Neurosci 34:13096–13109. 10.1523/JNEUROSCI.1297-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, Tse MT (2007) Dopaminergic regulation of inhibitory and excitatory transmission in the basolateral amygdala-prefrontal cortical pathway. J Neurosci 27:2045–2057. 10.1523/JNEUROSCI.5474-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholizadeh S, Sun N, De Jaeger X, Bechard M, Coolen L, Laviolette SR (2013) Early versus late-phase consolidation of opiate reward memories requires distinct molecular and temporal mechanisms in the amygdala-prefrontal cortical pathway. PLoS One 8:e63612. 10.1371/journal.pone.0063612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Jiang Q, Yuen EY, Yan Z (2006) Activation of dopamine D4 receptors induces synaptic translocation of Ca2+/calmodulin-dependent protein kinase II in cultured prefrontal cortical neurons. Mol Pharmacol 69:813–822. 10.1124/mol.105.018853 [DOI] [PubMed] [Google Scholar]
- Hauger RL, Olivares-Reyes JA, Dautzenberg FM, Lohr JB, Braun S, Oakley RH (2012) Molecular and cell signaling targets for PTSD pathophysiology and pharmacotherapy. Neuropharmacology 62:705–714. 10.1016/j.neuropharm.2011.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotler M, Cohen H, Segman R, Gritsenko I, Nemanov L, Lerer B, Kramer I, Zer-Zion M, Kletz I, Ebstein RP (1997) Excess dopamine D4 receptor (D4DR) exon III seven repeat allele in opioid-dependent subjects. Mol Psychiatry 2:251–254. 10.1038/sj.mp.4000248 [DOI] [PubMed] [Google Scholar]
- Lauzon NM, Laviolette SR (2010) Dopamine D4 receptor modulation of cortical neuronal network activity: implications for neuropsychiatric disorders. Behav Brain Res 208:12–22. 10.1016/j.bbr.2009.11.037 [DOI] [PubMed] [Google Scholar]
- Lauzon NM, Bishop SF, Laviolette SR (2009) Dopamine D1 versus D4 receptors differentially modulate salient versus nonsalient emotional information in the medial prefrontal cortex. J Neurosci 29:4836–4845. 10.1523/JNEUROSCI.0178-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauzon NM, Ahmad T, Laviolette SR (2012) Dopamine D4 receptor transmission in the prefrontal cortex controls the salience of emotional memory via modulation of calcium calmodulin-dependent kinase II. Cereb Cortex 22:2486–2494. 10.1093/cercor/bhr326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauzon NM, Bechard M, Ahmad T, Laviolette SR (2013) Supra-normal stimulation of dopamine D1 receptors in the prelimbic cortex blocks behavioral expression of both aversive and rewarding associative memories through a cyclic-AMP-dependent signaling pathway. Neuropharmacology 67:104–114. 10.1016/j.neuropharm.2012.10.029 [DOI] [PubMed] [Google Scholar]
- Laviolette SR. (2007) Dopamine modulation of emotional processing in cortical and subcortical neural circuits: evidence for a common final pathway in schizophrenia? Schizophr Bull 33:971–981. 10.1093/schbul/sbm048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviolette SR, Grace AA (2006) Cannabinoids Potentiate Emotional Learning Plasticity in Neurons of the Medial Prefrontal Cortex through Basolateral Amygdala Inputs. J Neurosci 26:6458–6468. 10.1523/jneurosci.0707-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviolette SR, Lipski WJ, Grace AA (2005) A subpopulation of neurons in the medial prefrontal cortex encodes emotional learning through a dopamine D4 receptor-dependent basolateral amygdala input. J Neurosci 25:6066–6075. 10.1523/JNEUROSCI.1168-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YQ, Li FQ, Wang XY, Wu P, Zhao M, Xu CM, Shaham Y, Lu L (2008) Central amygdala extracellular signal-regulated kinase signaling pathway is critical to incubation of opiate craving. J Neurosci 28:13248–13257. 10.1523/JNEUROSCI.3027-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lintas A, Chi N, Lauzon NM, Bishop SF, Gholizadeh S, Sun N, Tan H, Laviolette SR (2011) Identification of a dopamine receptor-mediated opiate reward memory switch in the basolateral amygdala-nucleus accumbens circuit. J Neurosci 31:11172–11183. 10.1523/JNEUROSCI.1781-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lintas A, Chi N, Lauzon NM, Bishop SF, Sun N, Tan H, Laviolette SR (2012) Inputs from the basolateral amygdala to the nucleus accumbens shell control opiate reward magnitude via differential dopamine D1 or D2 receptor transmission. Eur J Neurosci 35:279–290. 10.1111/j.1460-9568.2011.07943.x [DOI] [PubMed] [Google Scholar]
- Lyons D, de Jaeger X, Rosen LG, Ahmad T, Lauzon NM, Zunder J, Coolen LM, Rushlow W, Laviolette SR (2013) Opiate exposure and withdrawal induces a molecular memory switch in the basolateral amygdala between ERK1/2 and CaMKIIα-dependent signaling substrates. J Neurosci 33:14693–14704. 10.1523/JNEUROSCI.1226-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malejko K, Abler B, Plener PL, Straub J (2017) Neural correlates of psychotherapeutic treatment of post-traumatic stress disorder: a systematic literature review. Front Psychiatry 8:85. 10.3389/fpsyt.2017.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeary J. (2009) The DRD4 exon 3 VNTR polymorphism and addiction-related phenotypes: a review. Pharmacol Biochem Behav 93:222–229. 10.1016/j.pbb.2009.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mel H, Horowitz R, Ohel N, Kramer I, Kotler M, Cohen H, Gritsenko I, Ebstein RP (1998) Additional evidence for an association between the dopamine D4 receptor (D4DR) exon III seven-repeat allele and substance abuse in opioid dependent subjects: relationship of treatment retention to genotype and personality. Addict Biol 3:473–481. 10.1080/13556219872029 [DOI] [PubMed] [Google Scholar]
- Mills KL, Lynskey M, Teesson M, Ross J, Darke S (2005) Post-traumatic stress disorder among people with heroin dependence in the Australian treatment outcome study (ATOS): prevalence and correlates. Drug Alcohol Depend 77:243–249. 10.1016/j.drugalcdep.2004.08.016 [DOI] [PubMed] [Google Scholar]
- Mills KL, Teesson M, Ross J, Darke S (2007) The impact of post-traumatic stress disorder on treatment outcomes for heroin dependence. Addiction 102:447–454. 10.1111/j.1360-0443.2006.01711.x [DOI] [PubMed] [Google Scholar]
- Nader K, van der Kooy D (1997) Deprivation state switches the neurobiological substrates mediating opiate reward in the ventral tegmental area. J Neurosci 17:383–390. 10.1523/JNEUROSCI.17-01-00383.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Takuma K, Kamei H, Ito Y, Nakamichi N, Ibi D, Nakanishi Y, Murai M, Mizoguchi H, Nabeshima T, Yamada K (2007) Dopamine D1 receptors regulate protein synthesis-dependent long-term recognition memory via extracellular signal-regulated kinase 1/2 in the prefrontal cortex. Learn Mem 14:117–125. 10.1101/lm.461407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Doherty DCM, Tickell A, Ryder W, Chan C, Hermens DF, Bennett MR, Lagopoulos J (2017) Frontal and subcortical grey matter reductions in PTSD. Psychiatry Res Neuroimaging 266:1–9. 10.1016/j.pscychresns.2017.05.008 [DOI] [PubMed] [Google Scholar]
- Onn SP, Wang XB, Lin M, Grace AA (2006) Dopamine D1 and D4 receptor subtypes differentially modulate recurrent excitatory synapses in prefrontal cortical pyramidal neurons. Neuropsychopharmacology 31:318–338. 10.1038/sj.npp.1300829 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1986) The rat brain in stereotaxic coordinates. Sydney: Academic. [DOI] [PubMed] [Google Scholar]
- Rosen LG, Sun N, Rushlow W, Laviolette SR (2015) Molecular and neuronal plasticity mechanisms in the amygdala-prefrontal cortical circuit: implications for opiate addiction memory formation. Front Neurosci 9:399. 10.3389/fnins.2015.00399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen LG, Zunder J, Renard J, Fu J, Rushlow W, Laviolette SR (2016) Opiate exposure state controls a D2-CaMKIIα-dependent memory switch in the amygdala-prefrontal cortical circuit. Neuropsychopharmacology 41:847–857. 10.1038/npp.2015.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham Y, Stewart J (1994) Exposure to mild stress enhances the reinforcing efficacy of intravenous heroin self-administration in rats. Psychopharmacology 114:523–527. 10.1007/BF02249346 [DOI] [PubMed] [Google Scholar]
- Shaham Y, Erb S, Stewart J (2000) Stress-induced relapse to heroin and cocaine seeking in rats: a review. Brain Res Brain Res Rev 33:13–33. 10.1016/S0165-0173(00)00024-2 [DOI] [PubMed] [Google Scholar]
- Shao C, Li Y, Jiang K, Zhang D, Xu Y, Lin L, Wang Q, Zhao M, Jin L (2006) Dopamine D4 receptor polymorphism modulates cue-elicited heroin craving in chinese. Psychopharmacology 186:185–190. 10.1007/s00213-006-0375-6 [DOI] [PubMed] [Google Scholar]
- Sripada RK, King AP, Welsh RC, Garfinkel SN, Wang X, Sripada CS, Liberzon I (2012) Neural dysregulation in posttraumatic stress disorder: evidence for disrupted equilibrium between salience and default mode brain networks. Psychosom Med 74:904–911. 10.1097/PSY.0b013e318273bf33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Laviolette SR (2012) Inactivation of the basolateral amygdala during opiate reward learning disinhibits prelimbic cortical neurons and modulates associative memory extinction. Psychopharmacology 222:645–661. 10.1007/s00213-012-2665-5 [DOI] [PubMed] [Google Scholar]
- Sun N, Chi N, Lauzon N, Bishop S, Tan H, Laviolette SR (2011) Acquisition, extinction and recall of opiate reward memory are signalled by dynamic neuronal activity patterns in the prefrontal cortex. Cereb Cortex 21:2665–2680. 10.1093/cercor/bhr031 [DOI] [PubMed] [Google Scholar]
- Tull MT, Gratz KL, Aklin WM, Lejuez CW (2010) A preliminary examination of the relationships between posttraumatic stress symptoms and crack/cocaine, heroin, and alcohol dependence. J Anxiety Disord 24:55–62. 10.1016/j.janxdis.2009.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HT, Han F, Gao JL, Shi YX (2010) Increased phosphorylation of extracellular signal-regulated kinase in the medial prefrontal cortex of the single-prolonged stress rats. Cell Mol Neurobiol 30:437–444. 10.1007/s10571-009-9468-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Han F, Wang HT, Shi YX (2011) Single-prolonged stress induces increased phosphorylation of extracellular signal-regulated kinase in a rat model of post-traumatic stress disorder. Mol Med Rep 4:445–449. 10.3892/mmr.2011.459 [DOI] [PubMed] [Google Scholar]
- Yuen EY, Yan Z (2011) Cellular mechanisms for dopamine D4 receptor-induced homeostatic regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. J Biol Chem 286:24957–24965. 10.1074/jbc.M111.221416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Zhong P, Yan Z (2010) Homeostatic regulation of glutamatergic transmission by dopamine D4 receptors. Proc Natl Acad Sci U S A 107:22308–22313. 10.1073/pnas.1010025108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong P, Yan Z (2016) Distinct physiological effects of dopamine D4 receptors on prefrontal cortical pyramidal neurons and fast-spiking interneurons. Cereb Cortex 26:180–191. 10.1093/cercor/bhu190 [DOI] [PMC free article] [PubMed] [Google Scholar]