

Abstract
Aging is known as a major risk factor for the structure and function of the nervous system. There is urgent need to overcome such deleterious effects of age-related neurodegeneration. Here we show that peripheral nerves of 24-month-old aging C57BL/6 mice of either sex show similar pathological alterations as nerves from aging human individuals, whereas 12-month-old adult mice lack such alterations. Specifically, nerve fibers showed demyelination, remyelination and axonal lesion. Moreover, in the aging mice, neuromuscular junctions showed features typical for dying-back neuropathies, as revealed by a decline of presynaptic markers, associated with α-bungarotoxin-positive postsynapses. In line with these observations were reduced muscle strengths. These alterations were accompanied by elevated numbers of endoneurial macrophages, partially comprising the features of phagocytosing macrophages. Comparable profiles of macrophages could be identified in peripheral nerve biopsies of aging persons. To determine the pathological impact of macrophages in aging mice, we selectively targeted the cells by applying an orally administered CSF-1R specific kinase (c-FMS) inhibitor. The 6-month-lasting treatment started before development of degenerative changes at 18 months and reduced macrophage numbers in mice by ∼70%, without side effects. Strikingly, nerve structure was ameliorated and muscle strength preserved. We show, for the first time, that age-related degenerative changes in peripheral nerves are driven by macrophages. These findings may pave the way for treating degeneration in the aging peripheral nervous system by targeting macrophages, leading to reduced weakness, improved mobility, and eventually increased quality of life in the elderly.
SIGNIFICANCE STATEMENT Aging is a major risk factor for the structure and function of the nervous system. Here we show that peripheral nerves of 24-month-old aging mice show similar degenerative alterations as nerves from aging human individuals. Both in mice and humans, these alterations were accompanied by endoneurial macrophages. To determine the pathological impact of macrophages in aging mice, we selectively targeted the cells by blocking a cytokine receptor, essential for macrophage survival. The treatment strongly reduced macrophage numbers and substantially improved nerve structure and muscle strength. We show, for the first time, that age-related degenerative changes in peripheral nerves are driven by macrophages. These findings may be helpful for treatment weakness and reduced mobility in the elderly.
Keywords: aging, biopsies, degeneration, macrophage, peripheral nerve, peripheral neuropathy
Introduction
Although generally considered as rather simply organized, a closer view on the peripheral nerve reveals an unexpected complexity: Schwann cells not only directly ensheath axons throughout their length, but non-neuronal cells, including endothelial cells, pericytes, fibroblasts, and macrophages, are important cellular players in peripheral nerves, particularly during nerve perturbation and disease. For instance, macrophages are pivotal players in the lesioned nerves during Wallerian degeneration, eventually facilitating axonal regrowth after phagocytosis of myelin (Griffin et al., 1993; Stoll and Müller, 1999; Martini et al., 2008, 2013; Scheib and Höke, 2013; Klein and Martini, 2016; Martini and Willison, 2016). Moreover, upon complete nerve transection, macrophages form a cellular bridge, mediating immigration of endothelial cells, followed by Schwann cells and eventually axons (Cattin et al., 2015). While these cellular reactions are of functional advantages and contribute to nerve repair, nerve macrophages can also fulfill detrimental functions. For instance, in the Guillain-Barré syndrome, subform “acute motor axonal neuropathy”, macrophages enter myelinating nerve fibers at the nodes of Ranvier and lead to axonal damage with potentially fatal consequences if untreated (Hafer-Macko et al., 1996). Another example is inherited peripheral neuropathies of the Charcot-Marie-Tooth (CMT)-type 1. In models of CMT1X and CMT1B, mice deficient for Cx32 and MPZ (P0), respectively, we identified unique pathomechanisms implicating endoneurial fibroblasts and macrophages (Klein and Martini, 2016; Martini and Willison, 2016). In this model, mutant Schwann cells interact with endoneurial fibroblasts by still unknown signals and cause the expression of CSF-1 that activates endoneurial macrophages, which damage nerve fibers (Klein and Martini, 2016; Martini and Willison, 2016). This disease mechanism is likely also present in CMT1 patients (Groh et al., 2012). Silencing this CSF-1–CSF-1R axis consequently leads to an alleviation of the primarily genetically caused disease (Klein and Martini, 2016), possibly paving the way for immunomodulatory treatment of the yet incurable disorders. Interestingly, primary inflammatory disorders, such as Guillain-Barré syndrome, and inherited neuropathies, appear not to be the only conditions in which macrophages fulfill detrimental functions. In the present study, we find macrophages readily detectable in nerve biopsies of elderly people displaying age-related nerve alterations. Moreover, in aging mice, we identify macrophages as causally linked to aging of peripheral nerves. These findings might have clinical consequences as silencing macrophages may be a treatment option. On the other hand, a detrimental low-grade inflammation in aging nerves may be a potential risk factor for further aggravation of neuropathy due to systemic infections, but also due to metabolic insults particularly emerging in the elderly and being associated with nerve inflammation, such as diabetes mellitus (Pop-Busui et al., 2016).
Materials and Methods
Nerve biopsies.
The nerve biopsy material used for the present study was retrieved from the diagnostic archives of the Institute of Neuropathology, RWTH University Hospital. The anonymous use of this material for scientific purposes is permitted according to the rulings of the Ethical Commission of the Medical Faculty, RWTH University, and to the laws of the State of North Rhine-Westphalia without further individual ethical ruling. The biopsies had been obtained from 1 patient of 39 years of age and 4 patients from 65 to 79 years of age (genders not known) during the course of examinations to determine the cause of neuromuscular symptoms. The patients included in the present study had undergone sural nerve biopsy surgery because of mild, unspecific sensory symptoms that had led to the suspicion of neuropathy. The biopsies then showed a normal appearing nerve, and no signs of neuropathy were reported later on. To our knowledge, no genetic testing was performed on these patients. The 65-year-old patient with chronic idiopathic axonal polyneuropathy (CIAP) whose biopsy is depicted as a disease control in Figure 1F was part of a recently published study (Hube et al., 2017). Images of the nerve biopsies were acquired with an Olympus Veleta camera system mounted to a CM10 electron microscope (Philips) and were processed with GNU Image Manipulation Program and ImageJ (National Institutes of Health).
Figure 1.
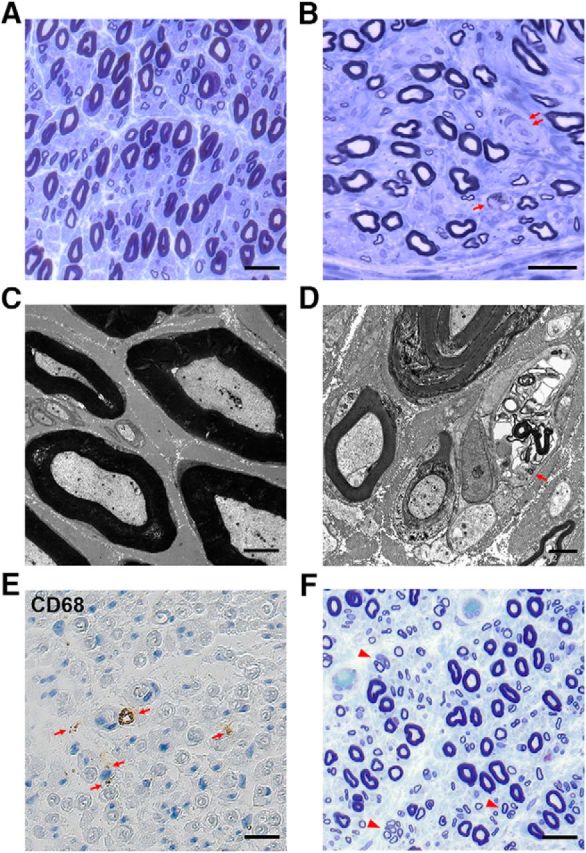
Pathological alterations in peripheral nerves of elderly humans. A, Sural nerve biopsy of a 39-year-old subject showing normal large and small myelinated nerve fibers without pathological alterations. Semithin resin section, toluidine blue. Scale bar, 40 μm. B, Myelinated large and small nerve fibers and a feature indicative of demyelination (double arrow) in a sural nerve biopsy from a 77-year-old patient. Note the presence of a macrophage (arrow). Semithin resin section, toluidine blue. Scale bar, 40 μm. C, Electron micrograph of myelinated and nonmyelinated axons of the normal nerve from the 39-year-old subject depicted in A. Scale bar, 3 μm. D, Macrophage profile (arrow) among several myelinated and unmyelinated axons in an electron micrograph of a nerve biopsy from a 70-year-old patient. Scale bar, 2 μm. E, CD68+ endoneurial macrophages (arrows) in the sural nerve biopsy of a 65-year-old patient. Paraffin section. Scale bar, 40 μm. F, Clusters of regenerated nerve fibers (arrows) in a sural nerve biopsy of a 65-year-old patient with CIAP. Semithin resin section, toluidine blue. Scale bar, 40 μm.
Mice.
All animal experiments performed in mice of either sex were in accordance with the German animal protection law and approved by the government of Lower Franconia, Germany.
Wild-type mice on a C57BL/6 background of either sex were investigated at the age of 12, 18 and 24 months. Mice were kept at the animal facility of the Department of Neurology, University Hospital of Würzburg, with a 12 h/12 h day/night rhythm (<300 lux) under barrier conditions.
CSF-1R inhibitor (PLX5622) treatment.
Rodent chow containing 300 mg/kg CSF-1R specific kinase (c-FMS) inhibitor (PLX5622) was provided by Plexxikon. Six mice were treated from 18 to 24 months and were analyzed afterward. During experiments, animals were monitored.
No animals had to be excluded from the study according to our standard criteria (Klein et al., 2015).
Dissection and tissue processing.
Tissue preparations were performed as previously reported (Klein et al., 2015). Animals were killed by asphyxiation with CO2 (according to guidelines by the State Office of Health and Social Affairs, Berlin). Blood was rinsed with PBS containing heparin. For immunohistochemistry of cross sections, femoral quadriceps nerves and flexor digitorum brevis muscles were freshly dissected, embedded in OCT medium (Sakura), and frozen in methylbutane cooled by liquid nitrogen. The harvested fresh frozen tissues were cut into 10-μm-thick cross sections on a cryostat (Leica) and stored at −20°C.
For whole-mount preparations, flexor digitorum brevis muscles were dissected from killed mice, then squeezed on a drop of 4% PFA in PBS between two glass slides and postfixed in 4% PFA as previously described (Groh et al., 2010).
For electron microscopy, killed mice were transcardially perfused with 4% PFA and 2% glutaraldehyde in 0.1 m cacodylate buffer. Dissected femoral nerves were subsequently postfixed in the same solution overnight at 4°C, followed by osmification, dehydration, and embedding in Spurr's medium. Ultrathin sections (80 nm) were mounted to copper grids and counterstained with lead citrate.
Immunohistochemistry.
Quantification of endoneurial macrophages was performed according to previously published protocols (Klein et al., 2015). Cross sections of femoral nerves were blocked with 5% BSA in 0.1 m PBS. Sections were incubated at 4°C with rat anti-mouse F4/80 primary antibody (1:300, MCAP497, Serotec) overnight. The primary antibody was omitted for negative controls. After washing with PBS, samples were incubated with Cy3-conjugated goat anti-rat IgG (1:300, 112-165-167, Dianova) secondary antibody for 1 h at room temperature. Nuclei were stained with DAPI (Sigma-Aldrich) and mounted with Aqua-Poly/Mount (Polysciences). Whole-nerve cross sections were analyzed, and the mean number of 7–10 consecutive sections per animal was calculated.
To determine muscle innervation, after blocking with 5% BSA with 0.3% Triton X-100, cross sections of the flexor digitorum brevis muscle were incubated with a primary antibody against synaptophysin (guinea-pig polyclonal, 1:500, 101004, Synaptic Systems) at 4°C overnight, followed by visualization of presynaptic terminals using Cy3-conjugated donkey anti-guinea-pig IgG secondary antibody (1:300, 706-165-148, Dianova). Postsynapses were labeled with AlexaFluor-488-conjugated α-bungarotoxin (1:300, B-13422, Invitrogen). At least 200 neuromuscular junctions (NMJs) per animal were quantified. Innervated NMJs were identified by the complete overlap of synaptophysin with α-bungarotoxin labeling. By contrast, partially or completely denervated NMJs showed postsynaptic labeling only incompletely covered with presynaptic staining or lacking presynaptic overlay, respectively. The latter two innervation patterns were collectively named as “abnormally innervated NMJs.”
To determine macrophages associated with NMJs, muscle cross sections were incubated at 4°C with rat anti-mouse F4/80 primary antibody (1:300, MCAP497, Serotec) overnight. Primary rat antibodies were detected by biotinylated goat anti-rat IgG (1:100, BA-4001, Vector Laboratories) and streptavidin-biotin-peroxidase (ABC-Kit, PK-4000, Vector Laboratories) complex using diaminobenzidine-HCl and H2O2. Afterward, presynapses and postsynapses were visualized as described above.
To visualize NMJs from whole-mount preparations, after blocking with 5% BSA with 0.3% Triton X-100, flexor digitorum brevis muscles were incubated with primary antibodies against synaptophysin (guinea-pig polyclonal, 1:500, 101004, Synaptic Systems) and βIII-tubulin (rabbit polyclonal, 1:500, AB18207, Abcam) at 4°C overnight. Primary antibodies were detected by Cy3-conjugated donkey anti-guinea-pig IgG secondary antibody (1:300, 706-165-148, Dianova) and AlexaFluor-488-conjugated goat anti-rabbit IgG secondary antibody (1:300, A11008, Invitrogen). Postsynapses were labeled with AlexaFluor-647-conjugated α-bungarotoxin (1:300, B35450, Invitrogen).
To identify macrophages, 3–4 μm paraffin sections of the formalin-fixed human sural nerve biopsy specimens were incubated for 1 h with the FLEX monoclonal antihuman CD68 antibody (1:50), followed by incubation with the EnVision FLEX+ mouse DAB visualization kit (all Dako).
Digital fluorescence microscopic images were acquired using an Axiophot 2 microscope (Carl Zeiss) equipped with a CCD camera (Visitron Systems) and afterward processed with Photoshop CS6 (Adobe) or ImageJ (National Institutes of Health).
Morphological analysis by electron microscopy of mouse peripheral nerves.
Multiple image alignments were acquired and pathological alterations were quantified in relation to the total number of axons in cross sections of the femoral quadriceps nerve (∼560 myelinated axons). Thinly myelinated axons, supernumerary Schwann cells (onion bulbs), fibers with undulated myelin, degenerating or degenerated axons, Schwann cell protrusions, regeneration clusters, periaxonal vacuoles, and myelin abnormalities were determined and then quantified. Foamy macrophages were counted, and their numbers were given as relative values per 100 axons within the nerve, which is a reliable method as axons remain nearly constant in aging mice. Fibers with undulated myelin were defined as fibers with >4 undulations on myelin. Myelin abnormalities were the sum of myelin infoldings, myelin outfoldings, and tomacular structures. Analysis was performed using a ProScan Slow Scan CCD camera mounted to a Leo 906E electron microscope (Carl Zeiss) with corresponding iTEM software (Olympus Soft Imaging Solutions).
RNA extraction and semiquantitative RT-PCR for investigating macrophage polarization.
RNA extraction and semiquantitative RT-PCR were performed as previously described (Fischer et al., 2008). After snap freezing in liquid nitrogen, femoral quadriceps nerves were homogenized (ART Labortechnik) in TRIzol reagent (Invitrogen). Total RNA of the nerves was isolated through phenol-chloroform extraction, phase separation, and subsequent precipitation. Before RNA precipitation, 20 μg of glycogen (Roche Diagnostics) was added to each sample. Precipitated RNA was solubilized in 10 μl DEPC-H2O and then incubated at 60°C for 10 min. Concentration and quality of RNA were measured subsequently with a photometer (Eppendorf).
Reverse transcription of 0.5 μg of RNA was performed in a 100 μl reaction tube with random hexamer primers and Moloney murine leukemia virus reverse transcriptase (Applied Biosystems) with the following steps and parameters: 10 min at 25°C, 60 min at 37°C, 5 min at 95°C; 1.5 μl of the product complementary DNA (cDNA) was analyzed in duplicates or triplicates by semiquantitative real-time PCR with TaqMan universal PCR master mix (Applied Biosystems) and predesigned TaqMan assays (Itgam, Mm00434455_m1; Arg1, Mm00475988_m1; Trem2, Mm00451744_m1; Adgre1, Mm00802529_m1; Nos2, Mm00440485_m1). Datasets were analyzed with the ΔΔCT method in relation to the corresponding 18s RNA content and were related to a 12 month (M) WT sample.
Neurographic recordings.
Mice were anesthetized by an intraperitoneal injection of a mixture of ketavet and xylavet (6–8 μl per g body weight), placed under a heating lamp to avoid hypothermia, and body temperature was controlled before and after measurements (34°C–36°C). Neurological properties of the left sciatic nerve were measured as described previously (Zielasek et al., 1996; Klein et al., 2015). Shortly after supramaximal stimulation of the tibial nerve at the ankle (distal) and at the sciatic notch (proximal) with monopolar needle electrodes, the compound muscle action potential amplitudes were recorded from the foot muscles using steel needle electrodes. Furthermore, distal and proximal latencies were measured together with the distance between stimulation sites, and the corresponding nerve conduction velocity was calculated. All neurographic recordings were performed on a digital Neurosoft-Evidence 3102 electromyograph (Schreiber & Tholen Medizintechnik).
Grip strength.
The strength of the hindlimbs was evaluated using an automated Grip Strength Meter (Columbus Instruments) as described previously (Groh et al., 2010). Briefly, with forelimbs supported, mice were trained to hold a grip bar properly and the maximum force (in Newtons) was measured when the mouse was pulled off the grip bar with constant strength. Ten measurements per day were performed on 3 consecutive days at approximately the same hour (before noon), and the average value of the measurements was calculated. Beforehand, investigated mice were gender- and weight-matched.
Experimental design and statistical analysis.
All experiments were performed in a blinded manner, with the investigators not aware of the age or the treatment status of the analyzed mice. Statistical analyses were performed using PASW Statistics 18 (SPSS, IBM) software. Differences among different mouse groups with normally distributed data were tested by one-way ANOVA, followed by Tukey's post hoc test or Bonferroni–Holm correction. In case of non-normally distributed datasets, the nonparametric Kruskal–Wallis test with Bonferroni–Holm correction was applied. Measurements and quantifications are shown as mean ± SD.
Results
As degenerative alterations in peripheral nerves have been reported to be an explanation for progressing weakness in the elderly (Ceballos et al., 1999; Verdu et al., 2000; Cowen et al., 2005; Leblhuber et al., 2011; Anish et al., 2015; Ward et al., 2015, 2016; Canta et al., 2016), we compared four sural nerve biopsies from individuals ranging in age from 65 to 77 years with biopsies from one 39-year-old subject (see Materials and Methods). After inspection, none of these individuals showed signs of a peripheral neuropathy. However, as opposed to the younger individual (Fig. 1A,C), we detected in the biopsies of the elderly subjects features of demyelination and axonal degeneration at the light (Fig. 1B) and electron microscopic levels, including thin myelin, incipient onion bulbs, and some few clusters of regenerating nerve fibers (Fig. 1D). In these biopsies, a considerable number of phagocytic endoneurial macrophages, often loaded with myelin debris, were present (Fig. 1B,D). Immunocytochemistry with antibodies to the lysosomal marker CD68 confirmed the presence of macrophages in the human biopsies (Fig. 1E). Compared with sural nerve biopsies of a previously published series of patients with so-called CIAP (Fig. 1F) (Hube et al., 2017), the sural nerve biopsies from the elderly subjects showed only minor signs of axonal regeneration and angiopathic changes.
In our previous studies in mice, we showed that macrophages are substantial amplifiers of hereditary peripheral neuropathies (Klein and Martini, 2016). Thus, as a next step, we aimed to study macrophages as a possible cause for pathological alterations in peripheral nerves during aging. We focused on peripheral nerves of normal wild-type mice at the age of 12 months (adult) and 24 months (aging). For histopathological analyses, we investigated the femoral quadriceps nerve. Previous studies from our laboratory in mouse models for inherited peripheral neuropathies proved its excellent suitability to analyze pathological changes quantitatively due to its easily manageable number of ∼560 (±40) myelinated axons from which 40%–50% are motor axons (Martini et al., 1995; Frei et al., 1999; Pereira et al., 2010; Klein et al., 2015; Groh et al., 2016). In 12-month-old adult mice, this nerve showed expectedly a regular organization of myelinated fibers with internodal myelin profiles displaying mostly an evenly bent, circular appearance in cross sections (Fig. 2A). In 24-month-old mice, myelin sheaths around axons showed an undulating appearance, likely due to shrinkage of axon caliber in the presence of a larger myelin sheath (Fig. 2B). Moreover, myelin aspects protruding into the axonal compartment and often appearing as isolated myelin corpuscles were frequently detectable (Fig. 2C). Most strikingly, hallmarks of a demyelinating neuropathy, as previously identified by others (Ceballos et al., 1999; Verdu et al., 2000), and similarly seen in the human biopsies (Lascelles and Thomas, 1966; Jacobs and Love, 1985), were detectable. These comprised thinly myelinated axons and the presence of supernumerary Schwann cells reminiscent of onion bulb cells (Fig. 2D). Further abnormities were related to axonal perturbation, such as periaxonal vacuoles, degenerating axons, and regeneration clusters (Fig. 2E–H). In addition to pathologically altered nerve fibers, we also detected phagocytic endoneurial macrophages often laden with myelin debris in femoral quadriceps nerves of 24-month-old mice (Fig. 2I), a feature highly reminiscent to the human nerves. Such profiles representing “foamy” macrophages were not detected in nerves of 12-month-old mice and rarely seen in 18-month-old mice (Fig. 3C). Only resident, nonactivated macrophages and endoneurial fibroblasts, both lacking a basement membrane, were detectable at this younger age (data not shown). Interestingly, the purely sensory, femoral saphenous nerve of 24-month-old mice lacked pathological alterations (Fig. 2J–L) as well as increased numbers of macrophages (data not shown), a phenomenon highly reminiscent of the so far unexplained preservation of sensory nerves in some mouse models of inherited neuropathies (Martini et al., 1995; Anzini et al., 1997; Scherer et al., 1998)
Figure 2.
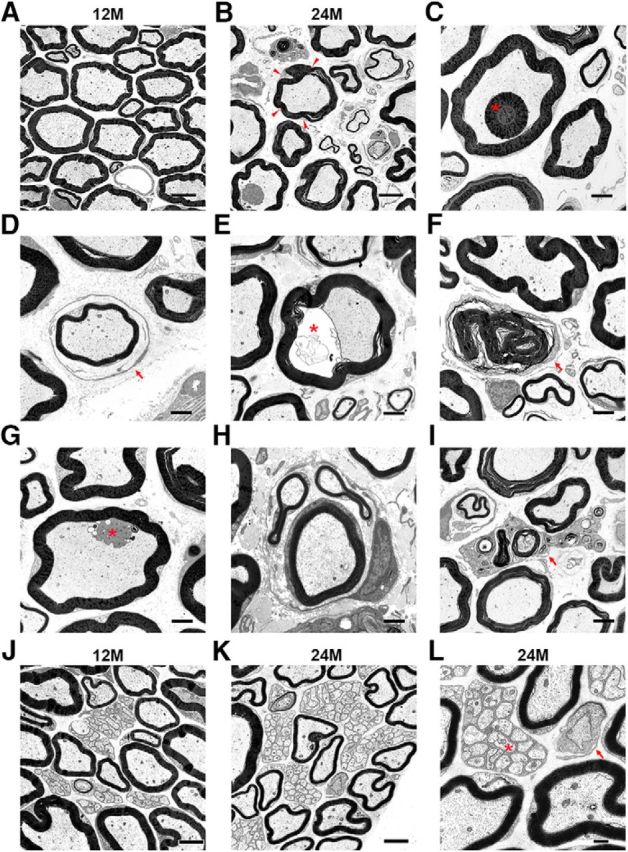
Pathological alterations in peripheral nerves of aged mice. A, B, Electron microscopy of representative ultrathin sections of femoral quadriceps nerves from 12-month-old (A) or 24-month-old (B) mice. Note the undulated myelin profiles in 24-month-old mice (arrowheads). C–I, Representative micrographs of the following: (C) a myelin inclusion (asterisk), (D) a thinly myelinated axon, surrounded by a layer of supernumerary Schwann cells (“onion bulb”; arrow), (E) a periaxonal vacuole (asterisk), (F) a Schwann cell–myelin profile devoid of an axon (arrow), (G) a Schwann cell protrusion (asterisk), (H) a regeneration cluster, and (I) a foamy macrophage (arrow). J–L, Electron microscopy of representative ultrathin sections of femoral saphenous nerves from 12-month-old (J) and 24-month-old (K,L) mice. L, Higher-magnification micrograph of K displaying a Remak bundle (asterisk) and a normal node of Ranvier (arrow). Scale bars: A, B, 5 μm; C–K, 2 μm; L, 0.5 μm.
Figure 3.

Elevated numbers of macrophages in peripheral nerves of aged mice and their reduction after CSF-1Ri treatment. A, Immunohistochemistry of F4/80+ macrophages (red) in cross sections of femoral quadriceps nerves from 12-month-old mice (12M), 18-month-old mice (18M), 24-month-old mice (24M), or 24-month-old mice treated with CSF-1Ri (24M + CSF-1Ri). Dashed circles represent the area of cross-sectioned femoral quadriceps nerves. Scale bar, 50 μm. B, Quantification of F4/80+ macrophages in quadriceps nerves from 12M, 18M, 24M, and 24M + CSF-1Ri mice. Values are mean ± SD; n = 4–7. ***p < 0.001, significance compared with 12M. ###p < 0.001, significance between 24M and 24M + CSF-1Ri mice. One-way ANOVA: 12M versus 18M, p = 0.701; 12M versus 24M, p < 0.001; 12M versus 24M + CSF-1Ri, p = 0.684; 18M versus 24M, p < 0.001; 18M versus 24M + CSF-1Ri, p = 0.998; 24M versus 24M + CSF-1Ri, p < 0.001. C, Quantification of foamy macrophages from ultrathin sections from 12M, 18M, 24M, or 24M + CSF-1Ri mice. n.d., Not detected. Values are mean ± SD; n = 4–7. *p < 0.05, significance compared with 12M. **p < 0.01, significance compared with 12M. #p < 0.05, significance among 18M, 24M, and 24M + CSF-1Ri mice. ##p < 0.01, significance among 18M, 24M, and 24M + CSF-1Ri mice. Kruskal–Wallis test: 12M versus 18M, p = 0.047; 12M versus 24M, p = 0.007; 12M versus 24M + CSF-1Ri, p = 0.019; 18M versus 24M, p = 0.008; 18M versus 24M + CSF-1Ri, p = 0.142; 24M versus 24M + CSF-1Ri, p = 0.011. D, Relative mRNA expression of M1/M2 macrophage polarization markers in femoral quadriceps nerves of 12M and 24M mice. Values are mean ± SD; n = 4. *p < 0.05, significance compared with values of the corresponding genes at 12M. **p < 0.01, significance compared with values of the corresponding genes at 12M. Student's t test: Itgam, p = 0.030; Arg1, p = 0.006; Adgre1, p = 0.002; Nos1, p = 0.935. Welch's t test: Trem2: p = 0.005.
After having shown that both in humans and mice comparable histopathological changes could be identified, including phagocytosing macrophages, we aimed to test the hypothesis that, similar to models for inherited peripheral neuropathies, endoneurial macrophages might mediate or amplify the aging-related degenerative changes in mouse peripheral nerves (Klein and Martini, 2016; Martini and Willison, 2016). As “foamy” macrophages comprise only a subpopulation of macrophages in diseased nerves, we at first quantified all macrophages in quadriceps nerves of 12-, 18-, and 24-month-old mice using the pan-macrophage marker F4/80 as a method independent from counting by electron microscopy. As expected and corroborating previous studies from our laboratory (Klein et al., 2015; Groh et al., 2016), an average of ∼5–6 F4/80+ macrophages was present/detectable in cross sections of femoral quadriceps nerves of 12- and 18-month-old mice (Fig. 3A,B). This number was nearly tripled in cross sections of femoral quadriceps nerves of 24-month-old mice (Fig. 3A,B). In addition and as opposed to 12- and 18-month-old mice, the F4/80+ macrophages in aging mice were predominantly rounded in shape, suggesting an activated phenotype. This is in line with the foamy, myelin-containing appearance of macrophages in aging mice as revealed by our electron microscopic study (see above).
In addition to macrophages within the nerve, we investigated F4/80+ macrophages associated with NMJs as the latter structures are altered during aging (see below). We found that 0.34% (±0.65%) and 1.07% (±0.95%) of the NMJ were associated with F4/80+ macrophages in 12- and 24-month-old mice, respectively.
We also profiled peripheral nerves of 12- and 24-month-old mice with regard to markers indicative of macrophage polarization. Although the proinflammatory markers, such as IFNγ and Il-12p40, were not detectable (data not shown), the low level of Nos2 (iNOS), a typical M1 marker, was not changed during aging (Fig. 3D). By contrast, Itgam (CD11b), Arg1 (Arginase 1), and Adgre1 (F4/80) were significantly, yet mildly, upregulated, potentially reflecting the increased number of macrophages. However, Trem2, another marker indicative for M2-polarization, was, compared with macrophage increase, disproportionally upregulated (Fig. 3D), suggesting that individual macrophages substantially may upregulate this molecule.
We targeted macrophages using the selective CSF-1R (c-FMS) inhibitor PLX5622. This inhibitor was applied in the chow at a concentration of 300 ppm, as previously performed in model mice for inherited neuropathies (Klein et al., 2015). To determine the optimal time point for the onset of treatment, we quantified F4/80+ macrophages in mice older than 12 but younger than 24 months. We found that, at 18 months of age, macrophage numbers were not yet elevated (6.26 ± 1.71 per section). Thus, this time point was chosen as the starting point for a 6-month-lasting treatment until 24 months of age.
CSF-1Ri treatment led to a 70% (±SD) reduction of macrophage numbers as revealed by immunohistochemical quantification of F4/80+ macrophages in femoral quadriceps nerve cross sections (Fig. 3A,B). Moreover, foamy macrophages were also significantly reduced at the electron microscopic level (Fig. 3C).
We then compared nerves from adult, aged, and CSF-1R inhibitor-treated aged mice by electron microscopy (Fig. 4A) and, on this base, quantified the pathological alterations. Interestingly, the most frequent abnormal profiles in nontreated aging mice, such as fibers associated with onion bulbs, and profiles with thin myelin sheaths were substantially reduced upon treatment (Fig. 4B,C). By contrast, abnormalities, such as fibers with undulated myelin (Fig. 4D), myelin inclusions, tomacular structures, and myelin outfoldings, were not influenced by macrophage targeting (Fig. 4I). The rarely observed Schwann cell protrusions and regeneration clusters (Fig. 4F,G) showed a nonsignificant trend toward reduction in treated aging mice, whereas periaxonal vacuoles (Fig. 4H) and the few profiles representing degenerated axons were not changed upon treatment (Fig. 4E).
Figure 4.
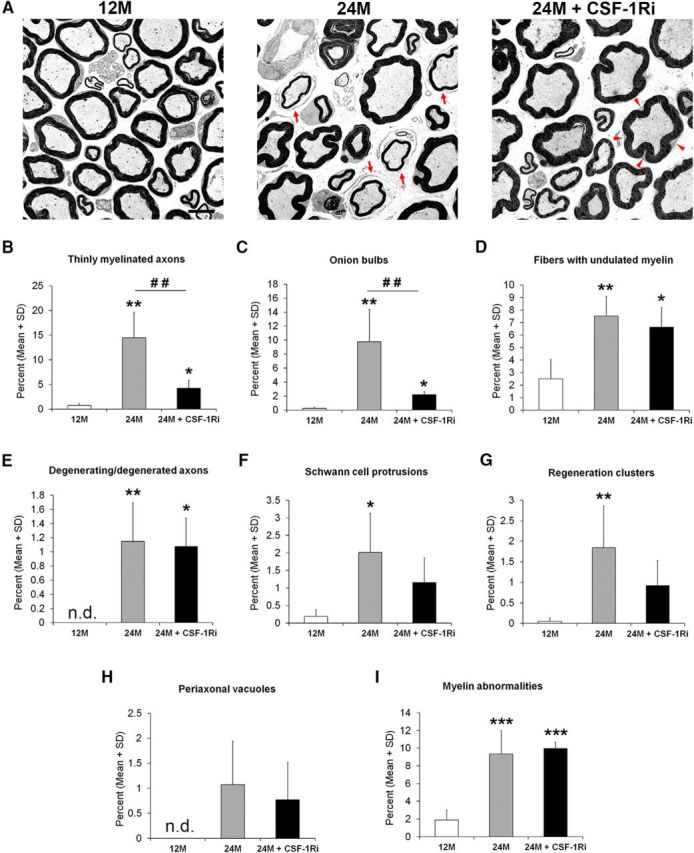
Pathological alterations characteristic for demyelination in peripheral nerves of aged mice were ameliorated after CSF-1Ri treatment. A, Electron microscopy of representative ultrathin sections of femoral quadriceps nerves from 12M, 24M, or 24M + CSF-1Ri mice. Features indicative of demyelination, as exemplified by thin myelin and onion bulbs (A, arrows, 24M), are lacking after CSF-1Ri treatment (24M + CSF-1Ri), whereas the undulating appearance of the myelin sheaths (arrowheads) is not changed in the 24-month-old treated mice. This feature is lacking in 12-month-old mice. Scale bar, 5 μm. B–I, Quantification of (B) thinly myelinated axons and (C) onion bulbs reflect a substantial alleviation of demyelination after treatment, whereas (D) fibers with undulated myelin, (E) degenerating or degenerated axons, (F) Schwann cell protrusions, (G) regeneration clusters, (H) periaxonal vacuoles, and (I) myelin abnormalities comprising myelin inclusion, myelin outfoldings, and tomacular structures are not influenced by the treatment. n.d., Not detected. Note different ranges of size (y-axis) of the different pathological features. Values are expressed as mean ± SD, n = 4–7. *p < 0.05, significance compared with 12M. **p < 0.01, significance compared with 12M. ***p < 0.001, significance compared with 12M. ##p < 0.01, significance between 24M and 24M + CSF-1Ri mice. B, Kruskal–Wallis test: 12M versus 24M, p = 0.008; 24M versus 24M + CSF-1Ri, p = 0.004; 12M versus 24M + CSF-1Ri, p = 0.014. C, Kruskal–Wallis test: 12M versus 24M, p = 0.008; 24M versus 24M + CSF-1Ri, p = 0.004; 12M versus 24M + CSF-1Ri, p = 0.014. D, One-way ANOVA: 12M versus 24M, p = 0.008; 24M versus 24M + CSF-1Ri, p = 0.777; 12M versus 24M + CSF-1Ri, p = 0.037. E, One-way ANOVA: 12M versus 24M, p = 0.008; 24M versus 24M + CSF-1Ri, p = 0.959; 12M versus 24M + CSF-1Ri, p = 0.018. F, One-way ANOVA: 12M versus 24M, p = 0.029; 24M versus 24M + CSF-1Ri, p = 0.265; 12M versus 24M + CSF-1Ri, p = 0.329. G, Kruskal–Wallis test: 12M versus 24M, p = 0.008; 24M versus 24M + CSF-1Ri, p = 0.062; 12M versus 24M + CSF-1Ri, p = 0.055. H, One-way ANOVA: 12M versus 24M, p = 0.079; 24M versus 24M + CSF-1Ri, p = 0.759; 12M versus 24M + CSF-1Ri, p = 0.280. I, One-way ANOVA: 12M versus 24M, p < 0.001; 24M versus 24M + CSF-1Ri, p = 0.850; 12M versus 24M + CSF-1Ri, p < 0.001.
We additionally investigated the functionally highly relevant NMJ in untreated and treated aging mice by immunocytochemistry. On muscle cross sections using the presynaptic and postsynaptic markers synaptophysin and α-bungarotoxin, respectively, nearly all NMJs of 12-month-old mice displayed complete colocalizations of the markers (Fig. 5A,D). In NMJs of 24-month-old mice, almost 25% of NMJ showed impaired colocalization of presynaptic and postsynaptic markers in the absence of treatment, comprising partially and totally denervated postsynapses (Fig. 5A,D). Such profiles were also detected in corresponding whole-mount preparations (Fig. 5B,C). Inclusion of a further presynaptic marker, βIII-tubulin, confirmed the presynaptic alterations. Strikingly, CSF-1R inhibitor treatment of 24-month-old mice led to a significant reduction of abnormally innervated NMJ (Fig. 5A,D), implicating both partially and completely.
Figure 5.
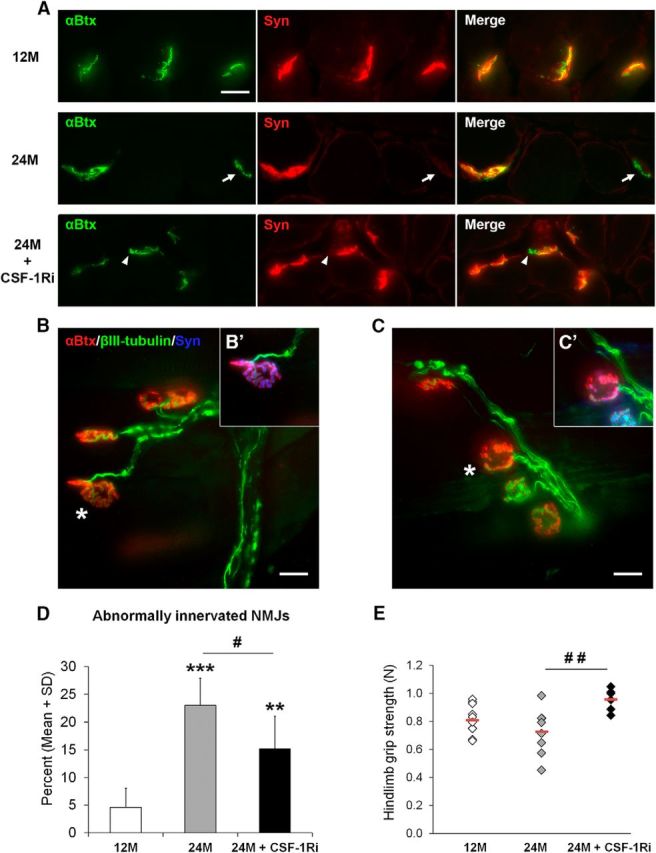
Ameliorated innervation of NMJs (A–D) and improved muscle function (E) in aged mice after CSF-1Ri treatment. A, Examples of completely innervated NMJs (top row, 12M), completely denervated NMJs (arrows, middle row, 24M), and partially denervated NMJs (arrowheads, bottom row, 24M + CSF-1Ri) as identified by double-immunolabeling with synaptophysin (red) and α-bungarotoxin (green) in cross sections of flexor digitorum brevis muscle of 12M, 24M, and 24M + CSF-1Ri mice. Right column represents merged double immunofluorescence. Scale bar, 20 μm. B, C, Examples of whole-mount preparations of flexor digitorum brevis muscles of 12M (B) and 24M (C) mice showing completely innervated and partially denervated NMJs, respectively, as identified by merged α-bungarotoxin (red) and βIII-tubulin (green) labeling. B′, C′, The same junctions as indicated in B, C (asterisks), with synaptophysin (blue) as an additional presynaptic marker. There is complete innervation in B, B′ and partial denervation in C, C′. D, Quantification of abnormally innervated NMJs from cross sections of flexor digitorum brevis muscle from 12M, 24M, or 24M + CSF-1Ri mice. Values are mean ± SD; n = 6 or 7. **p < 0.01, significance compared with 12M. ***p < 0.001, significance compared with 12M. #p < 0.05, significance between 24M and 24M + CSF-1Ri mice. ##p < 0.01, significance between 24M and 24M + CSF-1Ri mice. One-way ANOVA: 12M versus 24M, p < 0.001; 24M versus 24M + CSF-1Ri, p = 0.023; 12M versus 24M + CSF-1Ri, p = 0.003. E, Grip strength of 12M, 24M, or 24M + CSF-1Ri mice. Diamond represents one individual mouse. Red lines indicate the mean values of the corresponding group; n = 6–8. #p < 0.05, significance between 24M and 24M + CSF-1Ri mice. ##p < 0.01, significance between 24M and 24M + CSF-1Ri mice. One-way ANOVA: 12M versus 24M, p = 0.320; 24M versus 24M + CSF-1Ri, p = 0.006; 12M versus 24M + CSF-1Ri, p = 0.068.
As an outcome measure of neuromuscular integrity, we also performed grip strength analysis. In line with our observations regarding the NMJ, grip strength was reduced in nontreated aging mice, whereas it was maintained at the level of 12-month-old mice upon macrophage depletion (Fig. 5E).
We furthermore investigated the conduction properties of peripheral nerves of untreated and treated aging mice. Untreated mice showed a nonsignificant trend toward reduced motor conduction velocity compared with 12-month-old adult mice. This trend was reduced by macrophage targeting (data not shown).
Discussion
In agreement with the fact that aging is a risk factor for the structure and function of the nervous system, peripheral nerves of aging individuals show a plethora of pathological and physiological alterations (Ceballos et al., 1999; Verdu et al., 2000; Cowen et al., 2005; Leblhuber et al., 2011; Anish et al., 2015; Ward et al., 2015, 2016; Canta et al., 2016; Moldovan et al., 2016). In this study, we could discriminate two situations: (1) the most frequent structural myelin abnormalities, including foldings and undulations; and (2) features reflecting “degenerative” processes, such as remyelination and onion bulb formation. Interestingly, the latter substantially responded to macrophage depletion, leading to improved function, most impressively reflected by preserved grip strength.
Of note, neuroactive steroids, such as progesterone and its derivatives, also mitigate demyelination in aging nerves, possibly by upregulating important Schwann cell-related myelin proteins, such as P0 and PMP22 (Melcangi et al., 1998, 1999, 2000, 2003a, b; Azcoitia et al., 2003). Reciprocally, peripheral neuropathies in rats caused by pathogenic PMP22 overexpression can be mitigated by progesterone antagonists (Sereda et al., 2003). Based on our present observations in this study and because progesterone can also act as anti-inflammatory compound (Perez-Alvarez and Wandosell, 2016), it is conceivable that dampening demyelination in aging nerves by steroids could also work, at least in part, via mitigation of inflammation.
The reasons for morphological and functional alterations in aging nerves are not clear. Melcangi et al. (2003b) correlated age-related alterations in myelin sheaths with a decline of myelin proteins in aging mice. This could be the result of demyelination-causing inflammation, as we have observed in the present study. However, it is presently not clear which mechanisms let inflammation emerge in aging nerves. It is conceivable that, similar to lesioned nerves (Martini et al., 2008, 2013) or comparable with some CMT1 models (Klein and Martini, 2016; Martini and Willison, 2016), Schwann cells from aging nerves express proinflammatory cytokines. Indeed, aging Schwann cells have been identified to display a changed secretory profile, designated senescence-associated secretory phenotype (Saheb-Al-Zamani et al., 2013; Gonzalez-Freire et al., 2014), which is linked to increased IL-6 expression. As IL-6 is a potent activator of macrophages under lesion conditions (Tofaris et al., 2002) and is indeed upregulated in peripheral nerves of aging mice (X.Y., D.K., unpublished observations), IL-6 is one likely candidate molecule to initiate changes in the aging nerves. Alternatively, it is conceivable that aging axons show a dying-back neuropathy as observed in the CNS (Adalbert and Coleman, 2013). Indeed, aging axons may no longer properly interact with their associated glial cells and, by this way, may trigger a Wallerian degeneration-like neuroinflammatory reaction that may amplify the aging-induced perturbation (Martini et al., 2013). The well-established impaired axonal regeneration in aging nerves has initially been considered to be a result of reduced axonal transport (Pannese, 2011) or reduced clearance of myelin debris by Schwann cells and macrophages (Verdu et al., 2000; Scheib and Höke, 2016a, b). Along these lines, Painter et al. (2014) recently demonstrated that impaired axonal regeneration of sensory fibers in the aging PNS is most likely not related to axonal features, arguing in favor of impaired Schwann cell/macrophage interaction. An argument that macrophage-like cells can “age” is derived from observations in myelinated regions of the CNS where myelin debris “overloads” microglial cells during life span (Safaiyan et al., 2016), combined with an age-related decline of cholesterol clearance causing inflammasome stimulation and eventually maladaptive immune reactions (Cantuti-Castelvetri et al., 2018). On the other hand, aging microglia is reported to display a low-grade activation as found in models of mild CNS injury (Streit et al., 2014) and may adopt a neuroprotective phenotype (Colonna and Butovsky, 2017). Importantly, in the peripheral nerve, “aging” macrophages show an unwanted, destructive activity, reminiscent of that seen in models for inherited neuropathies (Martini et al., 2013; Klein and Martini, 2016; Martini and Willison, 2016). This pattern is associated with a disproportionally prominent upregulation of TREM-2, a molecule associated with a phagocytic, yet neuroprotective, phenotype (Colonna and Butovsky, 2017), as it is observed during Wallerian degeneration where TREM-2 has also been found to be a leading molecule indicative of M2 polarization (Ydens et al., 2012, 2013). It is not known why peripheral nerve macrophages in aging mice appear to be much more active than the microglial counterpart in the CNS. It is plausible to assume that the Wallerian degeneration-like phenotype of the peripheral nerve macrophages is related to a potential descent from “fresh” blood monocytes while replacement of burned-out parenchymal microglial cells in the CNS may be limited under aging conditions (Prinz et al., 2017; Rice et al., 2017). Further studies are necessary to understand this obvious auto-aggressive dysfunction of macrophages in the normal aging nerve.
Of note, age-dependent nerve damage could also be related to well-known vascular changes in the aging nerve that directly or indirectly activate macrophages (Leblhuber et al., 2011). Indeed, we occasionally recognized vascular changes in the aging nerve, including increased thickness of vascular epithelial walls in cross sections. However, we failed to see perivascular infiltrates and/or a gradient of pathology originating from blood vessels (Üçeyler et al., 2015), meaning lack of histopathological evidence for vasculitis. All in all, further studies are necessary to identify the mechanisms that eventually lead to the primary causes of nerve-destructive neuroinflammation mediated by macrophages.
Along with age-related alterations in the nerve, we also realized NMJ changes. Such changes in aging rodents have been extensively described previously (Valdez et al., 2010, 2012; Nishimune, 2012; Gonzalez-Freire et al., 2014; Jones et al., 2017; Rogers and Nishimune, 2017), while we show, for the first time, that these changes are at least partially mediated by inflammation. Some studies revealed that exercise appears to diminish age-related changes in NMJ (Valdez et al., 2010, 2012). Although the corresponding mechanisms are not yet completely understood, some of these exercise effects might be related to dampened inflammation via the “cholinergic anti-inflammatory pathway” (Woods et al., 2012).
To characterize the alterations in NMJ in more detail, we quantified macrophages in association with NMJ and found only 0.3% and 1% of the synapses associated with macrophages in 12- and 24-month-old mice, respectively. This rare association rate between synapses and macrophages rather argues against than in favor of synaptic macrophages being a major mediator of denervation of NMJ of aging mice, although further studies might be necessary to elucidate the role of synapse-associated macrophages. Thus, changes in the NMJ of aging mice might predominantly be the consequence of macrophage-mediated nerve damage “upstream” to the NMJ.
In conclusion, we could demonstrate that morphological and functional alterations in peripheral nerves of aging mice are mediated by a low-grade inflammation, implicating macrophages. As we saw analogous alterations in human biopsies, we assume a similar inflammatory process in nerves of aging people. This is of clinical and therapeutic interest. According to this study, age-related nerve damage is potentially treatable by immune modulation, either by CSF-1R inhibitors, as used here for showing macrophage implication, or by immune modulators developed for other neuroinflammatory diseases. However, future studies are necessary to investigate whether already ongoing age-related inflammation can be mitigated by CSF-1R inhibitors.
Based on the concept that disease-related inflammatory reactions in the nervous system are particularly susceptible to a secondary stimulus, such as systemic infections or other systemic perturbations associated with inflammation (Palin et al., 2008; Moreno et al., 2011; Perry and Holmes, 2014), the obviously inevitable inflammation in the “healthy” nerve during aging can be viewed as a risk factor for further aggravation and amplification upon additional insults. Interestingly, diabetic neuropathy, very common in the elderly, is most likely associated with inflammation (Vincent et al., 2011; Höke, 2012; Martini and Willison, 2016), and it is plausible to assume that subclinical inflammation in elderly persons may exacerbate upon development of diabetes. Reciprocally, diabetic neuropathy might be amplified when the respective patient is developing further inflammation due to aging. Further studies in aging neuropathy and inflammation may pave the way for ameliorating the reduction in life quality in a community, which is characterized by constantly increasing longevity.
Footnotes
This work was supported by German Research Foundation MA 1053/6-2 to R.M., German CMT-NET/Förderschwerpunkt des BMBF 01GM1511F, R4 to R.M. and 01GM1511D, R6, S3b to J.W. and I.K., and Plexxikon Inc. We thank Heinrich Blazyca, Silke Loserth, Bettina Meyer, E. Beck, and H. Mader for expert technical assistance; Helga Brünner, Anja Weidner, Jacqueline Schreiber, and Thomas Bimmerlein for attentive care of mice; and Dr. Janos Groh for valuable discussions.
B.L.W. is an employee of Plexxikon Inc. The remaining authors declare no competing financial interests.
References
- Adalbert R, Coleman MP (2013) Review: axon pathology in age-related neurodegenerative disorders. Neuropathol Appl Neurobiol 39:90–108. 10.1111/j.1365-2990.2012.01308.x [DOI] [PubMed] [Google Scholar]
- Anish L, Nagappa M, Mahadevan A, Taly AB (2015) Neuropathy in elderly: lessons learnt from nerve biopsy. Age Ageing 44:312–317. 10.1093/ageing/afu171 [DOI] [PubMed] [Google Scholar]
- Anzini P, Neuberg DH, Schachner M, Nelles E, Willecke K, Zielasek J, Toyka KV, Suter U, Martini R (1997) Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin 32. J Neurosci 17:4545–4551. 10.1523/JNEUROSCI.17-12-04545.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azcoitia I, Leonelli E, Magnaghi V, Veiga S, Garcia-Segura LM, Melcangi RC (2003) Progesterone and its derivatives dihydroprogesterone and tetrahydroprogesterone reduce myelin fiber morphological abnormalities and myelin fiber loss in the sciatic nerve of aged rats. Neurobiol Aging 24:853–860. 10.1016/S0197-4580(02)00234-8 [DOI] [PubMed] [Google Scholar]
- Canta A, Chiorazzi A, Carozzi VA, Meregalli C, Oggioni N, Bossi M, Rodriguez-Menendez V, Avezza F, Crippa L, Lombardi R, de Vito G, Piazza V, Cavaletti G, Marmiroli P (2016) Age-related changes in the function and structure of the peripheral sensory pathway in mice. Neurobiol Aging 45:136–148. 10.1016/j.neurobiolaging.2016.05.014 [DOI] [PubMed] [Google Scholar]
- Cantuti-Castelvetri L, Fitzner D, Bosch-Queralt M, Weil MT, Su M, Sen P, Ruhwedel T, Mitkovski M, Trendelenburg G, Lütjohann D, Möbius W, Simons M (2018) Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 359:684–688. 10.1126/science.aan4183 [DOI] [PubMed] [Google Scholar]
- Cattin AL, Burden JJ, Van Emmenis L, Mackenzie FE, Hoving JJ, Garcia Calavia N, Guo Y, McLaughlin M, Rosenberg LH, Quereda V, Jamecna D, Napoli I, Parrinello S, Enver T, Ruhrberg C, Lloyd AC (2015) Macrophage-induced blood vessels guide Schwann cell-mediated regeneration of peripheral nerves. Cell 162:1127–1139. 10.1016/j.cell.2015.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceballos D, Cuadras J, Verdú E, Navarro X (1999) Morphometric and ultrastructural changes with ageing in mouse peripheral nerve. J Anat 195:563–576. 10.1046/j.1469-7580.1999.19540563.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M, Butovsky O (2017) Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol 35:441–468. 10.1146/annurev-immunol-051116-052358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen T, Ulfhake B, King RH (2005) Aging in the peripheral nervous system. In: Peripheral neuropathy, Ed 4 (Dyck PJ, Thomas PK, eds), pp 483–507. Philadelphia, PA: Elsevier Saunders. [Google Scholar]
- Fischer S, Weishaupt A, Troppmair J, Martini R (2008) Increase of MCP-1 (CCL2) in myelin mutant Schwann cells is mediated by MEK-ERK signaling pathway. Glia 56:836–843. 10.1002/glia.20657 [DOI] [PubMed] [Google Scholar]
- Frei R, Mötzing S, Kinkelin I, Schachner M, Koltzenburg M, Martini R (1999) Loss of distal axons and sensory Merkel cells and features indicative of muscle denervation in hindlimbs of P0-deficient mice. J Neurosci 19:6058–6067. 10.1523/JNEUROSCI.19-14-06058.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Freire M, de Cabo R, Studenski SA, Ferrucci L (2014) The neuromuscular junction: aging at the crossroad between nerves and muscle. Front Aging Neurosci 6:208. 10.3389/fnagi.2014.00208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JW, George R, Ho T (1993) Macrophage systems in peripheral nerves: a review. J Neuropathol Exp Neurol 52:553–560. 10.1097/00005072-199311000-00001 [DOI] [PubMed] [Google Scholar]
- Groh J, Heinl K, Kohl B, Wessig C, Greeske J, Fischer S, Martini R (2010) Attenuation of MCP-1/CCL2 expression ameliorates neuropathy in a mouse model for Charcot-Marie-Tooth 1X. Hum Mol Genet 19:3530–3543. 10.1093/hmg/ddq269 [DOI] [PubMed] [Google Scholar]
- Groh J, Weis J, Zieger H, Stanley ER, Heuer H, Martini R (2012) Colony-stimulating factor-1 mediates macrophage-related neural damage in a model for Charcot-Marie-Tooth disease type 1X. Brain 135:88–104. 10.1093/brain/awr283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh J, Basu R, Stanley ER, Martini R (2016) Cell-surface and secreted isoforms of CSF-1 exert opposing roles in macrophage-mediated neural damage in Cx32-deficient mice. J Neurosci 36:1890–1901. 10.1523/JNEUROSCI.3427-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafer-Macko C, Hsieh ST, Li CY, Ho TW, Sheikh K, Cornblath DR, McKhann GM, Asbury AK, Griffin JW (1996) Acute motor axonal neuropathy: an antibody-mediated attack on axolemma. Ann Neurol 40:635–644. 10.1002/ana.410400414 [DOI] [PubMed] [Google Scholar]
- Höke A. (2012) Animal models of peripheral neuropathies. Neurotherapeutics 9:262–269. 10.1007/s13311-012-0116-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hube L, Dohrn MF, Karsai G, Hirshman S, Van Damme P, Schulz JB, Weis J, Hornemann T, Claeys KG (2017) Metabolic syndrome, neurotoxic 1-deoxysphingolipids and nervous tissue inflammation in chronic idiopathic axonal polyneuropathy (CIAP). PLoS One 12:e0170583. 10.1371/journal.pone.0170583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JM, Love S (1985) Qualitative and quantitative morphology of human sural nerve at different ages. Brain 108:897–924. 10.1093/brain/108.4.897 [DOI] [PubMed] [Google Scholar]
- Jones RA, Harrison C, Eaton SL, Llavero Hurtado M, Graham LC, Alkhammash L, Oladiran OA, Gale A, Lamont DJ, Simpson H, Simmen MW, Soeller C, Wishart TM, Gillingwater TH (2017) Cellular and molecular anatomy of the human neuromuscular junction. Cell Rep 21:2348–2356. 10.1016/j.celrep.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein D, Martini R (2016) Myelin and macrophages in the PNS: an intimate relationship in trauma and disease. Brain Res 1641:130–138. 10.1016/j.brainres.2015.11.033 [DOI] [PubMed] [Google Scholar]
- Klein D, Patzkó Á, Schreiber D, van Hauwermeiren A, Baier M, Groh J, West BL, Martini R (2015) Targeting the colony stimulating factor 1 receptor alleviates two forms of Charcot-Marie-Tooth disease in mice. Brain 138:3193–3205. 10.1093/brain/awv240 [DOI] [PubMed] [Google Scholar]
- Lascelles RG, Thomas PK (1966) Changes due to age in internodal length in the sural nerve in man. J Neurol Neurosurg Psychiatry 29:40–44. 10.1136/jnnp.29.1.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblhuber F, Schroecksnadel K, Beran-Praher M, Haller H, Steiner K, Fuchs D (2011) Polyneuropathy and dementia in old age: common inflammatory and vascular parameters. J Neural Transm (Vienna) 118:721–725. 10.1007/s00702-011-0579-8 [DOI] [PubMed] [Google Scholar]
- Martini R, Willison H (2016) Neuroinflammation in the peripheral nerve: cause, modulator, or bystander in peripheral neuropathies? Glia 64:475–486. 10.1002/glia.22899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R, Zielasek J, Toyka KV, Giese KP, Schachner M (1995) Protein zero (P0)-deficient mice show myelin degeneration in peripheral nerves characteristic of inherited human neuropathies. Nat Genet 11:281–286. 10.1038/ng1195-281 [DOI] [PubMed] [Google Scholar]
- Martini R, Fischer S, López-Vales R, David S (2008) Interactions between Schwann cells and macrophages in injury and inherited demyelinating disease. Glia 56:1566–1577. 10.1002/glia.20766 [DOI] [PubMed] [Google Scholar]
- Martini R, Klein D, Groh J (2013) Similarities between inherited demyelinating neuropathies and Wallerian degeneration: an old repair program may cause myelin and axon perturbation under nonlesion conditions. Am J Pathol 183:655–660. 10.1016/j.ajpath.2013.06.002 [DOI] [PubMed] [Google Scholar]
- Melcangi RC, Magnaghi V, Cavarretta I, Martini L, Piva F (1998) Age-induced decrease of glycoprotein Po and myelin basic protein gene expression in the rat sciatic nerve. repair by steroid derivatives. Neuroscience 85:569–578. 10.1016/S0306-4522(97)00628-3 [DOI] [PubMed] [Google Scholar]
- Melcangi RC, Magnaghi V, Cavarretta I, Zucchi I, Bovolin P, D'Urso D, Martini L (1999) Progesterone derivatives are able to influence peripheral myelin protein 22 and P0 gene expression: possible mechanisms of action. J Neurosci Res 56:349–357. [DOI] [PubMed] [Google Scholar]
- Melcangi RC, Magnaghi V, Galbiati M, Ghelarducci B, Sebastiani L, Martini L (2000) The action of steroid hormones on peripheral myelin proteins: a possible new tool for rebuilding of myelin? J Neurocytol 29:327–339. 10.1023/A:1007105121765 [DOI] [PubMed] [Google Scholar]
- Melcangi RC, Ballabio M, Cavarretta I, Gonzalez LC, Leonelli E, Veiga S, Martini L, Magnaghi V (2003a) Effects of neuroactive steroids on myelin of peripheral nervous system. J Steroid Biochem Mol Biol 85:323–327. 10.1016/S0960-0760(03)00228-0 [DOI] [PubMed] [Google Scholar]
- Melcangi RC, Azcoitia I, Ballabio M, Cavarretta I, Gonzalez LC, Leonelli E, Magnaghi V, Veiga S, Garcia-Segura LM (2003b) Neuroactive steroids influence peripheral myelination: a promising opportunity for preventing or treating age-dependent dysfunctions of peripheral nerves. Prog Neurobiol 71:57–66. 10.1016/j.pneurobio.2003.09.003 [DOI] [PubMed] [Google Scholar]
- Moldovan M, Rosberg MR, Alvarez S, Klein D, Martini R, Krarup C (2016) Aging-associated changes in motor axon voltage-gated Na(+) channel function in mice. Neurobiol Aging 39:128–139. 10.1016/j.neurobiolaging.2015.12.005 [DOI] [PubMed] [Google Scholar]
- Moreno B, Jukes JP, Vergara-Irigaray N, Errea O, Villoslada P, Perry VH, Newman TA (2011) Systemic inflammation induces axon injury during brain inflammation. Ann Neurol 70:932–942. 10.1002/ana.22550 [DOI] [PubMed] [Google Scholar]
- Nishimune H. (2012) Active zones of mammalian neuromuscular junctions: formation, density, and aging. Ann N Y Acad Sci 1274:24–32. 10.1111/j.1749-6632.2012.06836.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter MW, Brosius Lutz A, Cheng YC, Latremoliere A, Duong K, Miller CM, Posada S, Cobos EJ, Zhang AX, Wagers AJ, Havton LA, Barres B, Omura T, Woolf CJ (2014) Diminished Schwann cell repair responses underlie age-associated impaired axonal regeneration. Neuron 83:331–343. 10.1016/j.neuron.2014.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palin K, Cunningham C, Forse P, Perry VH, Platt N (2008) Systemic inflammation switches the inflammatory cytokine profile in CNS Wallerian degeneration. Neurobiol Dis 30:19–29. 10.1016/j.nbd.2007.11.012 [DOI] [PubMed] [Google Scholar]
- Pannese E. (2011) Morphological changes in nerve cells during normal aging. Brain Struct Funct 216:85–89. 10.1007/s00429-011-0308-y [DOI] [PubMed] [Google Scholar]
- Pereira JA, Baumann R, Norrmén C, Somandin C, Miehe M, Jacob C, Lühmann T, Hall-Bozic H, Mantei N, Meijer D, Suter U (2010) Dicer in Schwann cells is required for myelination and axonal integrity. J Neurosci 30:6763–6775. 10.1523/JNEUROSCI.0801-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Alvarez MJ, Wandosell F (2016) Stroke and neuroinflamation: role of sexual hormones. Curr Pharm Des 22:1334–1349. 10.2174/138161282210160304112834 [DOI] [PubMed] [Google Scholar]
- Perry VH, Holmes C (2014) Microglial priming in neurodegenerative disease. Nat Rev Neurol 10:217–224. 10.1038/nrneurol.2014.38 [DOI] [PubMed] [Google Scholar]
- Pop-Busui R, Ang L, Holmes C, Gallagher K, Feldman EL (2016) Inflammation as a therapeutic target for diabetic neuropathies. Curr Diab Rep 16:29. 10.1007/s11892-016-0727-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Erny D, Hagemeyer N (2017) Ontogeny and homeostasis of CNS myeloid cells. Nat Immunol 18:385–392. 10.1038/ni.3703 [DOI] [PubMed] [Google Scholar]
- Rice RA, Pham J, Lee RJ, Najafi AR, West BL, Green KN (2017) Microglial repopulation resolves inflammation and promotes brain recovery after injury. Glia 65:931–944. 10.1002/glia.23135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RS, Nishimune H (2017) The role of laminins in the organization and function of neuromuscular junctions. Matrix Biol 57:86–105. 10.1016/j.matbio.2016.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safaiyan S, Kannaiyan N, Snaidero N, Brioschi S, Biber K, Yona S, Edinger AL, Jung S, Rossner MJ, Simons M (2016) Age-related myelin degradation burdens the clearance function of microglia during aging. Nat Neurosci 19:995–998. 10.1038/nn.4325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saheb-Al-Zamani M, Yan Y, Farber SJ, Hunter DA, Newton P, Wood MD, Stewart SA, Johnson PJ, Mackinnon SE (2013) Limited regeneration in long acellular nerve allografts is associated with increased Schwann cell senescence. Exp Neurol 247:165–177. 10.1016/j.expneurol.2013.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheib J, Höke A (2013) Advances in peripheral nerve regeneration. Nat Rev Neurol 9:668–676. 10.1038/nrneurol.2013.227 [DOI] [PubMed] [Google Scholar]
- Scheib J, Höke A (2016a) Impaired regeneration in aged nerves: clearing out the old to make way for the new. Exp Neurol 284:79–83. 10.1016/j.expneurol.2016.07.010 [DOI] [PubMed] [Google Scholar]
- Scheib JL, Höke A (2016b) An attenuated immune response by Schwann cells and macrophages inhibits nerve regeneration in aged rats. Neurobiol Aging 45:1–9. 10.1016/j.neurobiolaging.2016.05.004 [DOI] [PubMed] [Google Scholar]
- Scherer SS, Xu YT, Nelles E, Fischbeck K, Willecke K, Bone LJ (1998) Connexin32-null mice develop demyelinating peripheral neuropathy. Glia 24:8–20. [DOI] [PubMed] [Google Scholar]
- Sereda MW, Meyer zu Hörste G, Suter U, Uzma N, Nave KA (2003) Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat Med 9:1533–1537. 10.1038/nm957 [DOI] [PubMed] [Google Scholar]
- Stoll G, Müller HW (1999) Nerve injury, axonal degeneration and neural regeneration: basic insights. Brain Pathol 9:313–325. 10.1111/j.1750-3639.1999.tb00229.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Xue QS, Tischer J, Bechmann I (2014) Microglial pathology. Acta Neuropathol Commun 2:142. 10.1186/s40478-014-0142-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofaris GK, Patterson PH, Jessen KR, Mirsky R (2002) Denervated Schwann cells attract macrophages by secretion of leukemia inhibitory factor (LIF) and monocyte chemoattractant protein-1 in a process regulated by interleukin-6 and LIF. J Neurosci 22:6696–6703. 10.1523/JNEUROSCI.22-15-06696.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Üçeyler N, Geng A, Reiners K, Toyka KV, Sommer C (2015) Non-systemic vasculitic neuropathy: single-center follow-up of 60 patients. J Neurol 262:2092–2100. 10.1007/s00415-015-7813-5 [DOI] [PubMed] [Google Scholar]
- Valdez G, Tapia JC, Kang H, Clemenson GD Jr, Gage FH, Lichtman JW, Sanes JR (2010) Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc Natl Acad Sci U S A 107:14863–14868. 10.1073/pnas.1002220107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez G, Tapia JC, Lichtman JW, Fox MA, Sanes JR (2012) Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLoS One 7:e34640. 10.1371/journal.pone.0034640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdu E, Ceballos D, Vilches JJ, Navarro X (2000) Influence of aging on peripheral nerve function and regeneration. J Periph Nerv Syst 5:191–208. 10.1046/j.1529-8027.2000.00026.x [DOI] [PubMed] [Google Scholar]
- Vincent AM, Callaghan BC, Smith AL, Feldman EL (2011) Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat Rev Neurol 7:573–583. 10.1038/nrneurol.2011.137 [DOI] [PubMed] [Google Scholar]
- Ward RE, Boudreau RM, Caserotti P, Harris TB, Zivkovic S, Goodpaster BH, Satterfield S, Kritchevsky S, Schwartz AV, Vinik AI, Cauley JA, Newman AB, Strotmeyer ES (2015) Sensory and motor peripheral nerve function and longitudinal changes in quadriceps strength. J Gerontol A Biol Sci Med Sci 70:464–470. 10.1093/gerona/glu183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward RE, Caserotti P, Cauley JA, Boudreau RM, Goodpaster BH, Vinik AI, Newman AB, Strotmeyer ES (2016) Mobility-related consequences of reduced lower-extremity peripheral nerve function with age: a systematic review. Aging Dis 7:466–478. 10.14336/AD.2015.1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods JA, Wilund KR, Martin SA, Kistler BM (2012) Exercise, inflammation and aging. Aging Dis 3:130–140. [PMC free article] [PubMed] [Google Scholar]
- Ydens E, Cauwels A, Asselbergh B, Goethals S, Peeraer L, Lornet G, Almeida-Souza L, Van Ginderachter JA, Timmerman V, Janssens S (2012) Acute injury in the peripheral nervous system triggers an alternative macrophage response. J Neuroinflammation 9:176. 10.1186/1742-2094-9-176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ydens E, Lornet G, Smits V, Goethals S, Timmerman V, Janssens S (2013) The neuroinflammatory role of Schwann cells in disease. Neurobiol Dis 55:95–103. 10.1016/j.nbd.2013.03.005 [DOI] [PubMed] [Google Scholar]
- Zielasek J, Martini R, Toyka KV (1996) Functional abnormalities in P0-deficient mice resemble human hereditary neuropathies linked to P0 gene mutations. Muscle Nerve 19:946–952. [DOI] [PubMed] [Google Scholar]