

Abstract
The onset of the headache phase during attacks of migraine with aura, which occur in ∼30% of migraineurs, is believed to involve cortical spreading depression (CSD) and the ensuing activation and sensitization of primary afferent neurons that innervate the intracranial meninges, and their related large vessels. The mechanism by which CSD enhances the activity and mechanosensitivity of meningeal afferents remains poorly understood, but may involve cortical metabolic perturbations. We used extracellular single-unit recording of meningeal afferent activity and monitored changes in cortical blood flow and tissue partial pressure of oxygen (tpO2) in anesthetized male rats to test whether the prolonged cortical hypoperfusion and reduction in tissue oxygenation that occur in the wake of CSD contribute to meningeal nociception. Suppression of CSD-evoked cortical hypoperfusion with the cyclooxygenase inhibitor naproxen blocked the reduction in cortical tpO2, but had no effect on the activation of meningeal afferents. Naproxen, however, distinctly prevented CSD-induced afferent mechanical sensitization. Counteracting the CSD-evoked persistent hypoperfusion and reduced tpO2 by preemptively increasing cortical blood flow using the ATP-sensitive potassium [K(ATP)] channel opener levcromakalim did not inhibit the sensitization of meningeal afferents, but prevented their activation. Our data show that the cortical hypoperfusion and reduction in tpO2 that occur in the wake of CSD can be dissociated from the activation and mechanical sensitization of meningeal afferent responses, suggesting that the metabolic changes do not contribute directly to these neuronal nociceptive responses.
SIGNIFICANCE STATEMENT Cortical spreading depression (CSD)-evoked activation and mechanical sensitization of meningeal afferents is thought to mediate the headache phase in migraine with aura. We report that blocking the CSD-evoked cortical hypoperfusion and reduced tissue partial pressure of oxygen by cyclooxygenase inhibition is associated with the inhibition of the afferent sensitization, but not their activation. Normalization of these CSD-evoked metabolic perturbations by activating K(ATP) channels is, however, associated with the inhibition of afferent activation but not sensitization. These results question the contribution of cortical metabolic perturbations to the triggering mechanism underlying meningeal nociception and the ensuing headache in migraine with aura, further point to distinct mechanisms underlying the activation and sensitization of meningeal afferents in migraine, and highlight the need to target both processes for an effective migraine therapy.
Keywords: cortical spreading depression, hypoperfusion, hypoxia, migraine, primary afferent, trigeminal
Introduction
Migraine is a multifactorial, episodic neurological disorder and the second leading cause of disability worldwide (Vos et al., 2017). It is now well accepted that the onset of migraine headache requires the activation and increased mechanosensitivity of trigeminal afferent neurons that innervate the intracranial meninges and their related large blood vessels (Strassman et al., 1996; Messlinger, 2009; Olesen et al., 2009; Levy, 2012). However, the endogenous processes underlying these afferent responses during a migraine attack are incompletely understood.
Migraine with aura, the second-most prevalent migraine subtype, is thought to involve cortical spreading depression (CSD), an abnormal wave of neuronal and glial activity (Ayata and Lauritzen, 2015). CSD has been suggested to mediate the aura phase (Pietrobon and Moskowitz, 2013), and also contributes to the onset of the headache phase, by promoting prolonged activation and increased mechanosensitivity of meningeal afferents (Zhang et al., 2010; Zhao and Levy, 2015, 2016) and the ensuing activation and sensitization of central trigeminal dorsal horn neurons that receive their input (Zhang et al., 2011b; Melo-Carrillo et al., 2017). The precise mechanisms by which CSD promotes the activation and sensitization of meningeal afferents are unclear, although dural neurogenic inflammation (Bolay et al., 2002; but see Zhao and Levy, 2017) and Pannexin-1-related signaling (Karatas et al., 2013) have been hypothesized.
Migraine with aura is associated with cortical hemodynamic changes, in particular a sustained reduction in cortical blood flow (CBF) contemporaneous with the onset of the headache phase (Olesen et al., 1981, 1982; Lauritzen, 1984; Hadjikhani et al., 2001; Hansen and Schankin, 2017). In animal studies, CSD has been shown to promote a similar prolonged cortical hypoperfusion (Fabricius and Lauritzen, 1993; Fabricius et al., 1995; Piilgaard and Lauritzen, 2009). CSD also leads to transient cortical hypoxia followed by prolonged and milder reduction in cortical tissue partial pressure of oxygen (tpO2) concomitant with the cortical hypoperfusion phase (Takano et al., 2007; Piilgaard and Lauritzen, 2009).
Reduced tissue oxygenation during acute exposure to high altitude can promote headache and trigger a migraine attack (Appenzeller, 1994; Broessner et al., 2016). Normobaric hypoxia has also been shown to trigger migraine in susceptible individuals (Schoonman et al., 2006; Arngrim et al., 2016). These clinical observations together with the notion that reduced blood flow and tissue oxygenation can lead to enhanced activity and responsiveness of primary afferent nociceptors that innervate the cornea, viscera, muscle, and skin (Haupt et al., 1983; Mense and Stahnke, 1983; Longhurst et al., 1991; MacIver and Tanelian, 1992; Hillery et al., 2011) prompted us to examine whether the cortical hypoperfusion and associated reduction in tpO2 that occur following CSD might contribute to the prolonged activation and sensitization of meningeal afferents. Recent work has shown that the inhibition of cortical cyclooxygenase abrogates CSD-evoked cortical hypoperfusion (Gariepy et al., 2017). Here, we investigated whether cyclooxygenase inhibition might also ameliorate the CSD-evoked decreases in tpO2 and interfere with the associated meningeal afferent responses. Having identified such effects, we further tested whether a preemptive increase in CBF to counteract the CSD-evoked hypoperfusion and reduced tpO2, using the ATP-sensitive potassium [K(ATP)] channel opener levcromakalim (Kleppisch and Nelson, 1995; Shin et al., 2003) might also inhibit the enhanced responses of meningeal afferents. Our data suggest that while cyclooxygenase inhibition and K(ATP) channel opening can counteract the CSD-related cortical hypoperfusion and reduced tpO2, they exert distinct inhibitory effects on the associated activation and sensitization of meningeal afferents, thus questioning the direct contribution of these cortical metabolic perturbations to meningeal nociception and the onset of headache in migraine with aura.
Materials and Methods
Animals, anesthesia, and surgical preparation.
The current study used Sprague Dawley rats (males; 250–350 g; Taconic). Rats were housed 3 per cage under a 12 h light/dark cycle, in a temperature- and humidity-controlled room in the animal care facility of the Beth Israel Deaconess Medical Center (Boston, MA). Food and water were available ad libitum. All experimental procedures followed the Guide for Care and Use of Laboratory Animal Resources (NIH publication No. 85–23, revised 1996), were approved by the Animal Care and Use Committee of the Beth Israel Deaconess Medical Center, and were in compliance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines. Animals were deeply anesthetized with urethane (1.5 g/kg, i.p.) and mounted on a stereotaxic frame (Kopf Instruments). Core temperature was kept at 37.5–38°C using a homoeothermic control system. Animals were intubated and breathed spontaneously room air enriched with O2. Physiological parameters were collected throughout the experiments using PhysioSuite (Kent Scientific) and CapStar-100 (CWE). Data used in this report were obtained from animals exhibiting physiological levels of oxygen saturation (>95%), heart rate (350–450 beats/min), and end-tidal CO2 (3.5–4.5%). A saline-cooled dental drill was used to perform three separate craniotomies (Fig. 1A). One craniotomy was used to expose the left transverse sinus and the posterior part of the superior sagittal sinus, as well as the adjacent cranial dura, extending ∼2 mm rostral to the transverse sinus. Another small craniotomy (1 × 1 mm) was made in the calvaria over the right hemisphere, and was centered 2 mm caudal and 2 mm lateral to bregma to allow insertion of the recording electrode. An additional small burr hole (diameter, 0.5 mm) was drilled to expose a small area of the frontal cortex to induce CSD (Zhao and Levy, 2015, 2016). The exposed dura was bathed with a modified synthetic interstitial fluid containing 135 mm NaCl, 5 mm KCl, 1 mm MgCl2, 5 mm CaCl2, 10 mm glucose, and 10 mm HEPES, pH 7.2. The femoral vein was cannulated to allow for intravenous administration.
Figure 1.

A, Experimental setup: three skull openings (red ovals) were made. A small burr hole was made over the left frontal cortex to elicit a single CSD event using a pinprick (PP) stimulation. Meningeal afferent activity was recorded in the left trigeminal ganglion (TG) using a tungsten microelectrode inserted through a craniotomy made over the contralateral hemisphere. An ipsilateral craniotomy was made to expose a part of the left transverse sinus (TS) and superior sagittal sinus (SSS) and their vicinity to search for mechanosensitive meningeal afferents. Quantitative mechanical stimuli were delivered to the receptive field of afferents using a feedback-controlled mechanical stimulator. A Laser Doppler Flowmeter (LDF) probe was placed over the cortex near the receptive field of afferents to record changes in CBF and validate the induction of CSD noninvasively. In some animals, we also recorded the induction of CSD by monitoring multiunit activity (MUA) of cortical neurons. An oxygen microelectrode was placed in the superficial cortex to record CSD-evoked changes in cortical tpO2. B, Example of raw data recording depicting the activation of a C afferent meningeal afferent (with an overdrawn spike waveform used for data analysis) following the elicitation of CSD with a PP during the prolonged reduction in CBF and tpO2. The afferent recording trace represents a peristimulus time histogram (1 s bin-size). Average ongoing activity rate (spikes/s) are denoted in parentheses. The activation phase and associated cortical hypoperfusion and reduced tpO2 are colored red. LDF values are in arbitrary units (AU). stim, Stimulus.
Recording of meningeal afferent activity.
Single-unit activity of meningeal afferents (1 afferent/rat) was recorded from their cell body in the ipsilateral (left) trigeminal ganglion using a contralateral approach, as described previously (Zhao and Levy, 2014, 2015, 2016; Fig. 1A) in which a platinum-coated tungsten microelectrode (impedance, 50–100 kΩ; FHC) is advanced through the right hemisphere into the left trigeminal ganglion using an angled (22.5°) trajectory. The insertion of the recording electrode using this approach does not produce CSD in the left cortical hemisphere (Zhao and Levy, 2015, 2016). Meningeal afferents were identified by their constant latency response to electrical stimuli applied to the dura above the ipsilateral transverse sinus (0.5 ms pulse, 5 mA, 0.5 Hz). The response latency was used to calculate conduction velocity (CV), based on a conduction distance to the trigeminal ganglion of 12.5 mm (Strassman et al., 1996). Neurons were classified as A-δ (1.5 ≤ CV ≤ 5 m/s) or C afferents (CV < 1.5 m/s). Neural activity was digitized and sampled at 10 KHz using power 1401/Spike 2 interface (CED). A real-time waveform discriminator (Spike 2 software, CED) was used to create and store a template for the action potential evoked by electrical stimulation, which was used to acquire and analyze afferent activity.
Assessment of changes in meningeal afferent mechanosensitivity.
Mechanical receptive fields of meningeal afferents were first identified by probing the exposed dura with a 2.0 g von Frey filament (Stoelting). The site of lowest mechanical threshold was further determined using monofilaments that exert lower forces (≥0.02 g). For quantitative determination of mechanical responsiveness, we applied mechanical stimuli to the site with the lowest threshold, using a servo force-controlled mechanical stimulator (Series 300B Dual Mode Servo System, Aurora Scientific), controlled via the power 1401 interface. The stimulus was delivered using a flat-ended cylindrical plastic probe that was attached to the tip of the stimulator arm. One of three probe diameters (0.5, 0.8, or 1.1 mm) was selected for each neuron, depending on the sensitivity of the neuron (Levy and Strassman, 2002). Stimulus trials for testing the mechanosensitivity of the afferent were made using a custom-written script for Spike 2 and consisted of “ramp and hold” stimuli (rise time, 100 ms; stimulus width, 2 s; interstimulus interval, 120 s). Each trial included a threshold stimulus (which normally evoked at baseline 1–3 Hz responses) followed by a suprathreshold stimulus (usually X2 of the threshold pressure). To minimize response desensitization, stimulus trials were delivered every 15 min throughout the experiment (Zhang et al., 2013; Zhao and Levy, 2016). Ongoing afferent discharge was recorded continuously between the stimulation trials. Responses to mechanical stimuli were determined during at least four consecutive trials before the elicitation of CSD. Data were analyzed only from afferents that exhibited consistent responses (variation of <0.5 Hz for threshold responses and <1.5 Hz for suprathreshold responses) during baseline recordings (Zhao and Levy, 2016).
Induction of CSD.
A single CSD episode was induced by quickly inserting a glass micropipette (diameter, 50 μm) ∼2 mm deep in the cortex for 2 s (Zhang et al., 2010). CSD was triggered in the left frontal cortex to avoid potential damage to the meningeal tissue near the tested receptive field of the afferent under study, which could have led to their activation and/or sensitization (Zhao and Levy, 2015).
Measurement of CSD-evoked changes in CBF using laser Doppler flowmetry.
Changes in CBF (Gariepy et al., 2017) were documented using a needle probe (diameter, 0.8 mm) connected to a laser Doppler flowmeter (Vasamedic). The probe was positioned just above the intact dura, ∼1 mm from the mechanical receptive field of the recorded afferent in an area devoid of large pial blood vessels (>100 μm). Such a system (using a near-infrared 785 nm probe and fiber separation of 0.25 mm) is expected to record blood flow changes at a cortical depth of ∼1 mm (Fabricius et al., 1997). The laser Doppler signal was digitized (100 Hz) and recorded using the power 1401/Spike 2. Laser Doppler recordings were obtained with the ambient lights turned off. Baseline data were based on recordings conducted for at least 30 min before CSD.
Measurement of cortical partial pressure of oxygen.
Cortical tpO2 was recorded using a modified Clark-type polarographic oxygen microelectrode (OX-10, Unisense) with a guard cathode. The oxygen electrode was connected to a high-impedance pico-amperometer (PA2000; Unisense) that sensed the currents of the oxygen electrodes. The tpO2 signal was digitized (100 Hz) and recorded using the Power 1401/Spike 2 interface. The microelectrode responded linearly to changes in oxygen concentration and was calibrated before and after each experiment in both air-saturated saline and in an oxygen-free solution consisting of 0.1 m of sodium-ascorbate and 0.1 m NaOH dissolved in saline. This approach has been used previously to record changes in cortical tpO2 in response to CSD (Takano et al., 2007; Piilgaard and Lauritzen, 2009; Thrane et al., 2013). Changes in tpO2 values were recorded at a cortical depth of 100–200 μm, with the electrode placed <0.5 mm away from the laser Doppler flowmetry (LDF) probe. Baseline tpO2 values were recorded for at least 30 min before CSD.
Electrophysiological recording of CSD.
A tungsten microelectrode (impedance, 1–2Ω; FHC) was used to record multiunit activity at a depth of 200–400 μm in the left visual cortex (V1). A silver ground electrode was placed in the neck muscle. Signal was amplified and filtered (20K x, 300 Hz low frequency; 10 KHz high frequency; DAM-80/BMA400, WPI/CWE), and then digitized and sampled at 10 KHz. Cortical activity was recorded simultaneously with CBF changes, with the recording electrode placed <0.5 mm away from the LDF probe.
Drugs.
Naproxen sodium and levcromakalim were purchased from Tocris Bioscience. Naproxen sodium was made in 0.9% immediately before administration (10 mg/kg, i.v). A levcromakalim stock solution (10 mm) was made in DMSO and further diluted with synthetic interstitial fluid (final concentration, 500 μm) immediately before its application. The concentration of levcromakalim expected to be attained at the superficial cortex was estimated to be 100–1000 times lower (Moore et al., 1982; Roth et al., 2014; Zhao et al., 2017).
Data analyses and statistics.
All data were analyzed and plotted using Prism 7 software and are presented as median (IQR). Group data in graphs show all data points and are plotted as box-and-whisker plots with minimum and maximum values. For CSD-related changes in CBF and tpO2, raw data were processed with a custom-written script made for Spike 2. Baseline values were based on the average of the values collected during the 600 s before the induction of CSD. Off-line analyses for afferent responses were conducted using template matching in Spike 2. Criteria used to consider meningeal afferent activation and sensitization responses were based on our previous studies (Zhao and Levy, 2015, 2016). In brief, prolonged increase in the ongoing activity of afferents was considered if the firing rate increased above the upper end point of the 95% CI calculated for the baseline mean for >10 min. Acute afferent activation during the CSD wave was considered as an increased ongoing discharge rate that occurred ≥30 s following the pinprick and lasted ≤120 s. Mechanical sensitization was considered only if the afferent responses changed according to the following criteria: threshold and/or suprathreshold responses increased to a level greater than the upper end point of the 95% CI calculated for the baseline mean; increased responsiveness began during the first 60 min post-CSD; and sensitization lasted for at least 30 min. Changes in CSD-evoked responses of meningeal afferents in the presence of naproxen or levcromakalim were compared with data obtained in control animals using the same surgical approach and CSD induction method. Control data were obtained from experiments in animals treated intravenously with the naproxen vehicle (0.9% saline, 60 min before CSD induction, n = 24), or locally (i.e., continuous dural bathing) with synthetic interstitial fluid (n = 36). The two datasets were comparable and therefore combined to increase power. Differences in CSD-evoked afferent activation and sensitization propensities were analyzed using two-tailed χ2 tests. Within-group and between-group comparisons were made using the Wilcoxon test and Mann–Whitney U test, respectively. Results were considered to be significant at p < 0.05.
Results
CSD-evoked prolonged activation of meningeal afferents coincides with cortical hypoperfusion and reduced tpO2
Protracted cortical hypoperfusion and reduced cortical tpO2 were observed previously (Piilgaard and Lauritzen, 2009; Fordsmann et al., 2013) at time points corresponding to the prolonged activation of meningeal afferents (Zhang et al., 2010; Zhao and Levy, 2015). To examine whether the CSD-evoked cortical metabolic perturbations indeed coincide with prolonged meningeal afferent activation, we used simultaneous recording of meningeal afferent activity with CBF and tpO2 (n = 6). As the example in Figure 1B demonstrates, persistent meningeal afferent activation could be observed during the prolonged post-CSD cortical hypoperfusion and reduced tpO2 stage.
Suppression of CSD-evoked cortical hypoperfusion and decreased tpO2 by naproxen is not associated with reduced activation of meningeal afferents
We initially verified that systemic administration of naproxen, which was used to ensure cortical COX (cyclooxygenase) inhibition also under the nerve endings of meningeal afferents branches localized to areas not exposed by the craniotomy, could also block the CSD-evoked cortical hypoperfusion, as we previously have shown using topical administration (Gariepy et al., 2017). Because cortical oxygenation level depends in large part on cortical perfusion (Offenhauser et al., 2005), we further examined whether blocking the CSD-evoked cortical hypoperfusion with naproxen might inhibit the reduction in cortical tpO2. When compared with control animals (n = 22), naproxen treatment (n = 22) augmented the CSD-related acute cortical hyperemic response and blocked, as expected, the protracted cortical hypoperfusion (Fig. 2). Using simultaneous recording of CBF and tpO2, we observed that the inhibitory effect of naproxen on the CSD-evoked cortical hypoperfusion was associated with altered tpO2 responses to CSD (Fig. 3). In the presence of naproxen (n = 6), CSD evoked an initial increase in tpO2 [Δ10.03 mmHg (5.22); W = 6.0; p = 0.03; Fig. 3B, phase 1 and arrowhead in the insert] that was smaller than in vehicle-control animals [n = 11; Δ14.18 mmHg (3.87); U = 7.0; p = 0.007; Fig. 3C]. While in control animals the hyperoxia phase was followed by acute hypoxia [bottom values: 17.66 mmHg (18.64); W = −66.0; p = 0.001], which lasted 110.0 s (70.30; Fig. 3A; phase 2), naproxen blunted this hypoxic response, and instead tpO2 values increased [113.4 mmHg (90.24); W = 21.0; p = 0.015; Fig. 3B, phase 2]. This hyperoxia phase returned to baseline levels at 15–25 min [59.02 mmHg (39.63); W = 7.0; p = 0.29; phase 3, Fig. 3B]. Overall, when compared with control animals, naproxen inhibited both the acute hypoxia (phase 2; U = 0; p < 0.001; Fig. 3D) and prolonged decrease in tpO2 (phase 3; U = 3; p = 0.005; Fig. 3E).
Figure 2.
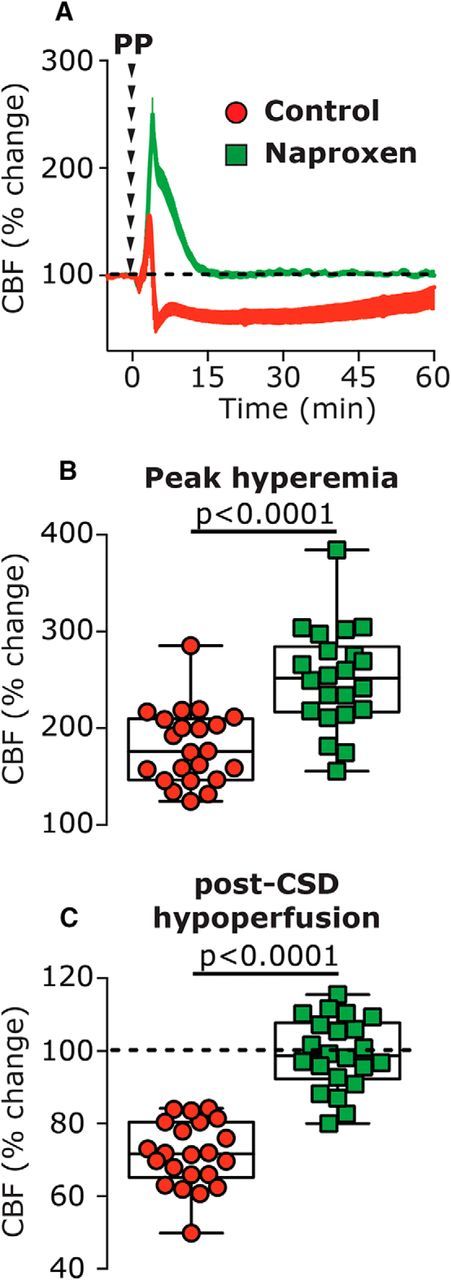
The effect of systemic naproxen on CSD-induced changes in CBF. A, Changes in CBF following induction of CSD in vehicle- and naproxen-treated animals. B, CBF changes during the hyperemia stage, showing the enhancing effect of naproxen. C, CBF changes during the hypoperfusion phase, 10–60 min following CSD, depicting the inhibitory effect of naproxen. p Values are based on a Mann–Whitney U test. PP, Pinprick.
Figure 3.

Naproxen inhibits CSD-induced decreases in cortical tpO2. A, B, Time course of changes in tpO2 following CSD induction in control (A) and naproxen-treated animals (B). C–E, Comparisons between CSD-evoked changes in control and naproxen-treated animals during the hyperoxia phase (phase 1), the acute hypoxia phase (phase 2, in control animals), and at 15–25 min post-CSD (phase 3). p Values are based on a Mann–Whitney U test. PP, Pinprick.
We next examined whether the suppressing effects of naproxen on CSD-evoked cortical hypoperfusion and decreased tpO2 might be associated with reduced activation of meningeal afferents. A-δ afferents recorded from naproxen-treated animals (n = 15) had baseline ongoing activity similar to that in control vehicle-treated animals [n = 26; 0.04 spikes/s (0.20) vs 0.05 spikes/s (0.25); U = 189.0; p = 0.87]. Baseline ongoing activity of C afferents was also not different between the naproxen group (n = 16) and control group [n = 34; 1.08 spikes/s (1.58) vs 0.57 spikes/s (1.35); U = 215.0; p = 0.24]. In naproxen-treated animals, CSD evoked acute activation in 5 of 15 A-δ afferents and 6 of 16 C afferents, ratios that were not significantly different from those observed in control animals [9 of 26 A-δ afferents, 14 of 34 C afferents; χ2 = 0.007; p = 0.99 for the A-δ afferent population; χ2 = 0.06; p = 0.80 for the C afferent populations; χ2 = 0.07; p = 0.79 for the combined population]. When compared with the control group, naproxen also did not affect the magnitude of the immediate activation in either A-δ afferents [4.2-fold (3.50) vs 3.6-fold (2.10); U = 22.0; p = 0.99] or in C afferents [1.6-fold (0.50) vs 2.1-fold (1.40); U = 30.0; p = 0.35, Fig. 4C].
Figure 4.

Naproxen treatment does not inhibit CSD-evoked acute activation of meningeal afferents. A, B, Examples of experimental trials depicting the acute activation of an A-δ afferent (A) and a C afferent (B) following the induction of CSD using a pinprick (PP) stimulus in animals treated with naproxen. Traces represent a post-stimulus time histogram (1 s bin size). The average ongoing activity rate (spikes/s) is denoted in parentheses. Red areas represent the acute phase of afferent activation during CSD. Raw LDF traces are provided below each recording showing the CSD-evoked increase in CBF. C, D, Comparisons of the magnitude of change in ongoing activity between afferents recorded in control and naproxen-treated animals depicting activated (C) and nonactivated afferents (D). p Values are based on a Mann–Whitney U test.
The likelihood of developing a prolonged increase in ongoing afferent activity following CSD in naproxen-treated animals (Fig. 5A,B, examples) was also not different when compared with that observed in control animals (A-δ afferents: 7 of 15 vs 13 of 26, χ2 = 0.04; p = 0.84; C afferents: 7 of 16 vs 22 of 34, χ2 = 1.96; p = 0.16; combined population: χ2 = 1.427; p = 0.23). Naproxen treatment also did not affect any of the CSD-evoked afferent activation parameters: persistent activation developed in A-δ afferents with a delay of 25 min (40), which was no different than in control animals [10 min (5.0); U = 33.0; p = 0.34, Fig. 5C]. The activation delay observed in C afferents in naproxen-treated animals [15 min (31.25)] was also not different than that in controls [5 min (17.5); U = 67.5; p = 0.58, Fig. 5C]. The duration of CSD-evoked prolonged activation was also unaffected by naproxen: A-δ afferents were activated for 15 min (15) vs 30 min (22.5) in the control group (U = 22.0; p = 0.07, Fig. 5D); C-afferents were activated for 30 min (31.25) vs 35 min (30.0) in the control group (U = 58.0; p = 0.34; Fig. 5D). Finally, when compared with controls, naproxen treatment did not reduce the magnitude of the afferent activation observed in either population [A-δ afferents: 2.3-fold (3.0)] vs [3-fold (3.8); U = 39.0, p = 0.64; C afferents: 1.8-fold (3.0) vs 1.5-fold (0.9); U = 51.0; p = 0.18, Fig. 5E]. In afferents deemed not activated by CSD, the changes in ongoing activity following CSD were also not different between the naproxen and control groups, in both the A-δ afferent (U = 41.0; p = 0.8) and C afferent (U = 35.0; p = 0.2) populations (Fig. 5F).
Figure 5.

Naproxen treatment does not inhibit CSD-evoked prolonged activation of meningeal afferents. A, B, Experimental trials depicting the CSD-evoked prolonged activation of an A-δ afferent (A) and a C afferent (B) in animals treated with naproxen. C–E, Naproxen treatment did not affect the onset latency of the afferent activation (C), its duration (D), or its magnitude (E). Changes in ongoing activity rate in nonactivated afferents in naproxen-treated animals were not different than those in controls (F). p Values are based on a Mann–Whitney U test. PP, Pinprick.
Naproxen inhibits CSD-evoked meningeal afferent mechanosensitization
We previously proposed that the activation and mechanosensitization of meningeal afferents that occur following CSD are mediated via distinct mechanisms (Zhao and Levy, 2016). We therefore examined next in 8 A-δ and 7 C afferents whether the inhibitory effects of naproxen on the cortical hypoperfusion and decreased tpO2 might be associated with amelioration of the post-CSD mechanosensitization response. Because the characteristics of the CSD-evoked sensitizations of the A-δ and C afferents are similar (Zhao and Levy, 2016), we combined the data from the two afferent populations for comparisons with the data obtained from vehicle-treated animals (10 A-δ afferents and 13 C afferents). At baseline, the number of dural mechanosensitive receptive fields and the mechanical responsiveness of afferents recorded in naproxen-treated animals were similar to those in the control group (Table 1). Unlike its ability to inhibit the CSD-evoked activation of meningeal afferents, naproxen blocked the afferent sensitization responses, at both the threshold level [3 of 15 vs 13 of 23; χ(2) = 4.97; p = 0.03; Fig. 6C] and the suprathreshold level [2 of 15 vs 13 of 23; χ(2) = 7.09; p = 0.008; Fig. 6]. When sensitization was defined as enhanced responsiveness at either stimulus level, there were also fewer sensitized afferents in the naproxen group [4 of 15 vs 17 of 23; χ(2) = 9.084; p < 0.003]. When the responses of all sensitized and nonsensitized units were combined, naproxen also had an overall inhibitory effect on the changes in threshold responses [Δ0 spike/s (0.6) vs Δ0.39 spikes/s (1.43); U = 90.5; p = 0.01, Fig. 6G] and suprathreshold responses [0.86-fold (0.5) vs 1.2-fold (0.5); U = 22.0; p = 0.004, Fig. 6H].
Table 1.
Baseline mechanical response properties of meningeal afferents in animals treated with vehicle, naproxen, or levcromakalim
| Receptive fields* | Threshold responses** | Suprathreshold responses*** | |
|---|---|---|---|
| Control | 2 (1) | 1.3 (0.7) | 7.3 (7.0) |
| Naproxen | 2 (1) | 0.5 (1.8) | 8.0 (6.5) |
| Levcromakalim | 2 (1) | 1.7 (1.5) | 9.0 (4.0) |
Data are expressed as the median (IQR). Threshold and suprathreshold neural responses (spikes/s) are based on activity recorded during a 2 s mechanical stimulus, and represent the average of three to four trials conducted prior to the elicitation of CSD in the presence of the vehicle, naproxen, or levcromakalim. Group differences were analyzed using Kruskal–Wallis test and were not statistically significant for the three parameters examined. H stands for “Kruskal-Wallis H test”; H(2) = H test with 2 degrees of freedom.
*H(2) = 3.85, p = 0.145;
**H(2) = 4.70, p = 0.095;
***H(2) = 3.24, p = 0.198.
Figure 6.
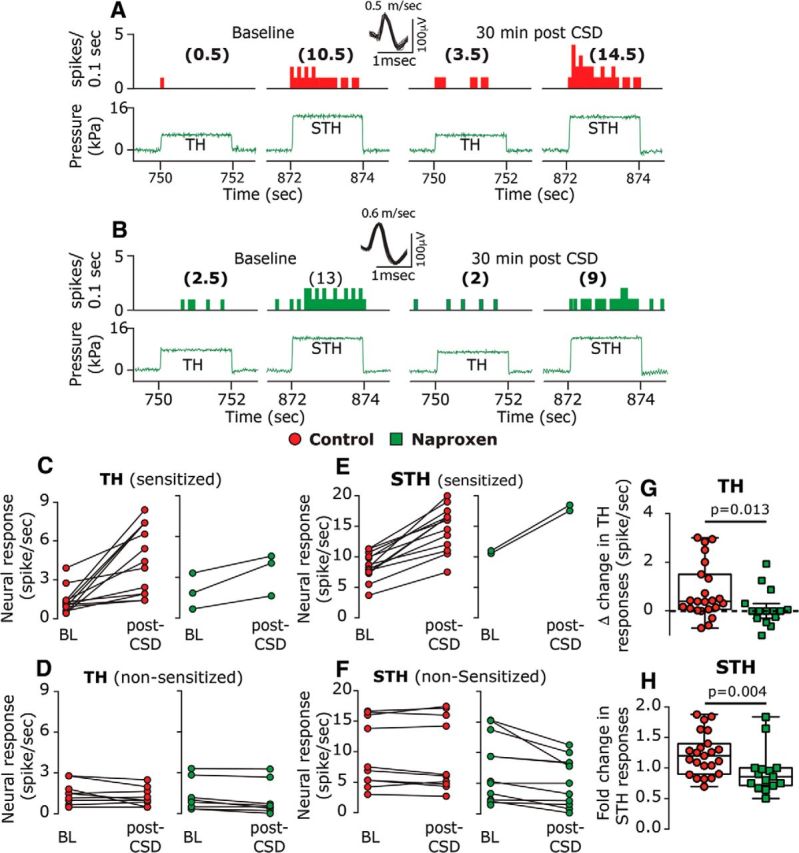
Naproxen inhibits CSD-induced mechanical sensitization of meningeal afferents. A, B, Examples of experimental trials depicting the responses (post-stimulus time histogram; 0.1 s) of two C afferents to threshold (TH) and suprathreshold (STH) mechanical stimuli (green traces) applied to the dural receptive field during baseline recording and then at 30 min following CSD elicitation in control (A) and naproxen-treated animals (B). C–F, Note the lack of sensitization following naproxen treatment. Individual threshold and suprathreshold responses of sensitized and nonsensitized afferents in control and naproxen-treated animals. G, H, Magnitudes of CSD-related threshold (G) and suprathreshold (H) changes in mechanosensitivity depicting the inhibition of sensitization by naproxen. p Values are based on a Mann–Whitney U test.
The K(ATP) channel opener Levcromakalim counteracts CSD-evoked cortical hypoperfusion and reduced tpO2
Naproxen may inhibit the CSD-evoked afferent mechanosensitization indirectly, by blocking the cortical hypoperfusion and reduced tissue oxygenation. However, it is also possible that the inhibition of sensitization is independent of these cortical metabolic effects, since prostanoids (elaborated by CSD), whose synthesis is blocked by naproxen, exert a direct sensitizing effect on meningeal afferents (independent of metabolic effects; Zhang et al., 2007), and naproxen also inhibits afferent sensitization induced by a mixture of inflammatory mediators (Levy et al., 2008). We therefore asked next whether opposing the CSD-evoked cortical hypoperfusion and the associated decrease in tpO2 using an alternative, cyclooxygenase-independent approach might also inhibit the afferent sensitization response. We postulated that local application of a potent cortical vasodilator could serve this purpose and tested the effects of levcromakalim, a selective K(ATP) channel opener with strong cortical vasorelaxation properties (Reid et al., 1995). In preliminary experiments, we observed that the maximum cortical vasodilatory effect of levcromakalim occurs within 15–25 min. Therefore, to counteract the CSD-evoked persistent hypoperfusion, we used a pretreatment approach and started levcromakalim treatment 30 min before CSD elicitation. Using this protocol, before CSD induction, levcromakalim (n = 28) increased basal CBF [196.10% (72.7); W = 231.0; p < 0.0001, Fig. 7A]. Levcromakalim did not inhibit the elicitation of CSD (n = 3, Fig. 7C), which was associated with a significant cortical hyperemic response, albeit smaller in magnitude when compared with the response in control animals [105.3% (1.9) vs 180.3% (61.6); U = 0.0; p < 0.0001; Fig. 7E]. In the presence of levcromakalim, there was a decrease in CBF following CSD, but values did not attain the hypoperfusion level observed in control animals (Fig. 7F; U = 51.0; p < 0.0001); up to the 45 min post-CSD period, values were not different from those observed during the predrug baseline period [99.56% (39.67); W = −8.0; p > 0.99] and increased afterward [119.1% (54.9)] at 60 min post-CSD (W = 366.0; p < 0.0001).
Figure 7.

Effects of the K(ATP) channel opener levcromakalim on CSD elicitation and CSD-evoked CBF changes. A, Levcromakalim increased basal CBF. B, C, Examples of the combined recording of intracortical neural multiunit activity (MUA; post-stimulus time histogram; 1 s in middle panels) and CBF (bottom panels) showing a similar induction of CSD in control (B) and levcromakalim-treated animals (C). Note the initial depolarization (increased cortical activity; arrows) followed by the suppression and gradual recovery of neural activity. Also note the CSD-evoked cortical hyperemic response in the control animal and the diminished response in levcromakalim-treated animal (arrowheads). D, Time course changes of CBF in control and levcromakalim-treated animals following PP-induced CSD. Note that despite the decrease in CBF in levcromakalim-treated animals, values remained near predrug baseline values (denoted as 100%) unlike the marked decrease in control animals. E, CBF changes in control and levcromakalim-treated animals during the hyperemia stage. F, CBF changes during the hypoperfusion phase, 10–60 min following CSD. p Values are based on a Mann–Whitney U test. PP, Pinprick.
We next tested whether levcromakalim might also affect the CSD-evoked changes in cortical tpO2 (n = 8). Before CSD elicitation, levcromakalim increased the basal tpO2 [Δ43.55 mmHg (35.3); W = 36.0; p = 0.008; Fig. 8A]. In the presence of levcromakalim, CSD gave rise to a further, brief increase in tpO2 [peak, 120.5 mmHg (56.3); W = 36.0; p = 0.008; Fig. 8A, phase 1] with a magnitude similar to that observed in control animals [Δ18.22 mmHg (10.32) vs Δ14.18 mmHg (3.87); U = 24.0; p = 0.11]. While the duration of this hyperoxic phase was shorter than that in control animals [70.0 s (30.0) vs 100 s (50.0); U = 11.0; p = 0.05], overall the hyperoxia phase [area under the curve (AUC)] was not different between the two groups (U = 33.0; p = 0.39; Fig. 8C). Following the brief hyperoxia, tpO2 values decreased transiently, reaching hypoxic values [18.5 mmHg (26.36)] that were not different from those in control animals [17.66 mmHg (18.64); U = 32.0; p = 0.35]. However, when compared with control animals, levcromakalim shortened this hypoxic phase [20 s (27.5) vs 110 s (70.0); U = 3.5; p = 0.0002], yielding a reduced hypoxic response [246.6 mmHg*sec (676.0) vs 2001.0 mmHg*sec (4883.9); U = 7.0; p = 0.002; Fig. 8D]. Levcromakalim also prevented the prolonged reduction in tpO2 when compared with control animals (U = 15.0; p = 0.001; Fig. 8B, Phase 3, E), with values not different than baseline at the 5–10 min time point [49.49 mmHg (43.2); U = 26.0; p = 0.46] and the 10–20 min time point [47.16 mmHg (44.51); U = 31.0; p = 0.64].
Figure 8.

Levcromakalim inhibits CSD-evoked cortical hypoxia. A, Time course changes (median and IQR) in cortical tpO2 values at baseline (BL) and during the acute hyperoxia and hypoxia phases in response to CSD induction with pinprick (phases 1 and 2, respectively) in control and levcromakalim-treated animals. Note the shorter duration of the hypoxia phase following levcromakalim treatment. B, The prolonged, post-CSD decrease in tpO2 (phase 3) in control animals and its amelioration by levcromakalim are shown. Dashed line represents predrug baseline values. C–E, Differences between control and levcromakalim-treated animals in the hyperoxic response (AUC, phase 1; C), acute hypoxia (AUC, phase 2; D), and the prolonged decrease in tpO2 at 5–25 min post-CSD (AUC, phase 3; E). p Values are based on a Mann–Whitney U test. Levcro, levcromakalim; PP, pinprick.
Levcromakalim inhibits CSD-evoked activation but not sensitization of meningeal afferents
Given its ability to counteract the cortical metabolic perturbations evoked by CSD, we next determined the effect of levcromakalim treatment on the development of CSD-evoked afferent activation in 9 A-δ and 11 C afferents. In the presence of levcromakalim, baseline ongoing activity in A-δ afferents [0.13 spikes/s (0.31)] and in C afferents [0.28 (0.89)] were not different from that in control animals (U = 91.0, p = 0.32 and U = 156.0, p = 0.41, respectively). Levcromakalim treatment, however, significantly inhibited the acute afferent activation [control group: A-δ afferents, 0 of 9 vs 9 of 26; χ(1) = 4.19, p = 0.04; C afferents, 1 of 11 vs 14 of 34; χ(2) = 3.85, p = 0.049); combined population: 1 of 20 vs 23 of 60; χ(2) = 7.937; p = 0.005; Fig. 9C]. Levcromakalim also inhibited the prolonged afferent activation when compared with control group [A-δ afferents, 1 of 9 vs 13 of 26; χ(2) = 4.21; p = 0.04; C afferents, 3 of 11 vs 22 of 34; χ(2) = 4.717; p = 0.03; combined population: 4 of 20 vs 35 of 60; χ(2) = 8.82; p = 0.003; Fig. 9D].
Figure 9.
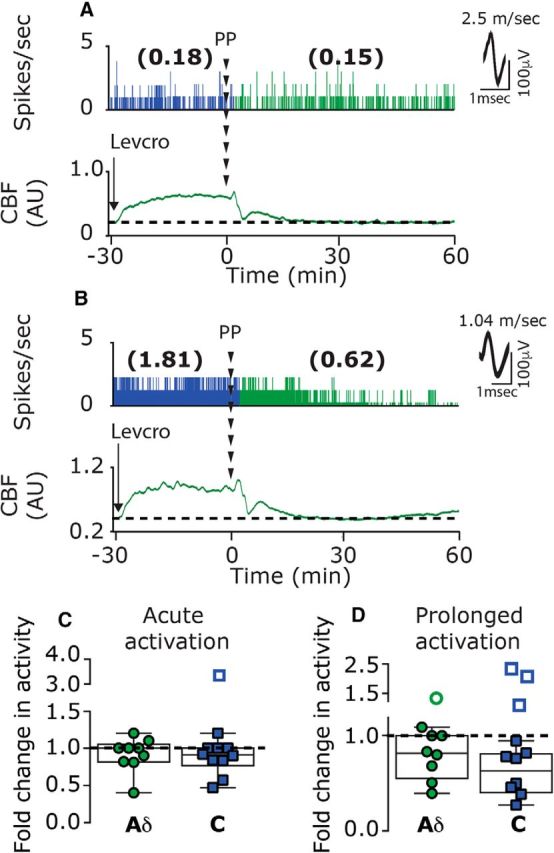
Levcromakalim inhibits CSD-evoked acute and prolonged meningeal afferent activation. A, B, Examples of single-unit recording of an A-δ afferent (A) and a C afferent (B), combined with laser Doppler flowmetry, depicting the lack of acute and prolonged afferents following the elicitation of CSD with a pinprick in animals treated with levcromakalim. C, In the presence of levcromakalim, only one C afferent became acutely activated (open square outside the minimum/maximum box). D, Prolonged activation was noted only in one A-δ and three C afferents (open circle and squares outside the minimum/maximum box). PP, Pinprick.
Finally, we tested the effect of levcromakalim on the development of CSD-evoked mechanical sensitization in 7 A-δ afferents and 10 C afferents, which had a similar number of identified dural mechanosensitive receptive fields and baseline mechanical responsiveness as the afferents tested in the control group (Table 1). Unlike its effect on the CSD-evoked afferent activation, levcromakalim treatment did not inhibit the sensitization response (Fig. 10A, example). When compared with control experiments, levcromakalim did not reduce the relative number of afferents that were deemed sensitized when using the threshold [10 of 17 vs 13 of 23 affected afferents; χ(2) = 0.04; p = 0.85] or the suprathreshold [6 of 17 vs 14 of 23 affected afferents; χ(2) = 1.1, p = 0.29] stimuli. When sensitization was defined as enhanced responsiveness at either stimulus level, the ratio of sensitized/nonsensitized was also not different when compared with that observed in control animals [11 of 17 vs 17 of 23; χ(2) = 0.2, p = 0.66]. When compared with controls, levcromakalim also did not decrease the magnitude of the sensitization responses to threshold stimuli [Δ1.73 spikes/s (3.36) vs Δ1.3 spikes/s (2.18); U = 38.5; p = 0.10; Fig. 10D] or suprathreshold stimuli [1.39-fold (0.5) vs 1.37-fold (0.54); U = 34.0; p = 0.54; Fig. 10E].
Figure 10.
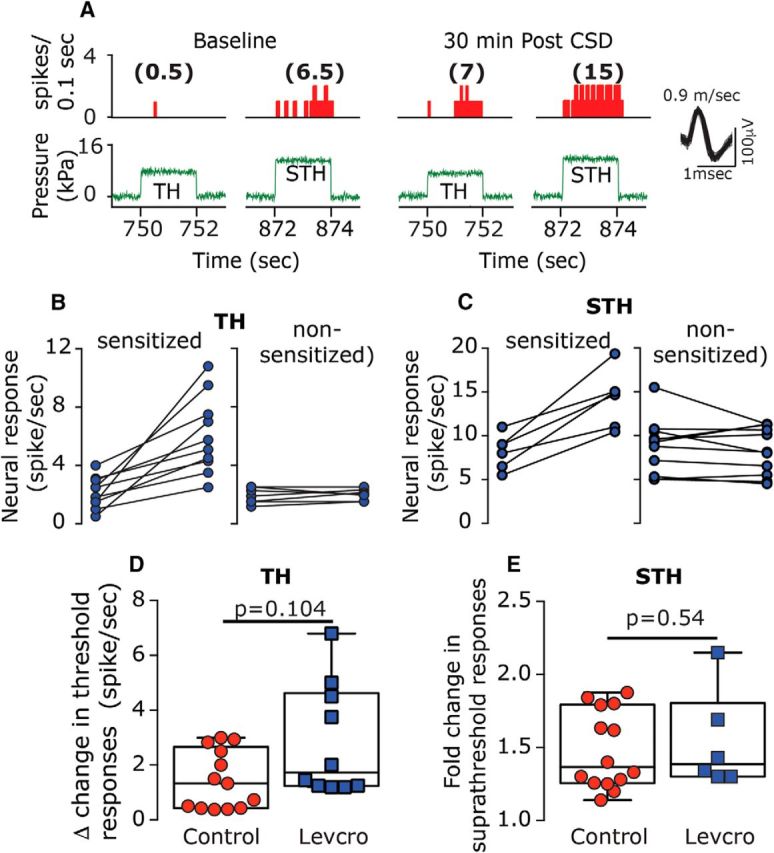
Levcromakalim does not inhibit CSD-evoked mechanical sensitization of meningeal afferents. A, An example of an experimental trial depicting the CSD-evoked increase in mechanical responsiveness of a C afferent recorded from a levcromakalim-treated animal. B, C, Note that CSD-evoked sensitization at both the threshold and suprathreshold levels (poststimulus time histogram; 0.1 s). Individual threshold (B) and suprathreshold (C) responses of sensitized and nonsensitized afferents. D, E, Comparisons between the magnitudes of CSD-related changes in mechanosensitivity at the threshold (D) and suprathreshold levels (E) depicting similar CSD-evoked sensitization responses in control and levcromakalim-treated animals. p Values are based on a Mann–Whitney U test. AU, Arbitrary units; Levcro, levcromakalim; STH, suprathreshold; TH, threshold.
Discussion
Spreading cortical hypoperfusion has been demonstrated during the early stage of migraine with aura and likely reflects a CSD-related vascular event (Ayata and Lauritzen, 2015), and decreased cortical oxygenation has been suggested to trigger migraine headache (Schoonman et al., 2006; Arngrim et al., 2016). In our CSD rodent model, the cortical hypoperfusion and decreased tpO2 coincided with the emergence of the prolonged activation of meningeal afferents, suggesting a potential interaction. However, a key finding of this study was that despite exerting a powerful inhibitory effect on the protracted cortical hypoperfusion and the related decrease in oxygenation, naproxen did not ameliorate the activation of meningeal afferents in response to CSD. Our data thus point to a dissociation between these CSD-related cortical metabolic perturbations, or downstream processes, and the mechanism responsible for driving meningeal afferent activity. The ineffectiveness of naproxen was somewhat surprising in light of its ability to reduce the persistent activation of meningeal afferents following application of a mixture of inflammatory mediators to their meningeal receptive field (Levy et al., 2008)—a model of meningeal nociception (Strassman et al., 1996) and migraine pain (Oshinsky and Gomonchareonsiri, 2007; Edelmayer et al., 2009; De Felice et al., 2013). This discrepancy suggests that the prolonged activation of meningeal afferents in the wake of CSD occurs via a distinct mechanism, one that does not involve local nociceptive actions of these classical inflammatory mediators (Strassman et al., 1996) or vasoconstricting prostanoids that mediate the cortical hypoperfusion following CSD (Shibata et al., 1992; Gariepy et al., 2017).
The cortical hyperemia that occurs during the CSD wave is likely driven in part by the acute activation of meningeal afferents, and the ensuing release of vasodilatory neuropeptides such as CGRP (Busija et al., 2008). We found that naproxen also increased the initial cortical hyperemia, which likely mediated the conversion of the acute hypoxia phase to hyperoxia, but these effects did not influence the acute activation of the afferents. These data support the view that cortical vasodilation and its related increases in tissue oxygenation do not drive meningeal afferent activity (Levy and Burstein, 2011), and that during the hyperemic phase of the CSD there is a concurrent release of opposing vasoconstricting prostanoids.
During the prolonged cortical hypoperfusion phase, CSD also gives rise to persistent vasodilatation of the middle meningeal artery (Bolay et al., 2002), a vascular response proposed to be mediated by prolonged activation of dural afferents and downstream signaling via a central parasympathetic reflex (Bolay et al., 2002). Importantly, a recent study (Karatas et al., 2013) demonstrated that this CSD-evoked meningeal vasodilatory response could be blocked by systemic administration of naproxen. That naproxen did not inhibit the CSD-evoked activation of meningeal afferents together with previous data showing its inability to block parasympathetic outflow to the cranium (Akerman et al., 2012) suggests therefore that CSD-evoked dural vasodilatation may not be driven by enhanced ongoing meningeal afferent activity. Our present results thus call into question the use of dural artery vasodilation as a surrogate of increased meningeal afferent activity and the activation of the migraine pain pathway in preclinical studies.
We found that counteracting the protracted cortical hypoperfusion and prolonged decrease in tpO2 by preemptively increasing CBF using levcromakalim was associated with inhibition of the acute and prolonged afferent activation phases. We propose, however, that the inhibitory effect of levcromakalim on these afferent responses did not involve the normalization of CBF or tpO2 given that naproxen, which abrogated the cortical metabolic perturbations following CSD, failed to inhibit the afferent activation. We cannot exclude the possibility that the cortical vasodilation and increased tpO2 induced by levcromakalim, before the onset of CSD, contributed somehow to the blockade of the post-CSD afferent activation. However, the finding that levcromakalim did not affect the basal ongoing activity of afferents suggests that the vascular effects of levcromakalim were unlikely to mediate to the inhibition of the CSD-evoked afferent activation.
Cortical neurons also express K(ATP) channels (Karschin et al., 1997), and their prolonged activation by levcromakalim could have resulted in diminished cortical excitability (Soundarapandian et al., 2007), potentially affecting the elicitation of CSD. However, levcromakalim did not inhibit the induction of CSD, and particularly not the initial strong depolarization before the prolonged suppression of cortical neural activity. K(ATP) activation in cortical neurons could, nonetheless, reduce the release of pronociceptive transmitters, such as glutamate (Zhao et al., 2013), which may contribute to the acute activation, and potentially to the prolonged activation, of meningeal afferents. Cortical astrocytes also express K(ATP) channels (Thomzig et al., 2001), and their activation could potentially decrease the high extracellular K+ level that is attained during the passage of the CSD wave (Kraig and Nicholson, 1978) by enhancing K+ “spatial buffering” (Kofuji and Newman, 2004). In response to CSD, elaborated K+ in the cortical extracellular space has been suggested to diffuse into the leptomeninges and promote the acute activation of meningeal afferents and potentially also their prolonged activation, at least in part (Pietrobon and Moskowitz, 2013; Zhao and Levy, 2017). Activation of astrocytic K(ATP) channels, leading to decreased parenchymal K+, thus could have potentially inhibited the CSD-evoked afferent activation. Our recent finding that K+-evoked acute activation of meningeal afferents was not associated with mechanical sensitization (Zhao and Levy, 2017) is also in agreement with the observation that levcromakalim did not inhibit the CSD-evoked mechanical sensitization response. Finally, activation of K(ATP) channels expressed at the peripheral nerve endings of meningeal afferents could have also contributed to the inhibition of the afferent activation by reducing their overall excitability (Chi et al., 2007). However, the finding that levcromakalim did not reduce basal ongoing discharge or inhibit the mechanical sensitization argues against such a mechanism.
We found that naproxen distinctly inhibited the CSD-evoked mechanical sensitization of meningeal afferents. That levcromakalim treatment greatly reduced the acute hypoxia phase and normalized the prolonged cortical hypoperfusion and decreased tpO2, but did not inhibit the enhanced mechanical responsiveness, suggests, however, that the mechanism underlying the mechanical sensitization following CSD is unrelated to the changes in cortical perfusion and tpO2. It should be noted that while levcromakalim treatment normalized CBF and tpO2 (to values not different than those observed before CSD), it did not inhibit the process that led to their decreases following CSD. We therefore propose that the mediators that contribute to these metabolic changes (likely cyclooxygenase derived; see below) are responsible for the afferent sensitization rather than the cortical hypoperfusion and abnormally reduced tpO2 per se. It is possible that the acute hypoxic phase that occurred in levcromakalim-treated animals (albeit for only a fraction of the time noted in control animals) may have contributed somehow to the afferent sensitization process. However, we also observed brief cortical hypoxia in some naproxen-treated animals, further suggesting a dissociation between the acute hypoxic phase and the afferent changes.
The discrepancy between the effects of naproxen and levcromakalim on the CSD-evoked afferent activation and mechanical sensitization responses supports the view that these two migraine-related nociceptive responses are mediated by distinct mechanisms (Levy and Strassman, 2004; Zhang et al., 2011a, 2013; Zhao and Levy, 2016). The site of action of naproxen and the nature of the cyclooxygenase-derived factors that distinctly promote the sensitization of meningeal afferents following CSD require further studies. The release of cyclooxygenase-derived sensitizing mediators from cortical neurons and glial limitans astrocytes that interface with the meninges (Xu et al., 2004) could contribute to the afferent sensitization. The mode of cortex-to-meninges transport of cyclooxygenase-derived prostanoids could involve diffusion or bulk flow via the glymphatic system (Iliff et al., 2012; Schain et al., 2017). The transport of sensitizing factors from the parenchymal interstitial space into the subarachnoid CSF could influence primarily leptomeningeal afferents and meningeal afferents that terminate within arachnoid granulations (von Düring and Andres, 1991; Fricke et al., 1997). Additional flow of sensitizing mediators from the subarachnoid CSF into the dural lymphatics system (Raper et al., 2016; Louveau et al., 2017) could affect the responses of dural afferents. Naproxen treatment could have also inhibited the CSD-evoked meningeal afferent sensitization by targeting constitutively expressed meningeal cyclooxygenase in meningeal vascular and immune cells (Zhang et al., 2009) and the ensuing release of sensitizing prostanoids (Zhang et al., 2011a).
In summary, our findings point to a dissociation between the CSD-evoked cortical metabolic perturbations and the prolonged activation and mechanical sensitization of meningeal afferents. The findings that K(ATP) activation and cyclooxygenase inhibition have distinct effects on the CSD-evoked activation and sensitization of meningeal afferents may explain the ineffectiveness of cyclooxygenase inhibitors in many cases of migraine (Suthisisang et al., 2010). A combination therapy, which includes a cyclooxygenase inhibitor and a K(ATP) channel opener, may be explored for treating the headache in migraine with aura.
Footnotes
The study was supported by National Institutes of Health Grants NS-086830, NS-078263, and NS-101405 to D.L. We thank Dr. A. M. Strassman for helpful comments on the manuscript.
The authors declare no competing financial interests.
References
- Akerman S, Holland PR, Summ O, Lasalandra MP, Goadsby PJ (2012) A translational in vivo model of trigeminal autonomic cephalalgias: therapeutic characterization. Brain 135:3664–3675. 10.1093/brain/aws249 [DOI] [PubMed] [Google Scholar]
- Appenzeller O. (1994) High-altitude headache. Cephalalgia 14:317–318. [DOI] [PubMed] [Google Scholar]
- Arngrim N, Schytz HW, Britze J, Amin FM, Vestergaard MB, Hougaard A, Wolfram F, de Koning PJ, Olsen KS, Secher NH, Larsson HB, Olesen J, Ashina M (2016) Migraine induced by hypoxia: an MRI spectroscopy and angiography study. Brain 139:723–737. 10.1093/brain/awv359 [DOI] [PubMed] [Google Scholar]
- Ayata C, Lauritzen M (2015) Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 95:953–993. 10.1152/physrev.00027.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA (2002) Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med 8:136–142. 10.1038/nm0202-136 [DOI] [PubMed] [Google Scholar]
- Broessner G, Rohregger J, Wille M, Lackner P, Ndayisaba JP, Burtscher M (2016) Hypoxia triggers high-altitude headache with migraine features: a prospective trial. Cephalalgia 36:765–771. 10.1177/0333102415610876 [DOI] [PubMed] [Google Scholar]
- Busija DW, Bari F, Domoki F, Horiguchi T, Shimizu K (2008) Mechanisms involved in the cerebrovascular dilator effects of cortical spreading depression. Prog Neurobiol 86:379–395. 10.1016/j.pneurobio.2008.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi XX, Jiang X, Nicol GD (2007) ATP-sensitive potassium currents reduce the PGE2-mediated enhancement of excitability in adult rat sensory neurons. Brain Res 1145:28–40. 10.1016/j.brainres.2007.01.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice M, Eyde N, Dodick D, Dussor GO, Ossipov MH, Fields HL, Porreca F (2013) Capturing the aversive state of cephalic pain preclinically. Ann Neurol 74:257–265. 10.1002/ana.23922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmayer RM, Vanderah TW, Majuta L, Zhang ET, Fioravanti B, De Felice M, Chichorro JG, Ossipov MH, King T, Lai J, Kori SH, Nelsen AC, Cannon KE, Heinricher MM, Porreca F (2009) Medullary pain facilitating neurons mediate allodynia in headache-related pain. Ann Neurol 65:184–193. 10.1002/ana.21537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabricius M, Lauritzen M (1993) Transient hyperemia succeeds oligemia in the wake of cortical spreading depression. Brain Res 602:350–353. 10.1016/0006-8993(93)90701-N [DOI] [PubMed] [Google Scholar]
- Fabricius M, Akgoren N, Lauritzen M (1995) Arginine-nitric oxide pathway and cerebrovascular regulation in cortical spreading depression. Am J Physiol 269:H23–H29. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Akgören N, Dirnagl U, Lauritzen M (1997) Laminar analysis of cerebral blood flow in cortex of rats by laser-Doppler flowmetry: a pilot study. J Cereb Blood Flow Metab 17:1326–1336. 10.1097/00004647-199712000-00008 [DOI] [PubMed] [Google Scholar]
- Fordsmann JC, Ko RW, Choi HB, Thomsen K, Witgen BM, Mathiesen C, Lønstrup M, Piilgaard H, MacVicar BA, Lauritzen M (2013) Increased 20-HETE synthesis explains reduced cerebral blood flow but not impaired neurovascular coupling after cortical spreading depression in rat cerebral cortex. J Neurosci 33:2562–2570. 10.1523/JNEUROSCI.2308-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke B, von Düring M, Andres KH (1997) Topography and immunocytochemical characterization of nerve fibers in the leptomeningeal compartments of the rat. A light- and electron-microscopical study. Cell Tissue Res 287:11–22. [DOI] [PubMed] [Google Scholar]
- Gariepy H, Zhao J, Levy D (2017) Differential contribution of COX-1 and COX-2 derived prostanoids to cortical spreading depression-evoked cerebral oligemia. J Cereb Blood Flow Metab 37:1060–1068. 10.1177/0271678X16650217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, Kwong KK, Cutrer FM, Rosen BR, Tootell RB, Sorensen AG, Moskowitz MA (2001) Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A 98:4687–4692. 10.1073/pnas.071582498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JM, Schankin CJ (2017) Cerebral hemodynamics in the different phases of migraine and cluster headache. J Cereb Blood Flow Metab. Advance online publication. Retrieved April 26, 2018. 10.1177/0271678X17729783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt P, Jänig W, Kohler W (1983) Response pattern of visceral afferent fibres, supplying the colon, upon chemical and mechanical stimuli. Pflugers Arch 398:41–47. 10.1007/BF00584711 [DOI] [PubMed] [Google Scholar]
- Hillery CA, Kerstein PC, Vilceanu D, Barabas ME, Retherford D, Brandow AM, Wandersee NJ, Stucky CL (2011) Transient receptor potential vanilloid 1 mediates pain in mice with severe sickle cell disease. Blood 118:3376–3383. 10.1182/blood-2010-12-327429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 4:147ra111. 10.1126/scitranslmed.3003748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatas H, Erdener SE, Gursoy-Ozdemir Y, Lule S, Eren-Koçak E, Sen ZD, Dalkara T (2013) Spreading depression triggers headache by activating neuronal Panx1 channels. Science 339:1092–1095. 10.1126/science.1231897 [DOI] [PubMed] [Google Scholar]
- Karschin C, Ecke C, Ashcroft FM, Karschin A (1997) Overlapping distribution of K(ATP) channel-forming Kir6.2 subunit and the sulfonylurea receptor SUR1 in rodent brain. FEBS Lett 401:59–64. 10.1016/S0014-5793(96)01438-X [DOI] [PubMed] [Google Scholar]
- Kleppisch T, Nelson MT (1995) ATP-sensitive K+ currents in cerebral arterial smooth muscle: pharmacological and hormonal modulation. Am J Physiol 269:H1634–H1640. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Newman EA (2004) Potassium buffering in the central nervous system. Neuroscience 129:1045–1056. 10.1016/j.neuroscience.2004.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraig RP, Nicholson C (1978) Extracellular ionic variations during spreading depression. Neuroscience 3:1045–1059. 10.1016/0306-4522(78)90122-7 [DOI] [PubMed] [Google Scholar]
- Lauritzen M. (1984) Long-lasting reduction of cortical blood flow of the brain after spreading depression with preserved autoregulation and impaired CO2 response. J Cereb Blood Flow Metab 4:546–554. 10.1038/jcbfm.1984.79 [DOI] [PubMed] [Google Scholar]
- Levy D. (2012) Endogenous mechanisms underlying the activation and sensitization of meningeal nociceptors: the role of immuno-vascular interactions and cortical spreading depression. Curr Pain Headache Rep 16:270–277. 10.1007/s11916-012-0255-1 [DOI] [PubMed] [Google Scholar]
- Levy D, Burstein R (2011) The vascular theory of migraine: leave it or love it? Ann Neurol 69:600–601. 10.1002/ana.22422 [DOI] [PubMed] [Google Scholar]
- Levy D, Strassman AM (2002) Mechanical response properties of A and C primary afferent neurons innervating the rat intracranial dura. J Neurophysiol 88:3021–3031. 10.1152/jn.00029.2002 [DOI] [PubMed] [Google Scholar]
- Levy D, Strassman AM (2004) Modulation of dural nociceptor mechanosensitivity by the nitric oxide–cyclic GMP signaling cascade. J Neurophysiol 92:766–772. 10.1152/jn.00058.2004 [DOI] [PubMed] [Google Scholar]
- Levy D, Zhang XC, Jakubowski M, Burstein R (2008) Sensitization of meningeal nociceptors: inhibition by naproxen. Eur J Neurosci 27:917–922. 10.1111/j.1460-9568.2008.06068.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhurst JC, Rotto DM, Kaufman MP, Stahl GL (1991) Ischemically sensitive abdominal visceral afferents: response to cyclooxygenase blockade. Am J Physiol 261:H2075–H2081. [DOI] [PubMed] [Google Scholar]
- Louveau A, Plog BA, Antila S, Alitalo K, Nedergaard M, Kipnis J (2017) Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J Clin Invest 127:3210–3219. 10.1172/JCI90603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver MB, Tanelian DL (1992) Activation of C fibers by metabolic perturbations associated with tourniquet ischemia. Anesthesiology 76:617–623. 10.1097/00000542-199204000-00020 [DOI] [PubMed] [Google Scholar]
- Melo-Carrillo A, Noseda R, Nir R, Schain AJ, Stratton J, Strassman AM, Burstein R (2017) Selective inhibition of trigeminovascular neurons by fremanezumab—a humanized monoclonal anti-CGRP antibody. J Neurosci 37:7149–7163. 10.1523/JNEUROSCI.0576-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mense S, Stahnke M (1983) Responses in muscle afferent fibres of slow conduction velocity to contractions and ischaemia in the cat. J Physiol 342:383–397. 10.1113/jphysiol.1983.sp014857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messlinger K. (2009) Migraine: where and how does the pain originate? Exp Brain Res 196:179–193. 10.1007/s00221-009-1756-y [DOI] [PubMed] [Google Scholar]
- Moore RA, Bullingham RE, McQuay HJ, Hand CW, Aspel JB, Allen MC, Thomas D (1982) Dural permeability to narcotics: in vitro determination and application to extradural administration. Br J Anaesth 54:1117–1128. 10.1093/bja/54.10.1117 [DOI] [PubMed] [Google Scholar]
- Offenhauser N, Thomsen K, Caesar K, Lauritzen M (2005) Activity-induced tissue oxygenation changes in rat cerebellar cortex: interplay of postsynaptic activation and blood flow. J Physiol 565:279–294. 10.1113/jphysiol.2005.082776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen J, Burstein R, Ashina M, Tfelt-Hansen P (2009) Origin of pain in migraine: evidence for peripheral sensitisation. Lancet Neurol 8:679–690. 10.1016/S1474-4422(09)70090-0 [DOI] [PubMed] [Google Scholar]
- Olesen J, Larsen B, Lauritzen M (1981) Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann Neurol 9:344–352. 10.1002/ana.410090406 [DOI] [PubMed] [Google Scholar]
- Olesen J, Lauritzen M, Tfelt-Hansen P, Henriksen L, Larsen B (1982) Spreading cerebral oligemia in classical- and normal cerebral blood flow in common migraine. Headache 22:242–248. 10.1111/j.1526-4610.1982.hed2206242.x [DOI] [PubMed] [Google Scholar]
- Oshinsky ML, Gomonchareonsiri S (2007) Episodic dural stimulation in awake rats: a model for recurrent headache. Headache 47:1026–1036. 10.1111/j.1526-4610.2007.00871.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrobon D, Moskowitz MA (2013) Pathophysiology of migraine. Annu Rev Physiol 75:365–391. 10.1146/annurev-physiol-030212-183717 [DOI] [PubMed] [Google Scholar]
- Piilgaard H, Lauritzen M (2009) Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J Cereb Blood Flow Metab 29:1517–1527. 10.1038/jcbfm.2009.73 [DOI] [PubMed] [Google Scholar]
- Raper D, Louveau A, Kipnis J (2016) How do meningeal lymphatic vessels drain the CNS? Trends Neurosci 39:581–586. 10.1016/j.tins.2016.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid JM, Davies AG, Ashcroft FM, Paterson DJ (1995) Effect of L-NMMA, cromakalim, and glibenclamide on cerebral blood flow in hypercapnia and hypoxia. Am J Physiol 269:H916–H922. [DOI] [PubMed] [Google Scholar]
- Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB (2014) Transcranial amelioration of inflammation and cell death after brain injury. Nature 505:223–228. 10.1038/nature12808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schain AJ, Melo-Carrillo A, Strassman AM, Burstein R (2017) Cortical spreading depression closes paravascular space and impairs glymphatic flow: implications for migraine headache. J Neurosci 37:2904–2915. 10.1523/JNEUROSCI.3390-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonman GG, Sándor PS, Agosti RM, Siccoli M, Bärtsch P, Ferrari MD, Baumgartner RW (2006) Normobaric hypoxia and nitroglycerin as trigger factors for migraine. Cephalalgia 26:816–819. 10.1111/j.1468-2982.2006.01112.x [DOI] [PubMed] [Google Scholar]
- Shibata M, Leffler CW, Busija DW (1992) Pial arteriolar constriction following cortical spreading depression is mediated by prostanoids. Brain Res 572:190–197. 10.1016/0006-8993(92)90469-P [DOI] [PubMed] [Google Scholar]
- Shin HK, Lee JH, Kim CD, Kim YK, Hong JY, Hong KW (2003) Prevention of impairment of cerebral blood flow autoregulation during acute stage of subarachnoid hemorrhage by gene transfer of Cu/Zn SOD-1 to cerebral vessels. J Cereb Blood Flow Metab 23:111–120. 10.1097/01.WCB.0000036561.60552.63 [DOI] [PubMed] [Google Scholar]
- Soundarapandian MM, Zhong X, Peng L, Wu D, Lu Y (2007) Role of K(ATP) channels in protection against neuronal excitatory insults. J Neurochem 103:1721–1729. 10.1111/j.1471-4159.2007.04963.x [DOI] [PubMed] [Google Scholar]
- Strassman AM, Raymond SA, Burstein R (1996) Sensitization of meningeal sensory neurons and the origin of headaches. Nature 384:560–564. 10.1038/384560a0 [DOI] [PubMed] [Google Scholar]
- Suthisisang CC, Poolsup N, Suksomboon N, Lertpipopmetha V, Tepwitukgid B (2010) Meta-analysis of the efficacy and safety of naproxen sodium in the acute treatment of migraine. Headache 50:808–818. 10.1111/j.1526-4610.2010.01635.x [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Lovatt D, Hansen AJ, Kasischke KA, Nedergaard M (2007) Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci 10:754–762. 10.1038/nn1902 [DOI] [PubMed] [Google Scholar]
- Thomzig A, Wenzel M, Karschin C, Eaton MJ, Skatchkov SN, Karschin A, Veh RW (2001) Kir6.1 is the principal pore-forming subunit of astrocyte but not neuronal plasma membrane K-ATP channels. Mol Cell Neurosci 18:671–690. 10.1006/mcne.2001.1048 [DOI] [PubMed] [Google Scholar]
- Thrane AS, Takano T, Rangroo Thrane V, Wang F, Peng W, Ottersen OP, Nedergaard M, Nagelhus EA (2013) In vivo NADH fluorescence imaging indicates effect of aquaporin-4 deletion on oxygen microdistribution in cortical spreading depression. J Cereb Blood Flow Metab 33:996–999. 10.1038/jcbfm.2013.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Düring M, Andres KH (1991) Sensory nerve fiber terminals in the arachnoid granulations of non-human primates. Neurosci Lett 127:121–124. 10.1016/0304-3940(91)90909-D [DOI] [PubMed] [Google Scholar]
- Vos T, Abajobir A, Abbafati C, Abbas KM, Abate KH, Abd-Alla F, Abdulkader RS, Abdulle AM, Abebo TA, Abera SF, Aboyans V, Abu-Raddad LJ, Ackerman IN, Adamu AA, Adetokunboh O, Afarideh M, Afshin A, Agarwal SK, Aggarwal R, Agrawal A, et al. (2017) Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet 390:1211–1259. 10.1016/S0140-6736(17)32154-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HL, Koenig HM, Ye S, Feinstein DL, Pelligrino DA (2004) Influence of the glia limitans on pial arteriolar relaxation in the rat. Am J Physiol Heart Circ Physiol 287:H331–H339. 10.1152/ajpheart.00831.2003 [DOI] [PubMed] [Google Scholar]
- Zhang XC, Kainz V, Jakubowski M, Burstein R, Strassman A, Levy D (2009) Localization of COX-1 and COX-2 in the intracranial dura mater of the rat. Neurosci Lett 452:33–36. 10.1016/j.neulet.2009.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XC, Kainz V, Burstein R, Levy D (2011a) Tumor necrosis factor-α induces sensitization of meningeal nociceptors mediated via local COX and p38 MAP kinase actions. Pain 152:140–149. 10.1016/j.pain.2010.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XC, Strassman AM, Burstein R, Levy D (2007) Sensitization and activation of intracranial meningeal nociceptors by mast cell mediators. J Pharmacol Exp Ther 322:806–812. 10.1124/jpet.107.123745 [DOI] [PubMed] [Google Scholar]
- Zhang X, Kainz V, Zhao J, Strassman AM, Levy D (2013) Vascular extracellular signal-regulated kinase mediates migraine-related sensitization of meningeal nociceptors. Ann Neurol 73:741–750. 10.1002/ana.23873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Levy D, Noseda R, Kainz V, Jakubowski M, Burstein R (2010) Activation of meningeal nociceptors by cortical spreading depression: implications for migraine with aura. J Neurosci 30:8807–8814. 10.1523/JNEUROSCI.0511-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Levy D, Kainz V, Noseda R, Jakubowski M, Burstein R (2011b) Activation of central trigeminovascular neurons by cortical spreading depression. Ann Neurol 69:855–865. 10.1002/ana.22329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Levy D (2014) The sensory innervation of the calvarial periosteum is nociceptive and contributes to headache-like behavior. Pain 155:1392–1400. 10.1016/j.pain.2014.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Levy D (2015) Modulation of intracranial meningeal nociceptor activity by cortical spreading depression: a reassessment. J Neurophysiol 113:2778–2785. 10.1152/jn.00991.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Levy D (2016) Cortical spreading depression promotes persistent mechanical sensitization of intracranial meningeal afferents: implications for the intracranial mechanosensitivity of migraine. eNeuro 3:ENEURO.0287–0216.2016. 10.1523/ENEURO.0287-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Levy D (2017) The CGRP receptor antagonist BIBN4096 inhibits prolonged meningeal afferent activation evoked by brief local K+ stimulation but not cortical spreading depression-induced afferent sensitization. Pain Rep 3:e632. 10.1097/PR9.0000000000000632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Bree D, Harrington MG, Strassman AM, Levy D (2017) Cranial dural permeability of inflammatory nociceptive mediators: potential implications for animal models of migraine. Cephalalgia 37:1017–1025. 10.1177/0333102416663466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Wen R, Wang X, Pei L, Yang Y, Shang Y, Bazan N, Zhu LQ, Tian Q, Lu Y (2013) EPAC inhibition of SUR1 receptor increases glutamate release and seizure vulnerability. J Neurosci 33:8861–8865. 10.1523/JNEUROSCI.5686-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]