

Abstract
Leukocyte common antigen-receptor protein tyrosine phosphatases (LAR-RPTPs) are hub proteins that organize excitatory and inhibitory synapse development through binding to various extracellular ligands. Here, we report that knockdown (KD) of the LAR-RPTP family member PTPσ reduced excitatory synapse number and transmission in cultured rat hippocampal neurons, whereas KD of PTPδ produced comparable decreases at inhibitory synapses, in both cases without altering expression levels of interacting proteins. An extensive series of rescue experiments revealed that extracellular interactions of PTPσ with Slitrks are important for excitatory synapse development. These experiments further showed that the intracellular D2 domain of PTPσ is required for induction of heterologous synapse formation by Slitrk1 or TrkC, suggesting that interaction of LAR-RPTPs with distinct intracellular presynaptic proteins, drives presynaptic machinery assembly. Consistent with this, double-KD of liprin-α2 and -α3 or KD of PTPσ substrates (N-cadherin and p250RhoGAP) in neurons inhibited Slitrk6-induced, PTPσ-mediated heterologous synapse formation activity. We propose a synaptogenesis model in presynaptic neurons involving LAR-RPTP-organized retrograde signaling cascades, in which both extracellular and intracellular mechanisms are critical in orchestrating distinct synapse types.
SIGNIFICANCE STATEMENT In this study, we sought to test the unproven hypothesis that PTPσ and PTPδ are required for excitatory and inhibitory synapse formation/transmission, respectively, in cultured hippocampal neurons, using knockdown-based loss-of-function analyses. We further performed extensive structure–function analyses, focusing on PTPσ-mediated actions, to address the mechanisms of presynaptic assembly at excitatory synaptic sites. Using interdisciplinary approaches, we systematically applied a varied set of PTPσ deletion variants, point mutants, and splice variants to demonstrate that both extracellular and intracellular mechanisms are involved in organizing presynaptic assembly. Strikingly, extracellular interactions of PTPσ with heparan sulfates and Slitrks, intracellular interactions of PTPσ with liprin-α and its associated proteins through the D2 domain, as well as distinct substrates are all critical.
Keywords: LAR-RPTPs, presynaptic assembly, protein–protein interaction, PTPσ, synaptic adhesion molecule
Introduction
Synaptogenesis is defined as a continuous process encompassing stabilization of initial contacts between presynaptic axons and postsynaptic dendrites, and formation of specialized subcellular machineries near nascent synaptic sites (McAllister, 2007). Earlier studies indicated that individual presynaptic components are preassembled as small numbers of modular units and then transported through the axon via two different vesicle types, synaptic vesicle protein transport vesicles and Piccolo-bassoon transport vesicles, tethered together with microtubule-based motor proteins (Ziv and Garner, 2004; Maas et al., 2012). Both vesicle types are rapidly recruited to, and trapped at, nascent synaptic sites of synaptogenic signaling (Suarez et al., 2013; Bury and Sabo, 2016). Postsynaptic proteins are also packaged into modular transport packets that are gradually recruited to nascent postsynaptic sites, although postsynaptic protein assembly is relatively slow (Bresler et al., 2004). However, the cellular and molecular mechanisms responsible for coordinating presynaptic and postsynaptic assembly remain unclear (Ziv and Garner, 2004; Jin and Garner, 2008).
Synaptogenic signaling pathways mediated by multiple trans-synaptic adhesion molecules are thought to be involved in creating sites of preferential synapse formation (Missler et al., 2012; Um and Ko, 2013; Ko et al., 2015a). Presynaptic neurexins and postsynaptic neuroligins are arguably the most extensively studied pair of synaptogenic trans-synaptic adhesion molecules (Südhof, 2008). Neurexins and neuroligins also bind other synaptic membrane proteins to specify specific features of various types of synapses and neural circuits (Südhof, 2017). Strikingly, neuroligins require extracellular interactions with neurexins, although they do not require neurexin cytoplasmic sequences to induce heterologous synapse formation (Gokce and Südhof, 2013). Thus, neurexins likely use an unidentified presynaptic membrane coreceptor, which subsequently mediates the cytoplasmic signaling cascades required for synapse formation (Gokce and Südhof, 2013). Together with neurexins, leukocyte common antigen-related receptor tyrosine phosphatases (LAR-RPTPs) are also postulated to localize to the presynaptic active zone (Südhof, 2012). LAR-RPTPs, comprising three vertebrate members, LAR, PTPδ, and PTPσ, are evolutionarily conserved cell-surface proteins with tyrosine phosphatase activity that organize synaptic structure and function through binding to a multitude of postsynaptogenic adhesion molecules. Notably, these LAR-RPTP binding partners show no overlap with neurexin-binding proteins, suggesting the existence of distinct intracellular signaling complexes that differ from neurexin-based signaling complexes (Han et al., 2016). Indeed, LAR-RPTPs directly bind to distinct sets of intracellular proteins, including liprins and caskins (Han et al., 2016); however, it is plausible that LAR-RPTPs also indirectly associate via liprin-α with the PDZ-domain proteins, calcium/calmodulin-dependent serine protein kinase (CASK) and mammalian synapse-defective-1 protein (mSYD-1), which have been shown to directly interact with neurexins (Um and Ko, 2013). These studies suggest the tantalizing hypothesis that presynaptic neurexins and LAR-RPTPs organize synapse structure and function through a shared subset of molecular components.
Here, we systematically investigated the effects of LAR-RPTP loss of function on properties of the heterologous synapses formed between cultured neurons and variously transfected HEK293T cells induced by postsynaptic ligands of LAR-RPTPs. Rescue experiments using various deletion variants, point mutants, and splice variants of PTPσ revealed that interactions of PTPσ with various postsynaptic ligands through its three immunoglobulin (Ig) domains (Ig1–3) are required for PTPσ-mediated heterologous synapse-formation activity. Interestingly, reexpression of PTPσ phosphatase-inactive mutants incompletely restored heterologous synapse formation in PTPσ-deficient neurons, indicating that other mechanisms beyond the intracellular phosphatase activity of PTPσ contribute to this function. Nevertheless, KD of p250RhoGAP (p250GAP) or N-cadherin, but not β-catenin, blocked formation of heterologous synapses between presynaptic neurons and postsynaptic non-neuronal cells expressing Slitrk6, suggesting that specific PTPσ substrates may act downstream of PTPσ to orchestrate the assembly of excitatory presynaptic sites. Furthermore, the carboxy-terminal D2 domain is critical for mediating the synapse-inducing activity of PTPσ. Together, our results reveal intracellular signaling mechanisms that collectively establish a molecular model of PTPσ-mediated heterologous synapse formation that differs from that of neurexins.
Materials and Methods
Construction of expression vectors. 1. Short-hairpin constructs.
The indicated shRNA lentiviral expression constructs were generated by annealing, phosphorylating, and subcloning into XhoI/XbaI sites of a KD lentiviral vector (L-309 or L-315) (Pang et al., 2010; Ko et al., 2011). Sequences of oligonucleotides used for cloning of shRNA constructs are presented in Table 1. 2. Rescue constructs. Human PTPσ splice variants containing the mini exons encoding MeA and MeB sites (but excluding its own signal peptide) were cloned into the pDisplay vector (Invitrogen) via BglII/SalI sites. The PTPσ splice variants PTPσMeA+MeB+, PTPσMeA+MeB−, PTPσMeA−MeB+, and PTPσMeA−MeB− were constructed by amplifying the indicated full-length human PTPσ splice variants by PCR, digesting with NheI and BsrGI using the corresponding pDisplay constructs as templates, and subcloning the resulting products into the L-313 vector (lentiviral expression vector). pDisplay-PTPσ encoding the full-length human PTPσMeA+MeB+ splice variant (GenBank accession number BC143287.1) was used as a template, and all PTPσ mutant constructs (except ΔFN1–2, ΔCyto, ΔD2, and SWAP) were cloned into the pDisplay vector via BglII/SalI sites. shRNA sequences targeting rat PTPσ differ by four nucleotides from the corresponding human PTPσ (Yim et al., 2013). Lentiviral expression constructs for rescue experiments were generated by PCR amplification of wild-type (WT) and mutated PTPσ WT, followed by digestion with NheI and BsrGI and subcloning into the L-313 vector. The PTPσ/PTPδ Swap construct was created by PCR amplification of the entire extracellular region of human PTPσ (GenBank accession number BC143287.1; aa 30–890) and of mouse PTPδ (GenBank accession number NM_011211.3; aa 1296–1917) and sequentially cloning the individual PCR fragments into the L-313 vector using an In-Fusion HD cloning kit (Clontech). The original mouse PTPδ (mPTPδ) sequence, 5′-GTG CCG GCT AGA AAC TTG-3′, corresponding to the shRNA target site, was mutated to 5′-GTA CCG GCG AGG AAT TTG-3′ (altered residues indicated by underlining) in the pcDNA3.1 Myc/His-mPTPδ (isoform C) vector (a gift from Dr. Fumio Nakamura, Yokohama City University, Japan), and the resulting construct (encoding residues 28–1917 of mPTPδ) was PCR-amplified and cloned into XmaI/SacII sites in the pDisplay vector (Invitrogen) to create an shRNA-resistant PTPδ expression construct. An L-313 construct of this shRNA-resistant PTPδ variant was generated by PCR amplification using pDisplay-PTPδ as a backbone, and subcloning the resulting product into NheI/BsrGI sites in the L-313 vector. Sequences of oligonucleotides for cloning of various PTPσ and PTPδ constructs are presented in Table 2.
Table 1.
List of shRNA constructs
| shRNA construct | Virus type | Target nucleotide sequence (5′-3′) | Reference | Full name |
|---|---|---|---|---|
| L-309 sh-PTPσ | Lentivirus | GCCACACACCTTCTATAAT | Yim et al., 2013 | Protein tyrosine phosphatase, receptor type, S (Ptprs) |
| L-309 sh-PTPδ | Lentivirus | GTGCCGGCTAGAAACTTG | Yim et al., 2013 | Protein tyrosine phosphatase, receptor type, D (Ptprd) |
| L-309 sh-LAR | Lentivirus | GCCTACATAGCTACACAG | Yim et al., 2013 | Leukocyte common antigen-related phosphatase (LAR) |
| L-315 sh-β-catenin | Lentivirus | GCAATCAGCTGGCCTGGTTTG | Current study | β-Catenin (Ctnnb1) |
| L-315 sh-p250RhoGAP | Lentivirus | ACAAGAAGCACCAAGTA | Nakazawa et al., 2008 | Rho GTPase activating protein 32 (Arhgap32) |
| L-315 sh-Lipirin-α2 | Lentivirus | AGCCAGTCTGATTACAGAA | Current study | PTPRF-interacting protein α2 (Ppfia2) |
| L-315 sh-Lipirin-α3 | Lentivirus | GCTAACATGAAGAAGCTTCAA | Current study | PTPRF-interacting protein α3 (Ppfia3) |
| L-315 sh-N-cadherin | Lentivirus | GGACAACTGTCAGTCACAAAG | Current study | Cadherin 2 |
Table 2.
Overview of PTPσ expression constructs
| PTPσ constructs | Amino acid boundary | Oligo sequence (5′-3′) |
|---|---|---|
| PTPσMeA + MeB+ | PTPσ (30-1516) | For pDisplay vector cloning: |
| Forward: GCG AGATCT GAAGAGCCCCCCAGG | ||
| Reverse: GCG GTCGAC TTAGGTTGCATAGTGGTCAAAG | ||
| For L-313 vector cloning: | ||
| Forward: ATCCGAATTC GCTAGC ATGGAGACAGACACACTCCT | ||
| Reverse: CCGCTTTACT TGTACA TTAGGTTGCATAGTGGTCAAAGC | ||
| PTPδ | PTPδ (28-1917) | For pDisplay vector cloning: |
| Forward: GGCCAGATCT CCCGGG GAGACACCCCCCAGGT | ||
| Reverse: CGACCTGCAG CCGCGGC TACGTTGCATAGTGATCAAA | ||
| For L-313 vector cloning: | ||
| Forward: ATCCGAATTC GCTAGC ATGGAGACAGACACACTCCT | ||
| Reverse: CCGCTTTACT TGTACA CTACGTTGCATAGTGATCAAA | ||
| ΔIg | PTPσ (321-1516) | For pDisplay vector cloning: |
| Forward: GCG AGATCT CTCCCCAAAGCTCCC | ||
| Reverse: GCG GTCGAC TTAGGTTGCATAGTGGTCAAAG | ||
| For L-313 vector cloning: | ||
| Forward: ATCCGAATTC GCTAGC ATGGAGACAGACACACTCCT | ||
| Reverse: CCGCTTTACT TGTACA TTAGGTTGCATAGTGGTCAAAGC | ||
| ΔFN1-2 | PTPσ (30-335/526-1516) | For L-313 vector cloning: 1st fragment |
| Forward: ATCCGAATTC GCTAGC ATGGAGACAGACACACTCCT | ||
| Reverse: CCGCTTTACT TGTACA TTTGGGGAGAGATCTAGATTTCAC | ||
| For L-313 vector cloning: 2nd fragment | ||
| Forward: TCTCCCCAAA TGTACA GTGCCGGGCCAG | ||
| Reverse: CCGCTTTACT TGTACA TTAGGTTGCATAGTGGTCAAAGC | ||
| ΔEcto | PTPσ (856-1516) | For pDisplay vector cloning: |
| Forward: GCG AGATCT GACCCCCAGCCCA | ||
| Reverse: GCG GTCGAC TTAGGTTGCATAGTGGTCAAAG | ||
| For L-313 vector cloning: | ||
| Forward: ATCCGAATTC GCTAGC ATGGAGACAGACACACTCCT | ||
| Reverse: CCGCTTTACT TGTACA TTAGGTTGCATAGTGGTCAAAGC | ||
| ΔCyto | PTPσ (30-890) | For L-313 vector cloning: |
| Forward: ATCCGAATTC GCTAGC ATGGAGACAGACACACTCCT | ||
| Reverse: CCGCTTTACT TGTACA TTAGTAGAGCAGGATAGCAATGA | ||
| ΔD2 | PTPσ (30-1237) | For L-313 vector cloning: |
| Forward: ATCCGAATTC GCTAGC ATGGAGACAGACACACTCCT | ||
| Reverse: CCGCTTTACT TGTACA TTAGGCCAGCTTCTGGATGTAGGC | ||
| Swap | PTPσ (30-890)/PTPδ (1296-1917) | For L-313 vector cloning: 1st fragment |
| Forward: ATCCGAATTC GCTAGC ATGGAGACAGACACACTCCT | ||
| Reverse: CCGCTTTACT TGTACA GTAGAGCAGGATAGCAATGACA | ||
| For L-313 vector cloning: 2nd fragment | ||
| Forward: CCTGCTCTAC TGTACA AAAAGGAAGAGGGCAGAGT | ||
| Reverse: CCGCTTTACT TGTACA CTACGTTGCATAGTGATCAAA | ||
| C1157S | PTPσ (30-1516) | For mutagenesis: |
| Forward: CCATCGTGGTTCACAGCAGTGCCGGTGTG | ||
| Reverse: CACACCGGCACTGCTGTGAACCACGATGG | ||
| For L-313 vector cloning: | ||
| Forward: ATCCGAATTC GCTAGC ATGGAGACAGACACACTCCT | ||
| Reverse: CCGCTTTACT TGTACA TTAGGTTGCATAGTGGTCAAAGC | ||
| R781A | PTPσ (30-1516) | For mutagenesis: |
| Forward: GAGGCGCAGCCTGGCGCACTCGCGTCAG | ||
| Reverse: CTGACGCGAGTGCGCCAGGCTGCGCCTC | ||
| For L-313 vector cloning: | ||
| Forward: ATCCGAATTC GCTAGC ATGGAGACAGACACACTCCT | ||
| Reverse: CCGCTTTACT TGTACA TTAGGTTGCATAGTGGTCAAAGC | ||
| D1125A | PTPσ (30-1516) | For mutagenesis: |
| Forward: CGGCGTGGCCGGCCCATGGCGTGCC | ||
| Reverse: GGCACGCCATGGGCCGGCCACGCCG | ||
| For L-313 vector cloning: | ||
| Forward: ATCCGAATTC GCTAGC ATGGAGACAGACACACTCCT | ||
| Reverse: CCGCTTTACT TGTACA TTAGGTTGCATAGTGGTCAAAGC | ||
| AAAA | PTPσ (30-1516) | For mutagenesis: |
| Forward: GTCTCAAAGCGCTGAGAGTTGACCGCCGCGCCCGCCGCGTTCCAGGTCACTCGTGGCTTGGG | ||
| Reverse: CCCAAGCCACGAGTGACCTGGAACGCGGCGGGCGCGGCGGTCAACTCTCAGCGCTTTGAGAC | ||
| For L-313 vector cloning: | ||
| Forward: ATCCGAATTC GCTAGC ATGGAGACAGACACACTCCT | ||
| Reverse: CCGCTTTACT TGTACA TTAGGTTGCATAGTGGTCAAAGC | ||
| R97/100A | PTPσ (30-1516) | For mutagenesis: |
| Forward: CAGCCGCTGGCGACACCGGCGGATGAAAACGTGTACG | ||
| Reverse: CGTACACGTTTTCATCCGCCGGTGTCGCCAGCGGCTG | ||
| For L-313 vector cloning: | ||
| Forward: ATCCGAATTC GCTAGC ATGGAGACAGACACACTCCT | ||
| Reverse: CCGCTTTACT TGTACA TTAGGTTGCATAGTGGTCAAAGC | ||
| Y233S | PTPσ (30-1516) | For mutagenesis: |
| Forward: CACCTGCCAACCTCTCCGTGCGAGAGCTTCG | ||
| Reverse: CGAAGCTCTCGCACGGAGAGGTTGGCAGGTG | ||
| For L-313 vector cloning: | ||
| Forward: ATCCGAATTC GCTAGC ATGGAGACAGACACACTCCT | ||
| Reverse: CCGCTTTACT TGTACA TTAGGTTGCATAGTGGTCAAAGC | ||
| R235D | PTPσ (30-1516) | For mutagenesis: |
| Forward: CTGCCAACCTCTACGTGGACGAGCTTCGAGAAGTCCG | ||
| Reverse: CGGACTTCTCGAAGCTCGTCCACGTAGAGGTTGGCAG | ||
| For L-313 vector cloning: | ||
| Forward: ATCCGAATTC GCTAGC ATGGAGACAGACACACTCCT | ||
| Reverse: CCGCTTTACT TGTACA TTAGGTTGCATAGTGGTCAAAGC |
Antibodies.
The following antibodies were obtained commercially: mouse monoclonal anti-GAD67 (clone 1G10.2; Millipore, RRID:AB_2278725); rabbit polyclonal anti-vesicular glutamate transporter 1 (VGLUT1) (Synaptic Systems, RRID:AB_887880); mouse monoclonal anti-ELKS1 (clone ELKS-30; Sigma-Aldrich, RRID:AB_2100013), and rabbit polyclonal anti-hemagglutinin (HA) (Sigma-Aldrich, RRID:AB_260070); mouse monoclonal anti-CASK (clone K56A/50; NeuroMab, RRID:AB_2068730), mouse monoclonal anti-PSD-95 (clone K28/43; NeuroMab, RRID:AB_2307331), and mouse monoclonal anti-GluN2B (clone BWJHL; Millipore, RRID:AB_417391); mouse monoclonal anti-PTPσ (MediMabs, RRID:AB_1808357); goat polyclonal anti-EGFP (Rockland, RRID:AB_218182); mouse monoclonal anti-HA (clone 16B12; Covance, RRID:AB_2314672); mouse monoclonal anti-Caskin-2 (Santa Cruz Biotechnology, RRID:AB_2713992); and mouse monoclonal anti-β-actin (clone C4; Santa Cruz Biotechnology, RRID:AB_626632). Rabbit polyclonal anti-Synapsin (JK014, RRID:AB_2651124) was acquired as previously described (Han et al., 2016). The following antibodies were gifts from the indicated investigators: rabbit polyclonal anti-Liprin-α2 and rabbit polyclonal anti-Liprin-α3 (Dr. Susanne Schoch, University of Bonn Medical Center, Bonn, Germany); rabbit polyclonal anti-Caskin-1 antibody (Dr. Katsuhiko Tabuchi, Shinshu University, Nagano Prefecture, Japan); and rat polyclonal anti-PTPδ antibody (Dr. Fumio Nakamura, Yokohama City University, Yokohama, Japan).
Cell-surface binding assays.
Recombinant Fc-fusion proteins of NGL-3 (NGL-3-Fc) and GPC-4 (GPC-4-Fc) were produced in HEK293T cells as previously described (Boucard et al., 2012; Ko et al., 2015b). Soluble Fc-fusion proteins were purified using protein A-Sepharose beads (GE Healthcare). Bead-bound proteins were eluted with 0.1 m glycine, pH 2.5, and then neutralized with 1 m Tris-HCl, pH 8.0. Transfected HEK293T cells expressing HA-tagged PTPσ WT or various PTPσ variants were incubated with 0.2 μm NGL-3-Fc, GPC-4-Fc, or Fc alone (negative control), as indicated. Images were acquired using a confocal microscope (LSM700; Carl Zeiss).
Surface/intracellular protein staining assays in HEK293T cells.
HEK293T cells (RRID:CVCL_0063) were transfected with expression vectors for HA-PTPσ WT or its various mutants for 24 h. The cells were then washed twice with PBS, fixed with 4% PFA for 10 min at 4°C, blocked with 3% horse serum/0.1% BSA (crystalline grade) in PBS for 15 min at room temperature, and incubated with 1 mg/ml mouse anti-HA antibody at room temperature. After 1 h 30 min, cells were washed twice with PBS and incubated with Cy3-conjugated anti-mouse antibodies (1:500 in blocking solution) for 1 h at room temperature. Cells were then permeabilized with 0.2% Triton X-100 in PBS for 10 min at 4°C, washed, and then incubated with rabbit anti-HA antibody for 1 h 30 min at room temperature to label intracellularly expressed PTPσ WT or its mutants. Immunoreactive proteins were detected by incubation with FITC-conjugated anti-rabbit secondary antibodies (1:150 in blocking solution). All experiments were repeated three times.
qRT-PCR in cultured neurons.
Cultured rat cortical neurons plated on a 12-well plate were treated with the indicated lentiviruses at DIV4. Lentivirus-infected neurons were homogenized at DIV11 in TRIzol Reagent (Invitrogen), and total RNA was extracted according to the manufacturer's protocol. cDNA was prepared from total RNA by random priming using a cDNA synthesis kit (Takara Bio). qRT-PCR was performed using a CFX96 Real-Time PCR system (Bio-Rad) and SYBR Green reagents (Takara Bio). mRNA for the ubiquitously expressed β-actin was detected as an endogenous control. Sequences of oligonucleotides used for qRT-PCR are presented in Table 3.
Table 3.
Oligonucleotides for qPCRs
| Gene | Species | Oligo sequence (5′-3′) | Reference |
|---|---|---|---|
| PTPσ | Rattus norvegicus | GAACCGATACGCCAATGTCA | Yim et al., 2013 |
| PTPδ | Rattus norvegicus | GGCGGATTGCAGCATAGG | Yim et al., 2013 |
| LAR | Rattus norvegicus | CCCGATGGCTGAGTACAACA | Yim et al., 2013 |
| β-Catenin | Rattus norvegicus | GCACTATGGCAGACACCATC | Current study |
| p250RhoGAP | Rattus norvegicus | GACCTGGAAGGTGAACAGGT | Current study |
| Liprin-α2 | Rattus norvegicus | CTGCCTCTCTTGAGCCAGATAGA | Current study |
| Liprin-α3 | Rattus norvegicus | CTGCCCCAGTACCGAAGCT | Current study |
| N-cadherin | Rattus norvegicus | TGGAAGGCAATCCCACTTAC | Current study |
| Actin | Rattus norvegicus | CCTGGACTTCGAGAATGAGATG | Current study |
Production of recombinant lentiviruses.
HEK293T cells were transfected with three plasmids, lentivirus vectors, psPAX2, and pMD2G, at a 2:2:1 ratio using FuGene-6 (Roche Diagnostics), according to the manufacturer's protocol. After 72 h, lentiviruses were harvested by collecting the media from transfected HEK293T cells and briefly centrifuging at 1000 × g to remove cellular debris. The filtered media with 5% sucrose were centrifuged at ∼118,000 × g for 2 h, after which supernatants were removed and washed with ice-cold PBS. The virus pellet was resuspended in 80 μl of PBS.
Heterologous synapse-formation assays.
Heterologous synapse-formation assays were performed as previously described (Kang et al., 2016). HEK293T cells were transfected with the indicated expression vectors or EGFP alone (Control) using FuGene (Roche Diagnostics). After 48 h, transfected HEK293T cells were trypsinized, seeded onto cultured hippocampal neurons at DIV9, and then coimmunostained with antibodies against the indicated epitopes and synaptic marker proteins at DIV11. Images were acquired by confocal microscopy (LSM700, Carl Zeiss). For quantification purposes, the contours of transfected HEK293T cells were chosen as the ROI. The fluorescence intensities of synaptic marker puncta, normalized with respect to the area of each HEK293T cell, were quantified for both red and green channels using MetaMorph Software (Molecular Devices, RRID:SCR_002368).
Primary neuronal culture, infections, immunocytochemistry, image acquisition, and analyses.
Hippocampal and cortical rat neuron cultures were prepared from embryonic day 18 (E18) rat embryos. Rat cultured neurons at DIV4 were infected with KD lentiviruses alone or together with rescue viruses, and immunostained at DIV14. For immunocytochemistry, cultured rat neurons were fixed with 4% PFA/4% sucrose in PBS for 10 min at 4°C, and permeabilized with 0.2% Triton X-100 in PBS for 10 min at 4°C. Neurons were then blocked with 3% horse serum/0.1% BSA in PBS for 15 min at room temperature and incubated with the indicated primary and secondary antibodies in blocking solution for 90 min at room temperature. Images of randomly selected neurons were acquired using a confocal microscope (LSM700, Carl Zeiss) with a 63× objective lens; all image settings were kept constant during image acquisition. z stack images obtained through confocal microscopy were converted to maximal projections, and puncta size and density of the indicated presynaptic marker proteins were analyzed in a blinded manner using MetaMorph software (Molecular Devices).
Semiquantitative immunoblotting in cultured neurons.
Cultured cortical rat neurons were prepared and infected with the indicated lentiviruses at DIV4. Neuron lysates were prepared at DIV11 and analyzed by immunoblotting with the indicated primary antibodies. Western blot signals were detected using an enhanced chemiluminescence system (PerkinElmer Life Sciences) and quantified using ImageJ software (Fiji, RRID:SCR_002285). For quantification of Western blot signals, a series of dilutions of neuron lysates was loaded to ensure that the signal intensity of β-actin (used as an internal control for normalization) changed linearly and in proportion to the amount of loaded protein, and that the intensities of Western blot signals were not saturated.
Electrophysiology.
Cell culture electrophysiology was performed as previously described (Ko et al., 2011; Um et al., 2014b). For recording mEPSCs, neurons infected with the indicated lentiviruses were patched with the following internal solution: 90 mm Cs-gluconate, 10 mm CsCl, 10 mm HEPES, 10 mm eEGTA, 5 mm NaCl, 4 mm Mg-ATP, and 0.3 mm Na-GTP. For mIPSCs, the internal solution was 100 mm CsCl, 5 mm NaCl, 10 mm EGTA, 10 mm HEPES, 4 mm Mg-ATP, and 0.3 mm Na-GTP. The recordings were performed in aCSF with the following composition: 100 mm NaCl, 4 mm KCl, 1 mm NaH2PO4, 20 mm HEPES, 30 mm glucose, 2 mm CaCl2, and 1 mm MgCl2. mEPSCs and mIPSCs were recorded at a holding potential of −70 mV in the presence of 1 μm TTX (Ascent Scientific) and 100 μm picrotoxin (for mEPSCs) or 1 μm TTX and 10 μm CNQX (for mIPSCs). The experimenter was blinded to the identity of the infected neurons throughout data collection and analysis. All recordings were digitized at 10 kHz and filtered at 2 kHz. Recordings were monitored with an EPC10 double USB patch-clamp amplifier (HEKA Elektronik) and analyzed offline using the Mini Analysis Program (Synaptosoft, RRID:SCR_002184).
Experimental design and statistical analysis.
Synaptogenic activity in heterologous synapse-formation assays and synaptic puncta in cultured hippocampal neurons were analyzed using HEK293T cells or cultured neurons infected with the indicated lentiviruses (for details, see figure legends). Data analysis and statistical tests were performed using Prism7 software (GraphPad, RRID:SCR_002798). Data are expressed as mean ± SEM unless stated otherwise, and significance is indicated with asterisk (compared with a value from control group) or hashtag (compared with a value from experimental group). All experiments were performed using at least three independent cultures, and the normality of data distributions was evaluated using the Shapiro–Wilk test. Data were compared by Student's t test, one-way ANOVA using a nonparametric Kruskal–Wallis test, followed by Dunn's multiple-comparison test for post hoc group comparisons, t test, or Mann–Whitney U test; n values used are indicated in the figure legends. Numbers shown indicate replicates, and tests to determine statistical significance are stated in the text and figure legends depicting results of the respective experiments. A p value <0.05 was considered statistically significant, and individual p values are indicated in the respective figure legend.
Results
PTPσ and PTPδ are selectively required for presynaptic assembly triggered by various postsynaptogenic adhesion molecules
The three members of the LAR-RPTP protein family bind to a host of postsynaptic adhesion molecules, including netrin-G ligand 3 (NGL-3), TrkC, interleukin 1 receptor accessory protein-like 1 (IL1RAPL1), interleukin 1 receptor accessory protein (IL-1RAcP), Slitrks (Slitrk1–6), glypicans (GPC-1–6), SALM3, and SALM5 (Um and Ko, 2013; Han et al., 2016). Previous studies have indicated that (1) PTPδ is required for IL1RAPL1- and IL-1RAcP-triggered excitatory heterologous synapse formation; (2) PTPδ and PTPσ are required for Slitrk-induced inhibitory and excitatory heterologous synapse formation, respectively; (3) LAR, PTPδ, and PTPσ are required for SALM3-induced heterologous synapse formation; and (4) PTPσ is required for SALM5-induced heterologous synapse formation (Valnegri et al., 2011; Yoshida et al., 2011; Takahashi et al., 2012; Yim et al., 2013; Li et al., 2015; Choi et al., 2016). However, whether proteins from the LAR-RPTP family are also required for NGL-3- or TrkC-induced heterologous synapse formation has not been tested. Thus, we first sought to determine which LAR-RPTPs are critical for NGL-3- and TrkC-dependent heterologous synapse formation. To this end, we cocultured HEK293T cells expressing NGL-3 or TrkC with hippocampal neurons infected with lentiviruses expressing an empty shRNA vector (sh-Control) or an shRNA KD construct targeting LAR (sh-LAR), PTPδ (sh-PTPδ), or PTPσ (sh-PTPσ) for 2 d and performed a series of heterologous synapse formation assays using VGLUT1 as a marker to visualize excitatory presynaptic sites. We found that PTPσ KD (sh-PTPσ) impaired heterologous excitatory synapse formation induced by TrkC, consistent with the specific interaction of TrkC with PTPσ (Fig. 1A,B) (Takahashi et al., 2011). In addition, PTPσ KD (sh-PTPσ) specifically impaired heterologous excitatory synapse formation triggered by Slitrk1, whereas PTPδ KD (sh-PTPδ) specifically impaired heterologous excitatory synapse formation induced by IL1RAPL1 (Fig. 1A,B) (Valnegri et al., 2011; Yoshida et al., 2011; Yim et al., 2013). Remarkably, KD of individual LAR-RPTP family proteins (sh-PTPσ, sh-PTPδ, or sh-LAR) did not impair NGL-3-induced heterologous synapse formation (Fig. 1A,B). Surprisingly, KD of all three LAR-RPTPs (sh-PTPσ, PTPδ, LAR) had no effect on NGL-3-mediated heterologous excitatory synapse formation (Fig. 1). In control experiments, heterologous synapse formation induced by neuroligin-1 (NL-1) was not affected by KD of individual LAR-RPTPs (Fig. 1A,B). Together, these data indicate that PTPσ and PTPδ are distinctively required for heterologous excitatory synapse formation mediated by LAR-RPTP-interacting postsynaptic ligands.
Figure 1.
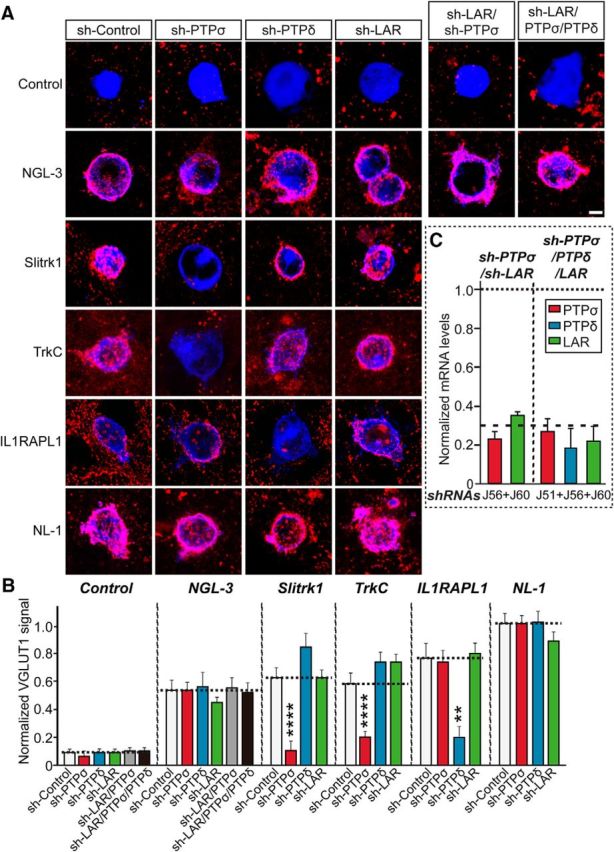
PTPσ and PTPδ are selectively required for the heterologous synapse-formation activities of distinct postsynaptogenic adhesion molecules. A, Representative images of the heterologous synapse-formation activities of various LAR-RPTP ligands. Neurons were infected at DIV4 with lentiviruses expressing sh-Control, sh-PTPσ, sh-PTPδ, sh-LAR, sh-LAR/sh-PTPσ, or sh-PTPσ/PTPδ/LAR as indicated, and then cocultured from DIV9 to DIV11 with HEK293T cells expressing various postsynaptic ligands. Neurons were stained with antibodies against HA or EGFP (blue) and VGLUT1 (red). Scale bar (all images), 10 μm. B, The synapse-forming activity in A was quantified by measuring the ratio of VGLUT1 staining intensity (red) to HA or EGFP immunoreactivity intensity (blue). Data are mean ± SEM. Mann–Whitney U test: ****p < 0.0001. n = number of neurons as follows: sh-Control/Control, n = 13; sh-PTPσ/Control, n = 15; sh-PTPδ/Control, n = 17; sh-LAR/Control, n = 15; sh-LAR/sh-PTPσ/Control, n = 11; sh-LAR/PTPσ/PTPδ/Control, n = 12; sh-Control/NGL-3, n = 18; sh-PTPσ/NGL-3, n = 17; sh-PTPδ/NGL-3, n = 16; sh-LAR/NGL-3, n = 16; sh-LAR/sh-PTPσ/NGL-3, n = 16; sh-LAR/PTPσ/PTPδ/NGL-3, n = 16; sh-Control/Slitrk1, n = 13; sh-PTPσ/Slitrk1, n = 15; sh-PTPδ/Slitrk1, n = 13; sh-LAR/Slitrk1, n = 11; sh-Control/TrkC, n = 15; sh-PTPσ/TrkC, n = 11; sh-PTPδ/TrkC, n = 12; sh-LAR/ TrkC, n = 11; sh-Control/IL1RAPL1, n = 11; sh-PTPσ/IL1RAPL1, n = 13; sh-PTPδ/IL1RAPL1, n = 14; sh-LAR/IL1RAPL1, n = 12; sh-Control/NL-1, n = 11; sh-PTPσ/NL-1, n = 12; sh-PTPδ/NL-1, n = 10; and sh-LAR/NL-1, n = 11. p values for Control condition: sh-Control vs sh-PTPσ, p = 0.439; sh-Control vs sh-PTPδ, p = 0.5604; sh-Control vs sh-LAR, p = 0.1835; sh-Control vs sh-LAR/sh-PTPσ, p = 0.9534; and sh-Control vs sh-LAR/PTPσ/PTPδ, p = 0.5641. p values for NGL-3 condition: sh-Control vs sh-PTPσ, p = 0.4129; sh-Control vs sh-PTPδ, p = 0.7706; sh-Control vs sh-LAR, p = 0.5832; sh-Control vs sh-LAR/sh-PTPσ, p = 0.8166; and sh-Control vs sh-LAR/PTPσ/PTPδ, p = 0.9921. p values for Slitrk1 condition: sh-Control vs sh-PTPσ, p < 0.0001; sh-Control vs sh-PTPδ, p = 0.3394; and sh-Control vs sh-LAR, p = 0.684. p values for TrkC condition: sh-Control vs sh-PTPσ, p < 0.0001; sh-Control vs sh-PTPδ, p = 0.1433; and sh-Control vs sh-LAR, p = 0.2958. p values for IL1RAPL1 condition: sh-Control vs sh-PTPσ, p = 0.7832; sh-Control vs sh-PTPδ, p < 0.01; and sh-Control vs sh-LAR, p = 0.5212. p values for NL1 condition: sh-Control vs sh-PTPσ, p = 0.9537; sh-Control vs sh-PTPδ, p = 0.8919; and sh-Control vs sh-LAR, p = 0.6262. C, Levels of PTPσ, PTPδ, and LAR mRNAs were measured at DIV12-DV13 by qRT-PCR in cultured cortical neurons infected at DIV4 with lentiviruses expressing the indicated shRNAs. Dashed line indicates 70% knockdown cutoff level for tests of biological effects.
PTPσ and PTPδ are selectively required for excitatory and inhibitory synapse structure/transmission, respectively, in cultured hippocampal neurons
Recombinant PTPσ and PTPδ were previously reported to localize to excitatory and inhibitory synaptic sites, respectively, in cultured hippocampal neurons (Takahashi et al., 2011, 2012); however, the subcellular localization of endogenous PTPδ in cultured neurons was not assessed (Takahashi et al., 2012). Therefore, we examined whether endogenous PTPδ specifically localizes to inhibitory synaptic sites. The PTPσ and PTPδ antibodies used in the current study exhibited no cross-reactivities, demonstrating specificity for the target antigen (Fig. 2A). The authenticity of these antibodies was further validated by showing that immunoreactivity to them was diminished in cultured hippocampal neurons infected with a KD virus targeting the corresponding protein (Fig. 2B,C). Using these validated antibodies, we found that PTPδ immunoreactivity was present in a punctate pattern decorating the dendrites of cultured hippocampal neurons at DIV14; these puncta overlapped with glutamate decarboxylase 1 (GAD67) by ∼48%, but with VGLUT1 by only ∼10% (Fig. 2D,E). PTPδ puncta that overlapped with GAD67 were also observed on axons that were not in contact with dendrites, suggesting an axonal localization (Fig. 2D,E). Thus, endogenous PTPδ is also specifically localized to inhibitory synaptic sites, consistent with the distribution pattern of recombinant PTPδ proteins (Takahashi et al., 2012). Endogenous PTPσ puncta showed a greater degree of colocalization with VGLUT1 than with vesicular GABA transporter (VGAT), as previously reported (Takahashi et al., 2011) (Fig. 2F,G).
Figure 2.

Specific localization of PTPσ and PTPδ at excitatory and inhibitory synaptic sites in cultured hippocampal neurons. A, Immunoblot analyses of PTPδ and PTPσ using lysates from HEK293T cells transfected with HA-tagged PTPδ (HA- PTPδ) or PTPσ (HA-PTPσ). The expression of HA-tagged PTP isoforms was confirmed by immunoblotting with anti-HA antibodies. Unt., Untransfected HEK293T cell lysates; Trans. (HA-PTPδ), lysates from HEK293T cells transfected with HA-PTPδ; Trans. (HA-PTPσ), lysates from HEK293T cells transfected with HA-PTPσ. B, Authenticity testing of anti-PTPδ and anti-PTPσ antibodies. Mature cultured hippocampal neurons (DIV14) infected with lentiviruses expressing sh-Control, sh-PTPδ (PTPδ KD), or sh-PTPσ (PTPσ KD) were labeled by single immunofluorescence staining for the indicated PTP antibodies (gray). Scale bar (all images), 10 μm. C, Summary graphs of B. Data are mean ± SEM. **p < 0.01 (Mann–Whitney U test). n = number of analyzed neurons as follows: sh-Control/PTPσ, n = 10; sh-PTPσ/PTPσ, n = 10; sh-Control/PTPδ, n = 10; and sh-PTPδ/PTPδ, n = 11. p value for PTPσ antibody: sh-Control vs sh-PTPσ, p = 0.0052. p value for PTPδ antibody: sh-Control vs sh-PTPδ, p = 0.0048. D, F, Immunolocalization of PTPδ (D) and PTPσ (F) in cultured hippocampal neurons, along with colocalization of GAD67, VGAT, or VGLUT1 as indicated. Scale bar (all images), 10 μm. E, G, Quantification of colocalization of endogenous PTPδ (E) and PTPσ (G) with synaptic markers.
We then asked whether KD of PTPσ or PTPδ changed the expression of specific proteins known to be functionally linked to LAR-RPTPs. To test this, we performed semiquantitative immunoblot analyses of 9 selected synaptic proteins (liprin-α2, liprin-α3, Caskin-1, Caskin-2, MIM-B, CASK, ELKS1, GIT1, and GluN2B) in cultured cortical neurons after shRNA-mediated KD of PTPσ or PTPδ. We found no alterations in any of these synaptic proteins (Fig. 3A,B). Interestingly, however, PTPσ KD decreased the levels of PTPσ mRNA by ∼75% (Fig. 1C) and PTPσ protein by ∼84% (Fig. 3A,B), and concurrently increased the levels of PTPδ proteins by 127%, without affecting PTPδ mRNA levels (Figs. 1C, 3A,B). Similarly, PTPδ KD reduced the levels of PTPδ mRNA by ∼80% (Fig. 1C) and PTPδ protein by ∼70% (Fig. 3A,B) while dramatically upregulating PTPσ protein levels by 235% (Fig. 3A,B). These results indicate that loss of PTPσ may activate a compensatory mechanism that upregulates PTPδ, and vice versa.
Figure 3.

Effects of PTPσ and PTPδ KD on the levels of selected synaptic proteins in cultured neurons. A, B, Representative semiquantitative immunoblot images (A) and quantitative summary (B) of the expression levels of the indicated synaptic proteins in cultured cortical neurons infected with sh-Control, sh-PTPσ, or sh-PTPδ lentiviruses. β-Actin was used as a loading control, and protein levels were expressed relative to those in the sh-Control group. Data are mean ± SEM. Parametric t test: *p < 0.05; ***p < 0.001; ****p < 0.0001; n = number of samples as follows: PTPσ, n = 4; PTPδ, n = 4; liprin-α2, n = 3; liprin-α3, n = 3; Caskin-1, n = 4; Caskin-2, n = 3; MIM-B, n = 3; CASK, n = 3; ELKS1, n = 3; GIT1, n = 3; GluN2B, n = 3; NL-1, n = 3; and β-actin, n = 6. p values for PTPσ blots: sh-Control vs sh-PTPσ, p < 0.0001; and sh-Control vs sh-PTPδ, p = 0.0003. p values for PTPδ blots: sh-Control vs sh-PTPσ, p = 0.0108; and sh-Control vs sh-PTPδ, p = 0.0005. p values for liprin-α2 blots: sh-Control vs sh-PTPσ, p = 0.6963; and sh-Control vs sh-PTPδ, p = 0.4269. p values for liprin-α3 blots: sh-Control vs sh-PTPσ, p = 0.524; and sh-Control vs sh-PTPδ, p = 0.437. p values for Caskin-1 blots: sh-Control vs sh-PTPσ, p = 0.643; and sh-Control vs sh-PTPδ, p = 0.6568. p values for Caskin-2 blots: sh-Control vs sh-PTPσ, p = 0.2875; and sh-Control vs sh-PTPδ, p = 0.8973. p values for MIM-B blots: sh-Control vs sh-PTPσ, p = 0.5808; and sh-Control vs sh-PTPδ, p = 0.4229. p values for CASK blots: sh-Control vs sh-PTPσ, p = 0.5549; and sh-Control vs sh-PTPδ, p = 0.4523. p values for ELKS1 blots: sh-Control vs sh-PTPσ, p = 0.0756; and sh-Control vs sh-PTPδ, p = 0.4072. p values for GIT1 blots: sh-Control vs sh-PTPσ, p = 0.0878; and sh-Control vs sh-PTPδ, p = 0.3826. p values for GluN2B blots: sh-Control vs sh-PTPσ, p = 0.4656; and sh-Control vs sh-PTPδ, p = 0.0672. p values for NL-1 blots: sh-Control vs sh-PTPσ, p = 0.4262; and sh-Control vs sh-PTPδ, p = 0.5466. p values for β-actin blots: sh-Control vs sh-PTPσ, p = 0.662; and sh-Control vs sh-PTPδ, p = 0.9238.
We next tested the effect of PTPσ or PTPδ KD on the density of synapses formed between neurons (Fig. 4A–E) and synaptic transmission in cultured hippocampal neurons (Fig. 4F–K). In line with our previous observation, immunocytochemical analyses showed that PTPσ KD specifically decreased the degree of excitatory, but not inhibitory, synapse density, an effect that was completely rescued by reexpression of full-length WT PTPσ (Ko et al., 2015b) (Fig. 4A–E). Conversely, PTPδ KD specifically reduced the degree of inhibitory, but not excitatory, synapse density, an effect that was similarly rescued by reexpression of full-length WT PTPδ (Fig. 4A–E). To corroborate the anatomical effects of PTPσ or PTPδ KD, we next infected cultured hippocampal neurons with lentiviruses expressing sh-PTPσ, sh-PTPδ, sh-LAR, or sh-Control at DIV3, and recorded mEPSCs or mIPSCs at DIV14-DIV15. PTPσ KD significantly decreased the frequency and amplitude of mEPSCs, consistent with our previous report (Ko et al., 2015b), whereas PTPδ KD specifically decreased the frequency and amplitude of mIPSCs (Fig. 4F–K). Collectively, these immunocytochemical and electrophysiological analyses reveal that PTPσ and PTPδ are required for excitatory and inhibitory synapse development, respectively, in cultured hippocampal neurons.
Figure 4.
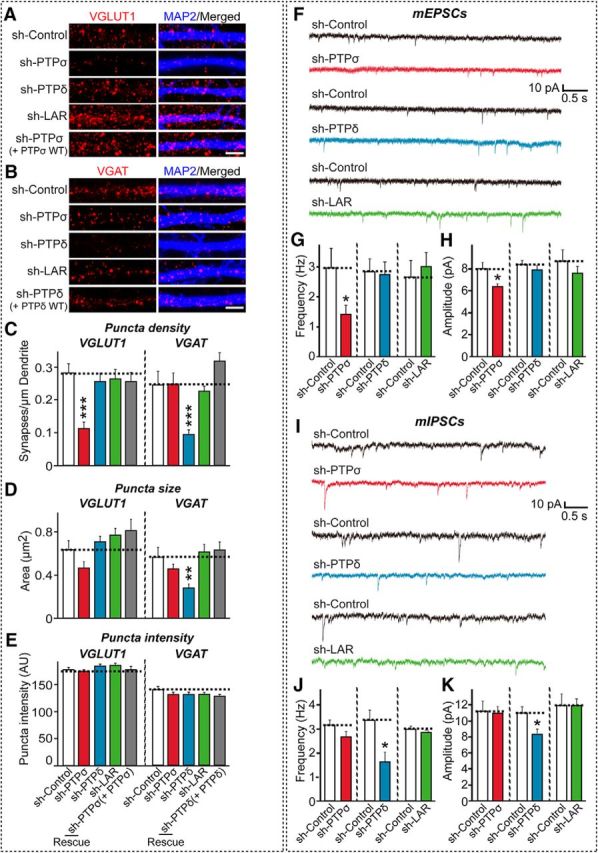
Effects of PTPσ and PTPδ KD on synapse density and synaptic transmission in cultured neurons. A, B, Cultured hippocampal neurons were infected with lentiviruses expressing sh-Control, sh-LAR, sh-PTPσ, or sh-PTPδ, or were coinfected with lentiviruses expressing sh-PTPσ or sh-PTPδ together with shRNA-resistant full-length PTPσ (+ PTPσ WT) or sh-PTPδ (+ PTPδ WT) at DIV4, and analyzed at DIV14 by double-immunofluorescence detection of MAP2 and VGLUT1 (A) or VGAT (B). Scale bar (all images), 10 μm. C–E, Summary graphs of A, B. Synaptic puncta density (C), synaptic puncta size (D), and synaptic puncta intensity (E) were measured using VGLUT1 and VGAT as excitatory and inhibitory synaptic markers, respectively. Two or three dendrites per infected neurons were analyzed and group-averaged. Data are mean ± SEM. Mann–Whitney U test: **p < 0.01; ***p < 0.001. n = number of neurons as follows: (E) sh-Control, n = 17; sh-PTPσ, n = 16; sh-PTPδ, n = 15; sh-LAR, n = 15; and sh-PTPσ+PTPσ WT, n = 13. F, sh-Control, n = 13; sh-PTPσ, n = 14; sh-PTPδ, n = 14; sh-LAR, n = 14; and sh-PTPδ+PTPδ WT, n = 14. p values for VGLUT1 puncta density: sh-Control vs sh-PTPσ, p < 0.001; sh-Control vs sh-PTPδ, p = 0.3322; sh-Control vs sh-LAR, p = 0.3765; and sh-Control vs sh-PTPσ (+PTPσ), p = 0.8210. p values for VGAT puncta density: sh-Control vs sh-PTPσ, p = 0.9781; sh-Control vs sh-PTPδ, p < 0.001; sh-Control vs sh-LAR, p = 0.1958; and sh-Control vs sh-PTPδ (+PTPδ), p = 0.8124. p values for VGLUT1 puncta size: sh-Control vs sh-PTPσ, p = 0.0820; sh-Control vs sh-PTPδ, p > 0.9999; sh-Control vs sh-LAR, p = 0.9999; and sh-Control vs sh-PTPσ (+PTPσ), p = 0.9212. p values for VGAT puncta size: sh-Control vs sh-PTPσ, p = 0.1248; sh-Control vs sh-PTPδ, p < 0.01; sh-Control vs sh-LAR, p > 0.9999; and sh-Control vs sh-PTPδ (+PTPδ), p = 0.8790. p values for VGLUT1 puncta intensity: sh-Control vs sh-PTPσ, p = 0.7176; sh-Control vs sh-PTPδ, p = 0.8392; sh-Control vs sh-LAR, p = 0.9115; and sh-Control vs sh-PTPσ (+PTPσ), p = 0.7938. p values for VGAT puncta intensity: sh-Control vs sh-PTPσ, p = 0.2190; sh-Control vs sh-PTPδ, p = 0.4771; sh-Control vs sh-LAR, p = 0.3198; and sh-Control vs sh-PTPδ (+PTPδ), p = 0.6787. F, I, Representative traces of mEPSCs (F) and mIPSCs (I) in neurons infected with control, shPTPσ, sh-PTPδ, or sh-LAR lentiviruses. Neurons were infected with lentiviruses at DIV4, and electrophysiological recordings were obtained at DIV14–16. G, H, Summary graphs of the frequencies (G) and amplitudes (H) of mEPSCs in neurons infected with control, shPTPσ, sh-PTPδ, or sh-LAR lentiviruses. Data are mean ± SEM. *p < 0.05 (Student's t test). n = number of neurons as follows: sh-Control 1, n = 16; sh-PTPσ, n = 21; sh-Control 2, n = 13; sh-PTPδ, n = 30; sh-Control 3, n = 11; and sh-LAR, n = 16. p values for mEPSC frequency: sh-Control 1 vs sh-PTPσ, p < 0.05; sh-Control 2 vs sh-PTPδ, p = 0.4237; and sh-Control 3 vs sh-LAR, p = 0.5211. p values for mEPSC amplitude: sh-Control 1 vs sh-PTPσ, p < 0.05; sh-Control 2 vs sh-PTPδ, p = 0.8921; and sh-Control 3 vs sh-LAR, p = 0.9783. J, K, Same as G, H, except that mIPSCs were measured. Data are mean ± SEM. *p < 0.05 (Student's t test). n = number of neurons as follows: sh-Control 1, n = 16; sh-PTPσ, n = 23; sh-Control 2, n = 11; sh-PTPδ, n = 20; sh-Control 3, n = 16; and sh-LAR, n = 23. p values for mIPSC frequency: sh-Control 1 vs sh-PTPσ, p = 0.3576; sh-Control 2 vs sh-PTPδ, p < 0.05; and sh-Control 3 vs sh-LAR, p = 0.6183. p values for mIPSC amplitude: sh-Control 1 vs sh-PTPσ, p = 0.7120; sh-Control 2 vs sh-PTPδ, p < 0.05; and sh-Control 3 vs sh-LAR, p = 0.8217.
The extracellular region of PTPσ is required for presynaptic assembly
To determine whether specific PTPσ splice variants containing MeA or MeB inserts responsible for Ig domain binding are involved in the maintenance of excitatory synapse development in neurons, we infected PTPσ-deficient neurons with lentiviruses expressing each of four different PTPσ splice variants (PTPσMeA+MeB+, PTPσMeA−MeB+, PTPσMeA+MeB−, or PTPσMeA−MeB−) and analyzed excitatory synapse density, as determined by VGLUT1 staining (Fig. 5). We found that expression of each of the four PTPσ splice variants rescued the deficit in excitatory synapse density observed in PTPσ-deficient neurons, indicating that PTPσ is able to mediate presynaptic assembly independent of binding partners that interact through its Ig domains.
Figure 5.
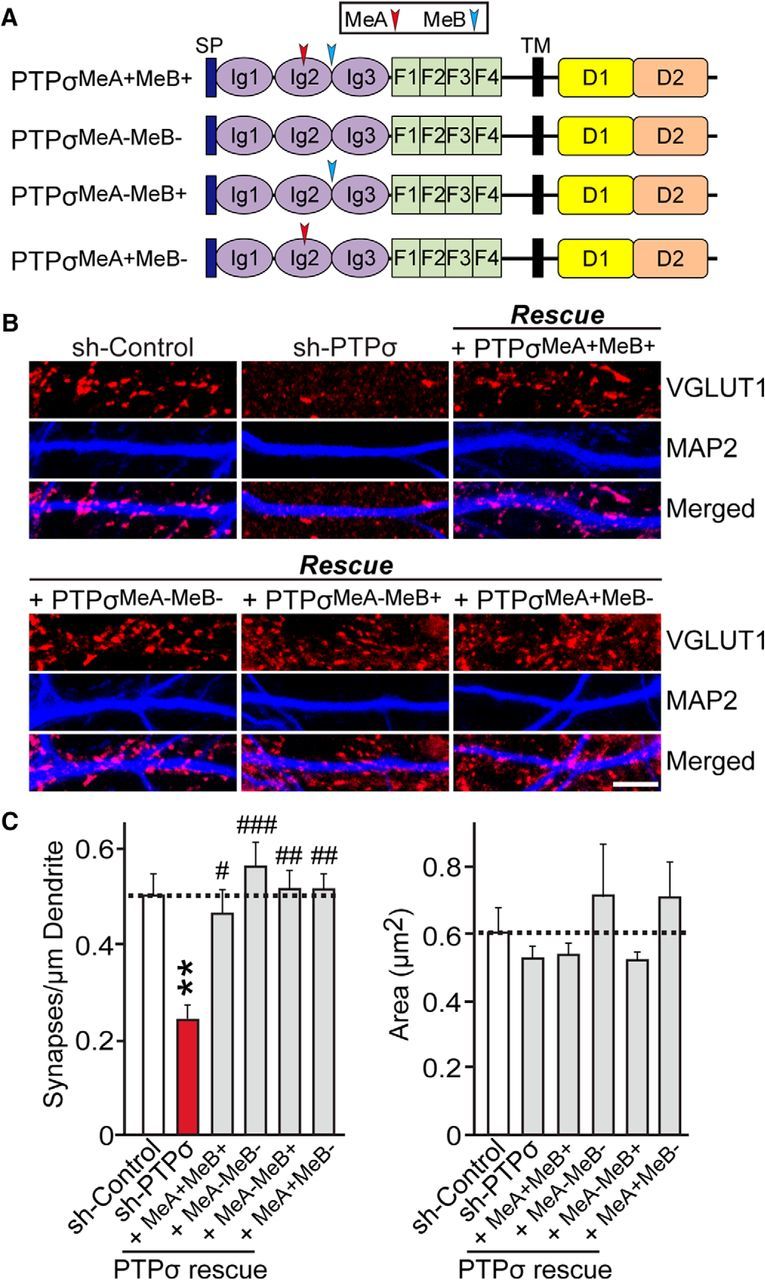
Effect of PTPσ alternative splicing on excitatory synapse development in cultured neurons. A, Schematic diagrams of PTPσ alternative splice variants used in the current study. D1, first catalytic domain of LAR-RPTPs; D2, second catalytic domain of LAR-RPTPs; F, fibronectin Type III repeat; Ig, Ig domain; MeA, mini-exon A; MeB; mini-exon B; SP, IgκB signal peptide contained in the pDisplay vector (Invitrogen); TM, transmembrane region. B, Representative images from cultured hippocampal neurons infected at DIV4 with lentiviruses expressing sh-Control, sh-PTPσ, or coinfected with lentiviruses expressing sh-PTPσ and the human PTPσ alternatively spliced variant rescue viruses, PTPσMeA+MeB+, PTPσMeA−MeB−, PTPσMeA−MeB+, or PTPσMeA+MeB−, and analyzed at DIV14 by double-immunofluorescence detection of MAP2 (blue) and the excitatory synaptic marker VGLUT1 (red). Scale bar (all images), 10 μm. C, Summary graphs of the effects of PTPσ splice variants in neurons on puncta density (left) and puncta size (right), measured using VGLUT1 as an excitatory synaptic marker. Two or three dendrites per infected neuron were analyzed and group-averaged. Data are mean ± SEM. ANOVA with a nonparametric Kruskal–Wallis test: **p < 0.01; #p < 0.05; ##p < 0.01; ###p < 0.001. n = number of neurons as follows: sh-Control/ VGLUT1, n = 14; sh-PTPσ/ VGLUT1, n = 15; PTPσMeA+MeB+ rescue/VGLUT1, n = 15; PTPσMeA−MeB− rescue/VGLUT1, n = 13; PTPσMeA−MeB+ rescue/VGLUT1, n = 15; and PTPσMeA+MeB− rescue/VGLUT1, n = 15. p values for puncta density: sh-Control vs sh-PTPσ, p = 0.0065; sh-Control vs PTPσMeA+MeB+ rescue, p > 0.9999; sh-Control vs PTPσMeA−MeB− rescue, p > 0.9999; sh-Control vs PTPσMeA−MeB+ rescue, p > 0.9999; sh-Control vs PTPσMeA+MeB rescue, p > 0.9999; sh-PTPσ vs PTPσMeA+MeB+ rescue, p = 0.0304; sh-PTPσ vs PTPσMeA−MeB− rescue, p = 0.0002; sh-PTPσ vs PTPσMeA−MeB+ rescue, p = 0.0015; and sh-PTPσ vs PTPσMeA+MeB rescue, p = 0.002. p values for puncta size: sh-Control vs sh-PTPσ, p > 0.9999; sh-Control vs PTPσMeA+MeB+ rescue, p > 0.9999; sh-Control vs PTPσMeA−MeB− rescue, p > 0.9999; sh-Control vs PTPσMeA−MeB+ rescue, p > 0.9999; sh-Control vs PTPσMeA+MeB rescue, p > 0.9999; sh-PTPσ vs PTPσMeA+MeB+ rescue, p > 0.9999; sh-PTPσ vs PTPσMeA−MeB− rescue, p > 0.9999; sh-PTPσ vs PTPσMeA−MeB+ rescue, p > 0.9999; and sh-PTPσ vs PTPσMeA+MeB rescue, p > 0.9999.
Presynaptic neurexins are required for induction of heterologous synapse formation by their postsynaptic ligands, neuroligin-1 or leucine-rich repeat transmembrane protein (LRRTM2), but only the extracellular regions of neurexins are required for neurexin-mediated signal transduction in presynaptic neurons (Gokce and Südhof, 2013). To examine whether presynaptic LAR-RPTPs mediate signal transduction in presynaptic neurons, we first generated lentiviral expression vectors for a variety of PTPσ deletion variants and point mutants containing a HA epitope tag, which we had introduced into most PTPσ rescue constructs to monitor expression level and surface transport (Fig. 6). Because we primarily used Slitrk1 (see Figs. 7–9) or TrkC (see Fig. 10) to induce heterologous synapse formation in the current study, we used a PTPσ splice variant containing both the MeA and MeB inserts (PTPσMeA+MeB+) to generate various PTPσ rescue vectors (Figs. 6–10, 12) (Coles et al., 2014; Um et al., 2014b). All PTPσ variants exhibited the expected molecular weights in immunoblot analyses (Fig. 6B), and, with the exception of a glycosylphosphatidylinositol (GPI)-anchored construct, was effectively targeted to the cell surface following expression in HEK293T cells (Fig. 6C) and bound well to known ligands (NGL-3 or GPC-4), as expected (Fig. 6D; see Fig. 8B).
Figure 6.

Characterization of total expression, surface trafficking, and ligand-binding properties of various PTPσ variants used in the current study. A, Overview of the PTPσ WT and various PTPσ variants (alternative splicing variants, deletion mutants, and point mutants). Alternatively spliced variants: PTPσMeA+MeB+, PTPσ containing an insert in both MeA and MeB splice sites; PTPσMeA−MeB−, PTPσ lacking an insert in both MeA and MeB splice sites; PTPσMeA−MeB+, PTPσ lacking an insert in the MeA splice site and containing an insert in the MeB splice site; and PTPσMeA+MeB−, PTPσ containing an insert in the MeA splice site and lacking an insert in the MeB splice site. Deletion variants: PTPσ ΔIg, PTPσ with a deletion of the three Ig domains; PTPσ ΔFN1–2, PTPσ with a deletion of the first two FNIII domains; PTPσ ΔEcto, PTPσ with a deletion of the entire extracellular region; PTPσ ΔCyto, PTPσ with a deletion of the entire intracellular region; and PTPσ ΔD2, PTPσ with a deletion of the intracellular D2 domain. Point mutants: PTPσ C1157S or D1125A, tyrosine phosphatase activity-defective mutants of PTPσ; PTPσ R781A, a proteolytic cleavage-defective mutant of PTPσ; PTPσ AAAA, a PTPσ mutant in which HS binding was abrogated by replacing four lysines of the first Ig domain (K68, K69, K71, and K72) with alanines; PTPσ R97/100A, a PTPσ mutant in which TrkC binding was abolished by replacing two arginine residues (R97 and R100) with alanines; PTPσ Y233S, a PTPσ mutant in which binding to both Slitrks and TrkC was eliminated by replacing a tyrosine residue (Y233) with serine; and PTPσ R235D, a PTPσ mutant in which binding to Slitrks was disrupted by replacing an arginine residue (R235) with aspartic acid. PTPσ Swap denotes a hybrid of the PTPσ extracellular region with a transmembrane segment and the PTPδ intracellular region. B, Representative immunoblot images from HEK293T cells transfected with HA-tagged PTPσ WT or the indicated PTPσ variants. Samples containing equal amounts of protein were resolved by SDS-PAGE and immunoblotted using anti-HA antibodies. PTPσ GPI denotes a full-length PTPσ in which a transmembrane segment was replaced with a GPI anchor sequence. C, Surface expression analysis of HEK293T cells expressing HA-PTPσ WT or the indicated HA-tagged PTPσ mutant variants. Transfected cells were immunostained with mouse anti-HA antibodies (green) and detected with FITC-conjugated anti-mouse secondary antibodies under nonpermeabilized conditions. Cells were then permeabilized and stained first with rabbit anti-HA antibodies (red) and subsequently with Cy3-conjugated anti-rabbit secondary antibodies. Scale bar (all images), 10 μm. D, Representative images of cell-surface binding assays. HEK293T cells expressing HA-tagged PTPσ WT or the indicated PTPσ variants were incubated with 10 μg/ml of control IgC (Fc), Ig-NGL-3 (NGL-3-Fc), or Ig-GPC4 (GPC4-Fc) and then analyzed by immunofluorescence imaging of Ig-fusion proteins (red) and HA antibodies (green). Scale bar (all images), 10 μm.
Figure 7.

Analysis of PTPσ extracellular mechanisms involved in heterologous synapse formation and excitatory synapse development in cultured neurons. A, Schematic diagrams of a series of PTPσ constructs for deletion variants of extracellular domains or point mutants at extracellular residues. AAAA, Quadruple alanine mutant; D1, first catalytic domain of LAR-RPTPs; D2, second catalytic domain of LAR-RPTPs; F, fibronectin Type III repeat; Ig, Ig domain; MeA, mini-exon A; MeB; mini-exon B; SP, IgκB signal peptide; TM, transmembrane region. B, Representative images of heterologous synapse-formation activities of PTPσ WT and the indicated extracellular domain variants and point mutants. Neurons were infected at DIV4 with lentiviruses expressing sh-Control or sh-PTPσ, or coexpressing sh-PTPσ and the various PTPσ mutant constructs (WT, deletion, or point mutants presented in A), and then cocultured from DIV9 to DIV11 with HEK293T cells expressing EGFP alone (Control) or HA-Slitrk1 (Slitrk1). Neurons were stained with antibodies against EGFP or HA (blue) and synapsin (red). Scale bar (all images), 10 μm. C, Synapse-formation activity in B was quantified by measuring the ratio of synapsin staining intensity (red) to HA/EGFP intensity (blue). Data are mean ± SEM. ANOVA with a nonparametric Kruskal–Wallis test: **p < 0.01; ***p < 0.001; #p < 0.05; ###p < 0.001. n = number of neurons as follows: sh-Control/Control, n = 9; sh-PTPσ/Control, n = 9; +WT/Control, n = 9; +ΔIg/Control, n = 9; +ΔFN1–2/Control, n = 15; +ΔEcto/Control, n = 10; +R781A/Control, n = 11; +AAAA/Control, n = 10; sh-Control/Slitrk1, n = 16; sh-PTPσ/Slitrk1, n = 18; +WT/Slitrk1, n = 16; +ΔIg/Slitrk1, n = 12; +ΔFN1–2/Slitrk1, n = 15; +ΔEcto/Slitrk1, n = 12; +R781A/Slitrk1, n = 15; and +AAAA/Slitrk1, n = 14. p values for Control condition: sh-Control vs sh-PTPσ, p > 0.9999; sh-Control vs +WT, p > 0.9999; sh-Control vs +ΔIg, p > 0.9999; sh-Control vs +ΔFN1–2, p > 0.9999; sh-Control vs +ΔEcto, p > 0.9999; sh-Control vs +R781A, p = 0.3835; sh-Control vs +AAAA, p = 0.7746; sh-PTPσ vs +WT, p > 0.9999; sh-PTPσ vs +ΔIg, p > 0.9999; sh-PTPσ vs +ΔFN1–2, p > 0.9999; sh-PTPσ vs +ΔEcto, p > 0.9999; sh-PTPσ vs +R781A, p = 0.9262; and sh-PTPσ vs +AAAA, p > 0.9999. p values for Slitrk1 condition: sh-Control vs sh-PTPσ, p < 0.0001; sh-Control vs +WT, p > 0.9999; sh-Control vs +ΔIg, p = 0.008; sh-Control vs +ΔFN1–2, p > 0.9999; sh-Control vs +ΔEcto, p = 0.0002; sh-Control vs +R781A, p > 0.9999; sh-Control vs +AAAA, p > 0.9999; sh-PTPσ vs +WT, p < 0.0001; sh-PTPσ vs +ΔIg, p > 0.9999; sh-PTPσ vs +ΔFN1–2, p = 0.0348; sh-PTPσ vs +ΔEcto, p > 0.9999; sh-PTPσ vs +R781A, p = 0.0341; and sh-PTPσ vs +AAAA, p = 0.0132. D, Representative images from cultured hippocampal neurons infected at DIV4 with lentiviruses expressing sh-Control or sh-PTPσ, or coinfected with lentiviruses expressing sh-PTPσ and the indicated rescue viruses for PTPσ WT and extracellular domain mutants and analyzed at DIV14 by double-immunofluorescence detection of MAP2 (blue) and the excitatory synaptic marker VGLUT1 (red). Scale bar (all images), 10 μm. E, F, Summary graphs of the effects of PTPσ molecular replacement in neurons on puncta density (E) and puncta size (F), measured using VGLUT1 as an excitatory synaptic marker. Two or three dendrites per transfected neuron were analyzed and group-averaged. Data are mean ± SEM. ANOVA with a nonparametric Kruskal–Wallis test: *p < 0.05; **p < 0.01; #p < 0.05; ##p < 0.01. n = number of neurons as follows: sh-Control, n = 28; sh-PTPσ, n = 20; +WT, n = 20; +ΔIg, n = 20; +ΔFN1–2, n = 21; +ΔEcto, n = 20; +R781A, n = 14; and +AAAA, n = 20. p values for puncta density: sh-Control vs sh-PTPσ, p = 0.0063; sh-Control vs +WT, p > 0.9999; sh-Control vs +ΔIg, p = 0.0178; sh-Control vs +ΔFN1–2, p > 0.9999; sh-Control vs +ΔEcto, p = 0.0493; sh-Control vs +R781A, p > 0.9999; sh-Control vs +AAAA, p = 0.0132; sh-PTPσ vs +WT, p = 0.0281; sh-PTPσ vs +ΔIg, p > 0.9999; sh-PTPσ vs +ΔFN1–2, p = 0.0073; sh-PTPσ vs +ΔEcto, p > 0.9999; sh-PTPσ vs +R781A, p = 0.0086; and sh-PTPσ vs +AAAA, p > 0.9999. p values for puncta sizes: sh-Control vs sh-PTPσ, p > 0.9999; sh-Control vs +WT, p > 0.9999; sh-Control vs +ΔIg, p > 0.9999; sh-Control vs +ΔFN1–2, p > 0.9999; sh-Control vs +ΔEcto, p > 0.9999; sh-Control vs +R781A, p > 0.9999; sh-Control vs +AAAA, p > 0.9999; sh-PTPσ vs +WT, p > 0.9999; sh-PTPσ vs +ΔIg, p > 0.9999; sh-PTPσ vs +ΔFN1–2, p > 0.9999; sh-PTPσ vs +ΔEcto, p > 0.9999; sh-PTPσ vs +R781A, p > 0.9999; and sh-PTPσ vs +AAAA, p > 0.9999.
Figure 9.

Analysis of PTPσ intracellular mechanisms involved in heterologous synapse formation and excitatory synapse development in cultured neurons. A, Schematic diagrams of a series of PTPσ constructs for deletion variants of intracellular domains or point mutants at intracellular residues. Cyto, Cytoplasmic; D1, first catalytic domain of LAR-RPTPs; D2, second catalytic domain of LAR-RPTPs; F, fibronectin Type III repeat; Ig, Ig domain; MeA, mini-exon A; MeB; mini-exon B; SP, IgκB signal peptide; TM, transmembrane region. B, Representative images of the heterologous synapse-formation activities of PTPσ WT and intracellular domain mutants. Neurons were infected at DIV4 with lentiviruses expressing sh-Control or sh-PTPσ, or coexpressing sh-PTPσ and the various PTPσ variants (WT, deletion variants, and point mutants presented in A), and then cocultured from DIV9 to DIV11 with HEK293T cells expressing EGFP alone (Control) or HA-Slitrk1 (Slitrk1). Neurons were stained with antibodies against EGFP or HA (blue) and synapsin (red). Scale bar (all images), 10 μm. C, The synapse-formation activity in B was quantified by measuring the ratio of synapsin staining intensity (red) to HA/EGFP intensity (blue). Data are mean ± SEM. ANOVA with a nonparametric Kruskal–Wallis test: ***p < 0.001; ****p < 0.0001; ##p < 0.01; ####p < 0.0001. n = number of neurons as follows: sh-Control/Control, n = 19; sh-PTPσ/Control, n = 15; +WT/Control, n = 16; +ΔCyto/Control, n = 9; +ΔD2/Control, n = 10; +Swap/Control, n = 11; +C1157S/Control, n = 10; and +D1125A/Control, n = 10; sh-Control/Slitrk1, n = 15; sh-PTPσ/Slitrk1, n = 15; +WT/Slitrk1, n = 15; +ΔCyto/Slitrk1, n = 16; +ΔD2/Slitrk1, n = 14; +Swap/Slitrk1, n = 21; +C1157S/Slitrk1, n = 21; and +D1125A/Slitrk1, n = 22. p values for Control condition: sh-Control vs sh-PTPσ, p > 0.9999; sh-Control vs +WT, p > 0.9999; sh-Control vs +ΔCyto, p = 0.2621; sh-Control vs +ΔD2, p = 0.0915; sh-Control vs +Swap, p > 0.9999; sh-Control vs +C1157S, p > 0.9999; sh-Control vs +D1125A, p > 0.9999; sh-PTPσ vs +WT, p > 0.9999; sh-PTPσ vs +ΔCyto, p > 0.9999; sh-PTPσ vs +ΔD2, p > 0.9999; sh-PTPσ vs +Swap, p > 0.9999; sh-PTPσ vs +C1157S, p > 0.9999; and sh-PTPσ vs +D1125A, p > 0.9999. p values for Slitrk1 condition: sh-Control vs sh-PTPσ, p < 0.0001; sh-Control vs +WT, p > 0.9999; sh-Control vs +ΔCyto, p < 0.0001; sh-Control vs +ΔD2, p = 0.002; sh-Control vs +Swap, p > 0.9999; sh-Control vs +C1157S, p = 0.0445; sh-Control vs +D1125A, p = 0.0045; sh-PTPσ vs +WT, p < 0.0001; sh-PTPσ vs +ΔCyto, p > 0.9999; sh-PTPσ vs +ΔD2, p > 0.9999; sh-PTPσ vs +Swap, p = 0.0023; sh-PTPσ vs +C1157S, p = 0.2585; and sh-PTPσ vs +D1125A, p = 0.6586. D, Representative images of cultured hippocampal neurons infected at DIV4 with lentiviruses expressing sh-Control or sh-PTPσ, or coinfected with lentiviruses expressing sh-PTPσ and the indicated rescue viruses for PTPσ WT and intracellular domain mutants. Images were obtained on DIV14 following double-immunofluorescence labeling of MAP2 (blue) and the excitatory synaptic marker VGLUT1 (red). Scale bar (all images), 10 μm. E, F, Summary graphs of the effects of PTPσ molecular replacement in neurons on puncta density (E) and puncta size (F), measured using VGLUT1 as an excitatory synaptic marker. Two or three dendrites per transfected neuron were analyzed and group-averaged. Data are mean ± SEM. ANOVA with a nonparametric Kruskal–Wallis test: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; #p < 0.05. n = number of neurons as follows: sh-Control, n = 17; sh-PTPσ, n = 21; +WT, n = 15; +ΔCyto, n = 15; +ΔD2, n = 14; +SWAP, n = 20; +C1157S, n = 25; and +D1125A, n = 26. p values for puncta density: sh-Control vs sh-PTPσ, p < 0.0001; sh-Control vs +WT, p > 0.9999; sh-Control vs +ΔCyto, p < 0.0001; sh-Control vs +ΔD2, p = 0.0184; sh-Control vs +Swap, p < 0.0001; sh-Control vs +C1157S, p = 0.0083; sh-Control vs +D1125A, p = 0.0147; sh-PTPσ vs +WT, p = 0.0105; sh-PTPσ vs +ΔCyto, p > 0.9999; sh-PTPσ vs +ΔD2, p > 0.9999; sh-PTPσ vs +Swap, p > 0.9999; sh-PTPσ vs +C1157S, p > 0.9999; and sh-PTPσ vs +D1125A, p > 0.9999. p values for puncta size: sh-Control vs sh-PTPσ, p > 0.9999; sh-Control vs +WT, p > 0.9999; sh-Control vs +ΔCyto, p < 0.0161; sh-Control vs +ΔD2, p > 0.9999; sh-Control vs +Swap, p = 0.0003; sh-Control vs +C1157S, p > 0.9999; sh-Control vs +D1125A, p > 0.9999; sh-PTPσ vs +WT, p > 0.9999; sh-PTPσ vs +ΔCyto, p > 0.9999; sh-PTPσ vs +ΔD2, p > 0.9999; sh-PTPσ vs +Swap, p = 0.266; sh-PTPσ vs +C1157S, p > 0.9999; and sh-PTPσ vs +D1125A, p > 0.9999.
Figure 10.
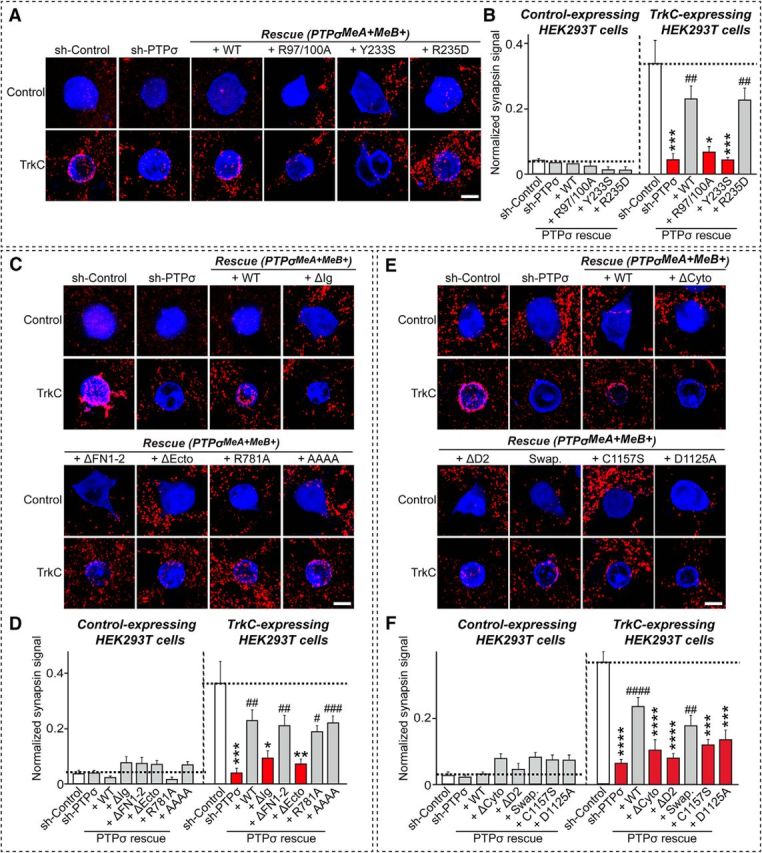
Analysis of PTPσ extracellular and intracellular mechanisms involved in heterologous synapse formation induced by TrkC. A, Representative images of the heterologous synapse-formation activities of PTPσ WT and the indicated specific ligand binding-defective point mutants. Neurons were infected at DIV4 with lentiviruses expressing sh-Control or sh-PTPσ, or coexpressing sh-PTPσ and various PTPσ mutant constructs presented in Figure 6, and then cocultured from DIV9 to DIV11 with HEK293T cells expressing EGFP alone (Control) or HA-TrkC (TrkC). Neurons were stained with antibodies against EGFP or HA (blue) and synapsin (red). Scale bar (all images), 10 μm. B, The synapse-formation activity in A was quantified by measuring the ratio of synapsin staining intensity (red) to HA/EGFP intensity (blue). Data are mean ± SEM. ANOVA with a nonparametric Kruskal–Wallis test: *p < 0.05; ***p < 0.001; ##p < 0.01. n = number of neurons as follows: sh-Control/Control, n = 8; sh-PTPσ/Control, n = 11; +WT/Control, n = 11; +R97/100A/Control, n = 9; +Y233S/Control, n = 9; and +R235D/Control, n = 12; sh-Control/TrkC, n = 13; sh-PTPσ/TrkC, n = 11; +WT/TrkC, n = 13; +R97/100A/TrkC, n = 11; +Y233S/TrkC, n = 16; and +R235D/TrkC, n = 13. p values for Control condition: sh-Control vs sh-PTPσ, p > 0.9999; sh-Control vs +WT, p > 0.9999; sh-Control vs +R97/100A, p = 0.2309; sh-Control vs +Y233S, p = 0.3307; sh-Control vs +R235D, p > 0.9999; sh-PTPσ vs +WT, p > 0.9999; sh-PTPσ vs +R97/100A, p = 0.0577; sh-PTPσ vs +Y233S, p = 0.0814; and sh-PTPσ vs +R235D, p = 0.0504. p values for TrkC condition: sh-Control vs sh-PTPσ, p = 0.0004; sh-Control vs +WT, p > 0.9999; sh-Control vs +R97/100A, p = 0.0153; sh-Control vs +Y233S, p = 0.0003; sh-Control vs +R235D, p > 0.9999; sh-PTPσ vs +WT, p = 0.002; sh-PTPσ vs +R97/100A, p > 0.9999; sh-PTPσ vs +Y233S, p > 0.9999; and sh-PTPσ vs +R235D, p = 0.001. C, Representative images of the heterologous synapse-formation activities of PTPσ WT and the indicated extracellular domain variants and point mutants. Neurons were infected at DIV4 with lentiviruses expressing sh-Control or sh-PTPσ, or coexpressing sh-PTPσ and various PTPσ mutant constructs (WT, deletion, or point mutants presented in Fig. 6), and then cocultured from DIV9 to DIV11 with HEK293T cells expressing EGFP alone (Control) or HA-TrkC (TrkC). Neurons were stained with antibodies against EGFP or HA (blue) and synapsin (red). Scale bar (all images), 10 μm. D, The synapse-formation activity in C was quantified by measuring the ratio of synapsin staining intensity (red) to HA/EGFP intensity (blue). Data are mean ± SEM. ANOVA with a nonparametric Kruskal–Wallis test: *p < 0.05; **p < 0.01; ***p < 0.001; #p < 0.05; ##p < 0.01; ###p < 0.001. n = number of neurons as follows: sh-Control/Control, n = 8; sh-PTPσ/Control, n = 11; +WT/Control, n = 11; +ΔIg/Control, n = 11; +ΔFN1–2/Control, n = 10; +ΔEcto/Control, n = 10; +R781A/Control, n = 10; and +AAAA/Control, n = 12; sh-Control/TrkC, n = 12; sh-PTPσ/TrkC, n = 11; +WT/TrkC, n = 13; +ΔIg/TrkC, n = 13; +ΔFN1–2/TrkC, n = 13; +ΔEcto/TrkC, n = 11; +R781A/TrkC, n = 14; and +AAAA/TrkC, n = 14. p values for Control condition: sh-Control vs sh-PTPσ, p > 0.9999; sh-Control vs +WT, p > 0.9999; sh-control vs +ΔIg, p > 0.9999; sh-Control vs +ΔFN1–2, p > 0.9999; sh-Control vs +ΔEcto, p > 0.9999; sh-Control vs +R781A, p > 0.9999; sh-Control vs +AAAA, p > 0.9999; sh-PTPσ vs +WT, p > 0.9999; sh-PTPσ vs +ΔIg, p > 0.9999; sh-PTPσ vs +ΔFN1–2, p > 0.9999; sh-PTPσ vs +ΔEcto, p > 0.9999; sh-PTPσ vs +R781A, p > 0.9999; and sh-PTPσ vs +AAAA, p > 0.9999. p values for TrkC condition: sh-Control vs sh-PTPσ, p = 0.0003; sh-Control vs +WT, p > 0.9999; sh-Control vs +ΔIg, p = 0.0374; sh-Control vs +ΔFN1–2, p > 0.9999; sh-Control vs +ΔEcto, p = 0.0085; sh-Control vs +R781A, p > 0.9999; sh-Control vs +AAAA, p > 0.9999; sh-PTPσ vs +WT, p = 0.0027; sh-PTPσ vs +ΔIg, p > 0.9999; sh-PTPσ vs +ΔFN1–2, p = 0.0082; sh-PTPσ vs +ΔEcto, p > 0.9999; sh-PTPσ vs +R781A, p = 0.0112; and sh-PTPσ vs +AAAA, p = 0.0006. E, Representative images of the heterologous synapse-formation activities of PTPσ WT and the indicated intracellular domain variants and point mutants. Neurons were infected at DIV4 with lentiviruses expressing sh-Control or sh-PTPσ, or coexpressing sh-PTPσ and the indicated PTPσ mutant constructs presented in Figures 9 and 11, and then cocultured from DIV9 to DIV11 with HEK293T cells expressing EGFP alone (Control) or HA-TrkC (TrkC). Neurons were stained with antibodies against EGFP or HA (blue) and synapsin (red). Scale bar (all images), 10 μm. F, The synapse-formation activity in E was quantified by measuring the ratio of synapsin staining intensity (red) to HA/EGFP intensity (blue). Data are mean ± SEM. ANOVA with a nonparametric Kruskal–Wallis test: ***p < 0.001; ****p < 0.0001; ##p < 0.01; ####p < 0.0001. n = number of neurons as follows: sh-Control/Control, n = 8; sh-PTPσ/Control, n = 11; +WT/Control, n = 11; +ΔCyto/Control, n = 11; +ΔD2/Control, n = 9; +Swap/Control, n = 8; +C1157S/Control, n = 9; and +D1125A/Control, n = 12; sh-Control/TrkC, n = 21; sh-PTPσ/TrkC, n = 28; +WT/TrkC, n = 21; +ΔCyto/TrkC, n = 12; +ΔD2/TrkC, n = 13; +Swap/TrkC, n = 17; +C1157S/TrkC, n = 20; and +D1125A, n = 15. p values for Control condition: sh-Control vs sh-PTPσ, p > 0.9999; sh-Control vs +WT, p > 0.9999; sh-Control vs +ΔCyto, p > 0.9999; sh-Control vs +ΔD2, p > 0.9999; sh-Control vs +Swap, p = 0.4602; sh-Control vs +C1157S, p = 0.6221; sh-Control vs +D1125A, p > 0.9999; sh-PTPσ vs +WT, p > 0.9999; sh-PTPσ vs +ΔCyto, p = 0.4431; sh-PTPσ vs +ΔD2, p = 0.6263; sh-PTPσ vs +Swap, p = 0.1718; sh-PTPσ vs +C1157S, p = 0.2347; and sh-PTPσ vs +D1125A, p > 0.9999. p values for TrkC condition: sh-Control vs sh-PTPσ, p < 0.0001; sh-Control vs +WT, p > 0.9999; sh-Control vs +ΔCyto, p < 0.0001; sh-Control vs +ΔD2, p < 0.0001; sh-Control vs +Swap, p = 0.0539; sh-Control vs +C1157S, p = 0.0002; sh-Control vs +D1125A, p = 0.0003; sh-PTPσ vs +WT, p < 0.0001; sh-PTPσ vs +ΔCyto, p > 0.9999; sh-PTPσ vs +ΔD2, p > 0.9999; sh-PTPσ vs +Swap, p = 0.0061; sh-PTPσ vs +C1157S, p = 0.3426; and sh-PTPσ vs +D1125A, p > 0.9999.
Figure 12.
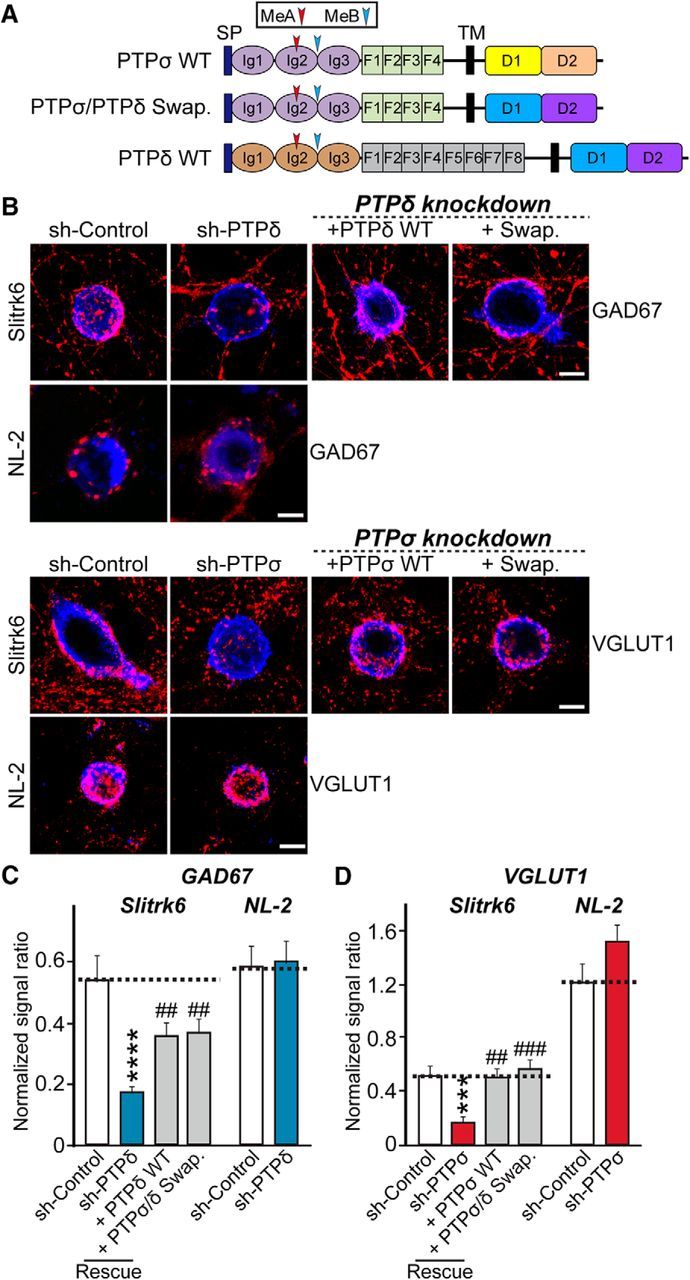
Effects of PTPσ extracellular domain and PTPδ intracellular domain on PTPσ-mediated heterologous synapse formation. A, Schematic illustration of PTPσ WT and mutants used in the experiments presented in B–D. B, Representative images of the heterologous excitatory or inhibitory synapse-formation activities of PTPσ WT and PTPσ/PTPδ Swap mutants. Neurons were infected at DIV4 with lentiviruses expressing sh-Control, sh-PTPσ, or sh-PTPδ, or coexpressing sh-PTPσ or sh-PTPδ with the indicated PTPσ or PTPδ expression vectors, and then cocultured from DIV9 to DIV11 with HEK293T cells expressing HA-Slitrk6 (Slitrk6) or neuroligin-2 fused to mVenus (NL-2). Neurons were stained with antibodies against HA (blue) and GAD67 or VGLUT1 (red). Scale bar (all images), 10 μm. C, D, The synapse-formation activity in B was quantified by measuring the ratio of GAD67 (C) or VGLUT1 (D) staining intensity (red) to HA/EGFP intensity (blue). Data are mean ± SEM. ANOVA with a nonparametric Kruskal–Wallis test: ***p < 0.001; ****p < 0.0001; ##p < 0.01; ###p < 0.001. n = number of neurons as follows: sh-Control/GAD67/Slitrk6, n = 16; sh-PTPδ/GAD67/Slitrk6, n = 15; + PTPδ/GAD67/Slitrk6, n = 16; + PTPσ Swap/GAD67/Slitrk6, n = 17; sh-Control/VGLUT1/Slitrk6, n = 15; sh-PTPσ/VGLUT1/Slitrk6, n = 15; + PTPσ WT/VGLUT1/Slitrk6, n = 12; + PTPσ Swap/VGLUT1/Slitrk6, n = 13; sh-Control/GAD67/NL-2, n = 21; sh-PTPδ/GAD67/NL-2, n = 23; sh-Control/VGLUT1/NL-2, n = 12; and sh-PTPσ/VGLUT1/NL-2, n = 11. p values for Slitrk6/GAD67 condition: sh-Control vs sh-PTPδ, p < 0.0001; sh-Control vs + PTPδ, p = 0.4456; sh-Control vs + PTPσ Swap, p = 0.4583; sh-PTPδ vs + PTPδ, p = 0.0064; and sh-PTPδ vs + PTPσ Swap, p = 0.0047. p values for Slitrk6/VGLUT1 condition: sh-Control vs sh-PTPσ, p = 0.0004; sh-Control vs + PTPσ, p > 0.9999; sh-control vs + PTPσ Swap, p > 0.9999; sh-PTPσ vs + PTPσ, p = 0.0019; and sh-PTPσ vs + PTPσ Swap, p = 0.0002. p values for NL-2/GAD67 condition: sh-Control vs sh-PTPδ, p > 0.9999. p values for NL-2/VGLUT1 condition: sh-Control vs sh-PTPσ, p > 0.9999.
Figure 8.

Analysis of the effects of PTPσ extracellular point mutants that selectively abolish interactions with TrkC or Slitrks on heterologous synapse formation. A, Schematic illustration of PTPσ WT and point mutants that exhibit defective binding to the specific postsynaptic ligands, TrkC or Slitrks. D1, First catalytic domain of LAR-RPTPs; D2, second catalytic domain of LAR-RPTPs; F, fibronectin Type III repeat; Ig, Ig domain; MeA, mini-exon A; MeB; mini-exon B; SP, IgκB signal peptide; TM, transmembrane region. B, Representative images of cell surface-binding assays. HEK293T cells expressing HA-tagged PTPσ WT or its various point mutants were incubated with 10 μg/ml control IgC (Control), Ig-GPC-4, or Ig-NGL-3, and then analyzed by immunofluorescence imaging of Ig-fusion proteins (red) and HA antibodies (green). Scale bar (all images), 10 μm. C, Representative images of the heterologous synapse-formation activities of PTPσ WT and specific ligand-binding-defective mutants. Neurons were infected at DIV4 with lentiviruses expressing sh-Control or sh-PTPσ, or coexpressing various PTPσ mutant constructs (WT and ligand-binding-defective mutants presented in A), and then cocultured from DIV9 to DIV11 with HEK293T cells expressing EGFP alone (Control) or HA-Slitrk1 (Slitrk1). Neurons were stained with antibodies against EGFP or HA (blue) and synapsin (red). Scale bar (all images), 10 μm. D, The synapse-formation activity in C was quantified by measuring the ratio of synapsin staining intensity (red) to HA/EGFP intensity (blue). Data are mean ± SEM. ANOVA with a nonparametric Kruskal–Wallis test: **p < 0.01; ***p < 0.001; ####p < 0.0001. n = number of neurons as follows: sh-Control/Slitrk1, n = 19; sh-PTPσ/Control, n = 15; +WT/Control, n = 16; +R97/100A/Control, n = 10; +Y233S/Control, n = 9; and +R235D/Control, n = 10; sh-Control/Slitrk1, n = 20; sh-PTPσ/Slitrk1, n = 25; +WT/Slitrk1, n = 25; +R97/100A/Slitrk1, n = 22; +Y233S/Slitrk1, n = 16; and +R235D/Slitrk1, n = 24. p values for Control condition: sh-Control vs sh-PTPσ, p > 0.9999; sh-Control vs +WT, p > 0.9999; sh-Control vs +R97/100A, p > 0.9999; sh-Control vs +Y233S, p = 0.3522; sh-Control vs +R235D, p = 0.1213; sh-PTPσ vs +WT, p > 0.9999; sh-PTPσ vs +R97/100A, p > 0.9999; sh-PTPσ vs +Y233S, p = 0.1596; and sh-PTPσ vs +R235D, p = 0.0504. p values for Slitrk1 condition: sh-Control vs sh-PTPσ, p < 0.0001; sh-Control vs +WT, p > 0.9999; sh-Control vs +R97/100A, p > 0.9999; sh-Control vs +Y233S, p = 0.0015; sh-Control vs +R235D, p = 0.0004; sh-PTPσ vs +WT, p < 0.0001; sh-PTPσ vs +R97/100A, p < 0.0001; sh-PTPσ vs +Y233S, p > 0.9999; and sh-PTPσ vs +R235D, p > 0.9999. E, Representative images of cultured hippocampal neurons infected at DIV4 with lentiviruses expressing sh-Control or sh-PTPσ, or coinfected with lentiviruses expressing sh-PTPσ and the indicated rescue viruses for human PTPσ alternative splicing variants. These images were taken on DIV14 following double-immunofluorescence labeling of MAP2 (blue) and the excitatory synaptic marker VGLUT1 (red). Scale bar (all images), 10 μm. F, Summary graphs of the effects of PTPσ molecular replacement in neurons on puncta density (left) and puncta size (right), measured using VGLUT1 as an excitatory synaptic marker. Two or three dendrites per transfected neuron were analyzed and group-averaged. Data are mean ± SEM. ANOVA with a nonparametric Kruskal–Wallis test: **p < 0.01; ***p < 0.001; #p < 0.05; ##p < 0.01. n = number of neurons as follows: sh-Control, n = 21; sh-PTPσ, n = 20; +WT, n = 16; +R97/100A, n = 21; +Y233S, n = 15; and +R235D, n = 15. p values for puncta density: sh-Control vs sh-PTPσ, p < 0.0001; sh-Control vs +WT, p > 0.9999; sh-control vs +R97/100A, p > 0.9999; sh-Control vs +Y233S, p = 0.0036; sh-Control vs +R235D, p = 0.0059; sh-PTPσ vs +WT, p = 0.0074; sh-PTPσ vs +R97/100A, p = 0.0179; sh-PTPσ vs +Y233S, p > 0.9999; and sh-PTPσ vs +R235D, p > 0.9999. p values for puncta size: sh-Control vs sh-PTPσ, p > 0.9999; sh-Control vs +WT, p > 0.9999; sh-Control vs +R97/100A, p > 0.9999; sh-Control vs +Y233S, p > 0.9999; sh-Control vs +R235D, p = 0.0177; sh-PTPσ vs +WT, p > 0.9999; sh-PTPσ vs +R97/100A, p > 0.9999; sh-PTPσ vs +Y233S, p > 0.9999; and sh-PTPσ vs +R235D, p = 0.2712.
Using this PTPσ “toolkit,” we performed heterologous synapse-formation assays using HEK293T cells expressing either Slitrk1 or TrkC, and cultured neurons infected with lentiviruses expressing the indicated PTPσ constructs. We found that heterologous synapse formation induced by Slitrk1 or TrkC was impaired in PTPσ-deficient neurons, a defect that was completely rescued by reintroduction of PTPσ WT (+WT) (see Figs. 7–10). Reexpression of PTPσ variants lacking Ig domains (+ΔIg) or the entire extracellular region (+ΔEcto) failed to rescue the impaired heterologous synapse formation, in accord with the requirement for direct interactions of PTPσ with Slitrk1 or TrkC through Ig domains (Fig. 7, 10C,D) (Um and Ko, 2013). In contrast, reexpression of a heparan sulfate (HS)-binding-defective PTPσ mutant (+AAAA) or one in which the first two fibronectin Type III repeats were deleted (+ΔFN1–2) effectively rescued the impaired heterologous synapse formation (see Figs. 7, 10C,D). Moreover, reexpression of a proteolytically uncleavable PTPσ mutant (+R781A) (Dunah et al., 2005) fully rescued the defective synaptogenic activity, indicating that cleavage by furin/subtilisin-like endoproteases is not necessary for the heterologous synapse formation activity (see Figs. 7, 10C,D). This contrasts with the requirement of endoprotease-mediated cleavage of PTPσ for the maintenance of excitatory neuronal synapse structures (Dunah et al., 2005).
We also examined whether the extracellular region of PTPσ is required to maintain excitatory synapse density in neuronal dendrites (Fig. 7). Extracellular interactions through PTPσ Ig-domains and HS-binding residues, but not FN1–2, were required for regulation of excitatory synapse development in neuronal synapses (Fig. 7) (Coles et al., 2011; Ko et al., 2015b). On the basis of these data, we conclude that PTPσ organizes heterologous synapse formation and neuronal synapse development through mechanisms involving the extracellular region.
PTPσ-mediated maintenance of excitatory synapse development in cultured hippocampal neurons requires interaction with Slitrks, but not with TrkC or GPC-4
The observation that extracellular interactions with various ligands are necessary for the excitatory synapse-promoting activity of PTPσ in neurons prompted us to test which PTPσ ligands are most important. Addressing this question requires information about the crystal structure of PTPσ complexes with ligands to pinpoint critical residues involved in the interactions with specific ligands. Among the known PTPσ ligands, structural information is currently available only for Slitrk1 and TrkC (Coles et al., 2014; Um et al., 2014a). Based on the published crystal structures of Slitrk1-PTPδ and TrkC-PTPσ complexes, we designed the following three point mutants: R97A/R100A, which specifically disrupts binding to TrkC, without affecting Slitrk1 binding; Y233S, which blocks binding to both Slitrk1 and TrkC; and R235D, which specifically disrupts bindings to Slitrk1, but not to TrkC (Fig. 8). All three PTPσ mutants were expressed well on the surface of transfected HEK293T cells and bound recombinant NGL-3-Fc proteins (Figs. 6C, 8B). PTPσ R97A/R100A, but not PTPσ Y233S or PTPσ R235D, showed impaired binding to recombinant GPC-4-Fc proteins (Fig. 8B), partly because of the fact that R97 and R100 residues of PTPσ also contribute to glycosaminoglycan binding (Aricescu et al., 2002; Coles et al., 2011). We next performed heterologous synapse-formation assays using HEK293T cells expressing either Slitrk1 (Fig. 8C,D) or TrkC (see Fig. 10), and cultured neurons infected with lentiviruses expressing sh-Control or sh-PTPσ, or coexpressing sh-PTPσ and the indicated PTPσ constructs (WT, R97/100A, Y233S, or R235D). We found that impaired Slitrk1-mediated heterologous synapse formation with PTPσ-deficient neurons was completely rescued by reexpression of PTPσ WT (+WT) or PTPσ R97A/R100A (+R97/100A) in neurons (Fig. 8C,D). Reexpression of PTPσ Y233S (+Y233S) or PTPσ R235D (+R235D) failed to rescue the impaired heterologous synapse formation elicited by Slitrk1, consistent with our previous report that these two PTPσ mutations abolish interactions with Slitrks (Fig. 8C,D) (Um et al., 2014a). Similarly, impaired TrkC-mediated heterologous synapse formation with PTPσ-deficient neurons was selectively rescued by reexpression of PTPσ WT (+WT) or PTPσ R235D (+R235D) (see Fig. 10A,B).
Using these characterized PTPσ point mutants, we then asked which PTPσ-interacting extracellular proteins primarily operate together with PTPσ in the maintenance of excitatory synapse development. Strikingly, the interaction of PTPσ with Slitrks rather than that with TrkC or GPC-4 appeared to be more critical, although the significance of TrkC or GPC-4 in PTPσ function cannot be discounted because the HS-binding property of PTPσ is also required for PTPσ-mediated excitatory synaptic transmission (Fig. 8E,F) (Ko et al., 2015b). In addition, it should be noted that contributions of other extracellular PTPσ-interacting proteins involved in PTPσ function, such as NGL-3 and SALMs, could not be assessed in the current study. Collectively, our data suggest that a set of specific extracellular ligand(s) is used for PTPσ-mediated excitatory synapse development.
Direct interaction of PTPσ with liprin-α through the PTPσ D2 domain is required for presynaptic assembly
As expected given the requirement for cytoplasmic regions of the PTPσ protein in mediating heterologous synapse formation (Um and Ko, 2013), reintroduction of a PTPσ deletion variant lacking the entire cytoplasmic region into PTPσ-deficient neurons also failed to rescue the impaired Slitrk1- and TrkC-mediated heterologous synapse formation activity, unlike the heterologous synapse formation activity induced by NL-1 (Figs. 9, 10E,F) (Gokce and Südhof, 2013). To further dissect the requirement of PTPσ cytoplasmic sequences in mediating presynaptic assembly, we performed heterologous synapse-formation assays using HEK293T cells expressing either Slitrk1 or TrkC and cultured neurons infected with lentiviruses expressing the indicated PTPσ deletion constructs and point mutants. We found that the impaired heterologous synapse formation activity observed in PTPσ-deficient neurons was not rescued by lentiviral expression of a PTPσ deletion variant that lacked the entire cytoplasmic region (+ΔCyto) or the D2 domain (+ΔD2), suggesting that interactions of PTPσ with intracellular proteins are necessary for excitatory presynaptic assembly (Fig. 9B,C, 10E, F). We also examined whether the intracellular region of PTPσ is required to maintain excitatory synapse density in neuronal dendrites (Fig. 9D–F). We found that PTPσ D2 domain-mediated protein interactions and tyrosine phosphorylation signaling pathways were required to regulate the development of excitatory synapses (Fig. 9).
To extend previous in vitro results identifying diverse LAR-RPTP-interacting proteins (Um and Ko, 2013), we sought further support for the requirement of the PTPσ D2 domain in presynaptic assembly in vivo by, first, conducting GST pull-down assays in mouse brain synaptosomal fractions using recombinant PTPσ D2 and PTPδ D2 proteins. Our mass spectroscopy analyses identified a subset of presynaptic proteins that had previously been reported to directly interact with LAR-RPTPs, including liprin-α3, ELKS1, CASK, and Caskin-1 (Fig. 11-1). The top priority among numerous putative candidate peptides was liprin-α family proteins because of the established functions of invertebrate orthologs in the CNS and their multiple interactions with other presynaptic active zone proteins and scaffolds, including RIM1, CASK, mSYD-1, GIT1 (G-protein coupled receptor interacting protein 1), and GRIPs (glutamate receptor-interacting proteins) (Spangler and Hoogenraad, 2007; Südhof, 2012; Han et al., 2016). Among four liprin-α isoforms, we focused on liprin-α2 and liprin-α3 because they are major liprin-α isoforms in the brain (Zürner et al., 2011). After screening liprin-α2 and liprin-α3 shRNAs for the most effective sequences, we constructed an sh-liprin-α2/α3-expressing lentiviral RNAi vector (L-309) (Pang et al., 2010) to simultaneously knock down both liprin-α2 and liprin-α3 (Fig. 11E). We then performed heterologous synapse-formation assays in cultured neurons infected with lentiviruses expressing sh-liprin-α2/α3. We found that KD of liprin-α2 and liprin-α3 impaired the heterologous synapse formation activity induced by Slitrk6 or NL-2 (Fig. 11A,B). These results reinforce the idea that the D2 domain of LAR-RPTPs acts an anchoring point for mediating intracellular signaling downstream of LAR-RPTPs for presynaptic assembly, notably through liprin-α and its multiprotein complexes, and that both LAR-RPTPs and neurexins orchestrate heterologous synapse formation through converging signaling pathways involving liprin-α2 and liprin-α3.
Figure 11.

Effects of PTPσ intracellular binding proteins on PTPσ-mediated heterologous synapse formation. A, Representative images of the heterologous synapse-formation activities of liprin-α2 and liprin-α3. Neurons were infected at DIV4 with lentiviruses expressing sh-Control or sh-liprin-α2/α3, and then cocultured from DIV9 to DIV11 with HEK293T cells expressing EGFP alone (Control), HA-Slitrk6 (Slitrk6), or HA-neuroligin-2 (NL-2). Neurons were stained with antibodies against EGFP or HA (blue) and synapsin (red). Scale bar (all images), 10 μm. Liprin-α3 and a subset of its associated proteins were isolated by mass spectroscopy (see Figure 11-1). B, The synapse-formation activity in A was quantified by measuring the ratio of synapsin staining intensity (red) to HA/EGFP intensity (blue). Data are mean ± SEM. Mann–Whitney U test: *p < 0.05; ***p < 0.001. n = number of neurons as follows: sh-Control/Control, n = 14; sh-liprin-α2/α3/Control, n = 10; sh-Control/Slitrk6, n = 16; sh-liprin-α2/α3/Slitrk6, n = 15; sh-Control/NL-2, n = 22; and sh-liprin-α2/α3/NL-2, n = 21. p values for Control condition: sh-Control vs sh-liprin-α2/α3, p = 0.2591. p values for Slitrk6 condition: sh-Control vs sh-liprin-α2/α3, p = 0.0003. p values for NL-2 condition: sh-Control vs sh-liprin-α2/α3, p = 0.0101. C, Representative images of the heterologous synapse-formation activities of three selected PTPσ substrates (β-catenin, N-cadherin, and p250GAP). Neurons were infected at DIV4 with lentiviruses expressing sh-Control, sh-β-catenin, sh-N-cadherin, or sh-p250GAP, and then cocultured from DIV9 to DIV11 with HEK293T cells expressing EGFP alone (Control), HA-Slitrk6 (Slitrk6), or neuroligin-2-mVenus (NL-2). Neurons were stained with antibodies against EGFP or HA (blue) and synapsin (red). Scale bar (all images), 10 μm. D, The synapse-formation activity in C was quantified by measuring the ratio of synapsin staining intensity (red) to HA/EGFP intensity (blue). Data are mean ± SEM. Mann–Whitney U test: *p < 0.05; ***p < 0.001. n = number of neurons as follows: sh-Control/Control, n = 14; sh-β-catenin/Control, n = 12; sh-N-cadherin/Control, n = 10; sh-p250GAP/Control, n = 13; sh-Control/Slitrk6, n = 16; sh-β-catenin/Slitrk6, n = 14; sh-N-cadherin/Slitrk6, n = 15; sh-p250GAP/Slitrk6, n = 16; sh-Control/NL-2, n = 22; sh-β-catenin/NL-2, n = 27; sh-N-cadherin/NL-2, n = 21; and sh-p250GAP/NL-2, n = 24. p values for Control condition: sh-Control vs sh-β-catenin, p > 0.9999; sh-Control vs sh-N-cadherin, p = 0.3904; and sh-Control vs sh-p250GAP, p > 0.9999. p values for Slitrk6 condition: sh-Control vs sh-β-catenin, p > 0.9999; sh-Control vs sh-N-cadherin, p < 0.0001; and sh-Control vs sh-p250GAP, p = 0.0121. p values for NL-2 condition: sh-Control vs sh-β-catenin, p > 0.9999; sh-Control vs sh-N-cadherin, p > 0.9999; and sh-Control vs sh-p250GAP, p > 0.9999. E, KD efficacies of shRNAs. Levels of target mRNAs were measured by qRT-PCR in cultured cortical neurons infected at DIV3 with lentiviruses expressing the indicated shRNAs. mRNAs were prepared at DIV12-DV13. Dotted line indicates the 70% knockdown cutoff level for tests of biological effects.
Detailed list of peptides obtained by mass spectroscopy analyses using GST-PTPδ D2 or GST-PTPσ D2 in mouse brain synaptosomal fractions. Download Figure 11-1, XLSX file (34.7KB, xlsx)
Specific PTPσ substrates are required for PTPσ-mediated presynaptic assembly
The intracellular region of PTPσ harboring its phosphatase activity partly appears to be critical for the presynaptic assembly process, as shown by the ability of lentiviral-mediated expression of PTPσ C1157S or PTPσ D1125A, catalytically inactive mutants of PTPσ, to partially rescue the impaired heterologous synapse-formation activity in PTPσ-deficient neurons (Figs. 9, 10) (Johnson and Van Vactor, 2003). Thus, we examined whether a subset of PTPσ substrates are involved in PTPσ-mediated presynaptic assembly. The currently known LAR-RPTP substrates include N-cadherin, β-catenin, Abelson kinase (Abl), Enabled (Ena), Trio, and p250GAP (Han et al., 2016). Of these putative substrates, three proteins, N-cadherin, β-catenin, and p250GAP, for which highly efficient shRNA sequences were available in our laboratory, were selected for further study (Fig. 11E). Remarkably, KD of p250GAP or N-cadherin, but not KD of β-catenin, abolished the heterologous synapse-formation activity induced by Slitrk6 (Fig. 11C,D), suggesting that a specific set of substrate proteins is involved in recruiting the presynaptic machinery involved in LAR-RPTP-mediated heterologous synapse formation. Together with our previous results, these data suggest that the recruitment of presynaptic machinery into nascent synaptic sites for heterologous synapse formation is accompanied by the concentration of intracellular scaffolds and substrates that directly or indirectly associate with PTPσ through the D2 domain.
The PTPσ D2 domain alone is insufficient to drive presynaptic assembly at excitatory synapses
Although our results unambiguously demonstrate a structural prerequisite for the action of LAR-RPTP family proteins in presynaptic assembly, how PTPσ specifically exerts its action toward the excitatory synapse type remains to be determined. LAR-RPTP family members are structurally similar, sharing 64% overall amino acid sequence identity (Pulido et al., 1995; Um and Ko, 2013). In particular, the D2 domain of human PTPσ and PTPδ exhibits 95%–99% similarity/identity (data not shown). Therefore, we were initially skeptical about the contribution of intracellular mechanisms to determining the specific actions of PTPσ and PTPδ in driving specific types of synapse assembly, especially given that the suggested role of PTPσ and PTPδ in driving excitatory and inhibitory synapse assembly, respectively, was still hypothetical. We thus expected that replacing the intracellular region of PTPσ with its PTPδ counterpart would not compromise the ability of PTPσ to mediate heterologous synapse formation. Using Slitrk6, a Slitrk isoform that induces inhibitory heterologous synapse formation more potently than Slitrk1, we performed heterologous synapse-formation assays in PTPσ- or PTPδ-deficient neurons. We previously reported that PTPδ KD specifically impaired the inhibitory, but not excitatory, heterologous synapse formation activity induced by Slitrk6, as we previously reported (Yim et al., 2013). Strikingly, reexpression of PTPσ WT or a PTPσ intracellularly swapped variant composed of the PTPσ extracellular region and PTPδ intracellular region (PTPσ Swap) rescued the defective synaptogenic activity of PTPσ-deficient neurons (Fig. 12B,D). Moreover, reintroduction of the PTPσ Swap mutant into PTPδ-deficient neurons also restored the impaired heterologous inhibitory synapse formation observed in PTPδ-deficient neurons (Fig. 12B,C). These data suggest that Slitrk6 induces presynaptic assembly of both excitatory and inhibitory synapses, provided that interactions with extracellular ligands (i.e., PTPσ or PTPδ) are permissive, regardless of the identity of intracellular signaling pathways. Notably, lentiviral expression of the PTPσ Swap mutant failed to maintain the density of excitatory synapse puncta at a level comparable with that supported by PTPσ WT (Fig. 9). In control experiments, neither PTPσ KD nor PTPδ KD affected the heterologous synapse-formation activity induced by NL-2 (Fig. 12B–D). Collectively, these data suggest that the PTPσ D2 domain per se does not dictate the identity of specific synapse types during heterologous synapse formation mediated by LAR-RPTPs, but that a combination of extracellular signals and intracellular signaling cascades may be critical for the maintenance of excitatory synapse density in neuronal dendrites.
Discussion
LAR-RPTPs control excitatory and inhibitory synapse development in an isoform-dependent manner
The three members of the LAR-RPTP protein family are structurally similar, but their expression profiles across brain regions differ markedly (Kwon et al., 2010). Importantly, individual members of this family bind to a distinct repertoire of extracellular ligands (Um and Ko, 2013). Given the high similarity of the Ig domains among all three LAR-RPTPs, it is puzzling why various extracellular ligands bind to only a subset of LAR-RPTPs. Nevertheless, several recent studies have suggested a converging molecular model, postulating that PTPσ is specifically involved in excitatory synapse development, whereas PTPδ is generally involved in inhibitory synapse development (Um and Ko, 2013).
However, previous studies did not demonstrate a requirement for PTPσ or PTPδ in the development of specific types of synapse in neurons. In addition, whether both synapse types involve similar vesicular packets (i.e., synaptic vesicle protein transport vesicles and/or Piccolo-bassoon transport vesicles) for the initial recruitment of nascent synaptic proteins has not been established, although both vesicular packets were reported to be similarly trapped and accumulated by trans-synaptic adhesion signaling (Bury and Sabo, 2014). In the present study, we demonstrated that PTPσ is specifically involved in the innervation of excitatory synaptic inputs and the regulation of excitatory synaptic transmission in cultured hippocampal neurons, consistent with our previous observations (Ko et al., 2015b). Similarly, PTPδ was shown to be specifically involved in the innervation of inhibitory synaptic inputs and the regulation of inhibitory synaptic transmission (Fig. 4). An exception to this scenario arises from the observation that presynaptic PTPδ is required for excitatory heterologous synapse formation induced by IL1RAPL1, which acts specifically to regulate excitatory synapse development (Valnegri et al., 2011; Yoshida et al., 2011) (Fig. 1). A more recent study appeared to resolve this conflict, showing that IL1RAPL1 regulates dendritic morphology in cultured neurons, independently of interactions with PTPδ (Montani et al., 2017), hinting at the possibility that a minor fraction of PTPδ proteins participates in excitatory synapse development. It remains to be determined when and how PTPδ is targeted to inhibitory or excitatory synaptic sites in presynaptic neurons.
Various extracellular and intracellular mechanisms are involved in LAR-RPTP-mediated presynaptic assembly
To reveal key synaptogenesis mechanisms underlying retrograde trans-synaptic signaling involving LAR-RPTPs, we used heterologous synapse-formation assays in conjunction with a lentiviral-mediated molecular replacement strategy using various PTPσ variants (Figs. 5–10). Although the heterologous synapse-formation approach used in the present study represents a reduced system, these analyses allowed us to dissect the detailed synaptogenesis mechanisms that occur in presynaptic or postsynaptic neurons during intercellular adhesion signaling. A similar approach was previously applied to identify the molecular mechanisms underlying neurexin-mediated heterologous synapse formation (Gokce and Südhof, 2013). The cytoplasmic regions of neurexins are dispensable for the recruitment of molecular machineries into nascent synaptogenic sites, although the C-terminal PDZ-binding motif of neurexins is essential for cell surface trafficking (Gokce and Südhof, 2013). These data suggest that neurexins require coreceptor(s) in presynaptic membranes to organize the trans-synaptic signaling cascades, likely through direct interactions of neurexin coreceptor(s) with intracellular proteins in presynaptic neurons. In the current study, we asked whether LAR-RPTPs act in a manner similar to that of neurexins. We elucidated four PTPσ regulatory mechanisms that collectively contribute to orchestrating PTPσ-mediated synaptogenic signaling at excitatory synapses (mechanisms that can possibly be extrapolated to PTPδ).
First, we evaluated the physiological significance of PTPσ alternative splicing in cultured neurons. Alternative splicing involving insertions at the MeA and MeB splice sites of PTPσ controls the affinity of PTPσ for most postsynaptic ligands, but there are exceptions (e.g., GPCs) (Han et al., 2016). Indeed, reexpression of PTPσMeA+MeB+ in PTPσ-deficient neurons rescued the abrogated heterologous synapse formation induced by Slitrk1 and TrkC (Figs. 7–10) (Um et al., 2014a). Strikingly, reexpression of PTPσ splice variants comparably rescued the decreased excitatory synaptic puncta density observed in PTPσ-deficient neurons to control levels, regardless of PTPσ splicing status. These findings suggest that alternative splicing of PTPσ primarily serves to select the postsynaptic ligand that organizes the specific trans-synaptic complex, whereas alternative splicing-independent mechanisms are involved in regulating PTPσ-mediated excitatory synapse development (Fig. 5).
Second, we assessed the effects of PTPσ interactions with a subset of ligands (Slitrks, TrkC, and GPCs). We found that reexpression of PTPσ Y233S or R235D (but not R97A/R100A) failed to rescue the impaired excitatory synapse development observed in PTPσ-deficient neurons, suggesting that PTPσ/Slitrk-mediated synaptic adhesion pathways may be more central than other PTPσ-mediated synaptic adhesion pathways in maintaining excitatory synapse development. Additional structural information obtained from structures of complexes involving PTPσ and other ligands (GPCs or SALMs) should provide additional mechanistic insights into PTPσ-mediated excitatory synapse development.
Third, we confirmed the contribution of the HS-binding feature of PTPσ in regulating excitatory synapse development (Fig. 7), as previously reported (Ko et al., 2015b). HS chains contribute to the assembly of PTPσ oligomers (Coles et al., 2011) and compete with TrkC for PTPσ binding, reflecting the partially overlapping binding interface of TrkC with that of HS (Coles et al., 2014). Thus, it is possible to postulate that HS-bound oligomeric PTPσ proteins form specific complexes with GPCs, and HS-unbound monomeric PTPσ proteins form specific associations with TrkC (Han et al., 2016). Indeed, reexpression of an HS-binding-defective PTPσ [AAAA] mutant in PTPσ-deficient neurons rescued impaired TrkC-induced heterologous synapse formation (Fig. 10). Reexpression of the PTPσ [AAAA] mutant in PTPσ-deficient neurons also rescued the impaired Slitrk1-induced heterologous synapse formation (Fig. 7), suggesting that HS chains serve a regulatory function that promotes preferential binding of monomeric PTPσ to TrkC and Slitrks instead of GPCs (Won et al., 2017).
Fourth, we identified a key intracellular mechanism underlying PTPσ-mediated synaptic signaling in presynaptic neurons (Figs. 9–11). We demonstrated a requirement for D2 domain-mediated recruitment of a subset of known PTPσ substrates in the vicinity of PTPσ. Although our data suggested that the phosphatase activity of PTPσ is not fully required for PTPσ-mediated heterologous synapse formation (Figs. 9, 10), p250GAP and N-cadherin, but not β-catenin, were necessary for PTPσ-mediated heterologous synapse formation (Fig. 11). Because postsynaptic β-catenin and N-cadherin are required for LAR-RPTP-mediated targeting of AMPA receptors to postsynaptic sites and regulating dendritic spines (Dunah et al., 2005; Saglietti et al., 2007), as well as controlling neurotransmitter release (Vitureira et al., 2011), both of which may involve tyrosine phosphorylation signaling pathways, differential sets of downstream mechanisms in both presynaptic and postsynaptic neurons may participate in distinct aspects of synaptogenesis. Other PTPσ substrates that were not tested in the current study include Abl, Ena, and Trio, all of which are known to be involved in actin polymerization, similarly to p250GAP (Lanier and Gertler, 2000; Bateman et al., 2001; Miller et al., 2013). Thus, it is tempting to speculate that regulation of the actin cytoskeleton by Rac signaling is an important step in PTPσ-mediated heterologous synapse formation in presynaptic neurons. This suggested role is unique in that actin is not enriched in the course of heterologous synapse formation mediated by presynaptic neurexins (Gokce and Südhof, 2013). Given that local assembly of an F-actin cytoskeleton at nascent presynaptic sites links synaptic adhesion events with axonal branch formation (Chia et al., 2014), subsequent studies are warranted to further elucidate LAR-RPTP-associated presynaptic assembly and their various postsynaptic ligands. In particular, how HS-binding-induced oligomerization of PTPσ might influence intracellular signaling cascades by triggering conformational changes, given the flexibility of the PTPσ structure (Coles et al., 2014), remains to be determined. In addition, proteomic analyses to identify the intracellular component of LAR-RPTP complex in vivo are required for accurately mapping of additional key components in presynaptic neurons, as recently attempted (Loh et al., 2016; Uezu et al., 2016) (Fig. 13). Considering the possibility that coreceptor(s) of neurexins converge on intracellular signaling pathways that overlap with components linked to LAR-RPTP-mediated signaling cascades (e.g., liprin-α2/3), profiling the complete collection of scaffolds in presynaptic neurons will facilitate our understanding of how trans-synaptic signals organize various aspects of presynaptic function.
Figure 13.

Molecular model of PTPσ signaling pathways in heterologous synapse formation. A, PTPσ triggers excitatory heterologous synapse formation through a combination of extracellular and intracellular signaling components. The PTPσ D2 domain binds intracellular adaptor proteins (e.g., liprin-α) and substrates (e.g., p250GAP and N-cadherin) to recruit the vesicular machinery for excitatory synapse development. This signal transduction model differs from that of neurexin (see below). B, Neurexins serve as anchor proteins that transduce postsynaptic signals from various ligands (e.g., neuroligins and LRRTM proteins) and transfer them to adjacent, but as yet unidentified, coreceptor protein(s) to mediate the signal transduction cascades necessary for full heterologous synapse formation activity (Gokce and Südhof, 2013).
Footnotes
This work was supported by Korea Healthcare Technology R & D Project, funded by the Ministry for Health and Welfare Affairs, Republic of Korea Grant HI17C0080 to J.K. We thank Dr. Ho Min Kim (KAIST, Korea) for input in designing various PTPσ point mutants.
The authors declare no competing financial interests.
References
- Aricescu AR, McKinnell IW, Halfter W, Stoker AW (2002) Heparan sulfate proteoglycans are ligands for receptor protein tyrosine phosphatase sigma. Mol Cell Biol 22:1881–1892. 10.1128/MCB.22.6.1881-1892.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman J, Reddy RS, Saito H, Van Vactor D (2001) The receptor tyrosine phosphatase dlar and integrins organize actin filaments in the Drosophila follicular epithelium. Curr Biol 11:1317–1327. 10.1016/S0960-9822(01)00420-1 [DOI] [PubMed] [Google Scholar]
- Boucard AA, Ko J, Südhof TC (2012) High affinity neurexin binding to cell adhesion G-protein-coupled receptor CIRL1/latrophilin-1 produces an intercellular adhesion complex. J Biol Chem 287:9399–9413. 10.1074/jbc.M111.318659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresler T, Shapira M, Boeckers T, Dresbach T, Futter M, Garner CC, Rosenblum K, Gundelfinger ED, Ziv NE (2004) Postsynaptic density assembly is fundamentally different from presynaptic active zone assembly. J Neurosci 24:1507–1520. 10.1523/JNEUROSCI.3819-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bury LA, Sabo SL (2014) Dynamic mechanisms of neuroligin-dependent presynaptic terminal assembly in living cortical neurons. Neural Dev 9:13. 10.1186/1749-8104-9-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bury LA, Sabo SL (2016) Building a terminal: mechanisms of presynaptic development in the CNS. Neuroscientist 22:372–391. 10.1177/1073858415596131 [DOI] [PubMed] [Google Scholar]
- Chia PH, Chen B, Li P, Rosen MK, Shen K (2014) Local F-actin network links synapse formation and axon branching. Cell 156:208–220. 10.1016/j.cell.2013.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Nam J, Whitcomb DJ, Song YS, Kim D, Jeon S, Um JW, Lee SG, Woo J, Kwon SK, Li Y, Mah W, Kim HM, Ko J, Cho K, Kim E (2016) SALM5 trans-synaptically interacts with LAR-RPTPs in a splicing-dependent manner to regulate synapse development. Sci Rep 6:26676. 10.1038/srep26676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles CH, Shen Y, Tenney AP, Siebold C, Sutton GC, Lu W, Gallagher JT, Jones EY, Flanagan JG, Aricescu AR (2011) Proteoglycan-specific molecular switch for RPTPsigma clustering and neuronal extension. Science 332:484–488. 10.1126/science.1200840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles CH, Mitakidis N, Zhang P, Elegheert J, Lu W, Stoker AW, Nakagawa T, Craig AM, Jones EY, Aricescu AR (2014) Structural basis for extracellular cis and trans RPTPsigma signal competition in synaptogenesis. Nat Commun 5:5209. 10.1038/ncomms6209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Hueske E, Wyszynski M, Hoogenraad CC, Jaworski J, Pak DT, Simonetta A, Liu G, Sheng M (2005) LAR receptor protein tyrosine phosphatases in the development and maintenance of excitatory synapses. Nat Neurosci 8:458–467. 10.1038/nn1416 [DOI] [PubMed] [Google Scholar]
- Gokce O, Südhof TC (2013) Membrane-tethered monomeric neurexin LNS-domain triggers synapse formation. J Neurosci 33:14617–14628. 10.1523/JNEUROSCI.1232-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han KA, Jeon S, Um JW, Ko J (2016) Emergent synapse organizers: LAR-RPTPs and their companions. Int Rev Cell Mol Biol 324:39–65. 10.1016/bs.ircmb.2016.01.002 [DOI] [PubMed] [Google Scholar]
- Jin Y, Garner CC (2008) Molecular mechanisms of presynaptic differentiation. Annu Rev Cell Dev Biol 24:237–262. 10.1146/annurev.cellbio.23.090506.123417 [DOI] [PubMed] [Google Scholar]
- Johnson KG, Van Vactor D (2003) Receptor protein tyrosine phosphatases in nervous system development. Physiol Rev 83:1–24. 10.1152/physrev.00016.2002 [DOI] [PubMed] [Google Scholar]
- Kang H, Han KA, Won SY, Kim HM, Lee YH, Ko J, Um JW (2016) Slitrk missense mutations associated with neuropsychiatric disorders distinctively impair slitrk trafficking and synapse formation. Front Mol Neurosci 9:104. 10.3389/fnmol.2016.00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Choii G, Um JW (2015a) The balancing act of GABAergic synapse organizers. Trends Mol Med 21:256–268. 10.1016/j.molmed.2015.01.004 [DOI] [PubMed] [Google Scholar]
- Ko JS, Pramanik G, Um JW, Shim JS, Lee D, Kim KH, Chung GY, Condomitti G, Kim HM, Kim H, de Wit J, Park KS, Tabuchi K, Ko J (2015b) PTPsigma functions as a presynaptic receptor for the glypican-4/LRRTM4 complex and is essential for excitatory synaptic transmission. Proc Natl Acad Sci U S A 112:1874–1879. 10.1073/pnas.1410138112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Soler-Llavina GJ, Fuccillo MV, Malenka RC, Südhof TC (2011) Neuroligins/LRRTMs prevent activity- and Ca2+/calmodulin-dependent synapse elimination in cultured neurons. J Cell Biol 194:323–334. 10.1083/jcb.201101072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SK, Woo J, Kim SY, Kim H, Kim E (2010) Trans-synaptic adhesions between netrin-G ligand-3 (NGL-3) and receptor tyrosine phosphatases LAR, protein-tyrosine phosphatase delta (PTPdelta), and PTPsigma via specific domains regulate excitatory synapse formation. J Biol Chem 285:13966–13978. 10.1074/jbc.M109.061127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LM, Gertler FB (2000) From abl to actin: abl tyrosine kinase and associated proteins in growth cone motility. Curr Opin Neurobiol 10:80–87. 10.1016/S0959-4388(99)00058-6 [DOI] [PubMed] [Google Scholar]
- Li Y, Zhang P, Choi TY, Park SK, Park H, Lee EJ, Lee D, Roh JD, Mah W, Kim R, Kim Y, Kwon H, Bae YC, Choi SY, Craig AM, Kim E (2015) Splicing-dependent trans-synaptic SALM3-LAR-RPTP interactions regulate excitatory synapse development and locomotion. Cell Rep 12:1618–1630. 10.1016/j.celrep.2015.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh KH, Stawski PS, Draycott AS, Udeshi ND, Lehrman EK, Wilton DK, Svinkina T, Deerinck TJ, Ellisman MH, Stevens B, Carr SA, Ting AY (2016) Proteomic analysis of unbounded cellular compartments: synaptic clefts. Cell 166:1295–1307.e21. 10.1016/j.cell.2016.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas C, Torres VI, Altrock WD, Leal-Ortiz S, Wagh D, Terry-Lorenzo RT, Fejtova A, Gundelfinger ED, Ziv NE, Garner CC (2012) Formation of Golgi-derived active zone precursor vesicles. J Neurosci 32:11095–11108. 10.1523/JNEUROSCI.0195-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK. (2007) Dynamic aspects of CNS synapse formation. Annu Rev Neurosci 30:425–450. 10.1146/annurev.neuro.29.051605.112830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MB, Yan Y, Eipper BA, Mains RE (2013) Neuronal rho GEFs in synaptic physiology and behavior. Neuroscientist 19:255–273. 10.1177/1073858413475486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missler M, Südhof TC, Biederer T (2012) Synaptic cell adhesion. Cold Spring Harb Perspect Biol 4:a005694. 10.1101/cshperspect.a005694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montani C, Ramos-Brossier M, Ponzoni L, Gritti L, Cwetsch AW, Braida D, Saillour Y, Terragni B, Mantegazza M, Sala M, Verpelli C, Billuart P, Sala C (2017) The X-linked intellectual disability protein IL1RAPL1 regulates dendrite complexity. J Neurosci 37:6606–6627. 10.1523/JNEUROSCI.3775-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa T, Kuriu T, Tezuka T, Umemori H, Okabe S, Yamamoto T (2008) Regulation of dendritic spine morphology by an NMDA receptor-associated Rho GTPase-activating protein, p250GAP. J Neurochem 105:1384–1393. 10.1111/j.1471-4159.2008.05335.x [DOI] [PubMed] [Google Scholar]
- Pang ZP, Cao P, Xu W, Südhof TC (2010) Calmodulin controls synaptic strength via presynaptic activation of calmodulin kinase II. J Neurosci 30:4132–4142. 10.1523/JNEUROSCI.3129-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido R, Serra-Pagès C, Tang M, Streuli M (1995) The LAR/PTP delta/PTP sigma subfamily of transmembrane protein-tyrosine-phosphatases: multiple human LAR, PTP delta, and PTP sigma isoforms are expressed in a tissue-specific manner and associate with the LAR-interacting protein LIP.1. Proc Natl Acad Sci U S A 92:11686–11690. 10.1073/pnas.92.25.11686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saglietti L, Dequidt C, Kamieniarz K, Rousset MC, Valnegri P, Thoumine O, Beretta F, Fagni L, Choquet D, Sala C, Sheng M, Passafaro M (2007) Extracellular interactions between GluR2 and N-cadherin in spine regulation. Neuron 54:461–477. 10.1016/j.neuron.2007.04.012 [DOI] [PubMed] [Google Scholar]
- Spangler SA, Hoogenraad CC (2007) Liprin-alpha proteins: scaffold molecules for synapse maturation. Biochem Soc Trans 35:1278–1282. 10.1042/BST0351278 [DOI] [PubMed] [Google Scholar]
- Suarez F, Thostrup P, Colman D, Grutter P (2013) Dynamics of presynaptic protein recruitment induced by local presentation of artificial adhesive contacts. Dev Neurobiol 73:98–106. 10.1002/dneu.22037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. (2008) Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455:903–911. 10.1038/nature07456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. (2012) The presynaptic active zone. Neuron 75:11–25. 10.1016/j.neuron.2012.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. (2017) Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell 171:745–769. 10.1016/j.cell.2017.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Arstikaitis P, Prasad T, Bartlett TE, Wang YT, Murphy TH, Craig AM (2011) Postsynaptic TrkC and presynaptic PTPsigma function as a bidirectional excitatory synaptic organizing complex. Neuron 69:287–303. 10.1016/j.neuron.2010.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Katayama K, Sohya K, Miyamoto H, Prasad T, Matsumoto Y, Ota M, Yasuda H, Tsumoto T, Aruga J, Craig AM (2012) Selective control of inhibitory synapse development by Slitrk3-PTPdelta trans-synaptic interaction. Nat Neurosci 15:389–398, S1–S2. 10.1038/nn.3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uezu A, Kanak DJ, Bradshaw TW, Soderblom EJ, Catavero CM, Burette AC, Weinberg RJ, Soderling SH (2016) Identification of an elaborate complex mediating postsynaptic inhibition. Science 353:1123–1129. 10.1126/science.aag0821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, Ko J (2013) LAR-RPTPs: synaptic adhesion molecules that shape synapse development. Trends Cell Biol 23:465–475. 10.1016/j.tcb.2013.07.004 [DOI] [PubMed] [Google Scholar]
- Um JW, Kim KH, Park BS, Choi Y, Kim D, Kim CY, Kim SJ, Kim M, Ko JS, Lee SG, Choii G, Nam J, Heo WD, Kim E, Lee JO, Ko J, Kim HM (2014a) Structural basis for LAR-RPTP/Slitrk complex-mediated synaptic adhesion. Nat Commun 5:5423. 10.1038/ncomms6423 [DOI] [PubMed] [Google Scholar]
- Um JW, Pramanik G, Ko JS, Song MY, Lee D, Kim H, Park KS, Südhof TC, Tabuchi K, Ko J (2014b) Calsyntenins function as synaptogenic adhesion molecules in concert with neurexins. Cell Rep 6:1096–1109. 10.1016/j.celrep.2014.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valnegri P, Montrasio C, Brambilla D, Ko J, Passafaro M, Sala C (2011) The X-linked intellectual disability protein IL1RAPL1 regulates excitatory synapse formation by binding PTPdelta and RhoGAP2. Hum Mol Genet 20:4797–4809. 10.1093/hmg/ddr418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitureira N, Letellier M, White IJ, Goda Y (2011) Differential control of presynaptic efficacy by postsynaptic N-cadherin and beta-catenin. Nat Neurosci 15:81–89. 10.1038/nn.2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won SY, Kim CY, Kim D, Ko J, Um JW, Lee SB, Buck M, Kim E, Heo WD, Lee JO, Kim HM (2017) LAR-RPTP clustering is modulated by competitive binding between synaptic adhesion partners and heparan sulfate. Front Mol Neurosci 10:327. 10.3389/fnmol.2017.00327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim YS, Kwon Y, Nam J, Yoon HI, Lee K, Kim DG, Kim E, Kim CH, Ko J (2013) Slitrks control excitatory and inhibitory synapse formation with LAR receptor protein tyrosine phosphatases. Proc Natl Acad Sci U S A 110:4057–4062. 10.1073/pnas.1209881110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Yasumura M, Uemura T, Lee SJ, Ra M, Taguchi R, Iwakura Y, Mishina M (2011) IL-1 receptor accessory protein-like 1 associated with mental retardation and autism mediates synapse formation by trans-synaptic interaction with protein tyrosine phosphatase delta. J Neurosci 31:13485–13499. 10.1523/JNEUROSCI.2136-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv NE, Garner CC (2004) Cellular and molecular mechanisms of presynaptic assembly. Nat Rev Neurosci 5:385–399. 10.1038/nrn1370 [DOI] [PubMed] [Google Scholar]
- Zürner M, Mittelstaedt T, tom Dieck S, Becker A, Schoch S (2011) Analyses of the spatiotemporal expression and subcellular localization of liprin-alpha proteins. J Comp Neurol 519:3019–3039. 10.1002/cne.22664 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed list of peptides obtained by mass spectroscopy analyses using GST-PTPδ D2 or GST-PTPσ D2 in mouse brain synaptosomal fractions. Download Figure 11-1, XLSX file (34.7KB, xlsx)