

Abstract
The human 16p11.2 microdeletion is one of the most common gene copy number variations linked to autism, but the pathophysiology associated with this chromosomal abnormality is largely unknown. The 593 kb deletion contains the ERK1 gene and other genes that converge onto the ERK/MAP kinase pathway. Perturbations in ERK signaling are linked to a group of related neurodevelopmental disorders hallmarked by intellectual disability, including autism. We report that mice harboring the 16p11.2 deletion exhibit a paradoxical elevation of ERK activity, cortical cytoarchitecture abnormalities and behavioral deficits. Importantly, we show that treatment with a novel ERK pathway inhibitor during a critical period of brain development rescues the molecular, anatomical and behavioral deficits in the 16p11.2 deletion mice. The ERK inhibitor treatment administered to adult mice ameliorates a subset of these behavioral deficits. Our findings provide evidence for potential targeted therapeutic intervention in 16p11.2 deletion carriers.
SIGNIFICANCE STATEMENT The ERK/MAPK pathway is genetically linked to autism spectrum disorders and other syndromes typified by intellectual disability. We provide direct evidence connecting the ERK/MAP kinases to the developmental abnormalities in neurogenesis and cortical cytoarchitecture associated with the 16p11.2 chromosomal deletion. Most importantly, we demonstrate that treatment with a novel ERK-specific inhibitor during development rescues aberrant cortical cytoarchitecture and restores normal levels of cell-cycle regulators during cortical neurogenesis. These treatments partially reverse the behavioral deficits observed in the 16p11.2del mouse model, including hyperactivity, memory as well as olfaction, and maternal behavior. We also report a rescue of a subset of these deficits upon treatment of adult 16p11.2del mice. These data provide a strong rationale for therapeutic approaches to this disorder.
Keywords: 16p11, autism, cortical development, ERK MAP kinases, neurodevelopment
Introduction
Autism spectrum disorders (ASDs) are complex, highly heritable neurodevelopmental disorders affecting ∼1 in 100 children. Copy number variations (CNVs) and other chromosomal rearrangements are associated with ∼10–20% of ASDs. CNV of human chromosome 16p11.2 is one of the most common genetic linkages to autism and deletion of this region accounts for ∼1% of ASDs (Levy et al., 2011). Individuals heterozygous for the 16p11.2 deletion exhibit a range of clinical symptoms including ASD, language impairment, intellectual disability (ID), anxiety, attention deficit hyperactivity disorder and epilepsy (Ghebranious et al., 2007; Zufferey et al., 2012; Hanson et al., 2015). The human 16p11.2 locus contains 27 genes, which includes the MAPK3 gene (encoding ERK1) and the major vault protein gene (MVP), both of which converge onto the ERK/MAP kinase pathway (Kumar et al., 2008).
The extracellular signal-regulated kinases, ERK1 and ERK2, are central elements of one of the most prominent intracellular signaling cascades, the mitogen activated protein kinase (MAPK) pathway. The ERKs play critical roles in brain development and synaptic plasticity (Sweatt, 2004) and are activated in response to a broad range of stimuli including growth factors, neurotransmitters, morphogens and transient increases in synaptic Ca2+ (Roskoski, 2012). Importantly, they are genetically linked to ASDs and other syndromes typified by ID (Marshall et al., 2008; Wen et al., 2016; Borrie et al., 2017; Mitra et al., 2017). Mutations in elements of the ERK/MAPK pathway alter the activity of the ERKs, resulting in a group of genetic disorders collectively known as “RASopathies”. These syndromes are typified by ID, developmental and language deficits, ASD, and psychiatric disease (Tidyman and Rauen, 2009; Fasano and Brambilla, 2011).
We have previously reported that a murine model of the 16p11.2 human microdeletion (16p11.2del) exhibits a reduction in brain size and perturbations in cortical cytoarchitecture, which are postulated to be due to impaired ERK-mediated regulation of neural progenitor proliferation (Newbern et al., 2008; Pucilowska et al., 2015). The 16p11.2del mice exhibit a paradoxical increase in ERK signaling coincident with aberrant cortical neurogenesis, ultimately resulting in behavioral deficits analogous to the 16p11.2 microdeletion carriers (Portmann et al., 2014; Hanson et al., 2015). Therefore, we postulated that treatment with brain-permeable Ras-ERK pathway inhibitors may correct the pathophysiology associated with the 16p11.2 deletion. Recently, we have validated two novel cell-permeable peptides (CPPs), RB1 and RB3, that efficiently inhibit Ras-ERK signaling in the postnatal developing brain and rescue morphological impairments in a severe mouse model of RASopathies (Papale et al., 2017). In addition, the RB1 and RB3 peptides are able to block cocaine-mediated ERK activation and the associated behavioral response (Papale et al., 2016).
In this study, we report that prenatal treatment with cell permeant RB1/RB3 peptides rescues developmental deficits in neurogenesis in the embryo and subsequently restores normal neuronal numbers and cortical cytoarchitecture in the 16p11.2del mice. Specifically, we report restoration of hippocampal-based memory function, anxiety, olfaction, maternal behavior as well as hyperactivity in the 16p11.2del mice. Furthermore, postnatal drug treatment of adult 16p11.2del mice results in partial amelioration of the behavioral deficits, suggesting a broader window for pharmacologic intervention. To assess the macroscale anatomical substrates affected by the drug treatment, we used high-resolution morphoanatomical MRI mapping to show partial restoration of gray matter volume in ventral hippocampal and lateral septal regions, which we found to be reduced in the 16p11.2del mice. We conclude that treatment with ERK pathway inhibitors may represent a potential therapeutic intervention in 16p11.2del carriers, as has been suggested for RASopathies (Tidyman and Rauen, 2009). This is the first example of the rescue of development abnormalities in this ASD model.
Materials and Methods
Animals
A mouse line carrying a microdeletion on chromosome 7qF3, the syntenic region of human chromosome 16p11.2, was generated by A. Mills and purchased from Jackson Laboratories (Horev et al., 2011). This mouse line has been maintained on a fixed ratio, mixed (129/C57) background with large numbers of mice examined to minimize any genetic background-associated variation. Embryonic and adult treatments were 5 d long and administered by subcutaneous injections of ERK inhibitor resuspended in PBS at a dose of 10 mg/kg. Pregnant dams were used with a plug date designated as 0.5 d.
Drugs
RB1 and RB3 CPPs have been recently described (Papale et al., 2016). Briefly, RB1 was designed around residues 59–73 within the KIM sequence of the ERK-specific phosphatase MKP3 (Liu et al., 2006) that interacts with a docking motif required for interactions of ERK1/2 with both its regulators and substrates. RB3 was designed, using the MOE software package (Molecular Operating Environment v10.10, Chemical Computing Group, ) by aligning and superposing the CDC25 domain of Ras-GRF1 (Freedman et al., 2006) the published crystal structure of a ternary Ras:SOS:Ras*GDP complex (Sondermann et al., 2004) using the default settings. The interacting surface between the two proteins was then visually analyzed and the portion of the CDC25 domain between residues 1173–1203 was selected for the preparation of the final peptide. RB1 (MGRKKRRQRRRPPQAPGIMLRRLQKGNLPVSRYPYDVPD), SCR RB1 (MGRKKRRQRRRPPQALSLKRLRSRGMNRTSATQSRYPYD), RB3 (GRKKRRQRRRPPCVPYLGMYLTDLVFIEEGTPNYTEDGLVN), and SCR RB3 (GRKKRRQRRRPPCFEVYPDSGDYTYEGELNGTLMVVPTN) were custom synthesized by GENECUST EUROPE.
For in vivo experiments, batches of 200 mg, highly purified by high-performance liquid chromatography (≥ 95%) with C-terminal amino acid (last) in D form and acetylated N-terminal (first) amino acid were used. The peptides were dissolved in PBS 1× and injected 10 mg/kg (10 mg/kg each peptide, i.p.).
Sample preparation and MRI acquisition
High-resolution morphoanatomical T2-weighted MR imaging of P90 ex vivo mouse brains was performed in paraformaldehyde fixed specimens. Standard sample preparation and MRI acquisition have been recently described in detail (Cutuli et al., 2016). Briefly, 16p11.2del mice and age-matched control littermates (treated with ERK inhibitor or vehicles) were deeply anesthetized and their brains were perfused in situ via cardiac perfusion. The perfusion was performed with PBS followed by 4% paraformaldehyde (PFA; 100 ml). Both perfusion solutions included a gadolinium chelate (Prohance) at a concentration of 10 and 5 mm, respectively, to shorten longitudinal relaxation times. Brains were imaged inside intact skulls to avoid postextraction deformations. A multichannel 7.0 tesla MRI scanner (Bruker) was used to acquire anatomical images of the brain, using a 72 mm birdcage transmit coil and a custom-built saddle-shaped solenoid coil for signal reception, with the following imaging parameters: FLASH 3D sequence with TR = 17 ms, TE = 10 ms, α = 30°, matrix size of 260 × 180 × 180, field-of-view of 1.82 × 1.26 × 1.26 cm and voxel size of 0.07 mm (isotropic).
Tensor-based morphometry automated anatomical labeling and structural covariance MRI network mapping
Inter-group morphoanatomical differences in local volumes were mapped with tensor-based morphometry (TBM) using ANTs (Avants et al., 2009). The registration-based TBM procedure used has been thoroughly described previously (Pagani et al., 2016). First, all the high-resolution T2-weighted images were corrected for intensity non-uniformity and skull stripped to remove extra-brain tissue. A study-based template was then created aligning preprocessed images to a common reference space using affine and diffeomorphic registrations. Individual images of 16p11.2del and control mice (treated with ERK inhibitor or vehicle) were registered to the study-based template and the Jacobian determinants of the deformation fields were calculated at each voxel, giving the voxel's relative expansion or contraction in the space of the study-based template. Jacobian determinants were also normalized by the total intracranial volume to further eliminate overall brain volume variations. We measured non-normalized intracranial brain volume in all the four treatment groups at P90. The results we obtained showed the presence of smaller brain volume in 16p11.2 mutants, a finding previously reported in mice modeling 16p11.2 deletion (Portmann et al., 2014). The treatment did not affect total intracranial volume in either WT or 16p11.2 mutants. The resulting maps were smoothed using a Gaussian kernel with a σ of three voxel width and used for voxelwise statistics. Regional volume differences between 16p11.2del and control mice were mapped by fitting a GLM (t > 2.3) followed by a cluster correction using a significant cluster threshold of p = 0.01 (Worsley et al., 1992) as implemented in FSL. We also used preprocessed images to independently calculate volumes of brain regions via automated anatomical labeling (Pagani et al., 2016), using two neuroanatomically parcellated reference MRI atlases for cortical (Ullmann et al., 2013) and subcortical areas (Dorr et al., 2008).
Behavioral analysis
Three-month-old male and female mice were used for standardized behavioral analyses including the elevated plus maze, open-field, novel object recognition, olfaction, maternal behavior, and fear conditioning tests in order as stated. All tests were conducted in a designated behavior room during the light cycle between 9:00 A.M. and 6:00 P.M. A maximum of five mice of mixed genotypes were housed together with ad libitum access to food and water with a 12 h light/dark cycle. All equipment was cleaned with 70% ethanol after each use to remove odor cues. The tester was blinded to the genotype of each animal. All tests were performed at the Case Western Reserve University Rodent Behavior Core. Three different cohorts (each with at least 50 mice divided to include both genotypes and treatment paradigms) were evaluated in three independent experiments. The data represents the pooled analysis of all three cohorts.
Elevated plus maze.
The maze consisted of two open and two closed arms crossing each other ∼1 m above the floor. The maze was fitted with infrared grid and video tracking system (Med Associates). Individual test mice were placed in the center facing the open arm and their activity was recorded for 5 min. The percentage time spent in open and closed arms, the number of entries into each arm, number of head dips, and frequency of urination/defecation were scored.
Open-field test.
A box (40 × 40 cm) was placed in a dimly lit environment. EthoVision XT 5.0 (Noldus) was used to digitally subdivide the box area into a 20 cm Å∼ 20 cm center area and a periphery. The peripheral area was further divided into middle (inner 10 cm) and an outer area (outer 10 cm) to determine thigmotaxic behavior. Mice were placed in the center and allowed to explore the area freely for 15 min. Locomotion parameters such as total distance, velocity, angular velocity, and immobility were measured. Frequency and time duration in the center, periphery, and outer area were recorded to determine anxiety-like behavior. In addition, data were nested into 5 min bins and distance moved during each of these three periods was recorded to evaluate habituation differences across groups.
Novel object.
This assay was conducted 24 h following the open field test in the same arena. Each subject mouse was placed in the chamber containing two identical objects and allowed free exploration for 10 min. Following a 3 h delay, one of the familiar objects was replaced with a novel object and the test mice were returned to the same arena. The time spent sniffing each object was measured. Tester was blinded to the genotypes of tested mice. We scored and reported raw data of time spent sniffing novel versus habituated objects. Mice that did not sniff either object and/or did not sniff for a total of 4 s were excluded from the final analysis.
Olfactory assay: buried food retrieval.
Mice were food deprived overnight and places in standard clean cages with normal cage bedding (3 cm). Mice were allowed to acclimate for 5 min, then were removed to another clean cage at which point a uniform piece of food (Teddy Graham) was placed in a random corner under ∼1 cm of normal bedding. Next, each mouse was returned to its cage and the time to retrieve the food source (latency) was recorded.
Contextual and cued fear conditioning.
Fear-conditioning test was conducted as the last behavioral test in the series of all behavioral assessments. Experiments used two standard conditioning chambers, each housed in an isolation cubicle and equipped with a stainless-steel grid floor connected to a solid-state shock scrambler. Each scrambler delivered an electronic constant-current shock source that was controlled via an interface connected to a Windows XP computer running FreezeFrame software (Coulbourn Instruments). A digital camera was mounted to the side of each chamber, and video signals were sent to the same computer for analysis. During training, mice were placed in the conditioning chamber for 12 min and then received four footshocks [conditioned stimulus: 85 dB sound at 2800 Hz for 30 s; unconditioned stimulus (US) 0.56 mA]. Retention test was performed 18 h later for 5 min in the absence of a shock.
Analysis of progenitor proliferation
Mice received a single intraperitoneal injection of BrdU (50 mg/kg, Sigma-Aldrich B5002) at E14.5 and killed exactly 30 min later [vehicle (veh)-treated: nWT = 8,nDel = 6; inhibitor (inh)-treated: nWT = 9,nDel = 9]. Sections were immunostained with anti-BrdU antibody (rat-anti-BrdU; 1:100; Abcam), or mouse anti-BrdU (1:100; BD Biosciences). The number of BrdU+ cells per cortical sections (proliferative fraction) was established using stereological technique and all positive cells within a 100 μm segment along the ventricular surface, extending from the ventricle to the pial surface, were counted. All data collection was blinded to the genotype and randomized. In embryonic brains all analyses were performed at the rostrocaudal axis of fully emerged ganglionic eminences (LGE as well as MGE). These structures as well as the presence of choroid plexus were noted and the images were taken just above the pallial/subpallial boundary in the VZ as well dorsomedial area of the VZ. In adult mice we evaluated BrdU staining in two different areas: (1) above the pallial/subpallial boundary (dorsolateral), and the other (2) at the level of dorsomedial cortex. For postnatal analyses we evaluated both motor and somatosensory cortex.
Microscopy and image analysis
All sections were imaged using a Zeiss LSM 510 confocal laser microscope equipped with argon and helium–neon lasers and analyzed with LCS confocal software, Prism and Photoshop (Adobe). All counts were performed on blinded sections and two-way ANOVA with Bonferroni post hoc was used to establish statistical significance for each experiment.
Western analysis
Cortices were dissected from E14.5 embryos and washed with ice-cold HBSS. The lysates were sonicated in lysis buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 1% NP-40, 10% glycerol, 1 mm EDTA, 1.5 mm MgCl2, 20 mm NaF, and 20 mm β-glycerophosphate) in the presence of protease inhibitors (1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 mm PMSF, and 1 mm Na3VO4). Samples were centrifuged and protein concentrations were established with bicinchoninic acid assay (Pierce) using a BSA standard. Equal amounts of protein were boiled in sample buffer, separated by SDS-PAGE gels, and transferred to Immobilon-P polyvinylidene difluoride membranes (Millipore). Membranes were blocked in 3% BSA (or 5% skim milk) in TBS and 0.1% Tween 20 (TBS-T) for 2 h at room temperature and incubated with primary antibodies overnight at 4°C. The primary antibodies used were as follows: rabbit anti-pERK (Cell Signaling Technology; 1:1000), mouse anti-ERK2 (BD Biosciences Discovery Labware; 1:3000), mouse anti-ERK1 (Zymed Laboratories; 1:1000), anti-CoxIV, and (1:3000; Cell Signaling Technology). Membranes were washed with TBS-T, incubated with HRP-conjugated secondary antibodies: goat anti-mouse or anti-rabbit (1:5000; GE Healthcare) in TBS-T with 5% milk for 2 h at room temperature. Detection was performed using Millipore chemiluminescence using BioMax MR x-ray film (Eastman Kodak). Densitometry was performed using Adobe Photoshop histogram function, and statistical analysis was done with GraphPad Prism software.
ELISA
The oxytocin (OT) ELISA was performed using ENZO Oxytocin kit (ADI-901-153A, Enzo Life Sciences) according to the manufacturer's instructions. Whole-brain homogenates of P90 WT and 16p11.2del brains were used and the calometric readout was reported in pictograms per milliliter.
Immunohistochemistry
The E14.5 brains were dissected in cold PBS and fixed by immersion in 4% PFA for 1 h or overnight at 4°C. P10 and adult mouse brains were fixed in 4% PFA/1× PBS at 4°C overnight and serially incubated in 10, 20 and 30% sucrose. The brains were sectioned (10 μm), then rehydrated in PBS for 10 min. Antigen retrieval using 1× Reveal Decloaker (Biocare) was performed for 10 min at 95°C. Sections were blocked in 10% (embryonic) and 2% (postnatal) normal goat or donkey serum for 1 h at room temperature with 0.1% (v/v) Triton X-100 in PBS. Slides were incubated with primary antibodies overnight at 4°C, rinsed with PBS and incubated with corresponding secondary antibodies for 1–2 h at room temperature. The primary antibodies used were as follows: polyclonal rabbit anti-pERK (Cell Signaling Technology; 1:100), rabbit polyclonal anti-Pax6 (Covance; 1:300), mouse anti-PH3 (Millipore; 1:250), rabbit anti-PH3 (Millipore; 1;500), rabbit anti-Tbr1 (Millipore Bioscience Research Reagents;1:1000), rabbit anti-Tbr2 (1:300), chicken anti-Tbr2 (1:250), goat anti-Brn1 (Santa Cruz Biotechnology; 1:50), rat anti-Ctip2 (Abcam; 1:500), rabbit anti-Cux1 (Santa Cruz Biotechnology; 1:100), and mouse anti-SatB2 (Abcam; 1:100). Secondary antibodies used were AlexaFluor 488 (1:1000), 546 or 593 (1:1000) conjugated to goat or donkey anti-mouse, anti-rabbit or anti-goat (Invitrogen). DNA was stained with 4′,6′-diamidino-2-phenylindole for 5 min (Invitrogen). Immunohistochemical staining was performed on all four groups simultaneously.
Embryonic brains.
Anatomically matched sections of littermate WT and mutant mice were analyzed. At least three consecutive sections per animal were analyzed alongside the mid-rostrocaudal axis of the dorsal telencephalon. Embryonic coronal sections were evaluated at the level of the ganglionic eminences and analyzed by counting all cells in standard 100 μm bins extending from the ventricle to the pial surface. Quantification was performed blind to genotype as previously described (Glickstein et al., 2009). Three to four litters were examined per experiment.
Adult brains.
We counted at least three consecutive tissue slices per slide and at least four slides per animal. In postnatal brains, coronal sections were used to count cells in 400 μm boxes in somatosensory and motor cortex. Slides were picked at random and the investigator was blinded to genotypes and treatments. All data from a single experiment, incorporating all four experimental groups, was averaged. The number of mice evaluated is indicated in figure legends, with three to four litters examined per experiment.
Statistical analysis
All data analyzed followed normal distribution according to the Kolmogorov–Smirnov test. Statistical significance was determined using two-way ANOVA followed by Bonferroni's post hoc test for multiple comparisons. Data analysis was performed using GraphPad Prism and presented as means ± SEM.
All experiments in this study were blinded and randomized. All mice bred for the experiments were used for preplanned experiments and randomized to experimental groups. Visibly sick animals were excluded before data collection and analysis. Data were collected, processed and analyzed randomly. The experimental design, treatments and handling of mice were identical across experiments. Littermates were used as controls with multiple litters (3–4) examined per experiments. All mice (including the MRI study) were bred in the Case Western Reserve Animal Core.
Results
Pharmacological normalization of ERK activity with an ERK pathway inhibitor in 16p11.2del mice
To test our hypothesis that there are ERK-mediated pathologic processes occurring in the 16p11.2del mice, we took advantage of two recently developed brain penetrant peptides, RB1/RB3, which act in concert to attenuate Ras-ERK activity in the brain(Papale et al., 2016, 2017). These brain penetrant peptides exhibit IC50 values in the micromolar range (Papale et al., 2016). Importantly, these peptides have been successfully used in vivo for early postnatal developmental treatments, manifesting a remarkable high degree of tolerability and low toxicity. Based on these promising results, we treated with the RB1/RB3 mix (hereafter termed “ERK inhibitor”) the pregnant 16p11.2del carrier dams for 5 consecutive days starting at E10.5, encompassing a critical period of cortical neurogenesis. We observed that the enhanced ERK activity in dorsomedial cortex of 16p11.2del mice at E14.5 was normalized in the inhibitor-treated mice (Fig. 1a–c). At E14.5 two-way ANOVA analysis of ERK1 activity showed a significant effect with respect to genotype (F(1,59) = 21.7, p < 0.0001) and inhibitor treatment (F(1,59) = 10.12, p = 0.002), but with an interaction between both variables (F(1,59) = 4.05, p = 0.049). Bonferonni post hoc analysis showed a twofold increase in ERK1 activity (p < 0.0001) in the vehicle-treated 16p11.2del mice compared with vehicle-treated WT mice that normalized to WT levels following inhibitor treatment. Similarly, ERK2 activity was also significantly affected by both genotype (F(1,77) = 4.31, p = 0.041) and inhibitor treatment (F(1,77) = 6.76, p = 0.011) when analyzed by two-way ANOVA, however a significant interaction between genotype and drug treatment was present (F(1,77) = 5.08, p = 0.027). A Bonferonni post hoc analysis showed a 49% (p = 0.017) increase in ERK2 activity in the vehicle-treated 16p11.2del mice compared with vehicle-treated WT mice that normalized to WT levels with inhibitor treatment. Importantly, total ERK1 levels were reduced in the 16p11.2del mice in both vehicle and inhibitor-treated animals, with a significant genotype effect observed by two-way ANOVA (p < 0.0001). We observed significant interactions between the inhibitor and genotype because, interestingly, the dose of the ERK inhibitor (10 mg/kg, i.p.) used in our studies did not alter ERK activity in the WT littermates. In addition, when ERK1/2 activity was analyzed in P10 embryonically-treated mice (Fig. 1d,e), there was an inhibitor effect (pERK1: p = 0.005, pERK2: p = 0.017) by two-way ANOVA, with a Bonferonni post hoc analysis showing a 75% (p < 0.01) and 62% (p < 0.01) increase in ERK1 and ERK2, respectively, in the 16p11.2del vehicle-treated mice that normalized to WT levels. Thus, a prenatal treatment of embryonic mice resulted in abrogation of abnormal ERK hyperactivity in the developing cortex when examined at E14.5 (Fig. 1b,c) or postnatally at P10 (Fig. 1d,e).
Figure 1.

Rescue of ERK activity with an ERK pathway inhibitor in 16p11.2del mice at E14.5 and P10. a, IHC of E14.5 coronal sections from mice treated with vehicle or ERK inhibitor. Veh-treated 16p11.2del mice shows upregulation of ERK activity in the dorsomedial cortex * (anti-pERK; green). This is abrogated after 5 d of inhibitor treatment starting at E10.5. b, Western analysis of E14.5 veh- or inh-treated cortical lysates. c, Quantification of Western analysis showing a significant increase in ERK1 and ERK2 activity in 16p11.2del animals (pERK1: ****p < 0.0001, pERK2: *p < 0.05), which is restored to normal level after inhibitor treatment (##p = 0.0087; #p < 0.05; veh-treated: nWT = 19, nDel = 13; inh-treated: nWT = 19, nDel = 12). ERK1 total levels are decreased in vehicle deletion animals (**p < 0.01) and inhibitor animals (****p < 0.0001; veh-treated: nWT = 8, nDel = 7; inh-treated: nWT = 10, nDel = 9). D, Western analysis of P10 veh- or inh-treated cortical lysates, and (e) Western analysis of P10 veh or inh prenatally treated cortical lysates, quantified in (e) ERK1 and ERK2 activity are elevated in deletion animals at P10 (pERK1: **p < 0.01, pERK2: p**<0.01), which is normalized in embryonic inhibitor treatment (#p < 0.05, ###p < 0.001). All values normalized to loading control GAPDH or CoxIV and reported as a fold-change. p Values are from Bonferroni post hoc analysis (*compares WT to deletion; #compares vehicle deletion to inhibitor deletion).
ERK inhibitor treatment rescues embryonic and postnatal cortical defects in the 16p11.2del Mice
Next, we investigated whether the embryonic treatment with the ERK pathway inhibitor could correct aberrant progenitor proliferation dynamics in the developing cortex (Fig. 2a,b) and rescue deficits in cortical neurogenesis (Fig. 2c,d). We evaluated the number of proliferating, BrdU+ progenitors following a single, 30 min IP BrdU injection at E14.5. A two-way ANOVA analysis showed no genotype or inhibitor effect, but Bonferroni post hoc analysis showed a 30% increase (p < 0.05) in BrdU+ progenitors in the vehicle-treated 16p11.2del mice compared with vehicle-treated WT mice. This increase in progenitor proliferation was abrogated in ERK inhibitor-treated 16p11.2del mice and returned to WT levels (Fig. 2a). Furthermore, Western blot analysis and immunohistochemistry (IHC) demonstrate that the number of Tbr2+ intermediate progenitor cells residing in the SVZ can be restored to normal levels after the inhibitor treatment (Fig. 2b). Western blot analysis of Tbr2 by two-way ANOVA showed a genotype and inhibitor interaction, but post hoc analysis revealed a 30% decrease in Tbr2 levels (p < 0.0001) in the vehicle 16p11.2del mice, which returned to normal following treatment compared with vehicle WT mice. This correlated with a 37% decrease (post hoc Bonferroni, p = 0.0012) in the number of Tbr2+ cells by IHC in the vehicle-treated 16p11.2del mice, that corrected to WT levels in the inhibitor-treated mice compared with vehicle WT animals (two-way ANOVA showed significant inhibitor and genotype interaction). Additionally, using Western analysis and IHC we also show that treatment with ERK inhibitor ameliorates the aberrant elevation of the number of early born cortical neurons marked by Ctip2+ and Tbr1+ that populate cortical layer V and VI, respectively (Fig. 2c,d). Again two-way ANOVA analyses of IHC and Western blots of Tbr1 and Ctip2 showed interactions between genotype and inhibitor, but the post hoc Bonferroni analysis showed a 50% increase in Ctip2 levels (p = 0.012) and a 30% increase in Ctip2+ cells by IHC (p = 0.0005) in the 16p11.2del vehicle-treated mice, which returned to WT levels compared with WT vehicle-treated animals. Tbr1 analysis showed a 35% increase in Tbr1 protein levels (p = 0.038) and a 20% increase in Tbr1+ cells via IHC (p = 0.025) in the 16p11.2del vehicle-treated mice, which returned to WT levels in the inhibitor-treated mice compared with WT vehicle-treated animals.
Figure 2.

Reversal of deficits in cortical neurogenesis in the 16p11.2del mice after treatment with ERK pathway inhibitor at E14.5. IHC of coronal sections and Western analyses of cortical lysates at E14.5 a, IHC with proliferation marker, BrdU injected 30 min. before kill. a′, The number of BrdU+ progenitors was analyzed (veh-treated: nWT = 8, nDel = 6; inh-treated: nWT = 9, nDel = 9; *p < 0.05, #p < 0.05). b, IHC with intermediate progenitor marker, Tbr2 (green). b′, Quantification of Tbr2+ progenitors (veh-treated: nWT = 11, nDel = 4; inh-treated: nWT = 9, nDel = 16; **p < 0.0012, ##p < 0.0052). b″, Quantification of Western analysis (veh-treated: nWT = 29, nDel = 37; inh-treated: nWT = 23, nDel = 21; ****p < 0.00001, #p < 0.0388). c, IHC for layer V marker, Ctip2 (red). c′, Quantification of Ctip2+ neurons (veh-treated: nWT = 5, nDel = 4; inh-treated: nWT = 4, nDel = 6; ***p = 0.0005, ####p < 0.0001, *p = 0.026). c″, Quantification of Western analysis (veh-treated: nWT = 15, nDel = 9; inh-treated nWT = 8, nDel = 11; *p = 0.0186, ##p = 0.0073). d, IHC with layer VI marker, Tbr1 (green). d′, Quantification of Tbr1+ neurons (veh-treated: nWT = 9, nDel = 9; inh-treated: nWT = 5, nDel = 5; *p = 0.025, ###p = 0.0004). d″, Quantification of Western analysis (veh-treated: nWT = 15, nDel = 7; inh-treated: nWT = 11, nDel = 12; *p = 0.038, ##p = 0.0017). All Western analyses data represented as a fold-change, normalized to a loading control. p Values are from Bonferroni post hoc analysis (*compares WT to deletion; #compares vehicle deletion to inhibitor deletion).
Next, we tested whether prenatal ERK inhibitor treatment resulted in permanent rescue of cortical cytoarchitecture defects by evaluation of P2 mice. We show that the number of Brn1+ and Satb2+ layer II–III pyramidal neurons is restored to normal levels after prenatal ERK inhibitor treatment (Fig. 3a,b). These are corticocortical connections, some of which project to contralateral hemisphere across the corpus callosum (O'Rourke et al., 1995). Satb2 analysis shows significant genotype (p = 0.048) and treatment effect (p = 0.013) by two-way ANOVA. Post hoc analysis shows significant 13% reduction in Satb2+ cells in vehicle deletion animals compared with WT vehicle (p = 0.03), which is abrogated with inhibitor treatment of deletion animals, which show Satb2+ cells similar to vehicle WT animals and significant increase compared with vehicle-treated 16p11.2del mice (p = 0.01). Brn1 analysis shows no genotype or treatment effect by two-way ANOVA, but post hoc analysis shows decrease Brn1+ cells in vehicle-treated 16p11.2del mice compared with WT vehicle-treated animals (p < 0.05), but inhibitor-treated 16p11.2del mice show no difference in Brn1+ cells compared with WT vehicle-treated animals.
Figure 3.

Prenatal treatment with an ERK pathway inhibitor stably restores normal cortical cytoarchitecture 16p11.2del mice. a, Mice were evaluated at P2 by IHC with layer II–IV marker Brn1 (green). a′, Quantification of Brn1+ neurons shows a rescue in somatosensory cortex of 16p11.2del mice (veh-treated: nWT = 11, nDel = 13; inh-treated: nWT = 17, nDel = 17; *p < 0.05). b, IHC with layer 2–4 marker, Satb2 (red). b′, Quantification of Satb2+ neurons (veh-treated: nWT = 14, nDel = 13; inh-treated: nWT = 10, nDel = 15; *p = 0.033, #p = 0.0105). c, IHC with layer V marker, Ctip2 (red). c′, Quantification of Ctip2+ neurons (veh-treated: nWT = 21, nDel = 14; inh-treated: nWT = 9, nDel = 13; *p = 0.014, #p = 0.033). d, IHC with layer VI marker, Tbr1+ (green). d′, Quantification of Tbr1+ neurons (veh-treated: nWT = 8, nDel = 9; inh-treated: nWT = 12, nDel = 10; **p = 0.0076, #p = 0.0535). p Values are from Bonferroni post hoc analysis (*compares WT to deletion; #compares vehicle deletion to inhibitor deletion).
Analysis of Tbr1 cells by two-way ANOVA showed an interaction between genotype and inhibitor treatment, but post hoc analysis of vehicle-treated 16p11.2del animals showed a 25% increase in Tbr1+ cells compared with vehicle-treated WT animals (post hoc Bonferroni, p = 0.0075), which is rescued with treatment of 16p11.2del animals with ERK inhibitor (Fig. 3d), consistent with the effect of the inhibitor on these neurons observed at E14.5 (Fig. 2d). The number of Ctip2+ layer V neurons, known to project to subcortical targets including the thalamus, midbrain, pons, and spinal cord, is increased during mid-neurogenesis, but decreased postnatally in vehicle 16p11.2del mice compared with WT vehicle-treated mice (post hoc Bonferroni, p = 0.014) and normalized by inhibitor treatment of 16p11.2del animals, which have more Ctip2+ neurons compared with vehicle 16p11.2del animals (p = 0.033) and no difference compared with WT vehicle-treated animals (Fig. 2c, 3c). It is unknown why these projection neurons are ultimately lost in the 16p11.2del mice. These data demonstrate the developmental rescue of cortical deficits in progenitor proliferation and neurogenesis in the 16p11.2del after pharmacological intervention with the ERK pathway inhibitor.
ERK inhibitor treatment normalizes levels of the cell-cycle regulators p27 Kip1 and cyclin D1
To examine whether the aberrant generation of cortical neurons is due to deficits in cell-cycle dynamics, we examined two critical cell-cycle regulators: p27 Kip1 and cyclin D1 (Fig. 4), which are directly regulated by ERK signaling and play an important role in progenitor proliferation (Calegari and Huttner, 2003; Dehay and Kennedy, 2007; Lange et al., 2009; Pucilowska et al., 2012). We performed IHC and Western blot analysis and observed a significant decrease in p27Kip1 protein levels in the 16p11.2del cortex, consistent with our previous observations. Two-way ANOVA analysis of p27kip1 showed no genotype or inhibitor effect, but post hoc analysis showed a 31% decrease (p < 0.01) in p27kip1 levels in the 16p11.2del vehicle-treated mice compared with the WT vehicle-treated animals, which normalized to 92% of the vehicle WT level after inhibitor treatment (Fig. 4b′,b″). When cyclin D1 levels were analyzed by two-way ANOVA, there was only a inhibitor effect (F(1,46), p < 0.0001), but no genotype effect, with significant decrease in cyclin D1 in the inhibitor-treated 16p11.2del mice (p < 0.001) compared with the vehicle-treated 16p11.2del mice (Fig. 4a–a″). These data show that the 16p11.2del mice exhibit ERK-dependent changes in cell-cycle dynamics of neural progenitor cells that are ameliorated with the prenatal ERK inhibitor treatment.
Figure 4.
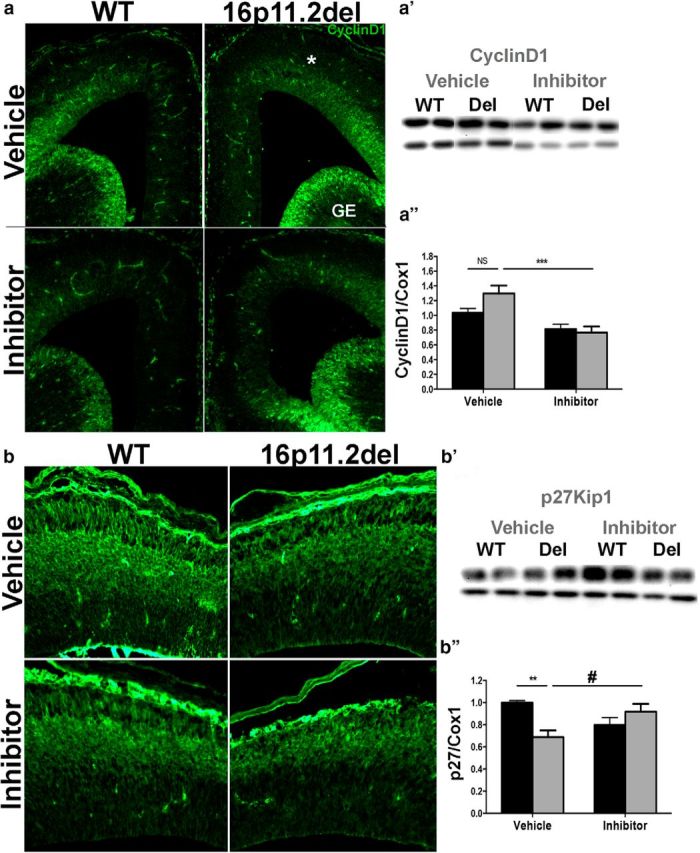
Treatment with ERK pathway inhibitor normalizes the levels of the downstream ERK effectors: p27 Kip1 and cyclin D1. IHC of coronal sections and Western analyses from WT and 16p11.2del mice at E14.5 a, Immunostaining against CyclingD1 antibody (green); a′, Western blot analysis, quantified in a″ (veh-treated: nWT = 16, nDel = 7; inh-treated: nWT = 16, nDel = 13; ***p < 0.001). b, Immunostaining against p27Kip1(green); b′, Western blot analysis, quantified in b″ (veh-treated: nWT = 8, nDel = 10; inh-treated: nWT = 8, nDel = 10; **p < 0.01, *p < 0.05). p Values are from Bonferroni post hoc analysis.
ERK inhibitor treatment partially rescues hippocampal and septal morphoanatomical abnormalities in 16p11.2del mice
To determine whether ERK inhibitor treatment would affect macroscale brain morphoanatomy in 16p11.2del mice, we applied voxelwise TBM and automated anatomical labeling to high-resolution MRI brain scans (Pagani et al., 2016). Consistent with previous reports (Horev et al., 2011), 16p11.del mice showed increases in the relative volume of the hypothalamus, superior colliculus and periaqueductal gray compared with control (WT) mice (Fig. 5a). Voxelwise TBM mapping also revealed foci of decreased volume in ventral hippocampal, amygdalar, entorhinal, and lateral septal areas in 16p11.2del mice compared with WT controls (Fig. 5a). Importantly, treatment with the ERK inhibitor partially-rescued ventral hippocampal and lateral septal volume in 16p11.2del mice compared with vehicle-treated controls (Fig. 5b,c). Interestingly, the ventral hippocampus is a region where pERK is highly expressed during mid-neurogenesis and a key substrate for anxiety-related behavior (Kjelstrup et al., 2002; Maren and Holt, 2004), which these mice exhibit.
Figure 5.

Prenatal ERK inhibitor treatment partially rescues ventral hippocampal and lateral septal volume in 16p11.2del mice. Mice were treated for 5 consecutive days starting at E10.5 and evaluated at P90. A, TBM analyses revealed significant increased volume of SC, PAG and Hypo in 16p11.2del mice compared with WT littermates (t test, p < 0.01 FWE cluster-corrected, with cluster defining threshold of |t| > 2.3). In 16p11.2 deletion mice, we also observed an increased volume of the RS ctx, as well as reduced volume of vHPC, LS, Ent ctx, and Amy (t test, p < 0.01 FWE cluster-corrected, with cluster defining threshold of |t| > 2.3). b, Comparison between treated and nontreated 16p11.2del mice shows that ERK-inhibitor produces bilateral foci of increased volume in the vHPC and LS (t test, p < 0.01 FWE cluster-corrected, with cluster defining threshold of |t| > 2.3). c, A composite illustration of a and b revealed that foci of increased gray matter volume (red/yellow; from b) are spatially located in the same hippocampal and septal regions exhibiting reduced gray matter volume in 16p11.2 del mice (blue/light blue; from a), suggesting a partial anatomical rescue of volumetric deficits upon treatment with ERK inhibitor. d, e, Consistent with TBM results, anatomical labeling revealed reduced relative volume in vHPC (t test: t(17) = 3.78, p = 0.001) and LS (t test: t(17) = 2.21, p = 0.041) in 16p11.2del mice compared with WT littermates (one-way ANOVA of vHPC: F(3,34) = 8.083, p < 0.001; one-way ANOVA of LS: F(3,34) = 1.692, p = 0.1872). Treatment with ERK inhibitor in 16p11.2del mice partially restored morphoanatomical volume in these brain regions (vHPC, t test: t(16) = 2.79, p = 0.013), although the effects in LS did not reach full statistical significance (t test, t(16) = 1.78, p = 0.078). Amy, Amygdala; Ent ctx, entorhinal cortex; Hypo, hypothalamus; LS, lateral septum; PAG, periaqueductal gray; RS, retrosplenial cortex; SC, superior colliculus; vHPC, ventral hippocampus. *p < 0.05,**p < 0.01.
Rescue of behavioral deficits in 16p11.2del mice after prenatal ERK inhibitor treatment
We, as well as others, have previously shown that the 16p11.2del mice are smaller than their WT littermates (Horev et al., 2011; Portmann et al., 2014; Pucilowska et al., 2015; Tidyman and Rauen, 2016). We report that prenatal treatment with ERK pathway inhibitor restored normal body weight in the 16p11.2del mice when examined at 3 months of age (WT veh-treated: 35.015 g; 16p11.2del veh-treated: 25.352 g; 16p11.2del inh-treated: 29.396 g).
The 16p11.2 del mice are reported to exhibit many behavioral deficits (Horev et al., 2011; Portmann et al., 2014; Pucilowska et al., 2015; Yang et al., 2015). We tested the 16p11.2del and WT control mice in a number of standard behavioral paradigms that are altered by the 16p11.2 deletion. Specifically, we assayed open field to evaluate hyperactivity and anxiety-like behaviors, novel object and fear conditioning to examine hippocampal-dependent memory, elevated-plus maze to test anxiety as well as maternal behavior and olfaction. We evaluated three large cohorts (n = 30/genotype) of 3-month-old male mice (except for maternal behavior and olfaction). Data were analyzed by two-way ANOVA, but unless otherwise indicated, significant interaction occurred between the genotype and inhibitor treatment, which resulted in reliance on post hoc analysis to show genotype or inhibitor effects.
We first conducted the EMP test, where examining entries into the closed arm, the post hoc analysis showed fewer closed arm entries by vehicle 16p11.2del mice compared with vehicle WT mice (p < 0.05), which was rescued in the inhibitor-treated deletion animals who showed more closed arm entries compared with vehicle-treated 16p11.2del animals (p < 0.05) and no difference with vehicle-treated WT animals. Furthermore, we observed statistical significance in time spent in the open arm (p < 0.05), which was only partially rescued by inhibitor treatment with no genotype or inhibitor effect by two-way ANOVA. We also noted an increase in open arm immobility, indicating freezing behavior (Fig. 6a). In the open-field test (Fig. 6b), we first examined level of activity, which showed that the 16p11.2del mice did not explore the field as much as the WT mice. Two-way ANOVA analysis of total distance traveled showed significant genotype (F(1,76), p = 0.005) and drug effect (F(1,76), p = 0.02) with no interaction, although post hoc tests did not show significance between vehicle WT and 16p11.2del animals or vehicle and inhibitor-treated 16p11.2del animals. However, in the open-field test (Fig. 6b), we observed that vehicle-treated 16p11.2del mice spent more time in the center compared with vehicle-treated WT mice (post hoc Bonferroni, p < 0.01), which was rescued by ERK inhibitor treatment, as inhibitor-treated 16p11.2del mice were similar to vehicle-treated WT mice and spent less time in the center compared with the vehicle-treated 16p11.2del mice (post hoc Bonferroni, p < 0.05). Interestingly, total distance traveled was also increased in the vehicle-treated 16p11.2del mice compared with vehicle-treated WT mice (post hoc Bonferroni, p < 0.05), which again normalized with inhibitor treatment of 16p11.2del mice, who showed significant decreased in distance traveled compared with vehicle-treated 16p11.2del n animals (p < 0.05) and no difference with vehicle-treated WT animals.
Figure 6.

Reversal of Behavioral Impairment of 16p11.2del mice after prenatal ERK pathway inhibitor treatment. WT or 16p11.2del 3-month-old male or female mice treated with Veh or Inh at E10.5 for 5 d. a, Elevated plus maze shows a no change in percentage of time in open arm, but decreased closed arm time in 16p11.2del mice that is rescued by inhibitor treatment (*p < 0.05, #p < 0.05; veh-treated: nWT = 25, nDel = 22; inh-treated: nWT = 27, nDel = 28). b, Open Field shows increased time spent in center in 16p11.2del mice that is rescued with inhibitor treatment (**p < 0.01, #p < 0.05; veh-treated: nWT = 23, nDel = 20; inh-treated: nWT = 19, nDel = 18). c, Fear conditioning shows increased freezing in 16p11.2del animals that improves with inhibitor treatment (**p < 0.01; veh-treated: nWT = 5, nDel = 6; inh-treated: nWT = 11, nDel = 11), conditioned stimulus: 85 dB sound at 2800 Hz for 30 s; US: 0.56 mA. Retention test performed 18 h later for 5 min in the absence of tone. d, NOR was evaluated in WT and 16p11.2del animals (#p < 0.05; veh-treated: nWT = 16, nDel = 12; inh-treated: nWT = 8, nDel = 16). e, Naive females were exposed to 3 WT pups placed in three corners of the cage, time to retrieve pups was recorded (first pup: **p < 0.01; second pup: **p < 0.01; third pup: ***p = 0.001; veh-treated: nWT = 21, nDel = 17; inh-treated: nWT = 6, nDel = 6). f, Mice were food deprived for 24 h, then placed in a cage containing one food pellet (Teddy Graham) buried under normal cage bedding; time to retrieve was recorded (**p < 0.01, ####p < 0.0001; veh-treated: nWT = 22, nDel = 22; inh-treated: nWT = 22, nDel = 7). p Values are from Bonferroni post hoc analysis (*compares WT to deletion; #compares vehicle deletion to inhibitor deletion).
Contextual fear conditioning showed significantly higher freezing percentage in the vehicle-treated 16p11.2del mice compared with the vehicle-treated WT cohort (post hoc Bonferroni, p < 0.01) indicating impaired contextual memory (Fig. 6c). This is improved by treatment of the 16p11.2del animals with the ERK inhibitor, which show no difference compared with vehicle-treated WT animals, but no significant difference was observed between vehicle and inhibitor-treated 16p11.2del animals.
Novel object recognition (NOR) did not reveal a significant genotype or inhibitor effect in post hoc analysis, but a trend showing impaired NOR in vehicle-treated 16p11.2del compared with vehicle-treated WT mice (p = 0.16), which improved with inhibitor treatment of 16p11.2del mice, who showed a trend toward improvement in NOR compared with vehicle-treated 16p11.2del mice (p = 0.11; Fig. 6d).
Both male and female 16p11.2del mice exhibited a significantly higher acuity in their sense of smell compared with WT mice (post hoc Bonferroni, p < 0.01), which was attenuated after the treatment (Fig. 6f). Inhibitor-treated 16p11.2del mice showed significant attenuation in olfaction compared with vehicle-treated 16p11.2del mice (p < 0.0001).
Additionally, females showed heightened maternal response, as measured in a pup retrieval assay that was reduced following ERK inhibitor treatment (Fig. 6e). Vehicle-treated 16p11.2del animals showed decreased latencies to retrieving all pups compared with vehicle-treated WT animals (pup1: p < 0.01, pup2: p < 0.01, pup3: p < 0.001). This effect was reverted with treatment of 16p11.2del animals with inhibitor as no difference in latencies was observed compared with WT vehicle-treated animals (post hoc Bonferroni, p < 0.01).
Altogether, the above evidence supports the notion that an early pharmacological intervention targeting ERK signaling in 16p11.2del mice is sufficient to reverse some of the behavioral alterations found in this model of ASD.
Postnatal treatment with ERK inhibitor partially restores behavioral deficits of adult 16p11.2del mice
We next questioned whether any of the behavioral deficits could be improved by ERK inhibitor treatment in adult mice. Therefore, we treated 3-month-old WT and 16p11.2del male mice with ERK inhibitor for 5 consecutive days at P90.
In the elevated plus maze, we observed that vehicle-treated 16p11.2del animals spent more time in the open arm compare to the vehicle WT cohort (Fig. 7a; post hoc Bonferroni, p < 0.05). This effect was abrogated in ERK inhibitor-treated 16p11.2del animals as no difference in open arm time was observed compared with vehicle-treated WT animals (two-way ANOVA showed significant interaction between genotype and inhibitor treatment). In the open-field test, we did not observe significant genotype effects between vehicle-treated 16p11.2del and WT animals in post hoc analysis, however, we did observe a trend toward more open entries in vehicle 16p11.2 del animals compared with WT (Fig. 7b). In addition, treatment of 16p11.2del animals with the ERK inhibitor showed significant reduction in entries into the center of the field compared with vehicle-treated 16p11.2del animals (post hoc Bonferroni, p < 0.01). Although the adult treated mice did not achieve statistical significance in the olfactory test, there was a trend toward heightened acuity in the 16p11.2del mice, which was at least partially recovered after the inhibitor treatment (Fig. 7d). Together our data suggest that postnatal treatment with the ERK inhibitor may result in partial rescue of some of the behavioral deficits seen in the 16p11.2del mice.
Figure 7.

Postnatal treatment with ERK pathway inhibitor partially restores behavioral deficits in adult 16p11.2del mice. WT or 16p11.2del 3-month-old male or female mice treated with veh or inh at P90 for 5 d. a, Elevated plus maze analysis of percentage of time in open arm (*p < 0.05), entries in open arm, immobility time (##p < 0.01), and total distance traveled (veh-treated: nWT = 15, nDel = 11; inh-treated: nWT = 12, nDel = 7). b, Open-field analysis of entries into center (##p < 0.01), time spent in center and total distance traveled (veh-treated: nWT = 22, nDel = 17; inh-treated: nWT = 23, nDel = 9). c, ELISA performed on whole brain lystaed of P90 mice (**p < 0.05; #p < 0.05; veh-treated: nWT = 10, nDel = 8; inh-treated: nWT = 7, nDel = 5). D, Mice were food deprived for 24 h, and then placed in a cage containing one food pellet (Teddy Graham) buried under normal cage bedding; time to retrieve (latency) was analyzed. p Values are from Bonferroni post hoc analysis (*compares WT to deletion; #compares vehicle deletion to inhibitor deletion).
Because other models linked to ASDs and the ERK pathway show aberrant levels of oxytocin (Hollander et al., 2007), we examined 3-month-old male mice and show that levels of oxytocin are elevated in the 16p11.2del male mice and importantly can be restored to normal after inhibitor treatment (post hoc Bonferroni, p < 0.01; Fig. 7c). We observed by two-way ANOVA a significant genotype (F(1,24) = 6.132, p = 0.0207) and drug effect (F(1,24) = 8.712, p = 0.0070) and significant interaction F(1,24) = 4.884, p = 0.0369.
The 16p11.2del mice do not exhibit typical social deficits that are common among other models of ASDs as well as 16p11.2del patients, thus, the enhanced oxytocin levels, which normally augment social interactions, could be masking the social impairment in these mice. Further studies using oxytocin inhibitors could clarify this phenomenon.
Discussion
ERK/MAPK pathway as a potential target for ASD therapy
The ERK/MAPK pathway has been extensively studied and found to play pivotal roles in neural development as well as in learning, memory, synaptic plasticity, and spine dynamics (Thomas and Huganir, 2004). The functional importance of signaling through this pathway is also reflected in the effects of activating mutations that alter the activity of the ERK1/2 kinases and result in a constellation of syndromic and nonsyndromic neurodevelopmental disorders including the RASopathies and the associated intellectual disability (Tidyman and Rauen, 2016; Borrie et al., 2017).
It has recently been appreciated that some forms of ASD are also associated with perturbations of several intracellular signaling cascades, including the Ras-ERK and the mTORC1 pathways (Kalkman, 2012; Adviento et al., 2014; Borrie et al., 2017). In addition to genetic linkages, pathway network analyses point to a convergence of a wide range of abnormalities associated with autism onto a few salient pathways, prominent among these is the ERK/MAPK pathway (Wen et al., 2016; Mitra et al., 2017). Importantly, many ASD mouse models with genetic alterations of Mecp2, FMR1, NF1, Syngap as well as, BTBR and 16p11.2del exhibit aberrant ERK signaling (Liang et al., 2010; Osterweil et al., 2010, 2013; Hamdan et al., 2011; Bhakar et al., 2012; Golzio et al., 2012; Y. Wang et al., 2012; Kelleher et al., 2012; C. C. Wang et al., 2013; Faridar et al., 2014; Pucilowska et al., 2015). These data argue that the abnormal ERK activity in the brain is central to pathology of many ASDs.
We and others have previously shown that the genetic deletion of the 16p11.2 interval results in a paradoxical increase in ERK activity that is associated with aberrant neural progenitor proliferation, which leads to dysregulation in the number of neurons generated within the cortex (Pucilowska et al., 2015) and subcortical structures (Portmann et al., 2014; Grissom et al., 2018). This results in altered volumes of the cortical lamina and subcortical nuclei.
These studies raise the possibility that ERK inhibitors can be used to normalize their activity and have therapeutic efficacy in 16p11.2 deletion carriers. We reasoned that normalization of ERK activity might rescue the CNS phenotypes observed in the 16p11.2del mice. It is noteworthy that Papale et al. (2017), using the newly developed ERK pathway inhibitor peptides, have recently shown that the treatment ameliorates defective synaptogenesis in a genetic model of RASopathy. Moreover, they have shown its effectiveness of ERK pathway inhibition by treatment of murine models of cocaine addiction, suppressing the behavioral phenotypes (Papale et al., 2016).
ERK inhibitor suppresses the increase in ERK activity in 16p11.2del mice
The 16p11.2del mice exhibit paradoxical increase in ERK activity in both the developing and mature brain. This finding was unexpected given that the deletion removes the Mapk3 gene encoding ERK1. The biological basis of dysregulation of the ERK pathway in this model is unknown. However, we and others have previously postulated that ERK1 acts a negative regulator of ERK2 (Mazzucchelli et al., 2002; Vantaggiato et al., 2006; Fasano and Brambilla, 2011; Trabalzini and Retta, 2014) and our findings are consistent with this hypothesis but basis of the elevated ERK1 activity remains unclear. We found that basal ERK activity in WT mice was not altered by drug treatment and this likely represents the intrinsic basal activity of the ERKs that is independent of upstream regulators, and thus not subject to further inhibition by the RB1 peptide.
The 16p11.2 deletion contains 27 genes. Mice lacking ERK1or ERK2 do not recapitulate the entire range of defects observed in the 16p11.2del mice, implicating the contribution of other genes within this interval to the ASD phenotypes. Importantly, at least two other genes (MVP and KCTD13) within the deleted region converge onto the MAPK pathway and affect cell proliferation, mGluR5 signaling, and protein turnover (Liang et al., 2010; Golzio et al., 2012; Tian et al., 2015). This suggests that multiple genes within the deletion interval converge on the ERK/MAPK pathway resulting in an overall increase in ERK activity.
ERK inhibitor rescues cortical defects in 16p11.2del mice
We provide mechanism-based evidence showing that treatment with ERK inhibitors during the peak of neurogenesis rescues the structural and behavioral deficits observed in the 16p11.2del mice. Specifically, we found that the aberrant generation of cortical neurons was restored to normal levels because of normalization of neurogenesis following developmental drug treatment. This was secondary to the ERK-dependent modulation of potent cell-cycle regulators cyclin D1 and p27Kip1. The retention of normal numbers of neural progenitors during critical neurogenic period allowed for generation of appropriate numbers of cells populating cortical lamina as well as normal circuitry. Importantly, we corroborated our molecular and biochemical findings with MRI data showing partial reversal of volumetric changes to the ventral hippocampus and lateral septum. It is of interest to note that the MRI analyses showed that the ERK inhibitor treatment did not rescue all of the anatomical abnormalities in the brain, which may be related to a short treatment window implemented in this study and the temporal variation in neurogenesis between different brain regions.
ERK inhibitor rescues behavioral deficits in 16p11.2del mice
One of the most striking findings from this study is the reversal of a wide range of behavioral deficits that arise from the 16p11.2 deletion. Our data show the first developmental rescue of ASD associated phenotypes in the 16p11.2del mouse model. The data from the elevated plus maze and open field did show statistical significance for changes in anxiety-like behaviors but not as strong as in previous studies, which is possibly due to increased sample size, longer handling habituation period and greater statistical rigor. However, we do observe significant improvement in contextual fear memories in the inhibitor-treated 16p11.2del mice. It is conceivable that the observed ventral hippocampal morphological rescue could explain the inhibitor effect on anxiety-like behaviors and contextual memory, given the established roles of these brain regions in emotional and stress coping responses (Kjelstrup et al., 2002; Bannerman et al., 2003; Maren and Holt, 2004; Adhikari et al., 2011).
The strongest effects we found were in olfaction and maternal behaviors, where we observed heightened responses in 16p11.2del mice, which were attenuated with inhibitor treatment. The decrease in latencies to retrieve pups and locate hidden food in the deletion mice may represent enhanced sensory perception or processing, but may also reflect changes to neural circuitry that govern motivational behavior.
Remarkably, we also report a partial rescue of the behavioral deficits by inhibitor treatment of adult mice. This effect is likely due to the fact that ERK dysregulation persists into adulthood in the 16p11.2del animals (Pucilowska et al., 2015) and potentially disrupts the normal role of ERK signaling in synaptic plasticity. At the synapse ERKs promote regulation of translational mechanisms promoting protein synthesis and thus cognition (Thomas and Huganir, 2004). These synaptic changes ultimately lead to refinement of neuronal connectivity. Furthermore, many mutations associated with ASDs converge on the ERK pathway impairing cognitive functioning and adaptive behavioral plasticity (Komiyama et al., 2002; Levitt and Campbell, 2009; Ebert and Greenberg, 2013). Correcting hyperactive ERK signaling in the adult 16p11.2del mice may thus lead to ameliorating of behavioral phenotypes that are mediated by the postnatal functions of ERK signaling. Importantly, these data suggest that dysregulation of ERK activity can be rescued during brain development through restoration of normal cortical proliferation dynamics, and in adult mice, presumably due to the synaptic actions of the ERKs previously described in this mouse model (Tian et al., 2015; Lu et al., 2018).
We show that normalization of ERK signaling in the 16p11.2del mice can reverse the molecular and behavioral phenotypes in the 16p11.2del mice. Although we observe significant effects on neuronal population sizes, our analysis was restricted to cortical neurons and, given that this is a germline hemideletion model, other areas of the nervous system are undoubtedly affected. This is further supported by our imaging analysis that shows multiple brain regions that are affected by the deletion. Although we observe some behavioral improvements from postnatal treatment with the ERK/MAPK inhibitor, further analysis of changes to neuronal morphology and synaptic physiology in the 16p11.2del mice is warranted to help characterize the postnatal effects of the 16p11.2 deletion. Finally, we have only examined a few behavioral phenotypes and gender differences in behaviors were not examined.
In conclusion, our findings lead to three important conclusions. Our data strongly supports the hypothesis that ASDs are neurodevelopmental disorders affecting global brain circuitry. Second, our work significantly strengthens the emerging hypothesis that multiple disorders of impaired cognition and ASDs converge onto a few fundamental pathways, such as the ERK/MAPK pathway, and affect a critical period of brain development. Finally, the cognitive and neuropsychiatric symptoms of 16p11.2 microdeletion disorder are amenable to targeted drug therapy, both during development and in the adult.
Footnotes
This work was supported by Grants from the Simons Foundation (SFARI 275316 to G.E.L.; SFARI 314688 and 400101 to A.G.), the Brain and Behavior Foundation (NARSAD to A.G.), and the Waterloo Foundation (I.M.).
The authors declare no competing financial interests.
References
- Adhikari A, Topiwala MA, Gordon JA (2011) Single units in the medial prefrontal cortex with anxiety-related firing patterns are preferentially influenced by ventral hippocampal activity. Neuron 71:898–910. 10.1016/j.neuron.2011.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adviento B, Corbin IL, Widjaja F, Desachy G, Enrique N, Rosser T, Risi S, Marco EJ, Hendren RL, Bearden CE, Rauen KA, Weiss LA (2014) Autism traits in the RASopathies. J Med Genet 51:10–20. 10.1136/jmedgenet-2013-101951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avants BB, Tustison N, Song G (2009) Advanced normalization tools (ANTS). Insight J 2:1–35. [Google Scholar]
- Bannerman DM, Grubb M, Deacon RM, Yee BK, Feldon J, Rawlins JN (2003) Ventral hippocampal lesions affect anxiety but not spatial learning. Behav Brain Res 139:197–213. 10.1016/S0166-4328(02)00268-1 [DOI] [PubMed] [Google Scholar]
- Bhakar AL, Dölen G, Bear MF (2012) The pathophysiology of fragile X (and what it teaches us about synapses). Annu Rev Neurosci 35:417–443. 10.1146/annurev-neuro-060909-153138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrie SC, Brems H, Legius E, Bagni C (2017) Cognitive dysfunctions in intellectual disabilities: the contributions of the ras-MAPK and PI3K-AKT-mTOR pathways. Annu Rev Genomics Hum Genet 18:115–142. 10.1146/annurev-genom-091416-035332 [DOI] [PubMed] [Google Scholar]
- Calegari F, Huttner WB (2003) An inhibition of cyclin-dependent kinases that lengthens, but does not arrest, neuroepithelial cell cycle induces premature neurogenesis. J Cell Sci 116:4947–4955. 10.1242/jcs.00825 [DOI] [PubMed] [Google Scholar]
- Cutuli D, Pagani M, Caporali P, Galbusera A, Laricchiuta D, Foti F, Neri C, Spalletta G, Caltagirone C, Petrosini L, Gozzi A (2016) Effects of omega-3 fatty acid supplementation on cognitive functions and neural substrates: a voxel-based morphometry study in aged mice. Front Aging Neurosci 8:38–52. 10.3389/fnagi.2016.00038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay C, Kennedy H (2007) Cell-cycle control and cortical development. Nat Rev Neurosci 8:438–450. 10.1038/nrn2097 [DOI] [PubMed] [Google Scholar]
- Dorr AE, Lerch JP, Spring S, Kabani N, Henkelman RM (2008) High resolution three-dimensional brain atlas using an average magnetic resonance image of 40 adult C57BL/6J mice. Neuroimage 42:60–69. 10.1016/j.neuroimage.2008.03.037 [DOI] [PubMed] [Google Scholar]
- Ebert DH, Greenberg ME (2013) Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493:327–337. 10.1038/nature11860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faridar A, Jones-Davis D, Rider E, Li J, Gobius I, Morcom L, Richards LJ, Sen S, Sherr EH (2014) Mapk/Erk activation in an animal model of social deficits shows a possible link to autism. Mol Autism 5:57. 10.1186/2040-2392-5-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasano S, Brambilla R (2011) Ras-ERK signaling in behavior: old questions and new perspectives. Front Behav Neurosci 5:79. 10.3389/fnbeh.2011.00079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman TS, Sondermann H, Friedland GD, Kortemme T, Bar-Sagi D, Marqusee S, Kuriyan J (2006) A ras-induced conformational switch in the ras activator son of sevenless. Proc Natl Acad Sci U S A 103:16692–16697. 10.1073/pnas.0608127103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghebranious N, Giampietro PF, Wesbrook FP, Rezkalla SH (2007) A novel microdeletion at 16p11.2 harbors candidate genes for aortic valve development, seizure disorder, and mild mental retardation. Am J Med Genet A 143A:1462–1471. 10.1002/ajmg.a.31837 [DOI] [PubMed] [Google Scholar]
- Glickstein SB, Monaghan JA, Koeller HB, Jones TK, Ross ME (2009) Cyclin D2 is critical for intermediate progenitor cell proliferation in the embryonic cortex. J Neurosci 29:9614–9624. 10.1523/JNEUROSCI.2284-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, Reymond A, Sun M, Sawa A, Gusella JF, Kamiya A, Beckmann JS, Katsanis N (2012) KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 485:363–367. 10.1038/nature11091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissom NM, McKee SE, Schoch H, Bowman N, Havekes R, O'Brien WT, Mahrt E, Siegel S, Commons K, Portfors C, Nickl-Jockschat T, Reyes TM, Abel T (2018) Male-specific deficits in natural reward learning in a mouse model of neurodevelopmental disorders. Mol Psychiatry 23:544–555. 10.1038/mp.2017.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Daoud H, Piton A, Gauthier J, Dobrzeniecka S, Krebs MO, Joober R, Lacaille JC, Nadeau A, Milunsky JM, Wang Z, Carmant L, Mottron L, Beauchamp MH, Rouleau GA, Michaud JL (2011) De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol Psychiatry 69:898–901. 10.1016/j.biopsych.2010.11.015 [DOI] [PubMed] [Google Scholar]
- Hanson E, Bernier R, Porche K, Jackson FI, Goin-Kochel RP, Snyder LG, Snow AV, Wallace AS, Campe KL, Zhang Y, Chen Q, D'Angelo D, Moreno-De-Luca A, Orr PT, Boomer KB, Evans DW, Kanne S, Berry L, Miller FK, Olson J, et al. (2015) The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol Psychiatry 77:785–793. 10.1016/j.biopsych.2014.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander E, Bartz J, Chaplin W, Phillips A, Sumner J, Soorya L, Anagnostou E, Wasserman S (2007) Oxytocin increases retention of social cognition in autism. Biol Psychiatry 61:498–503. 10.1016/j.biopsych.2006.05.030 [DOI] [PubMed] [Google Scholar]
- Horev G, Ellegood J, Lerch JP, Son YE, Muthuswamy L, Vogel H, Krieger AM, Buja A, Henkelman RM, Wigler M, Mills AA (2011) Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc Natl Acad Sci U S A 108:17076–17081. 10.1073/pnas.1114042108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkman HO. (2012) Potential opposite roles of the extracellular signal-regulated kinase (ERK) pathway in autism spectrum and bipolar disorders. Neurosci Biobehav Rev 36:2206–2213. 10.1016/j.neubiorev.2012.07.008 [DOI] [PubMed] [Google Scholar]
- Kelleher RJ 3rd, Geigenmüller U, Hovhannisyan H, Trautman E, Pinard R, Rathmell B, Carpenter R, Margulies D (2012) High-throughput sequencing of mGluR signaling pathway genes reveals enrichment of rare variants in autism Esteban FJ, ed. PLoS One 7:e35003. 10.1371/journal.pone.0035003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjelstrup KG, Tuvnes FA, Steffenach HA, Murison R, Moser EI, Moser MB (2002) Reduced fear expression after lesions of the ventral hippocampus. Proc Natl Acad Sci U S A 99:10825–10830. 10.1073/pnas.152112399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama NH, Watabe AM, Carlisle HJ, Porter K, Charlesworth P, Monti J, Strathdee DJ, O'Carroll CM, Martin SJ, Morris RG, O'Dell TJ, Grant SG (2002) SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J Neurosci 22:9721–9732. 10.1523/JNEUROSCI.22-22-09721.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH Jr, Dobyns WB, Christian SL (2008) Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet 17:628–638. 10.1093/hmg/ddm376 [DOI] [PubMed] [Google Scholar]
- Lange C, Huttner WB, Calegari F (2009) Cdk4/CyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 5:320–331. 10.1016/j.stem.2009.05.026 [DOI] [PubMed] [Google Scholar]
- Levitt P, Campbell DB (2009) The genetic and neurobiologic compass points toward common signaling dysfunctions in autism spectrum disorders. J Clin Invest 119:747–754. 10.1172/JCI37934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Ronemus M, Yamrom B, Lee YH, Leotta A, Kendall J, Marks S, Lakshmi B, Pai D, Ye K, Buja A, Krieger A, Yoon S, Troge J, Rodgers L, Iossifov I, Wigler M (2011) Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70:886–897. 10.1016/j.neuron.2011.05.015 [DOI] [PubMed] [Google Scholar]
- Liang P, Wan Y, Yan Y, Wang Y, Luo N, Deng Y, Fan X, Zhou J, Li Y, Wang Z, Yuan W, Tang M, Mo X, Wu X (2010) MVP interacts with YPEL4 and inhibits YPEL4-mediated activities of the ERK signal pathway. Biochem Cell Biol 88:445–450. 10.1139/O09-166 [DOI] [PubMed] [Google Scholar]
- Liu S, Sun JP, Zhou B, Zhang ZY (2006) Structural basis of docking interactions between ERK2 and MAP kinase phosphatase 3. Proc Natl Acad Sci U S A 103:5326–5331. 10.1073/pnas.0510506103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HC, Mills AA, Tian D (2018) Altered synaptic transmission and maturation of hippocampal CA1 neurons in a mouse model of human chr16p11.2 microdeletion. J Neurophysiol 119:1005–1018. 10.1152/jn.00306.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Holt WG (2004) Hippocampus and Pavlovian fear conditioning in rats: muscimol infusions into the ventral, but not dorsal, hippocampus impair the acquisition of conditional freezing to an auditory conditional stimulus. Behav Neurosci 118:97–110. 10.1037/0735-7044.118.1.97 [DOI] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, Thiruvahindrapduram B, Fiebig A, Schreiber S, Friedman J, Ketelaars CE, Vos YJ, Ficicioglu C, Kirkpatrick S, Nicolson R, Sloman L, et al. (2008) Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 82:477–488. 10.1016/j.ajhg.2007.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pagès G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouysségur J, Brambilla R, et al. (2002) Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34:807–820. 10.1016/S0896-6273(02)00716-X [DOI] [PubMed] [Google Scholar]
- Mitra I, Lavillaureix A, Yeh E, Traglia M, Tsang K, Bearden CE, Rauen KA, Weiss LA (2017) Reverse pathway genetic approach identifies epistasis in autism spectrum disorders. PLOS Genet 13:e1006516. 10.1371/journal.pgen.1006516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbern J, Zhong J, Wickramasinghe RS, Li X, Wu Y, Samuels I, Cherosky N, Karlo JC, O'Loughlin B, Wikenheiser J, Gargesha M, Doughman YQ, Charron J, Ginty DD, Watanabe M, Saitta SC, Snider WD, Landreth GE (2008) Mouse and human phenotypes indicate a critical conserved role for ERK2 signaling in neural crest development. Proc Natl Acad Sci U S A 105:17115–17120. 10.1073/pnas.0805239105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke NA, Sullivan DP, Kaznowski CE, Jacobs AA, McConnell SK (1995) Tangential migration of neurons in the developing cerebral cortex. Development 121:2165–2176. [DOI] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, Bear MF (2010) Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci 30:15616–15627. 10.1523/JNEUROSCI.3888-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil EK, Chuang SC, Chubykin AA, Sidorov M, Bianchi R, Wong RK, Bear MF (2013) Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 77:243–250. 10.1016/j.neuron.2012.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani M, Damiano M, Galbusera A, Tsaftaris SA, Gozzi A (2016) Semi-automated registration-based anatomical labelling, voxel based morphometry and cortical thickness mapping of the mouse brain. J Neurosci Methods 267:62–73. 10.1016/j.jneumeth.2016.04.007 [DOI] [PubMed] [Google Scholar]
- Papale A, Morella IM, Indrigo MT, Bernardi RE, Marrone L, Marchisella F, Brancale A, Spanagel R, Brambilla R, Fasano S (2016) Impairment of cocaine-mediated behaviours in mice by clinically relevant Ras-ERK inhibitors. eLife 5:e17111. 10.7554/eLife.17111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papale A, d'Isa R, Menna E, Cerovic M, Solari N, Hardingham N, Cambiaghi M, Cursi M, Barbacid M, Leocani L, Fasano S, Matteoli M, Brambilla R (2017) Severe intellectual disability and enhanced gamma-aminobutyric acidergic synaptogenesis in a novel model of rare RASopathies. Biol Psychiatry 81:179–192. 10.1016/j.biopsych.2016.06.016 [DOI] [PubMed] [Google Scholar]
- Portmann T, Yang M, Mao R, Panagiotakos G, Ellegood J, Dolen G, Bader PL, Grueter BA, Goold C, Fisher E, Clifford K, Rengarajan P, Kalikhman D, Loureiro D, Saw NL, Zhengqui Z, Miller MA, Lerch JP, Henkelman M, Shamloo M, et al. (2014) Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell Rep 7:1077–1092. 10.1016/j.celrep.2014.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucilowska J, Puzerey PA, Karlo JC, Galán RF, Landreth GE (2012) Disrupted ERK signaling during cortical development leads to abnormal progenitor proliferation, neuronal and network excitability and behavior, modeling human neuro-cardio-facial-cutaneous and related syndromes. J Neurosci 32:8663–8677. 10.1523/JNEUROSCI.1107-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucilowska J, Vithayathil J, Tavares EJ, Kelly C, Karlo JC, Landreth GE (2015) The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J Neurosci 35:3190–3200. 10.1523/JNEUROSCI.4864-13.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R., Jr (2012) ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res 66:105–143. 10.1016/j.phrs.2012.04.005 [DOI] [PubMed] [Google Scholar]
- Sondermann H, Soisson SM, Boykevisch S, Yang SS, Bar-Sagi D, Kuriyan J (2004) Structural analysis of autoinhibition in the ras activator son of sevenless. Cell 119:393–405. 10.1016/j.cell.2004.10.005 [DOI] [PubMed] [Google Scholar]
- Sweatt JD. (2004) Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14:311–317. 10.1016/j.conb.2004.04.001 [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL (2004) MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 5:173–183. 10.1038/nrn1346 [DOI] [PubMed] [Google Scholar]
- Tian D, Stoppel LJ, Heynen AJ, Lindemann L, Jaeschke G, Mills AA, Bear MF (2015) Contribution of mGluR5 to pathophysiology in a mouse model of human chromosome 16p11.2 microdeletion. Nat Neurosci 18:182–184. 10.1038/nn.3911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidyman WE, Rauen KA (2009) The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 19:230–236. 10.1016/j.gde.2009.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidyman WE, Rauen KA (2016) Pathogenetics of the RASopathies. Hum Mol Genet 25:R123–R132. 10.1093/hmg/ddw191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabalzini L, Retta SF (2014) Ras signaling: methods and protocols. New York: Humana. [Google Scholar]
- Ullmann JF, Watson C, Janke AL, Kurniawan ND, Reutens DC (2013) A segmentation protocol and MRI atlas of the C57BL/6J mouse neocortex. Neuroimage 78:196–203. 10.1016/j.neuroimage.2013.04.008 [DOI] [PubMed] [Google Scholar]
- Vantaggiato C, Formentini I, Bondanza A, Bonini C, Naldini L, Brambilla R (2006) ERK1 and ERK2 mitogen-activated protein kinases affect ras-dependent cell signaling differentially. J Biol 5:14. 10.1186/jbiol38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CC, Held RG, Hall BJ (2013) SynGAP regulates protein synthesis and homeostatic synaptic plasticity in developing cortical networks. PLoS One 8:e83941. 10.1371/journal.pone.0083941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kim E, Wang X, Novitch BG, Yoshikawa K, Chang LS, Zhu Y (2012) ERK inhibition rescues defects in fate specification of Nf1-deficient neural progenitors and brain abnormalities. Cell 150:816–830. 10.1016/j.cell.2012.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Alshikho MJ, Herbert MR (2016) Pathway network analyses for autism reveal multisystem involvement, major overlaps with other diseases and convergence upon MAPK and calcium signaling. PLoS One 11:e0153329. 10.1371/journal.pone.0153329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worsley KJ, Evans AC, Marrett S, Neelin P (1992) A three-dimensional statistical analysis for CBF activation studies in human brain. J Cereb Blood Flow Metab 12:900–918. 10.1038/jcbfm.1992.127 [DOI] [PubMed] [Google Scholar]
- Yang M, Mahrt EJ, Lewis F, Foley G, Portmann T, Dolmetsch RE, Portfors CV, Crawley JN (2015) 16p11.2 deletion syndrome mice display sensory and ultrasonic vocalization deficits during social interactions. Autism Res 8:507–521. 10.1002/aur.1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey F, Sherr EH, Beckmann ND, Hanson E, Maillard AM, Hippolyte L, Macé A, Ferrari C, Kutalik Z, Andrieux J, Aylward E, Barker M, Bernier R, Bouquillon S, Conus P, Delobel B, Faucett WA, Goin-Kochel RP, Grant E, Harewood L, et al. (2012) A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J Med Genet 49:660–668. 10.1136/jmedgenet-2012-101203 [DOI] [PMC free article] [PubMed] [Google Scholar]