

Abstract
In addition to mechanisms promoting protein-synthesis-dependent long-term memory (PSD-LTM), the process appears to also be specifically constrained. We present evidence that the highly conserved receptor tyrosine kinase dAlk is a novel PSD-LTM attenuator in Drosophila. Reduction of dAlk levels in adult α/β mushroom body (MB) neurons during conditioning elevates LTM, whereas its overexpression impairs it. Unlike other memory suppressor proteins and miRNAs, dAlk within the MBs constrains PSD-LTM specifically but constrains learning outside the MBs as previously shown. Dendritic dAlk levels rise rapidly in MB neurons upon conditioning, a process apparently controlled by the 3′UTR of its mRNA, and interruption of the 3′UTR leads to enhanced LTM. Because its activating ligand Jeb is dispensable for LTM attenuation, we propose that postconditioning elevation of dAlk within α/β dendrites results in its autoactivation and constrains formation of the energy costly PSD-LTM, acting as a novel memory filter.
SIGNIFICANCE STATEMENT In addition to the widely studied molecular mechanisms promoting protein-synthesis-dependent long-term memory (PSD-LTM), recent discoveries indicate that the process is also specifically constrained. We describe a role in PSD-LTM constraint for the first receptor tyrosine kinase (RTK) involved in olfactory memory in Drosophila. Unlike other memory suppressor proteins and miRNAs, dAlk limits specifically PSD-LTM formation as it does not affect 3 h, or anesthesia-resistant memory. Significantly, we show conditioning-dependent dAlk elevation within the mushroom body dendrites and propose that its local abundance may activate its kinase activity, to mediate imposition of PSD-LTM constraints through yet unknown mechanisms.
Keywords: Alk, Drosophila, memory constraint, mushroom bodies, PSD-LTM
Introduction
Since its identification, the receptor tyrosine kinase (RTK) anaplastic lymphoma kinase (ALK) has emerged as a key player in the physiology and pathology of the CNS (Hallberg and Palmer, 2013). ALK belongs to the insulin receptor subfamily of monomeric RTKs. It features a large extracellular ligand-binding domain, a short transmembrane-spanning region, and an intracellular tyrosine kinase (TK) domain. Upon activation by its recently identified ALKAL ligands (Guan et al., 2015; Reshetnyak et al., 2015), ALK initiates the activation of several intracellular signaling pathways, including the canonical Ras/ERK cascade. Unsurprisingly, ALK has been linked to multiple human cancers (Hallberg and Palmer, 2013) characterized by constitutive kinase activation due to point mutations, chromosomal translocations, or overexpression by gene amplification, which confer ligand independence. However, the normal role of ALK in the CNS remains poorly documented.
ALK is highly conserved in vertebrates, Caenorhabditis elegans and Drosophila melanogaster (Lorén et al., 2001; Hallberg and Palmer, 2013). Drosophila Alk (dAlk) and its secreted activating ligand Jelly Belly (Jeb) are essential for gut muscle differentiation, axon targeting in the retina, organ and body size growth control, and synapse growth at the larval neuromuscular junction (Hallberg and Palmer, 2013). Recent studies indicate that dAlk activity regulates ethanol sensitivity (Lasek et al., 2011), sleep (Bai and Sehgal, 2015), and learning/short-term memory (STM) (Gouzi et al., 2011) in adult flies. Jeb or dAlk overexpression precipitates associative olfactory learning deficits, whereas genetic or pharmacological inhibition of dAlk kinase activity enhances learning performance (Gouzi et al., 2011). dAlk-dependent learning, spatial memory, and novel object recognition inhibition were subsequently confirmed in mice also (Weiss et al., 2012).
dAlk is widely expressed in the adult brain (Gouzi et al., 2011), accumulating preferentially in the dendrites of the mushroom bodies (MBs), neuroanatomical structures essential for olfactory learning and memory (de Belle and Heisenberg, 1994), sleep (Joiner et al., 2006; Pitman et al., 2006), and ethanol sensitivity (King et al., 2011). However, whereas dAlk-dependent regulation of sleep and ethanol sensitivity require its expression within the MBs (Lasek et al., 2011; Bai and Sehgal, 2015), regulation of learning does not (Gouzi et al., 2011). However, dAlk is present within MBs; and because these neurons are also essential for intermediate/middle-term memory (MTM), anesthesia-resistant memory (ARM), and protein-synthesis-dependent long-term memory (PSD-LTM) (Tully et al., 1994; Guven-Ozkan and Davis, 2014), we hypothesized that dAlk could serve one or more of these forms of labile and consolidated olfactory memories.
Materials and Methods
Drosophila culture and strains.
Drosophila were cultured on standard wheat-flour-sugar food supplemented with soy flour and CaCl2, at 25°C in 50%–70% relative humidity in a 12 h light/dark cycle (Gouzi et al., 2011). Alk1 mutants (Lorén et al., 2003) were a kind gift from R. H. Palmer (University of Gothenburg, Gothenburg, Sweden). AlkMB06458 flies were obtained from the Bloomington Stock Center (BL25509). Transgenic fly strains used in this work were as follows: UAS-AlkWT (Lorén et al., 2001), UAS-AlkRNAi (v11446 and v107083, Vienna Drosophila Resource Centre), UAS-Jeb (Englund et al., 2003), UAS-JebRNAi (v30799 and v103047, Vienna Drosophila Resource Centre), UAS-mCD8::GFP (Lee and Luo, 1999), and TubGal80ts (McGuire et al., 2004). The Gal4 driver lines used in this work were as follows: Alk(38)-Gal4 (Gouzi et al., 2011), repo-Gal4 (BL715), nSyb-Gal4, Ras2-Gal4, Elav[C155]-Gal4 (BL458), TH-Gal4, MB247-Gal4, c739-Gal4, OK107-Gal4 (BL854), LeoMB-Gal4 (Messaritou et al., 2009), 17d-Gal4 (BL51631), NP1131-Gal4, and 1471-Gal4 (BL9465; Isabel et al., 2004). The c739-Gal4,TubGal80ts line was obtained from G. Roman (University of Mississippi, Oxford, MS). The GH146-Gal4,TubGal80ts line was from M. Ramaswami (Trinity College Dublin, Dublin, Ireland). The MB-specific Gal80 (MBGal80), which drives expression predominantly in the MBs, was introduced into the Elav-Gal4 strain through standard genetic crosses. All strains were backcrossed into the resident Cantonised-w1118 control isogenic strain for six generations to normalize their genetic background.
Drug feeding.
The selective Alk inhibitor NPV-TAE684 (Galkin et al., 2007) was dissolved in DMSO, and serial dilutions of stock solutions were prepared following the previously described method (Gouzi et al., 2011). Briefly, the solution was mixed into 10 ml Brewers-yeast paste and was fed for 16 h to flies previously starved for 6–8 h and transferred into normal fly-food vials 1 h before behavioral conditioning.
Western blot analysis.
For detection of Jeb levels, 10 adult heads or 5 larval CNSs were homogenized in standard Laemmli buffer supplemented with protease and phosphatase inhibitors. Extract equivalent to one adult head was loaded per lane on 10% acrylamide gels, transferred to PVDF membranes, and probed with primary antibodies, which were used at 1:1000 for guinea-pig anti-JEB (Englund et al., 2003) and at 1:2000 for mouse anti-Syntaxin (8C3, Developmental Studies Hybridoma Bank), which was used to normalize sample loading.
Immunohistochemical analysis and confocal imaging.
Whole-mount adult brains were dissected in cold PBS, fixed in 4% PFA for 20 min, and permeabilized using 1% Triton X-100 in PBS. The primary antibodies used were as follows: rabbit anti-dALK (1:1000) (Lorén et al., 2001), guinea-pig anti-JEB (1:1000) (Englund et al., 2003), mouse anti-DLG1 (1:1000) (4F3), and mouse anti-ChAT (1:1000) (4B1) (both from the Developmental Studies Hybridoma Bank, University of Iowa). The following secondary antibodies were used: goat anti-mouse, or anti-rabbit conjugated with AlexaFluor secondary antibodies (1:400, all from Invitrogen). Confocal laser microscopy was performed using a TCS SP5 Confocal system (Leica Microsystems) equipped with the LAS AF image acquisition analysis software suite (Leica Microsystems). To quantify dAlk expression levels in the MBs, we used an adapted semiquantitative immunofluorescence protocol detailed previously (Liu et al., 2007). Whole-mount brains were dissected and prepared as described above and were stained with a rabbit anti-dALK antibody and counterstained with rhodamine-conjugated phalloidin (Invitrogen; 1:100) to mark the neuropil. Single confocal plane images of MB calyces were captured at the same section level corresponding to the middle part of the calyx and using constant optical acquisition settings (laser power, gain, pinhole, offset, zoom) and examined within a single session to allow comparison between control and experimental samples. dAlk expression levels in the calyces were estimated by subtracting from the measured mean grayscale intensity of a ROI (delimited by hand around a calyx), the measured mean grayscale intensity of an identical area adjacent to the calyx (background region). Fluorescence intensities were measured with ImageJ 1.51k software (National Institutes of Health).
Behavioral analyses and conditioning.
Olfactory learning and memory in the Pavlovian negatively reinforced conditioning paradigm, coupling aversive odors as conditioned stimuli (CS+ and CS−) with the electric shock unconditioned stimulus (US), were performed essentially as described previously, for 3 min/learning (Gouzi et al., 2011) and for 24 h memory/LTM (Pavlopoulos et al., 2008). Briefly, all experiments were performed in a genotype-balanced manner, with the experimenter blind to genotype and under dim red light, at 25°C and 70% humidity. Groups of ∼75, 2- to 3-d-old flies were transferred to fresh vials 1 h before training and then trained in a T-maze apparatus. The aversive odors used were benzaldehyde and 3-octanol, diluted in isopropyl myristate (Fluka). Training for 3 min/learning or 3 h/mid-term memory experiments consisted of 1 single session of 12 US/CS pairings of 90 V electric shocks (US) with one odor (CS+) over 1 min, followed after a 30 s purge with air, by the presentation of the second odor (CS−) without shocks for 1 min. For 24 h memory, animals were subjected to five such training sessions, either massed together or spaced by a 15 min rest interval. For testing, flies were allowed to choose between the two odors presented in the two arms of the T-maze apparatus for 1.5 min. Performance was measured by calculating a performance index (PI) as follows: the fraction of flies that avoided the shock-associated odor (CS+) minus the fraction that avoided the control odor (CS−) represented half of the PI. One PI was calculated as the average of the half-learning indexes for each of the two groups of flies trained to complementary conditioning stimuli. To validate consolidated LTM, flies were placed for 16 h at 25°C in empty vials containing a piece of Whatman filter paper (2 × 2 cm) soaked with 200 μl of 5% sucrose solution (Fisher Scientific) containing 35 mm cycloheximide (Sigma-Aldrich). Flies were then transferred to standard food vials 30 min before training. After training, flies were kept in standard food vials for 24 h at 18°C until testing. To assess 10 min and 3 h ARMs, flies were cold-shocked in prechilled glass vials on ice for 2 min after a single round of training. Complete anesthesia and recovery were controlled. After a 2 min recovery, they were transferred back to vials at 25°C in the dark and maintained until testing. For 10 min short-term ARM (ST-ARM), trained flies were cold-shocked immediately after training as described previously (Knapek et al., 2011; Bouzaiane et al., 2015). For 3 h middle-term ARM (MT-ARM), trained flies were cold-shocked 2 h after training, as described by Bouzaiane et al. (2015). Task-relevant sensory behavioral responses, such as odor avoidance and electric shock avoidance, were controlled and performed as described previously in detail (Gouzi et al., 2011).
Experimental design and statistical analysis.
For all experiments, controls and experimental genotypes were tested in the same session in balanced design. The order of training and testing these genotypes was randomized. We required an experimental result to be significantly different from both genetic controls. Data are shown as mean ± SEM. The final number of experiments per genotype is listed on the bars in all figures and represents data collected from at least two broods of the given genotypes.
Untransformed (raw) data were analyzed parametrically with the JMP 7.1 statistical software package (SAS Institute) as described previously (Gouzi et al., 2011). Following initial ANOVA, planned comparison contrast analyses (LSM) were performed between the experimental group (in black throughout all figures) and its genetic or treatment controls, using α = 0.05. The level of significance was adjusted for the experimentwise error rate. Detailed results of all ANOVA and planned comparisons are reported in the text.
Results
Pharmacological inhibition of dAlk enhances both STM and LTM
To investigate whether dAlk plays a role in negatively reinforced associative olfactory memory, we administered the dAlk selective kinase inhibitor TAE684 (Galkin et al., 2007) to control (w1118) flies (Fig. 1). In agreement with published data (Gouzi et al., 2011), 100 nm and 1 μm TAE684 enhanced 3 min memory by ∼12% and 20%, respectively, (STM: ANOVA F(2,26) = 8.3362, p = 0.0018; subsequent LSM: p = 0.0208 and p = 0.0005 for 100 nm and 1 μm, respectively, vs vehicle-fed w1118 controls), but significantly it also precipitated a very robust increase in 24 h memory of ∼20% and 36% (LTM: ANOVA F(2,31) = 11.6696, p = 0.0002; subsequent LSM: p = 0.0194 and p = 4.2 × 10−5 for 100 nm and 1 μm, respectively, vs vehicle). Together, these initial pharmacological results strongly suggest that dAlk inhibits olfactory memory in Drosophila in addition to its established role in inhibition of learning/3 min memory in the context of Nf1-regulated pathway(s) (Gouzi et al., 2011).
Figure 1.

Pharmacological inhibition of dAlk activity enhances STM and LTM. Mean PIs (bars) ± SEMs (error bars) are shown. The number of experimental replicates (n) are indicated within the bars. *Significant differences denoted by horizontal line pairs. Left, TAE684 administration 100 nm or 1 μm to w1118 flies enhanced their learning/3 min STM performance in a high-resolution limited training protocol (1 cycle of 3 CS/US pairings). Right, TAE684 administration to w1118 flies enhanced their 24 h LTM performance. LTM performance was obtained using the typical spaced training protocol of 5 cycles of 12 CS/US pairings, each cycle separated (spaced) by a 15 min rest interval.
Downregulation of dAlk specifically within MB neurons enhances LTM
To validate this memory enhancement genetically, we attenuated dAlk in cells endogenously expressing the RTK by RNAi-mediated abrogation with AlkRNAi(11446) under Alk(38)-Gal4 (Gouzi et al., 2011). Consistent with the TAE684 results, dAlk abrogation in Alk-expressing cells (Fig. 2A) elevated 24 h memory (Alk(38)-Gal4: ANOVA: F(2,31) = 12.8918, p < 0.0001; subsequent LSM vs controls: p = 0.0011 and p = 4.5 × 10−5, respectively), suggesting that the RTK mediates long-term memory attenuation. Task-relevant sensory responses were known to be normal (Gouzi et al., 2011).
Figure 2.
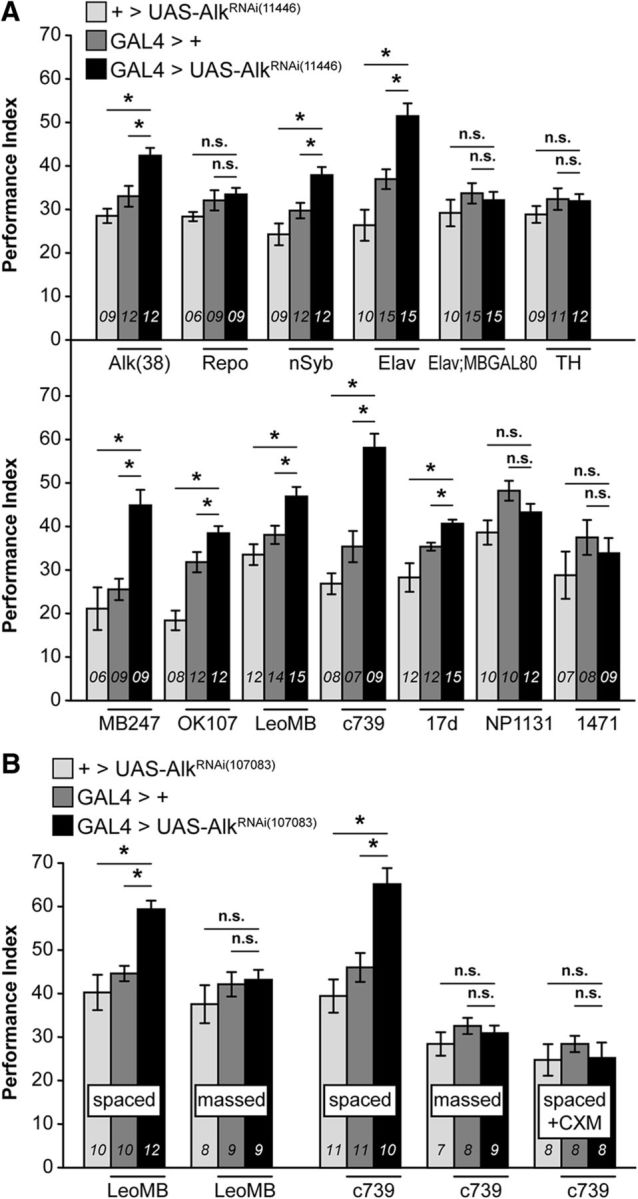
Downregulation of dAlk in intrinsic α/β MB neurons enhances PSD-LTM. Mean PIs (bars) ± SEMs (error bars) are shown. The number of experimental replicates (n) are indicated within the bars. Mean PIs are shown after typical spaced training of 5 cycles of 12 CS/US pairings, unless indicated otherwise. *Significant differences denoted by horizontal line pairs. n.s. means not significant. A, GAL4 screen performed with expression of a UAS-AlkRNAi transgene in defined GAL4-marked neuronal subsets of the fly brain. Neuronal and glial GAL4 drivers (top) and MB GAL4 drivers (bottom) represent LTM PI of flies expressing the UAS-AlkRNAi(11446) transgene driven by the indicated GAL4 (dark bars) and relevant genetic controls (light and dark gray bars). Significant LTM enhancement was revealed for Alk(38), nSyb, Elav, MB247, OK107, LeoMB, c739, and 17d driven dAlk abrogation. B, Validation of the specificity of the 24 h memory enhancement with the independent RNAi-mediating transgene UAS-AlkRNAi(107083). Attenuation of dAlk specifically in LeoMB and c739 marked MB neurons (black bars) improved 24 h memory performance after spaced training (spaced) and phenocopied the effect previously observed with AlkRNAi(11446). In contrast, 24 h memory performance observed after massed training (massed) or cycloheximide treatment (35 mm, spaced + CXM) was not enhanced, demonstrating that the enhanced memory consists entirely of bona fide PSD-LTM.
To determine the neurons requiring dAlk function to constrain 24 h memory, we abrogated the RTK in defined brain cell types (Fig. 2A, top). Attenuating dAlk in glia did not affect 24 h memory (Repo-Gal4: ANOVA: F(2,23) = 1.9385, p = 1688). In contrast, pan-neuronal dAlk abrogation (nSyb-Gal4: ANOVA: F(2,31) = 12.9304, p < 0.0001; subsequent LSM vs controls: p = 0.0026 and p = 3 × 10−5, respectively; and Elav-Gal4: ANOVA: F(2,39) = 19.9132, p < 0.0001; subsequent LSM vs controls: p = 0.0003 and p = 3.8 × 10−7) enhanced 24 h memory, mimicking the results with Alk(38)-Gal4, but not if MB neurons were spared (Elav-Gal4; MBGal80: ANOVA: F(2,39) = 0.9212, p = 0.4070). Hence, dAlk within MB neurons appears necessary for the 24 h memory enhancement. Dopaminergic neurons (TH-Gal4: ANOVA: F(2,31) = 0.9152, p = 0.4117) were not required, minimizing the possibility that enhanced memory is the consequence of attenuated forgetting (Berry et al., 2012).
To determine which MB neurons require dAlk for memory suppression, several specific drivers were used (Fig. 2A, bottom). dAlk abrogation under the panMB drivers MB247-Gal4, OK107-Gal4, and LeoMB-Gal4 (Messaritou et al., 2009), and c739-Gal4, 17d-Gal4 marking α/β neurons, yielded significantly elevated 24 h memory (MB247-Gal4: ANOVA: F(2,23) = 13.9858, p = 0.0001; subsequent LSM vs controls: p = 3.4 × 10−4 and p = 1.2 × 10−4, respectively; OK107-Gal4: ANOVA: F(2,31) = 23.3012, p < 0.0001; subsequent LSM: p = 0.0172 and p = 1.8 × 10−7 vs controls, respectively; LeoMB-Gal4: ANOVA: F(2,39) = 10.9654, p = 0.0002; subsequent LSM: p = 0.0034 and p = 6.1 × 10−5 vs controls, respectively; c739-Gal4: ANOVA: F(2,23) = 31.3183, p < 0.0001; subsequent LSM: p = 2.6 × 10−5 and p = 1.8 × 10−7 vs controls, respectively; 17d-Gal4: ANOVA: F(2,38) = 11.7483, p < 0.0001; subsequent LSM: p = 0.0257 and p = 2.4 × 10−5 vs controls, respectively). In contrast, attenuation within γ neurons did not affect memory (NP1131-Gal4: ANOVA: F(2,31) = 4.4173, p = 0.0212; 1471-Gal4: ANOVA: F(2,23) = 1.0385, p = 0.3715). The stronger memory improvement yielded by c739-Gal4 compared with 17d-Gal4 is consistent with previous reports (Akalal et al., 2006) that, although both mark MB α/β lobes, c739-Gal4 is expressed higher and in more than twice the number of Kenyon cells compared with 17d-GAL4. These results demonstrate that dAlk is required specifically in α/β neurons, known to be essential for 24 h memory (Isabel et al., 2004; Yu et al., 2006; Pavlopoulos et al., 2008). These results were further validated (Fig. 2B, spaced) with an independent RNAi-mediating transgene (UAS-AlkRNAi(107083)) (Gouzi et al., 2011), which also yielded elevated 24 h memory after spaced conditioning under LeoMB-Gal4 and c739-Gal4 (LeoMB-Gal4 spaced: ANOVA: F(2,31) = 14.6651, p < 0.0001; subsequent LSM vs controls: p = 0.0005 and p = 1.9 × 10−5, respectively; and c739-Gal4 spaced: ANOVA: F(2,31) = 13.8101, p < 0.0001; subsequent LSM vs controls: p = 0.0007 and p = 2 × 10−5).
Two forms of consolidated memory are detectable 24 h after training: PSD-LTM, formed after spaced training; and ARM, formed after massed training, which does not depend on de novo protein synthesis (Tully et al., 1994; Isabel et al., 2004). Significantly, MB-specific dAlk downregulation did not affect ARM (Fig. 2B, massed), suggesting that the 24 h memory enhancement is PSD-LTM-specific (LeoMB-Gal4 massed: ANOVA: F(2,25) = 0.8317, p = 0.4480; and c739-Gal4 massed: ANOVA: F(2,23) = 0.9291, p = 0.4105). Consistently, inhibition of protein synthesis before spaced training with cycloheximide (Tully et al., 1994) eliminated the 24 h memory enhancement (Fig. 2B, spaced + CXM: c739-Gal4: ANOVA: F(2,23) = 0.4472, p = 0.6454), demonstrating that it consists entirely of bona fide PSD-LTM.
Downregulation of dAlk in the MBs enhances PSD-LTM specifically
Are earlier memories affected by dAlk attenuation in α/β MB neurons? To increase resolution and assess all memory forms concurrently, we used 1 training cycle of 12 US/CS pairings. Under these conditions (Fig. 3A), dAlk abrogation did not affect 10 min memory (STM, 10 min cold shock: ANOVA: F(1,15) = 0.2659, p = 0.6142) as expected (Gouzi et al., 2011), or labile short-term ARM (Knapek et al., 2011; Bouzaiane et al., 2015) (10 min + cold shock: ANOVA: F(1,15) = 0.0717, p = 0.7928). Moreover, 3 h middle-term memory (MTM) (3 h − cold shock: ANOVA: F(1,15) = 0.3578, p = 0.5593) and labile middle-term ARM (3 h + cold shock: ANOVA: F(1,15) = 0.0313, p = 0.8622) remained unaffected (Fig. 3A). However, a mild enhancement was detectable at 6 h after training (ANOVA: F(1,15) = 8.6609, p = 0.0107), which was striking at 12 h (ANOVA: F(1,15) = 17.1629, p = 0.0010) and 24 h (24 h-CXM: ANOVA: F(1,15) = 19.2126, p = 0.0006). Even after one training cycle, the resultant 24 h memory was also bona fide PSD-LTM and not long-term ARM (Bouzaiane et al., 2015), as it was eliminated by cycloheximide treatment (24 h + CXM: ANOVA: F(1,15) = 0.1461, p = 0.7081) (Fig. 3A).
Figure 3.

Downregulation of dAlk in the MBs enhances PSD-LTM specifically. Mean PIs (bars) ± SEMs (error bars) are shown. The number of experimental replicates (n) are indicated within the bars. *Significant differences denoted by horizontal line pairs. n.s. means not significant. A, After a single round of 12 US/CS conditioning, memory retention was significantly enhanced at 6, 12, and 24 h upon attenuation of dAlk levels within c739-marked MB neurons (black bars) compared with the indicated relevant control (light gray bars). The improvement of 24 h memory consisted of bona fide PSD-LTM, as treatment with the protein synthesis inhibitor CXM totally eliminated the improvement. In contrast, 10 min and 3 h memories were not improved upon dAlk attenuation. ARM immediately after training (10 min + cold shock) and ARM after 3 h (3 h + cold shock) were also not elevated. B, Learning (3 min memory) was not significantly improved in flies with attenuated dAlk expression in c739-marked MB neurons (black bars) compared with control (light gray bars) under any US/CS pairing protocol (3, 6, 12 US/CS).
Because 10 min memory was maximal under the intense 12 US/CS training conditions, enhanced learning may not have been visible due to ceiling effect. Hence, to verify that dAlk attenuation within the MBs did not result in enhanced learning, we limited the number of US/CS pairings from 12 to 3 (Fig. 3B). Nevertheless, enhanced learning was not detectable even under limited training (3 US/CS: ANOVA: F(1,15) = 1.9861, p = 0.1806; 6 US/CS: ANOVA: F(1,15) = 0.5248, p = 0.4808; 12 US/CS: ANOVA: F(1,15) = 0.0428, p = 0.8392). In conclusion, a single training cycle upon dAlk abrogation in α/β MB lobes elicits bona fide PSD-LTM, which is not consequent of enhanced learning per se.
dAlk levels constrain PSD-LTM in adult MBs
Το ascertain that dAlk plays an acute role in PSD-LTM, we modulated dAlk levels specifically in the adult CNS (McGuire et al., 2004). Adult-restricted dAlk abrogation (Fig. 4A) yielded significant memory elevation whether it was pan-neuronal (Elav;Gal80ts: ANOVA: F(2,31) = 35.5212, p < 0.0001; LSM vs controls: p = 8 × 10−7 and p = 1.1 × 10−8, respectively), pan-MB (Leo;Gal80ts: ANOVA: F(2,38) = 8.1033, p = 0.0012; subsequent LSM vs controls: p = 0.0038 and p = 0.0008), or specifically in α/β neurons (c739;Gal80ts: ANOVA: F(2,31) = 15.7468, p < 0.0001; LSM vs controls: p = 0.0005 and p = 1 × 10−5). Hence, dAlk attenuation within α/β MB neurons appears sufficient for the 24 h PSD-LTM elevation, and developmental alterations within MBs cannot account for the enhancement. Although still formally possible, it is highly unlikely that adult-specific attenuation of dAlk outside of the MBs could lead to LTM enhancement as constitutive attenuation of dAlk under Elav-Gal4; MB-Gal80 did not elevate it (Fig. 2A).
Figure 4.
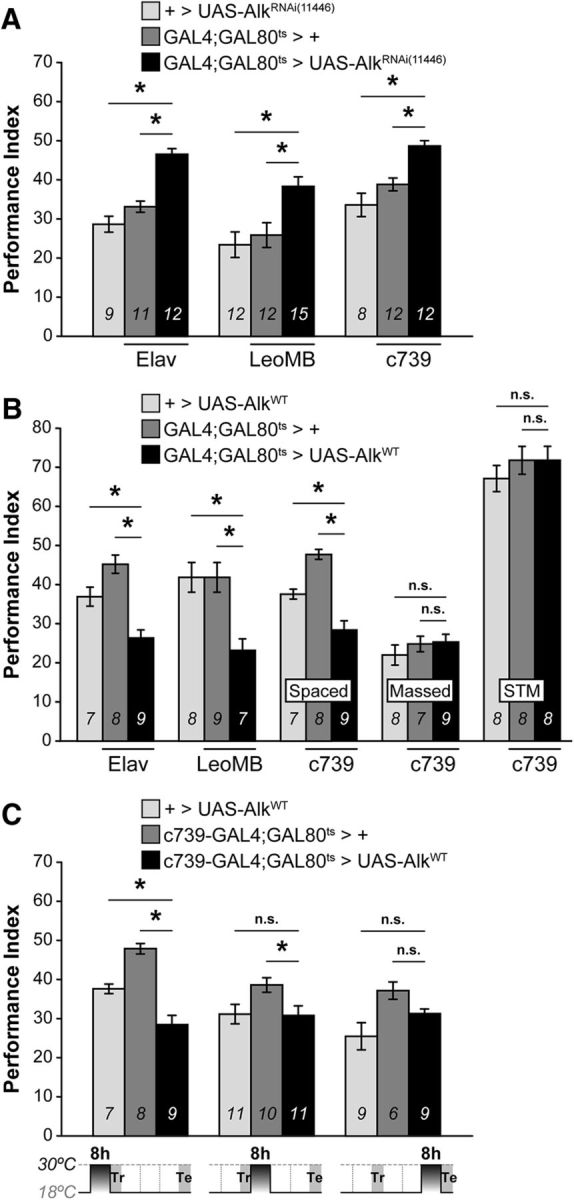
dAlk levels in the adult MBs during conditioning enhance or constrain LTM performance. Mean PIs (bars) ± SEMs (error bars) are shown. The number of experimental replicates (n) are indicated within the bars. *Significant differences denoted by horizontal line pairs. n.s. means not significant. A, Attenuation of dAlk expression levels with the dAlkRNAi(11446) transgene restricted specifically to the adult CNS for 48 h before training yielded significant LTM increase (black bars) compared with controls (light and dark gray bars) when the transgene was expressed pan-neuronally (Elav;G80ts), in all MB neurons (LeoMB;G80ts) and more specifically in α/β MB neurons (c739;G80ts). B, Conversely, adult-specific overexpression of the wild-type dAlkWT transgene using the same indicated G80ts drivers elicited significant LTM deficits. In contrast, ARM and learning/3 min memory (Massed and STM) were not affected. C, Induction of dAlkWT expression in adult c739;G80ts MB neurons for 8 h before conditioning (t−8 h-t0) resulted in deficient LTM; but in contrast, it left LTM intact if the transgene was induced for 8 h immediately following conditioning (t0-t+8 h), or 8 h before testing (t+16 h-t+24 h), as indicated in the diagram below.
Collectively, dAlk appears to constrain PSD-LTM formation and its abrogation enhances it, even after minimal training. It follows then that acute dAlk elevation within α/β neurons could suppress PSD-LTM. Notably, increasing the levels of dAlk in adult neurons marked by commonly used drivers resulted in lethality within 24 h but was circumvented by reducing transgene induction to 8 h. Acute adult-specific overexpression of dAlk precipitated a highly significant LTM deficit (Fig. 4B), whether pan-neuronal (Elav;Gal80ts: ANOVA: F(2,23) = 19.2806, p < 0.0001; subsequent LSM vs controls: p = 3.9 × 10−6 and p = 0.0031), pan-MB (LeoMB;Gal80ts: ANOVA: F(2,23) = 6.6558, p = 0.0058; LSM vs controls: p = 0.0037 and p = 0.0048, respectively), or only in α/β neurons (c739;Gal80ts: ANOVA: F(2,23) = 30.1603, p < 0.0001; LSM vs controls: p = 1.3 × 10−7 and p = 0.0021). This deficit seems specific to LTM as sensory responses under Elav were normal (Gouzi et al., 2011), the flies were viable, normal externally, without apparent locomotor deficits (data not shown), and both their ARM after massed training and 3 min memory (Fig. 4B, massed and STM) were normal (massed: ANOVA: F(2,23) = 0.7627, p = 0.4789; STM: ANOVA: F(2,23) = 0.6109, p = 0.5522). Thus, the deficit is not consequent of disabled or unfit flies, and increased dAlk levels impair LTM, whereas its attenuation improves it.
We used the fact that 8 h of dAlkWT induction suffice to yield deficits, to dissect the requirement for this RTK in PSD-LTM formation, consolidation, or retrieval (Tully et al., 1994) (Fig. 4C). Inducing dAlkWT in adult MB neurons 8 h before conditioning manifested in deficient LTM ([t−8 h-t0]: ANOVA: F(2,23) = 30.1603, p < 0.0001; LSM vs controls: p = 1.3 × 10−7 and p = 0.0021, respectively). However, elevating dAlk for 8 h post-training or 8 h pre-testing did not result in LTM deficits ([t0-t+8 h]: ANOVA: F(2,31) = 3.8911, p = 0.0318; [t+16 h-t+24 h]: ANOVA: F(2,23) = 4.6712, p = 0.0210). Therefore, to constrain PSD-LTM formation, dAlk activity is required during conditioning; therefore, it acts specifically as a negative regulator of its formation, not of its consolidation or retrieval.
dAlk is confined to the dendritic postsynaptic active zones of Kenyon cells
Is the distribution of dAlk within the MBs consistent with its proposed role in LTM inhibition? As shown in Figure 5A1, intense anti-dAlk staining was observed in the calyces and absent in other parts of MB neurons, such as the pedunculus and the lobes (Fig. 5A2–A4) in a manner reminiscent of another memory suppressor protein in Drosophila, SLC22A (Gai et al., 2016).
Figure 5.
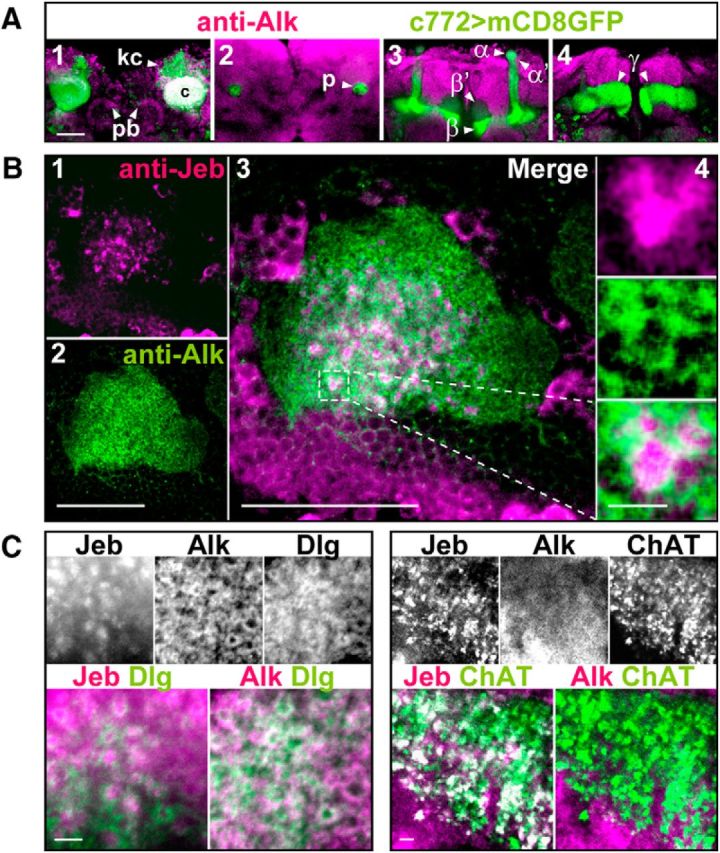
dAlk and its ligand Jeb are enriched in synapses of projection neurons and Kenyon cells. A, dAlk is enriched in MB calyces but is absent in the MB lobes. Confocal images of single optical sections of the fly brain from posterior to anterior (A1–A4) were acquired at the level of MB calyces (A1), pedunculus (A2), α/β lobes (A3), and γ lobe (A4). Confocal images were acquired at the same section levels with identical settings. dAlk was visualized with an anti-dAlk antibody (purple). Fly brain structures were marked with a membrane GFP (green) encoded by the mCD8:GFP transgene expressed with the neuronal c772-GAL4 driver. White represents colocalization of dAlk and GFP immunofluorescence. dAlk protein accumulates within the dendrites (A1: calyces, c) and cell bodies (A1: Kenyon cells, kc, arrowhead) of MB neurons and in the protocerebral bridge (A1: pb, arrowheads). It is also expressed widely in the neuropil, above background. In contrast, MB axons (A2: pedunculus, p, arrowhead) (A3: α, α′, β, and β′ lobes, arrowheads) (A4: γ lobes, arrowheads) are devoid of dAlk staining. Scale bar, 50 μm. B, dAlk and Jeb display a complementary pattern of expression within MB calyces. Confocal images of a single optical transverse section of an MB calyx were acquired using identical settings and at the same section level corresponding approximately to its middle section (B1–B3). Jeb protein was visualized with an anti-Jeb antibody (B1, purple), and dAlk protein was visualized with an anti-dAlk antibody (B2, green). Scale bar, 50 μm. Colocalization of Jeb and dAlk immunofluorescence (B3, merge, white) is shown at the same magnification. Scale bar, 50 μm. Inset, Higher magnification of the hatched box, showing in a single synaptic microglomerulus (B4), from top to bottom, Jeb, dAlk, and their complementary pattern. Scale bar, 10 μm. C, dAlk is expressed at the postsynaptic dendritic active zones of the MB calyces, whereas Jeb is expressed presynaptically in the synaptic buttons of apparent projection neurons. Confocal images of single optical sections of MB calyces were acquired at similar levels, using identical settings and displaying triple staining of Jeb, dAlk, and membrane GFP. Scale bars, 10 μm. Left, White represents signals of anti-Jeb, anti-dAlk, and anti-Dlg, a marker of the postsynaptic active zones. Colored images represent lack of colocalization of Jeb (purple) and Dlg (green), and colocalization (white) of dAlk (purple) and Dlg (green). Right, White represents signals of anti-Jeb, anti-dAlk, and anti-ChAT, a marker of presynaptic terminal buttons. Colored images represent complete colocalization of Jeb (purple) and ChAT (green), and lack of colocalization (white) between dAlk (purple) and ChAT (green).
Further examination at higher magnification and single optical sections revealed that dAlk is broadly distributed throughout the calyces (Fig. 5B2) in contrast to its ligand Jeb, whose distribution appeared discrete, punctate, and granular (Fig. 5B1), proximal to dAlk, but without obvious colocalization (Fig. 5B3). Indeed, dAlk appeared to surround areas of Jeb immunoreactivity (Fig. 5B4), whose size and morphology are characteristic of MB calycal microglomeruli (Leiss et al., 2009). These postsynaptic microstructures are formed by the claw-like dendritic specializations of the KCs, and presynaptically by the enlarged terminal specializations/boutons of projection neurons (PNs). This notion was validated with typical presynaptic and postsynaptic MB markers (Fig. 5C). dAlk is extensively colocalized with the postsynaptic active zone marker DLG (discs large), whereas Jeb colocalized within characteristic large puncta with ChAT (Choline Acetyl Transferase), a PN presynaptic marker (Leiss et al., 2009). Thus, dAlk and Jeb are components of the calycal PN-MB synapses, sites of structural plasticity underlying olfactory learning and memory.
The dAlk-activating ligand Jeb does not affect LTM
Based on the complementarity of dAlk and Jeb patterns in MB calyces, we predicted that reducing Jeb levels would phenocopy dAlk attenuation and enhance PSD-LTM. However, developmental (Fig. 6A) or adult-specific (Fig. 6B) downregulation of Jeb by pan-neuronal (Elav, Ras2, and nSyb), glial (Repo), or PN-specific (GH146) expression of two different JebRNAi transgenes did not enhance LTM (Fig. 6A; JebRNAi(103047): Elav-Gal4: ANOVA: F(2,29) = 6.6866, p = 0.0044; LSM vs controls: p = 0.0011 and p = 0.0988, respectively; Ras2-Gal4: ANOVA: F(2,30) = 8.7923, p = 0.0011; LSM vs controls: p = 0.0002 and p = 0.0563, respectively; nSyb-Gal4: ANOVA: F(2,30) = 0.3647, p = 0.6976; Repo-Gal4: ANOVA: F(2,31) = 2.1992, p = 0.1291; Fig. 6B; JebRNAi(103047): Elav-Gal80ts: ANOVA: F(2,37) = 0.1693, p = 0.8449; Repo-Gal80ts: ANOVA: F(2,55) = 2.5336, p = 0.0890; GH146-Gal80ts: ANOVA: F(2,31) = 0.4709, p = 0.6291; Fig. 6B; JebRNAi(30799): Elav-Gal80ts: ANOVA: F(2,68) = 0.5738, p = 0.5662). Jeb overexpression also did not alter LTM (Fig. 6B, JebWT: GH146-Gal80ts: ANOVA: F(2,31) = 1.1982, p = 0.3162). We are certain that the tools used for Jeb abrogation are functional because they dramatically reduce its level (Fig. 6D) and recapitulated established Jeb-dependent phenotypes (Gouzi et al., 2011) of increased pupal size (Elav-Gal80ts: ANOVA: F(2,103) = 85.3173, p < 0.0001; LSM vs controls: p = 5.9 × 10−17 and p = 1.3 × 10−20, respectively) and enhanced 3 min memory (Elav-Gal80ts: ANOVA: F(2,35) = 13.0861, p < 0.0001; LSM vs controls: p = 0.0026 and p = 1.6 × 10−5, respectively) (Fig. 6C). Therefore, the dAlk-dependent LTM inhibition is Jeb independent.
Figure 6.

The dAlk-activating ligand Jeb is not involved in LTM. Mean PIs (bars) ± SEMs (error bars) are shown. The number of experimental replicates (n) are indicated within the bars. *Significant differences denoted by horizontal line pairs. n.s. means not significant. A, Attenuation of Jeb expression levels with the JebRNAi(103047) transgene (black bars) did not affect 24 h LTM performance compared with controls (light and dark gray bars) when the transgene was expressed pan-neuronally or in glial cells only using the indicated Gal4 drivers. B, Attenuation of Jeb expression levels with the JebRNAi(103047) transgene restricted to the adult CNS for 48 h before training also did not affect 24 h LTM when the transgene was expressed pan-neuronally, in glial cells or in projection neurons using the indicated G80ts drivers. Lack of LTM effects was verified with pan-neuronal expression (Elav;Gal80ts) of a second independent JebRNAi(30799) transgene. Jeb overexpression (JebWT) in projection neurons (GH146,Gal80ts) also did not affect 24 h LTM. C, Pan-neuronal Jeb attenuation with the JebRNAi(103047) transgene yielded a substantial increase of pupal size compared with controls when the transgene was expressed throughout development and yielded a substantial increase of learning/3 min memory (STM, 1 cycle of 3 CS/US pairings) when the transgene was expressed only during adulthood for 2 d before conditioning. D, Representative semiquantitative immunoblot showing a dramatic reduction of endogenous Jeb levels upon pan-neuronal expression of both independent JebRNAi transgenes (UAS-JebRNAi(103047) and UAS-JebRNAi(30799)). Jeb protein (Jeb) was revealed with the anti-Jeb antibody (Jeb), and Syntaxin (Syx) was used as loading control.
dAlk levels increase rapidly in KC dendrites upon conditioning
Because excessive dAlk in the MBs suppressed LTM, we wondered whether, under physiological conditions, its levels change within these neurons consequent to conditioning. Therefore, we used a well-established quantification protocol (Liu et al., 2007) to monitor dAlk levels in MB calyces after conditioning with 1 round of 12 US/CS, which is sufficient to trigger elevated LTM upon its attenuation (Fig. 3A). Given the time required to condition and sacrifice the flies, we monitored its levels at 20 min and 3 h after conditioning.
Significantly, dAlk levels were substantially elevated in conditioned flies (US/CS paired), in contrast to animals after sham conditioning (US/CS unpaired) with the US preceding the CS without coincidence (Fig. 7A). Quantification of dAlk levels (Fig. 7B, left) revealed that US/CS pairing resulted in a significant ∼1.5-fold elevation (ANOVA: F(3,50) = 7.9802, p = 0.0002; LSM 20′ paired vs 20′ unpaired: p = 0.005; and p = 0.0003 for 3 h paired vs 20′ unpaired). Because each group of flies experienced both stimuli, this result indicates that dendritic dAlk elevation emerges specifically upon stimulus pairing. In confirmation, dAlk levels after sham training were found equivalent to those in naive flies (LSM 20′ naive vs 20′ unpaired: p = 0.8114). This difference cannot be attributed to positioning or to the size of the flies because female flies of similar age were used for all quantifications and the calycal areas quantified were statistically identical (ANOVA F(3,50) = 0.3589, p = 0.7829) in the 51 individuals evaluated (Fig. 7E, left). Finally, dAlk levels were not found further elevated upon increasing the number of conditioning rounds from 1× to 5× (spaced conditioning) (Fig. 7B, right, ANOVA F(1,31) = 0.0000, p = 0.9976). Therefore, the first round of conditioning appears sufficient for dAlk to attain maximal accumulation levels in the early phase of memory encoding. This is consistent with the notion that dAlk elevation attenuates or blocks PSD-LTM formation upon a single round of conditioning, and congruent with PSD-LTM formation with a single round of conditioning when dAlk was abrogated (Fig. 3A).
Figure 7.

Dendritic dAlk levels in the MBs increase rapidly upon conditioning to constrain LTM. A, Representative grayscale images of anti-dAlk immunofluorescence in control w1118 flies. Scale bar, 50 μm. Single confocal plane images of MB calyces were captured at the same section level corresponding to the middle part of the calyx, using constant optical acquisition settings, and examined in a single session to allow comparison between control (unpaired 20 min, 1 round, 1×) and experimental samples (paired 20 min, 1 round, 1×). B, Semiquantitative analysis of dAlk immunoreactivity (IR) in the calyces of w1118 control flies was achieved by subtracting from the mean grayscale intensity of the signal within a calyx (dashed circle 1, delimited by hand in A), the mean grayscale intensity of the signal in an adjacent area (dashed circle 2, same size and shape as circle 1). The mean intensity differences ± SEM for the indicated number (n) of calyces are shown. Control w1118 flies were either trained with the typical conditioning protocol of 12 US/CS pairings (US/CS: paired) or trained with a modified protocol in which the US preceded the CS+ without coincident pairing (US/CS: unpaired) or untrained (UC/CS: −). Flies were trained using either a single (1×) round or 5× spaced rounds of 12 US/CS pairings. Their brains were dissected and fixed either immediately (20′) or 3 h after training (Time PT, Time post-training). In control w1118 flies, pairing the US with the CS+ significantly increased calycal dAlk levels 20 min after conditioning, which apparently perdured at least for 3 h. Training flies either with 1× round or with 5× spaced rounds did not alter increased calycal dAlk levels. In contrast, dAlk levels in flies submitted to unpaired stimuli (unpaired) were not different from in naive flies (−). C, Single confocal plane images of a MB calyx (left) and lobes (right) in AlkMB06458 homozygote fly brains stained with the anti-dAlk antibody (top row) and counterstained with rhodamine-conjugated phalloidin (bottom row) to mark the neuropil. In AlkMB06458, dAlk retained its calycal localization, remaining excluded from the lobes (arrowheads). Scale bar, 50 μm. D, Semiquantitative analysis of dAlk immunoreactivity (IR) in the calyces of w1118 and AlkMB06458 flies. The mean dAlk intensity differences (calculated as indicated above) ± SEM for the indicated number of calyces (n) are shown. Differences in steady-state levels of dendritic dAlk were not detected in AlkMB06458 mutants and w1118 control flies (unpaired). However, in flies submitted to 1 round of conditioning (paired), the pairing-dependent increase of dendritic dAlk observed in w1118 controls was absent in AlkMB06458 homozygotes. Time PT, Time post-training. E, Mean areas (in square pixels ± SEM) of the calyces used in B and D to obtain the intensity measurements. No significant differences between calycal areas were detected that could explain the differences observed in dAlk intensities. Each bar corresponds to its equivalent in B and D. −, Naive flies; u, unpaired; p, paired; 1× and 5×, number of rounds. Brains were all dissected/fixed 20 min after training, except where specified. F, AlkMB06458 homozygotes present enhanced LTM (spaced) but not 3 min memory (STM, 3 US/CS) or ARM (massed). Mean PIs (bars) ± SEMs (error bars) are shown. The number of experimental replicates (n) are indicated within the bars. *Significant differences denoted by horizontal line pairs. n.s. means not significant. G, Task-relevant olfactory and shock reactivity responses in AlkMB06458 homozygotes (black bars) were indistinguishable from controls (white bars). H, The dosage of dAlk protein in MB dendrites after conditioning calibrates the level of LTM performance. Mean PIs (bars) ± SEMs (error bars) are shown. The number of experimental replicates (n) are indicated within the bars. *Significant differences denoted by horizontal line pairs. n.s. means not significant. Alk1/AlkMB06458 heteroallelics present even higher LTM than AlkMB06458 homozygotes. The fact that heteroallelics present LTM elevation indicates that the mutations do not complement and are indeed allelic.
Importantly, dAlk elevation in MB dendrites was detected 20 min after conditioning and remained elevated for at least 3 h (Fig. 7B; LSM 3 h paired vs 20′ paired: p = 0.3305). The short time required for a rather large transmembrane protein to increase its dendritic levels fits the temporal requirement for locally translated proteins (Steward and Schuman, 2001). Considering the role of dAlk in PSD-LTM formation, its restricted dendritic distribution, and its rapid increase upon US/CS pairing, we suggest that its mRNA is likely transported to dendrites and translated upon conditioning. Furthermore, we hypothesized that dendritic targeting and local translation of dAlk mRNA may be conferred by sequences in its 3′UTR, as for several other synaptic proteins (Steward and Schuman, 2001; Ashraf et al., 2006). Inspection of the dAlk 3′UTR with TargetScanFly 6.2 (Lewis et al., 2005) and RBPmap 1.1 (Paz et al., 2014) revealed that it contains numerous elements potentially controlling mRNA stability and translation, such as AU-rich elements and cytoplasmic polyadenylation elements, but also sites for RNA binding proteins (RBPs). Disruption of the spatial arrangement of such sequences could reduce or eliminate its translation and keep local dAlk levels low, in effect mimicking its attenuation.
To test this possibility, we capitalized on flies carrying a P-element insertion in the 3′UTR of dAlk (AlkMB06458), which disrupts the spatial continuity of the putative translational control sequences. These mutants present increased responses to ethanol, are otherwise normal, and their total dAlk levels remain comparable with controls (Lasek et al., 2011). Consistent with these observations, we did not detect differences in the steady-state levels of dendritic dAlk in mutants and controls (Fig. 7D, unpaired) (ANOVA: F(3,53) = 18.2206, p < 0.0001; LSM unpaired w1118 vs unpaired mutant: p = 0.9219). However, after conditioning, the pairing-dependent dAlk increase in controls (LSM unpaired w1118 vs paired w1118: p = 2.3 × 10−7) was absent from the mutants (LSM unpaired mutant vs paired mutant: p = 0.5769). Therefore, in AlkMB06458 mutant homozygotes, MB dendritic levels of dAlk remain low even after conditioning (LSM paired w1118 vs paired mutant: p = 3.5 × 10−7), in effect mimicking its transgenic attenuation. Again, size and positioning cannot account for the effect, as the calycal areas quantified were identical (ANOVA: F(3,53) = 0.4121, p = 0.7450) in controls and mutants (Fig. 7E, right). These results suggest that the conditioning-dependent modulation of dAlk levels in MB dendrites is conferred by the spatial arrangement of control sequences in the 3′UTR of its mRNA. Notably, dAlk retained its dendritic localization in the mutants, suggesting that the insertion does not disrupt signals targeting the mRNA to the calyces (Fig. 7C).
The attenuated conditioning-dependent dAlk levels in MB calyces predict that the AlkMB mutants will present enhanced LTM. Indeed, 3 min memory (Fig. 7F; 3 US/CS) of controls, mutant heterozygotes, and homozygotes were indistinguishable (ANOVA: F(2,32) = 0.5853, p = 0.5632). In contrast, mutant homozygotes exhibited substantially enhanced 24 h memory (Fig. 7F; spaced: ANOVA: F(2,37) = 12.5306, p < 0.0001; subsequent LSM vs controls p = 3.9 × 10−5 and p = 7.8 × 10−4, respectively), which was PSD-LTM specific, as it was not elicited by massed training (Fig. 7F; massed: ANOVA F(2,31) = 0.1495, p = 0.8618). Task-relevant sensory responses were normal in the mutants (Fig. 7G) (reactivity to shock: ANOVA: F(1,21) = 0.4616, p = 0.5047; octanol avoidance: ANOVA: F(1,19) = 0.0846, p = 0.7745; benzaldehyde avoidance: ANOVA: F(1,19) = 1.7238, p = 0.2057).
To ascertain that the elevated PSD-LTM phenotype was consequent to disruption of the dAlk 3′UTR and not of a neighboring gene, we generated heteroallelics with the lethal (Lorén et al., 2003) null Alk1 allele (Fig. 7H). Significantly, Alk1/AlkMB heteroallelics presented a substantial LTM enhancement over that of either Alk1 or AlkMB heterozygotes (ANOVA: F(3,39) = 18.5255, p < 0.0001; subsequent LSM Alk1/AlkMB vs Alk1/+: p = 1.9 × 10−7; vs AlkMB06458/+: p = 4.7 × 10−7), confirming that the mutations are allelic. Moreover, PSD-LTM in the heteroallelics was even higher than that of AlkMB homozygotes (LSM Alk1/AlkMB06458 vs AlkMB06458: p = 0.0092) as hypothesized, given that Alk1 is a null allele and conditioning-dependent upregulation of dAlk levels is blocked in AlkMB. This is consistent with the notion that PSD-LTM formation is sensitive to the precise postconditioning levels of dAlk in MB dendrites.
Discussion
Our findings reveal a novel role for dAlk in regulation of PSD-LTM formation in addition to its established role in learning (Gouzi et al., 2011). dAlk constrains both processes, but whereas learning attenuation requires its activity outside the MBs, suppression of PSD-LTM formation requires its elevation within the dendrites of α/β MB neurons. Moreover, although its activating ligand Jeb is required for learning attenuation, it is dispensable for PSD-LTM constraint.
Global pharmacological inhibition of dAlk activity resulted in both STM and LTM enhancement as expected (Fig. 1) because it addressed all neurons expressing this RTK. dAlk activity outside the MBs is known to be required for learning/3 min memory suppression (Gouzi et al., 2011), and these neurons are clearly affected by TAE684, as also are the MBs yielding enhanced PSD-LTM. In contrast, dAlk levels were specifically abrogated within the MBs where converging studies (Isabel et al., 2004; Yu et al., 2006; Pavlopoulos et al., 2008; Bouzaiane et al., 2015) established that STM and LTM engage different MB neuron types, the γ and the α/β, respectively. dAlk attenuation in γ MB neurons did not affect 3 min memory (Gouzi et al., 2011) or PSD-LTM, strongly suggesting that dAlk is not expressed therein.
The notion of memory suppression almost invariably refers to forgetting, broadly defined as a decay of memory that either actively dissipates in time or undergoes interference by additional learning of unrelated or irrelevant information (Wixted, 2004; Dewar et al., 2007). Forgetting an odor/shock association in Drosophila (Davis and Zhong, 2017) requires the small G-protein Rac (Shuai et al., 2010), or dopamine (DA) signaling predominantly through the DAMB receptor (Berry et al., 2012), and its suppression appears as an enhancement of all types of 3–24 h memories. However, 24 h memory enhancement resulting from Rac attenuation appears distinct from PSD-LTM (Shuai et al., 2010), and inhibition of DA signaling in the MB-afferent DAN neurons does not enhance 16–24 h memories (Berry et al., 2012). Hence, Rac and/or DA signaling inhibit recently acquired labile memories rather than consolidated forms. dAlk also acts during the labile stage of memory formation, but not its dissipation, and is specific to PSD-LTM, not 3 h memory or ARM (Figs. 2B, 3A). Furthermore, dAlk is not required (Fig. 2A) within the essential for forgetting dopaminergic neurons (Davis and Zhong, 2017). Therefore, dAlk-mediated LTM inhibition is distinct from dissipation of labile memories.
A number of memory suppressor genes have been recently described in Drosophila (Walkinshaw et al., 2015) and mice (Lee and Silva, 2009), indicating that, although its exact role is unclear, memory restraint is evolutionarily conserved. Constraining memory may limit the conditioned associations processed toward the energetically demanding PSD-LTM (Plaçais et al., 2017), ensure the fidelity of associations that progress toward consolidation, or inhibit proactive or retroactive interference (Wixted, 2004; Dewar et al., 2007; Davis and Zhong, 2017).
The role of all apparent memory suppressor proteins and miRNAs identified to date in Drosophila (Busto et al., 2015; Walkinshaw et al., 2015) has not been fully delineated, but some mechanistic aspects emerge. Drosophila memory suppressor miRNAs ostensibly regulate translation of postsynaptic proteins involved in MB excitability (Li et al., 2013; Guven-Ozkan et al., 2016); hence, attenuation of their levels and the resultant hyperexcitability could underlie enhanced memory. Accordingly, loss of the apparent signal-tempering acetylcholine transporter DmSLC22A from MB neurons enhances their excitability and elevates memory (Gai et al., 2016). Interestingly, like dAlk, DmSLC22A is found in calycal microglomeruli (Gai et al., 2016). Therefore, in conjunction with our results, it appears that memory-constraining mechanisms depend on the level of postsynaptic proteins that limit the amplitude or duration of MB neuronal excitation. In contrast to other memory suppressor proteins and miRNAs (Busto et al., 2015, 2017; Walkinshaw et al., 2015), dAlk elevation in the MBs is not required for 3 h memory (Fig. 3) but appears specific to PSD-LTM. The temporal specificity of dAlk suggests that its activity may not constrain MB excitability, but rather LTM consolidation mechanisms, a hypothesis under investigation.
Conditioning-dependent dAlk elevation in MB dendrites appears to result via local translation regulated by the 3′UTR of its mRNA. This 3′UTR-conferred property is shared with multiple dendritic proteins (Steward and Schuman, 2001), including another RTK involved in memory formation, the BDNF receptor trkB (Nagappan and Lu, 2005). Similarly, 3′UTR sequences direct the mRNA of Drosophila CaMKII, a kinase also implicated in memory, to be translated in the postsynaptic zones of MB calyces (Ashraf et al., 2006). Significantly, the 3′UTR of dAlk mRNA contains more numerous regulatory elements than those on CaMKII transcripts, including several stabilizing AU-rich elements. The putative miRNA binding sites include those for miR-305 and miR-932, both implicated in memory formation (Cristino et al., 2014; Busto et al., 2015) and possibly in dAlk local translation. Translational regulation may also involve identified putative RBP binding sequences at the dAlk 3′UTR. Some, such as pumilio (pum) and the cytoplasmic polyadenylation element binding protein (CPEB) Orb2 (Dubnau et al., 2003; Keleman et al., 2007), are translation suppressors with known function in memory. Whether others, such as Rox8, the ortholog of the stress granule-associated vertebrate protein TIA1, play a role in memory formation is currently unknown. Whether miRNAs and RBPs interact with dAlk mRNA upon conditioning to regulate its dendritic levels will be the focus of forthcoming work.
The PSD-LTM constraint depends on dAlk activity and the increased levels per se, as demonstrated by the elevated memory upon treatment with the inhibitor TAE684. How is calycal dAlk activated to constrain LTM formation since Jeb is dispensable for LTM attenuation (Fig. 6A)? Presently, we cannot exclude the possibility that a yet unidentified ligand may activate dAlk upon spaced conditioning. However, another explanation we currently favor is that, upon spaced training, dAlk can autoactivate in response to its local elevation in the calyx. Level-dependent autoactivation has been reported for human ALK-positive cancers (Hallberg and Palmer, 2013), or neurons transfected with ALK (Moog-Lutz et al., 2005), a feature shared by almost all RTKs (Lemmon and Schlessinger, 2010). Local elevation-dependent autoactivation of dAlk is in agreement with our own experimental data that acute dAlk elevation attenuates LTM (Fig. 4B) and that conditioning elevates the endogenous protein in MB dendrites (Fig. 7). Moreover, dAlk autoactivation is consistent with the independence of dAlk-dependent PSD-LTM attenuation from Jeb (Fig. 6). The conditioning-dependent dAlk elevation and autoactivation in MB dendrites are likely considerably slower than acute activation by Jeb of extant dAlk outside the MBs required to constrain learning/3 min memory formation (Gouzi et al., 2011) (Fig. 6C). Furthermore, pan-neuronal elevation of Jeb left PSD-LTM unaffected (Fig. 6B), consistent with the notion that the two methods of dAlk activation, Jeb-dependent activation and autoactivation, are operant in spatially distinct neurons (outside and inside the MBs, respectively) and of distinct functional consequences.
Hence, we propose that conditioning results in local elevation of unliganded dAlk monomers in MB dendrites, raising the probability of encounter, lateral dimerization, autophosphorylation, and activation of the kinase domain at the postsynaptic plasma membrane. Unfortunately, an antibody specific to phosphorylated, hence activated, dAlk is not currently available; therefore, it is not possible to test this prediction in situ.
Downstream mechanisms engaged by dAlk to restrain LTM are still unknown. In our previous study (Gouzi et al., 2011), we described dAlk outside the MBs as an upstream activator of a dNf1-regulated Ras/ERK signaling pathway responsible for learning/STM attenuation. Interestingly, dAlk and dNf1 colocalize extensively in MB calyces (Gouzi et al., 2011), suggesting that they could also interact to mediate PSD-LTM attenuation. However, unlike for dAlk abrogation, dNf1 loss results in PSD-LTM deficits restored by reexpression of the protein in α/β MB neurons under c739-Gal4 (Buchanan and Davis, 2010). Therefore, although possible that dAlk and dNf1 interact within these neurons, they are likely antagonistic with respect to PSD-LTM formation, a process potentially engaging and requiring suppression of Ras signaling, a hypothesis currently under investigation.
In conclusion, we have identified dAlk as a specific negative regulator of PSD-LTM formation. Thus far, dAlk appears unique among RTKs in that it constrains LTM formation, possibly acting as a memory filter. The nature of the specific signals engaged by dAlk and the downstream PSD-LTM-constraining mechanisms remains yet to be elucidated in future work.
Footnotes
This work was supported by Fondation Santé. We thank Seth Tomchik (Scripps Research Institute) for sharing Gal4 strains; Ruth H. Palmer (University of Gothenburg) for fly stocks and reagents, and providing critical assistance in manuscript preparation; the Developmental Hybridoma Studies Bank for antibodies; the Bloomington Drosophila Stock Center and the Vienna Drosophila RNAi Center for resources; and Flybase for essential information.
We declare no competing financial interests.
References
- Akalal DB, Wilson CF, Zong L, Tanaka NK, Ito K, Davis RL (2006) Roles for Drosophila mushroom body neurons in olfactory learning and memory. Learn Mem 13:659–668. 10.1101/lm.221206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf SI, McLoon AL, Sclarsic SM, Kunes S (2006) Synaptic protein synthesis associated with memory is regulated by the RISC pathway in Drosophila. Cell 124:191–205. 10.1016/j.cell.2005.12.017 [DOI] [PubMed] [Google Scholar]
- Bai L, Sehgal A (2015) Anaplastic lymphoma kinase acts in the Drosophila mushroom body to negatively regulate sleep. PLoS Genet 11:e1005611. 10.1371/journal.pgen.1005611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry JA, Cervantes-Sandoval I, Nicholas EP, Davis RL (2012) Dopamine is required for learning and forgetting in Drosophila. Neuron 74:530–542. 10.1016/j.neuron.2012.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzaiane E, Trannoy S, Scheunemann L, Plaçais PY, Preat T (2015) Two independent mushroom body output circuits retrieve the six discrete components of Drosophila aversive memory. Cell Rep 11:1280–1292. 10.1016/j.celrep.2015.04.044 [DOI] [PubMed] [Google Scholar]
- Buchanan ME, Davis RL (2010) A distinct set of Drosophila brain neurons required for neurofibromatosis type 1-dependent learning and memory. J Neurosci 30:10135–10143. 10.1523/JNEUROSCI.0283-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busto GU, Guven-Ozkan T, Fulga TA, Van Vactor D, Davis RL (2015) microRNAs that promote or inhibit memory formation in Drosophila melanogaster. Genetics 200:569–580. 10.1534/genetics.114.169623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busto GU, Guven-Ozkan T, Davis RL (2017) MicroRNA function in Drosophila memory formation. Curr Opin Neurobiol 43:15–24. 10.1016/j.conb.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristino AS, Barchuk AR, Freitas FC, Narayanan RK, Biergans SD, Zhao Z, Simoes ZL, Reinhard J, Claudianos C (2014) Neuroligin-associated microRNA-932 targets actin and regulates memory in the honeybee. Nat Commun 5:5529. 10.1038/ncomms6529 [DOI] [PubMed] [Google Scholar]
- Davis RL, Zhong Y (2017) The biology of forgetting: a perspective. Neuron 95:490–503. 10.1016/j.neuron.2017.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Belle JS, Heisenberg M (1994) Associative odor learning in Drosophila abolished by chemical ablation of mushroom bodies. Science 263:692–695. 10.1126/science.8303280 [DOI] [PubMed] [Google Scholar]
- Dewar MT, Cowan N, Sala SD (2007) Forgetting due to retroactive interference: a fusion of Müller and Pilzecker's (1900) early insights into everyday forgetting and recent research on anterograde amnesia. Cortex 43:616–634. 10.1016/S0010-9452(08)70492-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubnau J, Chiang AS, Grady L, Barditch J, Gossweiler S, McNeil J, Smith P, Buldoc F, Scott R, Certa U, Broger C, Tully T (2003) The staufen/pumilio pathway is involved in Drosophila long-term memory. Curr Biol 13:286–296. 10.1016/S0960-9822(03)00064-2 [DOI] [PubMed] [Google Scholar]
- Englund C, Lorén CE, Grabbe C, Varshney GK, Deleuil F, Hallberg B, Palmer RH (2003) Jeb signals through the alk receptor tyrosine kinase to drive visceral muscle fusion. Nature 425:512–516. 10.1038/nature01950 [DOI] [PubMed] [Google Scholar]
- Gai Y, Liu Z, Cervantes-Sandoval I, Davis RL (2016) Drosophila SLC22A transporter is a memory suppressor gene that influences cholinergic neurotransmission to the mushroom bodies. Neuron 90:581–595. 10.1016/j.neuron.2016.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin AV, Melnick JS, Kim S, Hood TL, Li N, Li L, Xia G, Steensma R, Chopiuk G, Jiang J, Wan Y, Ding P, Liu Y, Sun F, Schultz PG, Gray NS, Warmuth M (2007) Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci U S A 104:270–275. 10.1073/pnas.0609412103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouzi JY, Moressis A, Walker JA, Apostolopoulou AA, Palmer RH, Bernards A, Skoulakis EM (2011) The receptor tyrosine kinase Alk controls neurofibromin functions in Drosophila growth and learning. PLoS Genet 7:e1002281. 10.1371/journal.pgen.1002281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J, Umapathy G, Yamazaki Y, Wolfstetter G, Mendoza P, Pfeifer K, Mohammed A, Hugosson F, Zhang H, Hsu AW, Halenbeck R, Hallberg B, Palmer RH (2015) FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. Elife 4:e09811. 10.7554/eLife.09811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guven-Ozkan T, Davis RL (2014) Functional neuroanatomy of Drosophila olfactory memory formation. Learn Mem 21:519–526. 10.1101/lm.034363.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guven-Ozkan T, Busto GU, Schutte SS, Cervantes-Sandoval I, O'Dowd DK, Davis RL (2016) MiR-980 is a memory suppressor MicroRNA that regulates the autism-susceptibility gene A2bp1. Cell Rep 14:1698–1709. 10.1016/j.celrep.2016.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallberg B, Palmer RH (2013) Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer 13:685–700. 10.1038/nrc3580 [DOI] [PubMed] [Google Scholar]
- Isabel G, Pascual A, Preat T (2004) Exclusive consolidated memory phases in Drosophila. Science 304:1024–1027. 10.1126/science.1094932 [DOI] [PubMed] [Google Scholar]
- Joiner WJ, Crocker A, White BH, Sehgal A (2006) Sleep in Drosophila is regulated by adult mushroom bodies. Nature 441:757–760. 10.1038/nature04811 [DOI] [PubMed] [Google Scholar]
- Keleman K, Krüttner S, Alenius M, Dickson BJ (2007) Function of the Drosophila CPEB protein Orb2 in long-term courtship memory. Nat Neurosci 10:1587–1593. 10.1038/nn1996 [DOI] [PubMed] [Google Scholar]
- King I, Tsai LT, Pflanz R, Voigt A, Lee S, Jäckle H, Lu B, Heberlein U (2011) Drosophila tao controls mushroom body development and ethanol-stimulated behavior through par-1. J Neurosci 31:1139–1148. 10.1523/JNEUROSCI.4416-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapek S, Sigrist S, Tanimoto H (2011) Bruchpilot, a synaptic active zone protein for anesthesia-resistant memory. J Neurosci 31:3453–3458. 10.1523/JNEUROSCI.2585-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasek AW, Lim J, Kliethermes CL, Berger KH, Joslyn G, Brush G, Xue L, Robertson M, Moore MS, Vranizan K, Morris SW, Schuckit MA, White RL, Heberlein U (2011) An evolutionary conserved role for anaplastic lymphoma kinase in behavioral responses to ethanol. PLoS One 6:e22636. 10.1371/journal.pone.0022636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Luo L (1999) Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22:451–461. 10.1016/S0896-6273(00)80701-1 [DOI] [PubMed] [Google Scholar]
- Lee YS, Silva AJ (2009) The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci 10:126–140. 10.1038/nrn2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiss F, Groh C, Butcher NJ, Meinertzhagen IA, Tavosanis G (2009) Synaptic organization in the adult Drosophila mushroom body calyx. J Comp Neurol 517:808–824. 10.1002/cne.22184 [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J (2010) Cell signaling by receptor tyrosine kinases. Cell 141:1117–1134. 10.1016/j.cell.2010.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20. 10.1016/j.cell.2004.12.035 [DOI] [PubMed] [Google Scholar]
- Li W, Cressy M, Qin H, Fulga T, Van Vactor D, Dubnau J (2013) MicroRNA-276a functions in ellipsoid body and mushroom body neurons for naive and conditioned olfactory avoidance in Drosophila. J Neurosci 33:5821–5833. 10.1523/JNEUROSCI.4004-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Krause WC, Davis RL (2007) GABAA receptor RDL inhibits Drosophila olfactory associative learning. Neuron 56:1090–1102. 10.1016/j.neuron.2007.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorén CE, Scully A, Grabbe C, Edeen PT, Thomas J, McKeown M, Hunter T, Palmer RH (2001) Identification and characterization of DAlk: a novel Drosophila melanogaster RTK which drives ERK activation in vivo. Genes Cells 6:531–544. 10.1046/j.1365-2443.2001.00440.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorén CE, Englund C, Grabbe C, Hallberg B, Hunter T, Palmer RH (2003) A crucial role for the anaplastic lymphoma kinase receptor tyrosine kinase in gut development in Drosophila melanogaster. EMBO Rep 4:781–786. 10.1038/sj.embor.embor897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire SE, Mao Z, Davis RL (2004) Spatiotemporal gene expression targeting with the TARGET and gene-switch systems in Drosophila. Sci STKE 2004:pl6. 10.1126/stke.2202004pI6 [DOI] [PubMed] [Google Scholar]
- Messaritou G, Leptourgidou F, Franco M, Skoulakis EM (2009) A third functional isoform enriched in mushroom body neurons is encoded by the Drosophila 14-3-3ζ gene. FEBS Lett 583:2934–2938. 10.1016/j.febslet.2009.08.003 [DOI] [PubMed] [Google Scholar]
- Moog-Lutz C, Degoutin J, Gouzi JY, Frobert Y, Brunet-de Carvalho N, Bureau J, Créminon C, Vigny M (2005) Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. J Biol Chem 280:26039–26048. 10.1074/jbc.M501972200 [DOI] [PubMed] [Google Scholar]
- Nagappan G, Lu B (2005) Activity-dependent modulation of the BDNF receptor TrkB: mechanisms and implications. Trends Neurosci 28:464–471. 10.1016/j.tins.2005.07.003 [DOI] [PubMed] [Google Scholar]
- Pavlopoulos E, Anezaki M, Skoulakis EM (2008) Neuralized is expressed in the alpha/beta lobes of adult Drosophila mushroom bodies and facilitates olfactory long-term memory formation. Proc Natl Acad Sci U S A 105:14674–14679. 10.1073/pnas.0801605105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz I, Kosti I, Ares M Jr, Cline M, Mandel-Gutfreund Y (2014) RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res 42:W361–W367. 10.1093/nar/gku406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman JL, McGill JJ, Keegan KP, Allada R (2006) A dynamic role for the mushroom bodies in promoting sleep in Drosophila. Nature 441:753–756. 10.1038/nature04739 [DOI] [PubMed] [Google Scholar]
- Plaçais PY, de Tredern É, Scheunemann L, Trannoy S, Goguel V, Han KA, Isabel G, Preat T (2017) Upregulated energy metabolism in the Drosophila mushroom body is the trigger for long-term memory. Nat Commun 8:15510. 10.1038/ncomms15510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reshetnyak AV, Murray PB, Shi X, Mo ES, Mohanty J, Tome F, Bai H, Gunel M, Lax I, Schlessinger J (2015) Augmentor α and β (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: hierarchy and specificity of ligand-receptor interactions. Proc Natl Acad Sci U S A 112:15862–15867. 10.1073/pnas.1520099112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuai Y, Lu B, Hu Y, Wang L, Sun K, Zhong Y (2010) Forgetting is regulated through rac activity in Drosophila. Cell 140:579–589. 10.1016/j.cell.2009.12.044 [DOI] [PubMed] [Google Scholar]
- Steward O, Schuman EM (2001) Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci 24:299–325. 10.1146/annurev.neuro.24.1.299 [DOI] [PubMed] [Google Scholar]
- Tully T, Preat T, Boynton SC, Del Vecchio M (1994) Genetic dissection of consolidated memory in Drosophila. Cell 79:35–47. 10.1016/0092-8674(94)90398-0 [DOI] [PubMed] [Google Scholar]
- Walkinshaw E, Gai Y, Farkas C, Richter D, Nicholas E, Keleman K, Davis RL (2015) Identification of genes that promote or inhibit olfactory memory formation in Drosophila. Genetics 199:1173–1182. 10.1534/genetics.114.173575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JB, Xue C, Benice T, Xue L, Morris SW, Raber J (2012) Anaplastic lymphoma kinase and leukocyte tyrosine kinase: functions and genetic interactions in learning, memory and adult neurogenesis. Pharmacol Biochem Behav 100:566–574. 10.1016/j.pbb.2011.10.024 [DOI] [PubMed] [Google Scholar]
- Wixted JT. (2004) The psychology and neuroscience of forgetting. Annu Rev Psychol 55:235–269. 10.1146/annurev.psych.55.090902.141555 [DOI] [PubMed] [Google Scholar]
- Yu D, Akalal DB, Davis RL (2006) Drosophila α/β mushroom body neurons form a branch-specific, long-term cellular memory trace after spaced olfactory conditioning. Neuron 52:845–855. 10.1016/j.neuron.2006.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]