

Abstract
The greatest unmet therapeutic need in Parkinson's disease (PD) is a treatment that slows the relentless progression of the symptoms and the neurodegenerative process. This review highlights the utility of genetics to understand the pathogenic mechanisms and develop novel therapeutic approaches for PD. The focus is on strategies provided by genetic studies: notably via the reduction and clearance of α-synuclein, inhibition of LRRK2 kinase activity, and modulation of glucocerebrosidase-related substrates. In addition, the critical role of precompetitive public-private partnerships in supporting trial design optimization, overall drug development, and regulatory approvals is illustrated. With these great advances, the promise of developing transformative therapies that halt or slow disease progression is a tangible goal.
Keywords: Parkinson's disease, alpha-synuclein, LRRK2, GBA, disease-modifying therapies
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disorder and primarily involves worsening motor impairments. In addition, patients experience nonmotor symptoms during the course of the disease, including constipation, anosmia, sleep disturbances, cognitive decline, anxiety, and depression. The motor symptoms can be initially managed with dopaminergic therapies; however, the disease continues to progress. Thus, PD patients are in urgent need of disease-modifying therapies that can slow or halt the relentless progression of the symptoms and the neurodegenerative process.
This review summarizes a symposium held at the 2018 Society for Neuroscience Annual Meeting and highlights novel advances in basic and translational research that impact our understanding of PD pathophysiology and the path to novel treatments for this devastating disease. Furthermore, it addresses how initial clinical, genetic, and epidemiological findings contribute to the understanding of disease pathophysiology and help develop proof-of-concept clinical trials to address the critical unmet medical needs.
Over the last two decades, considerable progress has been made in the identification of mutations that cause and/or alter the risk for PD. The utility of genetics in therapeutics relies primarily on leveraging findings to understand the pathogenesis of PD. This new knowledge has enabled the development of therapies that are aimed at halting or slowing the underlying disease process, rather than just ameliorating symptoms. Current advances in therapeutics aimed at reduction of α-synuclein (α-syn), inhibition of LRRK2 kinase activity, and modulation of glucocerebrosidase (GBA)-related glycosphingolipids are discussed (Fig. 1).
Figure 1.
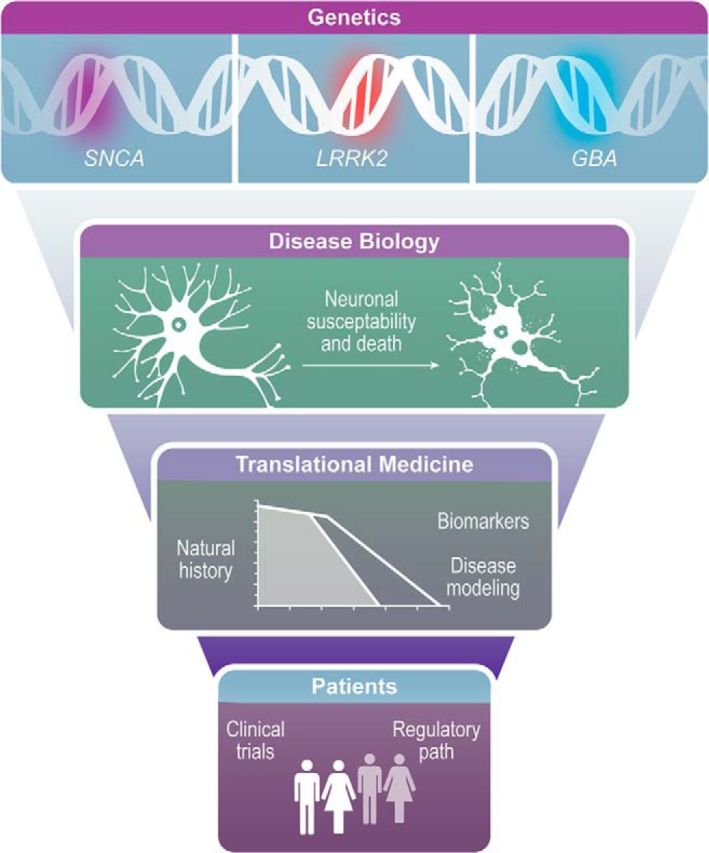
Targeted therapies for PD: from genetics to the clinic. Discovery of genes implicated in PD and subsequent understanding of their physiopathological roles provide the foundation for these genetic-based therapeutic approaches. Large natural history studies, novel biomarker discovery, and disease modeling can help design clinical trials targeting specific patient populations and suitable endpoints.
α-syn as therapeutic target in PD
Why target α-synuclein? α-syn is considered an important therapeutic target in PD for several reasons. First, aggregated α-syn is the major protein constituent of Lewy bodies and Lewy neurites, which are characteristic features of PD pathology. While this fact does not prove that the Lewy pathology is toxic, as opposed to an innocuous epiphenomenon or part of a protective response whereby the cell sequesters toxic misfolded protein, there is abundant evidence from experimental model systems indicating that α-syn aggregates trigger neuronal dysfunction and death. Second, point mutations in, and multiplications of, the α-syn gene lead to neurodegenerative conditions that display parkinsonism as a feature. Third, injections of α-syn fibrillar aggregates into animals trigger the misfolding of endogenous, natively unfolded α-syn with the spreading of α-syn pathology throughout the brain and progressive loss of nigral dopamine neurons in cases where the basal ganglia are targeted. This clearly demonstrates that α-syn can play a pivotal role in the gradual development of experimental neuropathology that mimics PD (Brundin et al., 2017; Sardi et al., 2018).
During the past decade, a range of experimental therapies that target α-syn in principally different ways have emerged. It is beyond the scope of this brief review to describe all current approaches, and more extensive accounts already exist in the literature (Brundin et al., 2017; Wong and Krainc, 2017). We will focus on the approaches that are either in clinical trials already or where clinical testing is imminent.
Because the simple overexpression of wild-type α-syn in neurons is associated with a heightened risk for α-syn aggregation and neurodegeneration, an approach gaining increased support currently focuses on reducing α-syn mRNA levels in PD. Building on the recent approval of antisense therapy for spinal muscular atrophy and the evidence of target engagement using antisense oligonucleotides to reduce the mutant huntingtin in Huntington disease (Wild and Tabrizi, 2017; Parente and Corti, 2018), there is an active program to develop antisense therapy targeting α-syn in PD (Cole et al., 2016). Another emerging approach to reduce α-syn expression uses β2-adrenergic agonists. The scientific premise for this approach is based on a large epidemiological study demonstrating reduced PD risk in individuals using β2-adrenergic agonists for asthma, coupled with a series of studies in cell cultures and animal models suggesting that β2-adrenergic agonists reduce α-syn mRNA expression through an epigenetic mechanism presumably involving Histone 3 lysine 27 acetylation (Mittal et al., 2017).
Another fundamentally different therapeutic approach is to reduce α-syn aggregation. The small-molecule NPT200–11 (or its chemical analog tool compound NPT100–18A) interferes with the interaction between α-syn and lipid membranes, which is deemed a crucial step in the oligomerization and further aggregation of α-syn. This molecule reduced behavioral and pathological aberrations in animal models of synucleinopathies (Wrasidlo et al., 2016), and a Phase I safety clinical trial was recently completed (www.ClinicalTrials.gov, Identifier NCT02606682).
Different forms of cellular autophagy, also involving lysosomal proteolysis, are considered major pathways through which α-syn is degraded by cells. There is abundant evidence from experimental models that mutations and pharmacological inhibitors that disrupt autophagy lead to accumulation and aggregation of α-syn. Conversely, genetic modifications and small molecules that enhance autophagy can reduce α-syn pathology. A class of inhibitors of the tyrosine kinase c-Abl that were developed as therapeutics in oncology have been found to reduce α-syn pathology in experimental models, and enhanced autophagy has been suggested to play a key role in mediating the beneficial effects (Karuppagounder et al., 2014). A safety trial with the c-Abl inhibitor Nilotinib in 11 patients with PD dementia and Lewy body dementia received widespread attention in 2016 (Pagan et al., 2016). While the trial was not designed or powered to detect efficacy, highly debated anecdotal reports suggested that patients improved dramatically (Wyse et al., 2016). In the wake of this trial, and a growing body of scientific evidence that c-Abl inhibitors are effective in experimental PD (Brahmachari et al., 2017), a series of Phase IIa trials are now underway in both PD and other synucleinopathies (www.ClinicalTrials.gov, Identifier NCT03205488).
One decade ago, the notion that α-syn could act as a prion-like protein was presented (Kordower et al., 2008; Li et al., 2008). The hypothesis posits that intraneuronal α-syn aggregates are secreted into the extracellular space, taken up by neighboring neurons, seed aggregation of endogenous, natively unfolded α-syn in the cells that they enter, and are transported between brain regions along axons. All of these features have been demonstrated in cell culture and experimental animal models. The concept of prion-like propagation of α-syn pathology can explain why Lewy pathology in PD appears to spread following a stereotypic pattern involving peripheral nerves and brain regions that are anatomically interconnected (Brundin and Melki, 2017). A key component of this model is that pathogenic α-syn is present in the extracellular space, which makes it amenable to targeting using antibody therapy. There are now several clinical programs that use either active (immunizing the patient with modified α-syn) or passive (injecting antibodies targeting α-syn at regular intervals) immunotherapy approaches to target different conformers of α-syn aggregates (www.ClinicalTrials.gov, Identifiers NCT03100149, NCT03318523, NCT03272165, and NCT02267434). These programs have been described in some detail in other articles, and it is beyond the scope of this mini-review to compare the different approaches (Sardi et al., 2018). However, it is notable that the first report of safety and target engagement using one of the passive immunotherapy approaches in patients with PD, assessed as a reduction of α-syn in blood, has now been published (Jankovic et al., 2018).
The number and diversity of experimental therapies targeting α-syn that are now entering the clinical arena are astounding. The scientific premise that α-syn plays a pivotal role in PD pathogenesis is strong, and the experimental data supporting the therapeutic strategies are often compelling. In light of this, there is cause for cautious optimism that one or more of the new strategies will eventually prove to be an effective disease-modifying therapy in PD. However, the path to a clinically proven therapy is going to be long, costly, and arduous.
LRRK2 as a therapeutic target in PD
The LRRK2 gene encodes for the enzyme leucine-rich repeat kinase 2 (LRRK2), a large protein kinase containing several conserved regions, including an armadillo repeat (ARM) region, an ankyrin repeat (ANK) region, a leucine-rich repeat (LRR) domain, a kinase domain, a RAS domain, a GTPase domain, and a WD40 domain. LRRK2 interacts with many key proteins implicated in PD, suggesting that LRRK2 may be a central player in the pathways underlying disease pathogenesis (Cookson, 2015).
Although missense mutations in LRRK2 are the most common cause of autosomal dominant PD, the LRRK2 locus also contains a polymorphic risk factor for idiopathic PD (Simón-Sánchez et al., 2009). Interestingly, genome-wide association studies implicated LRRK2 as a major susceptibility gene in chronic inflammatory bowel diseases (Barrett et al., 2008). These results prompted a growing body of research suggesting a role for LRRK2 in the regulation of chronic inflammatory responses in PD (Cook et al., 2017; Hui et al., 2018).
While it is generally believed that pathogenic mutations in LRRK2 confer a toxic gain of function, and increased LRRK2 kinase activity has been strongly implicated in pathogenesis (Greggio et al., 2006), the relative activation state of wild-type LRRK2 in idiopathic PD has largely been unknown. Nevertheless, there are hints that, independent of mutations, LRRK2 may play a role in this more common form of the disease. For example, genetic ablation of endogenous wild-type LRRK2 or pharmacological inhibition of its kinase activity in rats protects the nigrostriatal system from neurodegeneration caused by AAV2-mediated α-syn overexpression (Daher et al., 2014, 2015). Implicit in the interpretation of these results is the assumption that endogenous LRRK2 kinase activity must play a pathogenic role in the neurodegeneration caused by elevated levels of nigrostriatal α-syn. It has been somewhat difficult, however, to measure LRRK2 activity or its physiological regulation. Because LRRK2 is a large (288 kDa) multidomain protein that is expressed at relatively low levels, conventional assays often rely on immunoprecipitation, and they typically use artificial substrates or assess kinase autophosphorylation by autoradiography (Lee et al., 2012). As such, it has been difficult to examine the activity of LRRK2 in specific regions or cell types in the brain.
A novel approach has recently been developed to circumvent some of these limitations (Di Maio et al., 2018). This approach is based on the proximity ligation (PL) technology that allow direct detection of proteins, protein interactions, and modifications with high specificity and sensitivity. There is a growing consensus that autophosphorylation of LRRK2 at serine 1292 (pSer1292) correlates with kinase activity (Sheng et al., 2012). Therefore, the authors developed a PL assay using an antibody that recognizes pSer1292 and another that recognizes an epitope in the C-terminal domain of the protein. Only when the 2 antibodies both bind specifically to their epitopes on LRRK2 is a strong PL signal generated. In this way, off-target binding is filtered out and specific binding is amplified. Additionally, the pSer1292 PL assay can be coupled to quantitative confocal immunofluorescence measurement of the phosphorylation state of a LRRK2 substrate, the Rab GTPase, Rab10, using an antibody against pThr73-Rab10, which has independently been suggested as a surrogate index of LRRK2 activity (Thirstrup et al., 2017). Furthermore, because LRRK2 binds to 14–3-3 protein when it is in an inactive state; the authors also developed a second PL assay to measure the interaction of LRRK2 with 14–3-3. Thus, LRRK2 activity is associated with (1) strong pSer1292 PL signal, (2) robust pThr73-Rab10 immunofluorescence signal, and (3) loss of the LRRK2:14–3-3 PL signal. Conversely, low LRRK2 kinase activity is defined by strong LRRK2:14–3-3 PL signal and loss of the pSer1292 PL and pThr73-Rab10 signals. The assays were validated using CRISPR/cas9-edited cells and pharmacological kinase inhibitors. These assays demonstrated excellent cellular and subcellular resolution, enabling assessment of LRRK2 activity in specific cell types under various physiological conditions.
When the assays were applied to sections of substantia nigra from idiopathic PD brains, there was a marked activation of LRRK2 in dopamine neurons as shown by strong pSer1292 PL and pThr73-Rab10 signals and an absence of LRRK2:14–3-3 PL (Di Maio et al., 2018). In contrast, control brains were marked by strong LRRK2:14–3-3 signal and very little pSer1292 PL or pThr73-Rab10 immunofluorescence. Thus, these results support the contention that endogenous wild-type LRRK2 is activated in the nigrostriatal system in idiopathic PD.
Interestingly, the nigrostriatal activation of LRRK2 was reproduced in animal models of disease by systemic administration of the mitochondrial toxin, rotenone, and by AAV2-mediated overexpression of α-syn. Each of these models impacts mitochondrial function and leads to generation of reactive oxygen species, raising the possibility that LRRK2 may be activated by oxidative mechanisms. Indeed, treatment of cells with physiological concentrations of H2O2 caused LRRK2 activation and phosphorylation of Rab10, and both effects were blocked by an antioxidant (Di Maio et al., 2018).
In vivo experiments showed that rotenone-induced nigrostriatal activation of LRRK2 and phosphorylation of its substrate, Rab10, could be blocked by treatment with a brain-penetrant LRRK2 kinase inhibitor (Di Maio et al., 2018). Rotenone-induced accumulation of pSer129-α-syn was also blocked by the LRRK2 inhibitor. Because there is evidence that phosphorylation of α-syn may target it for autophagic degradation, this accumulation may suggest a problem with autophagy after rotenone exposure. Indeed, rotenone causes a marked loss of the lysosomal markers LAMP1 and LAMP2A, both of which were preserved by treatment with a LRRK2 kinase inhibitor (Di Maio et al., 2018).
In summary, these studies suggest that, independent of mutations, wild-type endogenous LRRK2 is activated by oxidative mechanisms in idiopathic PD and that it plays a pathogenic role in PD-associated neurodegeneration, likely by causing endolysosomal dysfunction. Treatment with a LRRK2 kinase inhibitor preserved endolysosomal markers and prevented the accumulation pSer129-α-syn. In this context, there is a strong rationale for the use of LRRK2 therapeutics, not just in mutation carriers, but in those with idiopathic disease. Several LRRK2 inhibitors are being pursued preclinically and in healthy human volunteers to assess drug dynamics, brain penetrance, safety, and tolerability (Alessi and Sammler, 2018).
Glucocerebrosidase as a therapeutic target in PD
Heterozygous mutations in the glucocerebrosidase gene (GBA) are the most common genetic risk factor for PD. Their discovery originated from astute observation of parkinsonsim in genetics clinics during the examination of patients with a rare lysosomal storage disease, Gaucher disease (Neudorfer et al., 1996). This initial suggestion was validated by a large collaborative group that analyzed GBA mutations in a large cohort by sequencing the entire coding region (Sidransky et al., 2009). Numerous genetic studies have substantiated the increased frequency of mutations in GBA in patients with PD and dementia with Lewy bodies (DLB). Heterozygous carriers of GBA mutations have an increased frequency of PD, and ∼7%-10% of sporadic PD patients carry mutations in GBA, which confirm mutations in this gene as the most common genetic predisposing risk factor for PD identified to date. It is worth noticing that, while 7%–10% of PD patients carry a GBA mutation, most of the GBA mutation carriers will not develop the disease, suggesting the presence of additional genetic modifiers (Beavan and Schapira, 2013).
GBA-associated PD is clinically indistinguishable from idiopathic PD; however, the presence of GBA mutations accelerates the disease course and conveys a higher risk for nonmotor symptoms (Cilia et al., 2016; Davis et al., 2016; Liu et al., 2016). Dementia is a major complication in the late stages of PD that greatly affects quality of life and survival. GBA mutation carriers exhibit an earlier and more rapid cognitive decline compared with noncarriers. The increased risk for dementia in GBA-related PD is consistent with earlier genetic studies demonstrating increased frequency of GBA mutations in patients with PD dementia (PDD) and DLB (Setó-Salvia et al., 2012; Nalls et al., 2013).
The precise mechanisms of by which GBA mutations predispose to synucleinopathies and affect disease progression are still undefined. The inverse relationship between glucocerebrosidase activity and oligomeric α-syn led to the proposal of a pathogenic feedback loop (Mazzulli et al., 2011). This proposal is supported by a growing body of epidemiological, clinical, and basic science studies (Sardi et al., 2015; Aflaki et al., 2017). Decreased glucocerebrosidase activity by heterozygous GBA mutations can alter glycosphingolipid homeostasis and membrane composition. This sphingolipid membrane imbalance can result in compromised cellular function, including vesicular transport, lysosomal/endosomal dysfunction, α-syn aggregation, and selective neuronal susceptibility (Sardi et al., 2015; Galvagnion, 2017; Kim et al., 2018; Zunke et al., 2018).
Current therapeutic approaches for GBA-related PD are founded on the premise that GBA-mediated loss of function causes an abnormal glycosphingolipid environment which impairs cellular protein homeostasis (proteinopathy) and neuronal function. In animal models of disease, decreased glucocerebrosidase activity results in progressive CNS accumulation of α-syn/ubiquitin/tau aggregates and associated cognitive and motor deficits. These pathological and behavioral aberrations can be ameliorated (and even reversed) by adeno-associated viral overexpression of human glucocerebrosidase in the CNS, which could act by restoring membrane glycosphingolipid balance (Sardi et al., 2011, 2013; Rocha et al., 2015; Rockenstein et al., 2016). Based on these results, several groups are currently developing gene therapy and small-molecule approaches to increase glucocerebrosidase activity in the CNS.
The use of brain penetrant small molecules has been proposed to overcome the limited distribution of gene therapy approaches (Sardi et al., 2015). Brain penetrant, small molecular chaperones capable of increasing lysosomal glucocerebrosidase activity are being developed for PD and related synucleinopathies. Two novel noninhibitory chaperones (NCGC758 and NCGC607) reduced lipid substrate accumulation, increased glucocerebrosidase lysosomal translocation and activity, and reversed α-syn accumulation in human dopaminergic neurons from patients with Gaucher disease or PD (Aflaki et al., 2016; Mazzulli et al., 2016). Another chaperone being investigated for PD is ambroxol, a European Medicines Agency-approved mucolytic. At high concentrations, ambroxol reportedly displayed glucocerebrosidase chaperone activity improving lysosomal transport and reducing α-syn and S129-phosphorylated α-syn protein levels in mouse models of synucleinopathy (Migdalska-Richards et al., 2016). Based on these results, the safety, tolerability, and efficacy of ambroxol in PD is currently being tested in Canada and the United Kingdom (www.ClinicalTrials.gov, Identifiers NCT02941822 and NCT02914366).
An alternative approach to stabilize the abnormal glycosphingolipid accumulation is the antagonism of glucosylceramide synthase (GCS). This approach is referred to as substrate reduction therapy and does not target the mutant enzyme. Substrate reduction therapy offsets the buildup of lipid substrates, equilibrating biosynthesis with the reduced breakdown due to GBA mutations. GCS inhibitors restored glycosphingolipid balance and diminished α-syn pathological conformers in PD patient neurons with and without GBA mutations (Kim et al., 2018; Zunke et al., 2018). Importantly, a novel brain-penetrant GCS inhibitor improved α-syn processing and behavioral outcomes in mouse models of GBA-related synucleinopathy and α-syn overexpression (Sardi et al., 2017). These results have prompted the initiation of a multinational double-blind, placebo-controlled study to assess the efficacy and safety of a GCS inhibitor in PD patients carrying a GBA mutation (www.ClinicalTrials.gov, Identifier NCT02906020).
Maintaining glucocerebrosidase activity and/or modulation of glycosphingolipid levels are potential targets for certain forms of sporadic PD. Similarly to patients carrying GBA mutations, PD patients carrying nonmutated GBA alleles present decreased glucocerebrosidase in brain, CSF, and blood (Gegg et al., 2012; Parnetti et al., 2014; Alcalay et al., 2015), suggesting a role for GBA-related mechanisms in sporadic disease. It is conceivable that perturbations in glycosphingolipid homeostasis might result in exacerbation of the disease process. Further studies to better define patient populations that would benefit from these interventions are ongoing.
Critical path for Parkinson's: increasing efficiency, safety, and speed in clinical trials
The current landscape of drug development in PD is rapidly evolving with numerous novel therapeutics holding great promise for patients. This exciting pipeline is being fueled by an increased understanding of the underlying pathogenic mechanisms and by innovative technology developments (Espay et al., 2016; Sardi et al., 2018), which are paving the way to identifying new treatments with a vision for personalized medicines (Espay et al., 2017; Kingwell, 2017). Thus, PD therapeutic development is truly at a transformative time when the hope for halting or slowing disease progression is a tangible goal.
Despite this sense of hope and optimism, most therapeutic candidates will not be approved (Kesselheim et al., 2015). Trials aimed at neuroprotection in PD have provided extensive lessons over >15 years, with no treatment gaining successful regulatory approval for disease modification (Stoessl, 2017; Lang and Espay, 2018). The challenges for drug developers include the following: unpredictable placebo response, absence of biomarkers to enable go/no go decisions through stages of drug development, lack of understanding of disease progression (particularly in nonmotor manifestations of the disease), lack of diagnostic tools, and need for outcome measures that objectively measure clinically meaningful changes. Despite such common themes, it has not been possible to capitalize on failed clinical trials beyond conceptual learning. There is increased recognition of the value of open science and data sharing initiatives (Open Science Collaboration, 2015; Frasier, 2016), which will be key to developing disease-modifying therapies for slowly progressing and highly variable diseases, such as PD (Mursaleen et al., 2017).
Public-private partnerships (PPPs) provide a collaborative platform to advance goals that benefit all stakeholders. Shared goals for PPPs focus on enhancing consistency and scientific underpinnings of medical product development to decrease the time, risk, and cost associated with bringing new products to patients. There are a growing number of precompetitive consortia focused on PD. These include initiatives led by both government and advocacy organizations including Parkinson's Progression Marker Initiative (2011), Parkinson's Disease Biomarkers Program (Rosenthal et al., 2016), Innovative Medicines Initiative (IMI) (Hofmann-Apitius et al., 2015), Joint Programme Neurodegenerative Disease (JPND) (Lerche et al., 2015; Heinzel et al., 2017), International Parkinsonism Genetics Network (http://pdgenetics.org/), and more recently Accelerating Medicines Partnership-Parkinson's Disease (https://www.nih.gov/research-training/accelerating-medicines-partnership-amp/parkinsons-disease). Such efforts commonly focus on biomarker discovery and natural history studies with the goal of identifying novel biomarkers as well as novel risk genes (Chen-Plotkin et al., 2018).
Despite the impressive progress within individual consortia, more can be gained by integrating diverse data collected across different cohorts and clinical trials. The Critical Path Institute, a nonprofit PPP, orchestrates the sharing of data, expertise, and knowledge among industry, regulatory authorities, government, patient advocacy groups, and academia in the precompetitive space to improve the drug development pathway (Brumfield, 2014; Parekh et al., 2015; Maxfield et al., 2017).
The Critical Path for Parkinson's (CPP) consortium was launched in 2015 as a precompetitive initiative aimed at sharing resources and data and advancing novel drug development tools for regulatory endorsement (Stephenson et al., 2015). The goal was to replicate the success of Critical Path Institute's Alzheimer's disease consortium, including the following: development of open access consensus data standards (Neville et al., 2017), integrating data from global sources to produce a clinical trial placebo database (Neville et al., 2015), and achieving regulatory acceptance of biomarkers and disease progression modeling tools (Hill et al., 2014; Romero et al., 2015; Stoessl, 2017).
CPP is unique compared with other precompetitive initiatives in that its focus is on data-driven strategies to advance regulatory science; representatives from health authority agencies are participants in the consortium's activities to assure alignment. CPP acquires and integrates patient-level data from around the world. These data from ongoing, and completed PD trials and cohorts can offer key insights to guide and improve development of new therapies. The deep reservoir of organized data serves as the platform to formally bring the data to regulatory agencies for review. A key aim is to develop quantitative model-based tools based on patient-level data from cohorts and clinical trials to ensure confidence that the clinical trial designs are appropriate. Qualified biomarkers and regulatory-endorsed tools are made publicly available to benefit the community and to improve trial efficiency in the future.
Understanding the natural progression of PD is critical for research and therapeutic development. Thus, great focus has been placed on advancing genetically defined targets, biomarker-driven phenotypes, precision medicine strategies, and early diagnosis (Fereshtehnejad et al., 2017; Jennings et al., 2017; Postuma et al., 2017). Global databases consisting of integrated data are needed to define disease progression across the spectrum of such a heterogeneous disease. Multiple cohort studies have been designed globally which hold the potential to identify PD subtypes and factors influencing the disease course.
In <2 years, CPP has created a large, pooled database of deidentified, patient-level data that describes the disease progression of PD patients (>5000 individual patient records) beginning at the onset of motor symptoms. CPP is advancing PD modeling tools in formal paths set in place with FDA and European Medicines Agency to advance drug development with direct input from the regulatory authorities. Recently, CPP has also achieved letters of support from the FDA (FDA, 2018) and European Medicines Agency (European Medicines Agency, 2018) for the use of dopamine transporter imaging as an exploratory enrichment biomarker in PD clinical trials targeting early motor stages of the disease. CPP continues to develop modeling and simulation tools for use in Phase II and III PD clinical trials.
In conclusion, there is an urgent need for effective treatments to slow disease progression in PD. Two decades of intense basic and clinical research are launching a variety of novel therapeutics into the clinical arena. Recent advances in biomarkers, digital technology, and disease modeling are also prompting a great deal of optimism. This next generation of PD clinical studies incorporates genetic and biomarker information to define suitable patient populations and trial endpoints for disease progression. Despite this optimistic outlook, it is important to underscore that this is just the beginning of a long journey; there are many challenges ahead. The development of PD therapeutics is particularly difficult because of the slow progression of the disease, the variability in clinical course between patients, and the lack of validated biomarkers that reflect brain pathology. Therefore, it is imperative to continue to accrue clinical and observational data, discover additional molecular and biomarker endpoints, validate emerging technologies, and develop global collaborations to enable more efficient clinical trials. We remain optimistic that this next decade will witness great advances in the treatment for PD.
Footnotes
Critical Path for Parkinson's was supported by Parkinson's UK and industry member organizations, including Pfizer, AbbVie, Takeda, GSK, Merck, Denali, Sanofi, GE Healthcare, UCB, and Biogen. We thank Alison E. Schoeder (Sanofi Biomedical Media Services) for figure artwork; Kasey Jackson for critical reading; and Critical Path for Parkinson's members and funders.
L.S.S. and S.P.S are employees and stockholders of Sanofi. P.B. has received commercial support as a consultant from Renovo Neural, Inc., Roche, Living Cell Technologies, Teva Inc., Lundbeck A/S, AbbVie, Neuroderm, Fujifilm-Cellular Dynamics, ClearView Healthcare, FCB Health, IOS Press Partners, and Capital Technologies, Inc. P.B. has received commercial support for grants/research from Roche, Renovo, and Teva/Lundbeck. P.B. has ownership interests in Acousort AB and is on the steering committee of the NILO-PD trial. J.T.G. has received research grants from Merck and Biogen. J.T.G. was also an advisory board member at Pfizer. D.S. is an employee of the Critical Path Institute.
References
- Aflaki E, Borger DK, Moaven N, Stubblefield BK, Rogers SA, Patnaik S, Schoenen FJ, Westbroek W, Zheng W, Sullivan P, Fujiwara H, Sidhu R, Khaliq ZM, Lopez GJ, Goldstein DS, Ory DS, Marugan J, Sidransky E (2016) A new glucocerebrosidase chaperone reduces alpha-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and parkinsonism. J Neurosci 36:7441–7452. 10.1523/JNEUROSCI.0636-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aflaki E, Westbroek W, Sidransky E (2017) The complicated relationship between Gaucher disease and parkinsonism: insights from a rare disease. Neuron 93:737–746. 10.1016/j.neuron.2017.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, Mazzoni P, Pauciulo MW, Nichols WC, Gan-Or Z, Rouleau GA, Chung WK, Wolf P, Oliva P, Keutzer J, Marder K, Zhang X (2015) Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain 138:2648–2658. 10.1093/brain/awv179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Sammler E (2018) LRRK2 kinase in Parkinson's disease. Science 360:36–37. 10.1126/science.aar5683 [DOI] [PubMed] [Google Scholar]
- Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, et al. (2008) Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet 40:955–962. 10.1038/ng.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beavan MS, Schapira AH (2013) Glucocerebrosidase mutations and the pathogenesis of Parkinson disease. Ann Med 45:511–521. 10.3109/07853890.2013.849003 [DOI] [PubMed] [Google Scholar]
- Brahmachari S, Karuppagounder SS, Ge P, Lee S, Dawson VL, Dawson TM, Ko HS (2017) c-abl and Parkinson's disease: mechanisms and therapeutic potential. J Parkinsons Dis 7:589–601. 10.3233/JPD-171191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumfield M. (2014) The Critical Path Institute: transforming competitors into collaborators. Nat Rev Drug Discov 13:785–786. 10.1038/nrd4436 [DOI] [PubMed] [Google Scholar]
- Brundin P, Melki R (2017) Prying into the prion hypothesis for Parkinson's disease. J Neurosci 37:9808–9818. 10.1523/JNEUROSCI.1788-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundin P, Dave KD, Kordower JH (2017) Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol 298:225–235. 10.1016/j.expneurol.2017.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Albin R, Alcalay R, Babcock D, Bajaj V, Bowman D, Buko A, Cedarbaum J, Chelsky D, Cookson MR, Dawson TM, Dewey R, Foroud T, Frasier M, German D, Gwinn K, Huang X, Kopil C, Kremer T, Lasch S, et al. (2018) Finding useful biomarkers for Parkinson's disease. Sci Transl Med 10:eaam6003. 10.1126/scitranslmed.aam6003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cilia R, Tunesi S, Marotta G, Cereda E, Siri C, Tesei S, Zecchinelli AL, Canesi M, Mariani CB, Meucci N, Sacilotto G, Zini M, Barichella M, Magnani C, Duga S, Asselta R, Soldà G, Seresini A, Seia M, Pezzoli G, et al. (2016) Survival and dementia in GBA-associated Parkinson disease: the mutation matters. Ann Neurol 80:662–673. 10.1002/ana.24777 [DOI] [PubMed] [Google Scholar]
- Cole T, Paumier K, Zhao H, Weihofen A, Kordasiewicz HB, Swayze EE (2016) Snca targeted antisense oligonucleotides mediate progression of pathological deposition in alpha synuclein rodent transmission models of Parkinson's disease. Neurology 86 [Suppl 16]:P6.239. [Google Scholar]
- Cook DA, Kannarkat GT, Cintron AF, Butkovich LM, Fraser KB, Chang J, Grigoryan N, Factor SA, West AB, Boss JM, Tansey MG (2017) LRRK2 levels in immune cells are increased in Parkinson's disease. NPJ Parkinsons Dis 3:11. 10.1038/s41531-017-0010-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR. (2015) LRRK2 pathways leading to neurodegeneration. Curr Neurol Neurosci Rep 15:42. 10.1007/s11910-015-0564-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher JP, Volpicelli-Daley LA, Blackburn JP, Moehle MS, West AB (2014) Abrogation of alpha-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc Natl Acad Sci U S A 111:9289–9294. 10.1073/pnas.1403215111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher JP, Abdelmotilib HA, Hu X, Volpicelli-Daley LA, Moehle MS, Fraser KB, Needle E, Chen Y, Steyn SJ, Galatsis P, Hirst WD, West AB (2015) Leucine-rich repeat kinase 2 (LRRK2) pharmacological inhibition abates alpha-synuclein gene-induced neurodegeneration. J Biol Chem 290:19433–19444. 10.1074/jbc.M115.660001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MY, Johnson CO, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, Van Deerlin VM, Quinn JF, Chung KA, Peterson-Hiller AL, Rosenthal LS, Dawson TM, Albert MS, Goldman JG, Stebbins GT, Bernard B, Wszolek ZK, Ross OA, Dickson DW, Eidelberg D,et al. (2016) Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol 73:1217–1224. 10.1001/jamaneurol.2016.2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maio R, Hoffman EK, Rocha EM, Keeney MT, Sanders LH, De Miranda BR, Zharikov A, Van Laar A, Stepan AF, Lanz TA, Kofler JK, Burton EA, Alessi DR, Hastings TG,Greenamyre JT (2018) Activation of LRRK2 in idiopathic Parkinson disease. Sci Transl Med 10:eaar5429. 10.1126/scitranslmed.aar5429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espay AJ, Bonato P, Nahab FB, Maetzler W, Dean JM, Klucken J, Eskofier BM, Merola A, Horak F, Lang AE, Reilmann R, Giuffrida J, Nieuwboer A, Horne M, Little MA, Litvan I, Simuni T, Dorsey ER, Burack MA, Kubota K, et al. (2016) Technology in Parkinson's disease: challenges and opportunities. Mov Disord 31:1272–1282. 10.1002/mds.26642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espay AJ, Brundin P, Lang AE (2017) Precision medicine for disease modification in Parkinson disease. Nat Rev Neurol 13:119–126. 10.1038/nrneurol.2016.196 [DOI] [PubMed] [Google Scholar]
- European Medicines Agency (2018) Qualification opinion on dopamine transporter imaging as an enrichment biomarker for Parkinson's disease clinical trials in patients with early Parkinsonian symptoms. https://www.ema.europa.eu/documents/regulatory-procedural-guideline/qualification-opinion-dopamine-transporter-imaging-enrichment-biomarker-parkinsons-disease-clinical_en.pdf.
- FDA (2018) Biomarker letter of support. https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/BiomarkerQualificationProgram/UCM605357.pdf.
- Fereshtehnejad SM, Montplaisir JY, Pelletier A, Gagnon JF, Berg D, Postuma RB (2017) Validation of the MDS research criteria for prodromal Parkinson's disease: longitudinal assessment in a REM sleep behavior disorder (RBD) cohort. Mov Disord 32:865–873. 10.1002/mds.26989 [DOI] [PubMed] [Google Scholar]
- Frasier M. (2016) Perspective: data sharing for discovery. Nature 538:S4. 10.1038/538S4a [DOI] [PubMed] [Google Scholar]
- Galvagnion C. (2017) The role of lipids interacting with alpha-synuclein in the pathogenesis of Parkinson's disease. J Parkinsons Dis 7:433–450. 10.3233/JPD-171103 [DOI] [PubMed] [Google Scholar]
- Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW, Schapira AH (2012) Glucocerebrosidase deficiency in substantia nigra of Parkinson disease brains. Ann Neurol 72:455–463. 10.1002/ana.23614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis 23:329–341. 10.1016/j.nbd.2006.04.001 [DOI] [PubMed] [Google Scholar]
- Heinzel S, Lerche S, Maetzler W, Berg D (2017) Global, yet incomplete overview of cohort studies in Parkinson's disease. J Parkinsons Dis 7:423–432. 10.3233/JPD-171100 [DOI] [PubMed] [Google Scholar]
- Hill DL, Schwarz AJ, Isaac M, Pani L, Vamvakas S, Hemmings R, Carrillo MC, Yu P, Sun J, Beckett L, Boccardi M, Brewer J, Brumfield M, Cantillon M, Cole PE, Fox N, Frisoni GB, Jack C, Kelleher T, Luo F, et al. (2014) Coalition Against Major Diseases/European Medicines Agency biomarker qualification of hippocampal volume for enrichment of clinical trials in predementia stages of Alzheimer's disease. Alzheimers Dement 10:421–429.e3. 10.1016/j.jalz.2013.07.003 [DOI] [PubMed] [Google Scholar]
- Hofmann-Apitius M, Ball G, Gebel S, Bagewadi S, de Bono B, Schneider R, Page M, Kodamullil AT, Younesi E, Ebeling C, Tegnér J,Canard L (2015) Bioinformatics mining and modeling methods for the identification of disease mechanisms in neurodegenerative disorders. Int J Mol Sci 16:29179–29206. 10.3390/ijms161226148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui KY, Fernandez-Hernandez H, Hu J, Schaffner A, Pankratz N, Hsu NY, Chuang LS, Carmi S, Villaverde N, Li X, Rivas M, Levine AP, Bao X, Labrias PR, Haritunians T, Ruane D, Gettler K, Chen E, Li D, Schiff ER, et al. (2018) Functional variants in the LRRK2 gene confer shared effects on risk for Crohn's disease and Parkinson's disease. Sci Transl Med 10:eaai7795. 10.1126/scitranslmed.aai7795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic J, Goodman I, Safirstein B, Marmon TK, Schenk DB, Koller M, Zago W, Ness DK, Griffith SG, Grundman M, Soto J, Ostrowitzki S, Boess FG, Martin-Facklam M, Quinn JF, Isaacson SH, Omidvar O, Ellenbogen A, Kinney GG (2018) Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-alpha-synuclein monoclonal antibody, in patients with Parkinson disease: a randomized clinical trial. JAMA Neurol. Advance online publication. Retrieved Jun. 18, 2018. doi: 10.1001/jamaneurol.2018.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings D, Siderowf A, Stern M, Seibyl J, Eberly S, Oakes D, Marek K (2017) Conversion to Parkinson disease in the PARS hyposmic and dopamine transporter-deficit prodromal cohort. JAMA Neurol 74:933–940. 10.1001/jamaneurol.2017.0985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuppagounder SS, Brahmachari S, Lee Y, Dawson VL, Dawson TM, Ko HS (2014) The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson's disease. Sci Rep 4:4874. 10.1038/srep04874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesselheim AS, Hwang TJ, Franklin JM (2015) Two decades of new drug development for central nervous system disorders. Nat Rev Drug Discov 14:815–816. 10.1038/nrd4793 [DOI] [PubMed] [Google Scholar]
- Kim S, Yun SP, Lee S, Umanah GE, Bandaru VV, Yin X, Rhee P, Karuppagounder SS, Kwon SH, Lee H, Mao X, Kim D, Pandey A, Lee G, Dawson VL, Dawson TM, Ko HS (2018) GBA1 deficiency negatively affects physiological alpha-synuclein tetramers and related multimers. Proc Natl Acad Sci U S A 115:798–803. 10.1073/pnas.1700465115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingwell K. (2017) Zeroing in on neurodegenerative alpha-synuclein. Nat Rev Drug Discov 16:371–373. 10.1038/nrd.2017.95 [DOI] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med 14:504–506. 10.1038/nm1747 [DOI] [PubMed] [Google Scholar]
- Lang AE, Espay AJ (2018) Disease modification in Parkinson's disease: current approaches, challenges, and future considerations. Mov Disord 33:660–677. 10.1002/mds.27360 [DOI] [PubMed] [Google Scholar]
- Lee BD, Li X, Dawson TM, Dawson VL (2012) Measuring the activity of leucine-rich repeat kinase 2: a kinase involved in Parkinson's disease. Methods Mol Biol 795:45–54. 10.1007/978-1-61779-337-0_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerche S, Liepelt-Scarfone I, Alves G, Barone P, Behnke S, Ben-Shlomo Y, Berendse H, Burn D, Dodel R, Grosset D, Heinzel S, Hu M, Kasten M, Krüger R, Maetzler W, Moccia M, Mollenhauer B, Oertel W, Roeben B, Sünkel U, et al. (2015) Methods in neuroepidemiology characterization of European longitudinal cohort studies in Parkinson's disease: report of the JPND working group BioLoC-PD. Neuroepidemiology 45:282–297. 10.1159/000439221 [DOI] [PubMed] [Google Scholar]
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, Widner H, Revesz T, Lindvall O, Brundin P (2008) Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med 14:501–503. 10.1038/nm1746 [DOI] [PubMed] [Google Scholar]
- Liu G, Boot B, Locascio JJ, Jansen IE, Winder-Rhodes S, Eberly S, Elbaz A, Brice A, Ravina B, van Hilten JJ, Cormier-Dequaire F, Corvol JC, Barker RA, Heutink P, Marinus J, Williams-Gray CH, Scherzer CR (2016) Specifically neuropathic Gaucher's mutations accelerate cognitive decline in Parkinson's. Ann Neurol 80:674–685. 10.1002/ana.24781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield KE, Buckman-Garner S, Parekh A (2017) The role of public-private partnerships in catalyzing the critical path. Clin Transl Sci 10:431–442. 10.1111/cts.12488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D (2011) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146:37–52. 10.1016/j.cell.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Zunke F, Tsunemi T, Toker NJ, Jeon S, Burbulla LF, Patnaik S, Sidransky E, Marugan JJ, Sue CM, Krainc D (2016) Activation of beta-glucocerebrosidase reduces pathological alpha-synuclein and restores lysosomal function in Parkinson's patient midbrain neurons. J Neurosci 36:7693–7706. 10.1523/JNEUROSCI.0628-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migdalska-Richards A, Daly L, Bezard E, Schapira AH (2016) Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice. Ann Neurol 80:766–775. 10.1002/ana.24790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal S, Bjørnevik K, Im DS, Flierl A, Dong X, Locascio JJ, Abo KM, Long E, Jin M, Xu B, Xiang YK, Rochet JC, Engeland A, Rizzu P, Heutink P, Bartels T, Selkoe DJ, Caldarone BJ, Glicksman MA, Khurana V, et al. (2017) β2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson's disease. Science 357:891–898. 10.1126/science.aaf3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mursaleen LR, Stamford JA, Jones DA, Windle R, Isaacs T (2017) Attitudes towards data collection, ownership and sharing among patients with Parkinson's disease. J Parkinsons Dis 7:523–531. 10.3233/JPD-161045 [DOI] [PubMed] [Google Scholar]
- Nalls MA, Duran R, Lopez G, Kurzawa-Akanbi M, McKeith IG, Chinnery PF, Morris CM, Theuns J, Crosiers D, Cras P, Engelborghs S, De Deyn PP, Van Broeckhoven C, Mann DM, Snowden J, Pickering-Brown S, Halliwell N, Davidson Y, Gibbons L, Harris J, et al. (2013) A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol 70:727–735. 10.1001/jamaneurol.2013.1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E, Reches A, Bembi B, Zimran A (1996) Occurrence of Parkinson's syndrome in type I Gaucher disease. QJM 89:691–694. 10.1093/qjmed/89.9.691 [DOI] [PubMed] [Google Scholar]
- Neville J, Kopko S, Broadbent S, Avilés E, Stafford R, Solinsky CM, Bain LJ, Cisneroz M, Romero K, Stephenson D (2015) Development of a unified clinical trial database for Alzheimer's disease. Alzheimers Dement 11:1212–1221. 10.1016/j.jalz.2014.11.005 [DOI] [PubMed] [Google Scholar]
- Neville J, Kopko S, Romero K, Corrigan B, Stafford B, LeRoy E, Broadbent S, Cisneroz M, Wilson E, Reiman E, Vanderstichele H, Arneric SP, Stephenson D (2017) Accelerating drug development for Alzheimer's disease through the use of data standards. Alzheimers Dement (N Y) 3:273–283. 10.1016/j.trci.2017.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Open Science Collaboration (2015) Psychology: estimating the reproducibility of psychological science. Science 349:aac4716. 10.1126/science.aac4716 [DOI] [PubMed] [Google Scholar]
- Pagan F, Hebron M, Valadez EH, Torres-Yaghi Y, Huang X, Mills RR, Wilmarth BM, Howard H, Dunn C, Carlson A, Lawler A, Rogers SL, Falconer RA, Ahn J, Li Z, Moussa C (2016) Nilotinib effects in Parkinson's disease and dementia with Lewy bodies. J Parkinsons Dis 6:503–517. 10.3233/JPD-160867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh A, Buckman-Garner S, McCune S, O'Neill R, Geanacopoulos M, Amur S, Clingman C, Barratt R, Rocca M, Hills I, Woodcock J (2015) Catalyzing the critical path initiative: FDA's progress in drug development activities. Clin Pharmacol Ther 97:221–233. 10.1002/cpt.42 [DOI] [PubMed] [Google Scholar]
- Parente V, Corti S (2018) Advances in spinal muscular atrophy therapeutics. Ther Adv Neurol Disord 11:1756285618754501. 10.1177/1756285618754501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson's Progression Marker Initiative (2011) The parkinson progression marker initiative (PPMI). Prog Neurobiol 95:629–635. 10.1016/j.pneurobio.2011.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnetti L, Chiasserini D, Persichetti E, Eusebi P, Varghese S, Qureshi MM, Dardis A, Deganuto M, De Carlo C, Castrioto A, Balducci C, Paciotti S, Tambasco N, Bembi B, Bonanni L, Onofrj M, Rossi A, Beccari T, El-Agnaf O, Calabresi P (2014) Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson's disease. Mov Disord 29:1019–1027. 10.1002/mds.25772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postuma RB, Gagnon JF, Pelletier A, Montplaisir JY (2017) Insomnia and somnolence in idiopathic RBD: a prospective cohort study. NPJ Parkinsons Dis 3:9. 10.1038/s41531-017-0011-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EM, Smith GA, Park E, Cao H, Brown E, Hayes MA, Beagan J, McLean JR, Izen SC, Perez-Torres E, Hallett PJ, Isacson O (2015) Glucocerebrosidase gene therapy prevents alpha-synucleinopathy of midbrain dopamine neurons. Neurobiol Dis 82:495–503. 10.1016/j.nbd.2015.09.009 [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Clarke J, Viel C, Panarello N, Treleaven CM, Kim C, Spencer B, Adame A, Park H, Dodge JC, Cheng SH, Shihabuddin LS, Masliah E, Sardi SP (2016) Glucocerebrosidase modulates cognitive and motor activities in murine models of Parkinson's disease. Hum Mol Genet 25:2645–2660. 10.1093/hmg/ddw124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero K, Ito K, Rogers JA, Polhamus D, Qiu R, Stephenson D, Mohs R, Lalonde R, Sinha V, Wang Y, Brown D, Isaac M, Vamvakas S, Hemmings R, Pani L, Bain LJ, Corrigan B (2015) The future is now: model-based clinical trial design for Alzheimer's disease. Clin Pharmacol Ther 97:210–214. 10.1002/cpt.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal LS, Drake D, Alcalay RN, Babcock D, Bowman FD, Chen-Plotkin A, Dawson TM, Dewey RB Jr, German DC, Huang X, Landin B, McAuliffe M, Petyuk VA, Scherzer CR, Hillaire-Clarke CS, Sieber BA, Sutherland M, Tarn C, West A, Vaillancourt D, et al. (2016) The NINDS Parkinson's Disease Biomarkers Program. Mov Disord 31:915–923. 10.1002/mds.26438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM, Passini MA, Grabowski GA, Schlossmacher MG, Sidman RL, Cheng SH, Shihabuddin LS (2011) CNS expression of glucocerebrosidase corrects α-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy Proc Natl Acad Sci U S A 108:12101–12106. 10.1073/pnas.1108197108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardi SP, Clarke J, Viel C, Chan M, Tamsett TJ, Treleaven CM, Bu J, Sweet L, Passini MA, Dodge JC, Yu WH, Sidman RL, Cheng SH, Shihabuddin LS (2013) Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc Natl Acad Sci U S A 110:3537–3542. 10.1073/pnas.1220464110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardi SP, Cheng SH, Shihabuddin LS (2015) Gaucher-related synucleinopathies: the examination of sporadic neurodegeneration from a rare (disease) angle. Prog Neurobiol 125C:47–62. 10.1016/j.pneurobio.2014.12.001 [DOI] [PubMed] [Google Scholar]
- Sardi SP, Viel C, Clarke J, Treleaven CM, Richards AM, Park H, Olszewski MA, Dodge JC, Marshall J, Makino E, Wang B, Sidman RL, Cheng SH, Shihabuddin LS (2017) Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc Natl Acad Sci U S A 114:2699–2704. 10.1073/pnas.1616152114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardi SP, Cedarbaum JM, Brundin P (2018) Targeted therapies for Parkinson's disease: from genetics to the clinic. Mov Disord 33:684–696. 10.1002/mds.27414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setó-Salvia N, Pagonabarraga J, Houlden H, Pascual-Sedano B, Dols-Icardo O, Tucci A, Paisán-Ruiz C, Campolongo A, Antón-Aguirre S, Martín I, Muñoz L, Bufill E, Vilageliu L, Grinberg D, Cozar M, Blesa R, Lleó A, Hardy J, Kulisevsky J, Clarimón J (2012) Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson's disease course. Mov Disord 27:393–399. 10.1002/mds.24045 [DOI] [PubMed] [Google Scholar]
- Sheng Z, Zhang S, Bustos D, Kleinheinz T, Le Pichon CE, Dominguez SL, Solanoy HO, Drummond J, Zhang X, Ding X, Cai F, Song Q, Li X, Yue Z, van der Brug MP, Burdick DJ, Gunzner-Toste J, Chen H, Liu X, Estrada AA, et al. (2012) Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med 4:164ra161. 10.1126/scitranslmed.3004485 [DOI] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Dürr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, et al. (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 361:1651–1661. 10.1056/NEJMoa0901281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Krüger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, et al. (2009) Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 41:1308–1312. 10.1038/ng.487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson D, Hu MT, Romero K, Breen K, Burn D, Ben-Shlomo Y, Bhattaram A, Isaac M, Venuto C, Kubota K, Little MA, Friend S, Lovestone S, Morris HR, Grosset D, Sutherland M, Gallacher J, Williams-Gray C, Bain LJ, Avilés E, et al. (2015) Precompetitive data sharing as a catalyst to address unmet needs in Parkinson's disease. J Parkinsons Dis 5:581–594. 10.3233/JPD-150570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoessl AJ. (2017) Challenges and unfulfilled promises in Parkinson's disease. Lancet Neurol 16:866–867. 10.1016/S1474-4422(17)30334-4 [DOI] [PubMed] [Google Scholar]
- Thirstrup K, Dächsel JC, Oppermann FS, Williamson DS, Smith GP, Fog K, Christensen KV (2017) Selective LRRK2 kinase inhibition reduces phosphorylation of endogenous Rab10 and Rab12 in human peripheral mononuclear blood cells. Sci Rep 7:10300. 10.1038/s41598-017-10501-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild EJ, Tabrizi SJ (2017) Therapies targeting DNA and RNA in Huntington's disease. Lancet Neurol 16:837–847. 10.1016/S1474-4422(17)30280-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Krainc D (2017) α-Synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med 23:1–13. 10.1038/nm.4269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrasidlo W, Tsigelny IF, Price DL, Dutta G, Rockenstein E, Schwarz TC, Ledolter K, Bonhaus D, Paulino A, Eleuteri S, Skjevik ÅA, Kouznetsova VL, Spencer B, Desplats P, Gonzalez-Ruelas T, Trejo-Morales M, Overk CR, Winter S, Zhu C, Chesselet MF, et al. (2016) A de novo compound targeting alpha-synuclein improves deficits in models of Parkinson's disease. Brain 139:3217–3236. 10.1093/brain/aww238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyse RK, Brundin P, Sherer TB (2016) Nilotinib-differentiating the hope from the hype. J Parkinsons Dis 6:519–522. 10.3233/JPD-160904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, Toker NJ, Jeon S, Fredriksen K, Mazzulli JR (2018) Reversible conformational conversion of alpha-synuclein into toxic assemblies by glucosylceramide. Neuron 97:92–107.e10. 10.1016/j.neuron.2017.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]