

Abstract
Background
Majority of the species within Senecioneae are classified in Senecio, making it the tribe’s largest genus. Certain intergeneric relationships within the tribe are vaguely defined, with the genus Senecio being partly linked to this ambiguity. Infrageneric relationships within Senecio remain largely unknown and consequently, the genus has undergone continuous expansion and contraction over the recent past due to addition and removal of taxa. Dendrosenecio, an endemic genus in Africa, is one of its segregate genera. To heighten the understanding of species divergence and phylogeny within the tribe, the complete chloroplast genomes of the first five Senecio and six Dendrosenecio species were sequenced and analyzed in this study.
Results
The entire length of the complete chloroplast genomes was ~ 150 kb and ~ 151 kb in Dendrosenecio and Senecio respectively. Characterization of the 11 chloroplast genomes revealed a significant degree of similarity particularly in their organization, gene content, repetitive sequence composition and patterns of codon usage. The chloroplast genomes encoded an equal number of unique genes out of which 80 were protein-coding genes, 30 transfer ribonucleic acid, and four ribosomal ribonucleic acid genes. Based on comparative sequence analyses, the level of divergence was lower in Dendrosenecio. A total of 331 and 340 microsatellites were detected in Senecio and Dendrosenecio, respectively. Out of which, 25 and five chloroplast microsatellites (cpSSR) were identified as potentially valuable molecular markers. Also, through whole chloroplast genome comparisons and DNA polymorphism tests, ten divergent hotspots were identified. Potential primers were designed creating genomic tools to further molecular studies within the tribe. Intergeneric relationships within the tribe were firmly resolved using genome-scale dataset in partitioned and unpartitioned schemes. Two main clades, corresponding to two subtribes within the Senecioneae, were formed with the genus Ligularia forming a single clade while the other had Dendrosenecio, Pericallis, Senecio and Jacobaea. A sister relationship was revealed between Dendrosenecio and Pericallis whereas Senecio, and Jacobaea were closely placed in a different clade.
Conclusion
Besides emphasizing on the potential of chloroplast genome data in resolving intergeneric relationships within Senecioneae, this study provides genomic resources to facilitate species identification and phylogenetic reconstructions within the respective genera.
Electronic supplementary material
The online version of this article (10.1186/s40529-019-0265-y) contains supplementary material, which is available to authorized users.
Keywords: Senecio, Dendrosenecio, Endemic, Chloroplast genome, Codon usage, Microsatellites, DNA barcodes, Phylogenomics
Background
Senecioneae, the largest tribe in the family Asteraceae, has over 160 genera with more than 3000 species, and new genera continue to be added (Chen et al. 2011; Nordenstam et al. 2009). The tribe is prominent for its size, and rich morphological and ecological diversity. It is mostly dominated by annual and perennial herbs, while the rest constitute shrubs, vines, trees, and epiphytes. It has a near cosmopolitan distribution, with southern Africa being one of its key diversity hotspot zones (Pelser et al. 2007). Majority of the species in the tribe are placed in Senecio L., making it one of the largest genera of angiosperms, with over 1250 species (Nordenstam et al. 2009). Senecio is characteristically diverse in morphology, life-history, growth forms, and thus, it has been markedly linked to the incongruous phylogenetic relationships within the tribe (Pelser et al. 2007). Its members are generally distinguished by style branches truncate with short sweeping hairs, separated stigmatic lines and sometimes with a median hair pencil, and with ecaudate anther bases and balusterform filament collar (Nordenstam 2007; Pelser et al. 2007).
Over the years, the genus has been under constant re-evaluation and reclassification, and until now, comprehensive infrageneric relationships are yet to be established. Consequently, numerous species have in the past been segregated as new genera mostly based on morphological, anatomical, and chromosomal variations (Jeffrey and Chen 1984; Jeffrey et al. 1977). One of such segregate genera is Dendrosenecio (Hauman ex Hedb.) B. Nord., upgraded by Nordenstam (1978) to constitute the Afromontane pachycaul taxa. Dendrosenecio was initially classified in Senecio based on the striking similarities in floral characters. It is therefore not surprising that the elevation of Dendrosenecio was at first controversial (Jeffrey et al. 1977) as the genus exhibited substantial morphological resemblances to other African perennials of Senecio. Despite these remarkable morphological similarities, amplified fragment length polymorphism analysis revealed considerable divergence between Senecio and Dendrosenecio (Knox and Palmer 1995b). Afterwards, internal transcribed spacer (ITS) data identified Oresbia Cron & B. Nord. as the closely related genus to Dendrosenecio (Pelser et al. 2007).
Majority of the segregated groups are now accepted on the basis of molecular data obtained from markers such as ITS (Pelser et al. 2007). However, it is evident that more valuable diagnostic molecular sequences are needed to further understand the generic and intergeneric relationships in Senecioneae. The large number of species, considerable variation in species life-history and over-dependence on morphological characters, the majority of which overlap, have been pointed out as the causes of the systematic conflict observed within Senecio. Similar to Senecio, infrageneric relationships within Dendrosenecio are still debatable, especially in relation to specific and subspecific classifications. Species of Dendrosenecio exhibit ‘mosaic of morphological variation’ arising from divergence and convergence as they dispersed to various geographical regions with similar habitat conditions (Knox 2005; Mabberley 1973). Besides, frequent hybridization events between species within each genus have been evidenced resulting in allopolyploid species (Hedberg 1957; Hegarty et al. 2012; Milton 2009). It is therefore imperative that more molecular markers and divergent regions are identified to facilitate species identification, speciation and adaptive evolution studies on species of Senecio and Dendrosenecio.
Partial plastid markers, species-specific or universal, have in the past decades been used to resolve phylogenetic relationships and species delimitations. This inclination is progressively being substituted by the use of plastid genome-scale data, resulting in improved phylogenetic resolutions and detailed evolutionary information about species at all taxonomic levels. Typically chloroplast DNA is uniparentally, maternally in angiosperms and paternally in gymnosperms, inherited and exhibits homologous recombination (Marechal and Brisson 2010). This attribute can greatly benefit studies on taxa that are affected by hybridization, introgression and convergent evolution. Additionally, chloroplast genomes are justifiably conserved in terms of gene composition and arrangement permitting comparative genomics even at the generic level. However, they harbour key variations e.g., in the inverted repeat (IR) size and positioning of the IR junctions even among close relatives (Downie and Jansen 2015), and in specific lineages, massive rearrangements, gene duplications, loss or gain have been observed e.g. in Campanulaceae (Knox 2014). These variations provide sufficient unique attributes to reconstruct phylogenetic relationships with strong statistical support, and to investigate the origin and evolutionary patterns of plastids (Pouchon et al. 2018; Tonti-Filippini et al. 2017) through comparative genomics.
To date, only eight chloroplast genomes have been sequenced and reported from three genera in the Senecioneae tribe (Doorduin et al. 2011; Lee et al. 2016; Wang et al. 2019). In this study, the first five and six chloroplast genomes in Senecio and Dendrosenecio respectively were sequenced and analysed. The objectives were: to generate, characterize and analyse the complete chloroplast genomes of 11 species of Senecioneae; to identify highly variable regions that could be of phylogenetic utility within the tribe through comparative analyses and; to investigate the potential of chloroplast phylogenomics in resolving phylogenetic relationships among the species of Senecioneae, with key interest on the phylogenesis of Senecio and Dendrosenecio.
Methods
Plant material and genome sequencing
Fresh young leaves of 11 species of Senecioneae were collected from the tropical mountains in eastern Africa (Table 1). The species were identified according to the morphological descriptions given in the Flora of Tropical East Africa (Beentje et al. 2005; Knox 2005). Voucher specimen for each species was deposited in the Herbarium of Wuhan Botanical Garden, Chinese Academy of Sciences (HIB). Total genomic DNA was extracted from approximately 100 mg of leaf material for each sample using a modified 2 × cetyltrimethylammonium bromide (CTAB) method (Doyle 1987). The DNA quality was checked using Qubit 2.0 Fluorometer (Life Technologies, CA, USA). A DNA library was constructed for each species by shearing the genomic DNA into short fragments of ~ 350 bp. The DNA was sequenced based on the pair-end sequencing technique implemented on an Illumina Hiseq 2500™ platform (Illumina Inc., San Diego, CA, USA). An average of 22.75 million paired reads, at least 5 Gb of raw sequence data, were generated for each species.
Table 1.
Characteristics of complete chloroplast genomes of 14 species of the tribe Senecioneae (Asteraceae)
| General genome characteristic | Source | Herbarium accession numbers | GenBank accession number | Genome size (bp) [GC%] | LSC length (bp) | SSC length (bp) | IR length (bp) | PCGs (duplicated in the IR) | Trnas (duplicated in the IR) | Rrna (duplicated in the IR) |
|---|---|---|---|---|---|---|---|---|---|---|
| Dendrosenecio johnstonii (Oliv.) B. Nord. | Mt. Kilimanjaro | SAJIT-002716 | MG560050 | 150,607 [37.5] | 83,469 | 17,754 | 24,692 | 80 (8) | 30 (7) | 4 (4) |
| D. meruensis (Cotton & Blakelock) E.B. Knox | Mt. Meru | SAJIT-002556 | MG560049 | 150,587 [37.5] | 83,448 | 17,755 | 24,692 | 80 (8) | 30 (7) | 4 (4) |
| D. elgonensis subsp. elgonensis (T.C.E.Fr.) E.B. Knox | Mt. Elgon | SAJIT-003220 | KY434194 | 150,548 [37.5] | 83,403 | 17,771 | 24,687 | 80 (8) | 30 (7) | 4 (4) |
| D. keniodendron (R.E. Fr. & T.C.E.Fr) B. Nord. | Mt. Kenya | SAJIT-002100 | KY434193 | 150,555 [37.5] | 83,418 | 17,755 | 24,691 | 80 (8) | 30 (7) | 4 (4) |
| D. battiscombei (R.E. Fr. & T.C.E.Fr) E.B. Knox | Mt. Kenya | SAJIT-002802 | KY434195 | 150,556 [37.5] | 83,410 | 17,762 | 24,692 | 80 (8) | 30 (7) | 4 (4) |
| D. brassiciformis (R.E.Fr. & T.C.E.Fr) Mabb. | Aberdares Ranges | SAJIT-003289 | MG560051 | 150,551 [37.5] | 83,424 | 17,747 | 24,690 | 80 (8) | 30 (7) | 4 (4) |
| Senecio moorei R.E.Fr. | Mt. Kenya | SAJIT-201834 | MH483949 | 151,204 [37.2] | 83,278 | 18,300 | 24,813 | 80 (8) | 30 (7) | 4 (4) |
| S. keniophytum R.E.Fr. | Mt. Kenya | SAJIT-201831 | MH483946 | 151,413 [37.2] | 83,422 | 18,277 | 24,857 | 80 (8) | 30 (7) | 4 (4) |
| S. purtschelleri Engl. | Mt. Kenya | SAJIT-201832 | MH483947 | 151,191 [37.2] | 83,243 | 18,302 | 24,823 | 80 (8) | 30 (7) | 4 (4) |
| S. schweinfurthii O.Hoffm. | Mt. Kenya | SAJIT-201833 | MH483950 | 151,260 [37.2] | 83,255 | 18,389 | 24,823 | 80 (8) | 30 (7) | 4 (4) |
| S. roseiflorus R.E.Fr. | Mt. Kenya | SAJIT-201835 | MH483948 | 151,228 [37.2] | 83,329 | 18,279 | 24,810 | 80 (8) | 30 (7) | 4 (4) |
| Jacobea vulgaris Gaertn. | – | – | NC_015543 | 150,689 [37.3] | 82,855 | 18,277 | 24,777 | 81 (8) | 29 (7) | 4 (4) |
| Ligularia fischeri (Ladeb.) Turcz. | – | – | KT988070 | 151,133 [37.5] | 83,238 | 18,233 | 24,831 | 80 (8) | 29 (7) | 4 (4) |
| Pericallis hybrida (Regel) B.Nord | – | – | NC_031898 | 151,267 [37.3] | 85,751 | 18,331 | 23,591 | 79 (8) | 30 (7) | 4 (4) |
Genome assembly and annotation
The raw data were filtered and trimmed using Fastp software using the default settings (Chen et al. 2018); all low-quality reads were discarded. The de novo assembly of the filtered reads, into complete chloroplast genomes, was performed using NOVOPlasty (Dierckxsens et al. 2017) with default seed and K-mer = 31–39. The contigs were then mapped to the chloroplast genomes of Jacobaea vulgaris Gaertn. (NC_015543; Doorduin et al. 2011) and Pericallis hybrida (Regel) B. Nord. (NC_031898; Wang et al. 2019) using Geneious Prime 2019 (Biomatters Ltd., Auckland, New Zealand; https://www.geneious.com). Basic local alignment search tool ver. 2.2.18+ (Camacho et al. 2009) was used to ascertain the positions of the single copies and the inverted repeat regions by self-blasting the assembled sequences.
GeSeq (Tillich et al. 2017) was used to annotate each of the chloroplast genomes using the complete chloroplast genome sequences of Jacobaea vulgaris and Pericallis hybrida as references. Where necessary, manual corrections were performed in Geneious Prime 2019 (Biomatters Ltd., Auckland, New Zealand), to rectify the start and stop codons of the protein-coding genes (PCGs), based on the annotations of J. vulgaris and P. hybrida. A circular genome map for each species was generated using OGDraw v1.2 (Lohse et al. 2007). All annotated genome sequences were submitted to the GenBank under the accession numbers listed in Table 1.
Codon usage and microsatellite repeats identification
The level of codon usage bias was determined by analysing the Relative Synonymous Codon Usage (RSCU; Sharp and Li 1987), Effective Number of codon (ENc; Wright 1990) and the Codon Biased Index (CBI; Morton 1993) for all PCGs, in DnaSP 6.10 (Rozas et al. 2017). The frequency of amino acid was also considered. The MicroSAtellite Identification tool Perl Script (MiSa; Thiel et al. 2003), was used to mine for SSRs with the parameters set at 10 for mononucleotides, 5 for dinucleotides, 4 for trinucleotides and 3 for tetra-, penta- and hexa-nucleotides.
Genome comparative analyses and divergence hotspot identification
The available chloroplast genomes of Asteraceae species have been shown to harbour no major differences in their sizes, gene content and arrangement. The whole genome size, GC percentage, LSC, SSC, IR and number of gene in each of the 11 species, were therefore compared to three other species of Senecioneae. Preliminary comparative analyses among the species within each genus revealed highly conserved sequences with > 99% pairwise identity and > 98% identical sites. Consequently, one chloroplast genome sequence was randomly picked per genus to conduct further comparative studies against other chloroplast genomes within the tribe. The expansion/contraction of the IR regions was assessed by comparing the positions of SC/IR junctions and their adjacent genes using IRscope (Amiryousefi et al. 2018).
Further, to outline any significant sequence divergence spots and genome rearrangements, the chloroplast genomes were aligned and plotted in MAUVE (Darling et al. 2004), with Nicotiana tabacum L. (NC_001879; Shinozaki et al. 1986) being used as an external reference genome. Nucleotide diversity (Pi) in the non-coding regions (> 200 bp) of the five species of Senecioneae was analysed in DnaSP v.6.10 (Rozas et al. 2017). Potential primers for ten sites with the highest Pi values were designed using Primer3 (Untergasser et al. 2012) using default settings.
Phylogenetic analyses
A total of 75 species, representing 49 genera from 11 tribes of Asteraceae, were downloaded from the NCBI (Additional file 1: Table S1) for phylogenetic analyses. Besides, data for Adenophora divaricata Franch. & Sav. and A. stricta Miq. (Cheon et al. 2017) were downloaded and used as outgroups in this analysis. Sequences of 70 common PCGs were extracted from the 77 species. Each gene was separately aligned using MUSCLE (Edgar 2004) and then concatenated in Geneious Prime 2019 (Biomatters Ltd., Auckland, New Zealand). Phylogenetic reconstructions were carried out using Maximum Likelihood (ML) and Bayesian Inference (BI) methods. Each method was used twice in independent analyses based on unpartitioned and partitioned data. Before the ML analysis using unpartitioned data, the best-fit DNA substitution model was determined using ModelFinder (Kalyaanamoorthy et al. 2017) as implemented in IQ-TREE version 1.5.4 (Nguyen et al. 2015). Maximum Likelihood (ML) analysis was conducted using IQ-TREE with a bootstrap analysis of 5000 replications under the GTR + F + R6 nucleotide substitution model. MrBayes v3.2.6 (Ronquist et al. 2012) was used to implement the BI analyses based on the unpartitioned data set, using four independent Markov Chain Monte Carlo runs with three heated and one cold chain. The chains were run for 2 × 106 generations with sampling from the cold chain run after every 103 generations. The analysis was stopped after the average standard deviation of split frequencies as calculated by Mr. Bayes was below 0.01, an indication that convergence had been attained. The first 25% of all generations were excluded, and a consensus phylogenetic tree was obtained based on majority rule from the remaining trees. Branch support was indicated by posterior probability (PP) values. The data set was then partitioned by categorizing the nucleotides in each gene based on the position (first, second, or third) they occupy in a codon. The best partitioning scheme and substitution models were calculated using PartitionFinder2 (Lanfear et al. 2017). The generated phylogenetic trees were visualised and formatted in Interactive tree of life (iTOL) v3 (Letunic and Bork 2016).
Results
Chloroplast genome organization and content
An average of 22.6 million (95.2%) clean reads were generated for each species. The chloroplast genomes of the two genera were comparable in terms of structural organization, gene content, and arrangement. The overall chloroplast genome size varied slightly within each genus, but significantly between the genera ranging within 150 kb in Dendrosenecio and to 151 kb in Senecio. Each of the chloroplast genomes had four regions including a large single copy of ~ 83.5 kb in both genera, two inverted repeats ~ 24.7 kb, and a small single copy ~ 17 kb in Dendrosenecio and ~ 18 kb in Senecio. The GC percentage values of the entire genome and for each of the sections were identical in all species within a respective genus (Table 1). Each of the plastid genomes encoded a total of 114 unique genes of which 80 were PCGs, 30 tRNAs and four rRNAs. All the PCGs, except three, had the standard AUG start codon (Table 2). Seventeen genes; 11 PCGs and six tRNA genes contained either one or two introns. Eighteen genes were duplicated in the IR regions, with rps12 being uniquely positioned with its 5′ end exon at the LSC and the other is located in the IR regions. Both ycf1 and rps19 genes also had their 3′ ends duplicated on the IR regions (Fig. 1; Table 1).
Table 2.
List of genes identified in the studied chloroplast genomes of 11 species of Senecioneae
| Gene family | Gene |
|---|---|
| Transfer RNA | trnA-UGC*, trnfM-CAU, trnI-GAU*, trnM-CAU, trnR-ACG, trnS-UGA, trnC-GCA, trnG-GC, trnK-UUU*, trnN-GUU, trnW-CCA, trnT-GGU, trnD-GUC, trnV-UAC*, trnL-CAA, trnY-GUA, trnR-UCU, trnT-UGU, trnE-UUC, trnH-GUG, trnL-UAA*, trnP-UGG, trnS-GCU, trnV-GAC, trnF-GAA, trnI-CAU, trnL-UAG, trnQ-UUG, trnS-GGA, trnG-UCC* |
| Small ribosomal units | rps2,3,4,7,8,11,12a,14,15,16*,18,19b |
| Large ribosomal units | rpl2*,14,16*,20,22,23,32,33,36 |
| RNA polymerase sub-units | rpoA, rpoB, rpoC1*, rpoC2 |
| Translation initiation facto | infA |
| NADH dehydrogenase | ndhA*, ndhB*, ndhC, ndhDb, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK |
| Photosystem I | psaA, psaB, psaC, psaI, psaJ, ycf3**, ycf4 |
| Photosystem II | psbA,B,C,D,E,F,H,I,J,K,Lb,M,N,T,Z |
| Cytochrome b/f complex | petA, petB*, petD*, petG, petL, petN |
| ATP synthase | atpA, atpB, atpE, atpF*, atpH, atpI |
| Large subunit of rubisco | rbcL |
| Maturase | matK |
| Protease | clpP** |
| Acetyl-CoA-carboxylase sub-unit | accD |
| Envelope membrane protein | cemA |
| Component of TIC complex | ycf1 |
| c-Type cytochrome synthesis | ccsA |
| Hypothetical genes reading frames | ycf2, ycf15 |
| Ribosomal RNA | rrn4.5, rrn5, rrn16, rrn23 |
* Genes with a single intron, ** genes with two introns, a trans-spliced genes, b genes with an alternative start codon
Fig. 1.

A representative chloroplast genome map of a Dendrosenecio and b Senecio. Genes are color-coded based on their function as shown in the legend. The inner circle indicates the inverted repeat boundaries and the genome’s GC content. The arrows indicate the direction of gene transcription
Codon usage and microsatellite sequences
The total sequence size coding for protein genes was 78,879–79,146 bp in Dendrosenecio and 78,203–78,531 in Senecio. These protein sequences encoded 26,293–26,382 and 26,067–26,177 codons respectively, including stop codons. Leucine was encoded by the highest number (average = 10.81% and 10.9%) of codons, whereas cysteine (average = 1.14% and 1.13%) was the least encoded in Dendrosenecio and Senecio respectively. Except for Methionine (AUG) and Tryptophan (UGG), whose RSCU values were 1 in all species, the usage of the other codons was biased. Generally, the usage of seven codons, eight in Senecio, was overrepresented (RSCU > 1.6) while the majority had low representation RSCU < 0.6. The ENc ranged from 49.76 to 51.49, while CBI ranged between 0.308 and 0.356 (Additional file 2: Table S2). The average RSCU and amino acid frequency values for each species were plotted using R-script (Fig. 2; Zhang et al. 2018).
Fig. 2.

Details of codon usage biasness in the chloroplast genomes of five species of Senecio and six species of Dendrosenecio. The values at the top of each stack indicate the usage frequency of each amino acid, while the bars (colour coded) depicts the relative synonymous codon usage values for each codon
In each genus, the analyses of repetitive sequences revealed minimal variation in the total number and position of each repeat motif, most of which were shared among the chloroplast genome sequences. In Dendrosenecio, 340 microsatellites were discovered while the species of Senecio had 331 SSR repeats. On average, species of Senecio had the highest number of mono-, tri-, tetra- and hexa-nucleotides, while di- and penta-nucleotides were the most abundant repeats in Dendrosenecio (Fig. 3). Certain repeat motifs were genus-specific while a few e.g., C/G and AGCTAT/AGCTAT in D. johnstonii and AATCT/AGATT and AATTC/AATTG in S. keniophytum were species-specific (Additional file 3: Table S3). The present SSRs were classified based on the variation in repeat type, the number of repeats in each motif, presence or absence of the repeat, and the position of each repeat in the genome. Microsatellites were considered polymorphic if they: showed variation, were present in all plastid genomes and were positioned at homologous regions across all species in each genus. Based on this criterion, 25 polymorphic SSRs were discovered in Senecio and only five in Dendrosenecio (Tables 3, 4).
Fig. 3.

The average number of each type of microsatellites in the chloroplast genomes of Dendrosenecio and Senecio
Table 3.
Details of 25 potentially polymorphic microsatellite repeats in five species of Senecio
| Microsatellites | Senecio schweinfurthii | S. keniophytum | S. moorei | S. purtschelleri | S. roseiflorus | Location | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Start | End | Start | End | Start | End | Start | End | Start | End | ||
| (AAAT)3;(ATT)4 | 1849 | 1860 | 1881 | 1892 | 1860 | 1871 | 1848 | 1859 | 1881 | 1892 | Intron |
| (T)10,11;(T)16n(TTAGA)3 | 8685 | 8694 | 9134 | 9180 | 8697 | 8706 | 8689 | 8699 | 8317 | 8326 | IGS |
| (T)10;(A)10;(A)10tca(T)10 | 9738 | 9747 | 9720 | 9729 | 9738 | 9760 | 9731 | 9754 | 9695 | 9704 | IGS |
| (T)17,10,16, | 10,859 | 10,875 | 10,857 | 10,866 | 10,872 | 10,887 | 10,866 | 10,881 | 10,852 | 10,861 | IGS |
| (T)11,23,16 | 12,933 | 12,943 | 12,919 | 12,941 | 12,945 | 12,955 | 12,919 | 12,929 | 12,925 | 12,940 | IGS |
| (T)16,16,15 | 16,878 | 16,893 | 18,219 | 18,228 | 16,890 | 16,905 | 16,864 | 16,878 | 16,875 | 16,884 | Intron |
| (AT)6;(A)10(TA)6 | 26,849 | 26,860 | 26,787 | 26,798 | 26,861 | 26,872 | 26,834 | 26,845 | 26,776 | 26,797 | IGS |
| (T)10 g(A)11;(T)10c(A)10;(A)14 | 28,219 | 28,240 | 28,166 | 28179 | 28,231 | 28,252 | 28,204 | 28,225 | 28,154 | 28,174 | IGS |
| (ATAAA)3;(T)10;(CTA)5 | 32,007 | 32,021 | 30,007 | 30,016 | 32,019 | 32,033 | 31,992 | 32,006 | 31,008 | 31,022 | IGS |
| (AT)5,7;9 | 34,922 | 34,931 | 34,890 | 34,916 | 34,934 | 34,943 | 34,907 | 34,916 | 34,879 | 34,892 | IGS |
| (A)11;(T)14 | 41,864 | 41,874 | 41,844 | 41,857 | 41,876 | 41,886 | 41,849 | 41,858 | 41,840 | 41,849 | IGS |
| (A)17,14,20 | 46,644 | 46,660 | 46,666 | 46,679 | 46,656 | 46,672 | 46,629 | 46,645 | 46,621 | 46,640 | IGS |
| (T)10,11,12;(A)10n(T)11 | 48,099 | 48,109 | 48,125 | 48,136 | 48,111 | 48,120 | 48,084 | 48,093 | 48,063 | 48,103 | IGS |
| (A)11.12 | 59,324 | 59,334 | 59,388 | 59,398 | 59,335 | 59,346 | 59,308 | 59,318 | 59,021 | 59031 | IGS |
| (A)12 | 64,609 | 64,620 | 64,721 | 64,732 | 64,621 | 64,632 | 64,593 | 64,605 | 64,658 | 64,668 | IGS |
| (A)10;(ATTT)3n;(A)11n(A)11 | 70,356 | 70,365 | 70,409 | 70,428 | 70,368 | 70,377 | 70,343 | 70,352 | 70,266 | 70,407 | Intron |
| (T)14,13,12 | 70,577 | 70,590 | 70,699 | 70,710 | 70,589 | 70,601 | 70,564 | 70,577 | 70,619 | 70,630 | Intron |
| (A)11,12,14 | 76,913 | 76,923 | 77,044 | 77,055 | 76,925 | 76,935 | 76,900 | 76,910 | 76,964 | 76,977 | IGS |
| (T)10,17 | 77,160 | 77,169 | 77,292 | 77,308 | 77,172 | 77,181 | 77,147 | 77,156 | 77,215 | 77,231 | CDS |
| (T)14n(T)16,17;(T)10 | 79,597 | 79,716 | 79,745 | 79,754 | 79,609 | 79,728 | 79,584 | 79,704 | 79,668 | 79,677 | IGS |
| (A)14,17 | 80,181 | 80,194 | 80,303 | 80,319 | 80,193 | 80,206 | 80,169 | 80,182 | 80,221 | 80,237 | IGS |
| (A)10,11,15 | 108,867 | 108,877 | 109,796 | 109,805 | 108,880 | 108,890 | 108,855 | 108,865 | 109,680 | 109,694 | CDS |
| (A)12,16 | 110,440 | 110,451 | 110,599 | 110,614 | 110,453 | 110,464 | 110,428 | 110,439 | 110,483 | 110,494 | CDS |
| (GATT)3;(GATT)3n(TAAT)3 | 115,162 | 115,173 | 115,324 | 115,409 | 115,174 | 115,185 | 115,149 | 115,160 | 115,206 | 115,291 | Intron |
| (AT)5,6 | 115,559 | 115,570 | 115,973 | 115,982 | 115,571 | 115,582 | 115,546 | 115,557 | 115,947 | 115,957 | Intron |
n = varying number of base pairs between two repeat motifs
Table 4.
Details of potentially polymorphic microsatellite repeats in six species of Dendrosenecio
| Microsatellites | D. keniodendron | D. elgonensis subsp. elgonensis | D. battiscombei | D. meruensis | D. johnstonii | D. brassiciformis | Location | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Start | End | Start | End | Start | End | Start | End | Start | End | Start | End | ||
| A13,14,16;TnA29 | 42,047 | 42,059 | 42,044 | 42,057 | 42,051 | 42,064 | 42,044 | 42,072 | 42,053 | 42068 | 42,049 | 54,600 | Intron |
| (T)10,11,13 | 59,146 | 59,155 | 59,130 | 59,140 | 59,150 | 59,160 | 59,179 | 59,189 | 59,200 | 59,210 | 59,147 | 59,210 | IGS |
| (A)10,11,12 | 70,469 | 70,478 | 70,456 | 70,465 | 70,474 | 70,483 | 70,368 | 70,378 | 70,521 | 70,532 | 70,473 | 70,532 | IGS |
| (T)11,12,15 | 79,748 | 79,762 | 79,736 | 79,746 | 79,740 | 79,754 | 79,781 | 79,791 | 79,802 | 79,813 | 79,802 | 79,813 | IGS |
| (A,AgaaatattttttgtA,AgaaatattttttgtA) 11,16,39,40, | 80,327 | 80,342 | 80,311 | 80,327 | 80,319 | 80,334 | 80,333 | 80,372 | 80,355 | 80,393 | 80,355 | 80,393 | IGS |
Genome comparative analyses
In each genus, the newly annotated chloroplast genomes had no significant differences, except for the slight variations in size and gene positioning. The size difference between the largest and the smallest genome among the Dendrosenecios and Senecio was 59 bp and 222 bp respectively. Based on the currently available chloroplast genomes, Dendrosenecios had the smallest chloroplast genome size within the Senecioneae tribe, with a difference of 82 bp from the immediate largest cp genome (Jacobaea vulgaris; Table 1). The genes adjacent to the IR/SC junctions (trnH, rps19, ycf1 and ndhF) were similar in all species, except in Pericallis hybrida which had rpl2 at the LSC/IR junction. The IRb region expanded into the coding region of rps19, resulting into a pseudogene (ψ) of varying length at the IRa in all but in P. hybrida and S. moorei. In S. moorei, a 14 bp gap was observed between the rps19 gene and the JLB border. At the JSB junction, the IRb expanded into the coding region of ycf1 gene; thus ψycf1 appeared on the IRa region in all species. Two genes, ndhF and ψycf1, were positioned at varying points adjacent to the JSA junction. In J. vulgaris and Ligularia fischeri (Ladeb.) Turcz., the ndhF and ψycf1 genes overlapped. The JLA junction was uniformly flanked between ψrps19 and trnH genes, except in S. moorei and P. hybrida where rpl2 and trnH bordered the junction. Figure 4 shows the genes adjacent to the junctions and their order in representative genomes from each genus. In Senecio two species were used to show the differences recorded at JLA and JLB in S. moorei.
Fig. 4.

Comparison of the large single copy, inverted repeats, and small single copy junction positions in six species of Senecioneae (Asteraceae). Genes adjacent to the junctions are shown as blocks of different colours
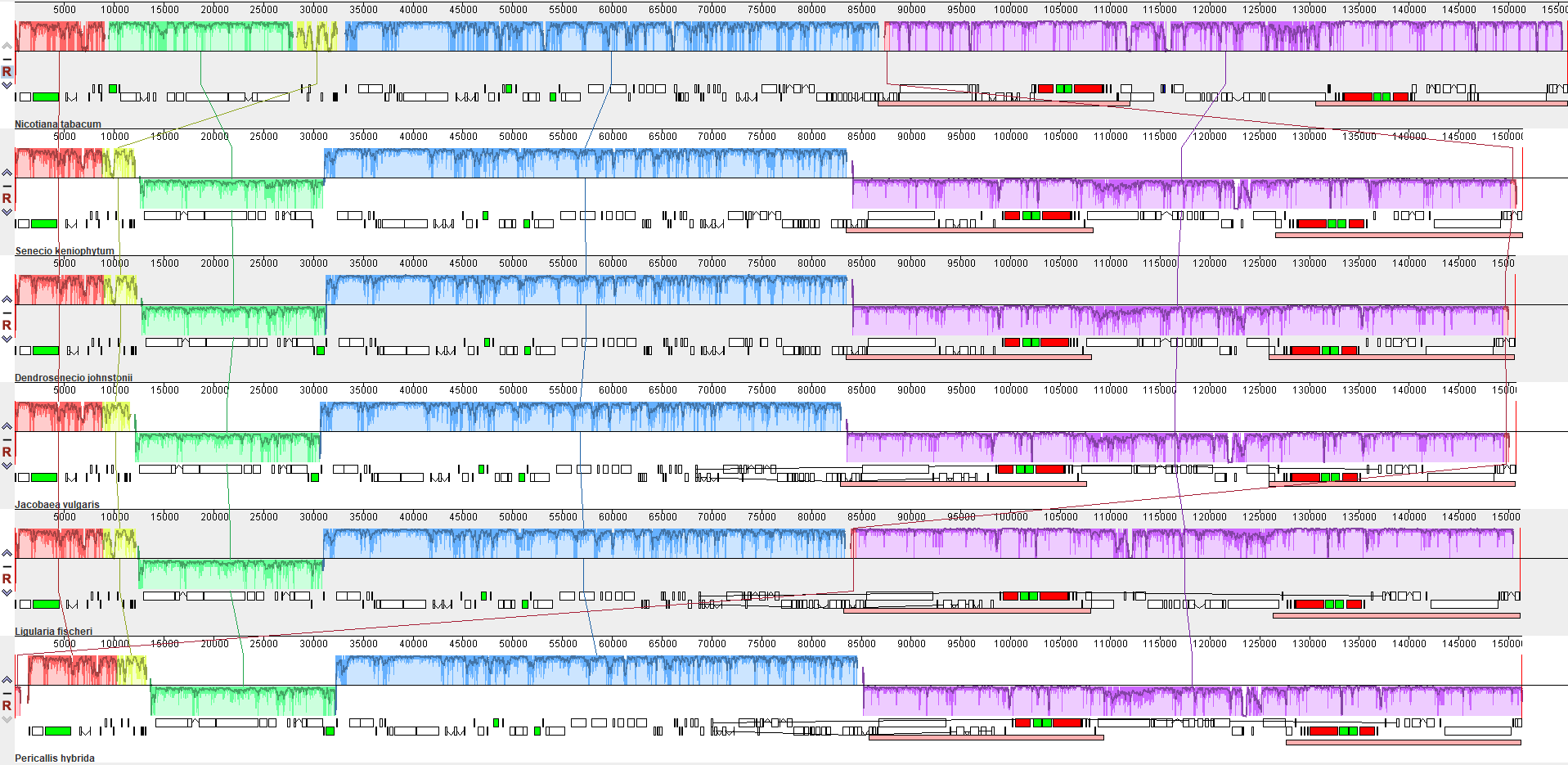
There were no major rearrangements detected among the newly sequenced chloroplast genomes, an indication that chloroplast genomes within these two genera could be much conserved (Fig. 5). However, the existence of two inversions in the LSC region in reference to Nicotiana tabacum was identified in all newly generated chloroplast genomes. The arrangement of genes in the SSC region was also different in the Asteraceae species, apart from L. fischeri, whose alignment was identical to that of N. tabacum (Additional file 4: Figure S1). The nucleotide polymorphism test identified 74 sites with Pi values ranging from 0.00089 (ndhB–ndhB) to 0.06852 (trnH-GUG-psbA). Figure 6 indicates the regions with high levels of intergeneric variation (Pi values > 0.01). Potential PCR primers were designed for the ten highly polymorphic sites (Table 5).
Fig. 5.

Comparison of sequence arrangement in the chloroplast genomes of 11 species of Senecioneae (Asteraceae). Conserved orthologs are indicated by locally collinear blocks. Similar blocks among the genomes are coded in one colour and joined by a line. The genes above the line are transcribed in a clockwise direction, those below the line are transcribed towards the counter-clockwise direction
Fig. 6.

Nucleotide variability (Pi) values of non-coding regions which were extracted from the chloroplast genomes of five species of Senecioneae
Table 5.
Details of ten primers that target the most divergent regions in the chloroplast genomes of Senecioneae species
| Orientation | Primers | Tm | Target region | Expected product size |
|---|---|---|---|---|
| F | AAATAGGAGGAAGCTGTGAC | 55 | trnH-psbA | 603 |
| R | GCACTATGGCTTTCAACCTA | 55 | ||
| F | CACGTCAGATGTTCTATGGA | 55 | ndhD-ccsA | 658 |
| R | TTGGGCCTTTATTACTTGGA | 55 | ||
| F | TTTGCGAAAAGAGGAAGACA | 55 | trnT-trnL | 812 |
| R | TCCATACCAAGGCTCAATTC | 55 | ||
| F | TGTGGTAATTGCGTTGAGTA | 55 | ndhI-ndhG | 759 |
| R | TACGTAAATAAGGAGCTGCG | 55 | ||
| F | CTTGCTTGTACCTACCCAAT | 55 | trnS-rps4 | 588 |
| R | TGACTCTTCAAGCATTCCAA | 54 | ||
| F | TTGACCTTGAAACAACAACG | 55 | rps18-rpl20 | 546 |
| R | ACAAGAGACAGTTGCTTCTT | 55 | ||
| F | GGAACTAAAATGAACAGTGCA | 54 | psbM-trnD | 880 |
| R | ATGTCTACGCTGGTTCAAAT | 55 | ||
| F | GCGGATTCTAAATTGTAACCA | 54 | ndhC-trnV | 959 |
| R | AGCCCTAGAGCCCTATAAAA | 55 | ||
| F | TAACGCTGCCAAATATCCTT | 55 | rpl32-ndhF | 1194 |
| R | AGAAGAAGTCCCAACCCTAT | 55 | ||
| F | ATTGCCACTTCATCAATCTT | 53 | psbZ-trnG- | 617 |
| R | ACTACACTATGACGGCTAAC | 54 |
Tm annealing temperature, F forward primer, R reverse primer
Phylogenetic relationships
The final sequence alignment of common protein-coding genes had 60,992 characters in 70 chloroplast genome loci for 77 taxa. Phylogenetic relationships among the 75 species representing ten tribes of Asteraceae were unveiled based on ML and BI analyses. The tribes were recovered as monophyletic clades each with significant statistical support in all the generated trees. Intergeneric relationships within tribe Senecioneae, to which the 11 newly sequenced chloroplast genomes belong, were clearly defined and strongly supported in all data schemes (100% BS and 1.0 PP). The phylogenetic analyses strongly supported three sub-clades within the tribe Senecioneae; one that comprised of the genus Ligularia, the second sub-clade contained both Senecio and Jacobaea while the third one had species from Dendrosenecio and Pericallis. The sister relationship between the species of Senecio was congruent in all the analyses, differing only in support values at the clade containing S. moorei and S. schweinfurthii which was highly supported (BS ≥ 92) in ML trees but gained weak support in BI analyses (PP ≤ 0.5). The six species of Dendrosenecio were split into two clades, distinctly separating species from Tanzania and species from Kenya (Fig. 7). However, the interspecific relationship within the Kenyan species differed in the different phylogenetic trees (Additional file 5: Figure S2).
Fig. 7.

Phylogenetic relationships of 75 species of Asteraceae inferred from an unpartitioned multi-gene dataset using a Maximum Likelihood (ML) and b Bayesian Inference methods
Discussion
Genome structure and content
It is typical of higher plants to possess a chloroplast genome that has a quadripartite structure and a relatively well-conserved gene content and arrangement (Ravi et al. 2008; Wicke et al. 2011; Yurina et al. 2017). However, minor to major variability in chloroplast genome structure has been observed in specific plant lineages (Guisinger et al. 2011). The current upsurge in the amount of chloroplast genome-scale data has played a crucial role in the enhancement of knowledge on the evolution and organization of chloroplast genomes. Both Senecio and Dendrosenecio are conspicuous genera in the tribe Senecioneae, the former for being the largest genus in the tribe and the latter for exhibiting an atypical growth habit. In this study, the first complete chloroplast genome sequences of five Senecio and six Dendrosenecio species were generated. The genome structures were comparable to those of higher plants each exhibiting four compartments; a pair of inverted repeats and a pair of single copies of unequal length. The overall chloroplast genome size of between 150 and 151 kb, was comparable to the majority of chloroplast genomes of species within the Asteraceae family (Salih et al. 2017). The GC content of species within each of the two genera was identical 37.2% in Senecio and 37.5% in Dendrosenecio (Table 1, Fig. 1). Usually, chloroplast genomes exhibit a high AT-GC content ratio (Ravi et al. 2008), which is a crucial factor in genome organization and stability (Niu et al. 2017).
Chloroplasts have undergone enormous changes since they evolved from cyanobacteria in over a billion years ago (Timmis et al. 2004). One of the notable transformations is the reduction in size, which is primarily accredited to considerable gene transfer from the chloroplast genome to the nuclear genome (Bock and Timmis 2008; Martin et al. 1998). Generally, the chloroplast genomes of higher plants currently encode 70–90 protein-coding genes, which is approximately 2% of the total PCGs found in cyanobacterium Synechocystis (Eckardt 2006). The number of unique genes encoded in the available chloroplast genomes of Asteraceae varies slightly between 110 and 115 (Dempewolf et al. 2010; Doorduin et al. 2011; Kumar et al. 2009; Lu et al. 2016; Walker et al. 2014). The 11 species reported here encoded an equal number of 114 unique genes, with 80 genes encoding for protein, similar to the chloroplast genomes of Taraxacum F.H. Wigg. (Asteraceae; Salih et al. 2017). The exon of some specific genes has been subjected to interruptions by either a single or several introns. In such cases, the entire sequence, containing both the exon and intron(s), is transcribed into a forerunner RNA and later the introns are detached for accurate production of a proper transcript (Eckardt 2007; Plant and Gray 1988). The rps12 gene is distinctively placed, with its 5′-end being positioned in the LSC region and the 3′-end is duplicated in the IR section, in a way similar to that of most other angiosperms including Nicotiana tabacum (Hildebrand et al. 1988). Seventeen genes, in each of the studied species, had either a single or several introns (Table 2). Excluding rps12 gene whose intron is exceptionally large, trnK-UUU had the largest single intron. Two genes, ycf3 and clpP, had two introns within their exons this arrangement has also been observed in other Asteraceae species including Artemisia annua L. (Shen et al. 2017). However, in Ageratina adenophora (Spreng.) R.M. King. & H. Rob., the rpoC1 gene has two introns (Nie et al. 2012), which is considered rare among other Asteraceae species.
Codon usage and repetitive sequences
Relative synonymous codon usage values of less than one indicate that the codons are less frequent, > 1 represents those that are more frequently used, whereas = 1 shows no bias (Uddin 2017). An identical trend in the manner in which the amino acids were encoded was discovered among the 11 species (Fig. 2, Additional file 2: Table S2). The usage frequency of leucine was higher than the rest, while cysteine had the least frequency which is congruent to most Asteraceae species e.g., (Salih et al. 2017; Shen et al. 2017). It was also observed that usage of synonymous codons was generally biased in favour of those ending with A/U bases. Consequently, some codons were over- (> 1.6) or under- (< 0.6) represented. In particular, only trnL-UAA, trnS-UCU, trnT-ACU, trnA-GCU, trnY-UAU, trnR-AGA, and stop codon UAA were over-represented, whereas the majority were under-represented (Additional file 2: Table S2). Methionine (AUG) and tryptophan (UGG) were uniformly used (RSCU = 1). Other indices of non-uniformity in codon usage include the Effective Number of codons (ENc), which ranges from 20 (one codon per amino acid) to 61 (equal use of synonymous codons; Wright 1990) and Codon Bias Index (CBI) which ranges from 0, no bias to 1 equal usage of all the synonymous codon (Morton 1993). The values for both ENc (49.76 to 51.49) and CBI (0.308 to 0.356) were insignificantly different among the species of Dendrosenecio, but similar to most species in Asteraceae (Nie et al. 2014). The common start codon for the protein coding genes is AUG (M) however, three genes, psbL, rps19, and ndhD deviated from the norm, and they had ACG, GUG, and GUG respectively.
Microsatellite repeats are abundantly distributed in the genome (Tautz and Renz 1984), and they display a high level of polymorphism, placing them among the most preferred genetic markers for genetic investigations. A total of 340 and 331 SSRs were discovered in six species of Dendrosenecio and five Senecio species. The majority were mononucleotides, followed by dinucleotides and tetranucleotides (Additional file 3: Table S3). Mononucleotides, usually A/T repeat types, are abundantly present in chloroplast genomes of Asteraceae species, e.g., Jacobaea vulgaris (Doorduin et al. 2011) Artemisia annua (Shen et al. 2017) and other families e.g., Paeonia ostii T. Hong & Z. X. Zhang (Paeoniaceae; Guo et al. 2018). In Dendrosenecio, a majority of the SSRs were located on homologous regions and except for five, the rest lacked any variations in terms of length and motif. On the contrary, 25 microsatellites in Senecio exhibited slight variations based on the same criteria. Being considered polymorphic, the identified microsatellites are therefore potential molecular markers for use in further studies within the respective genus.
Chloroplast genome comparison
There were no remarkable structural rearrangements among the taxa of these two genera. The chloroplast genomes are highly conserved with an identical structure and an equal number of genes, an indication that this could be the case in chloroplast genomes of most species of these genera. Comparative analyses against representatives of three other genera of Senecioneae revealed a similar trend of the well-preserved structure and organization. The inverted repeat region is present in a majority of angiosperms chloroplast genomes. Initially the IR was reported to serve as a whole-genome stabilizer by reducing recombination between the two SC regions; however, these reports lacked support as more chloroplast genomes revealed significant rearrangements even with both copies of the IR present (Jansen and Ruhlman 2012). Comparative analyses between plants of different plant lineages revealed that inverted repeats could contract/expand up to a few hundred base pairs even among closely related species (Goulding et al. 1996). In this study, the comparison of the IR/SC junctions showed a slight expansion of the IR in all except in Senecio moorei and Pericallis hybrida. In the novel chloroplast genomes, the same genes were found adjacent to the junctions, and only slight length variations were recorded in P. hybrida and S. moorei. In P. hybrida the LSC/IRb junction contracted into the rpl2 gene, whereas it extended into the rps19 in all the other analysed species. Two pseudogenes (ψrps19 and ψycf1) of varying length were generated in the IRa region, as a result of the expansion of the IR into the exons of rps19 and ycf1 genes respectively (Fig. 4). This pattern of expansion of the IR, and the introduction of partial copies of genes with non-coding abilities represents a familiar phenomenon in majority of Asteraceae species (Wang et al. 2015), and besides being a source of DNA barcodes, it can offer insights into the evolutionary processes of plastid genomes.
The entire sequences of the 11 chloroplast genomes generated here, lack any striking inversions or rearrangements and therefore were outlined as a single locally collinear block in our analyses. However, certain regions harboured divergent sites the majority of which were in the non-coding regions. Among the few sites with significant deviations include trnH-(GUG)-psbA, ndhD-ccsA, trnT(UGU)-trnL(UAA), ndhI-ndhG, and trnL-UAG-rpl32. Other regions including trnL(UAG)-rpl32 and the exons of ndhF and ycf1 were within a conserved block, but they had significant divergent points. These findings were supported by results obtained from the DNA polymorphism test based on genus representatives, as same regions were noted to have high nucleotide variability (Pi). Some of these regions have previously been reported in chloroplast genomes of other species (Salih et al. 2017; Wu et al. 2018) and used in phylogenetic studies of numerous taxa including Senecio (Kandziora et al. 2016). Non-coding regions in chloroplast genomes have shown high potential for use as molecular markers for phylogenetic studies at low taxonomic levels in Angiosperms (Shaw et al. 2005). Therefore, the regions identified herein are prospective sources of highly informative markers for phylogenetic utility in elucidating intergeneric relationships within the tribe. Subsequently, ten potential markers were developed, allowing for specific amplification of each of the ten most polymorphic sites.
We compared the Senecioneae chloroplast genomes against Nicotiana tabacum and detected the two inversions reported to be shared by all clades of Asteraceae family, apart from species of the Barnadesioideae subfamily (Kim et al. 2005). Six conserved gene blocks were identified among the chloroplast genomes indicating the most conserved regions of the genomes. The SSC region in Ligularia fischeri was differently oriented in relation to the other Asteraceae species. This re-inversion is considered an ordinary phenomenon among chloroplast genomes of higher plants, and it is not a product of any evolutionary event, as single-copy regions exist in two equimolar states (Palmer 1983; Walker et al. 2015).
Phylogenomics analyses
The rapid increase in the amount of complete chloroplast genome sequences during the past decade, provided essential data to elucidate further and resolve phylogenetic relationships among species. Consequently, in a move towards chloroplast phylogenomics, clarification of phylogenetic relationships at higher and lower taxonomic levels have been achieved (Lu et al. 2015; Ma et al. 2014; Wu et al. 2013). The tribe Senecioneae is often subdivided into three subtribes; Senecioninae, Tussilagininae, and Blennospermatinae (Chen et al. 2011). In this study, the multi-gene analysis resulted in a phylogenetic tree whose branches were strongly supported. The five genera of Senecioneae formed two distinct clades that corresponded to two of the three subtribes of Senecioneae including Senecioninae (Dendrosenecio, Senecio, Jacobaea and Pericallis) and Tussilagininae (Ligularia; Fig. 7, Additional file 5: Figure S2). The 11 newly generated species were well placed within the Senecioneae tribe by both ML and BI phylogenetic methods under partitioned and unpartitioned data schemes. The afro-alpine species of Senecio are classified in at least five clades of Senecio (Kandziora et al. 2016). The present phylogenetic study recovered a monophyletic group with two sub-clades which split S. keniophytum and S. roseiflorus from S. purtschelleri, S. moorei and S. schweinfurthii. The relationships among the species of Senecio was identical in all data schemes differing only in support of the S. moorei and S. schweinfurthii relationship (94% and 0.3 PP; Fig. 7). The genus Jacobaea, just like Dendrosenecio, was previously classified in Senecio under section Jacobaea (Pelser et al. 2002) but later segregated from Senecio based on new insights from molecular phylogeny (Pelser et al. 2006). A sister relationship between Senecio and Jacobaea was highlighted in this study.
Previously, genetic relationship within Dendrosenecio species was shown to be strongly correlated to geographic distance (Knox and Palmer 1995a) as geographically close species were genetically more related than distantly located species. In this study two clades were formed within a monophyletic group of Dendrosenecio, one contained D. johnstonii and D. meruensis, both from Tanzanian mountains. The other clade had species from Kenyan mountains; D. keniodendron, and D. battiscombei (Mt. Kenya), D. elgonensis subsp. elgonensis (Mt. Elgon) and D. brassiciformis, which was sampled from the Aberdare ranges (Fig. 7a). However, the sister relationships among the Kenyan species was conflicting between the ML and the BI phylogenetic reconstructions. In both ML trees a clear distinction is established concerning geographical (different mountains) and altitudinal (same mountain) variations (Fig. 7a; Additional file 5: Figure S2a), though this correlation is missing in the BI tree (Fig. 7b; Additional file 5: Figure S2b). Therefore, there is need to carry out further analyses, including more species of Dendrosenecio from all habitats in order to make a comprehensive conclusion. A majority of the intergeneric relationships defined here were significantly supported and congruent to most of the previous studies based on a few DNA fragments, including the position of Parastrephia quadrangularis (Meyen) Cabrera within the species of Diplostephium Kunth (Vargas et al. 2017). Therefore, this study strongly underscored the potential of chloroplast genome-scale data in outlining both inter- and intra-generic phylogenetic relationships within the tribe Senecioneae species and in the family at large. Nonetheless, interspecific relationships were weakly supported and therefore, further comprehensive studies that include more taxon sampling are necessary to enhance our understanding of the evolutionary histories of both Senecio and Dendrosenecio.
Conclusion
Dendrosenecio is a segregate genus to Senecio. Despite exhibiting some striking morphologic similarities, a few differences existed based on which the two genera were separated. Initially, controversies arose over the segregation although a consensus was later arrived at. Amplified fragment length polymorphism data distinctly separated the two genera, affirming the earlier decisions. However, lack of or limited molecular resources have impeded further studies on the respective genera. This study generated the first complete chloroplast genome sequences in each genus. Chloroplast genomes in both genera are highly similar in structure, gene composition and synteny, but they significantly differ in size. A chloroplast genome multi-gene dataset revealed three strongly supported clades within the tribe Senecioneae, markedly splitting Dendrosenecio from Senecio. Ten primers, targeting the ten highly divergent regions in the chloroplast genomes of Senecioneae species, were designed. Also, 25 polymorphic cpSSR in Senecio and five in Dendrosenecio were identified. The ten divergent hotspots could offer the much-needed DNA barcodes for species identification and phylogenetic reconstructions within the tribe, while the cpSSRs provides potential markers for future population-level research in each respective genus.
Additional files
Additional file 1: Table S1. Details of the Asteraceae species used in the phylogenetic analyses.
Additional file 2: Table S2. Details of Relative Synonymous Codon Usage in chloroplast genomes of 11 species of Senecioneae.
Additional file 3: Table S3. Number and type of microsatellite repeat motifs in each of the 11 complete chloroplast genomes.
{kind=link}
Additional file 4: Figure S1. Comparison of sequence arrangement in the chloroplast genomes of five species of Senecioneae (Asteraceae), against Nicotiana tabacum as an external reference genome. Conserved orthologs are indicated by locally collinear blocks. Similar blocks among the genomes are coded in one colour and joined by a line. The genes above the line are transcribed in a clockwise direction, those below the line are transcribed towards the counter-clockwise direction.
{kind=link}
Additional file 5: Figure S2. Phylogenetic relationships of 75 species of Asteraceae inferred from a partitioned chloroplast genome multi-gene dataset using (a) Maximum Likelihood (ML) and (b) Bayesian Inference (BI) methods.
Acknowledgements
We thank Elizabeth M. Kamande for her support in fieldwork, Josphat K. Saina for his assistance in data analyses, and Justus M. Mulinge for proofreading the manuscript.
Abbreviations
- ITS
internal transcribed spacer
- IR
inverted repeat
- SSC
small single copy
- LSC
large single copy
- SSR
simple sequence repeat
- CTAB
cetyltrimethylammonium bromide
- PCGs
protein-coding genes
- RNA
ribonucleic acid
- RSCU
relative synonymous codon usage
- CUB
codon usage bias
- ENc
effective number of codons
- ML
maximum likelihood
- BS
bootstrap support
- BI
Bayesian inference
- PP
posterior probability
- cpSSRs
chloroplast microsatellites
Authors’ contributions
QF, GH, and JC conceived and designed the experiment. AWG, SA, GH, and JC conducted fieldwork. AWG and SA performed the experiments. AG, SA, and ZL analysed the data. AG wrote the manuscript. ZL assisted in revising the manuscript. All authors read and approved the final manuscript.
Funding
This work was funded by Sino Africa Joint Research Center (Nos. Y323771W07 and SAJC201322) and the CAS-TWAS President’s Fellowship Program for developing countries.
Availability of data and materials
The datasets generated during the current study are available in the GenBank repository under the accession numbers KY434193–KY434195, MG560049–MG560051 and MH483946–MH483950. All the datasets used for phylogenetic and comparative analyses were downloaded from GenBank, and the accession numbers are provided in the additional files.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Andrew Wanyoike Gichira and Sheila Avoga contributed equally to this work
Contributor Information
Qingfeng Wang, Email: qfwang@wbgcas.cn.
Jinming Chen, Email: jmchen@wbgcas.cn.
References
- Amiryousefi A, Hyvönen J, Poczai P. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 2018 doi: 10.1093/bioinformatics/bty220. [DOI] [PubMed] [Google Scholar]
- Beentje HJ, Jeffrey C, Hind DJN. Compositae (Part 3) In: Beentje HJ, Ghazanfar SA, editors. Flora of tropical east Africa. London: Royal Botanic Gardens, KEW; 2005. pp. 547–869. [Google Scholar]
- Bock R, Timmis JN. Reconstructing evolution: gene transfer from plastids to the nucleus. BioEssays. 2008;30:556–566. doi: 10.1002/bies.20761. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinform. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YL, et al. Senecioneae. In: Wu ZY, Raven PH, Hong DY, et al., editors. Flora of China, vol 20–21. Beijing: Missouri Botanical Garden Press; 2011. pp. 371–544. [Google Scholar]
- Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheon K-S, Kim K-A, Yoo K-O. The complete chloroplast genome sequences of three Adenophora species and comparative analysis with Campanuloid species (Campanulaceae) PLoS ONE. 2017;12:e0183652–e0183652. doi: 10.1371/journal.pone.0183652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempewolf H, et al. Establishing genomic tools and resources for Guizotia abyssinica (L.f.) Cass.—the development of a library of expressed sequence tags, microsatellite loci, and the sequencing of its chloroplast genome. Mol Ecol Resour. 2010;10:1048–1058. doi: 10.1111/j.1755-0998.2010.02859.x. [DOI] [PubMed] [Google Scholar]
- Dierckxsens N, Mardulyn P, Smits G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017;45:e18. doi: 10.1093/nar/gkw955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorduin L, Gravendeel B, Lammers Y, Ariyurek Y, Chin-A-Woeng T, Vrieling K. The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs, microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 2011;18:93–105. doi: 10.1093/dnares/dsr002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downie SR, Jansen RK. A comparative analysis of whole plastid genomes from the Apiales: expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Syst Bot. 2015;40:336–351. doi: 10.1600/036364415X686620. [DOI] [Google Scholar]
- Doyle JJ. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull Bot Soc Am. 1987;19:11–15. [Google Scholar]
- Eckardt NA. Genomic Hopscotch: gene transfer from plastid to nucleus. Plant Cell. 2006;18:2865. doi: 10.1105/tpc.106.049031. [DOI] [Google Scholar]
- Eckardt NA. Chloroplast intron splicing mechanisms. Plant Cell. 2007;19:3838. doi: 10.1105/tpc.108.191210. [DOI] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding SE, Olmstead RG, Morden CW, Wolfe KH. Ebb and flow of the chloroplast inverted repeat. Mol Gen Genet. 1996;252:195–206. doi: 10.1007/BF02173220. [DOI] [PubMed] [Google Scholar]
- Guisinger MM, Kuehl JV, Boore JL, Jansen RK. Extreme reconfiguration of plastid genomes in the Angiosperm family Geraniaceae: rearrangements, repeats, and codon usage. Mol Biol Evol. 2011;28:583–600. doi: 10.1093/molbev/msq229. [DOI] [PubMed] [Google Scholar]
- Guo S, et al. Complete chloroplast genome sequence and phylogenetic analysis of Paeonia ostii. Molecules. 2018 doi: 10.3390/molecules23020246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedberg O. Afroalpine vascular plants: a taxonomic revision. Symb Bot Upsal. 1957;15:1–411. [Google Scholar]
- Hegarty MJ, Abbott RJ, Hiscock SJ. Allopolyploid speciation in action: the origins and evolution of Senecio cambrensis. In: Soltis PS, Soltis DE, editors. Polyploidy and genome evolution. Berlin: Springer; 2012. pp. 245–270. [Google Scholar]
- Hildebrand M, Hallick RB, Passavant CW, Bourque DP. Trans-splicing in chloroplasts: the rps 12 loci of Nicotiana tabacum. Proc Natl Acad Sci USA. 1988;85:372–376. doi: 10.1073/pnas.85.2.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen RK, Ruhlman TA. Plastid genomes of seed plants. In: Bock R, Knoop V, editors. Genomics of chloroplasts and mitochondria. Dordrecht: Springer Netherlands; 2012. pp. 103–126. [Google Scholar]
- Jeffrey C, Chen Y-l. Taxonomic studies on the tribe Senecioneae (Compositae) of Eastern Asia. Kew Bull. 1984;39:205–446. doi: 10.2307/4110124. [DOI] [Google Scholar]
- Jeffrey C, Halliday P, Wilmot-Dear M, Jones SW. Generic and sectional limits in Senecio (Compositae): I. Progress report. Kew Bull. 1977;32:47–67. doi: 10.2307/4117259. [DOI] [Google Scholar]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandziora M, Kadereit JW, Gehrke B. Frequent colonization and little in situ speciation in Senecio in the tropical alpine-like islands of eastern Africa. Am J Bot. 2016;103:1483–1498. doi: 10.3732/ajb.1600210. [DOI] [PubMed] [Google Scholar]
- Kim K-J, Choi K-S, Jansen RK. Two chloroplast DNA inversions originated simultaneously during the early evolution of the sunflower family (Asteraceae) Mol Biol Evol. 2005;22:1783–1792. doi: 10.1093/molbev/msi174. [DOI] [PubMed] [Google Scholar]
- Knox EB. Dendrosenecio. In: Beentje HJ, Jeffrey C, Hind DJN, editors. Flora of Tropical East Africa, Compositae (part 3) London: Kew, Royal Botanic Gardens; 2005. [Google Scholar]
- Knox EB. The dynamic history of plastid genomes in the Campanulaceae sensu lato; is unique among angiosperms. Proc Natl Acad Sci USA. 2014;111:11097. doi: 10.1073/pnas.1403363111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox EB, Palmer JD. Chloroplast DNA variation and the recent radiation of the giant senecios (Asteraceae) on the tall mountains of eastern Africa. Proc Natl Acad Sci USA. 1995;92:10349–10353. doi: 10.1073/pnas.92.22.10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox EB, Palmer JD. The origin of Dendrosenecio within the Senecioneae (Asteraceae) based on chloroplast DNA evidence. Am J Bot. 1995;82:1567–1573. doi: 10.2307/2446185. [DOI] [Google Scholar]
- Kumar S, Hahn FM, McMahan CM, Cornish K, Whalen MC. Comparative analysis of the complete sequence of the plastid genome of Parthenium argentatum and identification of DNA barcodes to differentiate Parthenium species and lines. BMC Plant Biol. 2009;9:131. doi: 10.1186/1471-2229-9-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 2017;34:772–773. doi: 10.1093/molbev/msw260. [DOI] [PubMed] [Google Scholar]
- Lee J, Lee H, Lee S-C, Sung SH, Kang JH, Lee TJ, Yang T-J. The complete chloroplast genome sequence of Ligularia fischeri (Ledeb.) Turcz. (Asteraceae) Mitochondrial DNA Part B. 2016;1:4–5. doi: 10.1080/23802359.2015.1137793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse M, Drechsel O, Bock R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2007;52:267–274. doi: 10.1007/s00294-007-0161-y. [DOI] [PubMed] [Google Scholar]
- Lu J-M, Zhang N, Du X-Y, Wen J, Li D-Z. Chloroplast phylogenomics resolves key relationships in ferns. J Syst Evol. 2015;53:448–457. doi: 10.1111/jse.12180. [DOI] [Google Scholar]
- Lu C, Shen Q, Yang J, Wang B, Song C. The complete chloroplast genome sequence of Safflower (Carthamus tinctorius L.) Mitochondrial DNA A DNA Mapp Seq Anal. 2016;27:3351–3353. doi: 10.3109/19401736.2015.1018217. [DOI] [PubMed] [Google Scholar]
- Ma PF, Zhang YX, Zeng CX, Guo ZH, Li DZ. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae) Syst Biol. 2014;63:933–950. doi: 10.1093/sysbio/syu054. [DOI] [PubMed] [Google Scholar]
- Mabberley DJ. Evolution in the giant groundsels. Kew Bull. 1973;28:61–96. doi: 10.2307/4117066. [DOI] [Google Scholar]
- Marechal A, Brisson N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010;186:299–317. doi: 10.1111/j.1469-8137.2010.03195.x. [DOI] [PubMed] [Google Scholar]
- Martin W, Stoebe B, Goremykin V, Hansmann S, Hasegawa M, Kowallik KV. Gene transfer to the nucleus and the evolution of chloroplasts. Nature. 1998;393:162. doi: 10.1038/30234. [DOI] [PubMed] [Google Scholar]
- Milton JJ. Phylogenetic analyses and taxonomic studies of Senecioninae: southern African Senecio section Senecio. St Andrews: University of St Andrews; 2009. [Google Scholar]
- Morton BR. Chloroplast DNA codon use: evidence for selection at the psb A locus based on tRNA availability. J Mol Evol. 1993;37:273–280. doi: 10.1007/bf00175504. [DOI] [PubMed] [Google Scholar]
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie X, et al. Complete chloroplast genome sequence of a major invasive species, Crofton weed (Ageratina adenophora) PLoS ONE. 2012;7:e36869. doi: 10.1371/journal.pone.0036869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie X, Deng P, Feng K, Liu P, Du X, You FM, Weining S. Comparative analysis of codon usage patterns in chloroplast genomes of the Asteraceae family. Plant Mol Biol Rep. 2014;32:828–840. doi: 10.1007/s11105-013-0691-z. [DOI] [Google Scholar]
- Niu Z, Xue Q, Wang H, Xie X, Zhu S, Liu W, Ding X. Mutational biases and GC-biased gene conversion affect GC content in the plastomes of Dendrobium genus. Int J Mol Sci. 2017;18:2307. doi: 10.3390/ijms18030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordenstam B. Taxonomic studies in the tribe Senecioneae (Compositae) Opera Bot Lund. 1978;44:1–84. [Google Scholar]
- Nordenstam B. Tribe Senecioneae. In: Kadereit JW, Jeffrey C, Kubitzki K, editors. The families and genera of vascular plants. Berlin: Springer; 2007. pp. 208–241. [Google Scholar]
- Nordenstam B, Pelser PB, Kadereit JW, Watson LE. Senecioneae. In: Funk VA, Susanna A, Stuessy TF, Bayer RJ, editors. Systematics, evolution, and biogeography of composiate. Vienna: International Association for Plant Taxonomy; 2009. [Google Scholar]
- Palmer JD. Chloroplast DNA exists in two orientations. Nature. 1983;301:92. doi: 10.1038/301092a0. [DOI] [Google Scholar]
- Pelser PB, Gravendeel B, van der Meijden R. Tackling speciose genera: species composition and phylogenetic position of Senecio sect. Jacobaea (Asteraceae) based on plastid and nrDNA sequences. Am J Bot. 2002;89:929–939. doi: 10.3732/ajb.89.6.929. [DOI] [PubMed] [Google Scholar]
- Pelser PB, Veldkamp JF, van der Meijden R. New combinations in Jacobaea Mill. (Asteraceae–Senecioneae) Compos Newsl. 2006;44:1–11. [Google Scholar]
- Pelser PB, Nordenstam B, Kadereit JW, Watson LE. An ITS phylogeny of tribe Senecioneae (Asteraceae) and a new delimitation of Senecio L. Taxon. 2007;56:1077–1104. doi: 10.2307/25065905. [DOI] [Google Scholar]
- Plant AL, Gray JC. Introns in chloroplast protein-coding genes of land plants photosynthesis. Research. 1988;16:23–39. doi: 10.1007/bf00039484. [DOI] [PubMed] [Google Scholar]
- Pouchon C, Fernández A, Nassar JM, Boyer F, Aubert S, Lavergne S, Mavárez J. Phylogenomic analysis of the explosive adaptive radiation of the Espeletia complex (Asteraceae) in the tropical Andes. Syst Biol. 2018;56:67. doi: 10.1093/sysbio/syy022. [DOI] [PubMed] [Google Scholar]
- Ravi V, Khurana JP, Tyagi AK, Khurana P. An update on chloroplast genomes. Plant Syst Evol. 2008;271:101–122. doi: 10.1007/s00606-007-0608-0. [DOI] [Google Scholar]
- Ronquist F, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sanchez-Gracia A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34:3299–3302. doi: 10.1093/molbev/msx248. [DOI] [PubMed] [Google Scholar]
- Salih M, Majeský Ľ, Schwarzacher T, Gornall R, Heslop-Harrison P. Complete chloroplast genomes from apomictic Taraxacum (Asteraceae): identity and variation between three microspecies. PLoS ONE. 2017;12:e0168008. doi: 10.1371/journal.pone.0168008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li W-H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J, et al. The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am J Bot. 2005;92:142–166. doi: 10.3732/ajb.92.1.142. [DOI] [PubMed] [Google Scholar]
- Shen X, et al. Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia annua. Molecules. 2017;22:1330. doi: 10.3390/molecules22081330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki K, et al. The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J. 1986;5:2043–2049. doi: 10.1002/j.1460-2075.1986.tb04464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D, Renz M. Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucleic Acids Res. 1984;12:4127–4138. doi: 10.1093/nar/12.10.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel T, Michalek W, Varshney R, Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.) Theor Appl Genet. 2003;106:411–422. doi: 10.1007/s00122-002-1031-0. [DOI] [PubMed] [Google Scholar]
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. GeSeq—versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017;45:W6–W11. doi: 10.1093/nar/gkx391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmis JN, Ayliffe MA, Huang CY, Martin W. Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat Rev Genet. 2004;5:123. doi: 10.1038/nrg1271. [DOI] [PubMed] [Google Scholar]
- Tonti-Filippini J, Nevill PG, Dixon K, Small I. What can we do with 1000 plastid genomes? Plant J. 2017;90:808–818. doi: 10.1111/tpj.13491. [DOI] [PubMed] [Google Scholar]
- Uddin A. Indices of codon usage bias. J Proteom Bioinform. 2017;10:e34. doi: 10.4172/jpb.1000e34. [DOI] [Google Scholar]
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas OM, Ortiz EM, Simpson BB. Conflicting phylogenomic signals reveal a pattern of reticulate evolution in a recent high-Andean diversification (Asteraceae: Astereae: Diplostephium) New Phytol. 2017;214:1736–1750. doi: 10.1111/nph.14530. [DOI] [PubMed] [Google Scholar]
- Walker JF, Zanis MJ, Emery NC. Comparative analysis of complete chloroplast genome sequence and inversion variation in Lasthenia burkei (Madieae, Asteraceae) Am J Bot. 2014;101:722–729. doi: 10.3732/ajb.1400049. [DOI] [PubMed] [Google Scholar]
- Walker JF, Jansen RK, Zanis MJ, Emery NC. Sources of inversion variation in the small single copy (SSC) region of chloroplast genomes. Am J Bot. 2015;102:1751–1752. doi: 10.3732/ajb.1500299. [DOI] [PubMed] [Google Scholar]
- Wang M, et al. Comparative analysis of Asteraceae chloroplast genomes: structural organization, RNA editing and evolution. Plant Mol Biol Rep. 2015;33:1526–1538. doi: 10.1007/s11105-015-0853-2. [DOI] [Google Scholar]
- Wang B, Li M, Yuan Y. The complete chloroplast genome of Pericallis hybrida (Asteridae) Mitochondrial DNA Part B. 2019;4:233–234. doi: 10.1080/23802359.2018.1542999. [DOI] [Google Scholar]
- Wicke S, Schneeweiss GM, dePamphilis CW, Müller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol. 2011;76:273–297. doi: 10.1007/s11103-011-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- Wu C-S, Chaw S-M, Huang Y-Y. Chloroplast phylogenomics indicates that Ginkgo biloba is sister to Cycads. Genome Biol Evol. 2013;5:243–254. doi: 10.1093/gbe/evt001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M-l, Li Q, Xu J, Li X-w. Complete chloroplast genome of the medicinal plant Amomum compactum: gene organization, comparative analysis, and phylogenetic relationships within Zingiberales. Chin Med. 2018;13:10. doi: 10.1186/s13020-018-0164-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurina NP, Sharapova LS, Odintsova MS. Structure of plastid genomes of photosynthetic eukaryotes. Biochemistry. 2017;82:678–691. doi: 10.1134/s0006297917060049. [DOI] [PubMed] [Google Scholar]
- Zhang D, Gao F, Li WX, Jakovlić I, Zou H, Zhang J, Wang GT. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. bioRxiv. 2018 doi: 10.1101/489088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Details of the Asteraceae species used in the phylogenetic analyses.
Additional file 2: Table S2. Details of Relative Synonymous Codon Usage in chloroplast genomes of 11 species of Senecioneae.
Additional file 3: Table S3. Number and type of microsatellite repeat motifs in each of the 11 complete chloroplast genomes.
Additional file 4: Figure S1. Comparison of sequence arrangement in the chloroplast genomes of five species of Senecioneae (Asteraceae), against Nicotiana tabacum as an external reference genome. Conserved orthologs are indicated by locally collinear blocks. Similar blocks among the genomes are coded in one colour and joined by a line. The genes above the line are transcribed in a clockwise direction, those below the line are transcribed towards the counter-clockwise direction.
Additional file 5: Figure S2. Phylogenetic relationships of 75 species of Asteraceae inferred from a partitioned chloroplast genome multi-gene dataset using (a) Maximum Likelihood (ML) and (b) Bayesian Inference (BI) methods.
Data Availability Statement
The datasets generated during the current study are available in the GenBank repository under the accession numbers KY434193–KY434195, MG560049–MG560051 and MH483946–MH483950. All the datasets used for phylogenetic and comparative analyses were downloaded from GenBank, and the accession numbers are provided in the additional files.