

Abstract
Dexmedetomidine (Dex) is an anesthetic widely used in lumbar discectomy, but its effect on chondrocytes remains unclear. Dex is speculated to promote cartilage degeneration by activating α-2 adrenergic receptor. However, the antioxidative and anti-inflammatory effects of Dex implied the potential chondrocyte protective effect under stress conditions. The present study aimed to determine the effect of Dex on chondrocytes under non-stress and stress conditions. Chondrocytes were isolated from human annulus fibrosus (AF) tissues and oxidative stress was induced by treatment with 1 mM hydrogen peroxide (H2O2). Chondrocytes were treated with Dex alone or in combination with H2O2. Treatment with Dex alone decreased mRNA expression of COL2A1 and increased that of MMP-3 and MMP-13, thus contributing to cartilage degeneration. However, Dex prevented H2O2-induced death and degeneration of chondrocytes partly by enhancing antioxidant capacity. Mechanistically, Dex attenuated H2O2-mediated activation of NF-κB and NACHT, LRR, and PYD domains-containing protein 3 (NLRP3), both of which play key roles in inflammation and inflammatory damage. Dex inactivated NLRP3 through the suppression of NF-κB and JNK signals. Co-treatment with Dex and H2O2 increased protein level of XIAP (X-linked inhibitor-of-apoptosis, an anti-apoptosis protein), compared with H2O2 treatment alone. H2O2 treatment increased the expression of neural precursor cell expressed developmentally down-regulated protein 4 (NEDD4) that is a ubiquitin ligase targeting XIAP. However, Dex decreased the amount of NEDD4 adhering to XIAP, thus protecting XIAP protein from NEDD4-mediated ubiquitination and degradation. Given that surgery inevitably causes oxidative stress and inflammation, the protective effect of Dex on chondrocytes during oxidative stress is noteworthy and warrants further study.
Keywords: annulus fibrosus chondrocytes, cartilage degeneration, Dexmedetomidine, NF-κB, NLRP3, XIAP
Introduction
The intervertebral disc (IVD) is the largest avascular organ in the human body, and metabolic exchange is predominantly reliant on the diffusion effect across cartilage endplate. Due to these structural and metabolic characteristics, chondrocytes within the IVD are sensitive to multiple etiological factors including aging, smoking, infection, abnormal mechanical stress, diabetes, trauma, and genetic predisposition [1]. Accumulative evidence indicates that overproduction of reactive oxygen species (ROS), which results in oxidative stress, plays a critical role in the damage of chondrocytes upon exposure to these etiological factors [1]. Excessive ROS reinforces inflammation and degradation of the extracellular matrix, changes cartilaginous phenotype, accelerates senescence of disc cells, and even triggers programmed cell death [2,3]. These pathological reactions are dependent on ROS acting as crucial intermediary factors in the signaling network and on the strong oxidation characteristics. These pathological changes consequently reduce the ability of IVD to bear weight causing several pathophysiological features, such as lower back pain (LBP), sciatica, and cauda equina symptoms. Each year in China, LBP affects millions of people, especially those over 50 years of age, which largely increases the social burden.
The adrenoreceptor (AR) family of proteins comprises α1, α2, and β subtypes and are constitutively expressed in chondrocytes. It has been identified that three subtypes of ARs are involved in the modulation of various physiological and pathological actions of chondrocytes. α1-AR mediates norepinephrine (NE)-induced activation of caspase-3 and 7 and subsequent apoptosis of chondrocytes, since α1-AR inhibitor, but not inhibitors of α2- and β-AR, abolished the apoptotic effect of NE [4]. Activation of α2-AR by NE decreases the expression of aggrecans but stimulates the production of MMP-3, MMP-13, and RANKL by chondrocytes, which suggests the potential role of α2-AR in cartilage degeneration [5]. However, these effects of α2-AR are dependent on the activation of the ERK1/2 and PKA pathways [5]. It is known that these signaling cascades are critical for cell proliferation and survival. Stimulation of β(2)-AR by isoproterenol inhibited mRNA and protein levels of Col II and Sox-6 by activating ERK1/2 and PKA [6]. Activation of β-AR by NE slows down the cell cycle and decreases cell proliferation [4]. Nevertheless, NE reverses IL-1β-induced gene expression of IL-8, MMP-13, glycosaminoglycan, and collagen II via β-AR activation [4], thereby exhibiting an anti-inflammatory effect.
Dexmedetomidine (Dex), a selective agonist of α2-AR, has been increasingly used during or after surgery for its sedative, analgesic, and sympatholytic effects. Theoretically, the activation of α2-AR by Dex likely induces cartilage degeneration by modulating the expression of aggrecans, MMP-3, MMP-13, and RANKL. However, substantial data indicate that Dex confers strong protection against cell damage caused by oxidative stress, inflammation, and hypoxia [7–9]. These protective effects of Dex are primarily due to its regulation of signaling molecules such as NACHT, LRR, and PYD domains-containing protein 3 (NLRP3), Nrf-2, NF-κB, ERK1/2, and PKA through the activation of α2-AR [7–9]. Since the protective effects of Dex has been reported in neurocytes, nephrocytes, and cardiomyocytes, we hypothesized that Dex could also exert protective effect on chondrocytes, especially when they are exposed to ROS. ROS are the key inducers of degeneration and death of chondrocytes. The effect of Dex against ROS is supposed to hinder this phenomenon. Thus, it is likely that Dex plays different roles in the degeneration of chondrocytes under non-stress and stress conditions.
Lumbar discectomy is a common surgical procedure for the management of LBP resulting from IVD degeneration, but this surgery inevitably causes tissue damage and thus triggers oxidative stress and inflammation. Given that Dex is widely used in lumbar discectomy to date, it is worthwhile to investigate the effect of Dex on chondrocytes under stress conditions. The present study aimed to determine the effect of Dex on IVD chondrocytes against oxidative stress induced by hydrogen peroxide (H2O2) and the underlying mechanism of Dex.
Materials and methods
Isolation and culture of human chondrocytes
Patients [four males and four females, mean age = 56 years (age: 43–72 years)] with LBP caused by disc herniation in the lumbar region, were enrolled in the present study. All participants provided written informed consent. The present study was approved by the Local Ethics Committee of Hunan People’s Hospital (Changsha, People’s Republic of China). These patients underwent lumbar discectomy by epiduroscopy, which is a minimally invasive technique, in Hunan People’s Hospital between January 2017 and March 2018. Human chondrocytes were isolated from annulus fibrosus (AF) tissues that were obtained from lumbar discectomy. The AF tissues were minced into small pieces (1 mm3) and digested with 0.25% trypsin for 30 min at 37°C. After three washes with PBS, the samples were further treated with 0.2% collagenase type II for 4 h at 37°C. Isolated chondrocytes were passed through a 70-mm nylon mesh (Falcon) to remove residual cartilage matrix fragments. The isolated cells were cultured in complete culture medium (DMEM/F12, Life Technologies, Carlsbad, CA, U.S.A.) supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, U.S.A.) and 1% penicillin/streptomycin (Life Technologies) in a humidified 5% CO2 incubator at 37°C.
Immunocytochemistry assay
Chondrocytes were cultured on coverslips in six-well plates. Cells were fixed with 4% paraformaldehyde for 10 min at room temperature and then with methanol at −20°C for 20 min. Normal goat serum (10%, HyClone, GE Healthcare Life Sciences, Shanghai, China) was added to the cells for 30 min to block non-specific binding sites. The fixed cells were immunostained with primary antibodies targeting collagen II (dilution 1:800, Abcam, Cambridge, United Kingdom) overnight at 4°C followed by biotin–conjugated secondary antibody (1:500 dilution, ZSGB-BIO, Beijing, China) for 1 h at 37°C. Finally, sections were lightly counterstained using Hematoxylin at 37°C for 1 min. Images were acquired using a high-resolution CoolSNAP™ CCD camera (Photometrics Inc., Tucson, AZ, U.S.A.) under the control of a computer using Leica FW4000 software, version 1.2 (Leica Microsystems, Ltd., Milton Keynes, U.K.).
Cell treatments
The chondrocytes were cultured in DMEM with 2% FBS for 12 h before and during the following treatments. Chondrocytes were treated with doses of Dex for different time periods to determine the optimum concentration and culture time. Oxidative stress was induced by treating the cells with 1 mM H2O2 (Sigma–Aldrich Chemical Co., U.S.A.) for 1 h. Afterward, cells were cultured in fresh medium or the medium with the supplementation of Dex (5 μM, Sigma Chemicals, St. Louis, MO, U.S.A.), tomatidine (10 μM, NF-κB inhibitor, Selleck, Shanghai, China), SP600125 (5 μM, JNK inhibitor, Selleck), or CY-09 (1 μM, NLRP3 inhibitor, Selleck) with further incubation for 24 h. The cells were washed twice after H2O2 treatment to avoid the potential chemical reaction between H2O2 and other agents. Cells in the control group received no above treatments.
Cell viability assay
Cells were seeded in 96-well plates (3 × 106 cells per well) for 24 h. After the cell treatments, cell viability was detected using cell counting kit-8 (CCK-8, Dojindo, Kyushu, Japan) according to the manufacturer’s instructions.
Flow cytometry analysis
Cell death rate was detected by flow cytometry. Cells were seeded in six-well plates. Apoptotic incidence was analyzed by the Annexin V-FITC/ Propidium Iodide (PI) detection kit (Beyotime Institute of Biotechnology, Shanghai, China) according to the manufacturer’s instructions. Briefly, cells were stained with Annexin V-FITC and PI for 15 min in the dark at room temperature. The rate of apoptosis was analyzed using a dual laser flow cytometer (Becton Dickinson; San Jose, CA, U.S.A.) and estimated using the ModFit LT software, version 1.0 (Verity Software House; Topsham, ME, U.S.A.).
Measurement of intracellular ROS levels
The production of intracellular ROS was monitored using a cell-permeable fluorogenic probe, 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA, Invitrogen). After the above indicated treatments, the cells were harvested and stained with 10 μM H2DCFDA (Sigma–Aldrich Chemical Co.) in the dark at 37°C for 15 min. The cells were then rinsed twice with PBS, and 10000 events were immediately analyzed using a flow cytometer (Becton Dickinson) with an excitation wavelength of 480 nm and an emission wavelength of 525 nm.
Antioxidant enzyme activity assay
The activities of antioxidant enzymes including catalase (CAT), superoxide dismutase (SOD), and glutathione peroxidase (GPx) were estimated by using the commercially available assay kits from Beyotime Bio-Corporation (Shanghai, China) as per the manufacturer’s instructions. To analyze the activity of CAT, cell homogenates were taken in a cuvette containing H2O2 at a known concentration and was catalyzed by CAT in our samples for an exact time between 1 and 5 min. The remaining H2O2 was coupled with a substrate and treated with peroxidase to generate a red product, N-4-antipyryl-3-chloro-5-sulfonate-p-benzoquinonemonoimine, which absorbs maximally at 520 nm. By estimating the remaining H2O2, we can calculate the amount of H2O2 that reacted with CAT and finally determine CAT activity. SOD present in our samples inhibited the process of superoxide transforming WST-8, a 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt, to a stable water-soluble WST-8 formazan. The latter can be evaluated by testing the optical density at 450 nm whereby SOD activity can be determined. The determination of GPx activity was based on the principle that nicotinamide adenine dinucleotide phosphate (NADPH) continually diminishes in the cycle of GPx transforming reduced glutathione to oxidized glutathione that is returned to reduced glutathione by glutathione reductase. Detecting reduced NADPH at an absorbance of 340 nm can indirectly estimate GPx activity.
RT-qPCR analysis
RNA was extracted using TRIzol/chloroform (15596-018, Invitrogen, Carlsbad, CA, U.S.A.) according to the manufacturer’s instructions. cDNA (1 μg) was reverse-transcribed from RNA using a reverse transcription kit (Applied Biosystems, Foster City, CA, U.S.A.) and mixed with the primers (Table 1) and Fast Universal Master Mix (Applied Biosystems). Gene expression was examined by RT-qPCR. Data were analyzed by the comparative 2−ΔΔCt method, with the housekeeping gene GAPDH as an internal control. Results were presented as gene expression relative to control (fold change).
Table 1. Primers used in PCR assay.
| Name | Sequence (5′–3′) | Tm (°C) | Amplicon size (bp) | |
|---|---|---|---|---|
| Aggrecan | Forward | ACTCTGGGTTTTCGTGACTCT | 61 | 81 |
| Reverse | ACACTCAGCGAGTTGTCATGG | |||
| Col2A1 | Forward | TGGACGCCATGAAGGTTTTCT | 62 | 183 |
| Reverse | TGGGAGCCAGATTGTCATCTC | |||
| MMP-3 | Forward | CTGGACTCCGACACTCTGGA | 62 | 79 |
| Reverse | CAGGAAAGGTTCTGAAGTGACC | |||
| MMP-13 | Forward | ACTGAGAGGCTCCGAGAAATG | 61 | 103 |
| Reverse | GAACCCCGCATCTTGGCTT | |||
| ADAMTS1 | Forward | TTCCACGGCAGTGGTCTAAAG | 62 | 100 |
| Reverse | CCACCAGGCTAACTGAATTACG | |||
| NLRP3 | Forward | GATCTTCGCTGCGATCAACAG | 61 | 81 |
| Reverse | CGTGCATTATCTGAACCCCAC | |||
| XIAP | Forward | AATAGTGCCACGCAGTCTACA | 61 | 103 |
| Reverse | CAGATGGCCTGTCTAAGGCAA | |||
| GAPDH | Forward | GGAGCGAGATCCCTCCAAAAT | 61 | 197 |
| Reverse | GGCTGTTGTCATACTTCTCATGG | |||
| miR-223-3p | Forward | UGUCAGUUUGUCAAAUACCCCA | 61 | |
| Reverse | CAGTGCGTGTCGTGGAGT | |||
| miR-302-3p | Forward | TAAGTGCTTCCATGTTTTGGTGA | 61 | |
| Reverse | CAGTGCGTGTCGTGGAGT | |||
| miR-520-3p | Forward | ACACTCCAGCTGGGAAAGTGCTTCCC | 61 | |
| Reverse | CTCAACTGGTGTCGTGGA | |||
| U6 | Forward | GCTTCGGCAGCACATATA | 61 | |
| Reverse | AACGCTTCACGAATTTGCGT |
Abbreviation: XIAP, X-linked inhibitor-of-apoptosis.
Western blot assay
Western blot assay was performed to examine the expression levels of indicated proteins in AF chondrocytes. Protein extracts were separated using 10–12% SDS/polyacrylamide gel electrophoresis and transferred on to nitrocellulose membranes. The membranes were incubated with the following primary antibodies, phospho (p)-p65 (1:1000; ab76302, Abcam), NLRP3 (1:500; ab214185, Abcam), caspase-1 (1:500; ab207802, Abcam), IL-1β (1:500; ab9722, Abcam), X-linked inhibitor-of-apoptosis (XIAP; 1:500; ab28151, Abcam), neural precursor cell expressed developmentally down-regulated protein 4 (NEDD4; 1:500; ab46521, Abcam), and GAPDH (1:1000; ab181602, Abcam) at 4°C overnight. The primary antibodies were visualized by adding biotin–conjugated secondary antibodies followed by an avidin/biotin/peroxidase enzyme complex (Vectastain ABC Elite kit; Vector Laboratories Inc, Burlingame, CA, U.S.A.) and an appropriate substrate (Vector Nova RED, Vectastain).
Immunoprecipitation assay
Cells were lysed with immunoprecipitation assay lysis buffer (RIPA, Sigma–Aldrich). Cell lysates with equal amount of protein (500 μg) were incubated with nickel beads conjugated to anti-XIAP antibody (Abcam) for 3 h, followed by washing with IP buffer (50 mM Tris, pH 7.5, 5 mM EDTA, 150 mM NaCl, and 0.5% NP-40). Bound proteins were detected by Western blotting using primary antibody against NEDD4 (Abcam) and HRP–conjugated secondary antibody.
Statistical analysis
Results were presented as mean ± standard deviation. Statistical analysis was performed using SPSS software, version 11.0 (SPSS, Chicago, IL, U.S.A.). One-way analysis of variance (ANOVA) was used for data analysis, followed by least significant difference test (Fisher’s test) and the unpaired Student’s t test was used for comparison between two means. P-values less than 0.05 (P>0.05) were considered as statistically significant.
Results
Dex improved the proliferation of AF chondrocytes in the presence or absence of H2O2
The isolated AF chondrocytes were identified by immunocytochemistry (ICC) assay (Figure 1A). The cells were stained with collagen II antibody–conjugated dye, but the staining was relatively weak suggesting some degree of degradation of the chondrocytes. As determined by CCK-8 assay, the viability of AF chondrocytes was increased by Dex at doses of 1, 5, and 25 μM at 12, 24, 36, or 48 h (P<0.05, Figure 1B). Treatment with 5 μM Dex showed a better effect on cell viability than at other dosages at 24 and 36 h. Cell viability was notably decreased by H2O2 (P<0.01, Figure 1C), but treatment with 1 and 5 μM Dex significantly reversed cell viability (P<0.05 vs. H2O2 group). Flow cytometry analysis showed that the death rate of AF chondrocytes was not significantly changed by Dex at the tested range of concentrations (Figure 1D). A dramatic increase in the rate of cell death was observed after H2O2 treatment (P<0.01). Treatment with 1 and 5 μM Dex lowered the death rate with more profound effect at 5 μM Dex (P<0.05 vs. H2O2 group).
Figure 1. Dex improved the proliferation of AF chondrocytes in the presence or absence of H2O2.

The isolated AF chondrocytes were identified by ICC assay (A). Chondrocytes were treated with doses of Dex for different time periods to determine the optimum concentration and culture time. Then cell viability was tested (B). Oxidative stress was induced by treating the cells with 1 mM H2O2 for 1 h. Afterward, cells were cultured in fresh medium or the medium with the supplementation of Dex with further incubation for 24 h. Then cell viability (C) and apoptosis rate (D) were tested. *P<0.05 and **P<0.01 vs. control group; #P<0.05 vs. the group that was treated with H2O2 alone.
Dex affected the degeneration of AF chondrocytes
The mRNA expression of COL2A1, but not aggrecan, was decreased with Dex treatment (P<0.05, Figure 2). Dex conversely increased the expression of MMP-3 and MMP-13 (P<0.05), but not ADAMTS5. Stimulation by H2O2 caused the dramatic reduction in COL2A1 (P<0.01) and aggrecan (P<0.01) expression and increase in MMP-3 (P<0.01), MMP-13 (P<0.01), and ADAMTS (P<0.05) expression in AF chondrocytes. Treatment with Dex after H2O2 blocked the reduction in COL2A1 and aggrecan as well as the increase in MMP-3, MMP-13, and ADAMTS expression (P<0.05 vs. H2O2 group).
Figure 2. Dex regulated the degeneration of AF chondrocytes.

Chondrocytes were treated with 5 μM Dex for 24 h. In addition, chondrocytes were treated with 1 mM H2O2 for 1 h. Afterward, cells were cultured in fresh medium or the medium with supplementation of 5 μM Dex with further incubation for 24 h. The relative expression levels of indicated mRNA were tested by PCR. *P<0.05 and **P<0.01 vs. control group; #P<0.05 vs. the group that was treated with H2O2 alone.
Dex prevented H2O2 induced increase in ROS in AF chondrocytes
The ROS levels in AF chondrocytes were notably increased by H2O2 (P<0.01, Figure 3A), but only marginally increased by 5 μM Dex. Dex in turn suppressed the H2O2-mediated increase in ROS levels (P<0.05 vs. H2O2 group). We also tested the activities of antioxidant enzymes including SOD, CAT, and GPx in AF chondrocytes. Dex increased the activities of SOD, CAT, and GPx in AF chondrocytes. H2O2 increased CAT activity (P<0.05, Figure 3B), but significantly decreased SOD (P<0.01) and GPx (P<0.05) activities in these cells. Compared with treatment with H2O2 alone, Dex treatment after H2O2 addition increased the SOD (P<0.05 vs. H2O2 group) and GPx (P<0.05 vs. H2O2 group) activities.
Figure 3. Dex regulated the antioxidative homeostasis in AF chondrocytes.

Chondrocytes were treated with 5 μM Dex for 24 h. In addition, chondrocytes were treated with 1 mM H2O2 for 1 h. Afterward, cells were cultured in fresh medium or the medium with supplementation of 5 μM Dex with further incubation for 24 h. Intracellular ROS was monitored using a cell-permeable fluorogenic probe, H2DCFDA by a flow cytometer (A). Activities of antioxidative enzymes were tested using the detection kits (B). *P<0.05 and **P<0.01 vs. control group; #P<0.05 vs. the group that was treated with H2O2 alone.
Dex inhibited the activation of NF-κB and NLRP3 caused by H2O2
As indicated by Western blot analysis, treatment with Dex alone decreased the phosphorylation level of JNK (P<0.05, Figure 4), but had no effect on phosphorylation level of p65 and protein levels of NLRP3 and its downstream targets caspase-1 and IL-1β. The phosphorylation level of p65 was increased by H2O2 (P<0.01), but Dex prevented the increase in p65 phosphorylation caused by H2O2 (P<0.05). Furthermore, the protein levels of NLRP3, caspase-1, and IL-1β were also increased when treated with H2O2 (P<0.01), while Dex reversed the increase in these proteins (P<0.05). Dex also blocked the increase in JNK phosphorylation induced by H2O2 (P<0.05 vs. H2O2 group).
Figure 4. Dex regulated the NF-kB, JNK and NLRP3 pathways in AF chondrocytes.

(A) Chondrocytes were treated with 5 μM Dex for 24 h. Western blot assay was performed to detect the protein levels of p-p65, NLRP3, Caspase-1, IL-1β, and p-JNK. (B) Chondrocytes were treated with 1 mM H2O2 for 1 h. Afterward, cells were cultured in fresh medium or the medium with supplementation of 5 μM Dex with further incubation for 24 h. Western blot assay was performed to detect the protein levels of p-p65, NLRP3, Caspase-1, IL-1β, and p-JNK. *P<0.05 and **P<0.01 vs. control group; #P<0.05 vs. the group that was treated with H2O2 alone.
We further performed a series of analysis to elucidate the mechanism by which Dex inhibited NLRP3 signaling. PCR analysis showed that NLRP3 mRNA was increased by H2O2, and this increase was abolished by Dex (P<0.01, Figure 5A). This suggested that Dex impaired H2O2-mediated pre-transcriptional regulation of NLRP3. Bioinformatics analysis (http://www.genome.ucsc.edu/) indicated that the expression of NLRP3 mRNA was affected by several transcription factors (such as AP-1, ΔCREB, NF-κB), while it was seldom affected by histone methylation and acetylation (Figure 5B). The 3′UTR of NLRP3 mRNA was predicted to bind to a few miRNAs, such as miR-223-3p, miR-302-3p, and miR-520-3p (Figure 5C, as indicated by http://www.targetscan.org/vert_72/). Considering that the activity of AP-1 transcription factor is predominantly regulated by JNK signaling pathway, we added a JNK inhibitor to investigate its effect on NLRP3 protein level. Both inhibitors of JNK and NF-κB inhibited the increase in NLRP3 protein level caused by H2O2 (P<0.05, Figure 5D). The expression levels of miR-223-3p, miR-302-3p, and miR-520-3p in AF chondrocytes were evaluated using PCR. Treatment with H2O2 decreased the expression of these miRNAs (P<0.05, Figure 5E), and Dex only marginally increased their expression.
Figure 5. Dex inhibited H2O2-induced activation of NLRP3 in AF chondrocytes probably through inhibition of NF-κB and JNK pathways.

Chondrocytes were treated with 5 μM Dex for 24 h. In addition, chondrocytes were treated with 1 mM H2O2 for 1 h. Afterward, cells were cultured in fresh medium or the medium with the supplementation of 5 μM Dex with further incubation for 24 h. The mRNA level of NLRP3 was evaluated using PCR assay (A). Bioinformatics analysis (http://www.genome.ucsc.edu/) indicated that the expression of NLRP3 mRNA was affected by several transcription factors, while it was seldom affected by histone methylation and acetylation (B). Bioinformatics analysis (http://www.targetscan.org/vert_72/) indicated that NLRP3 mRNA was targeted by a few miRNAs (C). Chondrocytes were treated with 1 mM H2O2 for 1 h. Afterward, cells were cultured in fresh medium or the medium with the supplementation of tomatidine (10 μM, NF-κB inhibitor) or SP600125 (5 μM, JNK inhibitor) with further incubation for 24 h. The protein levels were evaluated using Western blot assay (D). Chondrocytes were treated with 1 mM H2O2 for 1 h. Afterward, cells were cultured in fresh medium or the medium with the supplementation of 5 μM Dex with further incubation for 24 h. The relative expression levels of indicated mRNA were tested by PCR (E). *P<0.05 and **P<0.01 vs. control group; #P<0.05 vs. the group that was treated with H2O2 alone. NF-κB Inh: administration of NF-κB inhibitor after H2O2 treatment; JNK Inh: administration of JNK inhibitor after H2O2 treatment.
We found that treatment with NF-κB inhibitor alone suppressed the chondrocyte viability (P<0.05, Figure 6A), while inhibitors of JNK and NLRP3 did not significantly affect cell viability. All the inhibition of NF-κB, JNK, and NLRP3 elevated the cell viability that was suppressed by H2O2 (P<0.05, Figure 6A). Inhibitors of NF-κB and JNK increased apoptosis rate (P<0.05, Figure 6B), but both of them plus NLRP3 inhibitor lowered the death rate that was increased by H2O2 (P<0.05, Figure 6B). In non-stress condition, inhibition of NF-κB, JNK, and NLRP3 had no effect on expression of COL2A1 and aggrecan mRNA (Figure 7A), but they suppressed expression of MMP-3, MMP-13, or ADAMTS (P<0.05). Adding NF-κB, JNK, and NLRP3 inhibitors after H2O2 treatment increased COL2A1 (P<0.05 or P<0.01) and aggrecan (P<0.05) expression but decreased MMP-3 (P<0.05 or P<0.01), MMP-13 (P<0.05 or P<0.01), and ADAMTS (P<0.05) compared with treatment with H2O2 alone (Figure 7B).
Figure 6. Cell viability and apoptosis rate after the blockage of NF-κB, JNK, and NLRP3 signals.

Tomatidine (10 μM, NF-κB inhibitor), SP600125 (5 μM, JNK inhibitor) or CY-09 (1 μM, NLRP3 inhibitor) was added to chondrocytes that were treated with 1 mM H2O2 for 1 h or not. The cell viability (A) and apoptosis rate (B) were tested after treatments with these inhibitors for 24 h. *P<0.05 vs. control group; #P<0.05 vs. the group that was treated with H2O2 alone.
Figure 7. Influence of the degeneration of AF chondrocytes after the blockage of NF-κB, JNK, and NLRP3 signals.
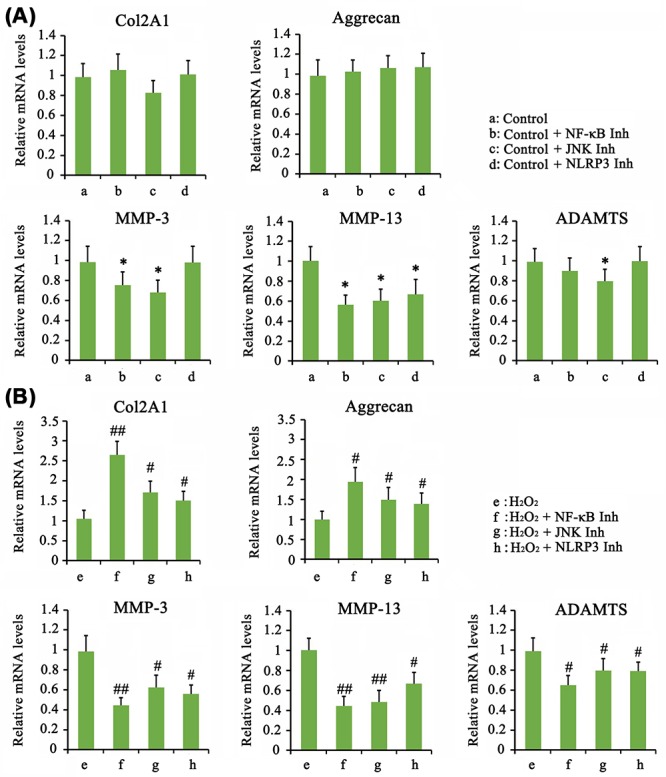
(A) Chondrocytes were treated with Tomatidine (10 μM, NF-κB inhibitor), SP600125 (5 μM, JNK inhibitor) or CY-09 (1 μM, NLRP3 inhibitor) for 24 h. (B) In addition, chondrocytes were treated with Tomatidine (10 μM, NF-κB inhibitor), SP600125 (5 μM, JNK inhibitor) or CY-09 (1 μM, NLRP3 inhibitor) for 24 h, followed by treatment with 1 mM H2O2 for 1 h. PCR was performed to detect the expression of COL2A1, Aggrecan, MMP-3, MMP-13, and ADAMTS1 in AF chondrocytes after treatments with these inhibitors for 24 h. *P<0.05 vs. control group; #P<0.05, ##P<0.01 vs. the group that was treated with H2O2 alone.
Dex regulated XIAP expression in H2O2-treated AF chondrocytes
Dex had no significant effect on the mRNA level of XIAP (Figure 8A). H2O2 also moderately increased the mRNA level of XIAP, while co-treatment with H2O2 and Dex significantly reduced the XIAP mRNA level compared with treatment with H2O2 alone (P<0.05). Western blot analysis showed that XIAP protein level was not changed by treatment with Dex and H2O2 alone (Figure 8B). Co-treatment with H2O2 and Dex conversely increased the XIAP protein level compared with treatment with H2O2 alone (P<0.05). Bioinformatics analysis (http://ubibrowser.ncpsb.org/ubibrowser/) showed that XIAP protein was most likely targeted by NEDD4 in addition to FBXW7 (Figure 8C). Both XIAP and NEDD4 were transcriptionally regulated by NF-κB according to bioinformatics analysis. NEDD4 protein level was increased by H2O2 (P<0.05, Figure 8B), while the effect of H2O2 was attenuated by Dex (P<0.05 vs. H2O2 group). As indicated by the immunoprecipitation assay, the amount of NEDD4 adhering to XIAP was decreased by Dex (P<0.05) but increased by H2O2 (P<0.05, Figure 8D). In addition, the increased amount of NEDD4 caused by H2O2 was attenuated by Dex (P<0.05 vs. H2O2 group).
Figure 8. The mechanisms underlying Dex regulating XIAP in AF chondrocytes.

Chondrocytes were treated with 5 μM Dex for 24 h. In addition, chondrocytes were treated with 1 mM H2O2 for 1 h. Afterward, cells were cultured in fresh medium or the medium with the supplementation of 5 μM Dex with further incubation for 24 h. The mRNA and protein levels of XIAP were evaluated using PCR (A) and Western blot assays (B), respectively. Bioinformatics analysis (http://ubibrowser.ncpsb.org/ubibrowser/) showed XIAP protein was regulated by many ubiquitination enzymes (C). Immunoprecipitation assay was performed to evaluate the amount of NEDD4 adhering to XIAP (D). *P<0.05 vs. control group; #P<0.05 vs. the group that was treated with H2O2 alone.
Discussion
Dex is commonly used in lumbar discectomy, but its effects on physiological and pathological functions of IVD chondrocytes have never been investigated. Since Dex is an activator of α2-AR and activation of α2-AR by NE is generally associated with rapid cartilage degeneration, it is possible that Dex also accelerates the progression of cartilage degeneration. Under non-stress conditions, Dex indeed decreased the mRNA expression levels of COL2A1 and increased those of MMP-3 and MMP-13 in AF chondrocytes. The reduction in COL2A1 expression is an important hallmark of cartilage degeneration. MMP-3 and MMP-13 are responsible for the decomposition of extracellular matrix, which impairs the cartilage structure and characteristics. Therefore, our results suggested that Dex promoted cartilage degeneration. Nevertheless, the viability of AF chondrocytes was improved by Dex. It has been found that Dex had no effect on cell viability, but presented potential chondrotoxicity at very high dosages (0.175 and 0.25 mg/ml) in a study on articular chondrocytes isolated from healthy equine articular cartilage of the metacarpo/metatarsophalangeal joints [10]. These chondrocytes probably have different characteristics from the chondrocytes isolated from AF tissues in the present study, which could explain the difference in the results. Study herein found that Dex inhibited JNK signal but had no effect on NF-κB and NLRP3 cascades. Treatment with JNK inhibitor increased the apoptosis rate, though decreasing MMP-3 and MMP-13 expression. These data suggest that the effect of Dex on the cartilage degeneration is probably associated with molecular mechanisms independent of JNK signal. Inhibition of JNK under non-stress status probably affects the normal physiological function of chondrocytes, resulting in the increase in apoptosis.
The effect of Dex on AF chondrocytes under non-stress conditions could not be similar to that effect of Dex when it is used during and after lumbar discectomy surgery. Considering that oxidative stress is easily induced during lumbar discectomy, it is more reasonable to investigate the effect of Dex on IVD cartilage under oxidative stress conditions. The present study indicated that Dex did not exert a synergetic or additive effect with H2O2, but conversely attenuated the detrimental actions of H2O2. These data were not surprising, since Dex has been confirmed to have antioxidative and anti-inflammatory properties, by numerous previous studies. Sha et al. [7] reported that Dex attenuated lipopolysaccharide-induced liver oxidative stress and cell apoptosis in rats by increasing the activity of GSK-3β/MKP-1/Nrf2 pathway via the α2 adrenergic receptor. An in vitro study showed that the oxidative stress induced by H2O2 was diminished by Dex, thereby preventing the apoptosis of lung alveolar epithelial cells [11]. With respect to chondrocytes, H2O2 not only induces cell death, but also contributes to its degeneration, because H2O2 is a strong inducer of ROS production, and ROS is further involved in signaling pathways resulting in chondrocyte degeneration [12]. In the present study, Dex decreased the intracellular levels of ROS partly by enhancing the activities of antioxidant enzymes and thereby, to some extent, attenuating the detrimental effects of H2O2. An interesting finding in the present study is that H2O2 increased CAT activity, but decreased SOD and GPx activity. The increase in CAT activity in cells is probably a response to H2O2, because CAT can directly remove H2O2. However, a large amount of ROS derived from H2O2 may impair other antioxidant defenses.
H2O2-activated inflammatory signals play important roles in the degeneration of chondrocytes. For example, H2O2 is able to induce the activation of NF-κB signal that can promote the expression and secretion of enzymes such as MMP-3, MMP-13, and ADAMTS [13–15]. In the present study, Dex blocked the activation of NF-κB caused by H2O2. This outcome is consistent with observations in most previous studies, regarding the anti-inflammatory effect of Dex [13–15]. Therefore, it is possible that Dex protection against H2O2 is mainly due to its antioxidant activity, which in turn is responsible for NF-κb signal modulation. In addition to NF-κB, the activation of NLRP3 by H2O2, was also attenuated by Dex treatment. NLRP3 inflammasome is the most well-studied inflammasome that is recognized to play a crucial part in the initiation and continuance of inflammation in various diseases [9,16–18]. Activation of NLRP3 inflammasome is involved in microglial cell activities in hippocampus, thus causing damage in a rat model of traumatic brain injury [9]. The blockage of NLRP3 by Dex administration inhibited microglial activation and increased neuronal viability and cognitive function. Moreover, Dex attenuated pancreatic inflammatory response in mice with pancreatitis by reduction in NLRP3 activation [16]. In lipopolysaccharide-induced acute lung injury, Dex inhibited NLRP3 activation through the up-regulation of miR-381 and miR-381-mediated degeneration of NLRP3 [17]. Lv et al. [18] reported that Dex promotes liver regeneration in mice after 70% partial hepatectomy by suppressing NLRP3 inflammasome and not TLR4/NF-κB, since NLRP3 inhibition is associated with better liver regeneration and liver function recovery, while NF-κB inhibition conversely diminished liver regeneration. In the present study, the inhibition of NLRP3 by Dex occurred likely through the suppression of NF-κB, since NF-κB inhibitor decreased NLRP3 expression and NLRP3-activated caspase-1. We found that the inhibition of JNK signaling was also involved in the mechanism underlying the inhibition of NLRP3 by Dex.
The pathological role of NLRP3 in the degeneration of chondrocytes has also been reported. Activated NLRP3 recruits ASC and caspase-1 to form a protein complex that is essential for caspase-1 activation. Activated caspase-1 leads to the maturation of pre-IL-1β and pre-IL-18. The NLRP3/caspase-1/IL-1β axis has been found to be active in human lumbar cartilaginous endplate degeneration and their expression levels are positively associated with the grades of disc degeneration [19,20]. A study reported that nucleus pulposus cells treated with H2O2 caused cartilage degeneration as indicated by the changes in expression levels of inflammatory mediators (Interleukin-6, COX-2, and iNOS), major matrix degrading proteases (MMP-3, MMP-13, ADAMTS5, and ADAMTS4), and cartilaginous mark proteins (COL2A1 and SOX9). Honokiol, a low molecular weight natural product, reversed H2O2-triggered cartilage degeneration, and this protective effect of honokiol is primarily associated with the inhibition of TXNIP/NLRP3/caspase-1/IL-1β signaling axis [21]. However, Bougault et al. [22] suggested that cartilage degeneration does not depend on NLRP3 inflammasome according to the following evidences: (i) osteoarthritis cartilage was not able to produce active IL-1β; (ii) LPS, IL-1α, and TNFα dose-dependently increased MMP-3, MMP-9, and MMP-13 activity in cultured chondrocytes and in NLRP3(−/−) chondrocytes; (iii) these effects of LPS, IL-1α, and TNFα did not change by inhibition of caspase-1 or IL-1β. Although the role of NLRP3 in cartilage degeneration remains controversial, the present study at least confirmed that NLRP3 activation promoted cell death of AF chondrocytes. NLRP3 activation easily triggers pyroptosis and apoptosis by activating caspase family proteins, such as caspase-1 and caspase-3. In the present study, using an inhibitor of NLRP3, we showed that it attenuated H2O2-triggered cell death.
XIAP is a member of the inhibitor-of-apoptosis proteins (IAP) that represents a family of endogenous caspase inhibitors. Studies have confirmed that up-regulation of XIAP is able to block apoptosis in degenerative nucleus pulposus and osteoarthritic cartilage [23,24]. In addition, XIAP is also involved in the regulation of inflammatory response of cells. Loss of XIAP facilitates the proinflammatory effect of TNF-α and IL-1β, causing severe sterile inflammation in diverse types of cells [25]. Yet, NF-κB acts as an important transcription factor by positively regulating XIAP expression. In many cancer studies, NF-κB-mediated drug resistance is through up-regulation of XIAP [26]. However, herein, treatments with H2O2 and Dex alone had no effect of both mRNA and protein levels of XIAP. Co-treatment with Dex and H2O2 caused the reduction in XIAP mRNA level, but the increase in XIAP protein level, compared with treatment with and H2O2 alone. NEDD4 is an important ubiquitin ligase targeting XIAP. NEDD4-mediated ubiquitination of XIAP can induce the degeneration, thus decreasing total amount of XIAP protein in cells. We found that NEDD4 protein level in cells was increased by H2O2, but the increase in NEDD4 protein was abolished by Dex. H2O2-induced the increase in NEDD4 was associated with increased amount of NEDD4 attaching to XIAP, suggesting that NEDD4-mediated ubiquitination of XIAP was enhanced. However, treatment with Dex decreased the amount of NEDD4 attaching to XIAP, which suggested that Dex conferred protective effect from NEDD4-mediated ubiquitination of XIAP.
In summary, the present study demonstrated that Dex treatment is associated with rapid degeneration of chondrocytes under non-stressed conditions. Although both Dex and H2O2 accelerated the cartilage degeneration, their underlying mechanisms might be different. Dex inducing cartilage degeneration is likely through AR-mediated ERK1/2 and PKA pathways according to previous report [5], while the effect of H2O2 on cartilage degeneration is more likely through NF-κB/NLRP3, JNK/NLRP3, and NEDD4/XIAP pathways. Since, Dex disrupted the effects of H2O2 on these pathways, Dex prevented the death and degeneration of chondrocytes during oxidative stress (as indicated in Figure 9). These data suggested a potential protective effect of Dex in lumbar discectomy.
Figure 9. Dex exerts dual effects on the degeneration of human AF chondrocytes depending on the oxidative stress status.

Under non-stressed condition, Dex induces cartilage degeneration likely through AR-mediated ERK1/2 and PKA pathways according to previous reports [5]. The effect of H2O2 on cartilage degeneration is partly through NF-κB/NLRP3, JNK/NLRP3 and NEDD4/XIAP pathways. Since, Dex disrupted the effects of H2O2 on these pathways, Dex prevented the death and degeneration of chondrocytes during oxidative stress. Dashed arrow indicated that the mechanism required further identification.
Abbreviations
- AF
annulus fibrosus
- AR
adrenoreceptor
- CAT
catalase
- CCK-8
cell counting kit-8
- Dex
Dexmedetomidine
- FBS
fetal bovine serum
- GPx
glutathione peroxidase
- H2DCFDA
2′,7′-dichlorodihydrofluorescein diacetate
- H2O2
hydrogen peroxide
- IVD
intervertebral disc
- LBP
lower back pain
- NADPH
nicotinamide adenine dinucleotide phosphate
- NE
norepinephrine
- NEDD4
neural precursor cell expressed developmentally down-regulated protein 4
- NLRP3
NACHT, LRR, and PYD domains-containing protein 3
- PI
propidium iodide
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- XIAP
X-linked inhibitor-of-apoptosis
Author Contribution
Lin Zhou and Jinhua Zhou performed the study and drafted the manuscript. Bin Sheng took the responsible for the design of the work. Xin Li and Youchao Yuan took the charge of the acquisition, analysis, or interpretation of data of the study.
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
Funding
This work was supported by the Youth Doctoral Foundation of Hunan Provincial People’s Hospital and Nurturing Project of the National Natural Science Foundation of China [grant number BSJJ201814]; and the Project of Hunan Provincial Natural Science Foundation [grant number 2019-250].
References
- 1.Feng C., Yang M., Lan M., Liu C., Zhang Y., Huang B.. et al. (2017) ROS: crucial intermediators in the pathogenesis of intervertebral disc degeneration. Oxid. Med. Cell Longev. 2017, 5601593 10.1155/2017/5601593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng C., Zhang Y., Yang M., Lan M., Liu H., Wang J.. et al. (2017) The matrikine N-acetylated proline-glycine-proline induces premature senescence of nucleus pulposus cells via CXCR1-dependent ROS accumulation and DNA damage and reinforces the destructive effect of these cells on homeostasis of intervertebral discs. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 220–230 10.1016/j.bbadis.2016.10.011 [DOI] [PubMed] [Google Scholar]
- 3.Xu Y., Yao H., Wang Q., Xu W., Liu K., Zhang J.. et al. (2018) Aquaporin-3 attenuates oxidative stress-induced nucleus pulposus cell apoptosis through regulating the P38 MAPK pathway. Cell. Physiol. Biochem. 50, 1687–1697 10.1159/000494788 [DOI] [PubMed] [Google Scholar]
- 4.Lorenz J., Schäfer N., Bauer R., Jenei-Lanzl Z., Springorum R.H. and Grässel S. (2016) Norepinephrine modulates osteoarthritic chondrocyte metabolism and inflammatory responses. Osteoarthritis Cartilage 24, 325–334 10.1016/j.joca.2015.08.007 [DOI] [PubMed] [Google Scholar]
- 5.Jiao K., Zeng G., Niu L.N., Yang H.X., Ren G.T., Xu X.Y.. et al. (2016) Activation of α2A-adrenergic signal transduction in chondrocytes promotes degenerative remodelling of temporomandibular joint. Sci. Rep. 6, 30085 10.1038/srep30085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitchell J., Lai L.P., Peralta F., Xu Y. and Sugamori K. (2011) β2-adrenergic receptors inhibit the expression of collagen type II in growth plate chondrocytes by stimulating the AP-1 factor Jun-B. Am. J. Physiol. Endocrinol. Metab. 300, E633–E639 10.1152/ajpendo.00515.2010 [DOI] [PubMed] [Google Scholar]
- 7.Sha J., Zhang H., Zhao Y., Feng X., Hu X., Wang C.. et al. (2019) Dexmedetomidine attenuates lipopolysaccharide-induced liver oxidative stress and cell apoptosis in rats by increasing GSK-3β/MKP-1/Nrf2 pathway activity via the α2 adrenergic receptor. Toxicol. Appl. Pharmacol. 364, 144–152 10.1016/j.taap.2018.12.017 [DOI] [PubMed] [Google Scholar]
- 8.Meng L., Li L., Lu S., Li K., Su Z., Wang Y.. et al. (2018) The protective effect of dexmedetomidine on LPS-induced acute lung injury through the HMGB1-mediated TLR4/NF-κB and PI3K/Akt/mTOR pathways. Mol. Immunol. 94, 7–17 10.1016/j.molimm.2017.12.008 [DOI] [PubMed] [Google Scholar]
- 9.Zheng B., Zhang S., Ying Y., Guo X., Li H., Xu L.. et al. (2018) Administration of Dexmedetomidine inhibited NLRP3 inflammasome and microglial cell activities in hippocampus of traumatic brain injury rats. Biosci. Rep. 38, 10.1042/BSR20180892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mancini F., Nannarone S., Buratta S., Ferrara G., Stabile A.M., Vuerich M.. et al. (2017) Effects of xylazine and dexmedetomidine on equine articular chondrocytes in vitro. Vet. Anaesth. Analg. 44, 295–308 10.1016/j.vaa.2016.04.004 [DOI] [PubMed] [Google Scholar]
- 11.Cui J., Zhao H., Wang C., Sun J.J., Lu K. and Ma D. (2015) Dexmedetomidine attenuates oxidative stress induced lung alveolar epithelial cell apoptosis in vitro. Oxid. Med. Cell Longev. 2015, 358396 10.1155/2015/358396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bolduc J.A., Collins J.A. and Loeser R.F. (2019) Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic. Biol. Med. 132, 73–82 10.1016/j.freeradbiomed.2018.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ra H.J., Oh M.Y., Kim H.J., Lee S.Y., Eom D.W., Lee S.K.. et al. (2018) Effects of salmon DNA fraction in vitro and in a monosodium iodoacetate-induced osteoarthritis rat model. Korean J. Physiol. Pharmacol. 22, 163–172 10.4196/kjpp.2018.22.2.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang D., Lin N., Tang Y. and Lu H. (2019) Inhibition of P2Y11R ameliorated TNF-α-induced degradation of extracellular matrix in human chondrocytic SW1353 cells. Am. J. Transl. Res. 11, 2108–2116 [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu S., Gu Y., Wang W., Bai J., Ge G., Zhang W.. et al. (2019) Sitagliptin ameliorates advanced glycation end-product (AGE)-induced degradation of extracellular matrix in human primary chondrocytes. Am. J. Transl. Res. 11, 2775–2783 [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y., Pan Y., Gao L., Lu G., Zhang J., Xie X.. et al. (2018) Dexmedetomidine attenuates pancreatic injury and inflammatory response in mice with pancreatitis by possible reduction of NLRP3 activation and up-regulation of NET expression. Biochem. Biophys. Res. Commun. 495, 2439–2447 10.1016/j.bbrc.2017.12.090 [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y., Wang X., Liu Z. and Yu L. (2018) Dexmedetomidine attenuates lipopolysaccharide induced acute lung injury by targeting NLRP3 via miR-381. J. Biochem. Mol. Toxicol. 32, e22211 10.1002/jbt.22211 [DOI] [PubMed] [Google Scholar]
- 18.Lv M., Zeng H., He Y., Zhang J. and Tan G. (2018) Dexmedetomidine promotes liver regeneration in mice after 70% partial hepatectomy by suppressing NLRP3 inflammasome not TLR4/NFκB. Int. Immunopharmacol. 54, 46–51 10.1016/j.intimp.2017.10.030 [DOI] [PubMed] [Google Scholar]
- 19.Tang P., Zhu R., Ji W.P., Wang J.Y., Chen S., Fan S.W.. et al. (2016) The NLRP3/Caspase-1/Interleukin-1β axis is active in human lumbar cartilaginous endplate degeneration. Clin. Orthop. Relat. Res. 474, 1818–1826 10.1007/s11999-016-4866-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Z.H., Jin S.H., Wang M.Y., Jin X.L., Lv C., Deng Y.F.. et al. (2015) Enhanced NLRP3, caspase-1, and IL- 1β levels in degenerate human intervertebral disc and their association with the grades of disc degeneration. Anat. Rec. (Hoboken) 298, 720–726 10.1002/ar.23059 [DOI] [PubMed] [Google Scholar]
- 21.Tang P., Gu J.M., Xie Z.A., Gu Y., Jie Z.W., Huang K.M.. et al. (2018) Honokiol alleviates the degeneration of intervertebral disc via suppressing the activation of TXNIP-NLRP3 inflammasome signal pathway. Free Radic. Biol. Med. 120, 368–379 10.1016/j.freeradbiomed.2018.04.008 [DOI] [PubMed] [Google Scholar]
- 22.Bougault C., Gosset M., Houard X., Salvat C., Godmann L., Pap T.. et al. (2012) Stress-induced cartilage degradation does not depend on the NLRP3 inflammasome in human osteoarthritis and mouse models. Arthritis Rheum. 64, 3972–3981 10.1002/art.34678 [DOI] [PubMed] [Google Scholar]
- 23.Böhm B., Hess S., Krause K., Schirner A., Ewald W., Aigner T.. et al. (2010) ADAM15 exerts an antiapoptotic effect on osteoarthritic chondrocytes via up-regulation of the X-linked inhibitor of apoptosis. Arthritis Rheum. 62, 1372–1382 10.1002/art.27387 [DOI] [PubMed] [Google Scholar]
- 24.Cheng X., Zhang L., Zhang K., Zhang G., Hu Y., Sun X.. et al. (2018) Circular RNA VMA21 protects against intervertebral disc degeneration through targeting miR-200c and X linked inhibitor-of-apoptosis protein. Ann. Rheum. Dis. 77, 770–779 10.1136/annrheumdis-2017-212056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yabal M., Müller N., Adler H.. et al. (2014) XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 7, 1796–1808 10.1016/j.celrep.2014.05.008 [DOI] [PubMed] [Google Scholar]
- 26.Zhao C., Zhao Q., Zhang C., Wang G., Yao Y., Huang X.. et al. (2017) miR-15b-5p resensitizes colon cancer cells to 5-fluorouracil by promoting apoptosis via the NF-κB/XIAP axis. Sci. Rep. 7, 4194 10.1038/s41598-017-04172-z [DOI] [PMC free article] [PubMed] [Google Scholar]