

Abstract
Antiepileptic drug (AED) therapy starts with an accurate diagnosis of epilepsy and is followed by sequential drug trials. Seizure freedom is largely achieved by the first two drug trials; thus, epilepsy that cannot be controlled after appropriately conducted trials of the first two drugs is defined as drug-resistant epilepsy (DRE). It is still unclear which mode of pharmacotherapy, among monotherapy and polytherapy, shows better outcomes in cases of DRE. However, in a recent large hospital cohort study over past two decades, combination therapy was associated with a progressive increase in seizure-free rate than monotherapy in DRE. The benefits of polytherapy in the management of DRE might be related to the recent introduction of many new AEDs with different and novel mechanisms of action and better pharmacokinetic and tolerability profiles. These new AEDs were introduced to the market after they have proven their superiority over placebos in randomized controlled trials (RCTs) on add-on therapy in patients with DRE. Therefore, polytherapy including these new AEDs in the regimen is the approved mode of treatment for cases of DRE; this has prompted physicians to try various combinations of polytherapy to optimize the clinical outcomes. In addition, the significant discrepancies in AED responder rates between RCTs and real-world practice may support the importance of judicious use of new drugs in polytherapy by experienced epileptologists. Most experts now agree to the concept of “rational polytherapy” consisting of mechanistic combinations of AEDs exerting synergistic interactions and to the importance of continuing trials of different rational polytherapy regimens to improve the outcome of the core population of epilepsy patients in the long term.
Keywords: Drug resistant epilepsy, Monotherapy, Polytherapy, New antiepileptic drugs, Real world practice
Introduction
Antiepileptic drug (AED) therapy is the mainstay of epilepsy management, which starts with accurate diagnosis of epilepsy, seizure types, and epilepsy syndromes. AED therapy for epilepsy is complicated by unpredictable drug efficacy, adverse effects (AEs), and the lack of information regarding optimal doses in individual patients, necessitating systematic AED trials consisting of initial monotherapy with the first drug and subsequent trials of the second, third, and next drugs in either monotherapy or polytherapy, along with careful assessment of patient responses to each step of the drug trials (Fig. 1). In a hospital-based observational study,1 the first drug therapy was successful in 49.5% of patients and the second drug was successful in 36.7% of the patients. The success rates of drug trials after failure of the first two drugs were significantly lower but were not different among subsequent drug regimens, ranging from 12.5% to 22.2%. Therefore, the first and second drug trials are likely to be the major determinants of therapeutic outcomes of epilepsy and support the International League Against Epilepsy (ILAE) proposal of drug-resistant epilepsy (DRE), which is defined as the failure of seizure control by adequate trials of the first two AEDs.2
Figure 1.
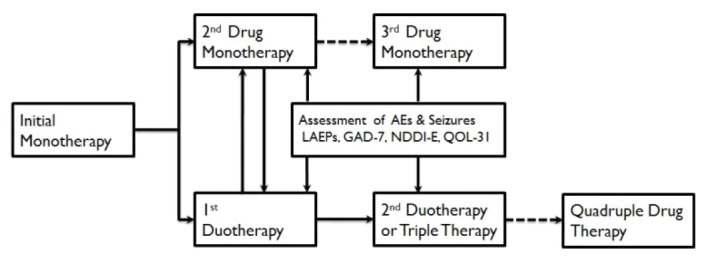
Sequential drug trials in pharmacotherapy of epilepsy. Initial monotherapy is the standard mode of therapy. If it failed, either substitution monotherapy or combination therapy is conducted according to the preference of physicians. During the era of conventional drugs, most epileptologists favored substitution monotherapy and diagnosed drug-resistant epilepsy after the failure of third drug monotherapy. In the era of new AEDs, duotherapy is increasingly undertaken after the failure of initial monotherapy, especially if the first drug was at least partially effective and well tolerable. If the second drug therapy failed, duotherapy is favored. AE, adverse effects; LAEPs, Liverpool Adverse Effect Profiles; GAD-7, Generalized anxiety disorder-7; NDDI-E, neurological disease depression inventories in epilepsy; QOL-31, quality of life-31; →, indicates options in favor; ⇢, suggests less favorable options.
The introduction of new AEDs starting with vigabatrin (VGB) in the UK and zonisamide (ZNS) in Japan in 1989 has yielded 16 new drugs already3 with significant impact on clinical practice for following reasons. First, the new drugs underwent rigorous randomized controlled clinical trials (RCTs) for add-on therapy in patients with DRE and showed significantly higher efficacy in comparison with placebo, because of which they were used primarily in combination therapy for DRE. If their performance as adjunctive agents for patients with DRE in real-world practice was considered appropriate, they underwent a head-to-head comparison with conventional drugs in monotherapy of newly diagnosed epilepsy. Although none of the new AEDs have proven their superiority over conventional AEDs in efficacy, some have shown better tolerability, and many of them are now promoted as first-line drugs for partial-onset seizures as well as the drugs of choice for specific seizure types or epilepsy syndromes (Fig. 2). Extensive RCTs of new AEDs either as add-on therapy or initial monotherapy as well as post-marketing clinical trials for diverse purposes have firmly established the era of evidence-based medicine (EBM) in pharmacotherapy of epilepsy. Second, the new AEDs are characterized by better pharmacological profiles, including linear pharmacokinetics, less potential for drug interactions, diverse mechanisms of action (MOAs), and better tolerability profiles, which are important advantages for polytherapy. The wide availability of the new AEDs and their EBM data has encouraged physicians to practice combination therapy of new AEDs in patients who did not respond to monotherapy; thus, polytherapy has slowly revived in the field of AED therapy. Third, the diversity of new AEDs in terms of MOAs, adverse effect profiles (AEPs), and therapeutic profiles, including their efficacy in various non-epileptic conditions, and improved clinical skills have changed the concept of pharmacotherapy to individual patient-oriented therapy4 from the disease (epilepsy)-oriented therapy in the past. In patient-oriented pharmacotherapy, appropriate drugs are chosen on the basis of multi-dimensional assessment of epilepsy (seizure types and epilepsy syndromes), individual drugs (EBM and pharmacological profiles), and the individual patient’s condition (demographic factors, physiological characteristics, comorbidities, and psychosocial conditions, etc.) under the slogan, “Drugs best fitting my patient” (Fig. 3). These major changes in the pharmacotherapy of epilepsy as well as the rapid progress in basic and clinical epileptology have significantly improved the outlook of patients with DRE, which was once considered as a grave epileptic condition.
Figure 2.

Clinical development process of conventional and new antiepileptic drugs. Conventional AEDs were introduced to the market without any rigorous RCTs and used in both monotherapy and polytherapy simultaneously. After many years of clinical experience, monotherapy was considered the preferred mode of therapy in both newly diagnosed and drug-resistant epilepsies, which was followed by comparative monotherapy trials of conventional AEDs, which promoted CBZ, PHT, and VPA as the major drugs. New AEDs were introduced to the market after rigorous RCTs of add-on therapy in patients with DRE taking 1 to 3 AEDs, which showed that the new AEDs were superior to placebo in adjunctive therapy. Thus, the indications of new AEDs were primarily for polytherapy in patients with DRE, which stimulated physicians to exercise various regimens of polytherapy resulting in the revival of polytherapy and the practice of rational polytherapy. New AEDs, after many years of use in polytherapy, underwent comparative monotherapy RCTs for their promotion as first-line drugs for partial-onset seizures, with several of them being recognized as the drugs of choice in a few specific epilepsy syndromes. Modified from reference 55 with permission. AED, antiepileptic drug; RCT, randomized controlled trial; AE, adverse effects; CBZ, carbamazepine; PHT, phenytoin; VPA, valproate; ESM, ethosuximide; TIG, tiagabine; PGB, pregabalin; FBM, felbamate; VGB, vigabatrin; LTG, lamotrigine; TPM, topiramate; OXC, oxcarbazepine; LEV, levetiracetam; GBP, gabapentin; ZNS, zonisamide; LCM, lacosamide; STR, stiripentol; LGS, Lennox-Gastaut syndrome; PER, perampanel; DRE, drug-resistant epilepsy.
Figure 3.

Pathways of epilepsy management. Epilepsy management starts with the accurate diagnosis of seizure types and epilepsy syndromes. Initial monotherapy is the standard mode of treatment resulting in 50% of prolonged seizure freedom (≥ 12 months). If initial monotherapy failed to control seizures, physicians chose a second drug for either substitution monotherapy or duotherapy with seizure-free rates of 10–20% (or 20–40% of patients who underwent second drug therapy). Thus, about 60% of patients may achieve seizure freedom by the first two drug trials. Failure to respond to adequate trials of the first two drugs satisfied the ILAE criteria for DRE, and physicians may re-evaluate the diagnostic precision of epilepsy to exclude the possibility of pseudo-pharmacoresistance or identify the cause of drug resistance as well as ensure accurate diagnosis of epilepsy syndromes. If patients were confirmed to have DRE and had a SRES, earlier referral to epilepsy surgery may carry a higher benefit-risk ratio in comparison with continuing drug trials. If patients did not have SRES, systematic trials of the third and next drugs in various combination therapies may be required with an expectation of about 30% seizure-free outcome. If the 5th to 6th drug trials failed to control seizures, chances of seizure remission by further drug trials are small and trials of alternative therapies, including VNS, DBS, or ketogenic diet therapy may be actively pursued. Modified from reference 55 with permission. Hx, history-taking; PEx/NEx, physical and neurological examination; MRI, magnetic resonance imaging of brain; EEG, electroencephalography; Dx, diagnosis; DRE, drug-resistant epilepsy; CCTV-EEG, closed circuit video-EEG recording; SRED, surgically remediable epilepsy syndromes; NSRED, not-surgically remediable epilepsy syndromes; VNS, vagal nerve stimulation; DBS, deep brain stimulation.
Polytherapy in patients with DRE
Conventional AEDs were limited in numbers and MOAs, and were either enzyme inducers or inhibitors with a high potential of causing adverse pharmacokinetic drug interactions and affecting the metabolism of endogenous substances crucial for normal physiology.5,6 Considering these pharmacological limitations of conventional drugs, although no RCTs were conducted to compare monotherapy with polytherapy, monotherapy was considered the best option in the era of conventional AEDs. A fair comparison of monotherapy and combination therapy requires balanced baseline patient characteristics; appropriate dose-titration schedules, including initial target dose (ITD), equivalent total drug load (TDL; ratio of the prescribed daily dose to the daily dose defined by World Health Organization guidelines), in the two groups; as well as appropriate selection of drugs for combined usage, preferably drugs carrying synergistic interactions.7,8 These requirements are difficult to meet in trials of patients who failed to respond to previous AED therapy, but may be feasible in newly diagnosed patients. Deckers et al.7 conducted a study comparing carbamazepine (CBZ) monotherapy with combination therapy of CBZ and valproate (VPA) as the initial treatment regimen in patients with untreated epilepsy, which was the only RCT comparing monotherapy with combination therapy with equivalent TDLs. The outcome measures in their study were numerically in favor of combination therapy, but the differences were not statistically significant. Criticisms against the study included that the combination of CBZ and VPA had significant pharmacokinetic drug interactions but no proven synergistic interactions. Recently, Lee et al.9 conducted an open randomized trial comparing CBZ-controlled release (CR) monotherapy and lamotrigine (LTG)-VPA combination therapy with equivalent TDLs in newly diagnosed or untreated patients with partial seizures. The completion rate, the primary outcome measure, was not different between the groups. However, secondary efficacy measures, which included the seizure-free rate during 52 weeks of the maintenance phase and the time to first seizure during the maintenance phase, were in favor of combination therapy, indicating the superiority of LTG-VPA combination therapy over CBZ-CR monotherapy as the initial drug regimen. Kwan and Brodie,10 in a systematic drug trial of 470 patients with newly diagnosed epilepsy, reported that the seizure-free rate was 47% in first-drug monotherapy and 13% in second-drug monotherapy, but after the failure of the first two drugs in monotherapy, it was only 1% in third-drug monotherapy and 3% in combination therapy. Thus, combination therapy seems to provide a better outcome than monotherapy in patients with DRE. A reassessment of an extended population from the same institution 10 years later and then 18 years later1,11 showed that the overall outcome of patients with newly diagnosed epilepsy was not much different, but more patients progressively did well on polytherapy, with seizure freedom in patients receiving polytherapy increasing from 3.0% to 6.4% in a first follow-up, and further increase to 8.4% in the second follow-up. This 20-year longitudinal hospital cohort observation study clearly indicated that polytherapy was associated with a progressive increase in seizure freedom by almost three-fold, whereas seizure freedom by monotherapy remained stagnant at around 60% over decades. Luciano and Shorvon12 reported that systematic trials of new drug combination therapy in 155 patients with chronic DRE achieved prolonged seizure freedom (≥ 1 year) in 28% of the patients and more than 50% seizure reduction in another 21%. In an Italian multicenter study,13 only 25% of 191 children and 21% of 933 adults with DRE were under monotherapy; thus, a majority of the patients with DRE were on polytherapy. They also showed that the burden of AEs assessed by AEP scores and quality of life (QOL) measures were similar between monotherapy and combination therapy.13,14 Lammers et al.15 reported that AEs were similar between monotherapy and polytherapy when TDL was ≤ 2, whereas none of patients under monotherapy and almost 60% of the patients under polytherapy were able to tolerate TDLs > 2. The study concluded that patients on polytherapy were able to tolerate higher TDLs than those on monotherapy, and the emergence of AEs in patients under polytherapy was not related to the number of drugs but a higher TDL than that in patients under monotherapy. In the Italian multicenter study,13 which was conducted in the era of new AEDs, a significant proportion of the patients under polytherapy were able to tolerate TDLs of 4 without any AEs. The authors hypothesized that AEs are determined more by individual susceptibility, type of AEDs used, and physician skill than the number of AEDs and AED load. Therefore, the previous assumption of monotherapy being associated with fewer AEs than polytherapy is probably not tenable anymore in the era of new AEDs. Although monotherapy, including both initial and alternative monotherapy, is still the prevailing mode of pharmacotherapy in epilepsy, the introduction of many new drugs over the past three decades has slowly attracted physicians’ interest in polytherapy, which has become the major mode of therapy at least in patients with DRE who failed to respond to monotherapy of two or more AEDs.
Rational polytherapy
“How to conduct the optimal polytherapy in patients with DREs?” is the most important question to be answered. Unfortunately, there are no specific guidelines yet, but only theoretical recommendations, which are often referred to as “rational polytherapy.”16,17
Drug interactions in combination therapy include both pharmacokinetic and pharmacodynamic interactions. Pharmacokinetic interactions do not change the therapeutic index of each drug under use, and they can be predicted and managed by appropriate dose adjustments. However, the use of enzyme-inducing AEDs (EI-AEDs) may increase the clearance of partner drugs and necessitate higher doses, which may also increase the concentration of metabolites to potentially toxic levels. These effects may also shorten the half-life of AEDs to cause larger fluctuations in effective serum concentrations, precipitating more frequent exposures to higher peak levels as well as lower trough levels and thereby increasing the chance of either neurotoxicity or breakthrough seizures. Therefore, it seems desirable to avoid the use of EI-AEDs or drugs exerting undesirable pharmacokinetic interactions in polytherapy. Pharmacodynamic interactions are primarily related to the MOAs of individual drugs and directly affect the therapeutic index of each drug, but they are difficult to measure in clinical practice; therefore, no RCTs investigating pharmacodynamic interactions in polytherapy have been undertaken yet. In animal experiments, assessments are performed by either isobolographic analysis18,19 or direct measurements of the therapeutic index in specific animal models20 and have revealed that combinations of drugs with different MOAs are usually associated with synergistic interactions, which may involve either supra-additive efficacy or infra-additive toxicity,19 thus supporting the principal concept of rational polytherapy. In clinical trials, combinations of drugs with sodium-channel blocking effects were found to lead to a higher incidence of AEs and premature withdrawal from the study than combinations of a sodium-channel blocker and a drug with a different MOA.21–23 Sake et al.23 reported that the addition of lacosamide (LCM; an inhibitor of slow inactivation of sodium channels) to drugs with a sodium–channel-blocking effect resulted in lower efficacy and more AEs than those obtained by the addition of LCM to non-sodium channel blockers. Margolis et al.24 analyzed a large insurance database and found that various combinations of drugs with same MOAs were associated with higher rates of discontinuation and a higher risk of inpatient admission and emergency department visits in comparison with combinations of drugs showing different MOAs, which supported the hypothesis of MOA-driven synergistic interactions. In clinical practice, several combination regimens have been reported to show synergistic interactions (Table 1). The synergistic interaction of LTG and VPA combination therapy was first reported by Brodie and Yuen,25 who found a much higher responder rate with LTG add-on therapy in patients under VPA monotherapy than in patients taking either CBZ or PHT monotherapy. Pisani et al.26 conducted a systematic, small-scale sequential drug trial of LTG and VPA to find that the LTG and VPA combination was highly effective in patients who were resistant to VPA and LTG monotherapy. The synergistic interactions of the LTG and VPA combination were further supported by many clinical investigations,27–29 which were also demonstrated in a comparative trial of LTG and VPA combination therapy with CBZ-CR monotherapy in patients with newly diagnosed partial epilepsy.9 For other combination regimens, clinical evidence is still limited and mostly at the level of case series or clinical observations (Table 1).30–36 Despite the lack of class 1 evidence, these combinations are the most frequently used combinations in daily practice.34
Table 1.
Combination regimens showing synergistic interactions in clinical studies
| Drug combination | Level of evidence |
|---|---|
| Valproate and lamotrigine25–29 | +++ |
| Valproate and ethosuximide30 | ++ |
| Lamotrigine and topiramate31 | + |
| Lacosamide and levetiracetam32,33 | ++ |
| Lamotrigine and levetiracetam35,36 | ++ |
| Valproate and levetiracetam34 | + |
| Valproate, clobazam and stiripentol37 | +++ |
| Valproate, lamotrigine and benzodiazepine38 | ++ |
Combinations containing enzyme-inducing drugs were excluded. +++, from controlled trials; ++, from case series or observational studies; +, case reports.
“How many drugs can we combine in polytherapy?” is another issue in real-world practice. Poolos et at.37 found that, in their cohort of intellectually impaired patients with severe DRE and static encephalopathy, duotherapy was more effective than monotherapy, but triple drug therapy did not provide any further improvement of seizures. However, the baseline seizure frequency was higher in patients undergoing triple drug therapy than in those receiving duotherapy, which was an important confounder for the lack of differences in efficacy. In fact, RCTs of most new AEDs were undertaken as either duotherapy or triple drug therapy, and, sometimes, even as quadruple-drug combination therapy because patients recruited to these trials were taking one to two, or sometimes three drugs before the commencement of add-on therapy of the study drugs, which resulted in regimens of two to four drugs. All of these patients showed significant improvement in seizure control in comparison with placebo, and subgroup analysis failed to show any different outcomes related to the number of drugs being taken by the study patients. Therefore, if patients’ seizures are not controlled by adequately conducted duo-therapies, it seems prudent to try triple drug therapy. A controlled trial of stiripentol (STR) add-on therapy in patients with Dravet syndrome38 who were taking VPA and clonazepam (CLZ) demonstrated an excellent outcome, and a conversion of various drug regimens to the triple drug combination of LTG, VPA, and clobazam (CLB) was highly effective in patients with severe epileptic encephalopathy.39 All of these clinical examples of synergistic interactions of combination therapy involved drugs with different MOAs, which is worthwhile to consider in real-world practice.
Another component of rational polytherapy is the combination of drugs with different AE profiles, which may be advantageous to avoid neurotoxicities from additive dose-related AEs, the most common AEs in clinical practice. This strategy may be important in explaining the better tolerability of polytherapy than monotherapy at equivalent TDLs.7,15
Polytherapy in real-world practice
Diagnostic re-evaluation of DREs
Treatment of DREs starts with the confirmation of the accurate diagnosis of drug resistance, seizure types, and epilepsy syndromes, which requires careful history-taking; a review of previous drug trials including assessment of compliance, AEs, and efficacy; and a thorough diagnostic evaluation often including epilepsy-dedicated magnetic resonance imaging and prolonged video-electroencephalography (EEG) monitoring. Pseudo-pharmacoresistance due to manageable causes (e.g., poor adherence to AED regimens, wrong drugs, or low doses), psychogenic non-epileptic seizures, or other non-epileptic events should be critically assessed and excluded before the commencement of further therapeutic attempts.40 Accurate diagnosis of epilepsy syndromes, assessment of patient QOL, comorbidities, seizure burden, and the psychosocial environment are all important components for the planning of optimal management. If patients have surgically remediable epilepsy syndromes, such as epilepsy caused by focal epileptogenic lesions that can be completely resected surgically without a high risk of neurological morbidities, they will be much better off with earlier referral to surgical treatment than with continuing long-term drug trials.41 If patients’ epilepsy is not surgically remediable or if patients refuse surgery, further systematic drug trials are mandatory to minimize seizure burden, improve QOL, and maximize control of comorbidities (Fig. 4). Although past clinical experiences suggest that the chance to achieve seizure freedom by further drug trials in patients with DRE is relatively small, it has become clear that continuing drug trials of regimens with previously unexposed AEDs are often effective prolonging seizure freedom in about 30% of patients with DRE,12,42 which may be even higher in patients with cryptogenic or non-lesional epilepsies.43,44
Figure 4.

Patient-oriented choice of antiepileptic drugs. The concept of choice of antiepileptic drugs has changed from “disease-oriented” in the past to “patient-oriented” in the era of new AEDs. Patient-oriented choice of drugs involves selecting the most suitable drug for the patient on the basis of comprehensive multi-dimensional assessment of epilepsy, AEDs, and the patient’s condition. Modified from reference 55 with permission. Wt, body weight; QoL: quality of life; LRE, localization-related epilepsy; GE, generalized epilepsy; LGS, Lennox-Gastaut syndrome; JME, juvenile myoclonic epilepsy; EEG, electroencephalography; EBM, evidence-based medicine; RCT, randomized controlled trial; AED, antiepileptic drug.
Practice of rational polytherapy
As the number of AEDs rapidly increases, the number of combination regimens has increased exponentially. At present, with 25 currently recognized AEDs, 300 combinations are possible for duotherapy and several thousands of possible combinations are possible for triple drug therapy. In the absence of any official guidelines, the numbers are simply too many to try by random selection; thus, selecting the best combination regimen for a given patient suffering from DRE would be a highly demanding task for the physicians and can be only accomplished by a thorough analysis of relevant clinical features and logical synthesis of orders or priorities by knowledgeable and experienced epileptologists.45 This is the reason why the ILAE-Task Force2 recommends the referral of patients to dedicated epilepsy centers if they fail to show adequate response in trials of the first two AEDs. The synthesis of combination regimens for trials consists of two steps (Table 2). The first step involves the selection of potential drugs considered useful to try on the given patients and listing them in the order of preference based on the following criteria: 1) no previous exposure, 2) in cases involving previous exposure, the drug should have been effective at least partly in monotherapy and was not associated with serious AEs, 3) desirable MOAs or MOAs different from those of the AEDs previously tried in a given patient, 4) non-EI AEDs or drugs with no or less pharmacokinetic interactions, 5) drugs effective for (or at least not adversely affecting) patients’ comorbidities, and 6) drugs with a higher therapeutic index. In the second step, a drug that best matches the partner drug is chosen from the list on the basis of the principles of rational polytherapy, i.e., a different MOA, no or minimal pharmacokinetic interactions, different side effect profiles, and belonging to known combination regimens carrying synergistic interactions. For example, if the partner drug was a drug having sodium–channel-blocking effects, choosing a drug with multiple MOAs or non-sodium– channel-blocking MOAs seems appropriate. If the partner drug is enzyme-inducing, the second drug of preference should be a non-hepatic metabolizing drug to avoid adverse pharmacokinetic interactions. Recognizing the various examples of combination regimens in Table 1 and consulting the recent report of the expert opinion survey46 may be helpful in choosing the drug under various clinical scenarios encountered in practice (case 1). Most patients with DREs usually take one or two first-line drugs matching for the given epilepsy syndrome. If the patient is on a single drug and the drug was considered at least partially effective and well tolerable, thus being considered an appropriate drug, physicians will retain the first drug and choose a second drug considered to show synergistic interactions with the first drug. If the first drug was not effective and poorly tolerable, it may be more appropriate to try a substitution monotherapy with the drug chosen.16
Table 2.
Process of selecting the second drug for duotherapy
| Step 1 | Step 2 |
|---|---|
| Candidate drugs of preference | Drugs matching the partner drug |
| Drugs with no previous exposure | Drugs with different MOAs |
| Drugs proven effective, at least partially, in previous exposure | Drugs with no or low potential of pharmacokinetic interactions |
| Drugs with desirable MOAs | Drugs showing different side effect profiles |
| Non-enzyme-inducing drugs or drugs showing no or low risk of pharmacokinetic interactions with concomitant drugs | Drugs known to have synergistic interactions in combination |
| Drugs effective for the patient’s comorbidities or at least not deleterious to the comorbidities | |
| Drugs with a high therapeutic index or good tolerability profile |
Therapeutic index: ratio of effective dose (ED50) to toxic dose (TD50). Modified from reference 55 with permission. MOA, mechanism of action.
Once the second drug was chosen for combination therapy, the next step is to start with a low dose and perform slow escalation of the dose up to the ITD, which is followed by careful assessment of the efficacy and tolerability of the chosen combination. ITD indicates the usual minimal dose of the drug being considered effective, which is based on clinical experience rather than the results of any specific clinical trials (Table 3). If patients tolerate the ITD well but seizures still recur, the drug’s dose will be slowly escalated to the point where full seizure control is achieved or patients start to manifest signs of AEs. If the second drug was effective but caused tolerable AEs, dose reduction of the baseline drug may be attempted to improve tolerability.47 Because patients under polytherapy are liable to be exposed to an excessive drug burden, a careful assessment of tolerability issues should be repeated at every clinical visit, preferably using an instrument like the Liverpool Adverse Effects Profile. Particularly, in patients taking a high TDL (i.e., TDL > 2), physicians should pay attention to the possibility of emerging AEs.15 However, it is likely that the introduction of new AEDs with higher therapeutic indices than conventional drugs might have improved the tolerability of polytherapy than that of conventional drugs to lessen the relationship between AEs and TDL. Canevini et al.13 found that AEs were not associated with TDL or the number of AEDs, but with the gender and depressed mood of the patient in the naturalistic settings of specialized clinical practice. At present, there is no consensus regarding the number of drugs that can be combined for polytherapy or the appropriate time to move to triple drug therapy from duotherapy. Since there are many drugs with different MOAs, it seems worthwhile to try them on the basis of careful assessments of duotherapy. If the first duotherapy was not effective, it may be better to switch with the third drug for the second duotherapy. If the second duotherapy failed or if the first duotherapy was effective but failed to achieve complete seizure freedom, it seems prudent to undertake first triple therapy by adding the drug of best preference.38,39 Clinical trials of add-on therapy with new AEDs48 often showed that the seizure-free rate was higher in patients with less frequent baseline seizures than in those with higher seizure frequencies before the commencement of the study drug, which may well be applicable to real-world practice. By analogy, if duotherapy reduced the patient’s seizure burden significantly, trials of triple drug therapy may have a better chance of seizure freedom. However, if duotherapy was not considerably effective, trials of alternative duotherapy by switching a drug with another chosen drug are preferred to find the best duotherapy regimen before moving to trials of triple drug therapy.
Table 3.
ITD and maintenance doses of antiepileptic drugs in adults
| Drug | ITD (mg/day) | MD (mg/day) | DDD* (mg) |
|---|---|---|---|
| Carbamazepine | 400–600 | 400–1,200 | 1000 |
| Clobazam | 10 | 10–40 | 8 |
| Eslicarbazepine | 800 | 800–1,200 | 800 |
| Ethosuximide | 500 | 500–1,500 | 1,250 |
| Gabapentin | 900 | 900–3,600 | 1,800 |
| Lacosamide | 200 | 200–400 | 300 |
| Lamotrigine | 150 (monotherapy) | 200–400 | 300 |
| Lamotrigine | 75 (with valproic acid) | 75–200 | |
| Lamotrigine | 300 (with enzyme inducer) | 200–500 | |
| Levetiracetam | 1,000 | 1,000–3,000 | 1,500 |
| Oxcarbazepine | 600 | 600–2,400 | 1,000 |
| Perampanel | 4 | 4–12 | 8 |
| Phenobarbital | 60 | 60–120 | 100 |
| Phenytoin | 200 | 200–400 | 300 |
| Pregabalin | 300 | 300–600 | 300 |
| Primidone | 500 | 500–1,500 | 1,250 |
| Rufinamide | 1,200 | 1,200–3,200 | 1,400 |
| Topiramate | 100 | 100–400 | 300 |
| Valproic acid | 500 | 500–2,000 | 1,500 |
| Vigabatrin | 1,000 | 1,000–3,000 | 2,000 |
| Zonisamide | 200 | 200–600 | 200 |
ITD, initial target dose; MD, maintenance dose; DDD, defined daily dose.
By World Health Organization.
Management of comorbidities
Pharmacotherapy of DREs is not restricted to the reduction of seizure burden but should include improvement of patient QOL by providing global healthcare to the patient, which includes control of comorbidities, psychosocial support, and control of drug-related AEs. The prevalence of comorbidities in patients with epilepsy is 2 to 8 times that in the general population, and about 50% of patients with epilepsy have at least one comorbidity, which is even higher in patients with DRE.49 Since many AEDs affect non-epileptic conditions, the choice of AEDs is not only directed to seizure types and epilepsy syndromes, but also to the various comorbidities carried by patients. Comorbidities present another concern related to the risk of potential drug interactions between concomitant drugs and AEDs, which should be considered for the choice of AEDs. Among the diverse comorbidities associated with epilepsy, depression and anxiety are the most common in patients with DRE; these two conditions are present in 30% to 60% of the patients and are responsible for their poor QOL, but are frequently under-recognized and under-treated.50–52 These affective disorders seem to have a bidirectional relationship with epilepsy; thus, they may possibly interact with each other to affect the clinical courses of DRE.52 Although no convincing evidence is available yet for the use of second-generation anti-depressants being responsible for improvements in seizure control, their use to manage affective disorders is an essential part of management to improve QOL and prevent suicidality.52 Depression is also responsible for the higher prevalence of diverse AEs of AEDs therapy, which may be responsible for the poor tolerability of AEDs and impair the adherence to AED therapy.14,52,53 Therefore, treatment of depression may help improve the outcomes of AED trials by improving the patient’s tolerability to the trial drugs. Selective serotonin reuptake inhibitors are also known to help prevent sudden unexpected death of epilepsy (SUDEP), an important cause of higher mortality in patients with DRE, in experimental models.54 However, this preclinical result has not been tested in clinical trials yet. Treatment of comorbidities in patients with DREs should be at a higher priority to prevent suicide, improve QOL, and improve the outcome of AED therapy. At present, we have a long list of drugs that are either preferred or avoided in relation to various comorbidities (Table 4). Other aspects of pharmacotherapy in patients with DREs may include the use of repurpose drugs based on a better knowledge of the epileptogenesis or molecular pathogenesis of epilepsy via genetic analysis of patients with DREs, which is the beginning of precision medicine or target-oriented epilepsy management. These are, however, beyond the scope of this review.
Table 4.
Choice of antiepileptic drugs related to comorbidities
| Choose | Avoid | |
|---|---|---|
| Obesity ± DM | TPM, ZNS | VPA, PGB, GBP, PER |
| Migraine | TPM, VPA, ZNS, PBG, GBP | |
| Skin rash | LEV, GBP, PGB, TPM, VPA, PER, LCM | LTG, OXC, CBZ, PHT, PB |
| Neuropathic pain | PGB, GBP, CBZ, OXC, PHT | |
| Depression ± Behav/Psych | LTG, CBZ, OXC, VPA, PGB | LEV, PB, PRM, TPM, ZNS, PER |
| Cognitive dysfunction | LTG, LEV, OXC | PB, TPM, ZNS |
| Concomitant drugs | GBP, LEV, PGB, VPA | EI-drugs |
| Restless legs syndrome | GBP, PGB, CZP | |
| Renal stone | TPM, ZNS | |
| Glaucoma | TPM | |
| Hematological disorder | CBZ, VPA | |
| Hyponatremia | OXC, ESL, CBZ | |
| Hepatic disease | New AEDs (not hepatic toxic, renal excretion) | VPA |
| Renal disease | Old AEDs (excreted by hepatic metabolism) | |
| Osteoporosis | LTG, LEV | EI-drugs, TPM, VPA, ZNS |
| Gait disturbances | CBZ, PHT, PER | |
| Tremor | TPM, PER | VPA |
| Parkinson disease | ZNS | |
| Cardiac arrhythmia | CBZ, LTG, LCM, and others SCB | |
| Cancer | VPA, LEV, PER | EI-drugs |
| Heat stroke | TPM, ZNS | |
| Atherosclerosis | EI-drugs |
Modified from reference 55 with permission.
DM, diabetes mellitus; TPM, topiramate; ZNS, zonisamide; VPA, valproic acid; PGB, pregabalin; GBP, gabapentin; PER, perampanel; LEV, levetiracetam; LCM, lacosamide; LTG, lamotrigine; OXC, oxcarbazepine; CBZ, carbamazepine; PHT, phenytoin; PB, 약어풀이; Behav/Psych, behavioral and/or psychiatric disorders; PRM, primidone; EI-drug, enzyme-inducing drugs; CZP, clonazepam; ESL, eslicarbazepine; AED, antiepileptic drug; SCB, sodium-channel blockers.
In conclusion, pharmacotherapy of epilepsy has become more diversified and sophisticated by introduction of many new AEDs with diverse MOAs, better pharmacokinetic profiles, and better tolerability profiles, which were instrumental for the concept of patient- tailored pharmacotherapy. This is a highly demanding model for the physicians and requires comprehensive assessment of patients with extensive knowledge of individual drugs and epilepsy syndromes. New AEDs were introduced to the market after extensive RCTs of combination therapy in patients with severe DRE, which has resulted in a gradual revival of polytherapy under the concept of rational polytherapy based on mechanistic combination principles. At present, polytherapy has become a major mode of therapy in patients with DRE. Although many experts are still skeptical about the impact of new AEDs on seizure outcome, the proportion of patients becoming seizure-free by polytherapy was tripled over the past two decades in a longitudinal study of a large hospital cohort. Through systematic add-on drug trials, seizure freedom in patients with DRE, which was once considered unlikely, has been accomplished in 30% of patients with DREs, and in even higher proportions of patients with cryptogenic or non-lesional disease. At present, most experts encourage continuing drug trials by introducing drugs with different MOAs among the previously tried drugs to obtain small but additional cases of seizure freedom.
Case
A 15-year-old female high school student developed a generalized tonic-clonic seizure (GTCS) and was brought to the emergency room (ER). When she arrived at the ER, she was awake and oriented, but she did not remember why she was in the hospital. Her perinatal and developmental history were unrevealing. There was no history of febrile convulsion or remote central nervous system insults. Family history was negative for any neurological disorders or epilepsy. Routine laboratory studies yielded essentially negative findings but showed markedly increased creatine phosphokinase levels (3,500 IU). The computed tomography scan of the brain was normal. EEG showed a few episodes of 4–6-Hz generalized spike and wave complexes. The patient was discharged from the ER under the diagnosis of first GTCS. At a scheduled outpatient clinic visit, careful history-taking revealed that the patient had experienced occasional episodes of brief jerks of shoulders and arms, usually in the morning, over the last 2 years. She denied having any episodes of blank staring or losing contact. She also reported that she had a few episodes of brief jerks before she lost consciousness on the day of ER visit.
A diagnosis of juvenile myoclonic epilepsy (JME) was determined by her physician, who prescribed VPA 500 mg twice a day. She remained seizure- and myoclonus-free without any side effects for more than 3 years. She entered to a college at 18 years of age and started to engage in various college activities. She skipped her AEDs occasionally and occasionally experienced a few episodes of shoulder jerks. On one morning during her first year in college, she developed a GTCS, which was followed by another episode of GTCS. She was brought again to the ER, where she developed another episode of GTCS, which was managed by intravenous injection of lorazepam (4 mg). She had fully recovered without any further recurrence. The blood level of VPA was checked and reported to be < 3.0 μg/mL. She had not taken her medications for 2 days and had stayed late at night with her friends at a party on the previous day with a few shots of hard liquor. She was referred to an epileptologist for further management of seizures, who explained her the problems with VPA related to teratogenesis and in-utero fetal exposure. He also recommended her to switch VPA to levetiracetam (LEV) monotherapy. Over a month, VPA was completely discontinued and switched to LEV monotherapy (500 mg × bid). She did well initially but started to experience a few myoclonic jerks, especially after sleep deprivation. The LEV dose was increased to 1,000 mg twice a day, but she continued to have occasional myoclonus and developed another episode of GTCS on a day after sleeping later at night with a few drinks. Her family was much concerned about the recurrent seizures and asked her doctor if she could use VPA again.
What are the options for treating this young woman?
Before the era of the new AEDs, the choice of drug for JME was VPA, and if VPA was not fully effective, CLZ was the drug of choice to add-on. In the era of new AEDs, VPA is still considered the most effective drug but is not considered as the first-line drug any longer for young woman at reproductive age, due to the high risk of teratogenicity and fetal cognitive and behavioral disturbances related to intrauterine exposure. VPA may also cause undesirable AEs in young women, e.g., weight gain, hair loss, and reproductive disorders including polycystic ovarian disease. Currently, most clinical guidelines recommend avoiding VPA in women of reproductive age. CLZ is effective but considered as a second-line drug because of its sedative AEs and the potential of tachyphylaxis. Among the new AEDs, LEV, LTG, topiramate (TPM), ZNS, and perampanel (PER) are broad-spectrum AEDs that are effective in both partial and generalized epilepsy and may be also effective in JME. However, none of them have been assessed in RCTs of initial monotherapy in JME and thus lack class 1 evidence. LEV was found to be effective in an RCT on add-on therapy in JME; it carried a low risk of teratogenicity and fetal malfunction and showed a lack of pharmacokinetic interactions with concomitant drugs, including oral contraceptives. It is also weight-neutral and is associated with low risks for reproductive health. Clearance of LEV during pregnancy increases by two-fold; thus, its serum concentration may decrease significantly to precipitate seizure recurrences during pregnancy, which can be prevented by dose adjustment. LTG is another drug that is effective in JME. However, LTG is associated with risks of skin rash and exacerbation of myoclonic seizures, necessitating a slow dose-escalation and careful patient monitoring during the early stage of LTG introduction. LTG has a high safety profile for its use in young women of reproductive age with less potential of pharmacokinetic interactions. LTG, like LEV, requires monitoring of plasma concentration during pregnancy because of a more than two-fold increase in its clearance. TPM was reported to be effective in uncontrolled trials of JME and may be preferred in patients with obesity or migraine. However, data from pregnancy registries suggested a higher rate of teratogenicity with TPM than with LTG and LEV. The role of ZNS and PER in young women of reproductive age with JME needs further data. If we consider that the patient is an intelligent, healthy, sexually active young college student, either LEV or LTG may be the first drug for initial monotherapy in this patient.
The first option is to increase the dose of LEV to 3,000 mg/day; however, the possibility of achieving seizure freedom with this dosage is very small. Subgroup analysis of LEV monotherapy trials in patients with partial-onset seizures revealed that most cases of seizure freedom (54.4% of study patients) were achieved at an LEV dose of ≤ 2,000 mg/day, and a dose-increase to 3,000 mg/day increased the seizure-free rate by only 2.2% (56.6% of study patients). Therefore, it is likely that a further dose-increase of LEV to 3,000 mg/day may carry a higher risk of side effects than the benefit of seizure freedom.
The second option is to conduct LEV and LTG combination therapy by introducing LTG with a slow dose-escalation. On the basis of careful assessment of the patient’s responses, the dosage of LTG and/or LEV can be adjusted to the optimal levels. In fact, a combination of LEV and LTG is the best match for rational polytherapy including different MOAs, no pharmacokinetic interactions, and different AE profiles. Synergistic interactions of LEV and LTG have been reported in both preclinical and clinical studies, and both drugs have high therapeutic indices indicating good tolerability. No evidence of a higher risk of teratogenicity or intrauterine exposure has been addressed in an analysis of multiple pregnancy registries.
The third option is a substitution monotherapy of LTG by introducing LTG and tapering of LEV simultaneously. However, LTG should be introduced slowly to lower the risk of skin rashes and the risk of GTCS recurrence as a result of rapid tapering of LEV, necessitating caution and a possible trial of transitional polytherapy preceding the substitution monotherapy. LTG may be slowly increased up to 150–200 mg/day while keeping LEV at the same dose. The patient will be observed on duotherapy of LTG and LEV for 3 months to assess the effectiveness of this regimen. If she is completely free of myoclonus and shows no side effects, she will remain on the same regimen for a longer period (> 1 year) before a trial of slow tapering off LEV. If she experiences marked improvement but still reports occasional myoclonus, it may be prudent to perform substitution monotherapy with LTG by increasing the dose of LTG until the myoclonus is completely controlled or she develops side effects, while tapering off the LEV. If LTG monotherapy fails to completely control myoclonus, retrial of LTG and LEV combination therapy would be a desirable option in this patient.
If LEV and LTG combination or alternative LTG monotherapy fail to completely control myoclonus but she does not show any further attacks of GTCS, lifestyle modification measures such as maintaining regular awake-sleep cycles and avoiding sleep deprivation, excessive stress, extreme tiredness, and alcohol consumption can be considered. It may also be feasible to try one of TPM, ZNS, PER, and CLZ as either duotherapy or triple drug therapy. Alternatively, a small dose of VPA (≤ 600 mg/day) may be added to LEV or LTG, because the risk of VPA-related teratogenicity or fetal malformations is dose-related but not related to the number of AEDs. Apparently both combinations, LEV + VPA and LTG + VPA, are well recognized to show synergistic interactions in previous clinical experiences and thus may be worthwhile in patients with refractory JME. Trials for all of these options should be undertaken only after thorough discussions with the patient and her family about the risk-benefit ratio for each option.
References
- 1.Brodie MJ, Barry SJE, Bamagous G, Norrie JD, Kwan P. Patterns of treatment response in newly diagnosed epilepsy. Neurology. 2012;78:1548–54. doi: 10.1212/WNL.0b013e3182563b19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51:1069–77. doi: 10.1111/j.1528-1167.2009.02397.x. [DOI] [PubMed] [Google Scholar]
- 3.French JA, Schachter SC, Sirven J, Porter R. The Epilepsy Foundation’s 4th Biennial Epilepsy Pipeline Update Conference. Epilepsy Behav. 2015;46:34–50. doi: 10.1016/j.yebeh.2015.02.033. [DOI] [PubMed] [Google Scholar]
- 4.Perucca E. Current trends in antiepileptic drug therapy. Epilepsia. 2003;44(Suppl 4):41–7. doi: 10.1046/j.1528-1157.44.s4.1.x. [DOI] [PubMed] [Google Scholar]
- 5.Mintzer S, Mattson RT. Should enzyme-inducing antiepileptic drugs be considered first-line agents? Epilepsia. 2009;50(Suppl 8):42–50. doi: 10.1111/j.1528-1167.2009.02235.x. [DOI] [PubMed] [Google Scholar]
- 6.Brodie MJ, Mintzer S, Pack AM, Gidal BE, Vecht CJ, Schmidt D. Enzyme induction with antiepileptic drugs: cause for concern? Epilepsia. 2013;54:11–27. doi: 10.1111/j.1528-1167.2012.03671.x. [DOI] [PubMed] [Google Scholar]
- 7.Deckers CL, Hekster YA, Keyser A, van Lier HJ, Meinardi H, Renier WO. Monotherapy versus polytherapy for epilepsy: a multicenter double-blind randomized study. Epilepsia. 2001;42:1387–94. doi: 10.1046/j.1528-1157.2001.30800.x. [DOI] [PubMed] [Google Scholar]
- 8.Barker-Haliski M, Sills GJ, White SH. What are the arguments for and against rational therapy for epilepsy? Adv Exp Med Biol. 2014;813:295–308. doi: 10.1007/978-94-017-8914-1_24. [DOI] [PubMed] [Google Scholar]
- 9.Lee BI, No SK, Yi SD, et al. Unblinded, randomized multicenter trial comparing lamotrigine and valproate combination with controlled-release carbamazepine monotherapy as initial drug regimne in untreated epilepsy. Seizure. 2018;55:17–24. doi: 10.1016/j.seizure.2017.12.008. [DOI] [PubMed] [Google Scholar]
- 10.Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–9. doi: 10.1056/NEJM200002033420503. [DOI] [PubMed] [Google Scholar]
- 11.Chen Z, Brodie MJ, Liew D, Kwan P. Treatment outcomes in patients with newly diagnosed epilepsy treated with established and new antiepileptic drugs: a 30-year longitudinal cohort study. JAMA Neurology. 2018;75:279–86. doi: 10.1001/jamaneurol.2017.3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luciano AL, Shorvon SD. Results of treatment changes in patients with apparently drug-resistant chronic epilepsy. Ann Neurol. 2007;62:375–81. doi: 10.1002/ana.21064. [DOI] [PubMed] [Google Scholar]
- 13.Canevini MP, De Sarro G, Galimberti CA, et al. Relationship between adverse effects of antiepileptic drugs, number of coprescribed drugs, and drug load in a large cohort of consecutive patients with drug-refractory epilepsy. Epilepsia. 2010;51:797–804. doi: 10.1111/j.1528-1167.2010.02520.x. [DOI] [PubMed] [Google Scholar]
- 14.Luoni C, Bisulli F, Canevini MP, et al. Determinants of health-related quality of life in pharmacoresistant epilepsy: results from a large multicenter study of consecutively enrolled patients using validated quantitative assessments. Epilepsia. 2011;52:2181–91. doi: 10.1111/j.1528-1167.2011.03325.x. [DOI] [PubMed] [Google Scholar]
- 15.Lammers MW, Hekster YA, Keyser A, Meinardi H, Renier WO, van Lier H. Monotherapy or polytherapy for epilepsy revisited: a quantitative assessment. Epilepsia. 1995;36:440–6. doi: 10.1111/j.1528-1157.1995.tb00484.x. [DOI] [PubMed] [Google Scholar]
- 16.French JA, Faught E. Rational polytherapy. Epilepsia. 2009;50(Suppl 8):63–8. doi: 10.1111/j.1528-1167.2009.02238.x. [DOI] [PubMed] [Google Scholar]
- 17.Brodie MJ, Sills GJ. Combining antiepileptic drugs--rational polytherapy? Seizure. 2011;20:369–75. doi: 10.1016/j.seizure.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Kwan P, Brodie MJ. Epilepsy after the first drug fails: substitution or add-on? Seizure. 2000;9:464–8. doi: 10.1053/seiz.2000.0442. [DOI] [PubMed] [Google Scholar]
- 19.Lasoń W, Dudra-Jastrzębska M, Rejdak K, Czuczwar SJ. Basic mechanisms of antiepileptic drugs and their pharmacokinetic/pharmacodynamics interactions: an update. Pharmacol Rep. 2011;63:271–92. doi: 10.1016/s1734-1140(11)70497-2. [DOI] [PubMed] [Google Scholar]
- 20.Kaminski RM, Matagne A, Patsalos PN, Klitgaard H. Benefit of combination therapy in epilepsy: a review of the preclinical evidence with levetiracetam. Epilepsia. 2009;50:387–97. doi: 10.1111/j.1528-1167.2008.01713.x. [DOI] [PubMed] [Google Scholar]
- 21.Besag FM, Berry DJ, Pool F, Newbery JE, Subel B. Carbamazepine toxicity with lamotrigine: pharmacokinetic or pharmacodynamic interaction? Epilepsia. 1998;39:183–7. doi: 10.1111/j.1528-1157.1998.tb01356.x. [DOI] [PubMed] [Google Scholar]
- 22.Barcs G, Walker EB, Elger CE, et al. Oxcarbazepine placebo-controlled, dose-ranging trial in refractory partial epilepsy. Epilepsia. 2000;41:1597–607. doi: 10.1111/j.1499-1654.2000.001597.x. [DOI] [PubMed] [Google Scholar]
- 23.Sake JK, Hebert D, Isojärvi J, et al. A pooled analysis of lacosamide clinical trial data grouped by mechanism of action of concomitant antiepileptic drugs. CNS Drugs. 2010;24:1055–68. doi: 10.2165/11587550-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 24.Margolis JM, Chu BC, Wang ZJ, Copher R, Cavazos JE. Effectiveness of antiepileptic drug combination therapy for partial-onset seizures based on mechanisms of action. JAMA Neurol. 2014;71:985–93. doi: 10.1001/jamaneurol.2014.808. [DOI] [PubMed] [Google Scholar]
- 25.Brodie MJ, Yuen AW. Lamotrigine substitution study: evidence for synergism with sodium valproate? 105 study group. Epilepsy Res. 1997;26:423–32. doi: 10.1016/s0920-1211(96)01007-8. [DOI] [PubMed] [Google Scholar]
- 26.Pisani F, Oteri G, Russo MF, Di Perri R, Perucca E, Richens A. The efficacy of valproate-lamotrigine comedication in refractory complex partial seizures: evidence for a pharmacodynamic interaction. Epilepsia. 1999;40:1141–6. doi: 10.1111/j.1528-1157.1999.tb00832.x. [DOI] [PubMed] [Google Scholar]
- 27.Kanner AM, Frey M. Adding valproate to lamotrigine: a study of their pharmacokinetic interaction. Neurology. 2000;54:588–91. doi: 10.1212/wnl.55.4.588. [DOI] [PubMed] [Google Scholar]
- 28.McCabe PH, NcNew CD, Michel NC. Effect of divalproex-lamotrigine combination therapy in frontal lobe epilepsy. Arch Neurol. 2001;58:1264–68. doi: 10.1001/archneur.58.8.1264. [DOI] [PubMed] [Google Scholar]
- 29.Moeller JJ, Rahey SR, Sadler RM. Lamotrigine-valproic acid combination therapy in medically refractory epilepsy. Epilepsia. 2009;50:475–79. doi: 10.1111/j.1528-1167.2008.01866.x. [DOI] [PubMed] [Google Scholar]
- 30.Rowan AJ, Meijer JW, de Beer-Pawlikowski N, van der Geest P, Meinardi H. Valproate-ethosuximide combination therapy for absence seizures. Arch Neurol. 1983;40:797–802. doi: 10.1001/archneur.1983.04050120047006. [DOI] [PubMed] [Google Scholar]
- 31.Stephen IJ, Sills GJ, Brodie MJ. Lamotrigine and topiramate may be useful combination. Lancet. 1998;351:958–59. doi: 10.1016/S0140-6736(05)60613-7. [DOI] [PubMed] [Google Scholar]
- 32.Brigo F, Ausserer H, Tezzon F, Nardone R. When one plus one makes three: the quest for rational antiepileptic polytherapy with supraadditive anticonvulsant efficacy. Epilepsy Behav. 2013;27:439–42. doi: 10.1016/j.yebeh.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 33.Chung S, Ben-Menachem E, Sperling MR, et al. Eamining the clinical utility of lacosamide: pooled analysies of three phase II/III clinical trials. CNS Drugs. 2010;24:1041–54. doi: 10.2165/11586830-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 34.Brodie MJ. Pharmacological treatment of drug-resistant epilepsy in adults: a practical guide. Curr Neurol Neurosci Rep. 2016;16:82. doi: 10.1007/s11910-016-0678-x. [DOI] [PubMed] [Google Scholar]
- 35.Kinirons P, McCarthy M, Doherty CP, Delanty N. Predicting drug-resistant patients who respond to add-on therapy with levetiracetam. Seizure. 2006;15:387–92. doi: 10.1016/j.seizure.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 36.Legge AW, Detyniecki K, Javed A, et al. Comparative efficacy of unique antiepileptic drug regimens in focal epilepsy: an exploratory study. Epilepsy Res. 2018;142:73–80. doi: 10.1016/j.eplepsyres.2018.03.011. [DOI] [PubMed] [Google Scholar]
- 37.Poolos NP, Warner LN, Humphreys SZ, Williams S. Comparative efficacy of combination drug therapy in refractory epilepsy. Neurology. 2012;78:62–8. doi: 10.1212/WNL.0b013e31823ed0dd. [DOI] [PubMed] [Google Scholar]
- 38.Chiron C, Marchand MC, Tran A, et al. Stiripentol in severe myoclonic epilepsy in infancy: a randomized placebo-controlled syndrome-dedicated trial. Lancet. 2000;356:1638–42. doi: 10.1016/s0140-6736(00)03157-3. [DOI] [PubMed] [Google Scholar]
- 39.Machado VH, Palmini A, Bastos FA, Rotert R. Long-term control of epileptic drop attacks with the combination of valproate, lamotrigine, and a benzodiazepine: a ‘proof of concept,’ open label study. Epilepsia. 2011;52:1303–10. doi: 10.1111/j.1528-1167.2011.03075.x. [DOI] [PubMed] [Google Scholar]
- 40.Dalic L, Cook MJ. Managing drug-resistant epilepsy: challeges and solutions. Neuropsychiatr Dis Treat. 2016;12:2605–16. doi: 10.2147/NDT.S84852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kwan P, Sperling MR. Refractory seizures: try additional antiepileptic drugs (after two have failed) or go directly to early surgery evaluation? Epilepsia. 2009;50(Suppl 8):57–62. doi: 10.1111/j.1528-1167.2009.02237.x. [DOI] [PubMed] [Google Scholar]
- 42.Choi H, Hayat MJ, Zhang R, et al. Drug-resistant epilepsy in adults: outcome trajectories after failure of two medications. Epilepsia. 2016;57:1152–60. doi: 10.1111/epi.13406. [DOI] [PubMed] [Google Scholar]
- 43.Berg AT, Levy SR, Testa FM, D’Souza R. Remission of epilepsy after two drug failures in children: a prospective study. Ann Neurol. 2009;65:510–97. doi: 10.1002/ana.21642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wirrell EC, Wong-Kisiel LC, Mandrekar J, Nickels KC. What predicts enduring intractability in children who appear medically intractable in the first 2 years after diagnosis? Epilepsia. 2013;54:1056–64. doi: 10.1111/epi.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santulli L, Coppola A, Balestrini S, Striano S. The challenges of treating epilepsy with 25 antiepileptic drugs. Pharmacol Res. 2016;107:211–9. doi: 10.1016/j.phrs.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 46.Shih JJ, Whitlock JB, Chimato N, Vargas E, Karceski SC, Frank RD. Epilepsy treatment in adult and aolescents: expert opinion, 2016. Epilepsy Behav. 2017;69:186–222. doi: 10.1016/j.yebeh.2016.11.018. [DOI] [PubMed] [Google Scholar]
- 47.Naritoku DK, Hulihan JF, Schwarzman LK, Kamin M, Olson WH. Effect of cotherapy reduction on tolerability of epilepsy add-on therapy: a randomized controlled trial. Ann Pharmacother. 2005;39:418–23. doi: 10.1345/aph.1E403. [DOI] [PubMed] [Google Scholar]
- 48.Heo K, Lee BI, Yi SD, et al. Efficacy and safety of levetiracetam as adjunctive treatment of refractory partial seizures in a multicentre open-label single-arm trial in Korean patients. Seizure. 2007;16:402–9. doi: 10.1016/j.seizure.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 49.Keezer MR, Sisodiya SM, Sander JW. Comorbidities of epilepsy: current concepts and future perspectives. Lancet Neurol. 2016;15:106–15. doi: 10.1016/S1474-4422(15)00225-2. [DOI] [PubMed] [Google Scholar]
- 50.Fiest KM, Dykeman J, Patten SB, et al. Depression in epilepsy: a systematic review and meta-analysis. Neurology. 2013;80:590–9. doi: 10.1212/WNL.0b013e31827b1ae0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boylan LS, Flint LA, Labovitz DL, Jackson SC, Starner K, Devinsky O. Depression but not seizure frequency predicts quality of life in treatment-resistant epilepsy. Neurology. 2004;62:258–61. doi: 10.1212/01.wnl.0000103282.62353.85. [DOI] [PubMed] [Google Scholar]
- 52.Kanner AM. Psychiatric comorbidities in new onset epilepsy: should they be always investigated? Seizure. 2017;49:79–82. doi: 10.1016/j.seizure.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 53.Kim SK, Park SP, Kwon OY. Impact of depression and anxiety on adverse event profiles in Korean people with epilepsy. Epilepsy Behav. 2015;46:185–91. doi: 10.1016/j.yebeh.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 54.Massey CA, Sowers LP, Dlouhy BJ, Richerson GB. Mechanisms of sudden unexpected death in epilepsy: the pathway to prevention. Nat Rev Neurol. 2014;10:271–82. doi: 10.1038/nrneurol.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee BI, Park KM, Kim SE, Heo K. Clinical opinion: Earlier employment of polytherapy in sequential pharmacotherapy of epilepsy. Epilepsy Res. 2019;156:106165. doi: 10.1016/j.eplepsyres.2019.106165. [DOI] [PubMed] [Google Scholar]