

Abstract
Glycopeptide-centric mass spectrometry has become a popular approach for studying protein glycosylation. However, current approaches still utilize fragmentation schemes and ranges originally optimized and intended for the analysis of typically much smaller unmodified tryptic peptides. Here, we show that by merely increasing the tandem mass spectrometry m/z range from 2000 to 4000 during electron transfer higher energy collisional dissociation (EThcD) fragmentation, a wealth of highly informative c and z ion fragment ions are additionally detected, facilitating improved identification of glycopeptides. We demonstrate the benefit of this extended mass range on various classes of glycopeptides containing phosphorylated, fucosylated, and/or sialylated N-glycans. We conclude that the current software solutions for glycopeptide identification also require further improvements to realize the full potential of extended mass range glycoproteomics. To stimulate further developments, we provide data sets containing all classes of glycopeptides (high mannose, hybrid, and complex) measured with standard (2000) and extended (4000) m/z range that can be used as test cases for future development of software solutions enhancing automated glycopeptide analysis.
Glycosylation is one of the most common and complex protein post-translational modifications that is involved in a myriad of biological processes,1 while changes in glycosylation have been observed in a number of pathological states.2−6 However, due to the heterogeneity of glycan structures associated with glycoproteins, accurate analysis of protein glycosylation still poses a significant analytical challenge. Currently, mass spectrometry (MS) is one of the most popular approaches used for characterization of protein glycosylation. MS analysis of released glycans (glycomics) is the best-established method for analyzing glycosylation. It provides great insight into fine variations of glycan structures and is amenable to high throughput and automated data analysis.7 Unfortunately, in glycomic analysis, information about the underlying protein carrier and its site-specific modification is not retained.
To determine the glycan structures and their site of attachment, analysis of intact glycopeptides is essential. However, glycopeptide analysis still poses a significant analytical challenge on multiple levels:8−10 (a) lower ionization efficiency of glycopeptides when compared to their nonmodified counterparts; (b) the heterogeneity of glycan structures associated with each modified site; (c) their generally more complicated tandem mass spectrometry (MS/MS) fragmentation spectra; and (d) the lack of adequate software tools for confident data analysis. For this reason, most of glycopeptide studies have been focused on the characterization of glycopeptides obtained from isolated glycoproteins. This was also due to reliance on collision induced dissociation fragmentation techniques that provide spectra rich in glycan fragments but produce very few peptide backbone cleavages. Recently, we introduced a hybrid fragmentation technique combining electron transfer dissociation and higher energy dissociation11 (EThcD) that has become a de facto method of choice for intact glycopeptide characterization.12−21 The benefit of EThcD is that it provides a rich series of c/z ions pinpointing the site of modification while also providing insight into the glycan composition. Additionally, there has been a surge of studies characterizing intact glycopeptides from complex mixtures such as plasma/serum,19,22−24 cell lysates,25,26 and various tissues.19,27−29 One of the main challenges observed in all of these studies is that the underlying data analysis still requires a great degree of manual validation. A possible cause of this is that in contrast to general proteomics and phosphoproteomic approaches in glycoproteomics, we, by using the standard instrument settings, almost never observe complementary ion fragments that would retain the glycan side chain, hampering confidence in site identification.
We here hypothesized that this is primarily due to the fact that most approaches for glycoproteomics are still based on well-established bottom-up proteomics workflows optimized for unmodified tryptic peptides, ignoring the fact that N-glycopeptides fall more within the realm of middle-down proteomics (3–10 kDa). In this work, we show that by merely extending the EThcD fragmentation range up to 4000 m/z (from the standard 2000), we observe several additional fragment ions and a rich series of complementary c and z ions that all together lead to a more complete sequence coverage and increased confidence in glycan annotation. We demonstrate the benefits of this workflow on (a) mannose-6-phosphate (M6P) glycopeptides enriched from a CHO cell lysate using Fe3+-IMAC, (b) complex sialylated glycopeptides enriched by strong anion exchange (SAX) from CHO cell lysate, and (c) human milk glycopeptides harboring complex and multiply fucosylated N-glycans enriched by HILIC. We provide exemplary standard and extended mass range EThcD fragmentation spectra for each glycan class and demonstrate that the extended m/z range provides a wealth of previously ignored informative fragment ions for each glycan class. Additionally, we noticed that the most commonly used software tool for the analysis of EThcD data, Byonic, underestimates the benefits of our extended range approach due to the arbitrary cut off of m/z 2500 in their scoring algorithm. Therefore, we make here publicly available the extensive data sets obtained both with standard and extended m/z range that cover all the classes of glycopeptides with the aim to use them as test sets for software development benefiting the rapidly growing field of glycoproteomics.
Experimental Procedure
The CHO cell sample (a kind gift from Henrik Clausen, University of Copenhagen) was lysed in the lysis buffer and sonicated. Next, methanol–chloroform protein precipitation was performed, and the protein precipitate was digested overnight. Fe3+-IMAC enrichment was performed as described previously.30 The flowthrough of the Fe3+-IMAC was loaded onto a Hypersep SAX cartridge (ThermoFisher Scientific, Germany) to enrich for glycopeptides missed by the initial Fe3+-IMAC enrichment. The human milk sample was subjected to centrifugation and ultracentrifugation to obtain the milk serum. After digestion, the human milk glycopeptides were enriched on a GlykoPrep cartridge (ProZyme, Denmark). Nanoflow liquid chromatography (LC)-MS/MS was performed by coupling an Agilent 1290 (Agilent Technologies, Middelburg, Netherlands) to an Orbitrap Fusion Lumos (Thermo Scientific, Bremen, Germany). The mass spectrometer was operated in data-dependent acquisition mode. We used a HCD-pd-EThcD method where EThcD was triggered upon observation of signature glycan oxonium ions during HCD fragmentation. All samples were run with two different methods: (a) the MS/MS range was set from 120 to 2000 or (b) the MS/MS range was set between 120 and 4000. Data were analyzed with Byonic (ver. 2.15.10) (Protein Metrics Inc., United States) and searched against the CHO UniProt database (34 962 entries) or focused human milk database (1259 entries). CHO samples were searched with a Byonic database of 182 glycans (with M6P glycoforms added in manually), while milk samples were searched with a database containing 309 glycans. A more detailed description of each part of the protocol can be found in the Supporting Information. Raw and fasta files have been deposited to MassIVE with the identifier: MSV000083710.
Results and Discussion
In recent years, there has been an increased interest in characterization of intact glycopeptides from complex mixtures.31,32 This has been mainly enabled by the development of hybrid fragmentation techniques such as EThcD, stepped HCD,33,34 and AI-ETD.27 Of note, all these recently reported comprehensive glycopeptide analyses seem to take the standard m/z 2000 as a cutoff in their analyses.
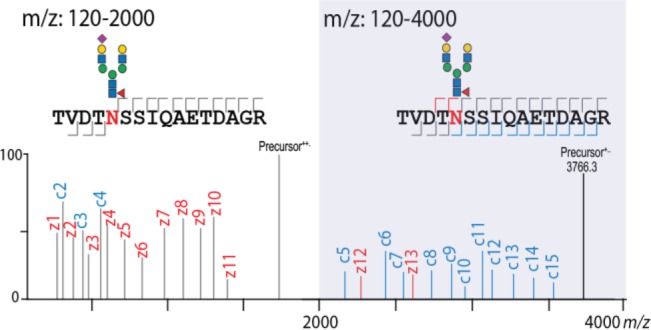
Series of complementary fragment ions (e.g., b/y and c/z) enhance peptide identification in standard proteomics and phosphoproteomics approaches. However, in the characterization of intact glycopeptides, such complementary fragment ions are (so far) hardly observed or remain unnoticed. In our previous work reporting on the characterization of mannose-6-phosphate glycopeptides from cell lysates, we observed highly abundant c ion series at the high end of our MS/MS m/z range that contained intact glycans still attached to the peptide backbone. We hypothesized that because glycopeptides, based on their average larger intact mass (3–10 kDa), fall within the range of middle-down proteomics, an extension of the MS/MS m/z range should give increased confidence in their identification. To test this hypothesis, we reanalyzed M6P glycopeptides extracted from a CHO cell lysate following an Fe3+-IMAC enrichment and analyzed them with the conventional (up to 2000) and extended m/z fragmentation range (up to 4000). To illustrate our point, we reproduce in Figure 1A the EThcD spectrum originating from a tryptic glycopeptide of palmitoyl-protein thioesterase 1, which has a M6P glycan attached at its N-terminus that produced c1, c2, and c3 ions with intact glycan attached that we published earlier.30 When this analysis was repeated using an extended range of up to 4000 m/z (Figure 1A), we observed a much fuller series of c ions spanning up to 3100 m/z covering the full sequence of the palmitoyl-protein thioesterase 1 M6P glycopeptide.
Figure 1.

Extended EThcD range results in the detection of additional fragment ions, increasing the sequence coverage of M6P glycopeptides. The EThcD fragmentation spectra of three different glycopeptides are shown. (A) GlcNAc2Man7P glycoform of palmitoyl-protein thioesterase 1, (B) GlcNAc2Man8PP glycoform of N-acetylglucosamine-6-sulfatase, and (C) GlcNAc2Man6P glycoform of cathepsin Z. Fragment ions are annotated and color coded (z/y red and c blue). Peptide sequences with corresponding glycoforms are depicted in the top right corner of each spectrum. The range from 2000 to 4000 m/z is shown in the shaded region magnified by a factor of 4. Green lines and shaded c/z ions connect fragment ion pairs containing the asparagine + intact glycan mass increment. Shaded glycan fragment ions represent signature M6P EThcD cleavage ions, facilitating confident glycan composition annotation. Lower case c in the peptide sequence indicates a carbamidomethylated cysteine.
Furthermore, when combined with the concomitant full series of z/y ions observed at lower m/z, we could assign every complementary fragment ion pair, enabling full peptide sequence coverage. Next, we focused on an M6P glycopeptide that had its modified asparagine near its N-terminus. As depicted in Figure 1B, the EThcD spectrum with extended m/z range is especially beneficial in this case as all c ion fragments containing the intact M6P glycan are found between 2400 and 3400 m/z range and thus would be completely missed when measured under the standard settings. More importantly, in this case, the extended range enabled unambiguous annotation of the glycan composition, as evidenced by the c4 and c5 fragment ions, which differ by an asparagine with a full glycan attached. This is further supported by a signature ion characteristic for a M6P composition arising from the core GlcNAc cleavage, as shown in the shaded region at 1660.44 m/z in Figure 1B. Also, when the N-glycosylation site is situated near the glycopeptide C-terminus, the extended m/z range setting performs favorably (Figure 1C). However, in this case, the range above 2000 m/z displays both c and z series, whereby the z3–z4 and c6–c7 fragment pairs nicely show the location of the asparagine and mass shift due to the intact glycan. Considering that one of the observed sources of incorrect glycopeptide annotation is due to mass coincidences between certain glycan compositions and amino acid combinations,35 we conclude that this issue can be avoided by having sequence coverage from both glycopeptide termini, as demonstrated in the extended range EThcD approach presented here.
Extended m/z Range Improves Identification Confidence for All Classes of N-Glycopeptides
After establishing that the extended m/z range results in an increased coverage of M6P glycopeptides, we next interrogated the performance of these settings on other classes of glycopeptides. For this purpose, we analyzed human milk glycopeptides, and two exemplary EThcD spectra of complex type N-glycopeptides are depicted in Figure 2. In short, all of the observations made above for the M6P glycopeptides also hold true for all other analyzed N-glycopeptides. Namely, in the case of glycopeptides modified with biantennary N-glycans containing branch fucose moieties (Figure 2A), we observed a series of insightful c ions in the 2200–3700 m/z range containing the intact glycan as well as a c3–c4 pair, indicative for the site of the asparagine and mass shift induced by the fucosylated biantennary N-glycan. A similar picture arose in analyzing a glycopeptide modified with monosialylated biantennary N-glycan (Figure 2B) where we could confidently assign the glycan composition based on the c1–c2 glycan fragment pair and determine the full sequence of the carrier peptide. Furthermore, various fucosylated and hybrid glycopeptides also show a similar behavior arguing for the use of the extended m/z range (Figure S1A–D.) In glycoproteomics, the analysis of multiply sialylated glycopeptides presents an even greater challenge due to the labile nature of the sialic acid group, especially under CID/HCD conditions. Thus, next, we analyzed multiply sialylated glycopeptides obtained via SAX enrichment performed on the flow-through resulting from the Fe3+-IMAC treated CHO cell sample. An illustrative example of an EThcD spectrum with extended m/z range of a disialylated biantennary N-glycopeptide is shown in Figure 3A. Also, for such glycopeptides, we did observe the full series of c and z ions, resulting in the confident determination of the glycan composition and peptide backbone sequence. However, in contrast to what we observed in previous examples, we noticed a series of highly abundant peaks above 1500 m/z that were not recognized by Byonic. Upon manual inspection, we determined that these peaks correspond to sequential losses of monosaccharides from the precursor ion (denoted by purple lines in Figure 3A). Manual inspection also resulted in the identification of low abundant c ion series (highlighted c ions in Figure 3A) resulting from sequential sugar losses (Figure 3A). This could create a potential bias against identifying sialylated glycopeptides as unexplained peaks lead to a decrease in their Byonic scores. Additionally, we also observed that by using the extended m/z range, we could get sufficient sequence coverage to confidently assign the glycan composition in some doubly N-glycosylated peptides (Figure S2A and B). This is especially significant considering that currently in most large-scale glycoproteomics studies, doubly glycosylated peptides are removed by default during processing steps, as the standard m/z range does not allow confident annotation of each of the modified sites.27 Finally, the standard m/z range spectra are usually populated with abundant oxonium ions, peptide fragments, and sequential sugar losses from the precursor resulting in very crowded fragmentation spectra that can result in many ambiguities. In contrast, the m/z range between 2000 and 4000 harbors fewer ions, but they are predominantly all c and z ions, making their identification a more straightforward task while still preserving the same oxonium ion abundances as observed at low m/z (Figure S3A–D).
Figure 2.

Extending the EThcD range enables confident assignment of complex fucosylated and sialylated glycopeptides. Depicted are two EThcD fragmentation spectra of (A) a monofucosylated biantennary glycopeptide from alpha-S1-casein and (B) a monosialylated biantennary glycopeptide originating from clusterin. Fragment ions are annotated and color-coded (z/y red and c blue). Peptide sequences with corresponding glycoforms are depicted in the top right corner of each spectrum. Green lines and shaded c ions connect fragment ion pairs containing asparagine + intact glycan mass increment. Lower case c in the peptide sequence indicates a carbamidomethylated cysteine.
Figure 3.

Extending the EThcD m/z range increases the confidence in glycopeptide identifications. (A) An example of a disialylated biantennary glycopeptide originating from Integrin is shown. Shaded c ions represent sequential sugar losses that are currently missed using automated annotation by Byonic. Precursor ions with sequential sugar losses are denoted with purple lines and were also missed by Byonic. (B) Distribution of LogP scores for M6P glycopeptides enriched by Fe3+-IMAC from CHO cells. (C) Distribution of LogP scores for glycopeptides, enriched by SAX, from a digested CHO cell lysate. (D) Distribution of LogP scores for glycopeptides, enriched by HILIC, from human milk. Numbers below the graphs denote the ranges used (2000 vs 4000 m/z). Two technical replicates were used for each range, and all forward glycopeptide identifications by Byonic were included, the number of which is indicated for each range (n).
To Get on Par with Standard Proteomics, Glycoproteomics Requires Further Development in Dedicated Software
After demonstrating the benefits of the extended m/z range for a whole array of glycopeptide classes, we were interested in a more global and direct comparison of the standard vs extended range. For this purpose, we searched large sets of glycopeptide EThcD MS/MS files with Byonic, which is one of the most popular software solutions used in large-scale glycopeptide analyses and to our knowledge the only tool that currently supports large scale annotation of glycopeptide EThcD spectra. For comparison purposes, we decided to use the log probability (LogP) score provided by Byonic, as this score takes into account 10 different features (Byonic score, delta, mass errors, p-values, etc.), making it a good measure of the identification confidence. A global comparison of the standard vs extended range analysis of EThcD glycopeptide spectra is shown in Figure 3B–D. It is immediately apparent that extending the m/z range from 2000 to 4000 leads to an increase of median LogP of about 0.4–1 (LogP is calculated as base 10 value, so this increase represents a 2.5–10-fold improvement) across all classes of glycopeptides measured here, clearly demonstrating the global benefits of extended mass range. However, it has to be noted that by using the current Byonic version, we could only take fragment ions up to 2500 m/z into account for score calculation, while in this work, we regularly see rich series of fragments ranging up to 4000 m/z. This, together with the observation that Byonic misses certain fragment ions which then decrease the score, suggests that the real benefit of the extended m/z range is probably substantially higher. Finally, we queried whether the substantial improvement we observed in EThcD-based analysis of glycopeptides using the extended m/z range was also observed when using solely HCD fragmentation (see Figure S4A–C). Notably, in the HCD-based analysis, the extended m/z range leads to hardly any improvement. We argue that this is mainly due to the commonly observed whole glycan loss from the peptide backbone induced by HCD fragmentation, which results in “typical” tryptic peptide fragmentation spectra (Figure S5A–C). Furthermore, in agreement with earlier reported studies,18,19 EThcD already outperformed HCD for glycopeptide analysis by using the standard m/z range but even more by using the extended m/z range. This difference was most striking when comparing the analysis of the M6P glycopeptides where the median LogP value in HCD was around 0.5, while with EThcD it was around 2.0 using 2000 as cutoff and rose to 3.1 by using 4000 m/z, which represent increases of 50- and 400-fold in annotation confidence, respectively. Clearly, EThcD with extended mass range is the optimal workflow for these glycopeptides.
Conclusion
Glycopeptide analysis has benefitted greatly from the improvements made in the more standard mass spectrometry-based proteomics. For instance, while EThcD was originally not developed for glycopeptide analysis, it is now clear that this hybrid fragmentation method is surely more advantageous compared to CID/HCD for glycopeptides,33 while its benefits for unmodified tryptic peptides are more marginal.11 Notwithstanding the fact that glycopeptide analysis has benefited from these and other proteomics advances (e.g., faster mass analyzers, more sensitive analyses at higher mass resolving power), it should not be forgotten that glycopeptides are inherently biochemically very distinct from unmodified tryptic peptides. Therefore, we argue that standard proteomic workflows should be critically evaluated for their performance in glycopeptide analysis. As we show here, very simple changes in these workflows such as the extension of the measured m/z range in MS/MS spectra can already boost the performance, substantially increasing the confidence of glycopeptide identification, but have surprisingly not yet been adopted in most recent comprehensive glycopeptide analyses.
To stimulate the much-needed further development of glycopeptide software solutions, we made publicly available all our raw files acquired with both standard and extended m/z ranges, containing EThcD spectra of a wide variety of glycopeptides that hopefully can serve as a benchmark for any future studies addressing the challenging issue of automated glycopeptide analysis.
Acknowledgments
The Netherlands Organization for Scientific Research (NWO) supported this research through funding of the large-scale proteomics facility Proteins@Work (project 184.032.201) embedded in The Netherlands Proteomics Centre. T.Č. and A.J.R.H. were further supported by the NWO Gravitation program Institute for Chemical Immunology. A.J.R.H. acknowledges further support by the NWO TOP-Punt Grant 718.015.003. We acknowledge additional funding through the European Union’s Horizon 2020 research and innovation program under Grant 686547 (MSMed). J.Z. acknowledges support from the Chinese Scholarship Council (CSC).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.9b02125.
Materials and methods; Figure S1, annotated EThcD spectra of various N-glycopeptides; Figure S2, annotated EThcD fragmentation spectrum of doubly glycosylated glycopeptides; Figure S3, comparison of oxonium ion intensities between standard vs extended m/z range; Figure S4, comparison of the distribution of LogP scores using HCD with a standard vs an extended m/z range; Figure S5, examples of extended m/z range HCD spectra (PDF)
Glycan compositions used in Byonic searches (XLSX)
Author Contributions
T.Č. designed the study. T.Č. and J.Z. performed the experiments and analyzed the data. T.Č. and A.J.R.H. wrote the manuscript.
Author Contributions
# These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Varki A. Biological Roles of Glycans. Glycobiology 2017, 27 (1), 3–49. 10.1093/glycob/cww086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowell S. R.; Ju T.; Cummings R. D. Protein Glycosylation in Cancer. Annu. Rev. Pathol.: Mech. Dis. 2015, 10, 473–510. 10.1146/annurev-pathol-012414-040438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S. S.; Reis C. A. Glycosylation in Cancer: Mechanisms and Clinical Implications. Nat. Rev. Cancer 2015, 15 (9), 540–555. 10.1038/nrc3982. [DOI] [PubMed] [Google Scholar]
- Ohtsubo K.; Marth J. D. Glycosylation in Cellular Mechanisms of Health and Disease. Cell 2006, 126 (5), 855–867. 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Yang W. H.; Heithoff D. M.; Aziz P. V.; Haslund-Gourley B.; Westman J. S.; Narisawa S.; Pinkerton A. B.; Millán J. L.; Nizet V.; Mahan M. J. Accelerated Aging and Clearance of Host Anti-Inflammatory Enzymes by Discrete Pathogens Fuels Sepsis. Cell Host Microbe 2018, 24 (4), 500–513. 10.1016/j.chom.2018.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W. H.; Heithoff D. M.; Aziz P. V.; Sperandio M.; Nizet V.; Mahan M. J.; Marth J. D. Recurrent Infection Progressively Disables Host Protection against Intestinal Inflammation. Science (Washington, DC, U. S.) 2017, 358 (6370), eaao5610. 10.1126/science.aao5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kailemia M. J.; Xu G.; Wong M. Y.; Li Q.; Goonatilleke E.; Leon F.; Lebrilla C. B. Recent Advances in the Mass Spectrometry Methods for Glycomics and Cancer. Anal. Chem. 2018, 90, 208. 10.1021/acs.analchem.7b04202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinneburg H.; Stavenhagen K.; Schweiger-Hufnagel U.; Pengelley S.; Jabs W.; Seeberger P. H.; Silva D. V.; Wuhrer M.; Kolarich D. The Art of Destruction: Optimizing Collision Energies in Quadrupole-Time of Flight (Q-TOF) Instruments for Glycopeptide-Based Glycoproteomics. J. Am. Soc. Mass Spectrom. 2016, 27 (3), 507–519. 10.1007/s13361-015-1308-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M.; Pioch M.; Pralow A.; Hennig R.; Kottler R.; Reichl U.; Rapp E. The Fine Art of Destruction: A Guide to In-Depth Glycoproteomic Analyses—Exploiting the Diagnostic Potential of Fragment Ions. Proteomics 2018, 18 (24), 1800282. 10.1002/pmic.201800282. [DOI] [PubMed] [Google Scholar]
- Narimatsu H.; Kaji H.; Vakhrushev S. Y.; Clausen H.; Zhang H.; Noro E.; Togayachi A.; Nagai-Okatani C.; Kuno A.; Zou X. Current Technologies for Complex Glycoproteomics and Their Applications to Biology/Disease-Driven Glycoproteomics. J. Proteome Res. 2018, 17 (12), 4097–4112. 10.1021/acs.jproteome.8b00515. [DOI] [PubMed] [Google Scholar]
- Frese C. K.; Altelaar A. F. M.; Van Den Toorn H.; Nolting D.; Griep-Raming J.; Heck A. J. R.; Mohammed S. Toward Full Peptide Sequence Coverage by Dual Fragmentation Combining Electron-Transfer and Higher-Energy Collision Dissociation Tandem Mass Spectrometry. Anal. Chem. 2012, 84 (22), 9668–9673. 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Yu Q.; Hao L.; Liu F.; Johnson J.; Tian Z.; Kao W. J.; Xu W.; Li L. Site-Specific Characterization and Quantitation of N-Glycopeptides in PKM2 Knockout Breast Cancer Cells Using DiLeu Isobaric Tags Enabled by Electron-Transfer/Higher-Energy Collision Dissociation (EThcD). Analyst 2018, 143 (11), 2508–2519. 10.1039/C8AN00216A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino F.; Bern M.; Mommen G. P. M.; Leney A. C.; Van Gaans-Van Den Brink J. A. M.; Bonvin A. M. J. J.; Becker C.; Van Els C. A. C. M.; Heck A. J. R. Extended O-GlcNAc on HLA Class-I-Bound Peptides. J. Am. Chem. Soc. 2015, 137 (34), 10922–10925. 10.1021/jacs.5b06586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Xie X.; Zhao X.; Tian F.; Lv J.; Ying W.; Qian X. Systems Analysis of Singly and Multiply O-Glycosylated Peptides in the Human Serum Glycoproteome via EThcD and HCD Mass Spectrometry. J. Proteomics 2018, 170, 14–27. 10.1016/j.jprot.2017.09.014. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Chen Z.; Zhang J.; An M.; Wu J.; Yu Q.; Skilton S. J.; Bern M.; Ilker Sen K.; Li L. Differential Quantitative Determination of Site-Specific Intact N-Glycopeptides in Serum Haptoglobin between Hepatocellular Carcinoma and Cirrhosis Using LC-EThcD-MS/MS. J. Proteome Res. 2018, 18 (1), 359–371. 10.1021/acs.jproteome.8b00654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darula Z.; Pap Á.; Medzihradszky K. F. Extended Sialylated O-Glycan Repertoire of Human Urinary Glycoproteins Discovered and Characterized Using Electron-Transfer/Higher-Energy Collision Dissociation. J. Proteome Res. 2018, 18 (1), 280–291. 10.1021/acs.jproteome.8b00587. [DOI] [PubMed] [Google Scholar]
- Medzihradszky K. F.; Kaasik K.; Chalkley R. J. Tissue-Specific Glycosylation at the Glycopeptide Level. Mol. Cell. Proteomics 2015, 14 (8), 2103–2110. 10.1074/mcp.M115.050393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover M. S.; Yu Q.; Chen Z.; Shi X.; Kent K. C.; Li L. Characterization of Intact Sialylated Glycopeptides and Phosphorylated Glycopeptides from IMAC Enriched Samples by EThcD Fragmentation: Toward Combining Phosphoproteomics and Glycoproteomics. Int. J. Mass Spectrom. 2018, 427, 35–42. 10.1016/j.ijms.2017.09.002. [DOI] [Google Scholar]
- Yu Q.; Wang B.; Chen Z.; Urabe G.; Glover M. S.; Shi X.; Guo L. W.; Kent K. C.; Li L. Electron-Transfer/Higher-Energy Collision Dissociation (EThcD)-Enabled Intact Glycopeptide/Glycoproteome Characterization. J. Am. Soc. Mass Spectrom. 2017, 28 (9), 1751–1764. 10.1007/s13361-017-1701-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst A. J.; Weidle C. E.; Gray M. D.; Frenz B.; Snijder J.; Joyce M. G.; Georgiev I. S.; Stewart-Jones G. B. E.; Kwong P. D.; McGuire A. T. Germline VRC01 Antibody Recognition of a Modified Clade C HIV-1 Envelope Trimer and a Glycosylated HIV-1 Gp120 Core. eLife 2018, 7, 1–32. 10.7554/eLife.37688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder J.; Ortego M. S.; Weidle C.; Stuart A. B.; Gray M. D.; McElrath M. J.; Pancera M.; Veesler D.; McGuire A. T. An Antibody Targeting the Fusion Machinery Neutralizes Dual-Tropic Infection and Defines a Site of Vulnerability on Epstein-Barr Virus. Immunity 2018, 48 (4), 799–811. 10.1016/j.immuni.2018.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollineni R. C.; Koehler C. J.; Gislefoss R. E.; Anonsen J. H.; Thiede B. Large-Scale Intact Glycopeptide Identification by Mascot Database Search. Sci. Rep. 2018, 8 (1), 1–13. 10.1038/s41598-018-20331-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totten S. M.; Feasley C. L.; Bermudez A.; Pitteri S. J. Parallel Comparison of N-Linked Glycopeptide Enrichment Techniques Reveals Extensive Glycoproteomic Analysis of Plasma Enabled by SAX-ERLIC. J. Proteome Res. 2017, 16 (3), 1249–1260. 10.1021/acs.jproteome.6b00849. [DOI] [PubMed] [Google Scholar]
- Parker B. L.; Thaysen-Andersen M.; Fazakerley D. J.; Holliday M.; Packer N. H.; James D. E. Terminal Galactosylation and Sialylation Switching on Membrane Glycoproteins upon TNF-Alpha-Induced Insulin Resistance in Adipocytes. Mol. Cell. Proteomics 2016, 15 (1), 141–153. 10.1074/mcp.M115.054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadlmann J.; Taubenschmid J.; Wenzel D.; Gattinger A.; Dürnberger G.; Dusberger F.; Elling U.; Mach L.; Mechtler K.; Penninger J. M. Comparative Glycoproteomics of Stem Cells Identifies New Players in Ricin Toxicity. Nature 2017, 549 (7673), 538–542. 10.1038/nature24015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo C. M.; Iavarone A. T.; Spiciarich D. R.; Palaniappan K. K.; Bertozzi C. R. Isotope-Targeted Glycoproteomics (IsoTaG): A Mass-Independent Platform for Intact N- and O-Glycopeptide Discovery and Analysis. Nat. Methods 2015, 12 (6), 561–567. 10.1038/nmeth.3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Hebert A. S.; Westphall M. S.; Coon J. J. Capturing Site-Specific Heterogeneity with Large-Scale N-Glycoproteome Analysis. Nat. Commun. 2019, 10 (1), 1311. 10.1038/s41467-019-09222-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noro E.; Togayachi A.; Sato T.; Tomioka A.; Fujita M.; Sukegawa M.; Suzuki N.; Kaji H.; Narimatsu H. Large-Scale Identification of N-Glycan Glycoproteins Carrying Lewis x and Site-Specific N-Glycan Alterations in Fut9 Knockout Mice. J. Proteome Res. 2015, 14 (9), 3823–3834. 10.1021/acs.jproteome.5b00178. [DOI] [PubMed] [Google Scholar]
- Parker B. L.; Thaysen-Andersen M.; Solis N.; Scott N. E.; Larsen M. R.; Graham M. E.; Packer N. H.; Cordwell S. J. Site-Specific Glycan-Peptide Analysis for Determination of N-Glycoproteome Heterogeneity. J. Proteome Res. 2013, 12 (12), 5791–5800. 10.1021/pr400783j. [DOI] [PubMed] [Google Scholar]
- Čaval T.; Zhu J.; Tian W.; Remmelzwaal S.; Yang Z.; Clausen H.; Heck A. J. R. Targeted Analysis of Lysosomal Directed Proteins and Their Sites of Mannose-6-Phosphate Modification. Mol. Cell. Proteomics 2019, 18 (1), 16–27. 10.1074/mcp.RA118.000967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Huang J.; Li L. Recent Advances in Mass Spectrometry (MS)-Based Glycoproteomics in Complex Biological Samples. TrAC, Trends Anal. Chem. 2018, 1–13. 10.1016/j.trac.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L.; Jia L.; Zhi Y.; Li P.; Zhao T.; Zhu B.; Lan R.; Hu Y.; Zhang H.; Sun S. Mapping Human N-Linked Glycoproteins and Glycosylation Sites Using Mass Spectrometry. TrAC, Trends Anal. Chem. 2019, 114, 143–150. 10.1016/j.trac.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiding K. R.; Bondt A.; Franc V.; Heck A. J. R. The Benefits of Hybrid Fragmentation Methods for Glycoproteomics. TrAC, Trends Anal. Chem. 2018, 108, 260–268. 10.1016/j.trac.2018.09.007. [DOI] [Google Scholar]
- Liu M. Q.; Zeng W. F.; Fang P.; Cao W. Q.; Liu C.; Yan G. Q.; Zhang Y.; Peng C.; Wu J. Q.; Zhang X. J.; et al. PGlyco 2.0 Enables Precision N-Glycoproteomics with Comprehensive Quality Control and One-Step Mass Spectrometry for Intact Glycopeptide Identification. Nat. Commun. 2017, 8 (1), 1–14. 10.1038/s41467-017-00535-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. W.; Pu T. H.; Viner R.; Khoo K. H. Novel LC-MS2 Product Dependent Parallel Data Acquisition Function and Data Analysis Workflow for Sequencing and Identification of Intact Glycopeptides. Anal. Chem. 2014, 86 (11), 5478–5486. 10.1021/ac500945m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.