

Abstract
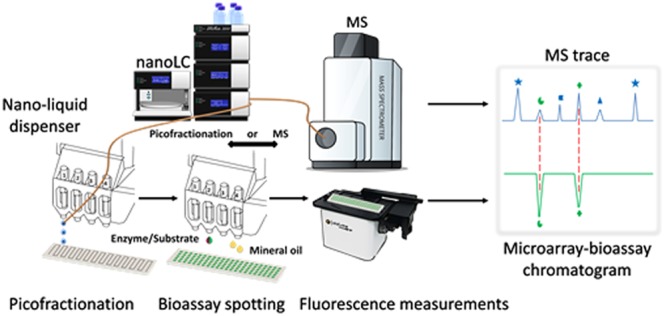
High-throughput screening platforms for the identification of bioactive compounds in mixtures have become important tools in the drug discovery process. Miniaturization of such screening systems may overcome problems associated with small sample volumes and enhance throughput and sensitivity. Here we present a new screening platform, coined picofractionation analytics, which encompasses microarray bioassays and mass spectrometry (MS) of components from minute amounts of samples after their nano liquid chromatographic (nanoLC) separation. Herein, nanoLC was coupled to a low-volume liquid dispenser equipped with pressure-fed solenoid valves, enabling 50-nL volumes of column effluent (300 nL/min) to be discretely deposited on a glass slide. The resulting fractions were dried and subsequently bioassayed by sequential printing of nL-volumes of reagents on top of the spots. Unwanted evaporation of bioassay liquids was circumvented by employing mineral oil droplets. A fluorescence microscope was used for assay readout in kinetic mode. Bioassay data were correlated to MS data obtained using the same nanoLC conditions in order to assign bioactives. The platform provides the possibility of freely choosing a wide diversity of bioassay formats, including those requiring long incubation times. The new method was compared to a standard bioassay approach, and its applicability was demonstrated by screening plasmin inhibitors and fibrinolytic bioactives from mixtures of standards and snake venoms, revealing active peptides and coagulopathic proteases.
Current approaches used for analyzing complex mixtures for bioactivity properties involve the use of screening platforms that commonly combine several analytical techniques.1−3 The so-called nanofractionation approach encompasses liquid chromatographic (LC) separation, using conventional-bore columns, online coupled to UV absorbance and mass spectrometric (MS) detection, as well as to a bioassay via continuous collection of microliter fractions of the column effluent.1 Fractions of 3- to 10-s are collected on microtiter well plates and subsequently centrifuge freeze-dried and exposed to a bioassay of choice. The overall bioassay readout is then constructed into a bioactivity chromatogram and correlated to the parallel obtained UV-absorbance and MS data with the objective to identify (unknown) bioactive compounds. This technology has been applied successfully to a wide range of complex mixtures, including snake venoms,4−6 plant extracts,7 environmental samples,8,9 and metabolic mixtures of new chemical entities (NCEs).10
Natural samples such as animal venoms or biofluids from small animals may be precious sources of new drugs. However, these samples are often only available in very small amounts and are therefore typically not suitable for most established screening platforms. Microfluidic high-resolution screening (HRS) platforms1 employing online homogeneous bioassays have shown potential to tackle this problem.11,12 However, these systems allow only short incubation times and require relatively high concentrations of a soluble target protein (e.g., receptor or enzyme) to attain satisfactory performance. An alternative to online microfluidic bioassaying could be the coupling of microarray bioassays, which can accommodate hundreds to thousands of minute-volume bioassay reactions, to nano liquid chromatography (nanoLC), which employs nL/min range flow rates and small sample injection volumes (nL to low μL). Small molecular microarrays (SMMs) have been successfully implemented in drug discovery processes,13−15 such as for drug–drug interaction screening of cytochrome P450 enzymes16−18 and drug cytotoxicity evaluation,19,20 but have also found application in the identification of pesticides in food21 and environmental samples22 and of bioweapon toxins.23 However, these microarray platforms are only suited for screening pure compound libraries or single-analyte samples, limiting their usefulness to analyze complex mixtures of bioactive compounds, such as venoms and natural extracts, as they would require preseparation.
Off-line chemical analysis of mixtures of components after nanoLC24 has been achieved via fractionation of nanoLC effluent using various deposition approaches.25−29 Pereira et al.28 and Küster et al.26,27 used a microfluidics T-junction for contact deposition generating droplets by shear force using perfluorodecalin and subsequent spotting onto plates for MALDI-MS. Küster et al.26 managed to deposit liquid droplets of ∼2.5 nL. Noncontact deposition of low-nL volume droplets requires application of some sort of force to dislodge droplets from a dispensing tip. Young and Li29 applied a pulsed voltage to a solenoid coil to deposit effluent droplets from a connected fused-silica capillary onto MALDI-plates in a noncontact fashion. This method was shown to be optimal for droplet sizes above 100 nL, but by decreasing the distance between the surface of the collecting plate and the capillary tip, spot volumes down to 20 nL could be achieved.
In the present study, we introduce a new fractionation platform for profiling bioactive components in small volumes of complex mixtures after separation by nanoLC. This “picofractionation” system analyzes nL to low-μL sample volumes and deposits 10-s fractions (50 nL each) of nanoLC effluent onto a coated glass slide. Our study for the first time demonstrates proof of concept analytics enabling nanoLC separations with nanoliter regime fraction collection followed by integrated postcolumn bioassaying. For this purpose, a nanoliter-volume liquid dispenser was used, providing contactless droplet array printing of the column effluent. The bioactivity of the fractionated samples was assessed by applying a fluorescence-based bioassay using a spot-on-spot approach. Microarray bioassay readout was performed kinetically using a modified fluorescence microscope. The resulting data were converted to microarray bioactivity chromatograms (MBCs), in which each point represents a slope of each bioassay reaction present on a glass slide. The MBCs were correlated with nanoLC-UV and -MS data for (structural) assignment of bioactive compounds. We evaluated the described picofractionation platform by employing a plasmin bioassay for the profiling of four snake venoms in order to detect the presence of plasmin inhibitors (antifibrinolytic agents) and proteases with fibrinolytic-like activities. The term “picofractionation” was chosen to continue the already existing nomenclature for fractionation platforms. Starting with microfractionation systems, the collected fractions often were found to be in the milliliter range.30−33 We found that this naming system was used already in publications dated as early as 1951.30 Successively, platforms working in the microliter range were called nanofractionation.34−36 Therefore, in this study we called the presented nanoliter range fractionation platform picofractionation.
Experimental Section
Chemicals and Snake Venoms
All chemicals used were purchased commercially, and the details can be found in Supporting Information (SI) section S1.
Lyophilized snake venom samples from Bothrops asper (Costa Rica), Deinagkistrodon acutus (captive bred of Chinese origin), Trimeresurus trigonocephalus (Sri Lanka), and Trimeresurus purpureomaculatus (Thailand) were sourced from historical venom samples stored in the Centre for Snakebite Research & Interventions at the Liverpool School of Tropical Medicine. For analysis, the venoms were diluted in Milli-Q water from a stock solution (5 mg/mL in water) to a concentration of 1 mg/mL. The venom samples were stored at −80 °C until use.
Preparation of Glass Slides and Coating
To ensure a hydrophobic surface, standard microscope glass slides (76 × 26 mm) with ground edges from Fisher Scientific (Loughborough, United Kingdom) were coated with 4 mL of a mixture comprising 16 mL of methyltrimethoxysilane (MTMOS), 6.4 mL of 5 mM HCl, and 16 mL of 25 mM potassium phosphate buffer at pH 8 according to the protocol of Lee et al.37 The slides were coated at least 4 days prior to nanoLC spotting. Detailed information on the coating procedure can be found in SI section S2.
NanoLC
NanoLC was performed with a Dionex Ultimate 3000 nanoLC system from Thermo Scientific equipped with an autosampler, two binary high pressure gradient pumps, one loading pump, and a variable wavelength UV absorbance detector set to 214 nm. Samples (1 μL unless otherwise stated) were first loaded onto a trapping column (0.3 mm ID; 5 mm length; packed with Acclaim PepMap 100 C18 from Thermo Fisher; 5 μm pore size) for 6 min using a loading pump delivering acetonitrile–water (2:98; v/v) with 0.1% TFA at a flow rate of 10 μL/min. After 6 min the trapping column was switched in line with the analytical column (C18; 75 μm ID; 150 mm length; 2 μm pore size) through a 6-port switch valve. All analyses were performed using a gradient elution with a mobile phase comprised of solvent A (water–acetonitrile–formic acid (98:2:0.1; v/v/v)) and solvent B (acetonitrile–water–formic acid (98:2:0.1; v/v/v). Further information on the nanoLC gradients can be found in SI section S3.
For sample analysis with MS detection, the nanoLC was coupled to a Bruker TIMS-TOF instrument (Bremen, Germany) using a CaptiveSpray source and a nanoBooster (Bruker Daltonics) employing acetonitrile. The MS source settings were as follows: capillary voltage, 1200 V; nanoBooster gas pressure, 0.25 bar; temperature, 150 °C. The range of m/z 50–3000 was covered. For nanoLC-MS analysis the injection volumes for D. acutus, T. trigonocephalus, and T. purpureomaculatus were 100 nL and for B. asper 500 nL. The chromatographic methods (i.e., gradients) used to separate the standard solution (a mixture of leupeptin diastereoisomers) and the dissolved snake venoms were the same as used for the postcolumn microarray bioassaying analyses.
Fractionation of nanoLC Effluent
Collection of nanoliter fractions of nanoLC effluent was facilitated by the nanovolume liquid dispenser Preddator from Red&Whyte Limited (London, UK) equipped with four (1–4) pressure fed solenoid valves (The presentation of valves 1–4 can be found in Figure S2). The nanoLC effluent was led to the Preddator by a ∼30 cm long fused-silica capillary (75 μm I.D. and 365 μm O.D.) which was connected to the 280-μm O.D. capillary exiting the UV absorbance detector using a Teflon sleeve connector (Thermo Scientific). The other end of the fused-silica capillary was inserted into a fluorinated ethylene propylene (FEP) tubing sleeve (length 5.8 cm; O.D. 1/16 in.; I.D. 0.0155 in.) which was mounted in a custom-made holder placed to the left side of the dispensing arm of the Preddator that moves in XY directions (Figure 1A). A schematic view of the platform is shown in Figure 1. The tip of the capillary inserted into the FEP tubing sleeve was protruding ∼5 mm outside the tubing sleeve and was placed centrally below the opening of the most left positioned solenoid valve (1) in the arm of the Preddator (Figure 1B).
Figure 1.
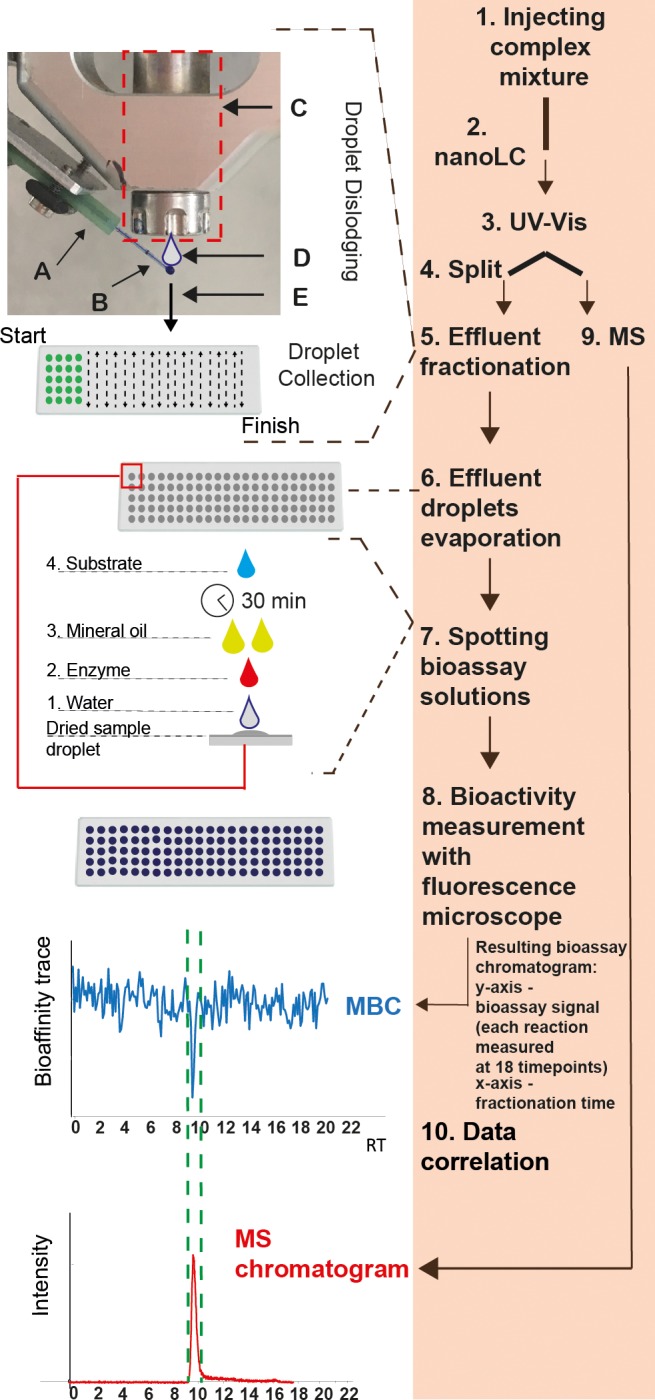
Schematic representation of a picofractionation platform. Under orange shading (right side of the figure) the main steps of the picofractionation method are outlined: (1) a complex mixture is injected; (2) mixture separation on nanoLC column; (3) the eluting sample components are first measured with an ultraviolet–visible detector [UV–vis]; (4) split to picofractionation and MS; (5) effluent droplets are picofractionated (10-s fractions) and collected on a coated glass slide (A: nanoLC effluent is delivered by a glass capillary that is inserted into a fluorinated ethylene propylene (FEP) tubing sleeve mounted to an arm of a low volume liquid dispenser; (B) the glass capillary is protruding outside the sleeve tube and is positioned directly below the nozzle of the solenoid valve; (C) solenoid valve dispensing 100 nL of water; (D) water droplets ejected from the solenoid valve hit and merge “in flight” with the nanoLC effluent droplet (50 nL), dispensing them on a coated glass slide (E); (6) evaporation of collected nanoLC effluent droplets; (7) spotting of the bioassay solutions; (8) bioactivity measurement with fluorescence microscope; (9) mass spectrometric (MS) measurement; (10) bioactivity data and chemical data correlation. MBC = Microarray Bioactivity Chromatogram generated by plotting bioassay activity (slopes) over 18 measurements of individual droplets on y-axis and fractionation time on x-axis; RT = retention time; MS = mass spectrometry.
Figure S3 presents a zoomed image of the interface between nanoLC and the nanovolume liquid dispenser. Collection of nanoliter effluent fractions was possible by using solenoid valve 1 to dispense water droplets of 100 nL that hit and merge “in flight” with the effluent droplets exiting the tip of the connecting capillary, providing contact-free deposition of LC effluent fractions onto the receiving glass slide (Figure 1D–E). Correct positioning of the capillary tip was essential in order to achieve repeatable and microarray-bioassay-compatible collection of the nanoLC effluent. To prevent “climbing” of the droplet along the outside of the capillary by adhesion forces, its tip was silylated with MTMOS coating solution (more detailed information on the capillary preparation can be found in SI, section S4). This treatment renders the tip more hydrophobic and lasted for ∼3–4 weeks. The picofractionation process was managed by the in-house developed software Ariadne v. 1.10, enabling spotting of effluent fractions ranging from 0.1 to 900 s. In this study, a fraction was spotted every 10 s.
Microarray Plasmin-Activity Assay with Fluorescence Readout
The deposited nanoliter fractions were exposed to a plasmin bioassay that was adapted from a previously developed 384 microtiter well-plate format as described by Zietek et al.38 Collected effluent fractions dried within ∼5 min at ambient atmosphere and were then covered with a 100-nL drop of water dispensed by solenoid valve 3 of the Preddator (spot-on-spot; Figure 1, Figure S2) at an overpressure of 200 mbar (i.e., atmospheric pressure +200 mbar). Next, 100 nL of a 200 ng/mL plasmin solution dissolved in 0.1 M Tris-HCl buffer enriched with 0.1% BSA (w/v) was spotted onto the water droplets on the glass slides using solenoid nozzle 2 at an overpressure of 200 mbar. Then, two droplets of mineral oil (200 nL each) were dispensed 0.2 mm left and right, respectively, of each spotted droplet of plasmin solution, in order to prevent disintegration of the enzyme solution droplet. The mineral oil was used to prevent evaporation of the bioassay liquids and was dispensed using solenoid valve 4 at an overpressure of 1650 mbar. Several other spotting sequences were tested prior to reaching the here described optimal procedure (see Figure S4). After depositing the mineral oil, the spots were incubated for 30 min at RT under a polystyrene black cover, without touching the surface of the glass slide. Finally, the bioassay was initiated by dispensing 100 nL of the fluorogenic substrate H-d-Val-Leu-Lys-AMC (10 μM) onto the incubation droplets, with solenoid valve 2 at an overpressure of 200 mbar. The prepared microarray glass slide was then directly placed in a glass slide holder of a Märzhäuser Wetzlar XY stage (type SCAN IM 130 × 100; Wetzlar, Germany) mounted on top of a Lumascope 620 (Etaluma, Carlsbad, CA, USA) inverted fluorescence microscope for fluorescence readout and image acquisition (Figure S5). For detection of the product of the enzymatic reaction (7-amino-4-methylcoumarin (AMC)) an excitation filter of 370–410 nm was used. In the Lumascope 620, fluorescence emission light was detected by a digital camera containing a high sensitivity monochrome complementary metal oxide semiconductor (CMOS) image sensor. An apochromatic microscope objective with 4× magnification (numerical aperture (NA), 0.13; working distance (WD), 17.2 mm, type number MA1012) from Meiji Techno (San Jose, California, USA) was used for the measurements. To automate image acquisition of all microarray bioassay spots at fixed time intervals (i.e., kinetic measurements), the XY stage and the fluorescence microscope were controlled with the μManager (version 1.4) open source software39 that allowed synchronization of the hardware. Information on the image acquisition and processing of the microarray data can be found in SI, section S5.
Performance and Quality of the Plasmin Microarray Bioassay
The performance of the microarray bioassay was first evaluated by determining IC50 values of the plasmin inhibitors leupeptin and aprotinin. Details on this procedure can be found in SI S6. The Z′-factor, a standardized statistical tool described by Zhang et al.40 was used to determine the assay quality (see SI S7).
Results and Discussion
We present the development and validation of a so-called picofractionation screening platform which combines nanoLC of bioactive samples with microarray bioassaying employing a fluorescence microscope via contactless fractionation of the nL/min column effluent. Parallel nanoLC-MS analyses of the same samples were performed for correlating bioactivity of components to their accurate mass.
Fractionation of nanoLC Effluent
To achieve noncontact deposition of 50-nL fractions of nanoLC effluent, a fused-silica capillary connected to the nanoLC column was positioned at an angle of 45° just underneath a solenoid valve of a dispensing arm of a high-accuracy low-volume dispenser (Figure S3). We chose a noncontact deposition because it offers a number of advantages over contact deposition, and these include a better repeatability and reproducibility of the deposited droplets, less chance of droplet contamination and memory effects, and no damage to the surface of the glass slide and/or nanoLC capillary (such as scratching the surface or crushing of the capillary). The latter is important in the case of fluorescence readouts as any damage to the surface of the glass slide would cause artifacts contributing to light scattering and increases in overall background signal which will affect the quality of the measurement. Finally, the noncontact spotting mode omits the use of a Z-stage in the spotting movement procedure and the hardware, which renders the system less technically complicated. In addition, X,Y stage spotting intrinsically is much faster since the Z-movement for every droplet deposition is not needed. Positioning, and the angle of the capillary with respect to the nozzle of the solenoid valve, along with the shape of the capillary tip, were important parameters to ensure reproducible and accurate deposition of effluent droplets on to the microarray glass slide. A capillary with a blunt end and a hydrophobic tip (silylated with MTMOS), prevented “climbing” of liquid along the outside of the capillary and yielded optimal detaching of nanoLC effluent by the ejected water droplets. The distance between the nanoLC effluent capillary and the spotter nozzle was kept small (∼2 mm), as was the distance between the capillary tip and the glass slide surface (∼2 mm). These parameters provided precise and consistent droplet deposition on the glass slide for mobile phases containing up to 67% solvent B. Above this percentage of solvent B, the position of the capillary had to be adjusted ∼0.5 mm further away from the spotter nozzle (decreasing the distance between the tip of the capillary and the glass slide to less than 1.5 mm) in order to ensure appropriate droplet deposition. As future technical advancement of the methodology, automatic adjustment of the needle will be incorporated into the system.
Microarray Bioassay Format
The bioactivity of individual compounds separated by nanoLC and deposited on the glass slide has to be measured with a bioassay suitable for microarray formats. Bioassaying in small volumes raises issues associated with unwanted evaporation of bioassay solvents and with redissolving of the dried fractions. To prevent water evaporation during the microarray assay incubation, we adapted the droplet-in-oil approach of Sun et al.41 In their approach, mineral oil was spotted first on a glass slide to generate droplet arrays into which analytes and the bioassay components were ejected for generation of bioassay-in-oil-droplets. However, in our case, after column effluent droplet spotting, the LC solvent evaporates leaving dry spots. Therefore, prior to spotting of the enzyme solution, a 100-nL droplet of water was dispensed on top of the dried fraction, in order to redissolve deposited components and thus to facilitate their mixing with the enzyme. The water droplet was partially evaporated (approximately 70% by visual assessment) prior to spotting of the enzyme solution to avoid additional dilution of the bioassay components. Then, mineral oil was spotted to the plasmin solution, followed by 30 min incubation at room temperature under a polystyrene cover. However, spot-on-spot printing of the oil resulted in disintegration of the previously spotted droplets of the plasmin solution. Therefore, different approaches of spotting mineral oil onto the bioassay reaction mix were evaluated (see Figure S4 for details). We found that spotting two oil droplets 0.2 mm adjacent (left and right) to an enzyme droplet maintained the enzyme droplet’s integrity, while the two oil droplets merged into one, covering the enzyme droplet. The bioassay reaction was subsequently initiated by spotting 100 nL of substrate solution through the oil cover into the enzyme and sample component-containing inner droplet. The mineral oil droplets were not removed from the reaction droplets. They were kept throughout the bioassay reactions as dome shaped cover shells to prevent the bioassay mixtures from evaporating. Considering the highly nonpolar nature of mineral oil, partitioning of the compounds relevant to this research (i.e., peptides, protein and relatively polar small molecules) is not an issue.
Even when taking an enzyme solution that was prepared freshly from a stock kept at −20 °C, a decrease of enzymatic activity over time was observed when the solution was dispensed (data not shown). This activity loss appeared to be due to exposure to ambient temperature solution and evaporation of the enzyme solution. Therefore, the enzyme solution was kept on ice as long as possible prior to dispensing and then dispensed quickly followed by coverage with mineral oil. This was achieved by dispensing not more than 100 droplets with one portion of cold enzyme solution. For the next 100 droplets, a new cold portion was used. Fast dispensing of the enzyme and the oil was achieved by time-optimizing the point-to-point movement of the liquid dispenser using the control software.
The biochemical conditions and reagent concentrations of the plasmin inhibition assay used in an earlier developed plate reader format38 were transferred to and evaluated with the microarray bioassay format. The microarray assay quality and dynamic range were assessed by determining the Z′-factor, which was 0.71, indicating an excellent bioassay according to Zhang et al.40 (Figure 2). Comparable results were obtained for the 384-well plate format, where the Z′-factor for the same bioassay was determined to be 0.74.38 The good agreement in the Z′-factors shows the straightforward transfer of a bioassay initially developed for the 384-well plate format (operating at 50 μL well volume) to the microarray format that requires only 200 nL of the bioassay mixture (i.e., 250 times lower volume). Although when considering the biological aspects only the two methods are expected to provide similar results, the difference lies in the technical difficulties. Performing a bioassay in for example 50 μL volume in wells of a well plate with readout using a standardized platereader is routine laboratory work. In contrast, the technical difficulties of preparing and measuring bioassays in 200 nL volumes are extremely high. Besides, all preparation steps (i.e., spotting), measuring procedures (i.e., the in-house developed confocal microscope bioassay spot reader), and data processing with automated spot recognition and integrated kinetic bioassay development monitoring per spot are complex and far from trivial.
Figure 2.
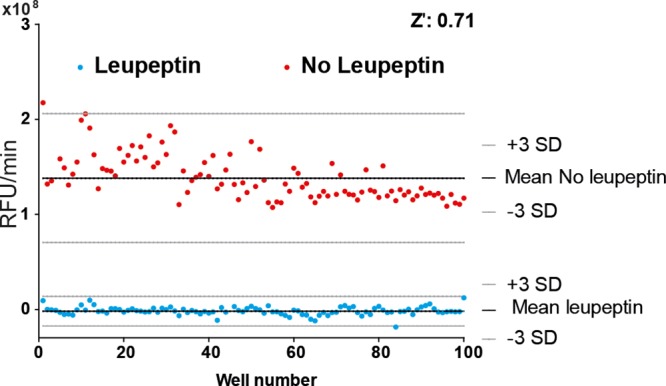
Bioassay evaluation with Z′-factor. Positive control (leupeptin at 200 μM; n = 100) and negative control (mobile phase A; n = 100). SD: standard deviation.
In order to evaluate whether the microarray bioassay can accurately determine inhibitory properties of bioactives, IC50 values of two potent plasmin inhibitors, leupeptin and aprotinin, were determined and compared with IC50 values obtained using the standard 384-well plate reader bioassay.38 Determination of IC50 values was performed with the microarray bioassay on dried inhibitor spots that were spotted on a MTMOS coated glass slide. Leupeptin and aprotinin were dispensed at ten different concentrations ranging from 0 to 400 μM and from 0 to 30 μM, respectively. Figure S6 shows the IC50 curve of leupeptin obtained with the microarray bioassay. The IC50 for leupeptin was found to be 4.7 μM (SD, 2.7 μM), which was approximately two times lower than the IC50 obtained using the 384-well plate reader format (8.9 μM; SD, 3.1 μM). For aprotinin an IC50 value of 51 nM (SD, 27 nM) was determined with the microarray bioassay, whereas the IC50 value was 35 nM (SD, 14) with the plate reader assay (Figure S6). Choo et al.42 reported an IC50 of 21.7 nM for aprotinin against plasmin. Hence, although we used different conditions for our plasmin bioassay—we used a different enzyme concentration and a fluorogenic instead of a chromogenic substrate as in the case of Choo et al.—the inhibitory potency of aprotinin toward plasmin found in our study agreed well with the value of Choo et al.
NanoLC-Microarray Bioassaying of Plasmin Inhibitors
In order to evaluate the performance of the picofractionation platform, in terms of repeatability and sensitivity, various concentrations of a mixture of leupeptin and aprotinin were analyzed by nanoLC and the column effluent was fractionated on MTMOS-coated glass slides. Figure 3 shows representative resulting microarray bioactivity chromatograms (MBCs) of leupeptin injected at concentrations ranging from 10 to 200 μM. The highest concentration injected induced full inhibition of plasmin, which is depicted in Figure 3 as a negative peak in the upper superimposed chromatogram. A gradually lower percentage of full inhibition was observed upon injection of lower leupeptin concentrations, as reflected by decreasing heights of the negative peaks. A concentration of 10 μM appeared to be the bioassay detection limit. Although comparable, in our previous study in which a nanofractionation system was used, the detection limit for leupeptin was 6.3 μM.38 It has to be noted that miniaturized bioassays do not lead to increased sensitivity as in the case of nanoLC-MS analysis of especially peptide mixtures (for proteomics analyses for example). Bioassay sensitivity depends on the bioassay and the detection system used for readout. Considering that in our current study a prototype custom-built new analytical device was used, a slightly lower fluorescence signal and thus sensitivity can be expected. The bioactivity peaks for leupeptin were relatively narrow, and no extensive dose-dependent peak broadening was observed. However, for aprotinin, a high-affinity plasmin inhibitor, extensive peak broadening was already observed at 30 μM, due to the sigmoidal dose–response relationship typical for bioactivity detection.10,43 Peak broadening in postcolumn bioassaying, such as high resolution screening43,44 and nanofractionation analytics,4,6,7,34 seems exaggerated compared to parallel UV or MS signals since bioassay responses are sigmoidal concentration–response in nature in contrary to chemical detector responses. Peak broadening is expected to be better visible after nanoLC separations than after normal bore LC separations because the column dimensions of the nanoLC result faster in column overloading and sample preconcentration at the start of the gradient resulting in higher concentrations of compounds when eluting.
Figure 3.

Superimposed microarray bioactivity chromatograms (MBCs) resulting from microarray bioassays of fractionated leupeptin (left) and aprotinin (right) injected at different concentrations to nanoLC. The MBCs are correlated with nanoLC-UV traces (bottom) obtained from measurement of leupeptin and aprotinin at 25 μM and 30 μM, respectively. For these analyses a short chromatographic gradient of 35 min was used.
The lowest concentration of aprotinin at which plasmin inhibition was still observed was an injected concentration of 44 nM. The retention time of the chromatographic peak and the bioactive peak was stable for both compounds at each concentration measured, demonstrating robustness of the method.
An important characteristic of the picofractionation system is the capability to maintain the chromatographic resolution obtained during separation. To test the picofractionation system for this characteristic, two leupeptin diastereoisomers were analyzed.45 Both diastereoisomers possess potent inhibitory activities toward plasmin and can be separated by RPLC. Previous analyses, however, were performed using a relatively fast gradient, which did not result in diastereoisomers separation (Figure 3). By applying a more shallow (60 min) gradient, the leupeptin diastereoisomers were separated with baseline resolution, allowing fractionation and microarray bioactivity testing of the individual diastereoisomers (Figure 4). When comparing picofractionation bioassay chromatograms with those given in the nanofractionation study of Zietek et al.,38 the picofractionation results do not give a better resolution. Improvement in separation resolution of venom proteins, however, was not the goal of the current study. Instead, the main goal of this study was to build and demonstrate an analytical methodology capable of postcolumn bioassaying complex bioactive mixtures using nanoliter range sample volumes. This offers the possibility to work with samples only available in low volumes such as snake venoms and insect venoms. As Figure 4 shows that the diastereoisomers of leupeptin can be separated and bioassayed without loss in separation efficiency from the nanoLC, higher resolutions in the system have to be achieved in the separation component of the system and not the picofractionation aspect. Improving the resolution of venom protein separations, however, is not the focus of this study.
Figure 4.

Analysis of a mixture of leupeptin diastereoisomers (25 μM) using the picofractionation platform (top). The microarray bioassay chromatogram (MBC) was correlated with the UV absorbance trace (middle) and MS chromatogram (bottom). For MS analysis, leupeptin at a concentration of 2 μM was used. BPC: base peak chromatogram. For this analysis a long chromatographic gradient of 60 min was used.
NanoLC-Microarray Bioassay Analysis of Snake Venoms
Venoms are complex mixtures containing multiple bioactive compounds with high potency and/or selectivity for a range of biological targets. Hence, bioactives derived from venoms are considered valuable entry points for drug discovery. Although attractive for these reasons, studying venoms can be challenging due to their complexity and, often, minute quantities of available material.46 To evaluate the usefulness of the nanoLC platform when coupled to both postcolumn microarray bioassaying and MS detection, we analyzed four venoms originating from the pit vipers Bothrops asper, Deinagkistrodon acutus, Trimeresurus trigonocephalus, and Trimeresurus purpureomaculatus, respectively. Figures 5 and S7 show MBCs of the four venoms correlated to UV chromatograms and separately obtained nanoLC-MS data. In the MBC of B. asper, also known as fer-de-lance (Figure 5), a broad positive signal between 28 and 33 min was observed, which suggests that the venom contains peptides and/or proteins with fibrinolytic activity. B. asper venom is known to contain a number of proteolytic enzymes, including class PI, PII, and PIII snake venom metalloproteinases (SVMPs), which are at least in part responsible for the devastating toxic effects in cases of snakebite envenoming.47 For further information on SVMP classification the reader is referred to a review on SVMPs by Takeda et al.48 Other reported literature on metalloproteinases isolated from B. asper describes hemorrhagic activity of SVMPs including the PI SVMP BaP1 (22.7 kDa)49 and PIII SVMP BaH4 (69 kDa).50 Considering the width of the positive MBC peak observed, most probably the observed bioactivity is due to multiple, partly coeluting, bioactive compounds present in the venom. Similar broad bioactive peaks were also observed during analyses of snake venom samples using the normal-bore chromatography based nanofractionation system.4 This implies that the picofractionation approach provides comparable separation efficiency. Upon correlating the MBC with the obtained MS data, the m/z values of a number of component ions were observed in the time frame of the positive MCB peak (Table S1). In Figure 5, the total ion chromatogram (TIC) and extracted ion chromatograms (XICs) of the relevant m/z values are plotted. Although a number of compounds with masses in the range of 20–30 kDa were observed, and which could be SVMPs or SVSPs (especially those eluting between 31 and 32 min), due to the coelution of these compounds it was not possible to determine which ion(s) were responsible for the bioactivity. Due to the very complex composition and high diversity of toxins in most venoms, a single separation usually does not suffice in obtaining baseline separation of the venom toxins. We have identified this limitation also in our previous studies in which the nanofractionation platform of high-resolution screening analytics was used. In order to address this limitation, an orthogonal separation using two different separation mechanisms (e.g., RP and HILIC) can be applied and was in fact demonstrated by us and others.6,7,38 Similar approaches are anticipated to be applied to the picofractionation platform, however, at later stages of its development.
Figure 5.

Analysis of venoms of B. asper (left) and T. trigonocephalus (right) using the picofractionation platform employing profiling of plasmin inhibition and protease activity. B. asper shows a positive peak between retention time 28 and 33 min, indicating the presence of multiple venom proteases and/or plasmin activators. From the parallel obtained MS data, multiple masses are found to match the positive peak area of the microarray bioassay chromatogram (MBC) as shown as extracted-ion chromatograms (XICs). For T. trigonocephalus, both positive and negative peaks were observed indicating the presence of proteases and/or plasmin activators as well as plasmin inhibitors. The XICs indicate multiple potential bioactive peptides or proteins per peak.
The venom of D. acutus, also known as “the hundred pacer”, exhibited similar fibrinolytic-like activities to those observed for B. asper. When analyzed using the picofractionation platform, a positive peak in the MBC between 23 and 28 min was visible (Figure S7). In addition, between 28 and 32 min, a negative peak representing plasmin inhibition was observed. While previous reports have described fibrinolytic metalloproteinases, e.g. FIIa (26 kDa) and Dacin (23 kDa) in the venom of D. acutus,51,52 to our knowledge antifibrinolytic activities have not yet been reported. The XICs of m/z values detected for components eluting between 28 and 32 min are shown in Figure S7 and originate from both singly and multiple charged species potentially representing peptides as well as SVMPs (∼20–30 kDa).
The other two snake venoms analyzed in this study were from the congeneric species T. trigonocephalus and T. purpureomaculatus, known as the Sri Lanka Pit Viper and the Mangrove Pit Viper, respectively. The MBCs of these two venoms showed similar profiles, both exhibiting fibrinolytic-like and antifibrinolytic activities (Figures 5 and S7). Venom from T. purpureomaculatus has been reported to contain a number of proteolytic enzymes, including for example, the thrombin-like enzyme (an SVSP) purpurase (35 kDa)53 and a proteolytic SVMP called hemorrhagin (72 kDa).54 Our nanoLC-MS setup was not able to effectively detect such high molecular masses due to a limited sensitivity for the higher mass proteases that are present at low concentration. The m/z values that were observed for the bioactivity peaks are listed in Table S1. The negative bioactivity peak observed in the MBC of T. purpureomaculatus between 29 and 33 min (Figure S7) represents antiplasmin (antifibrinolytic) activities. In the case of T. trigonocephalus venom (Figure 5), to our knowledge, the biochemical antiplasmin profile has not been reported yet, which can be due to the fact that the venom of this species is considered to be of low medical relevance55 in contrast to, for example, the venom of B. asper. Nevertheless, clinical features of envenomation by T. trigonocephalus have been reported to include coagulopathy and local hemorrhage,55 pathologies consistent with the bioactivities observed here and which are likely due to the action of proteolytic enzymes.
Conclusions
This study describes the development and application of a new picofractionation system which couples nanoLC to microarray bioassaying via noncontact spotting of nanoliter-range effluent fractions. A number of adaptations, such as spot-on-spot dispensing, hydrophobic coating of deposition capillary, and use of spot-adjacent oil droplets, allowed the effective transfer of a conventional microtiter well-plate-based bioassay to microarray format. Suitable bioassay performance was indicated by a good correlation of determined IC50 values for leupeptin and aprotinin obtained with a conventional bioassay format and with the new miniaturized method. The new method was evaluated by the analysis of different concentrations of the plasmin inhibitors leupeptin and aprotinin by the nanoLC-microarray bioassay system. The ability of the system to achieve high-resolution fractionation at nanoLC flow rates was shown by separating and distinctively depositing diastereoisomers of leupeptin followed by individually assessing their inhibiting activity. Furthermore, we validated the “real-world” applicability of the picofractionation system by screening minute amounts of snake venoms from Bothrops asper, Deinagkistrodon acutus, Trimeresurus trigonocephalus and Trimeresurus purpureomaculatus. The results indicated the presence of proteases as well as compounds with both antifibrinolytic and fibrinolytic-like properties. It should be noted, however, that some venom toxins can (partly) denaturate during LC separation rendering them inactive and thus not detectable in the plasmin bioassay. In general, the results obtained were in agreement with the available literature. The new method described herein will facilitate bioactivity profiling complex bioactive mixtures and consumes only very low sample volumes (i.e., in the range of 50 nL to 1 μL). Anticipated future developments of the picofractionation platform include further optimization of the chromatographic part of the system to achieve better separations for venoms, miniaturization of the microarray dimensions to allow bioassay multiplexing, and implementation of an effluent split after UV detection that will permit simultaneous parallel online MS analysis next to microarray spotting and bioassaying. The method presented in this study is anticipated to be generic in nature in the sense that other bioassay formats can be implemented in a straightforward manner. Preliminary data has for example shown that a fluorescence enhancement ligand binding assay functions in the methodology as well (data not shown). With minor modifications (such as effluent droplet ejections with MALDI matrix solution instead of the now used water) the current methodology is expected to be adaptable for integrated postcolumn MALDI-MS analysis as well.
Acknowledgments
The authors acknowledge ARIADME, a European FP7 ITN Community’s Seventh Framework Program under Grant Agreement No. 607517. Many thanks to Etaluma Inc. and Red&White for their scientific and technical support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.9b01261.
Details of experimental methods, supplementary figures, additional results and discussion, table of mass-to-charge values (PDF)
Author Contributions
○ B.M.Z. and K.B.M.S. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Kool J.; Giera M.; Irth H.; Niessen W. M. A. Advances in Mass Spectrometry-Based Post-Column Bioaffinity Profiling of Mixtures. Anal. Bioanal. Chem. 2011, 399 (8), 2655–2668. 10.1007/s00216-010-4406-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonker N.; Kool J.; Irth H.; Niessen W. M. A. Recent Developments in Protein-Ligand Affinity Mass Spectrometry. Anal. Bioanal. Chem. 2011, 399, 2669–2681. 10.1007/s00216-010-4350-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciesla L.; Moaddel R. Comparison of Analytical Techniques for the Identification of Bioactive Compounds from Natural Products. Nat. Prod. Rep. 2016, 33 (10), 1131–1145. 10.1039/C6NP00016A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mladic M.; Zietek B. M.; Iyer J. K.; Hermarij P.; Niessen W. M. A.; Somsen G. W.; Kini R. M.; Kool J. At-Line Nanofractionation with Parallel Mass Spectrometry and Bioactivity Assessment for the Rapid Screening of Thrombin and Factor Xa Inhibitors in Snake Venoms. Toxicon 2016, 110, 79–89. 10.1016/j.toxicon.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Still K.; Nandlal R.; Slagboom J.; Somsen G.; Casewell N.; Kool J. Multipurpose HTS Coagulation Analysis: Assay Development and Assessment of Coagulopathic Snake Venoms. Toxins 2017, 9 (12), 382. 10.3390/toxins9120382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mladic M.; de Waal T.; Burggraaff L.; Slagboom J.; Somsen G. W.; Niessen W. M. A.; Manjunatha Kini R.; Kool J. Rapid Screening and Identification of ACE Inhibitors in Snake Venoms Using At-Line Nanofractionation LC-MS. Anal. Bioanal. Chem. 2017, 409 (25), 5987–5997. 10.1007/s00216-017-0531-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otvos R. A.; Mladic M.; Arias-Alpizar G.; Niessen W. M. A.; Somsen G. W.; Smit A. B.; Kool J. At-Line Cellular Screening Methodology for Bioactives in Mixtures Targeting the A7-Nicotinic Acetylcholine Receptor. J. Biomol. Screening 2016, 21 (5), 459–467. 10.1177/1087057115625307. [DOI] [PubMed] [Google Scholar]
- Jonker W.; Lamoree M. H.; Houtman C. J.; Hamers T.; Somsen G. W.; Kool J. Rapid Activity-Directed Screening of Estrogens by Parallel Coupling of Liquid Chromatography with a Functional Gene Reporter Assay and Mass Spectrometry. J. Chromatogr. A 2015, 1406, 165–174. 10.1016/j.chroma.2015.06.012. [DOI] [PubMed] [Google Scholar]
- Jonker W.; Ballesteros-Gómez A.; Hamers T.; Somsen G. W.; Lamoree M. H.; Kool J. Highly Selective Screening of Estrogenic Compounds in Consumer-Electronics Plastics by Liquid Chromatography in Parallel Combined with Nanofractionation-Bioactivity Detection and Mass Spectrometry. Environ. Sci. Technol. 2016, 50 (22), 12385–12393. 10.1021/acs.est.6b03762. [DOI] [PubMed] [Google Scholar]
- Zietek B. M.; Mladic M.; Bruyneel B.; Niessen W. M. A.; Honing M.; Somsen G. W.; Kool J. Nanofractionation Platform with Parallel Mass Spectrometry for Identification of CYP1A2 Inhibitors in Metabolic Mixtures. SLAS Discovery Adv. Life Sci. R&D 2018, 23 (3), 283–293. 10.1177/2472555217746323. [DOI] [PubMed] [Google Scholar]
- Heus F.; Otvos R.; Aspers R.; van Elk R.; Halff J.; Ehlers A.; Dutertre S.; Lewis R.; Wijmenga S.; Smit A.; et al. Miniaturized Bioaffinity Assessment Coupled to Mass Spectrometry for Guided Purification of Bioactives from Toad and Cone Snail. Biology (Basel, Switz.) 2014, 3 (1), 139–156. 10.3390/biology3010139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer J. K.; Otvos R. A.; Kool J.; Kini R. M. Microfluidic Chip-Based Online Screening Coupled to Mass Spectrometry. J. Biomol. Screening 2016, 21 (2), 212–220. 10.1177/1087057115602648. [DOI] [PubMed] [Google Scholar]
- MacBeath G.; Koehler A. N.; Schreiber S. L. Printing Small Molecules as Microarrays and Detecting Protein-Ligand Interactions En Masse. J. Am. Chem. Soc. 1999, 121 (34), 7967–7968. 10.1021/ja991083q. [DOI] [Google Scholar]
- Fu J.; Xia A.; Qi X. Identification of Novel Peptoid Agonists of Fibroblast Growth Factor Receptor Using Microarray-Based Screening. MedChemComm 2016, 7, 1183–1189. 10.1039/C6MD00121A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H.; Bao K.; Frangioni J. V.; Choi H. S. Screening of Small Molecule Microarrays for Ligands Targeted to the Extracellular Epitopes of Living Cells. Microarrays 2015, 4 (1), 53–63. 10.3390/microarrays4010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran S. M.; Potsaid B.; Lee M. Y.; Clark D. S.; Dordick J. S. Development of a Fluorescence-Based, Ultra High-Throughput Screening Platform for Nanoliter-Scale Cytochrome P450 Microarrays. J. Biomol. Screening 2009, 14 (6), 668–678. 10.1177/1087057109336592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrabito G.; Galati C.; Castellano S.; Pignataro B. Luminometric Sub-Nanoliter Droplet-to-Droplet Array (LUMDA) and Its Application to Drug Screening by Phase I Metabolism Enzymes. Lab Chip 2013, 13 (1), 68–72. 10.1039/C2LC40948H. [DOI] [PubMed] [Google Scholar]
- Kwon S. J.; Lee D. W.; Shah D. A.; Ku B.; Jeon S. Y.; Solanki K.; Ryan J. D.; Clark D. S.; Dordick J. S.; Lee M. Y. High-Throughput and Combinatorial Gene Expression on a Chip for Metabolism-Induced Toxicology Screening. Nat. Commun. 2014, 5 (May 2013), 1–12. 10.1038/ncomms4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.-Y.; Kumar R. A.; Sukumaran S. M.; Hogg M. G.; Clark D. S.; Dordick J. S. Three-Dimensional Cellular Microarray for High-Throughput Toxicology Assays. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (1), 59–63. 10.1073/pnas.0708756105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.; Park C. B.; Dordick J. S.; Clark D. S. Metabolizing Enzyme Toxicology Assay Chip (MetaChip) for High-Throughput Microscale Toxicity Analyses. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (4), 983–987. 10.1073/pnas.0406755102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Liu N.; Ning B.; Liu M.; Lv Z.; Sun Z.; Peng Y.; Chen C.; Li J.; Gao Z. Simultaneous and Rapid Detection of Six Different Mycotoxins Using an Immunochip. Biosens. Bioelectron. 2012, 34 (1), 44–50. 10.1016/j.bios.2011.12.057. [DOI] [PubMed] [Google Scholar]
- Desmet C.; Blum L. J.; Marquette C. A. Multiplex Microarray ELISA versus Classical ELISA, a Comparison Study of Pollutant Sensing for Environmental Analysis. Environ. Sci. Process. Impacts 2013, 15 (10), 1876. 10.1039/c3em00296a. [DOI] [PubMed] [Google Scholar]
- Jenko K. L.; Zhang Y.; Kostenko Y.; Fan Y.; Garcia-Rodriguez C.; Lou J.; Marks J. D.; Varnum S. M. Development of an ELISA Microarray Assay for the Sensitive and Simultaneous Detection of Ten Biodefense Toxins. Analyst 2014, 139 (20), 5093–5102. 10.1039/C4AN01270D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noga M.; Sucharski F.; Suder P.; Silberring J. A Practical Guide to Nano-LC Troubleshooting. J. Sep. Sci. 2007, 30 (14), 2179–2189. 10.1002/jssc.200700225. [DOI] [PubMed] [Google Scholar]
- Hioki Y.; Tanimura R.; Iwamoto S.; Tanaka K. Nano-LC/MALDI-MS Using a Column-Integrated Spotting Probe for Analysis of Complex Biomolecule Samples. Anal. Chem. 2014, 86 (5), 2549–2558. 10.1021/ac4037069. [DOI] [PubMed] [Google Scholar]
- Küster S. K.; Fagerer S. R.; Verboket P. E.; Eyer K.; Jefimovs K.; Zenobi R.; Dittrich P. S. Interfacing Droplet Microfluidics with Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry: Label-Free Content Analysis of Single Droplets. Anal. Chem. 2013, 85, 1285–1289. 10.1021/ac3033189. [DOI] [PubMed] [Google Scholar]
- Küster S. K.; Pabst M.; Jefimovs K.; Zenobi R.; Dittrich P. S. High-Resolution Droplet-Based Fractionation of Nano-LC Separations onto Microarrays for MALDI-MS Analysis. Anal. Chem. 2014, 86 (10), 4848–4855. 10.1021/ac4041982. [DOI] [PubMed] [Google Scholar]
- Pereira F.; Niu X.; deMello A. J.. A Nano LC-MALDI Mass Spectrometry Droplet Interface for the Analysis of Complex Protein Samples. PLoS One 2013, 8 ( (5), ), e63087. 10.1371/journal.pone.0063087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young J. B.; Li L. An Impulse-Driven Liquid-Droplet Deposition Interface for Combining LC with MALDI MS and MS/MS. J. Am. Soc. Mass Spectrom. 2006, 17, 325–334. 10.1016/j.jasms.2005.11.018. [DOI] [PubMed] [Google Scholar]
- ZYGMUNTOWICZ A. S.; WOOD M.; CHRISTO E.; TALBOT N. B. Studies of Urinary 17-Ketosteroid Excretion by Means of New Micro-Chromatographic Fractionation Procedure. J. Clin. Endocrinol. Metab. 1951, 11 (6), 578–596. 10.1210/jcem-11-6-578. [DOI] [PubMed] [Google Scholar]
- Queiroz E. F.; Ioset J.-R.; Ndjoko K.; Guntern A.; Foggin C. M.; Hostettmann K. On-Line Identification of the Bioactive Compounds FromBlumea Gariepina by HPLC-UV-MS and HPLC-UV-NMR, Combined with HPLC-Micro-Fractionation. Phytochem. Anal. 2005, 16 (3), 166–174. 10.1002/pca.839. [DOI] [PubMed] [Google Scholar]
- Bohni N.; Cordero-Maldonado M. L.; Maes J.; Siverio-Mota D.; Marcourt L.; Munck S.; Kamuhabwa A. R.; Moshi M. J.; Esguerra C. V.; de Witte P. A. M.; et al. Integration of Microfractionation, QNMR and Zebrafish Screening for the In Vivo Bioassay-Guided Isolation and Quantitative Bioactivity Analysis of Natural Products. PLoS One 2013, 8 ( (5), ), e64006. 10.1371/journal.pone.0064006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila F. C.; Colombo R.; De Lira T. O.; Yariwake J. H. HPLC Microfractionation of Flavones and Antioxidant (Radical Scavenging) Activity of Saccharum Officinarum L. J. Braz. Chem. Soc. 2008, 19 (5), 903–908. 10.1590/S0103-50532008000500014. [DOI] [Google Scholar]
- Mladic M.; Scholten D. J.; Niessen W. M. A.; Somsen G. W.; Smit M. J.; Kool J. At-Line Coupling of LC–MS to Bioaffinity and Selectivity Assessment for Metabolic Profiling of Ligands towards Chemokine Receptors CXCR1 and CXCR2. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2015, 1002, 42–53. 10.1016/j.jchromb.2015.08.004. [DOI] [PubMed] [Google Scholar]
- Giera M.; Heus F.; Janssen L.; Kool J.; Lingeman H.; Irth H. Microfractionation Revisited: A 1536 Well High Resolution Screening Assay. Anal. Chem. 2009, 81 (13), 5460–5466. 10.1021/ac900622b. [DOI] [PubMed] [Google Scholar]
- Kool J.; De Kloe G.; Denker A. D.; Van Altena K.; Smoluch M.; Van Iperen D.; Nahar T. T.; Limburg R. J.; Niessen W. M. A.; Lingeman H.; et al. Nanofractionation Spotter Technology for Rapid Contactless and High-Resolution Deposition of LC Eluent for Further off-Line Analysis. Anal. Chem. 2011, 83 (1), 125–132. 10.1021/ac102001g. [DOI] [PubMed] [Google Scholar]
- Lee M.; Dordick J. S.; Clark D. S.. Metabolic Enzyme Microarray Coupled with Miniaturized Cell-Culture Array Technology for High-Throughput Toxicity Screening. In Microarray Methods for Drug Discovery. Methods in Molecular Biology (Methods and Protocols); Chittur S. V., Ed.; Humana Press: Totowa, NJ, 2010; Vol. 632, pp 221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zietek B. M.; Mayar M.; Slagboom J.; Bruyneel B.; Vonk F. J.; Somsen G. W.; Casewell N. R.; Kool J. Liquid Chromatographic Nanofractionation with Parallel Mass Spectrometric Detection for the Screening of Plasmin Inhibitors and (Metallo) Proteinases in Snake Venoms. Anal. Bioanal. Chem. 2018, 410, 5751–5763. 10.1007/s00216-018-1253-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein A. D.; Tsuchida M. A.; Amodaj N.; Pinkard H.; Vale R. D.; Stuurman N. Advanced Methods of Microscope Control Using MManager Software. J. Biol. Methods 2014, 1 (2), 10. 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.-H.; Thomas D. Y.; Oldenburg K. R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screening 1999, 4 (2), 67–73. 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Chen X.; Zhou X.; Zhu J.; Yu Y. Droplet-in-Oil Array for Picoliter-Scale Analysis Based on Sequential Inkjet Printing. Lab Chip 2015, 15 (11), 2429–2436. 10.1039/C5LC00356C. [DOI] [PubMed] [Google Scholar]
- Choo Y. M.; Lee K. S.; Yoon H. J.; Qiu Y.; Wan H.; Sohn M. R.; Da Sohn H.; Jin B. R. Antifibrinolytic Role of a Bee Venom Serine Protease Inhibitor That Acts as a Plasmin Inhibitor. PLoS One 2012, 7 (2), 1–5. 10.1371/journal.pone.0032269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool J.; De Kloe G. E.; Bruyneel B.; De Vlieger J. S.; Retra K.; Wijtmans M.; Van Elk R.; Smit A. B.; Leurs R.; Lingeman H.; et al. Online Fluorescence Enhancement Assay for the Acetylcholine Binding Protein with Parallel Mass Spectrometric Identification. J. Med. Chem. 2010, 53 (12), 4720–4730. 10.1021/jm100230k. [DOI] [PubMed] [Google Scholar]
- Falck D.; De Vlieger J. S. B.; Niessen W. M. A.; Kool J.; Honing M.; Giera M.; Irth H. Development of an Online P38α Mitogen-Activated Protein Kinase Binding Assay and Integration of LC-HR-MS. Anal. Bioanal. Chem. 2010, 398 (4), 1771–1780. 10.1007/s00216-010-4087-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saino T.; Someno T.; Miyazaki H.; Ishii S.-I. Semisynthesis of 14C-Labelled Leupeptin. Chem. Pharm. Bull. 1982, 30 (7), 2319–2325. 10.1248/cpb.30.2319. [DOI] [Google Scholar]
- Fry B. G.; Koludarov I.; Jackson T. N. W.; Holford M.; Terrat Y.; Casewell N. R.; Undheim E. A. B.; Vetter I.; Ali S. A.; Low D. H. W.; et al. Seeing the Woods for the Trees: Understanding Venom Evolution as a Guide for Biodiscovery. In Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics; The Royal Society of Chemistry, 2015; pp 1–36. [Google Scholar]
- Alape-Girón A.; Flores-Díaz M.; Sanz L.; Madrigal M.; Escolano J.; Sasa M.; Calvete J. J. Studies on the Venom Proteome of Bothrops Asper: Perspectives and Applications. Toxicon 2009, 54 (7), 938–948. 10.1016/j.toxicon.2009.06.011. [DOI] [PubMed] [Google Scholar]
- Takeda S.; Takeya H.; Iwanaga S. Snake Venom Metalloproteinases: Structure, Function and Relevance to the Mammalian ADAM/ADAMTS Family Proteins. Biochim. Biophys. Acta, Proteins Proteomics 2012, 1824 (1), 164–176. 10.1016/j.bbapap.2011.04.009. [DOI] [PubMed] [Google Scholar]
- Watanabe L.; Shannon J. D.; Valente R. H.; Rucavado A.; Alape-Girón A.; Kamiguti A. S.; Theakston R. D. G.; Fox J. W.; Gutiérrez J. M.; Arni R. K. Amino Acid Sequence and Crystal Structure of BaP1, a Metalloproteinase from Bothrops Asper Snake Venom That Exerts Multiple Tissue-Damaging Activities. Protein Sci. 2003, 12 (10), 2273–2281. 10.1110/ps.03102403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi A.; Rucavado A.; Mora N.; Gutiérrez J. M. Purification and Characterization of BaH4, a Hemorrhagic Metalloproteinase from the Venom of the Snake Bothrops Asper. Toxicon 2000, 38 (1), 63–77. 10.1016/S0041-0101(99)00127-0. [DOI] [PubMed] [Google Scholar]
- Xiuxia L.; Jiashu C.; Yingna Z.; Pengxin Q.; Guangmei Y. Purification and Biochemical Characterization of F IIa, a Fibrinolytic Enzyme from Agkistrodon Acutus Venom. Toxicon 2001, 39 (8), 1133–1139. 10.1016/S0041-0101(00)00206-3. [DOI] [PubMed] [Google Scholar]
- Liang X. X.; Zhou Y. N.; Chen J. S.; Qiu P. X.; Chen H. Z.; Sun H. H.; Wu Y. P.; Yan G. M. Enzymological Characterization of FIIa, a Fibrinolytic Enzyme from Agkistrodon Acutus Venom. Acta Pharmacol. Sin. 2005, 26 (12), 1474–1478. 10.1111/j.1745-7254.2005.00204.x. [DOI] [PubMed] [Google Scholar]
- Tan N. H. Isolation and Characterization of the Thrombin-like Enzyme from Cryptelytrops Purpureomaculatus Venom. Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol. 2010, 151 (1), 131–136. 10.1016/j.cbpc.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Khow O.; Chanhome L.; Omori-Satoh T.; Puempunpanich S.; Sitprija V. A Hemorrhagin as a Metalloprotease in the Venom of Trimeresurus Purpureomaculatus: Purification and Characterization. Toxicon 2002, 40 (4), 455–461. 10.1016/S0041-0101(01)00234-3. [DOI] [PubMed] [Google Scholar]
- Namal Rathnayaka R. M. M. K.; Ranathunga P. E. A. N.; Kularatne S. A. M. Epidemiology and Clinical Features of Green Pit Viper (Trimeresurus Trigonocephalus) Envenoming in Sri Lanka. Toxicon 2017, 137, 99–105. 10.1016/j.toxicon.2017.07.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.