

Abstract
Prader-willi syndrome (PWS) is the most common form of dysmorphic genetic obesity associated with mental retardation1,2. About 60% of cases have a cytological deletion of chromosome 15q11q13 (refs 2, 3). These deletions occur de novo exclusively on the paternal chromosome4,5. By contrast, Angelman syndrome (AS) is a very different clinical disorder and is also associated with deletions of region 15q11q13 (refs 6–8), indistinguishable from those in PWS6,8 except that they occur de novo on the maternal chromosome6. The parental origin of the affected chromo-somes 15 in these disorders could, therefore, be a contributory factor in determining their clinical phenotypes. We have now used cloned DNA markers specific for the 15q11q13 subregion5,9,10 to determine the parental origin of chromosome 15 in PWS individuals not having cytogenetic deletions; these individuals account for almost all of the remaining 40% of PWS cases. Probands in two families displayed maternal uniparental disomy for chromosome 15q11q13. This is the first demonstration that maternal heterodisomy—the presence of two different chromosome 15s derived from the mother—can be associated with a human genetic disease. The absence of a paternal contribution of genes in region 15q11q13, as found in PWS deletion cases4,5, rather than a mutation in a specific gene(s) in this region may result in expression of the clinical phenotype. Thus, we conclude that a gene or genes in region 15q11q13 must be inherited from each parent for normal human development.
Given the unusual parental origins of deletion chromosomes associated with PWS and AS, we attempted to determine the parental origins of chromosomes 15 in non-deletion cases of PWS. For each family, we performed restriction fragment length polymorphism (RFLP) analyses with nine RFLPs at seven loci specific for proximal chromosome 15q (refs 5,11). The first family that we studied has a proband with a phenotype typical of PWS (refs 1, 2), that is, hypotonia and growth delay in infancy, short stature, hyperphagia with consequent obesity, small hands and feet, hypogonadism and mild mental retardation. This family also displays inheritance of a balanced robertsonian translocation, t(13; 15) (Fig. 1a). Association of this translocation with the phenotype of PWS is highly unlikely because unaffected maternal relatives carry the same balanced translocation (Fig. 1a), and the translocation breakpoint is at the centromere of chromosome 15, some distance from the critical region at 15q11q13. In this family the PWS proband (PWS1) had inherited two maternal alleles but no paternal allele (Fig. 1b) specific for probe 3-21, which maps to the region absent in PWS patients with deletions5,9,10. The 8.2-kilobase (kb) TaqI fragment detected by probe 3-21 (Fig. 1b) is shared by the three individuals (PWS1, his mother M1, and half-sister) carrying the t(13; 15) translocation and is, therefore, a marker for this chromosome. The 8.9-kb TaqI fragment which hybridized to probe 3-21 is present in both PWS1 and his mother and is not present in the other individuals of the family, clearly indicating the maternal derivation of the non-translocated chromosome 15 in PWS1, at least for region 15q11q13 surrounding the probe 3-21 locus. We confirmed and extended this result by using CMW-1, a multiallelic DNA marker11 that maps distal to the region of deletions associated with PWS (unpublished result). The segregation pattern again showed two maternal alleles but no paternal allele for the proband (Fig. 1c). We excluded the possibility of non-paternity by analysis with a cloned fragment (3′HVR) from the 3′ hypervariable region of the α-globin locus on chromosome 16 (ref. 12, Fig. 1d).
FIG. 1.
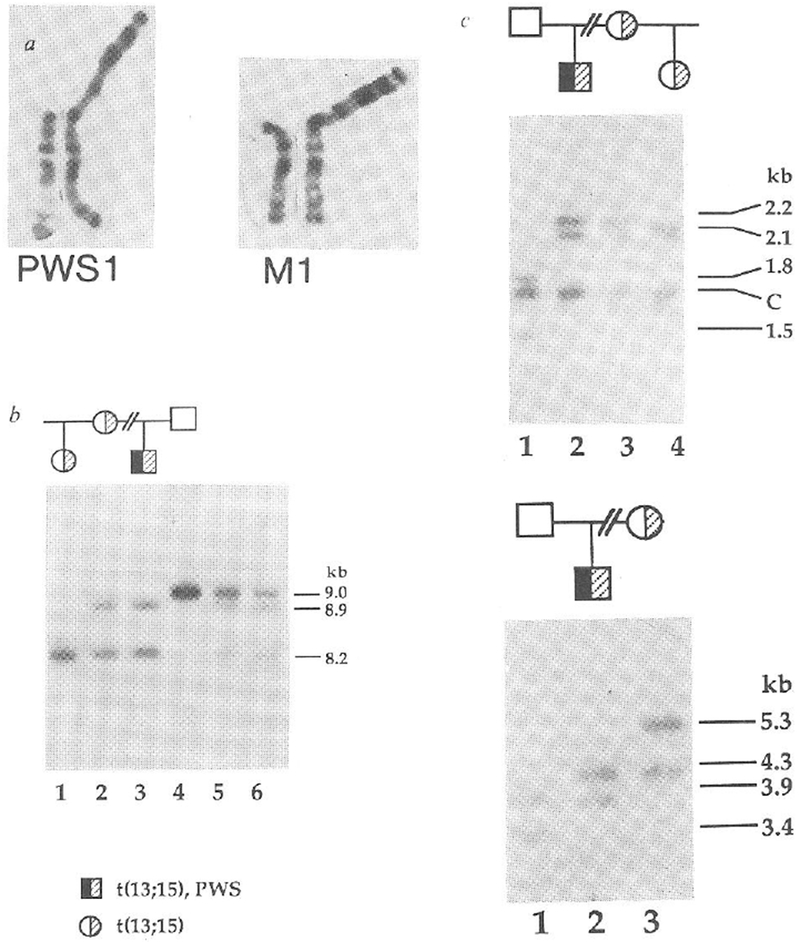
Maternal uniparental disomy in a PWS patient with a balanced Robertsonian translocation. a, Normal chromosome 15 and translocated homologue from patient PWS1 (left) and his mother M1 (right). Other unaffected maternal relatives, including the half-sister of PWS1 carry the same balanced chromosome 13q15q robertsonian translocation, b, Maternal origin of the two chromosomes 15 in patient PWS1. An 8.2-kb TaqI allele of probe 3–21 (D15S10) is shared by PWS1 (lane 3), his mother M1 (lane 2) and half-sister (lane 1), whereas an 8.9-kb band is shared by PWS1 and M1. PWS1 does not inherit a 9.0-kb paternal allele (F1, lane 4). Mixing experiments (lane 5, PWS1 and F1; lane 6, M1 and F1) show the identity of the three alleles in this new RFLP system, c, Presence of two maternal alleles only for CMW-1 (D15S24) in PWS1. PWS1 (lane 2) shares two TaqI restriction fragments, of 2.1 and 2.2 kb. with M1 (lane 3) but has neither of the 1.5- or 1.8-kb paternal alleles (F1, lane 1). The half-sister (lane 4) displays the maternal 2.1-kb and a 2.15-kb allele. The band at 1.7 kb is constant, C. d, Exclusion of non-paternity for PWS1. PvuII-digested DNA was hybridized to the α-globin 3’HVR marker (D16S85), one of the most highly polymorphic markers in the human genome12. PWS1 (lane 2) shows mendelian inheritance of one paternal 3.9-kb allele (lane 1) and one maternal 4.3-kb allele (lane 3). The half-sister inherits her mother’s 5.3-kb allele (result not shown).
METHODS. DNA was isolated from blood and/or cell lines from each individual as described previously5. Hybridizations were carried out as previously described5 High-resolution chromosome analyses from peripheral blood lymphocytes29 were performed by Giemsa-trypsin banding30.
The proband (PWS2) of the second family also has classical PWS (refs 13, 14). Cytogenetically, PWS2 has two intact chromosome 15s (Fig. 2a). We demonstrated the lack of paternal alleles for two loci by haplotype analysis (Fig. 2b, c). With probe 34, the father is homozygous for 6.5 kb alleles, whereas the mother is homozygous for 6.3 kb alleles (Fig. 2b). For the proband, we found only a 6.3-kb band (Fig. 2b), consistent with the maternal origin of both cytogenetically normal chromosomes 15. Like-wise, only maternal alleles specific for probe IR39d had been inherited (Fig. 2c). By dosage analyses we demonstrated that PWS2 has two copies of each of these maternally derived alleles (Fig. 2d and results not shown), indicating that loci detected by probes 34 and IR39d had not been deleted. These two loci are flanked by loci detected by probe IR4-3R (results not shown) and probe IR10-1, respectively, (Fig. 2b) that are heterozygous in the proband. To rule out the possibility of a submicroscopic deletion, we analysed the long-range structure of the 15q11q13 region by pulsed-field gel electrophoresis (PFGE). This showed that probe IR4-3R and probe 34 detect the same large unaltered 2,500-kb NotI fragment in PWS2 (Fig. 3), which provides strong evidence that the absence of a paternal allele specific for probe 34 does not represent a microdeletion. Furthermore, we detected a band corresponding to a fragment of the same size (2,500 kb) in the mother, despite differential methylation leading to partial NotI digestion in this region of the genome (Fig. 3) and in other normal individuals (results not shown). We again excluded the possibility of non-paternity using the 3′HVR fragment, which detected an entirely different set of alleles in the PWS2 family (Fig. 2e) from those that it detected in the PWS1 family (Fig. 1d).
FIG. 2.
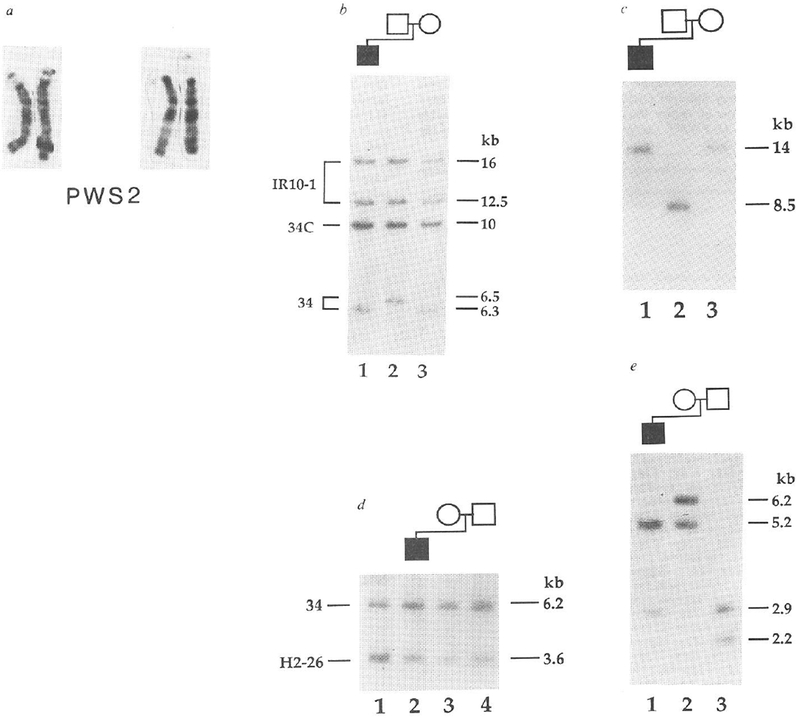
Maternal uniparental disomy in a PWS patient with apparently normal chromosomes 15. a, Two pairs of intact chromosomes 15 of PWS2. Previous reports have indicated these chromosomes as being intact’3 or deleted 15q (ref. 14); in the deletion report a polymorphic shortening of 15q11 (ref. 7) was probably detected, because we show here at a higher level of resolution that the chromosomes are intact. b, Maternal origin of the two chromosomes 15 in PWS2. DNA was digested with SeaI. PWS2 (lane 1) inherits a maternal 6.3-kb allele (lane 3) but no paternal 6.5-kb allele (lane 2) for probe 34 (D15S9). Each individual is heterozygous for IR10-1 (D15S12; 16- and 12.5-kb alleles), which is from the same 15q11q13 region as probe 34 (ref. 10). The band at 10 kb is constant. C. for probe 34 in all individuals5 c. PWS2 inherits maternal alleles only for probe IR39d (D15S18). DNA was digested with SacI and hybridized with the 15q11q13-specific probe IR39d. PWS2 (lane 1) inherits his mother’s (lane 3) 14-kb alleles and not his father’s (lane 2) 8.5-kb alleles. d, Two copies of the maternal allele for probe 34 in PWS2. HindIII-digested DNAs from PWS2 (lane 2), mother M2 (lane 3), father F2 (lane 4) and a PWS patient displaying both a cytological and molecular deletion of 15q11q13 (ref. 5; lane 1. HS2) were hybridized simultaneously with probes 34 and H2-26 (D13S28)31 . Probe H2-26 from chromosome 13 serves as a reference for quantitation of copy number10. Probe 34 shows a reduced hybridization intensity relative to H2-26 only in HS2, which is consistent with a deletion of 15q11q13 in this individual. No reduction in probe 34 hybridization intensity is observed in PWS2 or his parents. To quantitate the intensity of the hybridization bands, the autoradiogram was scanned with an LKB Ultrascan XL laser densitometer (Pharmacia LKB Biotechnology). The area ratio of the probe 34 to probe H2-26 peaks was calculated for each lane. The number of copies of probe 34 per genome was obtained by setting the ratio in lane 3 (M2, mother) to 2 and normalizing the values in the other lanes to this value. The number of copies of probe 34 per genome determined by this method were 1.0 in lane 1 (the HS2 deletion), 1.8 in lane 2 (PWS2) and 2.2 in lane 4 (F2, father). e, Exclusion of non-paternity for PWS2. One maternal 5.2-kb (lane 2) and one paternal 2.9-kb (lane 3) PvuII allele are inherited by PWS2 (lane 1) for the 3’HVR. METHODS. DNA and cytogenetic studies were as for Fig. 1. Probe IR39d is a subclone of IR39 (ref. 10), which lacks repetitive sequences. PWS2 has been described before as patient number 5 (ref. 14) and patient number 31 (ref. 13).
FIG. 3.
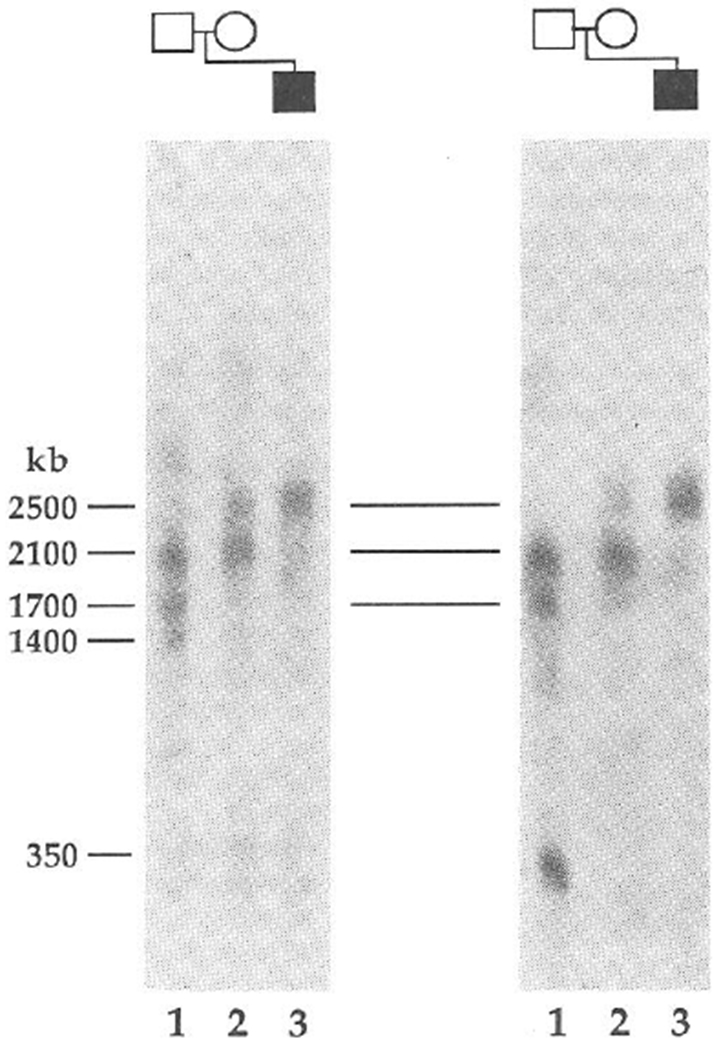
PFGE provides evidence for the lack of a submicroscopic deletion in PWS patients with uniparental disomy. NotI fragments are shown for probes IR4-3R (D15S11) (left) and 34 (right) in PWS2 (lane 3), M2 (lane 2) and F2 (lane 1). Multiple bands are indicative of partial cleavage at restriction sites when CpG is methylated32 and a similar pattern of multiple bands has been observed in other normal controls (results not shown). PWS2 shares a normal 2,500-kb band with his mother, although in the latter this results from partial digestion at a NotI site in this region of the genome. The same 2,500-kb band has been detected in other normal individuals. The larger three fragments are common to probes IR4-3R, 189-1 (results not shown) and 34, showing that these loci are closely linked in region 15q11q13. These probes and a 1,300-kb fragment detected with probes 3-21 and IR10-1 (unpublished data) are deleted in all PWS10 and AS6 patients with cytological deletions. Additional PFGE analysis (results not shown) also indicates that PWS2 displays PFGE restriction fragments of normal size for IR10-1 in a NotI digest (1,300 kb) and for IR39d in a BssHII digest (250 kb). PWS1 also seems to be intact for the probe 34 and 3-21 loci because the restriction fragments detected using NotI and BssHII are unaltered compared with those of normal individuals.
METHODS. PFGE analyses were performed as described previously33 using a modification of techniques for DNA preparation and digestion in agarose blocks34. Electrophoresis was performed in 0.8% agarose in TBE (89 mM Tris buffer, pH 8.0, 89 mM boric acid, 2 mM EDTA) at 22 °C and was divided into three 48-h intervals during which the forward polarity pulse durations were varied from 75-600 s, 600-2,500 s and 2,500-3,000 s and field intensities were set at +2, +1.7 and +1.3 V cm−1, respectively. The ratios of the duration and intensity of the inverse polarity pulse relative to the forward were 0.4 and 0.5, respectively.
The results presented here indicate that both PWS probands inherited two different, intact chromosome 15q11q13 regions from their mothers. Maternal heterodisomy—a newly defined form of uniparental disomy15,16—for at least the critical region of chromosome 15, could thus have an aetiological role in PWS. The extent of disomy remains to be determined by genetic analysis with other markers. The disomy extends beyond the PWS critical region identified from deletion studies because locus CMW-1, which maps distal to the deletion region, is disomic in PWS1. Several predictions can be made about an association between uniparental disomy and the aetiology of PWS. First, there should be no chromosome deletion, as is shown in this study by using both cytogenetic and molecular genetic techniques. Additional PFGE mapping data for PWS1 and PWS2 (Fig. 3 legend) confirmed that a large part (4-5 mega-bases) of the 15q11q13 region is intact in these two patients. Second, there should be no specific gene mutation. Although point mutations cannot be ruled out until the gene(s) responsible for PWS are isolated, these seem unlikely because each case displays two independent maternal contributions to region 15q11q13 and the disorder is genetically dominant, which would rule out mechanisms such as the uncovering of a recessive mutation. Finally, the frequency of maternal disomy should be high in PWS patients with normal chromosomes. Consistent with this prediction, our preliminary data on four additional families strongly indicates that all of the PWS patients show maternal uniparental disomy.
It seems that the clinical phenotype of the two PWS probands arises from the absence of a paternal contribution to region 15q11q13, rather than from a specific gene mutation. This implies functional differences in alleles of a gene or genes from this region of the genome that depend on the sex of the transmitting parent (genetic imprinting18–21). Under this scheme, normal human development would require genetic input from both parents, whereas the absence of a paternal contribution to region 15q11q13, whether by paternal deletion4,5 or maternal uniparental disomy (as demonstrated here), would result in PWS. Consistent with this hypothesis is our recent finding of differential transmission of parental alleles in PWS and AS (ref. 6). It is conceivable, therefore, that the absence of a maternal contribution to the same 15q11q13 region could result in a different disorder, AS.
Experimental evidence for genetic imprinting has been largely restricted to the mouse in which a requirement for a contribution of both the maternal and paternal genome for normal development has been demonstrated18–22. Transgenic studies have provided preliminary evidence that the chromatin alteration involved in the differential modification and expression of parental alleles is DNA methylation18–22, a process also associated with transcriptional regulation of gene activity23. In humans, evidence of a developmental requirement for both parental genomes is provided by the finding that only the paternal genome is present in complete hydatidiform moles24. Furthermore, differential transmission of parental alleles in several disorders such as Huntington’s chorea18,25, and somatic changes in childhood tumours such as Wilms tumour, osteosarcoma (reviewed in ref. 26) and rhabdomyosarcoma27 could involve genome imprinting. Growth failure in two individuals with isodisomy15 for chromosome 7 (refs 16, 28) could indicate genetic imprinting16, although the aetiology of cystic fibrosis (CF) in these patients is homozygosity for a mutant CF allele inherited from the mother16,28.
In conclusion, the association of PWS with maternal uniparental disomy for region 15q11q13 implicates a role for genetic imprinting in the aetiology of the PWS phenotype. Similar phenomena could contribute to other clinical syndromes. Isolation of the gene(s) responsible for PWS and other disorders in which genetic imprinting plays a part might be possible by identifying a gene that shows a differential modification of its parental alleles. Our findings for PWS represent a step towards understanding the developmental basis of this common human genetic disorder.
ACKNOWLEDGEMENTS.
We are grateful to Dr L. Kunkel for his critical reading of the manuscript, D. Shook and K. Glatt for technical assistance, B. Woolf for help in preparation of the manuscript, Dr D. Ledbetter for providing the CMW-1 probe and Dr C. Sapienza for communicating results and hypotheses before their publication. We also thank Dr S. Kapur for clinical evaluation of one of the patients. This work was supported in part by the NIH. M.L. is a senior associate of the Howard Hughes Medical Institute.
References
- 1.Cassidy SB. Curr. Problems Pediatr 14, 1–55 (1984). [DOI] [PubMed] [Google Scholar]
- 2.Butler. MG. Am. J. med. Genet (in the press). [Google Scholar]
- 3.Ledbetter DH, Greenberg F, Holm VA & Cassidy SB. Am. J. med. Genet 28, 779–790 (1987).3688016 [Google Scholar]
- 4.Butler MG, Meaney FJ & Palmer CG. Am. J. med. Genet 23, 793–809 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicholls RD et al. Am. J med. Genet 33, 66–77 (1989). [DOI] [PubMed] [Google Scholar]
- 6.Knoll JHM et al. Am. J. med Genet 32, 285–290 (1989). [DOI] [PubMed] [Google Scholar]
- 7.Pembrey M et al. J. Med. Genet 26, 73–77 (1989) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donlon TA Hum. Genet 80, 322–328 (1988). [DOI] [PubMed] [Google Scholar]
- 9.Donlon TA, Lalande M, Wyman A, Bruns G & Latt SA Proc. natn. Acad. Sci. U.S.A 83, 4408–4412 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tantravahi U et al. Am. J. med. Genet 33, 78–87 (1989). [DOI] [PubMed] [Google Scholar]
- 11.Rich DC, Witkowski CM, Summers KM, van Tuinen P & Ledbetter DH. Nucleic Acids Res 16, 8740 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Higgs DR et al. Proc. natn. Acad. Sci. U.S.A 83, 5165–5169 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butler MG & Meaney F Am. J. med. Genet 26, 445–455 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butler MG Am. J. hum. Genet 45, 140–146 (1989). [PMC free article] [PubMed] [Google Scholar]
- 15.Engel E Am. J. med. Genet 6, 137–143 (1980). [DOI] [PubMed] [Google Scholar]
- 16.Spence JE et al. Am. J. hum. Genet 42, 217–226 (1988). [PMC free article] [PubMed] [Google Scholar]
- 17.Warburton D Am. J. hum. Genet 42, 215–216 (1988). [PMC free article] [PubMed] [Google Scholar]
- 18.Solter DA Rev. Genet 22, 127–146 (1988). [DOI] [PubMed] [Google Scholar]
- 19.Sapienza C Annls N.Y. Acad. Sci (in the press). [Google Scholar]
- 20.Monk M Genes Dev 2, 921–925 (1988). [DOI] [PubMed] [Google Scholar]
- 21.Holliday R. Sci. Am 260, 60–73 (1989). [DOI] [PubMed] [Google Scholar]
- 22.Surani MA, Reik W & Allen ND. Trends Genet 4, 59–62 (1988). [DOI] [PubMed] [Google Scholar]
- 23.Cedar H Cell 53, 3–4 (1988). [DOI] [PubMed] [Google Scholar]
- 24.Kajii T & Ohama K Nature 268, 633–634 (1977) [DOI] [PubMed] [Google Scholar]
- 25.Reik W J. med. Genet 25, 805–808 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reik W & Surani MA Nature 338, 112–113 (1989). [DOI] [PubMed] [Google Scholar]
- 27.Scrable H et al. Proc. natn. Acad. Sci. U.S.A (in the press). [Google Scholar]
- 28.Voss R et al. Am. J. hum. Genet 45, 373–380 (1989). [PMC free article] [PubMed] [Google Scholar]
- 29.Yunis JJ Science 191, 1268–1270 (1976). [DOI] [PubMed] [Google Scholar]
- 30.Seabright M Lancet ii, 971–972 (1971). [DOI] [PubMed] [Google Scholar]
- 31.Lalande M et al. Cancer Genet. Cytogenet 13, 283–295 (1984). [DOI] [PubMed] [Google Scholar]
- 32.Fischel-Ghodsian N, Nicholls RD & Higgs DR Nucleic. Acids Res 15, 6197–6207 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lalande M, Noolandi J, Turmel C, Rousseau J & Slater GW Proc. natn. Acad. Sci. U.S.A 84, 8011–8015 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwartz DC, & Cantor CR Cell 37, 67–75 (1984). [DOI] [PubMed] [Google Scholar]