

Abstract
The implementation of next-generation sequence analysis of disease-related genes has resulted in a increasing number of genetic variants with an unknown clinical significance. The functional analysis of these so-called “variants of uncertain significance” (VUS) is hampered by the tedious and time-consuming procedures required to generate and test specific sequence variants in genomic DNA.
Here we describe an efficient pipeline for the generation of gene variants in a full-length human gene, BRCA2, using a bacterial artificial chromosome (BAC). This method permits the rapid generation of intronic and exonic variants in a complete gene through the use of an exon-replacement strategy based on simple site-directed mutagenesis and an effective positive-negative selection system in E. coli. The functionality of variants can then be assessed through the use of functional assays, such as complementation of gene-deficient mouse embryonic stem (mES) cells in the case of hBRCA2. Our methodology builds upon an earlier protocol and, through the introduction of a series of major innovations, now represents a practical proposition for the rapid analysis of BRCA2 variants and a blueprint for the analysis of other genes using similar approaches. This method enables rapid generation and reliable classification of VUS in disease-related genes, allowing informed clinical decision-making.
Introduction
The adoption of next-generation sequencing in research and clinical diagnostics has resulted in an enormous increase in the number of variants identified. Although the biological and clinical significance of some variants can be readily determined, a rapidly expanding number of variants of uncertain significance (VUS) are being identified in a wide range of disease-related genes. Most VUS are (extremely) rare and therefore family-based clinical analyses are often insufficient to make clinically inferences about their associated disease risk. The determination of the contribution of these variants to disease therefore requires rapid and reliable molecular methods to prepare variants for analysis, and efficient functional assays to define biological impact. While cDNA constructs are commonly used for the analysis of VUS, cDNA-based complementation assays lack the biological context of a complete gene, lack the ability to test intron variants and variants that may affect RNA splicing, and often hamper assessment of physiological relevance due to overexpression. A major step forward in the assessment of VUS in a physiological relevant context came in 2008 with an important publication by Kuznetsov and colleagues. These authors developed a VUS analysis pipeline using a bacterial artificial chromosome (BAC) that contains the entire human BRCA2 gene (MIM#600185) of over 80Kb, including all exon and intron sequences and the physiological promoter (Kuznetsov et al. 2008). The use of a BAC-derived gene allowed testing of variants in the context of an intact gene and the testing of intronic and exonic variants that could potentially affect RNA splicing.
Despite the clear biological advantages of the use of BAC as compared to cDNA constructs, a major drawback has been the speed and ease of manipulation of these often unwieldy constructs. We therefore set out to redesign and re-engineer the major steps of the Kuznetsov assay, with the aim of developing a pipeline and model system that would allow efficient analysis of gene-specific variants and provide a blueprint for the analysis of variants in other disease-related genes.
Using the human BRCA2 gene (hBRCA2) as a model, we increased the throughput of VUS generation using both direct recombineering and an exon-swapping/ cassette replacement strategy that required only a single round of BAC recombineering. We designed a positive-negative selection system in E. coli that dramatically increases the identification of genuine recombinants and achieved efficient selection of clones that express full-length mRNA and show physiological levels of protein expression. Finally, we showed efficient complementation of mBRCA2 deficiency and explored the physiological relevance and practical utility of several functional assays.
These innovations increased the efficiency of the original procedure 10-fold and together represent the maturing of this methodology to the stage of practical utility and wide applicability. In addition, each individual step is equally applicable to many other genes and can be taken as a blueprint for the design of similar systems for many other disease-related genes.
Materials and Methods
Generation of the hBRCA2 variants
All hBRCA2 variants were created in a bacterial artificial chromosome (BAC) (clone RP11–777I19, BACPAC) stably maintained in an rpsL-deficient DH10B E. coli strain. The first step involved removing two loxP sequences from the pBACe3.6 vector backbone of the BAC using a previously described Red/ET BAC recombineering system (Poser et al. 2008) with synthetic oligonucleotides containing sequences that flank the loxP sites. Next, a selection marker containing a neomycin/kanamycin (Neo) resistance gene, preceded by an internal ribosomal entry site (IRES) sequence, was introduced by Red/ET recombineering at the 3’ end of the hBRCA2 gene, immediately after the stop codon at exon 27. Correct integration of the IRES-Neo selection marker and removal of the loxP sites was confirmed by PCR and sequence analysis.
For positive-negative selection during BAC-recombineering in E. coli, the ribosomal protein S12 gene (rpsL) was fused to an ampicillin resistance gene (Amp). The rpsL gene, including promoter, was derived from the pSK+ rpsL-kana plasmid (a kind gift from Dr. J Zhu) and cloned upstream of the Amp gene in pUC19. To allow sequence-directed recombination, the rpsL-Amp selection cassette was amplified by PCR (KAPA HiFi) using primers with 50 bp sequence homology to the hBRCA2 gene region flanking the VUS to be analyzed. The rpsL-Amp selection cassette was subsequently integrated by Red/ET recombineering at the position of the VUS in the hBRCA2-IRES-Neo BAC clone. As an alternative approach, we replaced the complete exon 17 or exon 18 (plus 100–150 bp of flanking intron sequence on either side) of the hBRCA gene with the rpsL-Amp selection cassette. Following transformation into E. coli, correct integration of the dual selection cassette into ampicillin resistant clones was assessed by PCR.
Using Red/ET recombineering, specific hBRCA2 variants were then introduced into the BAC by replacing the rpsL-Amp selection cassette with a small DNA fragment containing the intended variant. This fragment was generated using 100-mer double-stranded (ds)DNA fragments containing the sequence variant that were created by annealing partially overlapping 60-mer oligonucleotides, followed by subsequent DNA synthesis as previously described (Yang and Sharan 2003). Alternatively, individual hBRCA2 exons (e.g. exon 17 or 18), including flanking intronic sequences, were amplified by PCR and cloned into pUC19, followed by modification to generate variants of interest using QuickChange site-directed mutagenesis (Stratagene) according to the manufacturer’s protocol. Variant-containing hBRCA2 exons were then amplified by PCR and used to replace the rpsL-Amp cassette in the respective hBRCA2 BAC with Red/ET recombineering. Clones in which a dsDNA fragment or complete exon replaced the rpsL-Amp cassette became streptomycin resistant and were selected for as described previously (Wang et al. 2009). Successful generation of the desired hBRCA2 variant base pair change was confirmed by sequence analysis of the complete replaced region. All primer sequences are summarized in Supplementary Table 1. BACs containing variants were also checked for the absence of gross genomic alterations by EcoR1, Xho1 and SalI restriction digestion analysis. BAC DNA containing the hBRCA2 variants was isolated using the nucleobond AX midiprep kit (Macherey-Nagel) according to manufacturer’s protocol.
mES cell culture and transfections
mES cell culture was performed as described previously (Hendriks et al. 2012). The mES cells were maintained in the presence of irradiated mouse embryonic fibroblasts as feeder cells in Knockout DMEM containing 10% fetal bovine serum, 2mM GlutaMAX, 1mM sodium pyruvate, 100 µM β-mercaptoethanol and leukemia inhibitory factor (LIF). For transfection and protein or RNA analysis, cells were cultured on gelatin-coated plates using buffalo rat liver cell (BRL)-conditioned mES cell medium. BACs carrying hBRCA2 variants were transfected into the Pl2F7 conditional mBrca2 knockout mES cell line (Kuznetsov et al. 2008) using Lipofectamine2000 (Invitrogen) as previously described (Hendriks et al. 2012). Transfected cells were selected with 200 µg/ml G418 for 2 weeks.
RT-PCR and Western blot
Expression of hBRCA2 variants in mES cells was confirmed by RNA and protein analysis. Total RNA was isolated from G418-selected monoclonal mES cell lines using TRIzol (Invitrogen). cDNA was synthesized using SuperScript III reverse transcriptase and oligo dT (12–18) primers (Invitrogen) according to the manufacturer’s protocol. Expression of the BAC-derived hBRCA2 gene was determined by PCR using primers to amplify exon 22 and exon 27 of the hBRCA2 gene and primers for the mAprt gene as internal standard. Protein expression of hBRCA2 was determined by Western blot analysis. Proteins were isolated using a nuclear extraction lysis buffer (20mM Tris pH 8.0, 300mM NaCl, 2% NP40, 20% glycerol, 10mM EDTA, complete mini protease inhibitor (Roche)). Cells were lysed for 20 min on ice and after nuclear protein extraction, nuclei were removed by centrifugation. Protein concentrations were determined by Bradford protein assay (Pierce, Thermo Fisher). Proteins were denatured at 50°C for 5 min and analysed on NuPage 4–12% BisTris PAGE gels in MOPS buffer (Invitrogen). Proteins were transferred to PVDF membrane (Millipore) and both human and mouse BRCA2 proteins were detected using mouse monoclonal antibodies against amino acids 1651–1821 (exon 11) of hBRCA2 (OP-95, Millipore). MSH2 antibodies (Ab-2 Calbiochem/Merck) were used as a protein loading control. Proteins were visualized using goat anti-mouse HRP secondary antibodies and chemiluminescence, and protein levels were quantified using ImageStudioLite software (LI-COR).
Functional testing of variants
The conditional KO mBrca2 allele in Pl2F7 mES cells expressing the hBRCA2 variants was removed by transient Cre expression and subsequent selection for restoration of an HPRT minigene using hypoxanthine-aminopterin-thymidine (HAT) (Gibco) as described previously (Kuznetsov et al. 2008). hBRCA2-expressing Pl2F7 cells were seeded at a density of 1.5 × 106 on gelatin-coated 60 mm cell culture dishes in BRL-conditioned mES cell culture medium 24h prior to transfection. After overnight transfection of a NLS-Cre expression (kind gift from C. Breukel) plasmid using Lipofectamine2000, cells were trypsinized and 0.1 × 106 cells were seeded in 90 mm culture dishes in the presence of HAT. After 10 days, hBRCA2-complemented mBrca2−/− clones were visualized by methylene blue staining. In addition, complemented mES clones were picked for functional analysis. Four independent mES cell clones were analyzed for each non-lethal hBRCA2 variant. The sensitivity of hBRCA2 expressing mBrca2−/− mES cells to DNA damaging agents was assessed by determining the percentage of viable cells after 24h continuous exposure to methyl methanesulfonate (MMS), mitomycin C (MMC) and a PARP inhibitor (KU-0058948, Astra Zeneca) using a flow cytometer (Guava, Millipore). The sensitivity of hBRCA2 complemented mES cells was confirmed by a clonal survival assay after exposure to MMS or MMC for 0.5h and 1h, respectively. on one of the clonal cell lines. Wild type 129/OLA mES cells (IB10) and mBRCA2-/loxP (Pl2F7) cells were used as controls.
Homologous recombination proficiency of clones expressing hBRCA VUS was determined by RAD51 foci formation 2h after exposure to 10 Gy ionizing radiation (IR) using rabbit anti-RAD51 (kind gift from Prof. R. Kanaar). Rabbit anti-γH2AX (Millipore) was used to visualize double stranded DNA breaks (DSB) 30 min after IR exposure. Both antibodies were visualized using Alexa488-labeled goat anti-rabbit secondary antibodies (Invitrogen). Immunofluorescence labelling and microscopy was performed as described previously (Vrouwe et al. 2011).
Results
Positive-negative selection for variants of uncertain significance
The method to generate hBRCA2 variants as originally described (Yang and Sharan 2003; Kuznetsov et al. 2008) involved two rounds of oligo targeting and multiple rounds of PCR testing of bacterial colonies to identify desired recombinants. To improve efficiency we designed a positive-negative selection system by combining an ampicillin (Amp) selection cassette for positive selection with the negative selectable marker rpsL that encodes the ribosomal protein S12. As the E. coli host strain containing the BAC is rpsL- deficient, loss of the rpsL-Amp selection cassette results in streptomycin resistance (strep). The first step is to integrate the rpsL-Amp positive-negative selection cassette in the hBRCA2 gene at the exact genomic position of the VUS under investigation (Figure 1A), followed by Amp selection and verification of correct rpsL-Amp integration in the hBRCA2 BAC by PCR analysis. In the negative selection step, the rpsL-Amp cassette is replaced by a synthetic oligonucleotide containing the VUS of interest using recombineering and streptomycin selection is then applied to identify bacteria that have lost the rpsL-Amp selection cassette. To test the efficiency of the positive-negative selection procedure we generated eight previously described hBRCA2 variants, five missense and three intronic variants (Table 1, Supp. Table 2). Recombineering with an oligonucleotide containing the wild type hBRCA2 sequence was used as control. PCR and sequence analysis of the resulting colonies indicated that approximately 80% of the Strr transformants contained the desired hBRCA2 sequence variant (8–12 clones analyzed for each variant, data not shown).
Figure 1: Generation of hBRCA2 variants in mES cells.
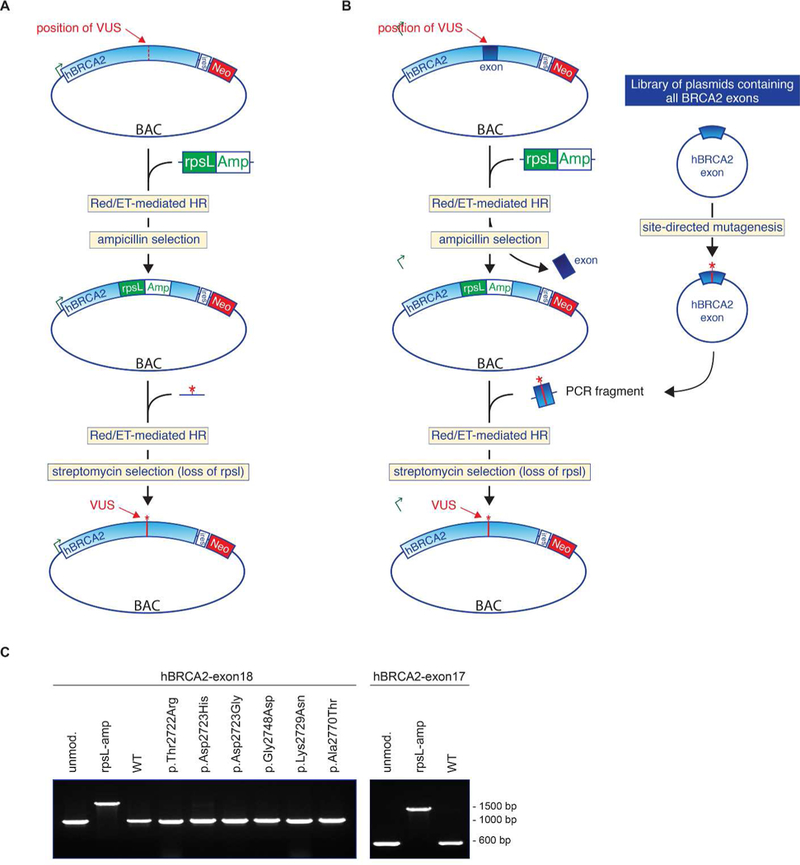
(A) hBRCA2 variants were generated by Red/ET mediated BAC recombineering in two steps. In the first step, an rpsL-Amp selection cassette was inserted in the hBRCA2 gene at the position of the desired VUS. The rpsL-Amp cassette confers ampicillin resistance and streptomycin sensitivity. In a second recombination reaction the rpsL-Amp cassette was replaced by an oligonucleotide that harbors the VUS, resulting in streptomycin resistance. (B) High throughput exon library approach for efficient introduction of VUS in the hBRCA2 gene. A complete exon of the hBRCA2 gene is replaced by the rpsL-Amp selection cassette by Red/ET mediated BAC recombineering. A library of plasmids each containing a single exon of hBRCA2 is generated. These plasmids can be used to easily introduce specific VUS using PCR-based site-directed mutagenesis. BAC recombineering is used to replace the rpsL-Amp cassette in the hBRCA2 gene by a VUS-containing hBRCA2 exon. (C) Replacement of hBRCA2 exon 18 with six exon 18 hBRCA2 variants. Reintroduction of the wild type hBRCA2 exon 18 was used as control. In addition, exon exchange of hBRCA exon 17 was performed. Successful replacement was confirmed by cDNA synthesis and PCR analysis. In total 50 bacterial clones were analyzed of which 50% were correctly modified.
Table 1.
Summary of the functional assay results
| Exon | Nucleotide*1 | Amino acid*1 | IARC Classification*2 | Previous assessment*3 | Method of variant generation | RNA expression level*4 | hBRCA2 protein levels*5 | RNA splicing*6 | Complementation of mBRCA2 deficiency*7 | Sensitivity to DNA damaging agents*8 | RAD51 foci*9 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | c.68–7T>A | p.? | Not available | Unlikely to affect function | Oligo targeting | + | + | Wildtype and ∆3 transcript from variant allele | Yes | No | + |
| 15 | c.7617+2T>G | p.? | Not available | Impairs protein function | Oligo targeting | + | Absent | Skip exon 15 | No | NA | NA |
| 17 | c.7878G>C | p.Trp2626Cys | Class 5 | Impairs protein function | Oligo targeting | + | + | ND | No | NA | NA |
| 17 | c.7879A>T | p.Ile2627Phe | Class 5 | Impairs protein function | Oligo targeting | + | + | ND | No | NA | NA |
| 18 | c.8165C>G | p.Thr2722Arg | Class 5 | Impairs protein function | Exon mutagenesis | + | + | ND | No | NA | NA |
| 18 | c.8167G>C | p.Asp2723His | Class 5 | Impairs protein function | Exon mutagenesis | + | + | ND | No | NA | NA |
| 18 | c.8168A>G | p.Asp2723Gly | Class 5 | Impairs protein function | Exon mutagenesis | + | + | ND | No | NA | NA |
| 18 | c.8187G>T | p.Lys2729Asn | Class 1 | Does not affect function | Exon mutagenesis/Oligo targeting | + | + | ND | Yes | No | + |
| 18 | c.8243G>A | p.Gly2748Asp | Class 5 | Impairs protein function | Exon mutagenesis | + | + | ND | No | NA | NA |
| 18 | c.8308G>A | p.Ala2770Thr | Class 2 | Does not affect function | Exon mutagenesis | + | + | ND | Yes | No | ND |
| 21 | c.8754+5G>A | p.? | Not available | Impairs protein function | Oligo targeting | + | Absent | Retention 46 nt from intron 21 | No | NA | NA |
| 24 | c.9154C>T | p.Arg3052Trp | Class 5 | Impairs protein function | Oligo targeting | + | + | ND | No | NA | NA |
| 24 | c.9155G>A | p.Arg3052Gln | Class 2 | Probably affects function | Oligo targeting | + | + | ND | Reduced number of clones | Increased senstivity | Reduced |
Nucleotide numbering reflects HGVS nomenclature where cDNA numbering +1 corresponding to the A of the ATG translation initiation codon in the reference sequence (BRCA2 NM_000059.3). The initiation codon is codon 1. nt: nucleotides.
IARC classification (Lindor et al., 2012); Class 1 non-pathogenic; Class 2 likely non-pathogenic; Class 5 pathogenic
Results from functional studies, see Supp Table S2
RNA expression levels (analysed with primers in exon 22–27) are comparable to wildtype hBRCA2 for all variants (+)
(+) hBRCA2 protein expression is similar to mBRCA2 protein expression
For intronic variants, RNA splicing was analyzed by PCR using primers spanning the exons that were previously shown to be affected by the variants.
“No”; no mESC clones were present; Yes, Equal number of clones were present compared to transfection with wt hBRCA2
“No” sensitivity is similar to wildtype hBRCA2
(+) hBRCA2 RNA expression level or hBRCA2 protein level similar to mBRCA2 or RAD51 foci per cell similar to wildtype
NA; not applicable since no clones were formed that could be tested
All variants are reported in public database (http://chromium.liacs.nl/LOVD2/cancer/home.php)
Exon-swapping strategy
Implementation of a positive-negative selection system (rpsL-Amp) during BAC recombineering strongly increased the efficiency of variant generation. As a further improvement, we tested the feasibility of exchanging a complete wild type exon in the BAC with a modified exon containing a specific variant. As proof of concept we replaced exon 18 of the hBRCA2 gene with the rpsL-Amp selection cassette. To allow the generation of multiple variants in parallel, we cloned exon 18 of hBRCA2 into pUC19, including approximately 100 bp of flanking intronic sequence at either side. This construct was subsequently used to generate six different sequence variants using a standard site-directed mutagenesis procedure (Figure 1B; Table 1). We then replaced the rpsL-Amp cassette with either the wild type hBRCA2 exon 18 or one of the six different VUS in exon 18 (Figure 1C). The same strategy was used to successfully generate variants in exon 15, exon 17 and exon 23 of the hBRCA2 gene (data not shown).
Insertion and physiological expression of a full-length gene in mES cells
Although mES cells entirely lacking BRCA2 are non-viable, heterozygous knockout cells remain viable and the remaining mouse allele can be replaced by a human allele (complementation). These cells can therefore be used to test the functionality of single hBRCA2 alleles that contain variants. However, complementation of mBrca2-/loxP conditional knock out in mES cells is challenging due to difficulties with integration and expression of the complete hBRCA2 gene due to its large size. We reasoned that placing a selectable marker gene after a full-length copy of the hBRCA2 gene would require full transcription of BRCA2 before transcription and production of the marker protein, thus favoring selection of a non-disrupted BRCA2 gene. We therefore inserted an internal ribosomal entry site (IRES) sequence followed by a neomycin (Neo) selection marker directly after the stop codon at the 3’ end of a BAC-derived hBRCA2 gene (Figure 2A). Following transfection into mES cells, expression of both the hBRCA2 and Neo resistance gene is controlled by the hBRCA2 promoter and both proteins are translated from the same mRNA molecule. Of the Neo resistant clones recovered (n=96), 65% expressed full-length hBRCA2 mRNA (Figure 2B). Comparison of hBRCA2 and endogenous mBRCA2 protein levels in Neo-selected monoclonal cell lines by Western blot analysis showed very similar expression levels for the mouse and human BRCA2 protein in all analyzed mES cell lines (Figure 2C–D). Together these data indicate that incorporation of hBRCA2 and the Neo selectable marker into a single bicistronic transcript provides an effective means to select for mES clones that express full-length human BRCA2 protein at physiological levels.
Figure 2: Expression of wild type hBRCA2 in mES cells.
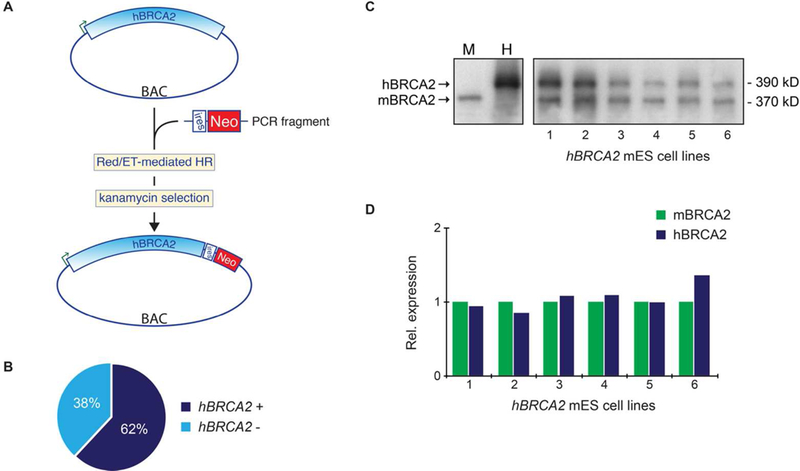
(A) A neomycin resistance marker (Neo) was introduced at the 3’-end of the hBRCA gene using BAC recombineering. An internal ribosomal entry site (IRES) sequence upstream of the Neo selection cassette places Neo under control of the hBRCA2 promoter and will favor the presence of an undisrupted hBRCA2 gene in G418-resistant mES transfectants. (B) Frequency of neomycin-resistant mES cell clones that expressed full length hBRCA2. Expression of hBRCA2 was determined by cDNA synthesis and PCR amplification of exon 22–27 of the hBRCA2 gene. Frequencies are based on analysis of 96 independent mES cell clones. (C) Western blot analysis of BAC-derived hBRCA protein expression in six independent mES cell lines. Wild type mES cells (M) and human TK6 cells (H) were used as control for the mouse and human BRCA2 proteins. (D) Quantification of mBRCA2 and hBRCA2 proteins levels in 6 independent monoclonal BRCA2-/loxP mES cell lines expressing BAC-derived hBRCA2. Expression of hBRCA2 is related to expression levels of the endogenous mBRCA2.
RNA splicing and protein expression levels
BACs containing either wild type or hBRCA2 variants were transfected into mBrca2−/loxP conditional knockout mES cells and after G418 selection, multiple monoclonal cell lines were isolated. PCR analysis of cDNA revealed that approximately 80% of the clones (n=80) expressed the full-length hBRCA2 gene (Figure 3A and data not shown). Importantly, BAC-derived hBRCA2 mRNA expression levels in the different cell lines were comparable for all analyzed variants.
Figure 3. RNA and protein expression of hBRCA2 variants in mES cells.
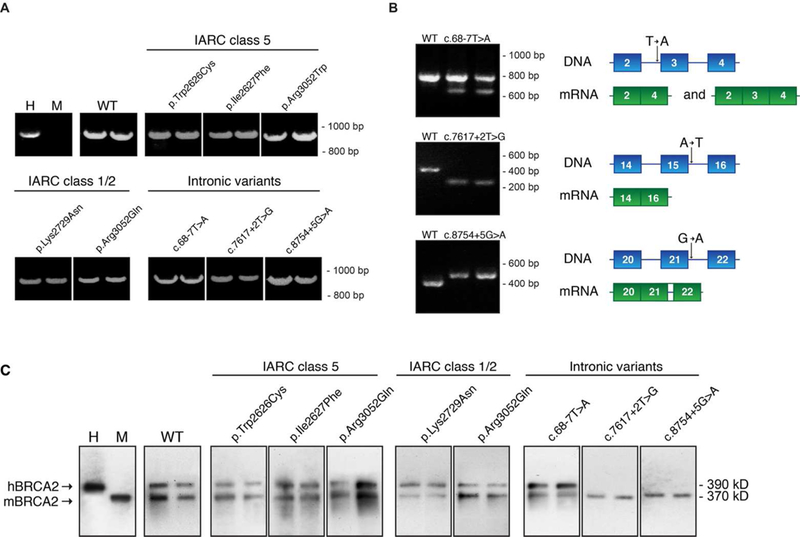
(A) RNA expression of wild type hBRCA2 (WT) and various missense and intronic variants of hBRCA2 in mES cells was determined by cDNA synthesis and PCR analysis. Primers for RNA analysis amplify exons 22–27 of the human BRCA2 gene. For each generated VUS, two independent cell clones were analyzed. hBRCA2 expression in human TK6 lymphoblast cells (H) and mouse ES cells (M) was determined as control. (B) For three intronic variants, RNA splicing was analyzed in detail by PCR using primers spanning the exons that were previously shown to be affected by the variants. Two independent ES cell clones were used for analysis and results were compared to that of a clone expressing wild type hBRCA2. A schematic representation of the observed splice defect is shown on the right panel. (C) Protein expression of wild type hBRCA2 (WT) and various missense and intronic variants of hBRCA2 in mES cells was determined by Western blot analysis. Lysates from human TK6 (H) or mES (M) cells were used as controls for human and mouse BRCA2 expression, respectively
As a test and demonstration of the advantages of BACS compared to cDNA constructs, we generated three constructs containing hBRCA2 intronic variants with a previously demonstrated effect on RNA splicing in primary patient fibroblasts (Vreeswijk et al. 2009). RNA splicing was analyzed by PCR using primers spanning the exons known to be affected by the variants. In accordance with the effects on RNA splicing observed in human BRCA2 carrier fibroblasts, partial skipping of exon 3 leading to an in-frame deletion (c.68–7T>A), complete loss of exon 15 (c.7617+2T>G) and retention of 46 nucleotides of intron 21 for c.8754+5G>A was observed in mES cells (Figure 3B). The concordance in RNA splicing patterns in mouse and human cells of hBRCA2 gene variants illustrates the usefulness of BAC-based functional testing for VUS effects on RNA splicing. Protein expression levels of hBRCA2 and mBRCA2 were similar for hBRCA2 missense variants and the intronic variant c.68–7T>A (Figure 3C). As expected, the variants in which aberrant RNA splicing resulted in an alteration of the reading frame during translation, i.e. c.7617+2T>G and c.8754+5G>A, completely lacked expression of the full-length hBRCA2 protein, congruent with the absence of wild type hBRCA2 RNA transcript.
Variant complementation and functionality in mBrca2-deficient mES cells
The functionality of generated hBRCA2 variants was determined by assessing their ability to complement mBrca2 deficiency. The conditional mBrca2 allele of mBRCA2−/loxP conditional knockout mES cells was excised from the genome by transient expression of the Cre recombinase. This excision concurrently leads to the restoration of an HPRT minigene, allowing cells to grow in the presence of HAT (Supp. Figure S1) (Kuznetsov et al. 2008). In agreement with their established functional impairment, none of the pathogenic hBRCA2 variants (p.Trp2626Cys, p.Ile2627Phe, p.Thr2722Arg, p.Asp2723His; p.Asp2723Gly, p.Gly2748Asp and p.Arg3052Trp, c.7617+2T>G, c.8754+5G>A) could complement the lethality of mBrca2-deficient mES cells, as shown by the absence of HATr clones or only low numbers of small, non-viable clones (Figure 4A; Table 1). In contrast, expression of a non-pathogenic variants (p.Lys2729Asn and p.Ala2770Thr) or a variant for which an effect on protein function is unlikely (c.68–7T>A) resulted in numbers of HAT resistant clones similar to those of wild type hBRCA2 expressing mES cells.
Figure 4. Functional analysis of hBRCA2 variants.
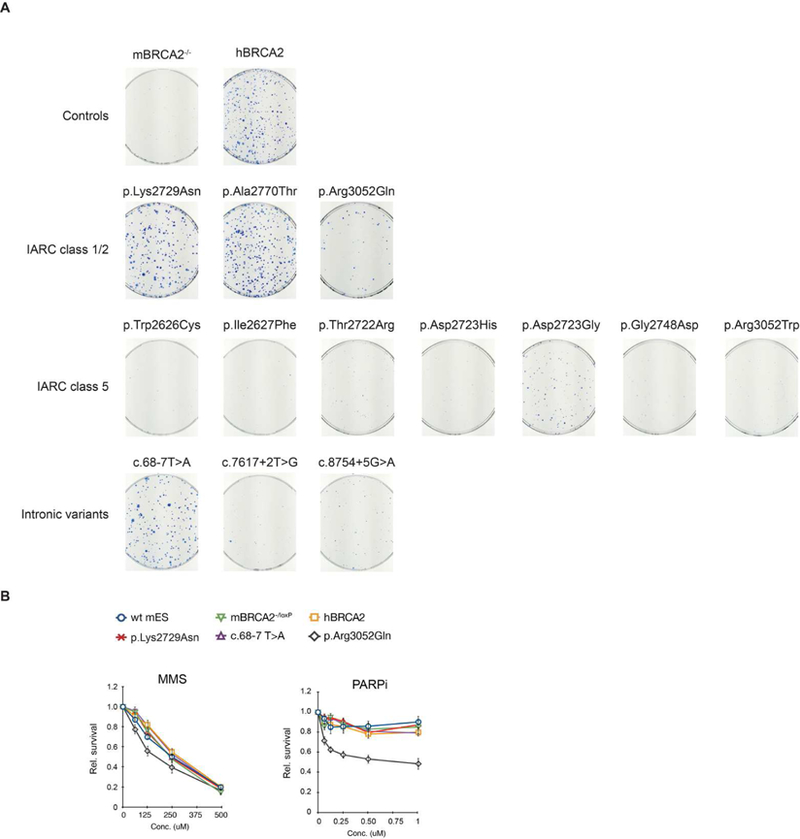
(A) Functionality of the hBRCA2 variants was determined by complementation of mBrca-deficient mES cells. mBrca2 conditional knockout mES cell lines expressing different hBRCA2 variants were transiently transfected with the Cre recombinase and selected for 10 days with HAT for successful deletion of the mBrca2 allele. (B) Sensitivity of mES cells expressing hBRCA2 variants p.Lys2729Asn, c.68–7T>A and p.Arg3052Gln. Wild type, mBrca2-/loxP conditional knockout and hBRCA2 complemented mES cells were used as control. Cell survival was determined by cell count after 24h exposure to the DNA damaging agent MMS and the PARP inhibitor KU-0058948.
To further establish the extent of functional complementation, we determined the sensitivity of mBRCA2-deficient cells complemented with either wild type hBRCA2 or the two hBRCA2 variants that complemented the lethality (p.Lys2729Asn, c.68–7T>A) to the DNA damaging agents MMS and MMC, cisplatin and to a synthetic PARP inhibitor. PARP inhibitors are currently in clinical trials as treatments for breast, ovary, and prostate cancers in BRCA1 and BRCA2 mutation carriers (https://clinicaltrials.gov/). The wild type hBRCA2-expressing mES cells showed similar sensitivity to DNA damage as wild type (IB10) and mBrca2-/loxP mES cells (Figure 4B, Supp. Figure S2A–B), indicating full functional complementation of mBrca2-deficient mES cells. In addition, mBrca2-deficient mES cells that were complemented with the IARC class 1/2 hBRCA variants displayed comparable sensitivity to DNA damaging agents and PARP inhibition as wild type hBRCA2-expressing mES cells (Figure 4B and Supp. Figure S2A–B).
The hBRCA2 variant p.Arg3052Gln could only partly complement the lethality of mBRCA2-deficient cells (Figure 4A). It not only showed increased cellular sensitivity to the DNA damaging agent MMS but also to a PARP inhibitor, in agreement with the reduced HR-efficiency of p.Arg3052Gln described by Kuznetsov et al. (2008) (Figure 4B). No difference in sensitivity to cross linking agents MMC and cisplatin was observed for this variant (Supp. Figure S2A–B).
Since BRCA2 is essential to the loading of RAD51 proteins on the single-stranded DNA required for DNA strand invasion during homologous recombination (HR) repair of DSBs, we assessed the ability of hBRCA2-complemented mES cells to form RAD51 foci in response to exposure to ionizing radiation (IR). Induction of IR-induced RAD51 foci per cell were similar for wild type mES cells and cells expressing non-pathogenic variants, p.Lys2729Asn or c.68–7 T>A hBRCA2. In agreement with reduced complementation and increased sensitivity to the PARP inhibitor, the variant p.Arg3052Gln showed reduced RAD51 foci formation after IR (Supp. Figure S2C).
Discussion
The increased genetic screening of disease-related genes has created a demand for reliable methods and assays that permit functional analysis of the rapidly increasing number of ‘variants of uncertain significance’. Current cDNA-based complementation assays often show overexpression of the investigated variant and lack the ability to test variants that may affect RNA splicing. In the present study we adopted various state-of-the-art molecular techniques that permit the efficient manipulation of a full-length human gene inserted into a bacterial artificial chromosome. The biological advantages of this system have already been clearly demonstrated by Kuznetsov and colleagues (Kuznetsov et al. 2008). However, as the original protocol included several highly inefficient steps that precluded its wide adoption by the research and clinical genetics community, we have now introduced a series of modifications to the original procedures.
We designed a positive-negative selection system in E. coli that involves insertion and positive selection of a selectable cassette at the VUS target site of interest. This cassette, containing a negatively selectable marker, can then be used to assess the integration by recombineering of VUS-containing oligos at the marker-cassette site. Loss of the cassette and subsequent sensitivity to streptomycin indicates successful integration of the VUS oligo. Compared to the labor intensive, hit-and miss approach in the original protocol, this procedure dramatically increases the identification of true recombinants. In addition to the introduction of VUS directly into a BAC in E. coli, we explored the in vitro generation of VUS as modified exons. This method has the advantage that it requires the generation of only a single recombineering construct for each exon, which can then be repeatedly modified using simple and rapid methods as needs demand. Using this method, a complete hBRCA2 exon was first replaced by an rpsL-Amp cassette. Variants in an exon were then generated by conventional site-directed mutagenesis in a plasmid library containing the hBRCA2 exons. Finally, the rpsL-Amp cassette in the BAC was replaced by the modified hBRCA2 exon in one round of BAC recombineering. An additional benefit of this approach is that the generation of variants does not require long, expensive synthetic oligonucleotides.
A third major innovation was the altered regulation of a mammalian selectable marker in such a way that it strongly selects for clones that express full-length mRNA. In fact, not only was full-length RNA recovered, in 60–70% of the clones investigated hBRCA2 mRNA and protein levels closely resembled expression of endogenous mBrca2.
The original mES BRCA2 functional assay allowed testing of 3–5 BRCA2 variants in 3 months, including five to seven weeks for the generation of mES cells expressing the hBRCA2 variants (Kuznetsov et al. 2008). Our improved procedures allow generation of eight to ten variants simultaneously. The current pipeline allows generation of two or three sets of variants side by side, permitting functional testing of 20–30 hBRCA2 variants within approximately 3 months (for details see Figure 5). The exon-swapping method in which the rpsl-Amp cassette is replaced by a complete exon containing the variant has the potential to yield even greater efficiency benefits. Once BAC constructs in which each exon is replaced by an rpsl-Amp cassette become available (n=27 in the case of BRCA2), a simple site-directed mutagenesis PCR and replacement of the rpsl-Amp cassette with the modified exon will suffice to generate a BAC with the desired variant. Taken together, our improvements to the original method increase throughput approximately 10-fold.
Figure 5:
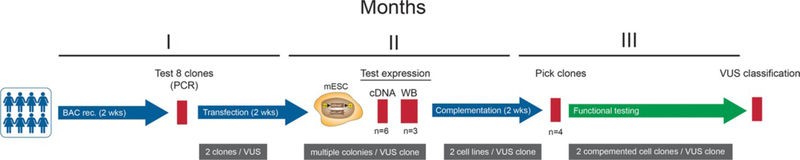
Pipeline for the generation and functional analysis of human BRCA2 variants. Eight to ten hBRCA2 variants are generated simultaneously by BAC recombineering. For each variant two independent clones are transfected into mES cells and for both BAC clones various mESC colonies are analyzed for RNA (n=6) and protein (n=3) expression. Two independent mES cell lines for each BAC clone are used for complementation testing. In case of complementation of mBRCA2-deficient mES cells, two complemented cell lines per BAC clone are used for further functional testing.
Assays assessing hBRCA2 function for generated VUS were in concordance with previously described results (supp. Table S2). BACs containing functionally impaired variants (n=9) could not rescue the lethal phenotype of mBrca2-deficient mES cells, whereas variants that do not affect protein function (n=3) showed full complementation, both on the ability to rescue the lethal phenotype of mBrca2-deficiency as well as on the functional level (e.g. sensitivity to MMS and PARP inhibitor). Additionally, the hBRCA2 variant p.Arg3052Gln that has been classified as “likely not pathogenic” (IARC Class 2) based on a multifactorial probability-based model for hBRCA2 (Lindor et al. 2012) but previously showed an intermediate phenotype in a functional assay was created and tested. In agreement with earlier findings, hBRCA2 p.Arg3052Gln showed a reduced ability to rescue the lethal phenotype of mBrca2-deficient mES cells (Kuznetsov et al. 2008). Furthermore, hBRCA2 p.Arg3052Gln expressing cells show increased sensitivity to MMS and PARP inhibitor, indicative of a reduced HR efficiency.
The efficient generation of large numbers of variants in hBRCA2 BACs now allows us to generate a large validation panel consisting of hBRCA2 variants (both pathogenic and non-pathogenic) for which the clinical significance has already been established. These variants can be used to establish the sensitivity, specificity, positive and negative predictive value of a given test, allowing reliable functional data on new variants to be generated and integrated into a multifactorial likelihood model. This model can then be used to generate an overall likelihood that a given variant is pathogenic or non-pathogenic with respect to cancer risk (Iversen et al. 2011).
Due to an exponential increase in the use of multigene panel testing, exome and whole-genome sequencing for DNA diagnostic purposes, the number of VUS identified in disease-predisposing genes is expected to increase strongly in the near future (Kingsmore and Saunders 2011). Since family-based clinical analyses will often be insufficient to make clinically meaningful inferences about the associated cancer risks of rare variants, the clinical interpretation is expected to rely significantly on functional testing. Validated functional assays will allow rapid and reliable classification of VUS and will facilitate informed clinical decision-making regarding cancer screening and targeted therapy.
Supplementary Material
Acknowledgements
We thank colleagues at the departments of Toxicogenetics and Human Genetics at the LUMC for support and ideas, Dr. I. Poser for help with the Red/ET BAC recombineering, C. Breukel, Dept. of Human Genetics, Leiden University Medical Center for the NLS-Cre plasmid, Dr. J. Zhu, Dept. of Cellular and Molecular Physiology, Pennsylvania State University for the pSK-rpsL-kana plasmid and Prof. R. Kanaar for the RAD51 antibody. This work was financially supported by the Dutch Cancer Society KWF (UL2012-5649).
References
- Antoniou A, Pharoah PDP, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, Pasini B, Radice P, et al. 2003. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am. J. Hum. Genet 72: 1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou AC, Cunningham AP, Peto J, Evans DG, Lalloo F, Narod SA, Risch HA, Eyfjord JE, Hopper JL, Southey MC, Olsson H, Johannsson O, et al. 2008. The BOADICEA model of genetic susceptibility to breast and ovarian cancers: updates and extensions. Br J Cancer 98: 1457–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, van der Gulden H, van der Heijden I, Drost R, Klijn CN, Prasetyanti P, Pieterse M, Wientjens E, Seibler J, Hogervorst FBL, Jonkers J. 2013. A high-throughput functional complementation assay for classification of BRCA1 missense variants. Cancer Discov 3: 1142–1155. [DOI] [PubMed] [Google Scholar]
- Chang S, Biswas K, Martin BK, Stauffer S, Sharan SK. 2009. Expression of human BRCA1 variants in mouse ES cells allows functional analysis of BRCA1 mutations. J Clin Invest 119: 3160–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidugli L, Carreira A, Caputo SM, Ehlen A, Galli A, Monteiro ANA, Neuhausen SL, Hansen TVO, Couch FJ, Vreeswijk MPG, ENIGMA consortium. 2014. Functional Assays for Analysis of Variants of Uncertain Significance in BRCA2. Hum Mutat 35: 151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks G, Atallah M, Morolli B, Calléja F, Ras-Verloop N, Huijskens I, Raamsman M, van de Water B, Vrieling H. 2012. The ToxTracker assay: novel GFP reporter systems that provide mechanistic insight into the genotoxic properties of chemicals. Toxicol. Sci 125: 285–298. [DOI] [PubMed] [Google Scholar]
- Iversen ES Jr, Couch FJ, Goldgar DE, Tavtigian SV, Monteiro AN. 2011. A computational method to classifiy variants of uncertain significance using functional assay data with application to BRCA1. Cancer Epidemiol Biomarkers Prev 20:1078–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsmore SF, Saunders CJ. 2011. Deep sequencing of patient genomes for disease diagnosis: when will it become routine? Sci Transl Med 3: 87ps23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov SG, Liu P, Sharan SK. 2008. Mouse embryonic stem cell-based functional assay to evaluate mutations in BRCA2. Nat. Med 14: 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindor NM, Guidugli L, Wang X, Vallée MP, Monteiro ANA, Tavtigian S, Goldgar DE, Couch FJ. 2012. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum Mutat 33: 8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavaddat N, Antoniou AC, Easton DF, Garcia-Closas M. 2010. Genetic susceptibility to breast cancer. Mol Oncol 4: 174–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchor L, Benítez J. 2013. The complex genetic landscape of familial breast cancer. Hum. Genet [DOI] [PubMed]
- Poser I, Sarov M, Hutchins JRA, Hériché J-K, Toyoda Y, Pozniakovsky A, Weigl D, Nitzsche A, Hegemann B, Bird AW, Pelletier L, Kittler R, et al. 2008. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat Methods 5: 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurdle AB, Healey S, Devereau A, Hogervorst FBL, Monteiro ANA, Nathanson KL, Radice P, Stoppa-Lyonnet D, Tavtigian S, Wappenschmidt B, Couch FJ, Goldgar DE, et al. 2012. ENIGMA--evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum Mutat 33: 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreeswijk MPG, Kraan JN, van der Klift HM, Vink GR, Cornelisse CJ, Wijnen JT, Bakker E, van Asperen CJ, Devilee P. 2009. Intronic variants in BRCA1 and BRCA2 that affect RNA splicing can be reliably selected by splice-site prediction programs. Hum Mutat 30: 107–114. [DOI] [PubMed] [Google Scholar]
- Vrouwe MG, Pines A, Overmeer RM, Hanada K, Mullenders LHF. 2011. UV-induced photolesions elicit ATR-kinase-dependent signaling in non-cycling cells through nucleotide excision repair-dependent and -independent pathways. J Cell Sci 124: 435–446. [DOI] [PubMed] [Google Scholar]
- Wang S, Zhao Y, Leiby M, Zhu J. 2009. A new positive/negative selection scheme for precise BAC recombineering. Mol Biotechnol 42: 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Sharan SK. 2003. A simple two-step, “hit and fix” method to generate subtle mutations in BACs using short denatured PCR fragments. Nucleic Acids Res 31: e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.