

Abstract
Anti-PD-1-based immunotherapy has had a major impact on cancer treatment but has only benefited a subset of patients. Among the variables that could contribute to interpatient heterogeneity is differential composition of the patients’ microbiome, which has been shown to affect antitumor immunity and immunotherapy efficacy in preclinical mouse models. We analyzed baseline stool samples from metastatic melanoma patients before immunotherapy treatment, through an integration of 16S ribosomal RNA gene sequencing, metagenomic shotgun sequencing, and quantitative polymerase chain reaction for selected bacteria. A significant association was observed between commensal microbial composition and clinical response. Bacterial species more abundant in responders included Bifidobacterium longum, Collinsella aerofaciens, and Enterococcus faecium. Reconstitution of germ-free mice with fecal material from responding patients could lead to improved tumor control, augmented T cell responses, and greater efficacy of anti-PD-L1 therapy. Our results suggest that the commensal microbiome may have a mechanistic impact on antitumor immunity in human cancer patients.
The therapeutic efficacy of immunotherapies targeting the PD-1/PD-L1 interaction is favored in patients who show evidence of a T cell-inflamed tumor microenvironment at baseline (1,2). Therefore, host and tumor factors that regulate the magnitude of endogenous immune priming and T cell infiltration into the tumor microenvironment are being sought as an opportunity to further expand therapeutic efficacy (3). Preclinical studies have indicated that the composition of the commensal microbiome could exert a major influence; mice with favorable microbiota showed far greater therapeutic activity of anti–PD-L1 treatment than did mice with an unfavorable microbiome, and this benefit could be transferred by cohousing or fecal transplant (4). These observations prompted an analogous analysis of the human microbiome with respect to therapeutic efficacy of anti–PD-1 in cancer patients.
To evaluate whether commensal bacterial composition might be associated with clinical efficacy of PD-1 blockade immunotherapy, stool samples were collected from 42 patients before treatment as part of a multidimensional biomarker analysis in metastatic melanoma. The majority of patients received an anti–PD-1 regimen; four patients received anti–CTLA-4 treatment, but the downstream data conclusions did not change with the removal of these subjects, so they were retained in the analysis. Clinical response rate was determined in a blinded manner from biomarker results by using Response Evaluation Criteria In Solid Tumors (RECIST) version 1.1. There were 16 responders (from here on, referred to as R) and 26 nonresponders (NR), yielding a response rate of 38%, which is in line with published clinical data of anti–PD-1 therapy in metastatic melanoma patients (5, 6). No major differences in patient characteristics were observed in R versus NR, except a borderline difference in prior (but not current) smoking history (table S1).
To determine whether the composition of the commensal microbiota is associated with clinical response, we integrated three methods for DNA sequence-based bacterial identification (fig. S1A). First, using 16S ribosomal RNA (rRNA) gene amplicon sequencing, we identified operational taxonomic units (OTUs) with taxonomic assignment present at different abundance in R versus NR (table S2). We used a Basic Local Alignment Search Tool (BLAST) search of the 16S sequences against the National Center for Biotechnology Information (NCBI) database to reveal potential species-level identities (table S3). Further level of confidence in species identification was gained by matching the OTUs from the 16S data set to species-level identities revealed through metagenomic shotgun sequencing (table S4). We used species-specific quantitative polymerase chain reaction (PCR) for those candidate species having previously validated primers (table S5). Compared with the 16S analysis, the metagenomic sequencing yielded a smaller number of species differentially represented in R versus NR, which overlapped with the 16S results (table S6). Treating these assays as a screen for maximizing the number of candidate species, we used the 16S sequencing method as a starting point in our analysis.
After removing OTUs present in less than 10% of the samples, the 16S sequencing revealed 62 OTUs of different abundance in R versus NR [P < 0.05, unadjusted, permutation test with Quantitative Insights Into Microbial Ecology (QIIME)] (table S2). Hierarchical clustering of samples based on relative abundance of these OTUs revealed that most patients were accurately grouped according to clinical response (fig. S2). Clustering of patients within each clinical group is depicted in Fig. 1A. Thirty-nine OTUs were more abundant in R, and 23 were more abundant in NR. One Bifidobacteriaceae OTU was significantly more abundant in R, and a second Bifidobacteriaceae OTU (559527) had borderline significance (P = 0.058, unadjusted) and was included in the analyses (total = 63 OTUs). This observation recapitulates our previous findings that associated Bifidobacteriaceae family members with improved immune-mediated tumor control and efficacy of anti–PD-L1 therapy in mice (4). A principal component analysis (PCA) of the 63 OTUs revealed separation of R from NR (Fig. 1B).
Fig. 1. Distinct commensal microbial communities in anti–PD-1 responding patients and nonresponding patients as assessed with 16S rRNA gene amplicon sequencing.
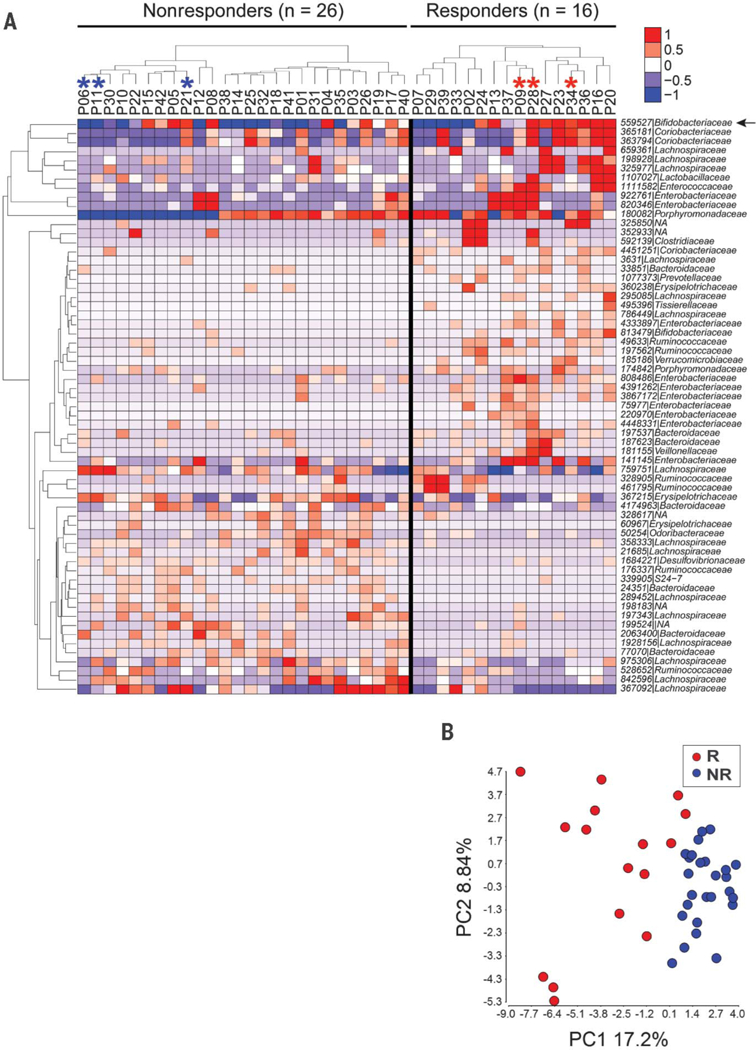
(A) Relative abundance of differentially abundant taxa in responders versus non-responders; 62 OTUs were identified as different with P < 0.05 (unadjusted, permutation test). An additional OTU 559527 (arrow) identified as Bifidobacteriaceae approached significance (P = 0.058). Hierarchical clustering of the samples was performed within each clinical group. Individual samples are organized in columns, labeled with patient identification number. Asterisks indicate samples used in further in vivo experiments. The ID of de novo assembled OTUs (new clean-up reference OTUs picked with QIIME) were abbreviated to show only the individual identifier digits, and the full OTU IDs are provided in table S4. (B) PCA of relative abundance of the 63 OTUs shown in Fig. 1A.
A BLAST search of the 63 OTUs against the NCBI database of bacterial sequences returned for most OTUs multiple species with ≥98% identity (table S3). To gain more accurate species-level characterization, the same samples were subjected to metagenomic shotgun sequencing (available for 39 of the 42 samples). Illumina pairedend reads were assigned to microbial clades and analyzed for closest matches to the 63 OTUs identified with 16S sequencing. Potential species matches were identified for 43 of the original 63 OTUs (table S4). Species-specific quantitative PCR assays were performed as an additional approach to assess the identity of species, for which sufficiently validated quantitative PCR primers were available (table S5). Thus, integration of the three methods led to the selection of 10 species differentially enriched in R versus NR. Eight of these were more abundant in R—Enterococcus faecium, Collinsella aerofaciens, Bifidobacterium adolescentis, Klebsiella pneumoniae, Veillonella parvula, Parabacteroides merdae, Lactobacillus sp., and Bifidobacterium longum—whereas two were more abundant in NR: Ruminococcus obeum and Roseburia intestinalis. As an example, the integrative analysis for B. longum (OTU 559627) is depicted in Fig. 2, A to C. Similar correlation analyses for the remaining nine species are depicted in figs. S3 and S4. Quantitative PCR results for these 10 species were integrated into a summation quantitative PCR score for each patient, which was significantly higher in responders (P = 0.004) (Fig. 2D).
Fig. 2. Identification of commensal bacterial species associated with patient clinical response to anti–PD-1 therapy.
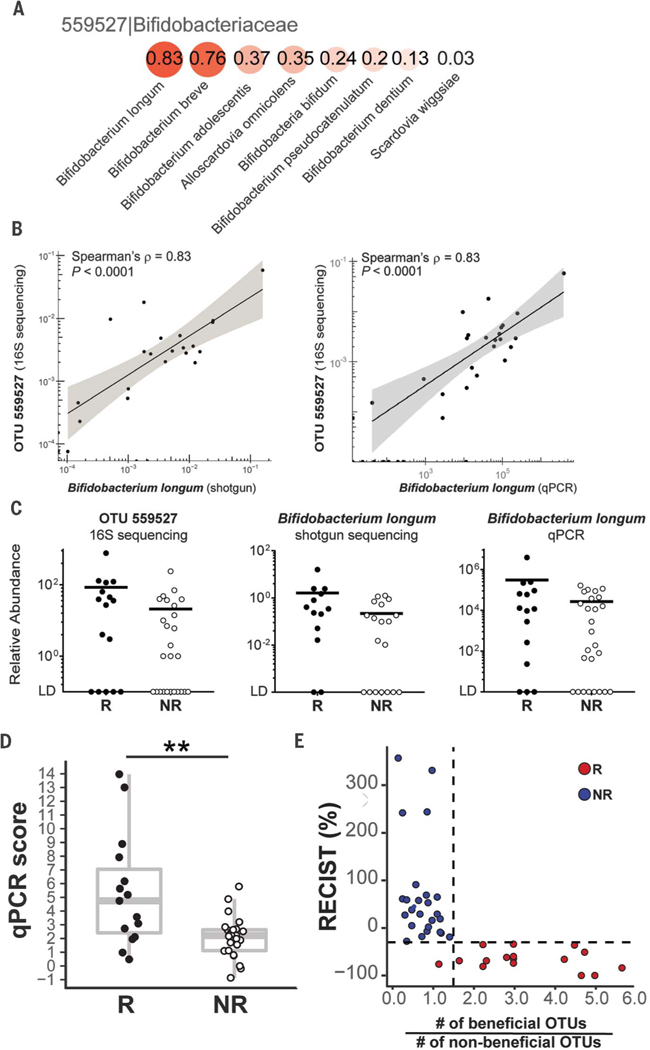
(A) Spearman’s correlation coefficients between the relative abundances of Bifidobacteriaceae OTU 559527 from the 16S data set and species-level identities suggested by shotgun sequencing. The species profiled with shotgun sequencing were compared with the taxonomy of OTUs generated from 16S sequencing at the family level. (B) Spearman’s correlation between abundance of OTU 559527 from the 16S data set and B. longum identified by means of metagenomics shotgun sequencing analysis (left) and quantitative PCR (right). Shaded band indicates 95% confidence interval (CI) of the values fitted by linear regression. (C) Relative abundance in responders (R) versus nonresponders (NR) of OTU 559527 (16S sequencing; left), B. longum (shotgun sequencing; middle), and B. longum (quantitative PCR; right). LD, limit of detection. (D) Quantitative PCR score representing aggregate data for the relative abundances of 10 species correlated to OTUs with differential abundance in R versus NR. Wilcoxon-Mann-Whitney test (nonparametric) was used to compare quantitative PCR score between R and NR groups. (E) Ratio of beneficial to nonbeneficial OTU numbers for each patient versus the patient’s RECIST aggregate tumor measurement change. Dashed lines label RECIST% = −30 and ratio = 1.5. Only the 43 16S OTUs confirmed with shotgun metagenomic sequencing were included. P < 0.05 was considered statistically significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
This list of species is likely an underestimate of the total number of entities showing differential abundance in R versus NR because of the stringency of this composite analysis. For example, Akkermansia muciniphila OTU (185186), in line with the study of anti–PD-1 efficacy in epithelial cancers by Routy et al. (7), was detected by means of 16S sequencing in four patients, and all were responders, but statistical analysis of the entire cohort is limited by the number of samples above the detection threshold. As an alternative way to represent the aggregate data toward development of a candidate predictive biomarker, the total numbers of potentially “beneficial” and “non-beneficial” OTUs were scored for each patient (fig. S5), and a ratio was calculated. When plotted against the absolute change in tumor size as assessed with RECIST, a clean correlation was observed so that patients with a ratio over 1.5 all showed clinical response (Fig. 2E). These results suggest that the commensal microbiota composition might be useful as a biomarker to predict response to checkpoint blockade therapy, which motivated comparison with other candidate predictive biomarkers. Archived pretreatment tumor specimens that passed quality control were available for 15 patients (5 R, 10 NR). Microbial composition remained significantly different in R versus NR for this subset (P < 0.01) (fig. S6, A and B). Exome sequencing followed by enumeration of nonsynonymous somatic mutations showed a trend of higher frequency in R, as did levels of PD-L1 and PD-1 mRNA (fig. S6, C to E) and enumeration of baseline CD8+ T cells by means of immunohistochemistry (fig. S6F). Although these trends not reaching statistical significance level of 0.05 were likely limited by sample size, the microbiota parameters still markedly separated responders and nonresponders.
The strong correlation between commensal bacteria and clinical response to immunotherapy suggested a potential causal effect, in light of data demonstrating an immune-potentiating impact of the microbiome in mouse tumor models (4,8–10). To investigate the capability of human commensal microbes to potentiate antitumor T cell responses, we used germ-free (GF) mice as recipients. We had previously reported that spontaneous immune-mediated tumor control in Taconic mice could be improved by means of fecal microbiota transfer from mice obtained from a different vendor, the Jackson Laboratories (4). In setting up the current model, we found that B16.SIY melanoma tumor growth in GF mice was similar to that in specific pathogen–free (SPF) mice (both from Taconic), and colonization of GF mice with feces from Taconic SPF mice did not affect this baseline growth rate (fig. S8). These results suggest a reduced spontaneous immune-mediated tumor control inherent to GF mice, which makes them suitable recipients for human-derived microbiota, with an opportunity to detect improved antitumor immunity depending on microbial composition. Fecal material was transferred from three R and three NR into cohorts of GF mice (Fig. 1A and figs. S2, S5, and S7), followed by implantation of B16.SIY melanoma cells 2 weeks later. The human microbiota-colonized mouse groups segregated into two phenotypes with respect to tumor growth rate: (i) a faster growing group and (ii) a slower growing group (Fig. 3A). Two of three mouse cohorts reconstituted with R fecal material had slower baseline tumor growth, and two of the three cohorts reconstituted from NR showed faster baseline tumor growth. Thus, the ability of the human microbiota to support improved tumor control in mice usually, but not always, paralleled the clinical response to anti–PD-1 seen in the donor patient. Achieving slower tumor growth with fecal transplant alone is similar to previous mouse studies, in which transfer of feces from Jackson into Taconic mice was sufficient for a partial therapeutic effect owing to a more favorable microbiome (4).
Fig. 3. Human commensal communities modulate antitumor immunity in a mouse melanoma model.
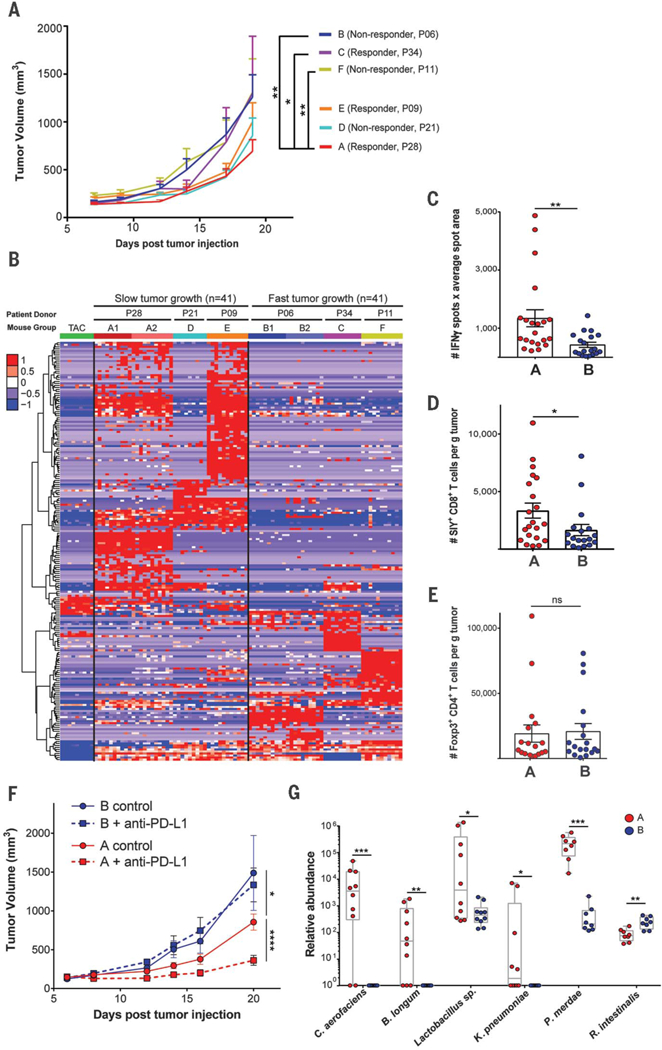
GF mice were gavaged with fecal material from three responder (P28, P34, and P09) and three nonresponder (P06, P21, and P11) patient donors. (A) B16. SIY melanoma was injected subcutaneously 2 weeks after gavage; tumor growth data are from one (groups C, D, E, and F) or two experiments (groups A and B) with 7 to 11 mice per group per experiment. Error bars represent mean + SEM. (B) Relative abundance of 207 OTUs from patient donors that colonized in mice and were differentially abundant between slow-and fast-tumor-growth groups. Columns depict individual mice arranged in groups A through F. Groups A1, B1, A2, and B2 are from two independent duplicate experiments. Rows indicate individual OTUs with exact reference ID match between human and mouse 16S rRNA data sets. (C) In groups A and B, ex vivo activation of splenocytes by SIY peptide was measured with IFN-γ ELISPOT 3 weeks after tumor injection. (D and E) Tumor-infiltrating SIY-specific CD8+ T cells (D) and FoxP3+ regulatory T cells (E) were enumerated with flow cytometry. (F) Efficacy of anti–PD-L1 therapy was determined in groups A and B. Data are from one experiment with 7 or 8 mice per group. (G) Relative abundance in mouse groups A and B of key species validated for quantitative PCR scoring. Six out of the 10 species are shown that gave positive PCR signals. The remaining four species were absent from these particular recipient groups. Tumor growth curves were analyzed with two-way analysis of variance by Tukey’s multiple comparisons post-test; flow cytometry and quantitative PCR data were analyzed by Wilcoxon-Mann-Whitney test (nonparametric). P < 0.05 was considered statistically significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Composition of bacterial taxa that successfully reconstituted mice and fidelity to the original human donor were assessed with 16S rRNA gene amplicon sequencing. Groups C and D, which did not show the same pattern of tumor control as the therapeutic outcome in the original human donors, showed a large degree of difference of microbiota composition from the original human donors (fig. S9). In agreement, a binary Bray-Curtis dissimilarity index for each donor/recipient pair was highest, at 0.7, for cohorts C and D versus 0.5 to 0.6 for the rest of the groups. We conclude that whereas reconstitution of GF mice with human fecal material often recapitulates the microbial composition and the phenotype of the human donor, in some cases there is a high degree of drift so that some bacteria expand and others contract to a degree that is sufficient to change phenotype. Nonetheless, for the reconstituted GF mice that do recapitulate the clinical outcome of the original donor, this model system may be useful for the ultimate isolation of specific bacteria that regulate antitumor immunity in vivo.
We focused on mouse groups A and B for further mechanistic studies. There was a high level of consistency between repeated experiments, both with respect to tumor growth rate and microbial colonization (Fig. 3B, group A1 versus A2 and B1 versus B2 comparisons). To determine whether the difference in tumor control could be attributed to host immunity, interferon-γ (IFN-γ) enzyme-linked immunosorbent spot (ELISPOT) of ex vivo SIY-stimulated splenocytes was performed and indicated an increased frequency of activated T cells from R microbiota-reconstituted mice 3 weeks after inoculation with B16.SIY melanoma cells (Fig. 3C). Analysis of the tumor microenvironment also showed a significantly greater number of SIY–specific CD8+ T cells, but not of FoxP3+CD4+ regulatory T cells, in these mice (Fig. 3, D and E), which is consistent with increased priming of tumor antigen-specific CD8+ T cells. Anti–PD-L1 was markedly efficacious in mice colonized with R microbiota yet completely ineffective in NR-derived mice (Fig. 3F), demonstrating a profound impact of the commensal microbiota on immunotherapy efficacy in vivo. Interrogation of fecal DNA from these mice by means of quantitative PCR recapitulated the results from our analysis of patients. Of the PCR reactions validated in patients, six were observed in re-constituted mice, with the same pattern of enrichment as was seen in patients (Fig. 2G).
Together, our data suggest that the composition of the commensal microbiota in patients is associated with therapeutic efficacy of anti–PD-1 monoclonal antibody (mAb). Although B. longum was one commensal identified in the current study that had also been found in mouse models to be associated with improved immune-mediated tumor control (4), it seems likely that multiple specific bacteria may contribute to improved antitumor immunity in patients. In addition to the panel of bacteria overrepresented in responders, several OTUs were overrepresented in nonresponders, and prior work in mice has indicated that some commensals have the potential to be immune-inhibitory—for example, through the induction of FoxP3+ regulatory T cells (11,12). In addition, our current cohort suggested that a ratio of “beneficial” OTUs to “nonbeneficial” OTUs was the strongest predictor of clinical response. This may indicate that a higher frequency of beneficial bacteria, together with a lower frequency of bacteria with negative impact, may combine for the most favorable clinical outcome.
Several of the bacterial species that were identified in the current study to be differentially abundant in responding versus nonresponding patients have been examined previously for mechanistic impact on host immune responses in GF mice in vivo (13). Monocolonization with several species found to be at increased frequency in our responders—including E.faecium, C. aerofaciens, B. adolescentis, and P. merdae—were reported to result in a decreased frequency of peripherally derived colonic regulatory T cells as compared with that of other bacterial species. An increased frequency of the Batf3-lineage dendritic cells (DCs) and greater T helper cell 1 (TH1) responses were also found with bacteria currently identified to be more abundant in responders (13). Decreased regulatory T cells, increased Batf3 DCs, and augmented TH1responses would all be expected to improve immune-mediated tumor control. Although care should be taken extrapolating these results in the setting of a complex microbiota, the data suggest that patient responder-associated bacteria may have a distinct effect on innate and adaptive immunity both locally and systemically.
Two additional studies have identified an association of the commensal microbiome with anti–PD-1 mAb efficacy in different solid cancers (7,14). Our approaches differ in the method of segregation of patients between R and NR groups and in some methods of analysis. Therefore, direct comparison of the differentially enriched commensal microbiota in R and NR patients across publications should be addressed with caution. Nonetheless, there is agreement with respect to a mechanistic impact of the microbiome on efficacy of anti–PD-1 immunotherapy because fecal transfer from R versus NR patients to GF mice was also able to recapitulate the patient phenotype.
In addition to the microbiome, it is apparent that additional tumor and host factors can affect the efficacy of antitumor immunity and cancer immunotherapy. Tumor-intrinsic activation of the Wnt/β-catenin pathway (15) and deletion or mutation of Pten (16) have been shown to lead to deficient T cell infiltration into the tumor microenvironment and resistance to checkpoint blockade immunotherapy. Germline polymorphisms in immune regulatory genes also have the potential to influence the magnitude of spontaneous anti-tumor T cell responses (17). Our results described here open the avenue for integrating commensal microbial composition, along with tumor genomics and germline genetics, into a multiparameter model with which to maximize the ability of predicting which patients are likely to respond to immunotherapies such as anti–PD-1.
Supplementary Material
ACKNOWLEDGMENTS
We thank E. Chang, A. Sivan, C. Nagler, L. Huang, P. Bleda-Ferre, N. Hubert, Z. Early, and B. Theriault for helpful discussions and M. Jarsulic, B. Choy, N. Martinec, S. Owens, S. Greenwald, H. Morrison, and J. Polinski for technical assistance. This work was supported by NlH grant R35 CA210098, a Team Science Award from the Melanoma Research Alliance, the American Cancer Society-Jules L. Plangere Jr. Family Foundation Professorship in Cancer Immunotherapy, funds from the University of Chicago Medicine Comprehensive Cancer Center, the University of Chicago Cancer Biology Training program (T32 CA009594), and The Center for Research Informatics of The University of Chicago Biological Science Division. The bioinformatics analysis was performed on Gardner High-Performance Computing clusters at Center for Research Informatics, Biological Sciences Division. Data reported in this study are tabulated in the main text and supplementary materials. The 16S and shotgun sequencing were performed at the Argonne National Laboratory and the University of Chicago-affiliated Marine Biological Laboratory, respectively; data files were deposited into The NCBI Sequence Read Archive (SRA) and are available under the accession no. SRP116709. Custom code and additional processed data used in this study are publicly available on GitHub at https://github.com/cribioinfo/sci2017_analysis. T.F.G. is an advisory board member for Roche-Genentech, Merck, Abbvie, Bayer, Aduro, and Fog Pharma. T.F.G. receives research support from Roche-Genentech, BMS, Merck, Incyte, Seattle Genetics, and Ono. T.F.G. is a shareholder/cofounder of Jounce Therapeutics. The University of Chicago holds a licensing arrangement with Evelo. T.F.G is an inventor on U.S. patent US20160354416 A1 submitted by the University of Chicago that covers the use the microbiota to improve cancer immunotherapy. J.J.L. is a consultant to and receives research funding from Bristol-Myers Squibb and Merck.
REFERENCES AND NOTES
- 1.Tumeh PC et al. , Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ayers M et al. , J. Clin. Invest. 127, 2930–2940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corrales L, Matson V, Flood B, Spranger S, Gajewski TF, Cell Res. 27, 96–108 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sivan A et al. , Science 350, 1084–1089 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topalian SL et al. , N. Engl. J. Med. 366, 2443–2454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robert C et al. , N. Engl. J. Med. 372, 2521–2532 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Routy B et al. , Science 359, 91–97 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Daillère R et al. , Immunity 45, 931–943 (2016). [DOI] [PubMed] [Google Scholar]
- 9.lida N et al. , Science 342, 967–970 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vètizou M et al. , Science 350, 1079–1084 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atarashi K et al. , Science 331, 337–341 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Round JL, Mazmanian SK, Proc. Natl. Acad. Sci. U.S.A. 107, 12204–12209 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geva-Zatorsky N et al. , Cell 168, 928–943.e11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gopalakrishnan V et al. , Science eaan4236 (2017). [Google Scholar]
- 15.Spranger S, Bao R, Gajewski TF, Nature 523, 231–235 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Peng W et al. , Cancer Discov. 6, 202–216 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ugurel S et al. , Cancer Immunol. Immunother. 57, 685–691 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.