

Abstract
Genomic Medicine, using DNA variation to individualize and improve human health, is the subject of this series of reviews. The idea that genetic variation can be used to individualize drug therapy – the topic addressed here – is often viewed as “low-hanging fruit” for Genomic Medicine. We review general mechanisms underlying variability in drug action, the role of genetic variation in mediating beneficial and adverse effects through variable drug concentrations (pharmacokinetics) and drug actions (pharmacodynamics), available data from clinical trials, and ongoing efforts to implement pharmacogenetics in clinical practice.
Introduction
It is a tenet of clinical medicine that patients vary in their response to drugs: doses effective in some patients will inevitably be ineffective or cause adverse drug reactions (ADRs) in others. Indeed, ADRs have been implicated as an important cause of hospital admissions, in one series accounting for 6.5% of all hospitalizations in two large UK hospitals.1 In the 1990s, a large survey suggested that ADRs occurring in hospital were the 4th-6th leading cause of in-hospital mortality in the US,2 and a follow up survey in 2010 showed no improvement.3 Fewer data are available on the consequences of the lack of efficacy, beyond recognizing that only a portion of a given patient population derives benefit from a given medication. The treatment of common diseases, such as hypertension, arrhythmias, or depression often involves a series of “therapeutic trials” among different drugs or different classes of drugs, and the healthcare burden imposed by lack of efficacy during these periods of trial-and-error may be considerable. It has been speculated, for example, that ineffective antidepressant therapy may increase risk for suicide.4
There are many reasons for variability in drug response. The failure of selected drug therapy to target the underlying disease mechanism (which may or may not be known), drug interactions, disease-related changes in drug concentrations or responsiveness, poor compliance, and system errors such as failure to deliver the correct drug or dose to the patient are commonly cited. In some instances, therapeutic non-responsiveness and ADRs vary by race/ethnicity and can contribute to disparities in clinical outcomes.5,6 This review will address how variation in the germline genome affects drug response. Tumor sequencing, identification of driver mutations, and implementation of mutation-specific therapy which are having a major impact in cancer have been reviewed in detail elsewhere and will not be addressed further here.7
Mechanisms underlying variable drug responses
Sir Archibald Garrod, who developed the concept of “inborn errors of metabolism”, speculated a century ago8 that aberrant metabolism of exogenous substances could account for unusual reactions to food or drugs. During and after World War II, the first instances of genetically-determined ADRs were described, including hemolytic anemia in African-American soldiers with G6PD deficiency exposed to antimalarials; malignant hyperthermia during anesthesia; and prolonged paralysis following succinylcholine in patients with pseudocholinesterase deficiency. The term pharmacogenetics (see Box 1) was coined by Motulsky9 at the University of Washington and Kalow10 at the University of Toronto.
Box 1. Comments on nomenclature.
The term “pharmacogenetics” was coined in the 1950s and captures the idea that large effect size DNA variants contribute importantly to variable drug actions in an individual. The term “pharmacogenomics” is now used by many to describe the idea that multiple variants across the genome and differing across populations affect drug response. The International Conference on Harmonisation (ICH), a world-wide consortium of regulatory agencies, has defined “pharmacogenomics” as the study of variations of DNA and RNA characteristics as related to drug response, and “pharmacogenetics” as the study of variations in DNA sequence as related to drug response.11
Pharmacogeneticists adopted a “star” nomenclature (e.g. CYP2C19*2) to describe variants in genes (sometimes termed “pharmacogenes”) underlying variability in drug response. Some star alleles may include more than one variant; for example, TPMT*3A designates an allele defined by the presence of two single nucleotide polymorphisms (SNPs), and distinguishing this allele from those carrying only one of the SNPs can be challenging.12 While the star nomenclature persists, as our understanding of the numbers of variants in important pharmacogenes increases, attempts are being made to reconcile the notation with alternate variant nomenclature such as the conventional “rs” designation.13,14 Most variants studied to date partially or completely inhibit function of the encoded protein. Occasionally, variants increase activity of drug-metabolizing enzymes; examples are noted in the text and include CYP2C19*17 and CYP2D6 duplications.
The field is also adopting a standard set of definitions of pharmacogenetic phenotypes; for pharmacokinetic genes these include “normal metabolizers” (NMs), “poor metabolizers” (PMs, carrying two loss-of-function alleles), “intermediate metabolizers” (IMs, carrying one loss-of-function allele), and “ultrarapid metabolizers” (UMs, carrying gain-of-function alleles or gene duplications), and for pharmacodynamic genes, designations such as positive or negative for high risk alleles.15 These are convenient shorthand designations and there is often some overlap in drug response (Figure 1A).
One review suggested that common genetic factors contribute to variable serious ADRs in a third of cases.16 The field of pharmacogenomics aims to define these genetic mechanisms, and ultimately to implement genetic testing to improve drug efficacy and reduce toxicity. Further, an understanding of the genetic basis of variable drug response can be used as a tool to expand the use of existing drugs to new indications and to develop new drugs. Well-recognized examples of genetically-determined variability in drug response, described further below, often involve single DNA variants common in a population, and associated with relatively large effect sizes and relatively clearly definable metabolizer phenotypes (Figure 1A; Box 1). As a result, the implementation of pharmacogenomic information into the clinical flow of medicine has been viewed as “low hanging fruit”. However, a number of barriers are now identified and need to be overcome in order to routinely use pharmacogenomic variant data in improving drug prescribing.
Figure 1:
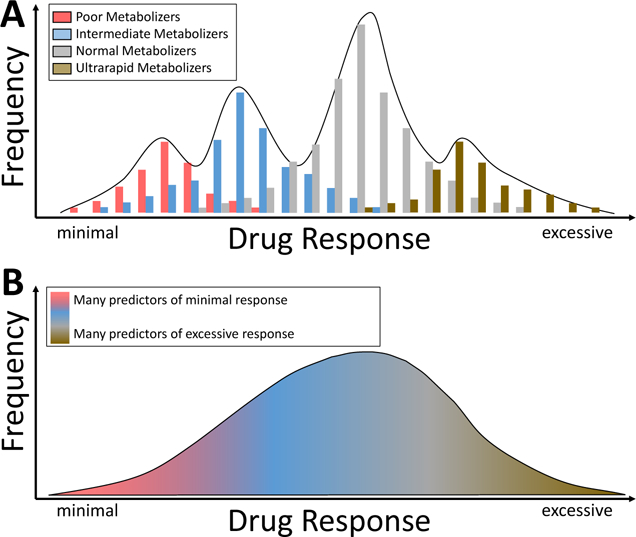
A. In some instances, variants in single genes (often those determining pharmacokinetics, as highlighted in Figure 2) have large effect sizes, and distinct metabolizer phenotypes can be predicted: poor metabolizers with two loss of function alleles, intermediate metabolizers with one functional allele, normal metabolizers with two functional alleles, and ultrarapid metabolizers with duplications or other variants conferring increased metabolic activity. In this situation, distinct genotype-dependent differences in drug response may be seen, although there may still be overlap. B. When variants in many pharmacogenes contribution to variability in drug action, the distribution of drug responses is not polymodal as (A), but rather a continuum.
Two conceptual pathways describe an organism’s overall response to drug exposure. Pharmacokinetics defines variability in the processes (absorption, distribution, metabolism, and elimination) modulating delivery of drug and active metabolites to and removal from their site(s) of action. Pharmacodynamics describes variability in drug action that is not attributable to variable drug concentrations: this can reflect variability in the interaction of active drug with its effector molecules, or other mechanisms such as variability in disease mechanisms. The earliest examples of pharmacogenomic variability involved variability in pharmacodynamic processes. With the development of robust methodologies to measure concentrations of drugs and their metabolites in plasma and other sites in the 1960s and 1970s came the ability to define pharmacokinetic outliers in whom unusually high or low plasma concentrations were associated with variable efficacy or ADRs. This in turn led to studies defining variants in key drug metabolizing or transport genes as the basis for these responses. More recently, agnostic methods such as the genome-wide association study (GWAS) paradigm have validated the role of these candidate genes and have identified new loci associated with variable drug responses.17 The majority of clinically actionable pharmacogenetic traits described to date have a pharmacokinetic basis (Table 1).
Table 1:
Drug-gene pairs with guidelines for use in clinical practice (from the Clinical Pharmacogenetics Implementation Consortium (CPIC) as of spring 2019*)
| Gene | Drugs |
|---|---|
| Pharmacokinetic mechanisms | |
| CYP2B6 | Efavirenz |
| CYP2C19 | Clopidogrel SSRIs, TCAs Voriconazole, proton pump inhibitors* |
| CYP2C9 | Celecoxib* phenytoin warfarin |
| CYP2D6 | codeine, oxycodone, tramadol SSRIs, TCAs ondansetron tamoxifen, atomoxetine* |
| CYP3A5 | tacrolimus |
| DPYD | 5-fluorouracil, capecitabine, tegafur |
| TPMT, NUDT15 | azathioprine, mercaptopurine, thioguanine |
| SLCO1B1 | simvastatin |
| UGT1A1 | atazanavir |
| Pharmacodynamic mechanisms | |
| CFTR | ivacaftor |
| CYP4F2 | warfarin |
| G6PD | rasburicase |
| HLA-B | abacavir allopurinol carbamazepine phenytoin |
| IFNL3 (IL28B) | interferon |
| RYR1, CACNA1S | inhaled anesthetics |
| VKORC1 | warfarin |
Guidelines published or
in process
CPIC: Clinical Pharmacogenetics Implementation Consortium (www.cpicpgx.org)
SSRI: selective serotonin uptake inhibitor
TCA: tricyclic antidepressant
Common genetic variants can produce large drug response effects
Pharmacokinetic variation:
Two scenarios illustrate how single gene variants affecting pharmacokinetics can have especially large effects. The first is with administration of a prodrug, a pharmacologically inactive substance that requires bioactivation by drug metabolism to achieve its therapeutic effects (Figure 2, top). Such bioactivation pathways usually involve a single drug metabolizing enzyme; genetic variants that result in loss-of-function of these enzymes can decrease or block drug action. Examples include codeine bioactivated to its major active metabolite morphine by CYP2D6 and the antiplatelet drug clopidogrel bioactivated by CYP2C19. While these effects are well-established and contribute to the perception that pharmacogenomic variants constitute “low hanging fruit” for implementation, it is important to recognize that there is a spectrum of even these large pharmacogenomic effects. Thus, in the case of clopidogrel, increasing the dose resulted in an antiplatelet effect in heterozygotes for CYP2C19*2 (the terminology for variants is further explained in Box 1), encoding a common loss-of-function variant, because they still have demonstrable CYP2C19 activity. On the other hand, a dosage increase did not generate an antiplatelet effect in individuals homozygous for the variant, because they completely lack CYP2C19 activity.18 A GWAS of clopidogrel inhibition in 429 subjects of ADP-related platelet activation resulted in very strong signals (P~10−13) at the CYP2C19 locus.19 Interestingly, while the pharmacologic effect of CYP2C19*2 is large, the total variability in clopidogrel antiplatelet effect attributable to this variant was only 12%.19 While this is a large effect for a single genetic variant, the finding also emphasizes that other genetic and/or environmental factors play a role in observed variability in clopidogrel drug action.
Figure 2:
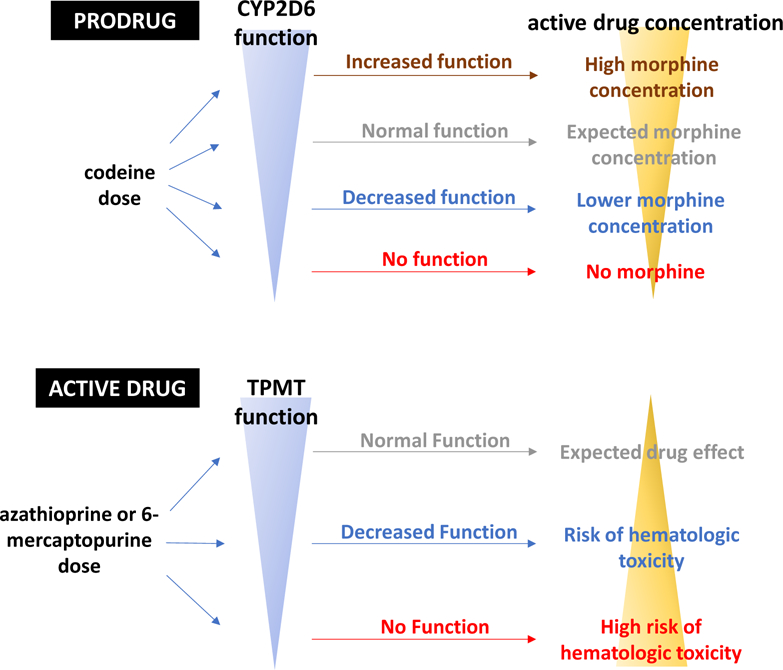
Two scenarios under which single variants in key pharmacokinetic genes can produce very large effects due to variability in active drug concentration. When a prodrug (top) such as codeine requires bioactivation to generate its active metabolite (morphine), increased enzymatic function can lead to morphine toxicity and decreased enzymatic function can lead to decreased analgesia. Similarly, variability in metabolism of an active drug such as azathioprine (bottom) can modulate risk of serious drug toxicity.
Most variants studied to date confer partial or complete loss of function. However, gain-of-function variants in bioactivation pathways have been described and can be associated with excess drug response. Examples include CYP2C19*17, which has been associated with bleeding during clopidogrel therapy20 and CYP2D6 duplications which have been associated with excess narcotic effect including respiratory arrest due to rapid and increased accumulation of morphine during codeine therapy (Figure 2, top).21
The second situation in which single pharmacokinetic variants can exert very large effects is during administration of an active drug with a narrow therapeutic range (i.e. a small margin between therapeutic and toxic doses) which undergoes elimination by a single drug metabolizing system (Figure 2, bottom). The antileukemic drug 6-mercaptoprine is bioinactivated by thiopurine S-methyltransferase (TPMT) and xanthine oxidase (XO). Loss-of-function TPMT variants result in decreased inactivation, higher parent drug concentrations, and increased generation of cytotoxic thioguanine nucleotide (TGN) metabolites; these TGNs are incorporated into DNA and associate with drug effect. Individuals homozygous for loss-of-function variants in TPMT will exhibit life threatening bone marrow toxicity with usual drug doses due to TGN accumulation.22 TGNs are themselves metabolized by NUDT15, and NUDT15 loss of function variants have also been associated with toxicity.22,23 The thiopurine immunosuppressant drug azathioprine is metabolized to 6-MP and variants in TPMT and NUDT15 are similarly associated with risk of hematologic toxicity.22
Similarly, variants in DPYD increase plasma concentrations, and toxicity risk, of 5-fluorouracil and other fluoropyrimidines such as capecitabine.24
Notably, loss-of-function variants can be mimicked by interactions with drugs that inhibit the same drug metabolism pathways; this is described as a “phenocopy”. Examples of phenocopies include: CYP2D6 inhibition by some selective serotonin reuptake inhibiters (SSRIs), CYP2C19 inhibition by many proton pump inhibitors, and XO inhibition by allopurinol which, by inhibiting an alternate pathway for azathioprine and 6-mercaptopurine metabolism, can increase generation of TGNs and thereby increase toxicity.
Drugs metabolized predominantly by a single enzyme and with wide therapeutic margins may display significant variability in pharmacokinetics due to pharmacogenomic variants, but because of the wide therapeutic margin, these differences may not drive clinically relevant variability in drug efficacy or toxicity. Similarly, drugs with narrow therapeutic margins that are inactivated by multiple enzymatic pathways are also less susceptible to unusual responses due to pharmacogenomic variants, unless there are multiple “hits” to individual pathways. For example, drug interactions or disease inhibiting one metabolic pathway combined with genetic variation inhibiting a second can account for unusual drug responses.25
Drug transport into and out of cells by specific drug transport molecules is another important potential mediator of variable drug concentrations at effector sites and thus drug action. The drug efflux transporter OATP1B1 encoded by SLCO1B1 is responsible for removal of simvastatin from the systemic circulation. The common loss-of-function SLCO1B1*5 variant has been associated with elevated simvastatin plasma concentrations and an increased risk for simvastatin myopathy,26,27 and also contributes to variability in methotrexate clearance in children treated for acute leukemia.28
Warfarin is a well-studied example of a drug whose variable actions are determined by both pharmacokinetic and pharmacodynamic variants, and variant frequency is highly dependent on ancestry. Warfarin is administered as a racemate and bioinactivation of the more active S-enantiomer is accomplished by CYP2C9. Variants that decrease CYP2C9 activity are therefore associated with an increase in S-warfarin plasma concentration and a resultant intensified pharmacologic effect, manifest as an increase in the international normalized ratio (INR) or bleeding risk. The *2 and *3 variants are commonest in European ancestry populations; *3 reduces CYP2C9 activity to a greater extent than does *2. Patients heterozygous for *2 may exhibit only a small pharmacogenomic effect, while those homozygous for *3 may exhibit drastic decreases in warfarin dose requirement, and may be difficult to anticoagulate because of day-to-day variability in INR.29,30 In African ancestry populations, these variants are rarer, and other variants have been reported.31,32 Traditional genetic linkage methods identified loss-of-function variants in VKORC1 as the cause of the rare syndrome of familial warfarin resistance, a failure of the INR to rise even with exposure to very large doses of warfarin;33 subsequent studies showed that VKORC1 encodes the warfarin target. A common promoter polymorphism in VKORC1 is associated with variability in hepatic mRNA levels and in warfarin dose requirement.34 Rarer reduction-of-function coding region variants in VKORC1, associated with increased warfarin dose requirements, have also been described and vary by ancestry: for example, a variant encoding D36Y is common (minor allele frequency (MAF) of 5%) in Ashkenazi populations.35 Multiple GWAS of variability in warfarin steady state dose requirements have yielded very strong signals at CYP2C9 and at VKORC1 as well as at CYP4F2, a gene responsible for bioinactivation of vitamin K.36–39 In African-American subjects, a GWAS identified a separate signal (whose specific function remains to be defined) near CYP2C8-CYP2C9.32 An estimated 50% of the variability in warfarin dose requirement has been attributed to common genetic variation identified in these studies.
Other pharmacodynamic variants:
As mentioned above, some of the earliest well-defined pharmacogenetic syndromes involve pharmacodynamic mechanisms. The risk of malignant hyperthermia on exposure to inhaled anesthetics or succinylcholine is mediated by variants in RYR1 or CACNA1S.40 Variants reducing G6PD function caused a high incidence of hemolytic anemia among African-American soldiers exposed to antimalarials during World War II and increase the risk for hemolytic anemia and methemoglobinemia with rasburicase, a recombinant urate oxidase used to treat hyperuricemia in cancer.41 Variants in IFNL3 (also termed IL28B) predict response to pegylated interferon-α and ribavirin in hepatitis C although the introduction of newer therapeutics has reduced the impetus for genotyping.42
ADRs described to this point are related to exaggerated drug effect, sometimes due to high plasma concentrations, such as bleeding with anticoagulants or hypotension with antihypertensives, and these have been termed “type A” ADRs. “Type B” ADRs are those unrelated to the drug’s known and intended pharmacologic effects and are often considered non-dose-dependent. Type B reactions include serious immunologically-mediated ADRs such as the Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis (SJS/TEN). Candidate gene and GWAS approaches using very small case numbers, often less than 100, and large numbers of drug-exposed controls, have implicated specific HLA variants in SJS/TEN. These studies also highlight the importance of ancestry in drug response. For example, HLA-B*15:02 confers risk of carbamazepine-related SJS/TEN in Southeast Asia where the allele is relatively common.43 In European ancestry populations, on the other hand, this allele is rare, and a different HLA risk allele (HLA-A*31:01) has been implicated.44 Importantly, in these cases, the HLA variant is judged necessary, but not sufficient to induce the immunologic response.45 Indeed, there is a very strong association between flucloxacillin-related hepatotoxicity and HLA-B*57:01,46 but it has been estimated that only one case will develop for each 13,000 genotyped positive patients exposed.45 For other drugs, this “number needed to test” (NNT) is smaller; for example, in the case of abacavir discussed further below,47 the NNT among patients with HLA-B*57:01 is 13. Variable susceptibility to type B reactions may also depend on plasma drug concentration. For example, HLA variants associate with ADRs caused by the anti-seizure medication phenytoin, a CYP2C9 substrate, and several studies have reported that risk is increased in subjects who also carry CYP2C9 loss of function alleles.48,49
Implementing pharmacogenomics: clinical trial data
Because preclinical and clinical mechanistic studies support the role of genetic variation as a contributor to variable drug responses, retrospective analyses and prospective trials have been mounted to test the hypothesis that pharmacogenomically-guided therapy will improve clinical drug outcomes.
After candidate gene studies identified HLA-B*57:01 as a strong risk factor for abacavir related SJS/TEN,50 a randomized clinical trial (RCT) was conducted in 1956 subjects to compare conventional antiretroviral regimens including abacavir to a pharmacogenomically-guided strategy in which abacavir was dropped from treatment if the HLA-B risk allele was present.47 A rash thought to be related to abacavir developed in 7.8% of controls and 3.4% of subjects in the pharmacogenomically-guided arm. However, subsequent protocol-mandated skin testing confirmed that the rash was abacavir-related in 2.7% of controls and in none of the patients in the pharmacogenomically-guided arm. This unambiguous outcome resulted in the FDA label requiring pre-prescription testing for HLA-B*57:01 in all individuals starting abacavir and not using the drug in genotype-positive subjects.
An RCT compared standard therapy to pharmacogenomically-guided dosing in 783 patients starting azathioprine or 6-mercaptopurine for inflammatory bowel disease.51 TPMT intermediate metabolizers (defined in Box 1) received 50% of the standard dose while poor metabolizers received 0–10% of the standard dose. Overall, there was no difference in serious ADRs or in disease progression in the genotype-guided vs standard therapy groups. However, among the 78 patients with TPMT loss-of-function variants (77 intermediate metabolizers and 1 poor metabolizer), there was a clear benefit of pharmacogenomically-guided therapy: the incidence of serious hematologic ADRs was 22.9% in the control group vs. 2.6% in the pharmacogenomically-guided group (relative risk 0.11, 95% confidence interval (CI): 0.01–0.85). These results highlight the fact that any benefit of pharmacogenomic testing will be confined to the subset in whom the target genetic variants are present, and that the apparent benefits will be diluted if testing is evaluated in the entire population comprising mostly low-risk patients. As discussed further below, the vast majority of patients harbor one or more functionally-important variants in key pharmacogenes, suggesting that pre-emptive testing of a panel of multiple pharmacogenes should be a strategy to be considered for pharmacogenetic implementation.
Retrospective analyses of the effect of common genetic variants on outcomes after clopidogrel was initiated for acute coronary syndrome have shown a consistent effect of genotype.5,52,53 Investigators in the IGNITE (Implementing Genomics in Practice) network summarized outcomes of genotyping to direct the choice of antiplatelet therapies between clopidogrel and alternate therapies in patients with CYP2C19 loss of function alleles. Among 1815 patients at 7 institutions, those with loss of function alleles (31.5%) had more cardiovascular events if treated with clopidogrel compared to treatment with alternate drugs (23.4 versus 8.7/100 patient-years, hazard ratio 2.26, 95% CI: 1.18 to 4.32; p = 0.013).54 One recent small prospective RCT reported a large decrease in late coronary events with a pharmacogenomically-driven strategy for clopidogrel.55 Nevertheless, to date, cardiovascular professional societies have not recommended genetic testing to guide clopidogrel therapy, despite the fact that some have argued the evidence is stronger than for other recommended tests.56
Multiple large RCTs have evaluated the effect of a pharmacogenomically-driven strategy including intensive INR monitoring versus a conventional clinical approach for warfarin. The first three large trials used a primary endpoint of time in therapeutic INR range or time required to achieve stable anticoagulation. Two studies used a clinical algorithm as the control,57,58 and one used a clinically-conventional fixed dose regimen.59 The fixed dose study showed a statistically significant improvement in the primary outcome, while there was no difference in outcome in the other two. The largest of these trials, the US-based COAG, included 27% African-American subjects, and the CYP2C9 variants interrogated are much more common in European ancestry individuals, while other CYP2C9 variants that play a role in subjects of African origin were not assayed.60 As a result, it has been speculated that the null result in COAG may reflect, in part, failure to consider ancestry-specific genetics.61
More recently, several other RCTs have reported that pharmacogenomically-guided warfarin therapy improves outcome. The Genetic Informatics Trial (GIFT) randomized 1650 patients following hip or knee replacement to a clinically-guided or a genotype-guided warfarin strategy and focused on the primary outcome of warfarin-related ADRs (major bleeding, INR >4, venous thromboembolism, and death) rather than time in therapeutic range.62 The primary endpoint occurred in 10.8% patients in the genotyped-guided group vs. 14.7% in the clinically-guided group (p=0.02). An RCT in Southeast Asia showed that a pharmacogenomically-guided strategy resulted in fewer dose titrations in the first two weeks of therapy (the primary endpoint for the trial).63
In all these warfarin trials, the frequency of serious bleeding was low, and none of the trials was powered to detect an effect of genotype on bleeding itself. Retrospective analyses of large numbers of patients presenting with warfarin-related bleeding, ascertained through administrative databases or electronic health records (EHRs), have reported a statistically significant effect of CYP4F2 V433M (odds ratio: 0.62; 95% CI: 0.43–0.91)64 and of CYP2C9*3 (adjusted odds ratio: 2.05; 95% CI: 1.04, 4.04).65 A smaller study of African Americans with bleeding attributed to warfarin at INR values <4 identified variants thought to regulate expression of EPHA7 in the vascular endothelium.66
An evaluation of the feasibility of a pharmacogenetically-driven strategy with dose adjustment based on 4 DPYD variants was conducted in 1103 patients receiving fluoropyrimidines. There were 85 variant carriers, and while they had a higher incidence of serious toxicity compared to non-carriers, the rates were lower than those seen in historical controls.24
There are a number of major lessons that these trials have identified to date (Table 2). A genetic testing strategy for an individual drug can only show benefit in those subjects with the variant genotype. In case of drug metabolizing enzymes and drug transporters, the pharmacogenomic effect size is much larger in homozygotes than in heterozygotes. While it is possible to mount trials using “surrogate” endpoints, such as time in therapeutic range, acceptance by the clinical practice community and thus the payer community is more likely if data are available on a “hard” outcome such as death. However, this may require very large studies even if only high risk populations are included. These issues likely contribute to slow uptake of genetic testing for warfarin and clopidogrel, as does increasing availability of alternate therapies which appear to be at least as effective without known major pharmacogenomic issues identified to date. On the other hand, when alternate drugs are not available or when ADRs are serious and clearly related to genetic variants, uptake is more likely particularly if a regulatory agency or professional society recommends testing, as in the case of abacavir.
Table 2:
Issues, obstacles, and potential solutions in pharmacogenomic implementation
| Category | Issue | Example | Perceived Obstacle | Potential Solution(s) |
|---|---|---|---|---|
| Pharmacogenes | Majority of individuals in most populations are “wild type” | Less than 1% of individuals are TPMT poor metabolizers111 | Very large numbers needed to test for successful prospective trials and for clinical benefit | • Prespecify plan to analyze subset with variant • Conduct trials of across multiple drugs and genes, which inform panel-based testing |
| Rare variants with uncertain effect | 46 of 64 haplotypes for CYP2C9 have unknown function83 | Insufficient data to ascertain phenotype with absolute certainty | • Assay only variants with known function • Include uncertainty on clinical reports • Functional studies |
|
| Spectrum of effects due to variants within one gene | Distinct variants in CYP2C19 confer complete loss of function, partial loss of function, or gain of function | Need to express genetic effect as quasi-continuous trait | • Use activity scores to annotate variant effect | |
| Complexity of gene assays | Different assay technologies required for CYP2C19, CYP2D6 and HLA | Lack of comprehensive local infrastructure for multiple laboratory developed tests | • Development of off-the-shelf assays for pharmacogenes • Reliance on send-out laboratories for some or all pharmacogenomic testing |
|
| Drug Effects | Hard endpoints are rare | There were no deaths in the 1650 randomized patients treated with warfarin in the GIFT trial62 | Robust methods to prove impact of genotype-guided therapy on hard endpoints not well-developed. | • Use surrogate, but clinically relevant, endpoints such as major bleeding, length of hospitalization, symptom control, or healthcare cost • Perform large retrospective analyses of hard endpoints using EHR-linked biobank data |
| Efficacy endpoints poorly defined outside of clinical trials | Serial assessment of depression symptoms inconsistently documented in EHR data | Cannot perform retrospective analyses on efficacy | • Prospective data collection with oversampling of participants with pharmacogenetic variants | |
| Healthcare institutions / Local health information technology | Results for each gene require interpretation to discrete clinical guidance | Clinical decision support for warfarin provides dosing calculation, not genetic test results | Lack of technological infrastructure for interpretation from gene test results to functional effect to dosing guidance | • Widespread sharing of technical solutions and clinical decision support across institutions |
| Functional predictions and clinical guidance evolve with new evidence | New evidence for the role of NUDT15 variants in thiopurine toxicity23 | Need to continually assess evidence, which is consistently expanding to include more drugs and more genes | • Continued support for development of guidelines to guide appropriate testing | |
| Provider resistance to receiving or using pharmacogenomic information | No agreement among healthcare providers about who should take responsibility for results85 | Limited ordering of pharmacogenomic testing and/or lack of use of pharmacogenomic guidance | • Identification and recruitment of clinical champions for specific drug-gene interactions • Increased provider education • Interruptive prescriber alerts making the pharmacogenomic-informed choices the default |
|
| Evolving EHR systems | EHR system changes or upgrades may cause loss of reporting or decision support functionality | Large ongoing costs of system maintenance | • Commitment from EHR vendors for continual support of pharmacogenomic implementation • Computable guidelines for pharmacogenomics |
|
| Healthcare systems | Patient movement across EHR systems | A patient’s pharmacogenomic results do not follow them when they receive care in another system | Loss of potential benefit of test and/or potential for repeat testing | • Provision of pharmacogenetic results to patients • Portability of results for transfer to other EHR systems |
| Diversity of pharmacogenomic assays | Depending on TPMT genotype interpretation, a patient may be labeled as poor or intermediate metabolizer | Lack of consistency of results across CLIA-approved tests | • Standardization of minimal test requirements • Standardization of interpretation of variant effects |
|
| Reimbursement challenges | Pharmacogenomic testing is variably reimbursed across clinical scenarios, states, genes/drugs, and payors | Pharmacogenomic testing is not cost-effective | • Increase data available on cost benefit and improve and standardize analyses to promote reimbursement • Develop comprehensive cost-effectiveness model as opposed to models for individual drug-gene pairs. |
Implementing pharmacogenomics: Current status
Experiments with implementing pharmacogenomics have used a “point of care” strategy or “pre-emptive” strategy. The point of care strategy uses genetic testing, generally with very rapid turnaround times, for a small number of individual variants, when a target drug such as clopidogrel is prescribed.54 The pre-emptive strategy, on the other hand, generates variant data for multiple pharmacogenes ideally prior to prescription of any target drug.67,68 Variant data are then embedded in EHRs and coupled to clinical decision support which delivers advice when a target drug is prescribed in a patient with variant genetics. Implementing such a pre-emptive strategy requires well-curated data relating individual genetic variants (and their combinations as haplotypes or diplotypes), designation of predicted metabolizer phenotype status (e.g. NM, PM, etc.; see Box 1), and advice on alternate therapeutic strategies in patients with genetic variants. Thus, a barrier to early adoption was the need for extensive curation of the pharmacogenomic evidence, expert design of the pharmacogenomic test, curation of predicted consequences of the genetic variants, clinical expertise regarding drug prescribing and alternatives, and technical expertise to support laboratory testing, reporting, and decision support. Many of these needs are now being met by evidence curation at pharmgkb.com and by the development of guidelines in the US and in Europe by the Clinical Pharmacogenetics Implementation Consortium (CPIC)69 and the Dutch Working Group (DWG)70 on pharmacogenetics. These largely independent efforts have generated similar guidelines across multiple drugs.71
Efforts to implement pharmacogenomics have also been supported by economic analyses for many of the common pharmacogenomic scenarios, such as CYP2C19 tailored selection of antiplatelet agents following percutaneous coronary intervention72 or selection of abacavir for HIV therapy.73 While most analyses find testing to be cost-effective when genetic test costs were minimized, they have not always led to changes in guideline recommendations or reimbursement policies.74 Indeed, lack of evidence for cost-effectiveness and thus lack of reimbursement has been identified as a major barrier for implementation of pharmacogenetic testing: one systematic review of cost-effectiveness studies in pharmacogenomics made the comment that “… these issues imply that cost-effectiveness analyses on their own cannot answer the question of whether or not a certain strategy should be used and funded, but should be considered in conjunction with other factors such as the available resources, the number of patients who benefit from the intervention and other ethical considerations.”74
Regulatory responses to pharmacogenomic variant data are evolving. While the US Food and Drug Administration includes pharmacogenomic information in over 100 drug labels,75 it has also included black box warnings against the use of certain drugs or dosages even when ADR risk is thought to be genetically-mediated. Thus, for example, the label limits simvastatin dosages to ≤40mg/day because higher dosages increase the risk of myopathy, although this risk is nearly confined to subjects with an SLCO1B1 risk variant.27 Similarly, codeine can produce respiratory depression particularly post-tonsillectomy and in young patients. The label now recommends against the use of the drug in this setting,21 although the risk seems confined to those with the ultra-rapid metabolizer (UM) phenotype.76 This labeling may result in prescription of more potent opioids with attendant risks of other adverse effects.77
While HLA-B*15:02, associated with carbamazepine SJS/TEN, is especially prevalent in Southeast Asia, there is controversy whether compulsory testing is cost-effective.78,79 In Hong Kong, implementation of a testing program resulted in a decrease in the prescription of carbamazepine (and a decrease in related SJS/TEN), but an increase in the prescription of other anti-seizure medications and no overall change in SJS/TEN.80 These data emphasize a need for implementation programs to include an educational component.
Thus, issues such as return on investment for adopter healthcare systems and reimbursement across payers remain unsettled. In oncology, adoption has been faster perhaps in part because tumor genetic testing allows definition of subsets of patients in whom therapy will not be effective thus placing a limit on widespread use of expensive therapies. By contrast, pharmacogenomic variants identifying patients at risk for ADRs during treatment with the older cheaper drugs like warfarin or clopidogrel may identify individuals who will benefit from a more expensive drug. The fragmented nature of healthcare reimbursement in the US represents a further barrier in that pharmacogenomic test results generated at one site may not be available should the patient move to a different provider in another health care or EHR system.
A number of reports have pointed out that when pharmacogenomic testing across multiple drug-gene pairs is performed, the vast majority of individuals have variant(s) that would be important were they to be prescribed specific target drugs.81–83 These data add to the appeal of the pre-emptive pharmacogenomic strategy. Identifying patients in whom the strategy is likely to be effective, i.e. those in whom target drugs are likely to be prescribed over the next several years, is one challenge.84 Another is practitioner reluctance to switch prescriptions in the face of pharmacogenomic variant data; reasons include individual preference, late delivery of genotype data, lack of familiarity with pharmacogenomic information, and expense or risk of alternate therapies.85
Engineering the EHR to accommodate pharmacogenomic data and to deliver clinical decision support (CDS) is another challenge. This includes developing and implementing robust methods for translating raw genetic data into predicted drug responses (e.g. by assignment of predicted pharmacogenetic phenotypes from variants in pharmacogenes). While single gene-based systems can accomplish this task using human interpretation or non-machine readable (often pdf format) reports, multiplexed programs increasingly rely on automated “omic ancillary systems”86 to integrate genomic data into EHR-based clinical workflows. Indeed, a survey of ten healthcare systems that adopted pharmacogenomic CDS identified non-specific barriers, such as staffing and coordination across multiple teams, rather than pharmacogenomic-specific ones.87 Maintenance and updating of variant translations and CDS recommendations is another EHR challenge shared with any use of genetic information in clinical care.
Role of genomics in the drug development process
Only a very small number of drug candidates entering clinical trials ultimately achieve regulatory approval. Available evidence strongly supports the idea that drugs with targets validated by human genetic studies have a much higher likelihood of successful marketing than those lacking such evidence.88,89 Thus, developing this evidence is becoming an increasingly important part of the drug development process. Approaches that are being explored include not only GWAS but also EHR-based phenome scanning, i.e. examination of the relationship between specific variants in candidate drug target genes and phenotypes across the EHR.90,91
The identification of rare sequence variants that appear to associate with important human phenotypes has also provided the basis for new drug development. Perhaps the most notable example to date is PCSK9, where gain-of-function variants were initially associated with striking elevation in LDL cholesterol and familial hypercholesterolemia (FH).92 Subsequently, the Dallas Heart Study showed that rare truncation (i.e. loss-of-function) variants, occurring largely in African-Americans, were associated with striking decreases in LDL cholesterol and in the Atherosclerosis Risk in Communities cohort a striking decrease in lifetime risk of coronary artery disease.93 These data propelled development of PCSK9 inhibitors to the market for treatment of elevated LDL cholesterol. Notably, the indications extend beyond FH itself, and while the drugs are indicated across ancestries, the original discovery was enabled by studying an African-American cohort. Other drug targets implicated or validated by identifying rare sequence variants associated with unusual phenotypes include APOC3 for hypertriglyceridemia,94 NPC1L1 (encoding the ezetimibe target) for cholesterol transport,95 SLC30A8 for prevention of obesity-related diabetes,96 ANGPTL4 for hyperlipidemia,97,98 and HSD17B13 for reduced risk of chronic liver injury.99
Another area in which human genetics is playing a major role in the development of new drugs is in the development of new therapies for rare Mendelian diseases. In cystic fibrosis, one relatively minor mechanism for dysfunction of the CFTR protein is altered conductance of channels that traffic normally to the cell surface. Ivacaftor, a conductance defect corrector, has been associated with improvement in functional status,100 and is now marketed for patients who carry specific germ-line variants that have been tested in clinical trials or show ivacaftor-mediated improvement in function in vitro. The commonest functional defect in cystic fibrosis is failure of channels to traffic to the cell surface, and lumacaftor has been developed and marketed (with ivacaftor) for this indication.101 A preliminary study suggests lumacaftor can also correct mistrafficking of cardiac potassium channels in one form of the long QT syndrome suggesting this drug or others correcting mistrafficking of cell proteins may have more widespread applicability.102
The Future
The field of pharmacogenomics has to date focused on a relatively small number of common, high effect size variants. The spectrum of effect sizes from pharmacogenomic variants varies from heterozygotes with reduction-of-function alleles to homozygotes for complete loss-of-function alleles in genes critical for the disposition of individual drugs. This spectrum of effect sizes has complicated the design and conduct of large clinical trials which often focus on individual drugs.
Genome science is providing new tools for understanding variability in drug response. One obvious area is the increasing use of exome or genome sequencing with the attendant recognition of very large numbers of rare missense variants in all genes. It is intuitively obvious that some variant drug responses must reflect the effect of such rare variants, alone or in combination, but the vast majority have not yet been characterized. Pharmacogenomics has focused on a small number of candidate genes, generally derived from a clear understanding of the mechanisms of underlying variability in drug action, notably in pharmacokinetics, pharmacodynamics, and immunopharmacogenomics. The extent to which an understanding of variability in drug action will be improved by moving beyond a candidate gene approach to considerations of the contribution of variants in multiple genes (Figure 1B) remains to be determined. One interesting example is the use of genetic risk scores (GRS), derived from multiple genetic variants which individually contribute a small amount to a variable phenotype but may confer larger effect sizes when present in combination. A GWAS identified no individual large effect size variants for drug-induced QT prolongation and associated polymorphic ventricular arrhythmias,103 but a subsequent analysis using a GRS derived from 61 individual variants identified in a GWAS of the QT interval itself readily separated cases from controls.104 Similarly, a GRS derived from baseline neuropsychiatric traits predicted response to antidepressant therapies.105 A set of 13 variants increased the area under the receiver operating curve from 0.64 to 0.81 in a clinical trial studying drug response in patients with advanced breast cancer.106 The extent to which these multigene markers can identify the genetic architecture of disease and its response to drugs remains an interesting but as yet largely unexplored area in the arena of drug response and toxicity. It may also be useful to intensively study individuals with clear outlier responses to drug exposure, for example to measure plasma drug and metabolite concentrations or to search for rare as-yet-uncharacterized variants in key pharmacogenes.
There are a number of trials that are ongoing that may further inform the field. TAILOR-PCI is comparing the effect of a pharmacogenomically-informed strategy to conventional strategies in the use of clopidogrel and other antiplatelet therapies. This trial aims to enroll 5270 patients and should report in 2020. The CETP inhibitor dalcetrapib was tested in 15,871 patients and failed to show any difference in a primary cardiovascular endpoint.107 However, a subsequent analysis of 5,749 subjects who provided DNA samples identified variants in ADCY9 as markers of a potentially beneficial response to drug therapy,108 and in vitro and animal studies have supported a role for ADCY9 in this drug’s action.109 A large trial, dal-GenE is underway to screen ~35,000 subjects to identify ~6,000 with the predicted response allele, and to then randomize these subjects to dalcetrapib or placebo. The study cohort has been accrued and is currently in follow-up.
The PREPARE (Preemptive Pharmacogenomic Testing for Preventing Adverse Drug Reactions) study of the European Union’s Ubiquitous Pharmacogenomics Study group is evaluating a pre-emptive pharmacogenomic testing strategy in 12 genes to reduce the incidence of ADRs related to 43 target drugs.110 PREPARE, which uses a crossover design, is being conducted at seven sites across Europe, and is randomizing subjects to a pharmacogenomically-guided strategy, with dose adjustments, compared to a conventional dosing strategy. The study was powered to detect a 30% decrease in severe ADRs, from 4 to 2.8%, and is scheduled to report in 2020. IGNITE is currently planning an evaluation of panel-based testing for management of depression, chronic pain, and acute post-operative pain.
Very large personalized medicine programs, that include extensive genotyping and/or sequencing, are being put in place across the globe. Some focus on single diseases, some are more broad-based but do not include a return of results capability, and others plan whole genome sequencing with return of results to participants and healthcare providers; the latter include Genome England that is aiming to sequence up to 5,000,000 whole genomes, and the US All of Us Program that is recruiting 1,000,000 participants.
Variability in response, and in particular in ADR risk, is a near-inevitable feature of contemporary drug therapy and includes a prominent genetic component. Defining that genetic component and understanding how best to apply that knowledge in a clinical context are ongoing challenges to pharmacogenomic science. The advent of inexpensive genotyping and sequencing and the development of increasingly sophisticated EHR systems holds the promise that implementing pharmacogenomic variant information will become a routine part of the practice of Genomic Medicine.
Acknowledgments
Grant support
This work was supported by NIH grants P50 GM115305 (DR), U01 HG006378 (DR, SVD), P50 GM 115279 (MR), U24 HG 010135 (MR), U01 HG010232 (JFP), R01 HG008701 (JFP), U01 HG009694 (JFP), U01 HG008679 (MW), R01 CA161608 (HLM) and grants from the Doris Duke Foundation (CSDA 2017075, SVD) and the Burroughs Wellcome fund (IRSA 1015006, SVD)
Footnotes
Competing interests
Roden None
McLeod Prof. McLeod is a member of the Board of Directors of Cancer Genetics Inc. and a Scientific Advisor to Pharmazam.
Relling none
Williams none
Mensah none
Peterson Dr. Peterson is a consultant for Color Genomics.
Van Driest Dr. Van Driest has received a speaking honorarium from Merck.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pirmohamed M, James S, Meakin S, et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. British Medical Journal 2004; 329: 15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. Journal of the American Medical Association 1998; 279: 1200–1205. [DOI] [PubMed] [Google Scholar]
- 3.Landrigan CP, Parry GJ, Bones CB, Hackbarth AD, Goldmann DA, Sharek PJ. Temporal Trends in Rates of Patient Harm Resulting from Medical Care. New England Journal of Medicine 2010; 363: 2124–2134. [DOI] [PubMed] [Google Scholar]
- 4.Friend WC, Weijer C. Re: CCNP position paper on the use of placebos in psychiatry. Journal of psychiatry & neuroscience : JPN 1996; 21: 354–359. [PMC free article] [PubMed] [Google Scholar]
- 5.Mega JL, Close SL, Wiviott SD, et al. Cytochrome P-450 Polymorphisms and Response to Clopidogrel. N Engl J Med 2009; 360: 354–362. [DOI] [PubMed] [Google Scholar]
- 6.Panattoni L, Brown PM, Te Ao B, Webster M, Gladding P. The cost effectiveness of genetic testing for CYP2C19 variants to guide thienopyridine treatment in patients with acute coronary syndromes: a New Zealand evaluation. PharmacoEconomics 2012; 30: 1067–1084. [DOI] [PubMed] [Google Scholar]
- 7.Nakagawa H, Fujita M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer science 2018; 109: 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garrod AE. Inborn errors of metabolism London: Henry Frowde and Hodder Stroughton; 1923. [Google Scholar]
- 9.Motulsky AG. Drug reactions, enzymes and biochemical genetics. Journal of the American Medical Association 1957; 165: 835–837. [DOI] [PubMed] [Google Scholar]
- 10.Kalow W Unusual responses to drugs in some hereditary conditions. Canadian Anaesthetists’ Society journal 1961; 8: 43–52. [DOI] [PubMed] [Google Scholar]
- 11.International Conference on Harmonisation; Guidance on E15 Pharmacogenomics Definitions and Sample Coding; Availability. Notice. Fed Regist 2008; 73: 19074–19076. [PubMed] [Google Scholar]
- 12.von Ahsen N, Armstrong VW, Oellerich M. Rapid, Long-Range Molecular Haplotyping of Thiopurine S-Methyltransferase (TPMT*) *3A, *3B, and *3C. Clinical Chemistry 2004; 50: 1528–1534. [DOI] [PubMed] [Google Scholar]
- 13.Kalman LV, Agundez J, Appell ML, et al. Pharmacogenetic allele nomenclature: International workgroup recommendations for test result reporting. Clin Pharmacol Ther 2016; 99: 172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaedigk A, Ingelman-Sundberg M, Miller NA, Leeder JS, Whirl-Carrillo M, Klein TE. The Pharmacogene Variation (PharmVar) Consortium: Incorporation of the Human Cytochrome P450 (CYP) Allele Nomenclature Database. Clin Pharmacol Ther 2018; 103: 399–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caudle KE, Dunnenberger HM, Freimuth RR, et al. Standardizing terms for clinical pharmacogenetic test results: consensus terms from the Clinical Pharmacogenetics Implementation Consortium (CPIC). Genet Med 2017; 19: 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pirmohamed M Personalized pharmacogenomics: predicting efficacy and adverse drug reactions. Annual review of genomics and human genetics 2014; 15: 349–370. [DOI] [PubMed] [Google Scholar]
- 17.Motsinger-Reif AA, Jorgenson E, Relling MV, et al. Genome-wide association studies in pharmacogenomics: successes and lessons. Pharmacogenet.Genomics 2013; 23: 383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mega JL, Hochholzer W, Frelinger AL 3rd, et al. Dosing clopidogrel based on CYP2C19 genotype and the effect on platelet reactivity in patients with stable cardiovascular disease. JAMA : the journal of the American Medical Association 2011; 306: 2221–2228. [DOI] [PubMed] [Google Scholar]
- 19.Shuldiner AR, O’Connell JR, Bliden KP, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. Journal of the American Medical Association 2009; 302: 849–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sibbing D, Koch W, Gebhard D, et al. Cytochrome 2C19*17 Allelic Variant, Platelet Aggregation, Bleeding Events, and Stent Thrombosis in Clopidogrel-Treated Patients With Coronary Stent Placement. Circulation 2010; 121: 512–518. [DOI] [PubMed] [Google Scholar]
- 21.Kuehn BM. Fda: No codeine after tonsillectomy for children. JAMA : the journal of the American Medical Association 2013; 309: 1100–1100. [DOI] [PubMed] [Google Scholar]
- 22.Relling MV, Schwab M, Whirl-Carrillo M, et al. Clinical Pharmacogenetics Implementation Consortium Guideline for Thiopurine Dosing Based on TPMT and NUDT15 Genotypes: 2018 Update. Clin Pharmacol Ther 2018: [DOI] [PMC free article] [PubMed]
- 23.Yang JJ, Landier W, Yang W, et al. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015; 33: 1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henricks LM, Lunenburg CATC, de Man FM, et al. DPYD genotype-guided dose individualization of fluoropyrimidine therapy: a prospective safety analysis on four relevant DPYD variants. Lancet Oncology 2018; Accepted for publication: [DOI] [PubMed]
- 25.Palmiere C, Lesta Mdel M, Sabatasso S, Mangin P, Augsburger M, Sporkert F. Usefulness of postmortem biochemistry in forensic pathology: illustrative case reports. Legal medicine (Tokyo, Japan) 2012; 14: 27–35. [DOI] [PubMed] [Google Scholar]
- 26.Pasanen MK, Neuvonen M, Neuvonen PJ, Niemi M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet.Genomics 2006; 16: 873–879. [DOI] [PubMed] [Google Scholar]
- 27.Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N Engl J Med 2008; 359: 789–799. [DOI] [PubMed] [Google Scholar]
- 28.Trevino LR, Shimasaki N, Yang W, et al. Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2009; 27: 5972–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steward DJ, Haining RL, Henne KR, et al. Genetic association between sensitivity to warfarin and expression of CYP2C9*3. Pharmacogenetics 1997; 7: 361–367. [DOI] [PubMed] [Google Scholar]
- 30.Ablin J, Cabili S, Eldor A, Lagziel A, Peretz H. Warfarin therapy is feasible in CYP2C9*3 homozygous patients. Eur.J Intern.Med 2004; 15: 22–27. [DOI] [PubMed] [Google Scholar]
- 31.Ramirez AH, Shi Y, Schildcrout J, et al. Predicting warfarin dosages in European-American and African-American subjects using DNA samples linked to an electronic health record Pharmagenomics 2012; in press: [DOI] [PMC free article] [PubMed]
- 32.Perera MA, Cavallari LH, Limdi NA, et al. Genetic variants associated with warfarin dose in African-American individuals: a genome-wide association study. The Lancet 2013; 382: 790–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rost S, Fregin A, Ivaskevicius V, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004; 427: 537–541. [DOI] [PubMed] [Google Scholar]
- 34.Rieder MJ, Reiner AP, Gage BF, et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. New England Journal of Medicine 2005; 352: 2285–2293. [DOI] [PubMed] [Google Scholar]
- 35.Scott SA, Edelmann L, Kornreich R, Desnick RJ. Warfarin Pharmacogenetics: CYP2C9 and VKORC1 Genotypes Predict Different Sensitivity and Resistance Frequencies in the Ashkenazi and Sephardi Jewish Populations. Am J Hum.Genet 2008; 82: 495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caldwell MD, Awad T, Johnson JA, et al. CYP4F2 genetic variant alters required warfarin dose. Blood 2008; 111: 4106–4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takeuchi F, McGinnis R, Bourgeois S, et al. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS.Genet 2009; 5: e1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teichert M, Eijgelsheim M, Rivadeneira F, et al. A genome-wide association study of acenocoumarol maintenance dosage. Hum.Mol Genet 2009; 18: 3758–3768. [DOI] [PubMed] [Google Scholar]
- 39.Cooper GM, Johnson JA, Langaee TY, et al. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood 2008; 112: 1022–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gonsalves SG, Dirksen RT, Sangkuhl K, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for the Use of Potent Volatile Anesthetic Agents and Succinylcholine in the Context of RYR1 or CACNA1S Genotypes. Clin Pharmacol Ther 2018: [DOI] [PMC free article] [PubMed]
- 41.Relling MV, McDonagh EM, Chang T, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for rasburicase therapy in the context of G6PD deficiency genotype. Clin Pharmacol Ther 2014; 96: 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muir AJ, Gong L, Johnson SG, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for IFNL3 (IL28B) genotype and PEG interferon-alpha-based regimens. Clin Pharmacol Ther 2014; 95: 141–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chung WH, Hung SI, Hong HS, et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature 2004; 428: 486. [DOI] [PubMed] [Google Scholar]
- 44.McCormack M, Alfirevic A, Bourgeois S, et al. HLA-A*3101 and Carbamazepine-Induced Hypersensitivity Reactions in Europeans. New England Journal of Medicine 2011; 364: 1134–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White KD, Chung WH, Hung SI, Mallal S, Phillips EJ. Evolving models of the immunopathogenesis of T cell-mediated drug allergy: The role of host, pathogens, and drug response. The Journal of allergy and clinical immunology 2015; 136: 219–234; quiz 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daly AK, Donaldson PT, Bhatnagar P, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat.Genet 2009; 41: 816–819. [DOI] [PubMed] [Google Scholar]
- 47.Mallal S, Phillips E, Carosi G, et al. HLA-B*5701 Screening for Hypersensitivity to Abacavir. The New England Journal of Medicine 2008; 358: 568–579. [DOI] [PubMed] [Google Scholar]
- 48.Chung WH, Chang WC, Lee YS, et al. Genetic variants associated with phenytoin-related severe cutaneous adverse reactions. JAMA : the journal of the American Medical Association 2014; 312: 525–534. [DOI] [PubMed] [Google Scholar]
- 49.Su SC, Chen CB, Chang WC, et al. HLA Alleles and CYP2C9*3 as Predictors of Phenytoin Hypersensitivity in East Asians. Clin Pharmacol Ther 2018: [DOI] [PubMed]
- 50.Mallal S, Nolan D, Witt C, et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 2002; 359: 727–732. [DOI] [PubMed] [Google Scholar]
- 51.Coenen MJ, de Jong DJ, van Marrewijk CJ, et al. Identification of Patients With Variants in TPMT and Dose Reduction Reduces Hematologic Events During Thiopurine Treatment of Inflammatory Bowel Disease. Gastroenterology 2015; 149: 907–917 e907. [DOI] [PubMed] [Google Scholar]
- 52.Simon T, Verstuyft C, Mary-Krause M, et al. Genetic determinants of response to clopidogrel and cardiovascular events. New England Journal of Medicine 2009; 360: 363–375. [DOI] [PubMed] [Google Scholar]
- 53.Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373: 309–317. [DOI] [PubMed] [Google Scholar]
- 54.Cavallari LH, Lee CR, Beitelshees AL, et al. Multisite Investigation of Outcomes With Implementation of CYP2C19 Genotype-Guided Antiplatelet Therapy After Percutaneous Coronary Intervention. JACC. Cardiovascular interventions 2017: [DOI] [PMC free article] [PubMed]
- 55.Notarangelo FM, Maglietta G, Bevilacqua P, et al. Pharmacogenomic Approach to Selecting Antiplatelet Therapy in Patients With Acute Coronary Syndromes: The PHARMCLO Trial. J Am Coll Cardiol 2018; 71: 1869–1877. [DOI] [PubMed] [Google Scholar]
- 56.Luzum JA, Cheung JC. Does cardiology hold pharmacogenetics to an inconsistent standard? A comparison of evidence among recommendations. Pharmacogenomics 2018; 19: 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Verhoef TI, Ragia G, de Boer A, et al. A Randomized Trial of Genotype-Guided Dosing of Acenocoumarol and Phenprocoumon. New England Journal of Medicine 2013; 369: 2304–2312. [DOI] [PubMed] [Google Scholar]
- 58.Kimmel SE, French B, Kasner SE, et al. A Pharmacogenetic versus a Clinical Algorithm for Warfarin Dosing. New England Journal of Medicine 2013; 369: 2283–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pirmohamed M, Burnside G, Eriksson N, et al. A Randomized Trial of Genotype-Guided Dosing of Warfarin. New England Journal of Medicine 2013; 369: 2294–2303. [DOI] [PubMed] [Google Scholar]
- 60.Ramirez AH, Xu H, Oetjens M, et al. Identifying genotype-phenotype relations in electronic medical record systems: application to warfarin pharmacogenomics. American Heart Association Annual Scientific Sessions 2010:
- 61.Limdi NA, Brown TM, Yan Q, et al. Race influences warfarin dose changes associated with genetic factors. Blood 2015; 126: 539–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gage BF, Bass AR, Lin H, et al. Effect of Genotype-Guided Warfarin Dosing on Clinical Events and Anticoagulation Control Among Patients Undergoing Hip or Knee Arthroplasty: The GIFT Randomized Clinical Trial. JAMA : the journal of the American Medical Association 2017; 318: 1115–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Syn NL, Wong AL, Lee SC, et al. Genotype-guided versus traditional clinical dosing of warfarin in patients of Asian ancestry: a randomized controlled trial. BMC medicine 2018; 16: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roth JA, Boudreau D, Fujii MM, et al. Genetic Risk Factors for Major Bleeding in Warfarin Patients in a Community Setting. Clin Pharmacol Ther 2014; 95: 636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kawai VK, Cunningham A, Vear SI, et al. Genotype and risk of major bleeding during warfarin treatment. Pharmacogenomics 2014; 15: 1973–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De T, Alarcon C, Hernandez W, et al. Association of Genetic Variants With Warfarin-Associated Bleeding Among Patients of African Descent. JAMA : the journal of the American Medical Association 2018; 320: 1670–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pulley JM, Denny JC, Peterson JF, et al. Operational Implementation of Prospective Genotyping for Personalized Medicine: The Design of the Vanderbilt PREDICT Project. Clin Pharmacol Ther 2012; 92: 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bielinski SJ, Olson JE, Pathak J, et al. Preemptive genotyping for personalized medicine: design of the right drug, right dose, right time-using genomic data to individualize treatment protocol. Mayo Clinic proceedings 2014; 89: 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Relling MV, Klein TE. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin Pharmacol Ther 2011; 89: 464–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Swen JJ, Wilting I, de Goede AL, et al. Pharmacogenetics: from bench to byte. Clin Pharmacol Ther 2008; 83: 781–787. [DOI] [PubMed] [Google Scholar]
- 71.Bank PCD, Caudle KE, Swen JJ, et al. Comparison of the Guidelines of the Clinical Pharmacogenetics Implementation Consortium and the Dutch Pharmacogenetics Working Group. Clin Pharmacol Ther 2018; 103: 599–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kazi DS, Garber AM, Shah RU, et al. Cost-effectiveness of genotype-guided and dual antiplatelet therapies in acute coronary syndrome. Ann Intern Med 2014; 160: 221–232. [DOI] [PubMed] [Google Scholar]
- 73.Schackman BR, Scott CA, Walensky RP, Losina E, Freedberg KA, Sax PE. The cost-effectiveness of HLA-B*5701 genetic screening to guide initial antiretroviral therapy for HIV. AIDS (London, England) 2008; 22: 2025–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Verbelen M, Weale ME, Lewis CM. Cost-effectiveness of pharmacogenetic-guided treatment: are we there yet? The pharmacogenomics journal 2017; 17: 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lesko LJ, Zineh I. DNA, drugs and chariots: on a decade of pharmacogenomics at the US FDA. Pharmacogenomics 2010; 11: 507–512. [DOI] [PubMed] [Google Scholar]
- 76.Gammal RS, Crews KR, Haidar CE, et al. Pharmacogenetics for Safe Codeine Use in Sickle Cell Disease. Pediatrics 2016; 138: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chung CP, Callahan ST, Cooper WO, et al. Outpatient Opioid Prescriptions for Children and Opioid-Related Adverse Events. Pediatrics 2018; 142: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rattanavipapong W, Koopitakkajorn T, Praditsitthikorn N, Mahasirimongkol S, Teerawattananon Y. Economic evaluation of HLA-B*15:02 screening for carbamazepine-induced severe adverse drug reactions in Thailand. Epilepsia 2013; 54: 1628–1638. [DOI] [PubMed] [Google Scholar]
- 79.Chong HY, Mohamed Z, Tan LL, et al. Is universal HLA-B*15:02 screening a cost-effective option in an ethnically diverse population? A case study of Malaysia. The British journal of dermatology 2017; 177: 1102–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen Z, Liew D, Kwan P. Effects of a HLA-B*15:02 screening policy on antiepileptic drug use and severe skin reactions. Neurology 2014; 83: 2077–2084. [DOI] [PubMed] [Google Scholar]
- 81.Van Driest SL, Shi Y, Bowton EA, et al. Clinically actionable genotypes among 10,000 patients with preemptive pharmacogenomic testing. Clin Pharmacol Ther 2014; 95: 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ji Y, Skierka JM, Blommel JH, et al. Preemptive Pharmacogenomic Testing for Precision Medicine: A Comprehensive Analysis of Five Actionable Pharmacogenomic Genes Using Next-Generation DNA Sequencing and a Customized CYP2D6 Genotyping Cascade. The Journal of molecular diagnostics : JMD 2016; 18: 438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bush WS, Crosslin DR, Owusu-Obeng A, et al. Genetic variation among 82 pharmacogenes: The PGRNseq data from the eMERGE network. Clin Pharmacol Ther 2016; 100: 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schildcrout JS, Denny JC, Bowton E, et al. Optimizing Drug Outcomes Through Pharmacogenetics: A Case for Preemptive Genotyping. Clin Pharmacol Ther 2012; 92: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Peterson JF, Field JR, Unertl KM, et al. Physician response to implementation of genotype-tailored antiplatelet therapy. Clin Pharmacol Ther 2016; 100: 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Starren J, Williams MS, Bottinger EP. Crossing the omic chasm: a time for omic ancillary systems. JAMA : the journal of the American Medical Association 2013; 309: 1237–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Herr TM, Bielinski SJ, Bottinger E, et al. Practical considerations in genomic decision support: The eMERGE experience. Journal of pathology informatics 2015; 6: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sanseau P, Agarwal P, Barnes MR, et al. Use of genome-wide association studies for drug repositioning. Nature biotechnology 2012; 30: 317–320. [DOI] [PubMed] [Google Scholar]
- 89.Nelson MR, Tipney H, Painter JL, et al. The support of human genetic evidence for approved drug indications. Nat Genet 2015; 47: 856–860. [DOI] [PubMed] [Google Scholar]
- 90.Pulley JM, Shirey-Rice JK, Lavieri RR, et al. Accelerating Precision Drug Development and Drug Repurposing by Leveraging Human Genetics. Assay and drug development technologies 2017; 15: 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rastegar-Mojarad M, Ye Z, Kolesar JM, Hebbring SJ, Lin SM. Opportunities for drug repositioning from phenome-wide association studies. Nature biotechnology 2015; 33: 342–345. [DOI] [PubMed] [Google Scholar]
- 92.Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003; 34: 154–156. [DOI] [PubMed] [Google Scholar]
- 93.Cohen JC, Boerwinkle E, Mosley TH Jr., Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354: 1264–1272. [DOI] [PubMed] [Google Scholar]
- 94.Crosby J, Peloso GM, Auer PL, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med 2014; 371: 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stitziel NO, Won HH, Morrison AC, et al. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med 2014; 371: 2072–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Flannick J, Thorleifsson G, Beer NL, et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet 2014; 46: 357–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stitziel NO, Stirrups KE, Masca NGD, et al. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N Engl J Med 2016: [DOI] [PMC free article] [PubMed]
- 98.Dewey FE, Gusarova V, O’Dushlaine C, et al. Inactivating Variants in ANGPTL4 and Risk of Coronary Artery Disease. N Engl J Med 2016; 374: 1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Abul-Husn NS, Cheng X, Li AH, et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N Engl J Med 2018; 378: 1096–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Whiting P, Al M, Burgers L, et al. Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost-effectiveness analysis. Health technology assessment (Winchester, England) 2014; 18: 1–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. New England Journal of Medicine 2015; 373: 220–231. [DOI] [PubMed] [Google Scholar]
- 102.Mehta A, Ramachandra CJA, Singh P, et al. Identification of a targeted and testable antiarrhythmic therapy for long-QT syndrome type 2 using a patient-specific cellular model. Eur Heart J 2018; 39: 1446–1455. [DOI] [PubMed] [Google Scholar]
- 103.Behr ER, Ritchie MD, Tanaka T, et al. Genome wide analysis of drug-induced torsades de pointes: lack of common variants with large effect sizes. PloS one 2013; 8: e78511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Strauss DG, Vicente J, Johannesen L, et al. Common Genetic Variant Risk Score Is Associated With Drug-Induced QT Prolongation and Torsade de Pointes Risk: A Pilot Study. Circulation 2017; 135: 1300–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ward J, Graham N, Strawbridge RJ, et al. Polygenic risk scores for major depressive disorder and neuroticism as predictors of antidepressant response: Meta-analysis of three treatment cohorts. PloS one 2018; 13: e0203896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rashkin SR, Chua KC, Ho C, et al. A Pharmacogenetic Prediction Model of Progression-Free Survival in Breast Cancer using Genome-Wide Genotyping Data from CALGB 40502 (Alliance). Clin Pharmacol Ther 2018: [DOI] [PMC free article] [PubMed]
- 107.Schwartz GG, Olsson AG, Abt M, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med 2012; 367: 2089–2099. [DOI] [PubMed] [Google Scholar]
- 108.Tardif JC, Rheaume E, Lemieux Perreault LP, et al. Pharmacogenomic determinants of the cardiovascular effects of dalcetrapib. Circulation. Cardiovascular genetics 2015; 8: 372–382. [DOI] [PubMed] [Google Scholar]
- 109.Rautureau Y, Deschambault V, Higgins ME, et al. Adenylate Cyclase Type 9 (ADCY9) Inactivation Protects from Atherosclerosis Only in the Absence of Cholesteryl Ester Transfer Protein (CETP). Circulation 2018: [DOI] [PubMed]
- 110.van der Wouden CH, Cambon-Thomsen A, Cecchin E, et al. Implementing Pharmacogenomics in Europe: Design and Implementation Strategy of the Ubiquitous Pharmacogenomics Consortium. Clin Pharmacol Ther 2017; 101: 341–358. [DOI] [PubMed] [Google Scholar]
- 111.Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther 2013; 93: 324–325. [DOI] [PMC free article] [PubMed] [Google Scholar]