

Abstract
The possibility of graphene oxide (GO) exposure to the environment has spurred several studies investigating the fate of this nanoparticle (NP). However, there is currently little or no data on the fate of GO in estuarine and marine waters. This study investigated the aggregation, sedimentation and transformation of GO in saline waters, considering the role of salinity (0–50 ‰), light (visible light and solar irradiation), aging, among others. The attachment efficiency of GO reached unity at 1.33 ‰. The sedimentation rate of GO increased with salinity up to 10 ‰ after which it decreased due to formation of ramified GO agglomerates and media density. Based on the sedimentation rate determined at 30 ‰ (0.121 m/d), the residence time of GO agglomerates in the euphotic zone of typical open oceans will exceed 500 days. Aging in the presence of visible light increased the relative abundance of the GO’s aromatic (C−C/C=C) fraction, reducing the NP. Reduction of GO in visible light was confirmed via UV-Vis and Raman spectroscopic techniques. Reduction of GO was faster under solar irradiation. This study demonstrates that when introduced into saline waters GO will undergo a range of transformations affecting its fate and potential effects to aquatic organisms.
Graphical Abstract
INTRODUCTION
Graphene oxide (GO) is composed of one or more sheets of graphenic carbon atoms that are covalently bonded to oxygen-based functional groups. The functional groups of GO are mainly epoxide (C−O−C) and hydroxyl (C−OH) at the basal plane, while the edges or defective sites are dominated by ester and carboxyl (O−C=O), and carbonyl (C=O) groups.1 GO has applications in consumer and industrial products: For instance, GO is bactericidal,2–4 which has led to its use as antibacterial coatings.2 GO-based coatings and nanocomposite matrices, with high corrosion-prevention efficiency, even in saline waters, have also been developed.5–8 In addition, several laboratory-scale studies have shown that GO is effective for water treatment.9
The fate of GO has been extensively studied in synthetic media such as pure water with or without salts,10–12 and less so in waters collected from natural freshwater sources including wastewater effluent,11 rivers,10, 11 and lakes.10, 13, 14 However, the fate of GO in saline waters, which constitutes most of the earth’s hydrosphere, is completely unknown. Existing studies have shown that the range of critical coagulation concentration (CCC) of NaCl and CaCl2 for GO is 38–188 mM and 0.9–2.6 mM, respectively, depending on the physicochemical properties of the NP and water chemistry.11, 14–18 A similar range of CCC was reported for radioactive elements.19 It is however unclear how these values can be used to predict the fate of GO in saline waters (natural waters with 0 ‰ < salinity ≤ 35‰), which typically contain multiple cations and anions.20 More so, natural waters contain biogenic and geogenic materials (such as suspended particles and macromolecules), and anthropogenic pollutants, which may be difficult to simulate in simple artificial systems. Thus, determining the fate of NPs in simple water systems is useful for understanding the basic mechanisms behind relevant processes but such knowledge cannot always be directly extrapolated to more complex natural waters. There is therefore a need for further studies conducted in natural waters.21–25
GO is not as chemically stable as graphene in aqueous media due to its functionalization. Transformation of GO can be caused by high intensity light (including sunlight),26–29 disinfectants,27, 30 biological processes and products,31 and high temperature. The environmental fate of transformed GO is likely different from that of the pristine material, as transformations may lead to changes in the morphology and functionalization of the NP.26, 27, 29, 32 The bactericidal property of GO has been correlated with its particle size and surface functionalization,2, 3 which suggests that changes to GO’s physicochemical properties will have impacts on its effects to organisms. It is therefore important to understand the physicochemical transformations of GO in natural waters and how the transformation of GO is affected by important natural media parameters such as salinity and light.
The objective of this study was to determine the colloidal stability and transformations of GO in saline waters. To our knowledge, this is the first study investigating the fate of GO in estuarine and marine systems, which are predicted as important sinks for engineered NPs.33
MATERIALS AND METHODS
GO characterization
Pristine, few-layer GO (2–4 sheets, purity = 99%) was obtained as dry powder from Cheap Tubes (Cambridgeport, VT). The GO powder and suspensions prepared in deionized (DI) water (0 ‰; Millipore Milli-Q Element, Billerica, MA) and seawater (30 ‰) were characterized as described in the supporting information (SI) section S1.
Marine waters and stocks preparation
Reconstituted saline waters (0–50 ‰) were prepared by combining 100 ‰ natural brine (prepared from 30 ‰ seawater, Narragansett Bay, RI) and DI water. The waters were filtered using 0.22 μm Millipore filter membrane before use to remove particulate materials that might have interfered with dynamic light scattering (DLS) analysis. The preparation of stocks of GO and natural organic matter (NOM) is fully described in SI sections S2 and S3. In this study, humic acid (HA) and extracellular polymeric substances (EPS) produced by a marine phytoplankton, Tetraselmis sp, were used as representatives of NOM.
Sedimentation and agglomeration kinetics
Sedimentation and agglomeration of GO were determined over a range of media salinity (0–50 ‰ and 0–30 ‰ respectively), NOM concentrations (1–10 mg/L dissolved organic carbon or DOC), and GO state (pristine and aged) using well-established methods.23, 34–37 Sedimentation experiments were conducted for 28 d, and sampling was carried out at 0, 2, 4, 6 h, and 1, 2, 3, 7, 14, 21, and 28 d. Full description of the sedimentation experiment is provided in SI section S4. Agglomeration kinetics of GO was studied at increasing salinity to determine if the typical reaction-limited (RLCA) and diffusion-limited (DLCA) clustering agglomeration regimes will be identified. The initial agglomeration rate constant (k) and attachment efficiencies (α) of GO at different salinities were determined as described in SI section S5.
Irradiation
Suspensions of GO prepared in 0 and 30 ‰ media were exposed to simulated sunlight obtained using an Atlas SunTest CPS solar simulator equipped with a 1 kW xenon arc lamp. The suspensions were placed in 35 mL quartz tubes submerged in a 20 °C thermostated water bath (Cole-Parmer Polystat recirculating bath) during irradiation. The incident light intensity at the tube surface, summed from 290 to 700 nm, was 0.07 W/cm2. Dark control samples were covered with aluminum foil and irradiated concurrently. After irradiation, samples were analyzed for zeta (ζ) potential, agglomeration and sedimentation kinetics, and chemical changes via X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy. To obtain samples for XPS and Raman analyses, irradiated GO suspension was filtered with the 0.22 μm Millipore membrane and the retained materials were lyophilized (Virtis Advantage Plus).
Statistical analyses
Studies were performed in triplicate unless otherwise noted. Normality and homogeneity of variance were checked for all analyses. ANOVA was performed, followed by post-hoc Tukey’s HSD test. A p value ≤ 0.05 was used to determine significance. Statistical analyses were performed using RStudio (Version 1.1.453).
RESULTS AND DISCUSSIONS
Physicochemical properties of GO
According to the manufacturer, the GO has a lateral size of 300–800 nm, which was confirmed via transmission electron microscopy (TEM) as shown in Figure 1A. The height profile obtained from atomic force microscopy (AFM) topography showed the GO had a thickness of about 0.5–1.5 nm (Figures 1B and S1), which corresponds to 1–2 GO layers.38 In the Fourier transform infrared (FTIR) spectrum of GO (Figure S2A), we observed OH stretch around 3200 cm−1 (due to the presence of adsorbed water and alcohol groups) superimposed on the OH stretch of the COOH group. The peaks at 1722 cm−1 and 1586 cm−1 were assigned to the C=O stretch of the undissociated COOH group and C=C stretches from unoxidized graphitic domain, respectively. The peak at 1259 cm−1 corresponds to C–OH stretch of a phenol group, while 1045 cm−1 was attributed to C–O stretching vibrations of the C–O–C group. The presence of these functional groups on GO was corroborated by XPS, which confirmed the presence of sp2/sp3 C (C=C/C−C), C−O−C/C−OH, C=O and O−C=O groups at binding energy (BE) of 284.8 eV (56.0%), 286.7 eV (37.6%), 287.8 eV (3.9%), and 288.8 eV (2.5%), respectively (Figure 1C). The characteristic D band (~1350 cm−1) and G band (~1580 cm−1) of carbonaceous materials were identified in the Raman spectrum of GO (Figure 1D). The ratio of the intensities of the D and G bands (ID/IG) was determined as 1.01. X-ray diffraction (XRD) showed the (002) diffraction peak of GO at 2θ = 9.8° (Figure S2B), indicating the presence of oxygen functional groups between the sheets of GO.
Figure 1.

Characterization of the graphene oxide (GO) used in this study via (a) transmission electron microscopy, (b) atomic force microscopy, (c) X-ray photoelectron spectroscopy, and (d) Raman spectroscopy
The average hydrodynamic size of GO was determined as 639.3 ± 50.4 nm at 0 ‰ but it increased to 1804 ± 139.4 nm at 30 ‰ due to fast agglomeration of the NP, as shown by the dominance of larger-sized particles at 30 ‰ (Figure S2C). Fast agglomeration at full strength seawater (30 ‰) is due to decreased electrostatic repulsion between the GO particles caused by elevated charge screening by seawater ions: The ζ potential of GO decreased in magnitude from −36.7 ± 1.3 mV at 0 ‰ to −5.3 ± 2.7 mV at 30 ‰. In media (0, 1, and 30 ‰), we observed the characteristic GO absorption peaks at 230 nm and 300 nm (Figure S2D), which correspond to π→π* transitions for the C=C bonding and n→π* transitions of the carbonyl groups (C=O), respectively.24, 28 An additional peak was observed around 210 nm when GO was dispersed in 30 ‰ media, possibly due to morphological changes or the agglomeration of GO during spectral measurements.
Influence of salinity
Agglomeration
Preliminary agglomeration kinetics experiments were conducted in simple media using NaCl and CaCl2 as electrolytes. The DLCA and RLCA regimes were identified for both conditions, and the corresponding CCC values were determined as 138 mM NaCl and 1.73 mM CaCl2 (Figure S3). These values fall within the range reported in the literature (38–188 mM NaCl and 0.9–2.6 mM CaCl2 11, 15–18). The CCC[Ca2+]/CCC[Na+] ratio yielded a proportionality fraction of z−6.3, where z is valency of Ca2+/valency of Na+. This agrees well with the classical Schulze-Hardy rule, which predicts a proportionality fraction of z−6. These CCC values cannot be used to accurately predict the fate of GO in estuarine and marine waters where multiple (monovalent and multivalent) ions co-occur. As a result, agglomeration kinetics was also studied in serially diluted natural brine.
As expected, GO agglomerated in saline waters and the agglomeration rate increased with salinity. Attachment efficiency (α) was determined for each salinity tested and are shown in Figure 2A. The presence of DLCA and RLCA regimes in the saline water tests indicated that the agglomeration of GO in marine waters is strongly affected by electrostatic interactions. Increasing salinity (ionic strength) reduced the electrostatic energy barrier between GO particles and deepens the secondary minimum, which promotes agglomeration and increases α. Critical coagulation salinity or CCS (analogous to CCC for simple salts), defined as the salinity at which α reached unity, was identified in a plot of log10[Salinity] versus log10α (Figure 2A). The CCS for GO was determined as 1.33 ‰, which implies that GO will be colloidally unstable at salinity ≥ 1.33 ‰.
Figure 2.

Colloidal stability of graphene oxide (GO) in saline waters. (a) Attachment efficiency of GO as a function of salinity (α reached unity at ~1.33 ‰), (b) sedimentation rate of GO as a function of salinity, (c) sedimentation rate of pristine and aged GO, (d) agglomeration rate constant of pristine and aged GO (box plot shows median, 25% and 75% quantiles and range)
Although the agglomeration rate of GO peaks at 1.33 ‰, further increase in salinity may still have observable influences on the colloidal stability of GO because of increasing concentrations of multivalent cations: In addition to charge screening, multivalent cations in saline waters such as Ca2+, Mg2+, Sr2+, etc. can cross the electric double layer and bind directly with GO’s graphitic C (via cation-π interactions) and functional groups (such as COOH and OH), leading to cation-bridging of GO particles.14, 16, 17, 39 One significance of cation-bridging is that edge-to-edge agglomeration of different GO particles may become important since there is abundant COOH at the edges.17 More so, these multivalent cations can lead to formation of 3D GO structures.16 We observed that the morphology of GO agglomerates formed at high salinities was different (larger and less compact) than the agglomerates formed at lower salinities (Figure S4).
Sedimentation
As the GO particles agglomerated to much larger sizes in saline waters, sedimentation becomes important for determining the fate of the NPs. The sedimentation rate of GO (Vs), determined over 28 d, at salinities between 0 and 50 ‰ is presented in Figure 2B. The Vs of GO was strongly dependent on salinity (p < 0.001); and GO agglomerates settled faster as salinity increased. However, at salinities above 10 ‰ (Vs = 0.145 m/d), Vs decreased and reached 0.108 m/d at 50 ‰. The decrease in the sedimentation rate of GO above 10 ‰ is due to the combined effects of GO morphology and media density at high salinities. In the presence of multivalent cations, GO forms large agglomerates that can grow into microstructures.16 Since GO is in the DLCA regime above 1.33 ‰, the agglomerates formed above 1.33 ‰ are not well-packed. With increase in salinity and thus, cation-bridged GO particles, the agglomerates formed are even more ramified (Figure S4), which can increase hydrodynamic drag and buoyancy much more than in compact agglomerates. In addition, the density of media increases with salinity, which can also reduce Vs, especially since ramified agglomerates have lower density than compact agglomerates of the same mass.
Based on studies previously conducted in simple media, the fate of GO would have been predicted to be the sediment phase of saline waters. But based on this study, more accurate predictions can be made about the fate of GO in estuarine and marine waters. For instance, given the Vs determined for GO in typical seawater conditions (0.121 m/d at 30 ‰), it will take GO agglomerates 536 d (or about 1.5 yr) to settle out of the euphotic zone of an ocean water column (assuming a depth of 65 m for the euphotic zone). The euphotic zone is the water layer that receives enough sunlight for photosynthesis to occur so primary production mainly occurs there.20 Hence, although GO will eventually settle out of water phase, organisms at the base of the pelagic food chain in estuarine and marine systems may still be exposed to the NP over a long period of time. It should be noted that the interactions of GO with aquatic organisms (which may increase or decrease Vs), suspended particles (which are likely to increase Vs), and other pollutants in natural waters were not considered in this studied and they may impact the Vs predicted here. In addition, the physicochemical properties of GO such as lateral size, oxygen/functionalization ratio, and layer thickness may also affect Vs, and should be tested in future studies.
Influence of aging
As discussed earlier, complete removal of GO from the water matrix by sedimentation will occur on the orders of months to years, or much longer, depending on their interactions with other biotic and abiotic factors. Hence, it is important to understand how the fate of GO may change while it is suspended or settling in aqueous media. In this study, we considered the fate of GO aged in low-intensity light conditions (white fluorescent lamps (16:8 light:dark cycle)), to simulate the irradiation scenario that may occur below the surface of natural waters.
Sedimentation
At the end of the sedimentation study performed to study the effect of salinity, the sample tubes containing GO in 0–50 ‰ media (for the previous 28 d) were shaken vigorously by hand to resuspend the NPs. Samples were then analyzed for the next 28 d to see the impact of (28-d) aging on the sedimentation of GO. Aging of GO significantly affected the rate at which the NP settled out of media (p < 0.05). As shown in Figure 2C, the sedimentation rate of aged GO was generally lower than that of pristine GO, especially in saline waters. At typical seawater conditions (that is, 30 ‰), Vs decreased from 0.121 m/d for pristine GO to 0.079 m/d for 28-d aged GO. This was regardless of the similarity in the ζ potential of GO before and after aging (Figure S5).
To determine if slower sedimentation of aged GO was due to slower agglomeration, the initial agglomeration rate constant (k) of pristine and aged GO was determined at 0.7 ‰ (Eq. S2). The selection of 0.7 ‰ was to ensure that GO was still in the RLCA regime where α < 1. The k of aged GO (92.2 nm/min) was significantly (p < 0.005) greater than that of pristine GO (49.8 nm/min) (Figure 2D). This agrees well with the increased C/O ratio of GO after aging, which implies decreased hydrophilicity. We therefore ascribed the decrease in the sedimentation rate of aged GO to the morphological changes in GO agglomerates in the media. As explained earlier, the agglomerates formed by GO in saline waters are large and ramified. More so, after interacting for 28 d, the bulk of the particles are likely in “deep primary minimum” (where agglomeration is irreversible) in the saline waters. As a result, the vigorous shaking introduced at the beginning of the aged GO sedimentation experiment did not re-disperse the agglomerates already formed. Slower sedimentation of aged GO thus resulted from the lower density of the ramified agglomerates relative to the individual particles/smaller-sized agglomerates that were present at the beginning of the sedimentation studies with pristine GO. The size of GO at 0, 1, and 30 ‰ over 7 d is shown in Figure S6, which clearly demonstrates size increase/ramification in the saline waters. This finding shows that the sedimentation rate of GO may vary over time and/or based on the agglomeration state of the NP.
Chemical transformation
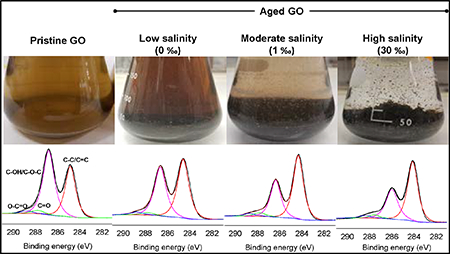
The elemental composition and functional groups of GO particles aged in 0, 1, and 30 ‰ media for 28 d was determined via XPS. Carbon (C) and oxygen (O) were observed in GO sheets aged in the waters, with slight differences in the C/O ratio (Figure 3). At 1 and 30 ‰, some of the C and O atoms may have originated from the media (e.g., NOM present in the brine used for saline water preparation) but this potential interference is at best minimal. Bias of GO atomic ratios arising from NOM adsorption was suppressed by ensuring the mass ratio of GO to media NOM was very high (70 or more) when suspensions were prepared for XPS analysis. In saline waters, sodium (Na) and chlorine (Cl) were also detected, which were likely adsorbed by GO from the waters. Overall, the relative abundance of the O atoms detected decreased slightly upon aging of GO in media. As shown in Figures 3 and S7, the relative abundance of sp2/sp3 C increased with aging, particularly at 1 and 30 ‰ while the C−OH/C−O−C ratio decreased in the same treatments. Although the abundance of C=O group did not change remarkably, O−C=O group increased with salinity upon aging. These results indicate that GO was reduced in the saline waters. The C−O (C−OH, C−O−C) group, which is located at the basal plane of GO, was the most-degraded species upon GO aging in saline media.
Figure 3.

High resolution XPS analysis of the C 1s peak of (a) pristine GO powder, and GO aged for 28 days in (b) 0 ‰, (c) 1 ‰, and (d) 30 ‰. A summary of the abundance of the functional groups in the C 1s peaks is shown in the table as well as the elemental composition of the pristine and aged GO particles
The reduction of GO with aging was also investigated via UV-Vis spectroscopy. The peak corresponding to π→π* transitions observed at 230 nm in pristine GO suspension and at the beginning of aging red-shifted to ~240 nm by day 28 (Figure S8) in all three media. This suggests that the electronic conjugation within GO sheets was restored. In addition, the peak due to n→π* transitions of the C=O groups was no longer observed after 28-d aging. These observations agree with XPS analysis that GO was reduced upon aging in the fresh and saline media. Raman spectroscopy was also used to analyze the samples (Figure S9). The ID/IG ratio indicates the degree of disorder or defects of GO.24, 28, 30, 40 After aging for 28 d, the ID/IG ratio of GO was 0.95, 0.97, and 1.13, in 0, 1, and 30 ‰, respectively. Increase in ID/IG ratio was also reported when GO was reduced using UV light, sunlight, and chemical methods.26, 41, 42 Increased ID/IG ratio upon the reduction of GO has been attributed to the formation of more defects/edges, and can be magnified by the fragmentation of the graphenic sheet during reduction.26, 41 The trend of the ID/IG ratio (and the relative abundance of sp2/sp3 C bonds from XPS analysis) suggest that salinity plays an important role in the reduction of GO in natural waters, and should be investigated further.
Previous studies have demonstrated the reduction of GO via high-energy irradiations, such as UV43 and sunlight.26, 27 Here, we showed that low-intensity irradiation from fluorescence lamps with negligible UV (UV intensity = 6.43 μW/cm2)22 can slowly reduce GO in aqueous media. Cool white fluorescent lamps mainly emit light in the visible region (400–700 nm), with spectral peaks around 560 nm. The energy per photon at 560 nm is 2.22 eV, which exceeds the activation energies for the photolysis of C−OH (0.7 eV) and C−O−C (1.9–2.1 eV) bonds.44, 45 The activation energy for the photolysis of C=O and O−C=O groups is 3–4 eV,44, 45 which largely exceeds the energy of most of the photons emitted by the fluorescent lamp (e.g. 3.10 eV at 400 nm). Photolysis may also cause the migration of OH within the basal plane of GO (activation energy = 0.32 eV), leading to eventual OH localization at defects/edges, evolution of water, and concomitant reduction of GO.45 The visible light emitted by the fluorescent lamps, which also constitutes a large fraction of light at the surface of natural waters, reduced GO by degrading the C−OH/C− O−C group. In the water column, attenuation of light energy further makes visible light more important, especially in the photic zone. This implies that over time, pelagic organisms will be exposed to photo-reduced forms of GO as the NP settles out of natural waters.
Influence of solar irradiation
We investigated the transformation of sunlight-irradiated GO at 0 and 30‰ for up to 24 h and determined the influence of solar irradiation on the physicochemical properties and stability of GO. Prolonged irradiation (that is, > 24 h) was not carried out as the amount of intense irradiation GO will be exposed to in natural waters will decrease over time as the NP settles below the water surface. For sedimentation studies, the GO stock suspension was irradiated (for 12 or 24 h) in media (0 or 30 ‰). After irradiation, the stock was diluted with the media of the same salinity (0 or 30 ‰) to achieve the desired initial concentration.
Chemical transformation
Solar irradiation decreased the oxygen functionalization of GO, marked by increase in (1) C/O ratio and (2) abundance of C−C/C=C group, relative to pristine GO. As shown in Figure 4A, at 0 ‰ C/O ratio was 3.1, 3.4, and 3.6 after solar irradiation for 6, 12, and 24 h, respectively. The ratio was lower when irradiated at 30 ‰; where the C/O ratio was 2.8, 2.9, and 3.2 after 6, 12, and 24 h, respectively. Consistent with the aging studies carried out under visible light, the most decrease was observed in the C−OH/C−O−C group, but also in the C=O group with solar irradiation. In all samples, the relative abundance of the O−C=O group increased (Table S1). The O−C=O group is very stable against photodegradation, particularly when in close proximity to an OH group (which stabilizes it via H-bonding).46 FTIR data, shown in Figure S10, corroborated the XPS results. The peak at ~1045 cm−1, which was attributed to C–O stretching vibrations of the C–O–C group decreased upon solar irradiation (with a clear trend at 0 ‰). The peak at 1259 cm−1 which corresponds to C–OH stretch of a phenol group also decreased, much more so at 30 ‰. Another important difference between the FTIR spectra obtained in the two media is the disappearance of the peak around 1722 cm−1 (assigned to the C=O stretch of the undissociated COOH) in the saline water. This is likely due to the dissociation of the COOH groups at 30 ‰ (pH 8.3) since the pKa of GO’s COOH group is 4.3 when there is an OH group present in the ortho position and 6.6 when the COOH group is isolated.29, 46 Only a partial dissociation of COOH groups must have occurred at 0 ‰ (unadjusted pH = 5.5). In addition, the cations in the 30 ‰ media (such as Na+, and Ca2+) can form COO-cation groups, which can lead to a shift in the position of the undissociated COOH peak.12 Unlike the fluorescent lighting, the irradiation from the solar simulator had a high intensity (0.07 W/cm2) and contained light in the UV (290–400 nm) and visible light (400–700) regions. This explains the rapid reduction of GO under solar irradiation such that the photoproducts (rGO) obtained after (less than) a day of irradiation had a higher C/O ratio than GO exposed to visible light for 28 d.
Figure 4.

(a) Carbon/oxygen ratio and (b) zeta potential of GO suspension prepared in 0 or 30 ‰ media and irradiated for up to 12 h. Sedimentation rate of GO suspension prepared in 0 or 30 ‰ media and irradiated for (c) 12 h and (d) 24 h
Reduction of GO (band gap = 2.7 eV)47 during UV and solar irradiation has been attributed to its photoreactivity, which causes electron-hole pair formation within the NP.26 The positive hole formed in the valence band (h+vb) and the electron promoted to the conduction band (e-cb) concurrently disproportionate GO to rGO, CO2, H2, hydroxylated and/or carboxylated polycyclic aromatic hydrocarbons (PAHs), reactive oxygen species (ROS), and H2O2.26–28, 40 Unlike the 0 ‰ media, the 30 ‰ media contains inorganic species such as Cl-, SO42-, HCO3-, Fe, Cu, etc. which can scavenge holes and/or electrons,48, 49 potentially reducing the rates of GO’s transformation. In addition, the suspended particles, ions and NOM present in the 30 ‰ media may absorb some of the irradiation, reducing the overall amount of light readily available for the transformation of GO in the saline waters (relative to the amount of light readily available for GO transformation in the 0 ‰ media). Thus, a slower reduction of GO may be expected in natural waters due to the presence of organic and inorganic scavengers and light absorbers. It should however be noted that indirect photochemical transformation may also be enhanced by the presence of NOM in natural waters.50
The ID/IG ratio of GO was 0.87–1.0 and 1.09–1.15 when exposed to solar irradiation time at 0 ‰ at 30 ‰, respectively (Figure S11). This is similar to the effect on ID/IG ratio observed when GO was aged in the presence of visible light in the two media. Increased This finding further reiterates the importance of salinity (ionic strength) in the photoreduction of GO. Although Increase ID/IG ratio of GO upon solar irradiation has been adduced to reduction of GO, the stark difference in the composition of both media will not allow us to confidently conclude that GO was reduced faster at 30 ‰ than at 0 ‰ without further systematic studies.
Physicochemical properties and stability
The changes in the lateral size of GO due to irradiation could not be clearly determined via microscopy due to the high salt content at 30 ‰. The ζ potential of GO in 0 ‰ media significantly (p < 0.05) decreased in magnitude after solar irradiation for 12 and 24 h from −36.7 ± 1.3 mV to −30.0 ± 3.4 mV and −30.4 ± 2.9 mV, respectively. At 30 ‰, there was no significant change in the ζ potential of GO after solar irradiation (Figure 4B). The overall changes in the surface charge of GO (as indicated by ζ potential) after solar irradiation is minimal compared to the overall changes in its chemical composition and functionalization. The surface charge of GO mainly originates from the ionization of the O−C=O and C−OH groups. The O−C=O group was not strongly affected by solar irradiation as mentioned earlier, hence, the surface charge of GO was not remarkably changed by solar irradiation. Regardless, irradiation led to increases in the agglomeration rate of GO, measured at 0.7 ‰ (to enable clear observation of differences in agglomeration as explained earlier). The measured k of GO increased from 49.8 nm/min to 89.9 nm/min and 102.1 nm/min after 12 h and 24 h irradiation, respectively (Figure S12). Despite the increased agglomeration rates, the sedimentation rate of GO at 0 and 30 ‰ decreased after 12 and 24 h solar irradiation (but the decrease was only significant for the sample irradiated for 24 h and measured at 30 ‰; Figure 4C–D). Like the irradiated samples, the settling rates of control samples (covered with aluminum foil during irradiation) were equivalent to or even lower than that of the pristine samples. Hence, irradiation only partly accounts for reduced sedimentation of photo-treated GO samples. We hypothesize that changes in the morphology of GO agglomerates during the 12 or 24 h irradiation time (as explained earlier) also played a role in the slower sedimentation of the samples.
Influences of NOM and initial GO concentration
Role of NOM
It should be noted that the natural brine used for this study contained some intrinsic NOM, as indicated by a DOC of 0.99 ± 0.02 mg-C/L when the brine was diluted with DI water to 30 ‰. The CCS of GO increased from 1.33 ‰ in the presence of only intrinsic NOM (Figure 2A) to 5.39 ‰ and 5.07 ‰ in the presence of additional 1 mg-C/L of HA and EPS, respectively (Figure 5A–B). The intrinsic NOM content at the determined CCS (~0.17 mg-C/L at ~5 ‰) was about an order of magnitude smaller than the added NOM (1 mg-C/L); therefore, we concluded that the increase in CCS was a result of the stabilizing ability of the added NOM. In a previous study conducted in artificial media, NOM did not substantially affect the surface charge of GO, thus, the mechanism of improved stability of GO in the presence of NOM was mainly due to steric repulsion.11 At 10 mg-C/L added NOM concentrations, the CCS of GO further increased to 7.10 ‰ in the presence of HA and 8.22‰ in the presence of EPS. As expected, the long-term stability of GO also improved in the presence of HA and EPS as indicated by decreases in sedimentation observed over 28 d (Figure 5C–D) compared to when there was no additional NOM (Figure 2B).
Figure 5.

Colloidal stability of GO in saline waters with added NOM. Attachment efficiency of GO as a function of salinity in the absence and presence of (a) humic acid (HA) or (b) extracellular polymeric substances (EPS). Sedimentation rate of GO as a function of salinity in the presence of (c) 1 mg-C/L HA and EPS, and (d) 10 mg-C/L HA and EPS
As shown in Figure 5C–D, the stabilizing capacity of the algal exudates (EPS) for GO compares to or is better than that of HA, a common surrogate for NOM. This agrees with other studies that have shown that microbial (bacterial and algal) EPS can stabilize engineered NPs, including CuO,51 Fe0,52 TiO2,36, 53 carbon nanotubes,23 etc. in aqueous media. Since EPS was freshly produced by microorganisms while HA was a product of biotic and abiotic degradation of diverse organic materials they varied in composition. FTIR analysis (Figure S13 with peak assignment in Tables S2–S3) showed that the HA contained a higher relative abundance of functional groups associated with amino acids (e.g. NH2 deformation in primary amines or C=O stretching in secondary amides around 1600 cm−1) than EPS. The occurrence of a higher amount of protein in HA was confirmed via calorimetry (modified Bradford assay), which showed that HA and EPS had an average of 5.01 and 2.15 mg-protein/mg-C, respectively. In addition, functional groups associated with nucleic acids were observed in the spectrum of EPS but not HA. For instance, the peak around 1200 cm−1 was assigned to the P=O stretch of phospholipids or nucleic acids and was only observed in the algal exudate. Similarly, using anthrone-sulfuric acid assay, carbohydrate content in EPS and HA was determined as 1.27 and 0.15 mg/mg-C, respectively. Further studies are needed to systematically correlate the composition of NOM (e.g., amount of carbohydrate, protein, etc.) to their GO-stabilizing potential.
Role of initial concentration
The measured k of GO (determined at 0.7 ‰) increased from 49.8 nm/min at an initial GO concentration of 30 mg/L to 141 and 593 nm/min when initial concentration increased to 50 and 100 mg/L, respectively (Figure S14A). This represents a linear increase in k with initial concentration of the NP (r2 = 0.98). As shown in Figure S14B, we found that higher initial concentration increased the settling velocity of GO at 1 ‰ (which is close to the CCS) and 30 ‰ (which is much higher than the CCS). Although the concentration of GO in natural waters is currently unknown, it is expected to be much less than the range of concentration studied here. The high levels studied in this work (10–100 mg/L) were chosen based on the sensitivity of analytical tools currently available for studying fate of the GO in conditions that are similar to the natural environment. We showed in this study that the initial concentration of GO will influence its short-term and long-term stability, but it is not clear how much influence initial concentration will have in natural waters where dispersive forces will continually decrease the NP concentration near the point of release.
ENVIRONMENTAL IMPLICATIONS
GO will agglomerate in saline waters due to charge screening and cation-bridging. Unlike several other NPs which settle faster at higher ionic strength, the settling velocity of GO decreases at salinities above 10 ‰ due to hydrodynamic drag and low particle density (relative to the density of saline water). As GO settles out of water, aging further enhances agglomerate ramification, and thus, buoyancy. This implies that the residence time of GO in natural waters will be much longer than would have been estimated from its agglomeration kinetics or studies conducted in artificial media. At or near the water surface, exposure to sunlight will photo-transform GO. Although the intensity of sunlight decreases below the surface, visible light can photo-reduce GO as it slowly settles out of the euphotic zone. Slow sedimentation of GO implies that pelagic organisms will be exposed to the NP for a relatively long time; and due to transformation and adsorption of ions present in water, the form of GO they will be exposed to will change over time. By the time GO reaches the benthic zone it is likely to be different from the pristine form of GO typically used for benthics exposure studies. Similar to several other studies,24 we observed no toxicity to mysids (Americamysis bahia) at GO concentrations up to 10 mg/L (Figure S15) but oxidative stress and epithelial inflammation have been reported in eastern oysters (Crassostrea virginica) at similar concentrations.54 It is thus paramount for future studies to investigate the potential effects of both pristine and GO transformed in diverse organisms under natural conditions.
Supplementary Material
ACKNOWLEDGEMENTS
The authors appreciate the insightful comments on the draft manuscript by the internal reviewers Dave Katz, Mark Cantwell and Rick McKinney. In addition, Bushra Khan and Stephen Rosso are thanked for help with the experiments. This is NHEERL Contribution ORD-028376. The views expressed in this article are those of the authors and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency. Any mention of trade names, products, or services does not imply an endorsement by the U.S. Government or the U.S. Environmental Protection Agency. The EPA does not endorse any commercial products, services, or enterprises.
Footnotes
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website. Section S1–S5 describe GO characterization, stock preparation, NOM preparation, colloidal stability studies. Tables SI shows XPS analysis of solar-irradiated GO. Tables S2 and S3 show the band assignments of NOM. Figures S1–S6 show the characterization of pristine GO and GO suspensions. Figures S7–S9 are the characterization of aged GO while Figures S10–12 are characterization of solar-irradiated GO. IR spectra of NOM are shown in Figure S13. Figure S14 is the effect of initial concentration on GO stability. Figure S15 is the effect of GO on the survival of mysids.
REFERENCES
- 1.Pei S; Cheng H-M , The reduction of graphene oxide. Carbon 2012, 50, (9), 3210–3228. [Google Scholar]
- 2.Li RB; Mansukhani ND; Guiney LM; Ji ZX; Zhao YC; Chang CH; French CT; Miller JF; Hersam MC; Nel AE; Xia T, Identification and Optimization of Carbon Radicals on Hydrated Graphene Oxide for Ubiquitous Antibacterial Coatings. Acs Nano 2016, 10, (12), 10966–10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perreault F; de Faria AF; Nejati S; Elimelech M, Antimicrobial Properties of Graphene Oxide Nanosheets: Why Size Matters. Acs Nano 2015, 9, (7), 7226–7236. [DOI] [PubMed] [Google Scholar]
- 4.Gao Y; Wu JC; Ren XM; Tan XL; Hayat T; Alsaedi A; Cheng C; Chen CL, Impact of graphene oxide on the antibacterial activity of antibiotics against bacteria. Environ Sci-Nano 2017, 4, (5), 1016–1024. [Google Scholar]
- 5.Krishnamoorthy K; Jeyasubramanian K; Premanathan M; Subbiah G; Shin HS; Kim SJ, Graphene oxide nanopaint. Carbon 2014, 72, 328–337. [Google Scholar]
- 6.Pourhashem S; Vaezi MR; Rashidi A; Bagherzadeh MR, Distinctive roles of silane coupling agents on the corrosion inhibition performance of graphene oxide in epoxy coatings. Progress in Organic Coatings 2017, 111, (Supplement C), 47–56. [Google Scholar]
- 7.Raza MA; Rehman ZU; Ghauri FA; Ahmad A; Ahmad R; Raffi M, Corrosion study of electrophoretically deposited graphene oxide coatings on copper metal. Thin Solid Films 2016, 620, (Supplement C), 150–159. [Google Scholar]
- 8.Mooss VA; Bhopale AA; Deshpande PP; Athawale AA, Graphene oxide-modified polyaniline pigment for epoxy based anti-corrosion coatings. Chem. Pap 2017, 71, (8), 1515–1528. [Google Scholar]
- 9.Adeleye AS; Conway JR; Garner K; Huang Y; Su Y; Keller AA, Engineered nanomaterials for water treatment and remediation: Costs, benefits, and applicability. Chemical Engineering Journal 2016, 286, 640–662. [Google Scholar]
- 10.Gao Y; Ren X; Tan X; Hayat T; Alsaedi A; Chen C, Insights into key factors controlling GO stability in natural surface waters. Journal of Hazardous Materials 2017, 335, 56–65. [DOI] [PubMed] [Google Scholar]
- 11.Chowdhury I; Duch MC; Mansukhani ND; Hersam MC; Bouchard D, Colloidal Properties and Stability of Graphene Oxide Nanomaterials in the Aquatic Environment. Environmental Science & Technology 2013, 47, (12), 6288–6296. [DOI] [PubMed] [Google Scholar]
- 12.Gao Y; Ren XM; Wu JC; Hayat T; Alsaedi A; Cheng C; Chen CL, Graphene oxide interactions with co-existing heavy metal cations: adsorption, colloidal properties and joint toxicity. Environ Sci-Nano 2018, 5, (2), 362–371. [Google Scholar]
- 13.Ren XM; Li J; Chen CL; Gao Y; Chen DY; Su MH; Alsaedi A; Hayat T, Graphene analogues in aquatic environments and porous media: dispersion, aggregation, deposition and transformation. Environ Sci-Nano 2018, 5, (6), 1298–1340. [Google Scholar]
- 14.Gao Y; Ren XM; Tan XL; Hayat T; Alsaedi A; Chen CL, Insights into key factors controlling GO stability in natural surface waters. Journal of Hazardous Materials 2017, 335, 56–65. [DOI] [PubMed] [Google Scholar]
- 15.Gudarzi MM, Colloidal Stability of Graphene Oxide: Aggregation in Two Dimensions. Langmuir 2016, 32, (20), 5058–5068. [DOI] [PubMed] [Google Scholar]
- 16.Yang KJ; Chen BL; Zhu XY; Xing BS, Aggregation, Adsorption, and Morphological Transformation of Graphene Oxide in Aqueous Solutions Containing Different Metal Cations. Environmental Science & Technology 2016, 50, (20), 11066–11075. [DOI] [PubMed] [Google Scholar]
- 17.Wu L; Liu L; Gao B; Muñoz-Carpena R; Zhang M; Chen H; Zhou Z; Wang H, Aggregation Kinetics of Graphene Oxides in Aqueous Solutions: Experiments, Mechanisms, and Modeling. Langmuir 2013, 29, (49), 15174–15181. [DOI] [PubMed] [Google Scholar]
- 18.Jiang Y; Raliya R; Fortner JD; Biswas P, Graphene Oxides in Water: Correlating Morphology and Surface Chemistry with Aggregation Behavior. Environmental Science & Technology 2016, 50, (13), 6964–6973. [DOI] [PubMed] [Google Scholar]
- 19.Gao Y; Chen K; Ren XM; Alsaedi A; Hayat T; Chen CL, Exploring the Aggregation Mechanism of Graphene Oxide in the Presence of Radioactive Elements: Experimental and Theoretical Studies. Environmental Science & Technology 2018, 52, (21), 12208–12215. [DOI] [PubMed] [Google Scholar]
- 20.Millero F; Sohn M, Chemical Oceanography . CRC Press Inc.: Boca Raton, FL, USA, 1992. [Google Scholar]
- 21.Conway JR; Adeleye AS; Gardea-Torresdey J; Keller AA, Aggregation, Dissolution, and Transformation of Copper Nanoparticles in Natural Waters. Environmental Science & Technology 2015, 49, (5), 2749–2756. [DOI] [PubMed] [Google Scholar]
- 22.Xiao Y; Ho KT; Burgess RM; Cashman M, Aggregation, Sedimentation, Dissolution, and Bioavailability of Quantum Dots in Estuarine Systems. Environmental Science & Technology 2017, 51, (3), 1357–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adeleye AS; Keller AA, Long-term colloidal stability and metal leaching of single wall carbon nanotubes: effect of temperature and extracellular polymeric substances. Water research 2014, 49, (0), 236–50. [DOI] [PubMed] [Google Scholar]
- 24.Goodwin D; Adeleye AS; Sung L; Ho KT; Burgess RM; Petersen EJ, Detection and Quantification of Graphene Family Nanomaterials in the Environment. Environmental Science & Technology 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao J; Wang Z; White JC; Xing B, Graphene in the Aquatic Environment: Adsorption, Dispersion, Toxicity and Transformation. Environmental Science & Technology 2014, 48, (17), 9995–10009. [DOI] [PubMed] [Google Scholar]
- 26.Hou W-C; Chowdhury I; Goodwin DG; Henderson WM; Fairbrother DH; Bouchard D; Zepp RG, Photochemical Transformation of Graphene Oxide in Sunlight. Environmental Science & Technology 2015, 49, (6), 3435–3443. [DOI] [PubMed] [Google Scholar]
- 27.Du T; Adeleye AS; Keller AA; Wu Z; Han W; Wang Y; Zhang C; Li Y, Photochlorination-induced transformation of graphene oxide: Mechanism and environmental fate. Water research 2017, 124, (Supplement C), 372–380. [DOI] [PubMed] [Google Scholar]
- 28.Mohandoss M; Gupta SS; Nelleri A; Pradeep T; Maliyekkal SM, Solar mediated reduction of graphene oxide. Rsc Adv 2017, 7, (2), 957–963. [Google Scholar]
- 29.Du TT; Adeleye AS; Zhang T; Jiang CJ; Zhang M; Wang HH; Li Y; Keller AA; Chen W, Influence of light wavelength on the photoactivity, physicochemical transformation, and fate of graphene oxide in aqueous media. Environ Sci-Nano 2018, 5, (11), 2590–2603. [Google Scholar]
- 30.Li Y; Yang N; Du T; Wang X; Chen W, Transformation of graphene oxide by chlorination and chloramination: Implications for environmental transport and fate. Water research 2016, 103, 416–423. [DOI] [PubMed] [Google Scholar]
- 31.Amieva EJC; López‐Barroso J; Martínez‐Hernández AL; Velasco‐Santos C, Graphene‐Based Materials Functionalization with Natural Polymeric Biomolecules In Recent Advances in Graphene Research, IntechOpen: 2016. [Google Scholar]
- 32.Chowdhury I; Hou W-C; Goodwin D; Henderson M; Zepp RG; Bouchard D, Sunlight affects aggregation and deposition of graphene oxide in the aquatic environment. Water research 2015, 78, 37–46. [DOI] [PubMed] [Google Scholar]
- 33.Keller AA; Lazareva A, Predicted Releases of Engineered Nanomaterials: From Global to Regional to Local. Environmental Science & Technology Letters 2014, 1, (1), 65–70. [Google Scholar]
- 34.Quik JTK; Velzeboer I; Wouterse M; Koelmans AA; van de Meent D, Heteroaggregation and sedimentation rates for nanomaterials in natural waters. Water research 2014, 48, 269–279. [DOI] [PubMed] [Google Scholar]
- 35.Adeleye AS Influence of Phytoplankton and Extracellular Polymeric Substances on the Fate of Engineered Nanomaterials in Natural Aquatic Systems. PhD Thesis, University of California, Santa Barbara, 2015. [Google Scholar]
- 36.Adeleye AS; Keller AA, Interactions between Algal Extracellular Polymeric Substances and Commercial TiO2 Nanoparticles in Aqueous Media. Environmental Science & Technology 2016, 50, (22), 12258–12265. [DOI] [PubMed] [Google Scholar]
- 37.Wang X; Adeleye AS; Wang H; Zhang M; Liu M; Wang Y; Li Y; Keller AA, Interactions between polybrominated diphenyl ethers (PBDEs) and TiO2 nanoparticle in artificial and natural waters. Water research 2018. [DOI] [PubMed] [Google Scholar]
- 38.Kwon S; Lee KE; Lee H; Koh SJ; Ko J-H; Kim Y-H; Kim SO; Park JY, The Effect of Thickness and Chemical Reduction of Graphene Oxide on Nanoscale Friction. Journal of Physical Chemistry B 2018, 122, (2), 543–547. [DOI] [PubMed] [Google Scholar]
- 39.Duan L; Hao R; Xu Z; He X; Adeleye AS; Li Y, Removal of graphene oxide nanomaterials from aqueous media via coagulation: Effects of water chemistry and natural organic matter. Chemosphere 2017, 168, 1051–1057. [DOI] [PubMed] [Google Scholar]
- 40.Adeleye AS; Wang X; Wang F; Hao R; Song W; Li Y, Photoreactivity of graphene oxide in aqueous system: Reactive oxygen species formation and bisphenol A degradation. Chemosphere 2018, 195, 344–350. [DOI] [PubMed] [Google Scholar]
- 41.Stankovich S; Dikin DA; Piner RD; Kohlhaas KA; Kleinhammes A; Jia Y; Wu Y; Nguyen ST; Ruoff RS, Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 2007, 45, (7), 1558–1565. [Google Scholar]
- 42.Konkena B; Vasudevan S, Engineering a Water-Dispersible, Conducting, Photoreduced Graphene Oxide. J. Phys. Chem. C 2015, 119, (11), 6356–6362. [Google Scholar]
- 43.Matsumoto Y, Simple photoreduction of graphene oxide nanosheet under mild conditions. Acs Appl. Mater. Interf 2010, 2, 3461–3466. [DOI] [PubMed] [Google Scholar]
- 44.Komeily Nia Z; Chen J-Y; Tang B; Yuan B; Wang X-G; Li J-L, Optimizing the free radical content of graphene oxide by controlling its reduction. Carbon 2017, 116, (Supplement C), 703–712. [Google Scholar]
- 45.McDonald MP; Morozov Y; Hodak JH; Kuno M, Spectroscopy and Microscopy of Graphene Oxide and Reduced Graphene Oxide In Graphene Oxide: Reduction Recipes, Spectroscopy, and Applications, Gao W, Ed. Springer International Publishing: Cham, 2015; pp 29–60. [Google Scholar]
- 46.Konkena B; Vasudevan S, Engineering a Water-Dispersible, Conducting, Photoreduced Graphene Oxide. Journal of Physical Chemistry C 2015, 119, (11), 6356–6362. [Google Scholar]
- 47.Velasco-Soto MA; Pérez-García SA; Alvarez-Quintana J; Cao Y; Nyborg L; Licea-Jiménez L, Selective band gap manipulation of graphene oxide by its reduction with mild reagents. Carbon 2015, 93, 967–973. [Google Scholar]
- 48.Cavicchioli A; Gutz IGR, Effect of scavengers on the photocatalytic digestion of organic matter in water samples assisted by TiO2 in suspension for the voltammetric determination of heavy metals. Journal of the Brazilian Chemical Society 2002, 13, 441–448. [Google Scholar]
- 49.Neppolian B; Choi HC; Sakthivel S; Arabindoo B; Murugesan V, Solar light induced and TiO2 assisted degradation of textile dye reactive blue 4. Chemosphere 2002, 46, (8), 1173–1181. [DOI] [PubMed] [Google Scholar]
- 50.Sawyer CN; McCarty PL; Parkin GF, Chemistry for environmental engineering and science. 5th ed.; McGraw-Hill: New York, 2003; p 752. [Google Scholar]
- 51.Adeleye AS; Conway JR; Perez T; Rutten P; Keller AA, Influence of Extracellular Polymeric Substances on the Long-Term Fate, Dissolution, and Speciation of Copper-Based Nanoparticles. Environ Sci Technol 2014, 48, (21), 12561–12568. [DOI] [PubMed] [Google Scholar]
- 52.Adeleye AS; Stevenson LM; Su Y; Nisbet RM; Zhang Y; Keller AA, Influence of Phytoplankton on Fate and Effects of Modified Zerovalent Iron Nanoparticles. Environ Sci Technol 2016, 50, (11), 5597–605. [DOI] [PubMed] [Google Scholar]
- 53.Lin D; Drew Story S; Walker SL; Huang Q; Cai P, Influence of extracellular polymeric substances on the aggregation kinetics of TiO2 nanoparticles. Water research 2016, 104, 381–388. [DOI] [PubMed] [Google Scholar]
- 54.Khan B; Adeleye AS; Burgess RM; Smolowitz R; Russo SM; Ho KT, A 72-h exposure study with eastern oysters (Crassostrea virginica) and the nanomaterial graphene oxide. Environmental Toxicology and Chemistry 2019, 38, (4), 820–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.