

Abstract
Selective, visible-light-driven conversion of CO2 to CO with a turnover frequency of 20 s−1 under visible light irradiation at 25 °C is catalyzed by an aqueous colloidal system comprising a pseudoternary complex formed among carbon monoxide dehydrogenase (CODH), silver nanoclusters stabilized by polymethacrylic acid (AgNCs-PMAA), and TiO2 nanoparticles. The photocatalytic assembly, which is stable over several hours and for at least 250000 turnovers of the enzyme’s active site, was investigated by separate electrochemical (dark) and fluorescence measurements to establish specific connectivities among the components. The data show (a) that a coating of AgNCs-PMAA on TiO2 greatly enhances its ability as an electrode for CODH- based electrocatalysis of CO2 reduction and (b) that the individual Ag nanoclusters interact directly and dynamically with the enzyme surface, most likely at exposed cysteine thiols. The results lead to a model for photocatalysis in which the AgNCs act as photosensitizers, CODH captures the excited electrons for catalysis, and TiO2 mediates hole transfer from the AgNC valence band to sacrificial electron donors. The results greatly increase the benchmark for reversible CO2 reduction under ambient conditions and demonstrate that, with such efficient catalysts, the limiting factor is the supply of photogenerated electrons.
Keywords: carbon monoxide dehydrogenase, silver nanoclusters, artificial photosynthesis, CO2 photoreduction, photocatalytic enzyme assembly
Graphic abstract
INTRODUCTION
There is significant economic and scientific interest in converting intermittent solar energy into storable fuels (artificial photosynthesis, AP) and intense efforts are underway to establish ways to produce H2 from water or reduced carbon compounds from CO2, in all cases using catalysts based on abundant elements.1–3 An inspirational approach to photo-H2 production has been to exploit the extremely high catalytic activity of hydrogenases by attaching them directly to light- harvesting systems.4–6 Direct reduction of a concentrated source of CO2 (as present in flue gas) is far more challenging than H2 production,7–13 but once again, insight has been derived from metalloenzymes such as carbon monoxide dehydrogenase (CODHs: Ni and Fe)14 or formate dehydrogenase (FRDs: Mo or W), each of which is highly active in catalyzing two-electron reduction of CO2 with extreme selectivity, avoiding the thermodynamically easier H2 evolution.15–18 Importantly, the activities of these enzymes are so high19 that the performance of the integrated photocatalytic systems in which they are incorporated is limited not by catalysis but by photophysical restrictions and charge-transport connections. An earlier dilemma was that, despite the high activity and efficiency expected for CODH on the basis of steady-state rates and experiments using protein film electrochemistry (PFE),20 the photocatalytic per-enzyme-molecule turnover frequency (TOF) was orders of magnitude lower than expected (the initial TOF being just 0.18 s−1).18 With such an active enzyme, it appeared that the shortcoming lay in low electron availability, as a result of either short excited-state lifetime and/or poor electronic coupling to the enzyme.
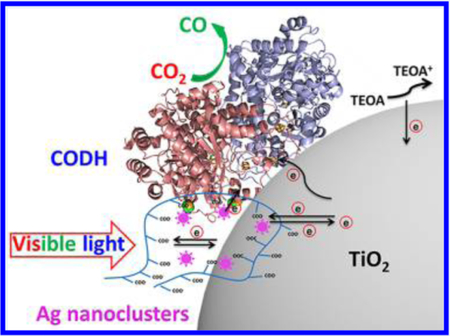
Here, we describe the profound effect of silver nanoclusters (AgNCs) on the CO2 photoreduction activity of CODH (specifically CODH I from Carboxydothermus hydrogenofor mans) upon attachment to TiO2 nanoparticles. Metal nano clusters have special optoelectronic properties due to quantum confinement and behave as molecular redox species: they are under investigation for analytical and energy-related applications, a particularly relevant one being their use as sensitizers for TiO2 solar cells.21 By templating in protective hosts such as polymethacrylic acid (PMAA), AgNCs containing <10 Ag atoms can be stabilized in water.22,23 The luminescent nanoclusters rapidly reduce methyl viologen upon illumination with visible light.24 Further, their emission properties are sensitive to interactions with specific ligands such as thiolates,25 which are often exposed on the surface of proteins. Considering that free carboxylates on PMAA are, in turn, suited for binding to the surface of TiO2,26,27 it was reasoned that PMAA-capped AgNCs (AgNCs-PMAA) should form stable connections between CODH and the surface of TiO2, thereby creating a soft assembly that simultaneously harvests visible light and directs electron and hole transfers to support rapid and efficient CO2 photoreduction.
EXPERIMENTAL SECTION
Preparation of Silver Nanoclusters (AgNCs).
Silver nanoclusters (AgNCs) were synthesized according to a previously published procedure.23 A freshly prepared aqueous solution of silver nitrate (0.1 M, 10 mL, Sigma-Aldrich, 99+%) was mixed with an equal volume of PMAA solution (5 mg/mL, adjusted with nitric acid to pH 3.0) at room temperature. After mixing, the colorless solution was purged by N2 bubbling for 15 min to remove O2 before exposing to laboratory sunlight. With increasing irradiation time the solution gradually changed from colorless to light pink and then to magenta. The solution was then mixed with tetrahydrofuran (17–30% w/w), and the deep magenta clusters that aggregated upon shaking were removed by centrifugation (3600 rpm, 15 min) and redissolved in water. The concentration of AgNCs was measured by inductively coupled plasma mass spectrometry (ICP-MS, Thermo Element 2). Matrix-assisted laser desorption time-of-flight mass spectra (MALDI-ToF-MS) were recorded on a Bruker Reflex III operated in reflectron mode using a N2 laser (337 nm) and an accelerating voltage of 20 kV. The sample was prepared by drop-coating the solution onto the target.
Electrochemistry Experiments.
The working electrode consisting of TiO2 deposited on indium tin oxide (ITO) (see the Supporting Information) was used in a single-compartment electrochemical cell with a Pt (wire) counter electrode, with a saturated calomel reference electrode held in a separate side arm and linked by a Luggin capillary. All potentials are quoted vs the standard hydrogen electrode (SHE) using the conversion relationship ESHE = ESCE + 241 mV at 25 °C. The electrolyte consisted of 0.1 M NaCl and 0.2 M MES at pH 6.0, and all solutions were prepared using purified water (Milli-Q, 18 MΩ cm). Electrochemical measurements were carried out with an Autolab potentiostat (PGSTAT30) controlled by Nova software (EcoChemie). Precise gas mixtures (BOC gases) were created using mass flow controllers (Sierra Instruments), and the electrochemical cell was constantly purged with the gas mixture throughout the experiments.
Quantitative Analysis of CO Formed in Photocatalytic Experiments.
An Agilent 7890A series gas chromatograph (GC) with electronic pneumatic control optimized for trace gas analysis was used to monitor CO production at regular intervals. Headspace gas samples (20 μL) were injected into the GC in pulsed splitless mode using a gastight Hamilton syringe. A 1.5 mm diameter split/splitless liner, a Restek ShinCarbon ST micropacked column, a micro thermal conductivity detector, and high-purity He carrier gas with a constant pressure of 25 psi were used. The column oven was heated from 40 to 200 °C at a rate of 10 °C min−1 during measurements.
RESULTS
Photocatalysis.
Figure 1 shows experiments in which an aqueous suspension of CODH/AgNCs-PMAA/TiO2 sealed under a CO2 atmosphere was illuminated with visible light. Details for the preparation of AgNCs-PMAA and AgNCs- PMAA/TiO2 nanomaterials are given in Figures S1–S3 in the Supporting Information. All suspensions were prepared in a glovebox under a N2 atmosphere before they were taken out for measurements. Briefly, a suspension of AgNCs-PMAA/TiO2 (5 mg) formed from 3.05 mg of TiO2 (Evonik Aeroxide P25, 21 nm diameter) and 1.95 mg of AgNCs templated in PMAA (resulting in a final Ag/Ti atomic ratio of 0.27 as determined by ICP-MS) was formed by sonicating the two components in a 5 mL aqueous solution of 0.1 M triethanolamine (TEOA), 0.1 M NaCl, and 25 mM EDTA, adjusted to pH 6.0, all contained in a Pyrex pressure vessel (total volume 22.5 mL). Then CODH (50 μL of 10 μM solution, see the Supporting Information) was added and the suspension was stirred gently for 3 min to allow adsorption of the enzyme, before the vessel was sealed tightly with a rubber septum and the headspace purged with CO2. The stirred suspension was irradiated with visible light using a 300 W arc lamp (Newport 67005) fitted with a 420 nm filter and held 5 cm from the vessel. The temperature was controlled by immersing the vessel in a water bath. Production of CO was monitored at regular intervals by removing small volumes (20 μL) of headspace gas for gas chromatography (GC) analysis. The amount of CO was quantified against calibration plots obtained with known amounts of CO (Figure S4). Turnover frequencies were obtained by dividing the quantity of CO formed in a given time by the total amount of CODH used.
Figure 1.
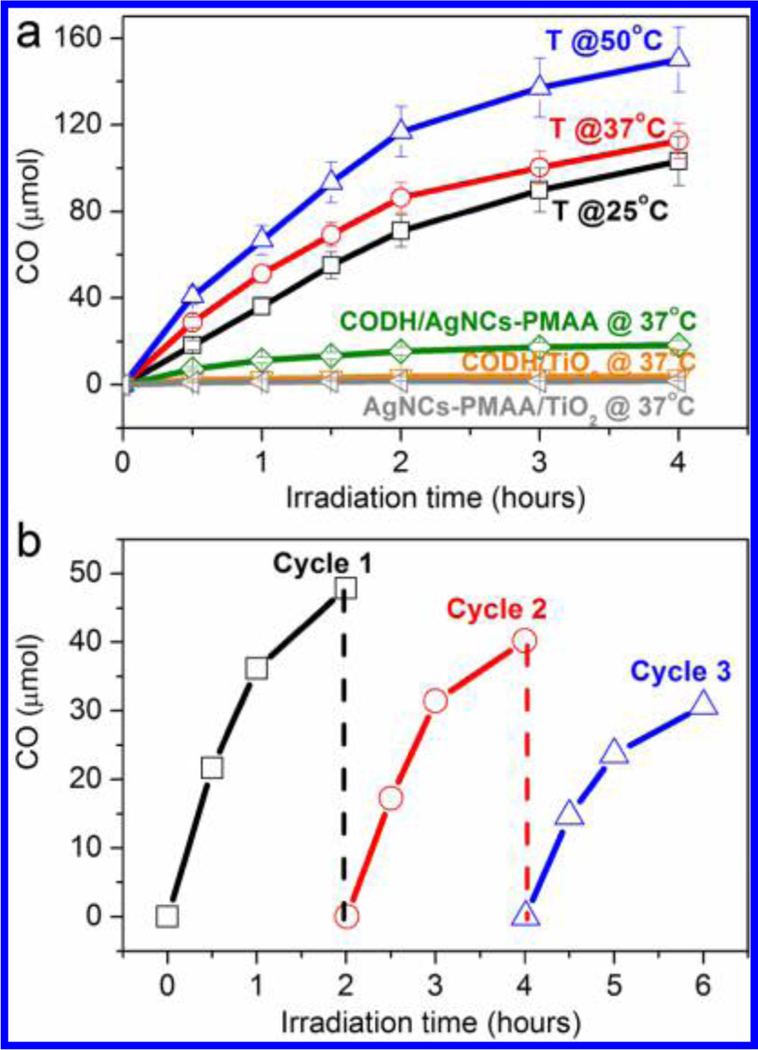
Photocatalytic CO2 reduction. (a) Time courses for photocatalytic CO production by the ternary CODH/AgNCs- PMAA/TiO2 complex and other combinations, along with various control experiments, under visible light irradiation, followed using GC. In all cases, the 5 mL of water in the vial contained 0.1 M TEOA, 0.1 M NaCl, and 25 mM EDTA at pH 6.0. For experiments with the ternary complex (T), the vial contained 5 mg of AgNCs-PMAA/TiO2 and 0.50 nmol of CODH. Experiments were carried out at 25 °C (black), 37 °C (red), and 50 °C (blue). Control experiments were carried out using CODH/AgNCs-PMAA (no TiO2), CODH/TiO2 (no AgNCs-PMAA) and AgNCs-PMAA/TiO2 (no CODH). (b) Production of CO over the course of 6 h, in which CO was flushed out at 2 h intervals and CO2 was restored to the initial level (98%).
The results obtained without one of the components present, CODH, TiO2 or AgNCs-PMAA, are included in Figure 1: the data suggest the requirement for a quasi-ternary complex containing all three components, although not necessarily in a stoichiometric manner (hence “quasi”). The initial turnover frequency (TOF) of 20 s−1 observed at 25 °C increases to 37 s−1 at 50 °C. At 25 °C the rate of CO production is steady during the first 1 h of irradiation before decreasing gradually to 14 s−1 over the next 3 h. Although the underlying stability is one factor, much of this decrease could be attributed to product inhibition by CO because the rate increased substantially each time the headspace was refreshed (Figure 1b). Taking cycle 1 as an example, the CO concentration reaches 6.7% in the headspace before flushing out: assuming a value of 1 mM for Henry’s constant, the corresponding CO concentration in the solution would be 67 μM, a value that exceeds the inhibition constant (46 μM) measured electrochemically under comparable conditions at −560 mV vs SHE.28 In no case was any H2 detected by GC: this important result is consistent with previous results29 showing that CODHs are highly selective Table 1. Visible-Light-Driven CO2 Reduction by CODH/ AgNCs-PMAA/TiO2 Assemblies under Different Conditions catalysts for CO2 reduction even though water reduction is favored thermodynamically. An interesting and obvious outcome of the experiments shown in Figure 1b is that, after 6 h, each CODH molecule has turned over about 250000 times. Results obtained under a wide range of conditions are shown in Table 1. The rate of CO production varies by less than a factor of 2 among different electron donors (TEOA, EDTA, and MES). No significant changes in CO yield after 30 min irradiation were observed for TEOA concentrations between 0.05 and 0.25 M (Figure S5). Using the filtered light intensity and illuminated facial area of the reaction vessel, we estimated that the quantum yield would be approximately 1.5% on the basis of a wavelength of 500 nm.
Table 1.
Visible-Light-Driven CO2 Reduction by CODH/AgNCs-PMAA/TiO2 Assemblies under Different Conditions
| COb (μmol h−1) | TOFc (s−1) | |
|---|---|---|
| standard conditiona | 36.12 ± 3.05 | 20 ± 2 |
| Enzyme | ||
| 0.25 nmol | 15.43 ± 2.52 | 17 ± 3 |
| 0.5 nmol | 36.12 ± 3.05 | 20 ± 2 |
| 1 nmol | 70.33 ± 5.55 | 19 ± 3 |
| AgNCs-PMAA/TiO2 Materials | ||
| 1 mg/mL | 36.12 ± 3.05 | 20 ± 2 |
| 2 mg/mL | 37.78 ± 4.10 | 21 ± 2 |
| 5 mg/mL | 35.03 ± 2.22 | 19 ± 1 |
| Temperature | ||
| 25 °C | 36.12 ± 3.05 | 20 ± 2 |
| 37 °C | 51.23 ± 2.32 | 28 ± 1 |
| 50 °C | 66.75 ± 4.85 | 37 ± 3 |
| pH | ||
| 6 | 36.12 ± 3.05 | 20 ± 2 |
| 7 | 33.72 ± 5.20 | 19 ± 3 |
| Control Group | ||
| AgNCs-PMAA/TiO2 | 0.34 ± 0.20 | Nd |
| CODH/TiO2 | 1.07 ± 0.52 | 0.06 ± 0.01 |
| CODH/AgNCs-PMAA | 11.36 ± 0.22 | 6 ± 0.3 |
| Electron Donor | ||
| TEOA | 30.37 ± 2.58 | 17 ± 1 |
| TEOA + EDTA | 36.12 ± 3.05 | 20 ± 2 |
| MESe | 23.33 ± 2.22 | 13 ± 1 |
Unless otherwise noted in the table, standard conditions were employed, which were as follows: CODH (50 μL of 10 μM solution) was added to a 5 mL dispersion of AgNCs-PMAA/TiO2 (5 mg, composed of 3.05 mg of TiO2 and 1.95 mg of AgNCs-PMAA, with Ag/Ti atom ratio of 0.27) in 0.1 M triethanolamine (TEOA), 25 mM ethylenediaminetetraacetic acid (EDTA), and 0.1 M NaCl at pH 6 and 25 °C. Irradiation was carried out with a Newport 67005 arc lamp (300 W) fitted with a 420 nm filter and located 5 cm from the solution.
CO production rate (μmol of CO h−1) based on the first hour of irradiation.
Turnover frequency (TOF) expressed as molecules of CO produced per second per molecule of CODH).
Value below 0.001 s−1.
2-(N-Morpholino)ethanesulfonic acid (MES).
The discovery that a quasi-ternary complex formulated as CODH/AgNCs-PMAA/TiO2 photoreduces CO2 far more quickly than any binary combination was followed up with studies designed to examine different aspects of the interaction. An estimation of the composition of AgNCs-PMAA (the “soft” and least well-de fined component) was carried out, on the basis of an average molecular weight of 5000 for PMAA. Taking, as a reference point, Ag5 to be the dominant nanocluster (Figure S1), the analytical mass ratio 1/0.53 determined by ICP-MS yielded 5 Ag5 per polymer molecule. Each PMAA has approximately 60 monomer units; therefore, numerous carboxylates should remain free to bind to the TiO2 surface.26
Electrocatalysis.
To investigate how AgNCs influence the “dark” electrocatalytic CO2/CO interconversion by CODH at a TiO2 surface, experiments were carried out in which an electrode consisting of TiO2 deposited on indium tin oxide (ITO) was modified with various combinations of AgNCs- PMAA, PMAA alone, and CODH. The results are shown in Figure 2.
Figure 2.
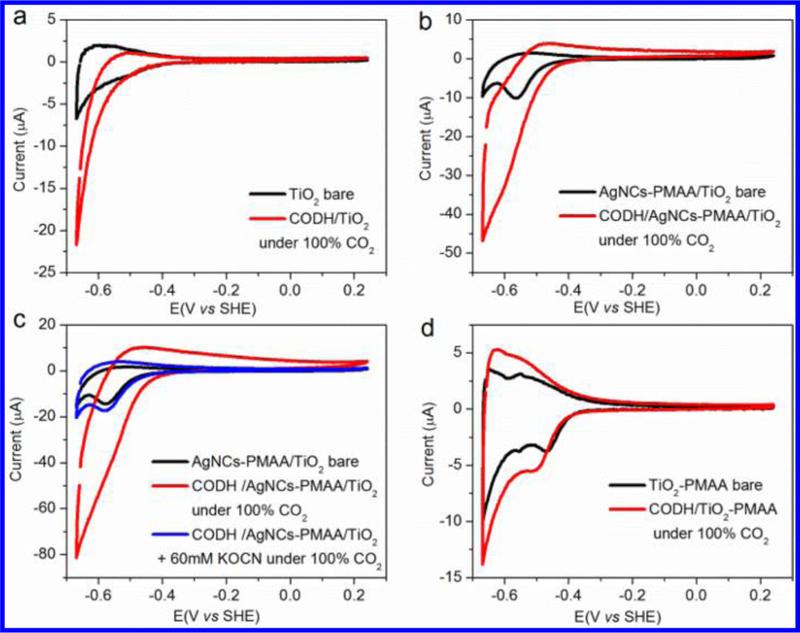
Cyclic voltammograms of CODH adsorbed on variously modi fied TiO2 electrodes under an atmosphere of 100% CO2. (a) Electrocatalytic activity of CODH/TiO2 and that of a bare TiO2 electrode. (b) Electrocatalytic activities of CODH/AgNCs-PMAA/TiO2 and AgNCs-PMAA/TiO2 electrodes. (c) Selective inhibition of CO2 reduction by CODH/AgNCs-PMAA/TiO2 electrode in the presence of 60 mM KOCN. (d) E ffect of PMAA alone on electrocatalytic activity, observed by comparing CODH/PMAA/TiO2 with PMAA/TiO2. All solutions contained 0.2 M MES (pH 6.0, 0.1 M NaCl), with a temperature of 25 °C and a scan rate of 10 mV s−1.
An unmodified TiO2 electrode displays a signature “trumpet shaped” voltammogram that may be interpreted, chemically, in terms of the electronic conductivity increasing at the most negative potential as Ti(IV) centers are converted to Ti(III).16 Comparisons between catalytic cyclic voltammograms (CVs) of CODH adsorbed at a TiO2 electrode (Figure 2a) and at an AgNCs-PMAA/TiO2 electrode (Figure 2b) reveal that CO2 reduction activity is greatly enhanced by the presence of AgNCs-PMAA, which causes the current to increase at a lower overpotential. A small current due to reoxidation of CO becomes detectable, and there is also a change in the “background” control voltammogram (no CODH) when AgNCs-PMAA is present. Proof that the catalytic current stems from enzymatic activity was obtained by injecting an aliquot of concentrated KOCN solution into the cell (to a final concentration of 60 mM). Cyanate (NCO−), which is isoelectronic with CO2, targets the Cred2 state of the enzyme and blocks CO2 reduction.28 Figure 2c depicts the selective inhibition of CO2 reduction on a CODH/AgNCs-PMAA/TiO2 electrode after adding KOCN. Comparison of the background traces in Figure 2a,b shows that modification of the TiO2 electrode with AgNCs-PMAA leads to a new reduction peak at −0.56 V (vs SHE), similar to that attributed to trap surface states of TiO2 introduced particularly by binding of oxygen atom donor ligands.30,31 Figure 2d shows that PMAA alone (without AgNCs) also gives rise to a change in the TiO2 background, introducing a small reduction peak similar to that observed with AgNCs-PMAA but shifted to more positive potential. Importantly, no catalytic current is observed when CODH is introduced, indicating that PMAA alone blocks the electrode with regard to electron transfer, enzyme binding, or both.
Experiments to determine the stability and temperature dependence of the catalytic current were carried out: C. hydrogenoformans is a thermophile, and CODH should be inherently stable at ambient temperatures.32 The temperature dependence of CO2 reduction activity at a CODH/AgNCs- PMAA/TiO2 electrode over the range 25–50 °C is shown in Figure S6, while Figures S7 and S8 show the stabilities determined by cyclic voltammetry and chronoamperometry, respectively. Cyclic voltammograms obtained under 100% CO2 showed only a small decrease in current over 24 h, and chronoamperometry at −0.66 V vs SHE revealed even a small improvement with time. Both results reinforce the observations of the photocatalytic time courses for CO production with colloidal CODH/AgNCs-PMAA/TiO2 and highlight the stark contrast with earlier results obtained with a RuP—TiO2 system.18,33 The fact that increasing the temperature from 25 to 50 °C yields only a modest (less than 2-fold) increase in rate suggests that CO2 reduction at the active site of the enzyme (a [Ni4Fe-4S] cofactor known as the “C-cluster”14,20) is still not a limiting factor.
Fluorescence.
The speci fic interaction between PMAA-stabilized AgNCs and CODH was studied by performing a photoluminescence (PL) titration along with nanosecond luminescence kinetics. As shown in Figure 3a, the emission intensity first decreases and then increases as CODH is added to a solution containing AgNCs-PMAA. The point at which emission is minimal should correspond to optimal formation of a CODH/AgNCs-PMAA complex, which in this case (Ag atom concentration 20 μM) occurs when the CODH concentration is 0.54 μM. However, due to the coexistence of different structures and compositions in AgNCs-PMAA solution (Figure S1), the binding stoichiometry and a meaningful dissociation constant (Kd) between AgNCs-PMAA and CODH cannot be determined.
Figure 3.
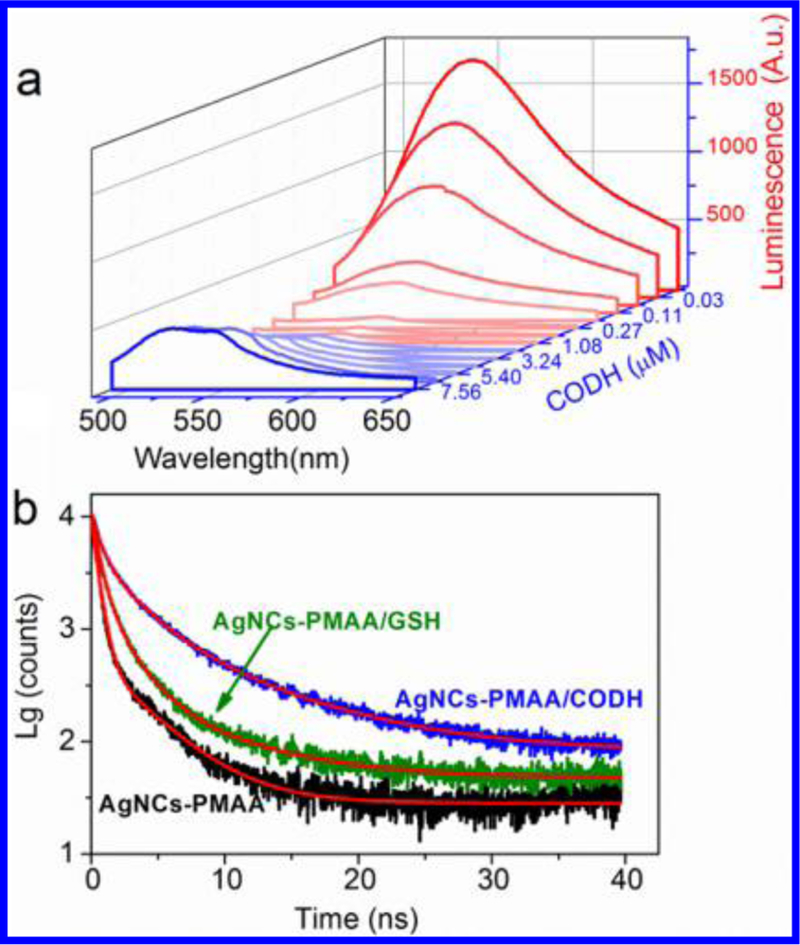
Interaction between AgNCs-PMAA and CODH. (a) Photoluminescence spectra of AgNCs-PMAA (Ag atom concentration 20 μM determined by ICP-MS) using CODH (0−7.56 μM) in 0.2 M MES bu ffer (0.5 mL, pH 6.0) with λex 460 nm. (b) Fluorescence decay traces of AgNCs-PMAA monitored at 460 nm. Black, green, and blue traces are for AgNCs-PMAA, AgNCs-PMAA/glutathione (GSH), and AgNCs-PMAA/CODH, respectively, in 0.2 M MES, pH 6, at room temperature. Fits are shown in red.
The mechanism of photoemission by metal nanoclusters is controversial,34–37 and independent studies have shown that the natures of the nanocluster core and anchoring ligands are both important. The quenching of emission that is observed as aliquots of CODH are added to AgNCs-PMAA resembles the quenching that is observed specifically in the presence of cysteine.25 The subsequent increase (Figure 3a and Figure S9 peak I′ at 544 nm) and appearance of a new blue-shifted emission peak (Figure S9 peak II′ at 522 nm) at higher CODH concentration suggests that further reactions occur beyond the binding of cysteine thiolate to Ag (see Figure S10).
Time-correlated single photon counting (TCSPC) provided further information about the charge-transfer dynamics underlying the AgNCs fluorescence quenching (Figure 3b and Table S1). The short lifetime for Ag nanoclusters is typical for metal core sp—d hole recombination (fluorescence originating from the recombination of electrons in the s−p band with holes in the d band).38 The longer emission lifetime observed in the presence of CODH suggests surface-state sensitivity:39 specifically, in this case, an interaction between the metal core and the thiolate-S.38,40
DISCUSSION
Although the structure of CODH I has not been solved, some idea of the availability of surface cysteines is obtained from the structure of CODH II (1SU7)41 and the CODH (CODH Rr) from Rhodospirillum rubrum (1JQK).42 A model for CODH I based on CODH-Rr using the SWISS-MODEL web program was simulated (Figure S11), and four possible surface cysteines were predicted for each half of the enzyme, these being cys162, cys271, cys346, and cys401. Of these possibilities, one cysteine (346) is conserved and surface exposed in the structure of CODH II and CODH Rr. Although the surface-exposed cysteine appears to be too remote to allow a direct electron transfer into the enzyme, it is likely that such a residue acts as an anchor point by which one Ag nanocluster is bound, the overall effect being to bring CODH, TiO2, and other PMAA- stabilized AgNCs into dynamic close contact.
Attempting to explain the interactions within a pseudoternary assembly in which the “soft” AgNCs-PMAA is a crucial component required an empirical approach. The connectivities among CODH, AgNCs, PMAA, and TiO2, in terms of all the experiments carried out, are therefore presented in Scheme 1. Whether measured by electrocatalysis (E) or photocatalysis (P), the ternary system outperforms the binary CODH/TiO2 system by about 2 orders of magnitude and the activity is much longer lived. The fact that the CODH/PMAA/TiO2 system (lacking AgNCs) is inactive electrochemically shows that any surface trap states that are introduced play no role the PMAA acting instead to block electron transfer.
Scheme 1.
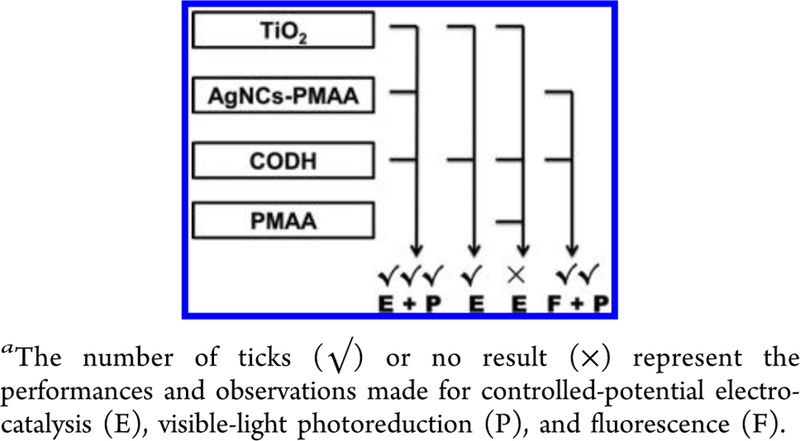
Connectivities between the Di fferent Components As Viewed in Various Experimentsa
Referring to Table 1, rates of photocatalysis scale with the amount of enzyme in the system, and notably, for each given quantity of CODH, the rate of CO production is independent of the amount of AgNCs-PMAA/TiO2.These results show that the Ag nanoclusters are not acting directly as reduction catalysts.43–45 Instead, since CO2 reduction is driven by visible light and not UV (as would be required for bandgap excitation of TiO2), the Ag nanoclusters must be operating as photosensitizers. The report that photoexcited AgNCs are highly proficient in reducing methylviologen is consistent with a calculation by Chen et al. indicating that the LUMO of a AgNC is 0.26 eV higher in energy than the conduction band of TiO2 (itself capable of reducing CO2).43 Although this energy gap means that electrons could pass from photoexcited AgNCs to TiO2, and then to enzyme, the electrochemical evidence discussed in more detail below suggests that electrons could instead transfer directly from photoexcited AgNCs to CODH and thence rapidly reduce CO2, the catalytic efficiency of the enzyme providing such an effective trap.
From the electrocatalysis experiments, AgNCs-PMAA adsorbed on TiO2 promotes CO2 reduction by CODH at a higher potential (lower overpotential) in comparison to TiO2 alone, whereas PMAA alone, which also produces a surface- state redox transition, completely blocks electrocatalysis. Therefore, the AgNCs contained in PMAA are acting as electron mediators to the coadsorbed enzyme. The AgNCs embedded in PMAA must therefore be capable of rapid motion within the polymer, fast intercluster electron transfer, or both. The specific binding of CODH to Ag nanoclusters that is observed by fluorescence (F) helps to account for the photocatalytic activity that is displayed (at a much lower level) by the binary system CODH/AgNCs-PMAA. On the basis of the selective interaction with cysteine reported by others,25 we expect that one or two surface-exposed cysteines are the primary targets, the interaction being dynamic and short-lived in order to account for the fact that the fluorescence is completely quenched during the titration. The surface- exposed cysteine thiolates on CODH are too remote to allow direct electron transfer to the active site from a transiently interacting AgNC, but a dynamic arrangement and/or fast electron transfer between AgNCs within the same PMAA particle would allow use of the surface-exposed D cluster.46
The increased catalytic activity observed for the quasi-ternary assembly that includes TiO2 in comparison to CODH/AgNCs- PMAA alone suggests that the TiO2 nanoparticle is more than just a supporting scaffold. An estimate indicates that on average at least two CODH molecules may be loaded onto a single AgNCs-PMAA/TiO2 nanoparticle (calculation given in the Supporting Information). On the basis of a proposal by Wang and co-workers,43 the most likely mechanistic role for TiO2 is to mediate hole transfer from the AgNC valence band to sacrificial electron donors reacting at its surface.
CONCLUSION
In summary, a stable, quasi-ternary complex of CODH coadsorbed on TiO2 nanoparticles with PMAA-stabilized Ag nanoclusters displays specific reduction of CO2 to CO under visible light, at a per-CODH rate that is 2 orders of magnitude higher and much more stable than that reported earlier when a photosensitizing RuP complex was used.18 The findings raise interesting questions about the overall mechanism, important details of which remain to be clarified. The results help to emphasize the value of using enzymes in investigations of colloidal photocatalysts: their superb abilities as fast, efficient, and selective electrocatalysts focuses attention instead on the limitations affecting the light-harvesting and charge-transfer components and the directions needed for major improvements, including capitalizing on what is already known about the interaction of CODH with CO2 and CO.14
Supplementary Material
ACKNOWLEDGMENTS
L. Z. thanks the Royal Society and Newton fund for a Newton international fellow award (DHR00490). F.A.A. is a Royal Society−Wolfson Research Merit Award holder: research in his laboratory has been supported by several grants from the UK Biotechnology and Biological Sciences Research Council (BBSRC) particularly BB/M005720/1. We thank Professor Stephen Faulkner for allowing us to use his fluorescence spectrometer.
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Gust D; Moore TA; Moore AL Solar Fuels via Artificial Photosynthesis. Acc. Chem. Res 2009, 42, 1890–1898. [DOI] [PubMed] [Google Scholar]
- (2).Kim D; Sakimoto KK; Hong DC; Yang PD Artificial Photosynthesis for Sustainable Fuel and Chemical Production. Angew. Chem., Int. Ed 2015, 54, 3259–3266. [DOI] [PubMed] [Google Scholar]
- (3).Lewis NS; Nocera DG Powering the Planet: Chemical Challenges in Solar Energy Utilization. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 15729–15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bachmeier A; Armstrong F Solar-driven Proton and Carbon Dioxide Reduction to Fuels Lessons from Metalloenzymes. Curr. Opin. Chem. Biol 2015, 25, 141–151. [DOI] [PubMed] [Google Scholar]
- (5).Brown KA; Wilker MB; Boehm M; Dukovic G; King PW Characterization of Photochemical Processes for H2 Production by CdS Nanorod−[FeFe] Hydrogenase Complexes. J. Am. Chem. Soc 2012, 134, 5627–5636. [DOI] [PubMed] [Google Scholar]
- (6).Woolerton TW; Sheard S; Chaudhary YS; Armstrong FA Enzymes and Bio-inspired Electrocatalysts in Solar Fuel Devices. Energy Environ. Sci 2012, 5, 7470–7490. [Google Scholar]
- (7).Wu Y; Rudshteyn B; Zhanaidarova A; Froehlich JD; Ding W; Kubiak CP; Batista VS Electrode-Ligand Interactions Dramatically Enhance CO2 Conversion to CO by the [Ni(cyclam)]- (PF6)2 Catalyst. ACS Catal 2017, 7, 5282–5288. [Google Scholar]
- (8).Urbain F; Tang P; Carretero NM; Andreu T; Gerling LG; Voz C; Arbiol J; Morante JR A Prototype Reactor for Highly Selective Solar-driven CO2 Reduction to Synthesis Gas Using Nanosized Earth-abundant Catalysts and Silicon Photovoltaics. Energy Environ. Sci 2017, 10, 2256–2266. [Google Scholar]
- (9).Schreier M; Heŕoguel F; Steier L; Ahmad S; Luterbacher JS; Mayer MT; Luo J; Graẗzel M Solar Conversion of CO2 to CO Using Earth-abundant Electrocatalysts Prepared by Atomic Layer Modification of CuO. Nat. Energy 2017, 2, 17087. [Google Scholar]
- (10).Liu X; Xiao J; Peng H; Hong X; Chan K; Nørskov JK Understanding Trends in Electrochemical Carbon Dioxide Reduction Rates. Nat. Commun 2017, 8, 15438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jovanov ZP; Hansen HA; Varela AS; Malacrida P; Peterson AA; Nørskov JK; Stephens IEL; Chorkendorff I Opportunities and Challenges in the Electrocatalysis of CO2 and CO Reduction Using Bifunctional Surfaces: A Theoretical and Experimental Study of Au−Cd Alloys. J. Catal 2016, 343, 215–231. [Google Scholar]
- (12).Wang WH; Himeda Y; Muckerman JT; Manbeck GF; Fujita E CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev 2015, 115, 12936–12973. [DOI] [PubMed] [Google Scholar]
- (13).Kortlever R; Balemans C; Kwon Y; Koper MTM Electrochemical CO2 Reduction to Formic Acid on a Pd-based Formic Acid Oxidation Catalyst. Catal. Today 2015, 244, 58–62. [Google Scholar]
- (14).Can M; Armstrong FA; Ragsdale SW Structure, Function, and Mechanism of the Nickel Metalloenzymes, CO Dehydrogenase, and Acetyl-CoA Synthase. Chem. Rev 2014, 114, 4149–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bachmeier A; Hall S; Ragsdale SW; Armstrong FA Selective Visible-Light-Driven CO2 Reduction on a p-Type Dye- Sensitized NiO Photocathode. J. Am. Chem. Soc 2014, 136, 13518–13521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bachmeier A; Wang VCC; Woolerton TW; Bell S; Fontecilla-Camps JC; Can M; Ragsdale SW; Chaudhary YS; Armstrong FA How Light-Harvesting Semiconductors Can Alter the Bias of Reversible Electrocatalysts in Favor of H2 Production and CO2 Reduction. J. Am. Chem. Soc 2013, 135, 15026–15032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chaudhary YS; Woolerton TW; Allen CS; Warner JH; Pierce E; Ragsdale SW; Armstrong FA Visible Light-driven CO2 Reduction by Enzyme Coupled CdS Nanocrystals. Chem. Commun 2012, 48, 58–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Woolerton TW; Sheard S; Pierce E; Ragsdale SW; Armstrong FA CO2 Photoreduction at Enzyme-modified Metal Oxide Nanoparticles. Energy Environ. Sci 2011, 4, 2393–2399. [Google Scholar]
- (19).Armstrong FA; Hirst J Reversibility and Efficiency in Electrocatalytic Energy Conversion and Lessons from Enzymes. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 14049–14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Parkin A; Seravalli J; Vincent KA; Ragsdale SW; Armstrong FA Rapid and Efficient Electrocatalytic CO2/CO Interconversions by Carboxydothermus hydrogenoformans CO Dehydrogenase I on an Electrode. J. Am. Chem. Soc 2007, 129, 10328–10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chen Y-S; Choi H; Kamat PV Metal-Cluster-Sensitized Solar Cells. A New Class of Thiolated Gold Sensitizers Delivering Efficiency Greater Than 2%. J. Am. Chem. Soc 2013, 135, 8822–8825. [DOI] [PubMed] [Google Scholar]
- (22).Xu H; Suslick KS Sonochemical Synthesis of Highly Fluorescent Ag Nanoclusters. ACS Nano 2010, 4, 3209–3214. [DOI] [PubMed] [Google Scholar]
- (23).Diez I; Pusa M; Kulmala S; Jiang H; Walther A; Goldmann AS; Muller AHE; Ikkala O; Ras RHA Color Tunability and Electrochemiluminescence of Silver Nanoclusters. Angew. Chem., Int. Ed 2009, 48, 2122–2125. [DOI] [PubMed] [Google Scholar]
- (24).Chen W-T; Hsu Y-J; Kamat PV Realizing Visible Photoactivity of Metal Nanoparticles: Excited-State Behavior and Electron-Transfer Properties of Silver (Ag8) Clusters. J. Phys. Chem. Lett 2012, 3, 2493–2499. [DOI] [PubMed] [Google Scholar]
- (25).Shang L; Dong S Sensitive Detection of Cysteine based on Fluorescent Silver Clusters. Biosens. Bioelectron 2009, 24, 1569–1573. [DOI] [PubMed] [Google Scholar]
- (26).Brennan BJ; Portoles MJL; Liddell PA; Moore TA; Moore AL; Gust D Comparison of Silatrane, Phosphonic Acid, and Carboxylic acid Functional Groups for Attachment of Porphyrin Sensitizers to TiO2 in Photoelectrochemical Cells. Phys. Chem. Chem. Phys 2013, 15, 16605–16614. [DOI] [PubMed] [Google Scholar]
- (27).Brown DG; Schauer PA; Borau-Garcia J; Fancy BR; Berlinguette CP Stabilization of Ruthenium Sensitizers to TiO2 Surfaces through Cooperative Anchoring Groups. J. Am. Chem. Soc 2013, 135, 1692–1695. [DOI] [PubMed] [Google Scholar]
- (28).Wang VC; Can M; Pierce E; Ragsdale SW; Armstrong FA A Unified Electrocatalytic Description of the Action of Inhibitors of Nickel Carbon Monoxide Dehydrogenase. J. Am. Chem. Soc 2013, 135, 2198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wang VC; Islam ST; Can M; Ragsdale SW; Armstrong FA Investigations by Protein Film Electrochemistry of Alternative Reactions of Nickel-Containing Carbon Monoxide Dehydrogenase. J. Phys. Chem. B 2015, 119, 13690–13697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Jankulovska M; Berger T; Wong SS; Gómez R; Lana-Villarreal T Trap States in TiO2 Films Made of Nanowires, Nanotubes or Nanoparticles: An Electrochemical Study. ChemPhy-sChem 2012, 13, 3008–3017. [DOI] [PubMed] [Google Scholar]
- (31).De la Garza L; Saponjic ZV; Dimitrijevic NM; Thurnauer MC; Rajh T Surface States of Titanium Dioxide Nanoparticles Modified with Enediol Ligands. J. Phys. Chem. B 2006, 110, 680–686. [DOI] [PubMed] [Google Scholar]
- (32).Svetlitchnyi V; Peschel C; Acker G; Meyer O Two Membrane-Associated NiFeS-Carbon Monoxide Dehydrogenases from the Anaerobic Carbon-Monoxide-Utilizing Eubacterium Carbox- ydothermus hydrogenoformans. J. Bacteriol 2001, 183, 5134–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Woolerton TW; Sheard S; Reisner E; Pierce E; Ragsdale SW; Armstrong FA Efficient and Clean Photoreduction of CO2 to CO by Enzyme-Modified TiO2 Nanoparticles Using Visible Light. J. Am. Chem. Soc 2010, 132, 2132–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Luo Z; Yuan X; Yu Y; Zhang Q; Leong DT; Lee JY; Xie J From Aggregation-Induced Emission of Au(I)−Thiolate Complexes to Ultrabright Au(0)@Au(I)−Thiolate Core−Shell Nano- clusters. J. Am. Chem. Soc 2012, 134, 16662–16670. [DOI] [PubMed] [Google Scholar]
- (35).Wu Z; Jin R On the Ligand’s Role in the Fluorescence of Gold Nanoclusters. Nano Lett 2010, 10, 2568–2573. [DOI] [PubMed] [Google Scholar]
- (36).Chen Y; Yang T; Pan H; Yuan Y; Chen L; Liu M; Zhang K; Zhang S; Wu P; Xu J Photoemission Mechanism of Water- Soluble Silver Nanoclusters: Ligand-to-Metal−Metal Charge Transfer vs Strong Coupling between Surface Plasmon and Emitters. J. Am. Chem. Soc 2014, 136, 1686–1689. [DOI] [PubMed] [Google Scholar]
- (37).Diez I; Ras RHA; Kanyuk MI; Demchenko AP On Heterogeneity in Fluorescent Few-atom Silver Nanoclusters. Phys. Chem. Chem. Phys 2013, 15, 979–985. [DOI] [PubMed] [Google Scholar]
- (38).Zhu M; Aikens CM; Hollander FJ; Schatz GC; Jin R Correlating the Crystal Structure of A Thiol-Protected Au25 Cluster and Optical Properties. J. Am. Chem. Soc 2008, 130, 5883–5885. [DOI] [PubMed] [Google Scholar]
- (39).Yau SH; Abeyasinghe N; Orr M; Upton L; Varnavski O; Werner JH; Yeh H-C; Sharma J; Shreve AP; Martinez JS; Goodson Iii T Bright Two-photon Emission and Ultra-fast Relaxation Dynamics in a DNA-templated Nanocluster Investigated by Ultra-fast Spectroscopy. Nanoscale 2012, 4, 4247–4254. [DOI] [PubMed] [Google Scholar]
- (40).Miller SA; Womick JM; Parker JF; Murray RW; Moran AM Femtosecond Relaxation Dynamics of Au25L18− Monolayer-Protected Clusters. J. Phys. Chem. C 2009, 113, 9440–9444. [Google Scholar]
- (41).Dobbek H; Svetlitchnyi V; Liss J; Meyer O Carbon Monoxide Induced Decomposition of the Active Site [Ni−4Fe−5S] Cluster of CO Dehydrogenase. J. Am. Chem. Soc 2004, 126, 5382–5387. [DOI] [PubMed] [Google Scholar]
- (42).Drennan CL; Heo J; Sintchak MD; Schreiter E; Ludden PW Life on Carbon Monoxide: X-ray structure of Rhodospirillum Rubrum Ni-Fe-S Carbon Monoxide Dehydrogenase. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 11973–11978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Chen HJ; Wang Q; Lyu MQ; Zhang Z; Wang LZ Wavelength-switchable Photocurrent in a Hybrid TiO2-Ag Nano- cluster Photoelectrode. Chem. Commun 2015, 51, 12072–12075. [DOI] [PubMed] [Google Scholar]
- (44).Attia YA; Buceta D; Blanco-Varela C; Mohamed MB; Barone G; Loṕez-Quintela, M. A. Structure-Directing and High- Efficiency Photocatalytic Hydrogen Production by Ag Clusters. J. Am. Chem. Soc 2014, 136, 1182–1185. [DOI] [PubMed] [Google Scholar]
- (45).Guo S-X; MacFarlane DR; Zhang J Bioinspired Electrocatalytic CO2 Reduction by Bovine Serum Albumin-Capped Silver Nanoclusters Mediated by [α-SiW12O40]4−. ChemSusChem 2016, 9, 80–87. [DOI] [PubMed] [Google Scholar]
- (46).Dobbek H; Svetlitchnyi V; Gremer L; Huber R; Meyer O Crystal Structure of a Carbon Monoxide Dehydrogenase Reveals a [Ni-4Fe-5S] Cluster. Science 2001, 293, 1281–1285. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.