

Summary
The energetic costs of duplicating chromatin are large and therefore likely depend on nutrient sensing checkpoints and metabolic inputs. By studying chromatin modifiers regulated by epithelial growth factor, we identified histone acetyltransferase 1 (HAT1) as an induced gene that enhances proliferation through coordinating histone production, acetylation and glucose metabolism. In addition to its canonical role as a cytoplasmic histone H4 acetyltransferase, we isolated a HAT1-containing complex bound specifically at promoters of H4 genes. HAT1-dependent transcription of H4 genes required an acetate-sensitive promoter element. HAT1 expression was critical for S-phase progression and maintenance of H3 lysine 9 acetylation at proliferation-associated genes, including histone genes. Therefore, these data describe a feed- forward circuit whereby HAT1 captures acetyl-groups on nascent histones and drives H4 production by chromatin binding to support chromatin replication and acetylation. These findings have important implications for human disease, since high HAT1 levels associate with poor outcomes across multiple cancer types.
Keywords: acetate, acetyl-Co-A, HAT1, nutrient sensing, cancer metabolism, chromatin replication, epigenetics, histone H4, H3K9Ac
Graphical Abstract
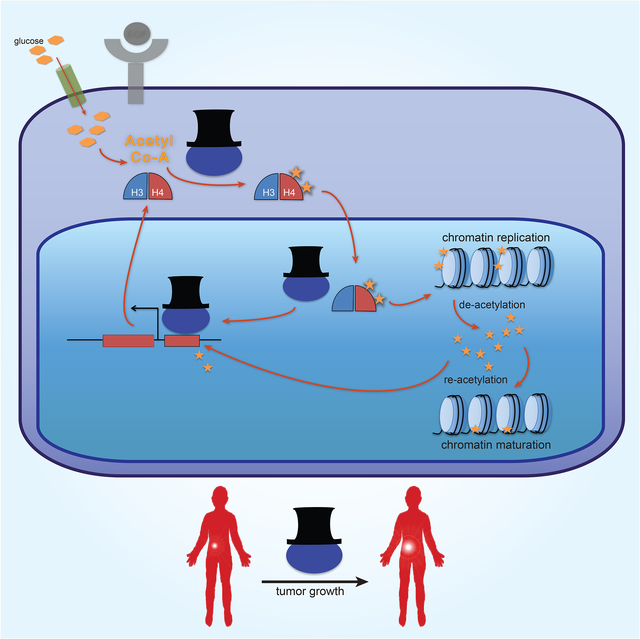
eTOC Blurb
Nascent histone H4 is acetylated by cytoplasmic histone acetyltransferase 1 (HAT1) then de-acetylated after chromatin insertion, releasing free acetate. Gruber et al. discover HAT1 also binds an acetate-sensitive promoter element in histone H4 genes. Therefore, histone production and acetylation are linked by HAT1 to drive cell division via acetyl-Co-A regeneration.
Introduction
There are approximately 3×107 nucleosomes per human diploid nucleus (Alberts, 2002), all of which must be duplicated to capture and compact newly replicated DNA during S phase, at an estimated bioenergetic cost of ~28,000 molecules of ATP per nucleosome (Lynch and Marinov, 2015). Thus, the cellular decision to commit to cell division implies access to sufficient biosynthetic resources to meet this demand. The ‘restriction point’ that governs the transition from G1 to S phase of the cell cycle is nutrient-sensitive (Pardee, 1974). During S phase, cyclin E-cdk2-induced phosphorylation of NPAT drives global and integrated histone gene transcription (Zhao et al., 2000). Prior work has observed metabolic integration of histone H2B biosynthesis with the glycolytic enzyme GAPDH (Zheng et al., 2003). However, direct metabolic inputs to other histone genes are less well understood.
Nucleosomes are composed of the core histone octomer (two copies each of histones H2A, H2B, H3, H4), wrapped by ~147 bases of DNA. Whereas histone protein isoforms exist for H2A, H2B and H3 (eg. macro-H2A, cenH3), there are no histone H4 protein isoforms in mice and humans (Marzluff et al., 2002), indicating that H4 is a core subunit of all nucleosomes and therefore may represent a key focal point for regulation. Indeed, newly synthesized histone H4 is di-acetylated on lysines 5 and 12 by the cytosolic histone acetyltransferase 1 (HAT1) enzyme (Parthun, 2013). However, the function of this histone mark remains enigmatic, in part because it is transient and removed 20–30 minutes after insertion into chromatin (Annunziato, 2013; Annunziato and Seale, 1983; Hammond et al., 2017; Nagarajan et al., 2013).
Recent work has integrated histone acetylation with the metabolic state of the cell. The majority of acetyl groups destined for incorporation into chromatin modifications are produced de novo from glucose (Evertts et al., 2013; Wellen et al., 2009), the primary nutrient utilized by proliferating cells. Two metabolic enzymes appear to control the fate of acetyl groups destined for incorporation into chromatin. ATP citrate lyase (ACLY) generates acetyl-Co-A from citrate, a product of mitochondrial metabolism exported to the cytosol (Zaidi et al., 2012). In addition, an acetyl-Co-A salvage pathway controlled by the Acyl-CoA short chain synthetase family member 2 (ACSS2) can regenerate acetyl-Co-A from acetate at chromatin (Li et al., 2017; Mews et al., 2017) and contributes to histone acetylation in the absence of ACLY (Zhao et al., 2016) or under hypoxic conditions (Bulusu et al., 2017; Kamphorst et al., 2014; Schug et al., 2015), and is required for tumor growth (Comerford et al., 2014). As all newly synthesized H4 is di-acetylated by HAT1, this enzyme may contribute to consumption of acetyl-Co-A during S-phase.
The current study is built on the observation that HAT1 is a growth factor-regulated HAT and is required for epithelial growth factor (EGF) and glucose-stimulated proliferation. This led to the identification of a nuclear role for HAT1 holoenzyme at H4 promoters, where HAT1 recruitment stimulates transcription of histone H4 genes. Because HAT1-induced histone acetylation is dependent on glucose availability, we conclude that HAT1 integrates nutrient availability by inducing production and acetylation of nascent histones. Furthermore, the data suggest that recycling of acetyl-groups derived from nascent histones could be an important source of substrate for chromatin acetylation.
Results
HAT1 is an EGF-stimulated transcript required for EGF-dependent proliferation
Previous work identified EGF as the most potent stimulator of cell division in mammary cell lines (Gruber, 2018). Human mammary epithelial cells immortalized with telomerase (hTert-HME1) were dependent on epithelial growth factor (EGF) for log-phase cell division (Fig. 1A). When EGF was removed from the culture medium cell division continued, but at a diminished rate (Fig. 1A). To determine epigenetic regulators contributing EGF-dependent growth transcriptome analysis by RNA sequencing was performed after 2 passages (6 days) in either EGF-stimulated versus EGF-free media. Overall, 7117 genes were differentially expressed in the presence or absence of EGF (Fig. 1B, FDR 0.1), of which 4313 were up-regulated and 2804 down-regulated upon EGF treatment.
Figure 1: HAT1 is an EGF-regulated gene required for EGF-induced proliferation.

A. hTert-HME1 cells were serially passaged in the presence or absence of EGF and population doublings +/− SD were measured. n=3 in each group.
B. Gene expression profiling by RNA-sequencing of hTert-HME1 cells grown in the presence of absence of EGF for two passages. Significantly regulated genes (FDR < 0.1) are red.
C. Gene set enrichment analysis was performed on differentially expressed genes identified in B.
D. GO terms were used to select genes annotated to contain histone acetyltransferase activity and gene expression values are represented by heatmap.
E. MCF10A cells were grown in the presence or absence of EGF for one passage then immunoblots were performed.
F. hTert-HME1 cells were infected with lentiviruses expressing either non-targeting control shRNA or three different shRNAs targeting the HAT1 transcript. Immunoblotting was performed.
G. Cells from 1F were used for proliferation assays in the presence or absence of EGF. Mean +/− SD. * p < 0.05.
Gene set enrichment analysis performed on the differentially expressed genes revealed multiple gene sets involved in chromatin biology among the top candidates (Fig. 1C). We specifically focused on epigenetic regulators as a mechanism to understand the broad differences in gene expression observed. Gene ontology analysis identified 14 confirmed or putative genes that encoded proteins with HAT activity; these were selected for further analysis. Of these candidates, histone acetyltransferase 1 (HAT1) was the most strongly differentially expressed between +/− EGF conditions (log2 fold change 1.3; FDR < 1 ×106; Fig. 1D). Immunoblotting confirmed that HAT1 protein levels were also stimulated by EGF and down-regulated in EGF- free conditions in an independent but similar cell line MCF10A (Fig. 1E).
As HATs in general confer positive effects on gene expression we tested whether HAT1 contributed to cell proliferation in EGF-stimulated conditions. Three independent shRNAs were used to reduce HAT1 protein levels in hTert-HME1 cells, compared to a control shRNA (Fig. 1F). Each shRNA targeted to HAT1 diminished proliferation (mean 1.8 +/− 0.44-fold) in EGF- stimulated conditions, compared to control shRNA-treated cells (Fig. 1G). In contrast, significant differences were not observed between HAT1 and control shRNAs in the EGF-free condition (Fig. 1G). These data indicate that HAT1 provides a critical function in EGF- stimulated cell proliferation but is not required for basal proliferation without EGF.
HAT1 holoenzyme binds histone H4 promoters
We sought to understand the molecular mechanism by which HAT1 contributed to cell proliferation. HAT1 is a cytoplasmic acetyl-transferase of newly synthesized histone H4, which it di-acetylates on lysines 5 and 12 (Parthun, 2013). It is subsequently imported into the nucleus together with the H3/H4 dimer and Rbap46/48 (Keck and Pemberton, 2012). In hTert-HME1 cells, immunofluorescence using specific antibodies showed strong nuclear localization for HAT1, but cytoplasmic localization was also observed (Fig. S1A). To assess if HAT1 has chromatin-binding properties we performed chromatin immunoprecipitation followed by high- throughput sequencing (ChIP-seq). Compared to input control, antibodies to HAT1 enriched only seven genomic loci (Fig. 2A, B; FDR < 0.01). Surprisingly, all of these loci mapped to a one megabase segment of chromosome 6, which is the Hist1 locus that encodes 35 replication- dependent histone genes. Examination of the location of the HAT1 ChIP-seq peaks showed that six were present within 400 bases of histone H4 gene transcription start sites (Fig. 2B, C). One localized downstream of the HIST1H2BC/H2AC locus, consistent with a potential enhancer site. Visual inspection also revealed a HAT1 ChIP-seq signal at the HIST4H4 promoter on chromosome 12, but it did not pass the statistical cut-off we used (FDR > 0.05; Fig. S1B). There was minimal HAT1 signal at HIST1H4F, HIST1H4G, HIST1H4J or HIST1H4K loci, which were weakly expressed and associated with a closed chromatin state in this cell line (Fig. S1C, arrows; and data not shown). In contrast, there was difficulty mapping reads to the HIST2H4A and HIST2H4B loci, preventing assessment of HAT1 recruitment to these loci.
Figure 2: HAT1 binds and regulates transcription of histone H4 genes.

A. Antibodies to HAT1 were used for ChIP-seq from hTert-HME1 cells and significant binding was computed compared to libraries made from input material. See also Figure S1A.
B. Table of the 7 loci identified that significantly enrich with HAT1 antibodies compared to input control by ChIP-seq. See also Figures S1B and S1C.
C. Signal track of ChIP-seq data using antibodies to HAT1 or Rbap46 at the hist1 locus on human chromosome 6. See also Figure S1D.
D. (upper) Schematics of Firefly luciferase constructs containing 500 bp (−1 to −500) of the HIST1H4E promoter and the indicated complete or partial deletions of the H4-box. H4-box indicates that the 17 bp H4-box sequence was deleted. H4–3’ indicates the 3’ portion of the H4-box was deleted. H4–5’ indicates that the 5’ portion of the H4-box was deleted. (lower) Normalized luciferase activity (Firefly/Renilla) for each luciferase construct after transfection to 293T cells. A Renilla luciferase construct driven by a TK promoter was co-transfected with the Firefly luciferase constructs to normalize for transfection efficiency. See also Figures S1E and S1F.
E. (upper) siRNAs targeting HAT1 or control were transfected to 293T cells and protein levels were assessed by immunoblot after three days. (lower) Two days after siRNA transfection, luciferase reporters with the 500 bp of promoter sequence from either the HIST1H4E wt promoter, the HIST1H4E- H4box were transfected and 16 hours later luciferase activity was measured. * p < 0.05. n= 3, +/− SD.
F. 293T cells were transfected with control or HAT1-targeted siRNAs and three days later cell cycle analysis was performed by propidium iodide staining and flow cytometry. The relative percentages of cells at each cell cycle phase are indicated.
As an independent confirmation that the HAT1 holoenzyme associates with histone H4 promoters, we examined other proteins components of the HAT1 complex. Rbap46 is a bona fide HAT1 interacting protein that also binds newly synthesized histone H4, likely presenting its N-terminus to HAT1 for acetylation (Murzina et al., 2008). ChIP-seq using antibodies to Rbap46 identified a much broader chromatin binding profile (30475 loci, FDR < 0.05; Fig. S1D) compared to HAT1 ChIP-seq, consistent with its known roles in other chromatin remodeling complexes including NuRD (Basta and Rauchman, 2015). However, histone H4 gene promoters were among the most highly enriched loci (Fig. S1D) and the Rbap46 ChIP-seq signal at the Hist1 locus overlapped tightly with the HAT1 ChIP-seq signal, showing specific enrichment at the H4 promoters (Fig. 2C). Therefore, the HAT1 holoenzyme localizes specifically to histone H4 gene promoters.
DNA sequences of the H4 promoters were examined to better understand the specific chromatin-binding pattern of the HAT1 holoenzyme complex. Sequence alignments of the H4 promoters identified a highly conserved genetic element of 17 base-pairs in length that is approximately 80 bases upstream of the ATG start codons (Fig. S1E) and contained within the previously described H4 subtype specific consensus sequence (Heintz, 1991; Osley, 1991) that binds the H4 transcription factor Hinfp (Mitra et al., 2003). We termed this genetic element the ‘H4 box.’ A position weight matrix using the H4 box was used to query for similar motif elements in the accessible DNA elements of the hTert-HME1 cell line derived from ATAC-seq data (Fig. S1F). This led to the identification of only H4 gene promoters (FDR < 0.01) indicating that this motif is restricted to H4 gene promoters throughout the accessible genome. Luciferase assays were used to confirm that the H4 box was critical for transactivation of the H4 promoter. Whereas the wild-type HIST1H4E promoter produced robust Firefly luciferase activity, complete or partial deletions of the H4-box strongly diminished this activity (Fig. 2D) when normalized to co-transfection of a control Renilla luciferase construct. Therefore, histone H4 promoters have a functional, unique DNA promoter element that may underlie the specific recruitment of HAT1 complexes to these loci.
HAT1 is critical for maintaining levels of newly-synthesized H4
Given that the HAT1 holoenzyme complex was specifically recruited to H4 promoters, the possibility of HAT1 directly regulating H4 gene transcription was tested. Short-interfering RNAs (siRNAs) were used to deplete HAT1 protein levels from 293T cells followed by transfection of luciferase reporters. Luciferase reporters bearing the HIST1H4E wild-type reporter were sensitive to HAT1 depletion, but deletion of the H4-box diminished sensitivity to HAT1 levels (Fig. 2E). Cell cycle analysis was performed to determine if this decrease in transcription activity was due to aberrant S phase progression. In cells depleted of HAT1 there was a modest accumulation of cells in G1 phase (5.2% increase) compared to control (Fig. 2F). This small change in cell cycle profile could not account for the 40% decrease in luciferase activity seen. These data indicate that HAT1 binding to HIST1H4E promoters may have a specific stimulatory effect on H4 gene transcription.
These observed effects of HAT1 on H4 transcription led us to examine the levels of newly synthesized core histones upon HAT1 depletion. Acute infection of hTert-HME1 cells with lentiviral shRNAs targeting HAT1 led to substantial depletion of newly synthesized histone H3 and H4 levels, compared to control shRNAs, with no effect on H2A and H2B levels (Fig. 3A), consistent with coordinated post-translation regulation of H3/H4 dimers (Cook et al., 2011). This effect was also true for the HAT1 holoenzyme as siRNAs targeting Rbap46 or Rbap48 both caused decreased H3/H4 dimers without a significant change in H2A or H2B levels, compared to control siRNA (Fig. 3B). Newly synthesized H3/H4 dimers, rather than nucleosomal H3/H4, appeared to be affected as determined by detergent and acid histone extraction, respectively (Fig. 3C). Additional experiments confirmed these newly synthesized histones were highly sensitive to detergent extraction, as compared to salt extraction (Fig. S2), consistent with previously published reports (Holden and Horton, 2009). Thus, HAT1 promotes accumulation of newly synthesized histone H3/H4 dimers without affecting levels of histones embedded in nucleosomes, which could be attributed to a defect in H4 synthesis or the ability of nascent H3/H4 dimers to bind with histone chaperones.
Figure 3: HAT1 is required for accumulation of newly synthesized histone H4.

A. hTert-HME1 cells were acutely infected with either control or two independent HAT1 shRNAs (A or C) and maintained in culture for six days, then SDS-PAGE and immunoblotting was performed after detergent-based protein extraction.
B. hTert-HME1 cells were transfected with the indicated siRNAs, then harvested by detergent extraction three days later and immunoblotting was performed.
C. (upper) hTert-HME1 cells were infected with control or HAT1-targeting shRNAs and maintained in culture for six days, then half of the cells were extracted with detergent then SDS- PAGE and immunoblotting was performed. (lower) Half of the cells were treated as in the upper panel then histones were acid-extracted, fractionated by SDS-PAGE and stained by memCode blue. See also Figure S2.
D. hTert-HME1 cells were infected with the indicated lentiviruses, cultured for six days, then fixed, stained with propidium iodide and analyzed by flow cytometry to distinguish cell cycle phases. See also Figure S3.
HAT1 is required for S phase entry and progression
Because the transcription of H4 genes is tightly coupled to S-phase, we performed cell cycle analysis after depletion of HAT1 with shRNAs in hTert-HME1 cells. A gene-dosage effect was identified whereby strong depletion of HAT1 (shHAT1-A; Fig. 3A) caused a strong block to S-phase entry with accumulation of cells in G1 (Fig. 3D), whereas a less robust depletion (shHAT1-C) caused more modest differences in the cell cycle profile, compared to control-treated cells. Therefore, HAT1 depletion caused both hTert-HME1 cells and 293T cells to accumulate in G1 phase, but the effect was more pronounced in hTert-HME1.
Replication-dependent histones are strongly induced during S-phase to support chromatin replication. To more precisely define the role of HAT1 in S-phase progression, double thymidine block was performed to arrest cells at the G1/S transition. After release from double-thymidine block cell cycle status was monitored by flow cytometry and immunoblots for cyclin A, E and phospho-histone H3, markers of G1, S and M phase, respectively (Fig. S3A, S3B). A similar percentage of control and HAT1-depleted cells arrested at the G1/S transition by double- thymidine block. However, the number of cells remaining in S-phase at 8 hours after release from block was increased in the HAT1-depleted cells compared to control (23.2% HAT1-depleted versus 18.4% control), consistent with delayed S-phase progression. Also, a greater percentage remained in G1 at 8 and 10 hours after release from the block, compared to control cells. Therefore, HAT1-depleted cells have less propensity to enter S phase or proceed more slowly through S phase compared to control cells.
A similar double-thymidine block and release was used to examine histone mRNA transcript levels during S-phase progression. Consistent with the block in S-phase progression observed, levels of all tested replication-dependent histone transcripts were diminished in HAT1- depleted cells (Fig. S2C). This likely reflects coordinated post-transcriptional effects mediated by common 3’ ends shared by all replication-dependent histone mRNAs (Marzluff et al., 2008).
HAT1-stimulated acetyl groups drive transcription via a H4 promoter element
To better understand how HAT1 affects transcription of histone genes, we examined the chromatin state of the Hist1 locus by mapping of the HAT1-dependent histone acetylation sites (H4 lysines 5 and 12) by ChIP-seq in six cell lines (3 shControl and 3 shHAT1 treated; Fig. 4A, B). ChIP-seq for the HAT1-dependent H4K12Ac and H4K5Ac marks identified 2590 +/− 3163 and 14012 +/− 9599 (mean +/− SD; FDR < 0.01) peaks, respectively. However, there was no significant alteration in genome-wide H4K12Ac or H4K5Ac ChIP-seq signal between control and HAT1-depleted cell lines (Fig. 4B). This was confirmed by immunoblots, which showed maintained H4K5 and H4K12 acetylation on nucleosomal histones after acid extraction, despite loss of H4K5 and H4K12 acetylation on newly synthesized H4 caused by HAT1 depletion (Fig. S4A). This is consistent with the reported high acetyltransferase activity of HAT1 towards free H3/H4 dimers, but lack of acetyltransferase activity for nucleosomal H4 (Parthun et al., 1996). Furthermore, there was no enrichment for H4K12Ac or H4K5Ac ChIP-seq signal at histone H4 promoters, despite identification of these marks at promoters elsewhere in the genome (representative examples of the HIST1H4E and ZBTB8OS/RBBP4 loci are shown in Fig. 4C). Therefore, it is unlikely that HAT1-induced acetylation of nucleosomal H4 at H4 promoters affects transcription from these promoters, however HAT1 may have other acetylation targets.
Figure 4: HAT1 promotes acetyl-transfer reactions to histone H3 at H4 promoters.

A. Stable cell lines expressing control shRNAs or three independent shRNAs to HAT1 were fractionated by SDS-PAGE, then immunoblotted.
B. ChIP-seq was performed with antibodies recognizing H4K12ac (left) or H4K5ac (right). Enriched loci were defined compared to input control sequencing. Loci that were significantly enriched or depleted are in red (FDR <0.05). See also Figure S4A.
C. Genome viewer tracks of read depth at the HIST1H4E (left) and the ZBTB8OS/RBBP4 (right) loci for HAT1 ChIP-seq, ATAC-seq, and histone modification ChIP-seq with antibodies that recognize acetylated lysines on H4K5, H4K12 and H3K9.
D. hTert-HME1 cells were treated with lentivirus expressing control or HAT1 shRNAs and also with lentiviruses expressing HAT1-flag, HAT1-E276Q-flag or vector control. (upper) Protein was fractionated by SDS-PAGE and immunoblotting was performed. (lower) Proliferation was measured by cell counting (mean +/− SD, n=3).
E. hTert-HME cells were infected with control or HAT1-targeted lentiviral shRNAs and maintained in culture for 6 days. Sodium acetate (5 mM) was added on day 4. Proteins were fractionated by SDS-PAGE and immunoblotted. See also Figure S4B.
F. Luciferase constructs were designed as indicated with 500 bp of the HIST1H4E promoter cloned upstream of Firefly luciferase. H4-box indicates that the 17 bp H4-box sequence was deleted. As a control 250 bp of the HIST1H2AC promoter was used. Constructs were co- transfected with a Renilla luciferase plasmid to 293T cells and luciferase activity (mean +/− SD) was measured 16 hours later. n=3 biological replicates. See also Figure S4C.
G. hTert-HME1 cells were infected with lentiviral control or HAT1-targeted shRNAs then six days later crosslinking and ChIP-qPCR was performed with antibodies to H3K9Ac and primer sets to the HIST1H4E promoter as indicated. Mean +/− SD, n = 3. The experiment has been independently replicated twice. See also Figure S4C.
As the HAT1-dependent histone modifications were not detected at the H4 promoters, we next tested if the acetyltransferase activity was important for cell proliferation. The HAT1- depleted cell lines were rescued with cDNAs encoding either wild-type HAT1 or HAT1 with an E276Q mutation that has been shown to impair acetyltransferase function (Wu et al., 2012). The HAT1 cDNAs carried silent mutations to disrupt the shRNA target site to allow expression after depletion of endogenous HAT1. Whereas wild-type HAT1 rescued proliferation of HAT1- depleted cells, the HAT1-E276Q mutant did not (Fig. 4D). This confirmed that the catalytic activity of HAT1 is critical to its role in proliferation.
Because the acetyltransferase function of HAT1 was required to support proliferation we tested whether exogenous acetate, the product of histone de-acetylation, could rescue histone H4 levels. Cells treated with control or HAT1-targeted shRNAs were cultured in the presence or absence of 5 mM sodium acetate. Exogenous acetate supplementation rescued histone H4 protein levels after HAT1 depletion and also increased H4 protein in control cells (Figs. 4E, S4B). Thus, although nucleosomal H4K5Ac and H4K12Ac are not associated with H4 promoters, free acetate is sufficient to boost H4 levels in the absence of HAT1. Acetate did not rescue acetylation of the HAT1-dependent histone marks H4 lysine 5 or 12 (Fig. 4E) indicating that acetate functioned downstream of HAT1 catalytic activity. Also, acetate supplementation was not sufficient to rescue proliferation or S-phase progression in the absence of HAT1 (data not shown). Therefore, HAT1 may contribute other essential functions for cell division beyond H4 stimulation. Taken together, these results suggest that acetate is likely regenerated to acetyl- Co-A and utilized to stimulate increased H4 protein levels, a process to which HAT1 may contribute through promoter binding.
Next, we sought to confirm that H4 promoters were indeed acetate sensitive. The HIST1H4E promoter fused upstream of firefly luciferase led to robust induction of luciferase activity when transfected to 293T cells, an effect that was further stimulated by treatment with exogenous sodium acetate (Fig. 4F). In contrast, when the H4-box was deleted from the HIST1H4E promoter (ΔH-box), there was diminished luciferase activity compared to the wild- type promoter and it was no longer responsive to acetate supplementation (Fig. 4F). As a control, the HIST1H2AC promoter, which does not contain an H4-box, was not acetate sensitive. Further analysis of chromatin accessibility at the reporter showed that the H4-box conferred increased chromatin accessibility after acetate treatment (Fig. S4C), an effect that was not evident if the H4-box was deleted. Therefore, histone H4 promoters are acetate sensitive and this property requires the H4-box.
Acetate supplementation has previously been described to stimulate acetylation of lysine residues on histone H3 (Gao et al., 2016). This motivated an assessment for changes in histone H3 lysine acetylation at histone H4 promoters. ChIP-qPCR identified that depletion of HAT1 led to a loss of H3 lysine 9 acetylation at the endogenous promoter of the HIST1H4E gene (Fig. 4G). This suggests that HAT1 stimulates histone H3 lysine 9 acetylation at H4 promoters, an effect that may stimulate increased chromatin accessibility.
HAT1 is required for locus-specific histone H3 acetylation
The identification of a defect in histone H3 lysine 9 acetylation at the HIST1H4E promoter motivated a more careful study of global histone acetylation after HAT1 depletion. ChIP-seq was performed for H3 lysine 9 acetylation in the six cell lines with stable HAT1 depletion and controls (Fig. 4A). This led to identification of 793 sites (FDR < 0.05) with decreased histone H3 lysine 9 acetylation in cell lines with stable HAT1 depletion compared to control cells (Fig. 5A). This indicates that HAT1 is required to maintain H3 lysine 9 acetylation levels at an array of genomic sites.
Figure 5: HAT1 is required for locus-specific histone H3 acetylation.

A. ChIP-seq was performed with antibodies to H3K9ac in six cell lines treated with either control or HAT1-targeted shRNAs (see Fig. 4A) and differential peak-calling was performed. Peaks differentially bound with FDR < 0.05 are indicated in pink. See also Figure S5A.
B. Pathway analysis of genes linked to altered H3 lysine 9 acetylation from Fig. 5A.
C. List of histone genes identified in the ‘nucleosome’ pathway from Fig. 5B to have significantly decreased H3 lysine 9 acetylation signal in the HAT1-depleted cells compared to control (FDR < 0.1).
D. Genome viewer track of H3 lysine 9 ChIP-seq signal at the HIST1H2BJ/HIST1H2AG locus in shControl and shHAT1-treated cells.
E. Genome viewer track of H3 lysine 9 ChIP-seq signal at the HIST1H4E locus in shControl and shHAT1-treated cells.
F. hTert-HME1 cells were transfected with control or KAT2A-targeted siRNAs and three days later proteins were harvested by detergent extraction and immunoblotting was performed. See also Figure S5B, C.
Next, pathway analysis was performed on the genes that had a H3 lysine 9 acetylation defect. This revealed a number of pathways associated with cell proliferation including cell cycle, DNA damage, RNA processing, nucleosome and kinetochore (Fig. 5B), suggesting that this acetylation defect could contribute to delayed growth and S phase progression in HAT1- depleted cells. Further examination of the genes in the ‘nucleosome’ pathway showed that many replication-dependent histone genes were affected including H4 genes, as well as genes for H1, H2A, H2B and H3 (Fig. 5C, D, E). Therefore, multiple genes contributing to cell proliferation have diminished H3 lysine 9 acetylation after HAT1 depletion.
Next, we sought to determine whether the H3 lysine 9 acetylation defect in HAT1- depleted cell lines was a primary defect or secondary to aberrant cell cycle progression. We compared global H3 lysine 9 acetylation ChIP-seq profiles between hTert-HME1 cells synchronized at the G1/S transition (early S phase) with cells synchronized in late S/G2. Overall, there were 24,937 differential H3 lysine 9 loci identified (FDR < 0.05; Fig. S5A), of which the vast majority (24,827) had increased signal at the G1/S transition (early S) compared to late- S/G2 phase. This is consistent with previous studies showing that histone H3 lysine 9 acetylation builds during G1 phase and diminishes during and after S-phase (Cai et al., 2011). Therefore, the observed HAT1-dependent decrease in H3 lysine 9 acetylation is unlikely to be secondary to a decrease proportion of HAT1-depleted cells in G2/M phase compared to control cells, but these data do not exclude a role for HAT1 in promoting G1/S transition, which could also lead to H3 lysine 9 acetylation.
As HAT1 does not acetylate H3 lysine 9 nor does it bind to the vast majority of genomic sites with diminished H3 lysine 9 acetylation we hypothesized that a dedicated H3 lysine 9 acetyltransferase may be responsible for H3 lysine 9 acetylation downstream of HAT1 activity. Examination of published ENCODE ChIP-seq data for H3 lysine 9 acetyltransferases showed that GCN5/KAT2A had extensive binding throughout the Hist1 locus (Fig. S5B). Therefore, HAT1-mediated acetylation of nascent histones may feed acetyl groups to GCN5 via de-acetylation of deposited nascent histones and regeneration of acetyl-Co-A by ACSS2. Indeed, depletion of GCN5/KAT2A led to reduction of nascent histone H4 levels, compared to control siRNA treatment (Fig. 5F), confirming that other histone acetyltransferases may work downstream of HAT1 to promote gene expression. A similar effect was seen when pools of siRNAs were used to deplete both GCN5 and the related acetyltransferase PCAF/KAT2B (Fig. S5C).
Elevated HAT1 levels are associated with poor outcomes in malignancies
The above data indicate that HAT1 is a positive regulator of EGF-dependent proliferation through its ability to stimulate histone acetylation and S-phase progression. Furthermore, growth factor signaling pathways are common driver mutations in human malignancy. This led to the hypothesis that HAT1 expression could be an important dependency of cell proliferation in human cancers. To determine whether HAT1 expression is associated with human cancer outcomes, overall survival was stratified by HAT1 expression levels in all tumor subsets in the available TCGA data. This led to the identification of six solid tumor subtypes (ACC, BLCA, KICH, LGG, LIHC, LUAD) wherein high HAT1 levels (upper quartile of expression) were significantly associated with inferior survival outcomes, compared to the low HAT1 expressing tumors (lowest quartile of expression; Fig. 6A). Therefore, HAT1 expression level may be an important predictor of patient outcomes in various human malignancies.
Figure 6: Elevated HAT1 levels are associated with poor patient outcomes in human cancer and maintain H4 acetylation during glucose stimulation.

A. Kaplan-Meier survival plots stratified by HAT1 expression levels (low = lowest quartile by z- score; high = highest quartile). Logrank p-values and total n in each group are indicated. TPM = transcripts per million (RNA-seq quantification). ACC=adrenocortical carcinoma; BLCA=bladder urothelial carcinoma; KICH=kidney chromophobe; LGG=brain lower grade glioma; LIHC=liver hepatocellular carcinoma; LUAD=lung adenocarcinoma.
B. Cancer cell lines A549 (left) and HepG2 (right) were treated with control or HAT1-targeted lentiviral shRNAs and population doublings were measured.
C. 4T1 mammary carcinoma cells expressing the indicated lentiviral shRNAs were orthotopically injected into bilateral mammary fat pads and tumor sizes were measured. n=5 mice in each group, each mouse bearing two tumors. Data are representative of two independent experiments, mean +/− SD is plotted.
D. Stable hTert-HME1 cell lines with controls or HAT1-targeted shRNAs were grown in media containing the indicated glucose concentrations and cells were counted after three days. n=3 biological replicates. * p < 0.05 compared to equimolar shControl condition.
E. Stable hTert-HME1 cell lines were grown in media with the indicated glucose concentrations for three days, then protein was fractionated by SDS-PAGE and immunoblotted. See also Figure S6A.
F. 293T cells were transfected with Firefly luciferase constructs containing either the HIST1H4E (H4E) promoter or the same promoter with the H4-box deleted (H4-box) in glucose containing (25 mM) or glucose-free (0 mM) media. Control renilla luciferase constructs were co- transfected and luciferase activity was measured 24 hours later. See also Figure S6B.
To confirm that HAT1 expression is important for human cancer cell proliferation, cell lines derived from LUAD and LIHC tumor types were treated control or HAT1 shRNAs. Depletion of HAT1 was associated with diminished proliferation in both A549 (LUAD) and HepG2 (LIHC) cancer cell lines (Fig. 6B). This suggests that HAT1 expression may be critical to support cancer cell proliferation in these cancer subtypes. In addition, Hat1 depletion impaired tumor formation in vivo as mice orthotopically injected with 4T1 mammary carcinoma cell lines with two independent shRNAs targeting Hat1 failed to form tumors, whereas the control shRNA-expressing 4T1 cells formed tumors robustly (Fig. 6C). Therefore, HAT1 expression engenders cancer cell proliferation and tumor formation.
Malignant cells that outgrow their local blood supply can experience nutrient limitation and metabolic stress. Glucose metabolism is a major anabolic pathway of proliferating cancer cells. To assess whether HAT1 contributed to glucose-dependent proliferation, cells were grown in varying levels of glucose-containing media. Cells with stable HAT1 depletion proliferated at a slower rate compared to control cells in both glucose-replete (25 mM) and glucose-limited (2.5 mM) conditions (Fig. 6D), whereas more profound glucose limitation equally impaired proliferation regardless of HAT1 levels. In addition, the HAT1-dependent acetylation marks on free H4 (H4K5 and H4K12) were strongly dependent on adequate glucose levels, and further reduced by HAT1 depletion (Fig. 6E). In contrast, the nucleosomal acetylation site on H3 lysine 9 was less sensitive to glucose limitation. These data indicate that HAT1 may support glucose- dependent proliferation through acetylation of free histone H4.
In contrast to the acute depletion of HAT1, which causes significant loss of histone H3/H4 dimers (Fig. 3A), stable cell lines with HAT1 depletion have normal H4 protein levels (Fig. 6E), consistent with previously published data (Nagarajan et al., 2013). Acetyl-Co-A can be generated from glutamine during periods of glucose withdrawal (Yang et al., 2009). Indeed, combined reduction of glucose and glutamine caused diminished nascent H4 levels in wild-type hTert-HME1 cells, an effect that was partially rescued by pyruvate (Fig. S6A).
To determine whether glucose stimulated nascent H4 production at the level of the histone H4 promoter luciferase assays were performed. This HIST1H4E promoter required glucose for full transcriptional activity (Fig. 6F). This effect depended on the presence of the H4-box because a HIST1H4E promoter luciferase construct with the H4-box deleted had decreased transcriptional activity and was no longer sensitive to glucose levels (Fig. 6F). In addition, ChIP-qPCR showed that H3 lysine 9 promoter acetylation of the HIST1H4C gene was sensitive to glucose levels (Fig. S6B). Taken together, these data suggest that H4 promoters are glucose sensitive and that HAT1 is required to maintain nascent histone H4 levels under conditions of glucose limitation, an effect which may depend both on the ability of HAT1 to acetylate nascent H4 and the ability of HAT1 to bind H4 promoters.
Discussion
Two lines of evidence support a role for HAT1 in a nutrient sensing pathway to integrate glucose metabolism with histone production. First, the HAT1-induced H4 di-acetylation marks are strongly dependent on adequate glucose levels (Fig. 6E). As glucose is the primary substrate for acetyl-Co-A generation in proliferating mammalian cells (Wellen et al., 2009) the data indicate that HAT1 captures glucose-derived acetyl groups on nascent H4. These are confirmed to be newly synthesized H4 di-acetylation, rather than nucleosomal acetylation marks, because HAT1 depletion did not cause a robust change in either H4K5 or H4K12 acetylation when measured by ChIP-seq or immunoblot (Figs. 4B, S4A). Taken together, the data suggest HAT1 likely senses acetyl-Co-A availability by placing available acetyl groups on nascent H4.
A second line of evidence that supports a nutrient sensing role for HAT1 in H4 production is that H4 promoters are themselves glucose and acetate responsive. We identify a genetic element in H4 promoters that confers acetate sensitivity, which is the likely mechanism for also sensing glucose availability, because glucose is metabolized to acetyl-Co-A in proliferating cells. HAT1 contributes to this process by stimulating H3 lysine 9 acetylation at H4 promoters. As HAT1 itself does not have biochemical activity towards H3 in nucleosomes (Parthun et al., 1996), HAT1 likely stimulates H3K9 acetylation through an indirect mechanism. Indeed, the H3 lysine 9 acetyltransferase GCN5 is bound throughout the Hist1 locus and is required for production of nascent H4. This suggests that nascent H4 acetylated by HAT1 could provide a source of acetate when these histones are de-acetylated upon insertion to chromatin leading to acetyl-Co-A regeneration through the Acss2 enzyme. Thereby, HAT1 may facilitate chromatin acetylation by passing acetyl-groups to downstream HATs.
The data indicate that HAT1 drives a gene-metabolite circuit to support H4 production during growth factor stimulation. HAT1 occupies two nodal points in the circuit, as an acetyltransferase for nascent H4 and as a chromatin-bound factor at H4 promoters. During growth factor stimulation HAT1 expression is induced and the enzyme acetylates nascent H4. The HAT1/Rbap46/H3/H4 complex is subsequently imported to the nucleus via importin 4 or other karyopherin members (Campos et al., 2010). HAT1/Rbap46 is dissociated from this complex upon deposition of H3/H4 dimers at the replication fork (Agudelo Garcia et al., 2017). Thereafter, HAT1 could accumulate at H4 promoters to stimulate further production of nascent H4 via promoter binding. As the H4 promoter requires HAT1-dependent acetyl-groups for high- output transcription, this circuit is also dependent on ongoing nutrient metabolism to support adequate acetyl-Co-A pools for H3 promoter acetylation. This feed-forward circuit likely operates to support rapid induction of histones during S-phase to fuel chromatin replication.
The requirement for the acetyltransferase activity of HAT1 for proliferation may point to HAT1-dependent acetylation of non-histone substrates, for example Hinfp, a factor also shown to bind H4 promoters (Medina et al., 2008; Mitra et al., 2003). Alternatively, HAT1 promoter binding is reminiscent of the Spt10 factor that specifically binds histone promoters in Saccharomyces cerevisiae (Eriksson et al., 2005) and is required for histone gene transcription and H3 lysine 9 acetylation at those promoters (Xu et al., 2005). Spt10 has a GNAT-type acetyltransferase domain, with homology to the acetyltransferase domain in HAT1. However, intriguingly, acetyltransferase activity for Spt10 has yet to be demonstrated despite efforts by multiple laboratories (Kurat et al., 2014b). Instead, Spt10 may recruit other acetyltransferases to induce transcription (Kurat et al., 2014a). Spt10 is poorly conserved in higher eukaryotes indicating that other acetyltransferase-containing proteins, perhaps HAT1, may have acquired its function in these organisms. However, mammalian cells also depend on post-transcriptional processing of histone mRNAs to maintain histone production (Harris et al., 1991; Marzluff et al., 2008; Stauber and Schumperli, 1988), an effect that is less important in yeast.
The purpose of the di-acetylation marks placed on nascent H4 has remained enigmatic since their discovery over four decades ago (Jackson et al., 1976; Ruiz-Carrillo et al., 1975). Similarly, the role of HAT1 in chromatin replication has been difficult to parse, given that nucleosome deposition can proceed without HAT1 or the HAT1-induced di-acetylation mark (Ma et al., 1998; Megee et al., 1990; Nagarajan et al., 2013; Shibahara et al., 2000; Zhang et al., 1998). Indeed, our data confirms that proliferation can proceed without HAT1, albeit at a diminished rate. Thus, di-acetylation of nascent histones may serve a metabolic purpose in providing additional nuclear acetyl-Co-A to downstream epigenetic reactions in growth-factor stimulated cells via action of the Acss2 enzyme. As a nucleo-cytoplasmic acetyl-transferase, HAT1 is ideally positioned to deposit newly produced acetyl-Co-A-derived acetyl groups onto nascent histones which are then shuttled to the nucleus and de-acetylated, yielding free acetate which can be converted to acetyl-Co-A. By linking acetyl-Co-A regeneration to histone H4 production, HAT1 may provide cells an elegant means to coordinate nutrient supply with chromatin replication in rapidly dividing cells.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Snyder (mpsnyder@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines:
Female human cell lines MCF10A (ATCC Cat# CRL-10317, RRID:CVCL_0598) and hTert-HME1 (ATCC Cat# CRL-4010, RRID:CVCL_3383) were obtained from ATCC and were maintained in MEGM media (Lonza or Promocell) in a humidified incubator at 37°C, with 5% CO2. Glucose and glutamine-free media was prepared with RPMI–glucose/glutamine formulation with addition of the MEGM growth factors with 1% penicillin-streptomycin. Male HepG2 (RRID:CVCL_0027) and male A549 (RRID:CVCL_0023) human cell lines were obtained from the ENCODE consortium and maintained in MEM and F-12K medias, respectively, supplemented with 10% FBS and 1% penicillin-streptomycin. The female 4T1 (ATCC Cat# CRL-2539, RRID:CVCL_0125) mouse cell line was purchased from ATCC and maintained in RPMI media with 10% FBS and 1% penicillin-streptomycin. ATCC performs routine cell line authentication and ENCODE maintains stocks of previously authenticated cell lines.
Animals:
Six to eight-week-old Balb/c female mice were purchased from Taconic Biosciences and housed in groups of up to 5 mice per cage in accordance with NIH and Stanford Administrative Panel on Laboratory Animal Care (APLAC) regulations. Studies were performed on mice aged 8–10 weeks. They were maintained with bedding, and freely available food pellets and water in a temperature and humidity-controlled room with a 12-hour light/dark cycle. Animals were randomly assigned to experimental groups.
METHOD DETAILS
Transfection, cell assays:
siRNA transfection was performed with 30 microliters of Lipofectamine RNAiMAX (Invitrogen), 3 mL OptiMEM media, 18 picomoles of siRNA and 5E5 cells in 10 cm plates. Cell proliferation was measured with a Biorad TC 10 with trypan blue. Population doublings were calculated using the formula: , where PD = population doublings, counted = number of cells counted, plated = number of cells plated. For luciferase assays, 500 bp of promoter sequence was cloned into the pGL4.23 vector (Promega) using gBlock synthesis (IDT) and Gibson assembly (NEB Hifi DNA Assembly Master Mix). Constructs were transfected to 293T cells with Xtremegene 9 (Roche). One tenth mass equivalent of pRL-TK plasmid was co-transfected. Luciferase activity was measured with Dual-Luciferase Reporter System (cat# E1910; Promega) 24 hours after transfection. Double- thymidine block was performed by addition of 2 mM thymidine for 18 hours, followed by 9-hour wash-out with fresh media, then a second block with thymidine (2 mM) for 15 hours, then wash- out and release at the G1/S transition.
RNAi and cDNAs:
HAT1 shRNAs were purchased from OriGene with the following sequences:
GATGGCACTACTTTCTAGTATTTGAGAAG;
AAGGATGGAGCTACGCTCTTTGCGACCGT;
TCCTACAGTTCTTGATATTACAGCGGAAG.
The HAT1 cDNA was purchased from OriGene (catalog# RC209571L1) and the 5’ end was extended by a Gibson assembly reaction to add the additional 255 nucleotides to obtain a full- length clone. Mutagenesis of this cDNA was performed with the QuikChange II XL Site- Directed Mutagenesis kit with the following primers to make the D276Q acetylation-dead form:
CAGTTCTTGATATTACAGCGCAAGATCCATCCAAAAGCTAT,
ATAGCTTTTGGATGGATCTTGCGCTGTAATATCAAGAACTG.
To mutate the shRNA-C target site mutagenesis was performed with the following primers: GGTCGCAAAGAGCGTAGCTCCGTCTTTGTTGTATTTCTCAAATACTAGAAAGTA GTGCCATCTTTCATC,
GATGAAAGATGGCACTACTTTCTAGTATTTGAGAAATACAACAAAGACGGAGC TACGCTCTTTGCGACC.
Silencer select (Ambion) siRNAs were purchased from Life Technologies (Fisher). siRNA sequences:
KAT2B: GGUACUACGUGUCUAAGAAtt
KAT2B: GGUGGUAUCUGUUUCCGUAtt
KAT2A: CCAUUUGAGAAACCUAAUAtt
KAT2A: AAUGGAACCUGUAAGUGUAtt
HAT1: GCAACACGCUAGAAGGGUUtt
Mouse tumor studies:
The 4T1 mammary carcinoma cell line was infected with lentiviral control or HAT1-targeted shRNAs (mA-GAAGCTACAGACTGGATATTA; mB-GAAGATCTTGCTGTACTATAT) in the pLKO-puro-IPTG-3xLacO vector (Sigma-Aldrich) then selected in puromycin and single cell clones were obtained by limiting dilution. To minimize clonal variation, two independent cell line clones from each shRNA were mixed in 1:1 ratio prior to injection of 50,000 cells to bilateral mammary fat pads of 8–10-week-old female Balb/c mice. At day 1 after cell injection 20 mM IPTG was added to the water bottles of all cages to induce shRNA expression and water was changed twice a week with repeat addition of IPTG. Tumor size was measured by calipers.
Immunoblots, qRT-PCR:
Protein extracts were made in RIPA buffer and quantitated by BCA assay and diluted to equal concentrations and mixed with 4× LDS sample buffer and sample reducing agent (Invitrogen). Polyacrylamide gel electrophoresis was performed on NuPAGE Novex gradient gels (Thermo Fisher) followed by wet transfer to nitrocellulose membranes. Blocking was briefly performed with 5% non-fat milk and primary antibody was incubated overnight at 4°C in 5% milk, followed by washing in PBST, then with HRP-conjugated secondary antibody (Cell Signaling) at room temperature for 1 hour followed by washing, then developed with ECL pico or femto (Thermo Fisher). For gene expression assays total RNA was isolated with All-prep (Qiagen), DNase treated, then reverse-transcribed with Superscript III (Invitrogen). qPCR was performed with 2× KAPA SYBR Fast Master Mix on a QuantStudio Flex 6 (Applied Biosystems).
ChIP-seq:
Log-phase growth cells were crosslinked in 1% formaldehyde at a density of 5×105 cells per milliliter for 10 minutes at room temperature, then quenched with 125 mM glycine for 5 minutes, washed in PBS and snap frozen. Cells were then thawed, and nuclei isolated by triton- X permeabilization (50 mM HEPES-KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, PIC) for 10 minutes at 4 deg C followed by washing in a low detergent buffer (200 mM NaCl, 1 mM EDTA pH 8, 0.5 mM EGTA, 10 mM Tris, pH 8, PIC) at room temperature for 10 minutes. Then RIPA with protease inhibitor cocktail (PIC) was added and sonication was performed by Branson (3 pulses of 30 seconds each with power output 4 W), followed by 14 cycles of sonication on the Bioruptor Pico. Chromatin extracts were then cleared by centrifugation and immunoprecipitation was performed with 5 micrograms of antibodies and protein A/G agarose beads overnight. The next day the beads were extensively washed with RIPA (10 mL washes for 10 minutes each × 3), then PBS once, then resuspended in TE with 1% SDS and crosslinks were reversed overnight at 65 degrees Celsius. The next day RNase A treatment and proteinase K treatment were performed, followed by recovery of DNA with Qiagen Qiaquick spin columns. High throughput sequencing libraries were then constructed by A tailing, Illumina Truseq adapter ligation and 10–15 cycles of PCR followed by library purification, removal of PCR primer-dimers and high-throughput sequencing by HiSeq 4000 (Illumina). Antibodies for ChIP-seq: Rbap46 (V415) from Cell Signaling (cat# 6882); HAT1 from Abcam (cat# ab194296); Histone H4 acetyl K12 from Abcam (cat# ab46983); acetyl- Histone H4 (Lys5) from Millipore (cat# 07–327), Anti-Histone H3 (acetyl K9) from Abcam (cat# 4441).
Chromatin accessibility:
A modified form of the published ATAC-seq protocol (Buenrostro et al., 2013) was used to assess chromatin accessibility on the luciferase reporter with specific primers (CAACACACCAACGAAAATAGCC, CGCTCGTTGTAGATGTCGTTAG). Cells were counted and 50,000 cells were used for nuclei isolation then transposition reaction with the Tn5 transposase (Illumina). Isolated DNA was then size purified by agarose electrophoresis to recover DNA of size 75–500 bp. Then qPCR was performed with the primers indicated. For deep sequencing, libraries were sequenced by HiSeq 4000 (Illumina).
Transcriptome profiling:
Total RNA was isolated with AllPrep from cell lines and rRNA depletion was performed with RiboZero (Illumina). rRNA depletion was confirmed by RNA Pico Bioanalyzer (Agilent). Approximately 25 ng of rRNA-depleted was used for library preparation with the ScriptSeq v.2 (Illumina) kit according to the manufacturer’s recommendation and sequencing was performed on HiSeq.
QUANTIFICATION AND STATISTICAL ANALYSIS
ChIP-seq and ATAC-seq peak calling and analysis:
Paired-end 100 bp reads were trimmed with cutadapt (Martin, 2011) version 1.8.1 with flags–u −50 –U −50 –a CTGTCTCTTATACACATCTCCGAGCCCACGAGAC -A CTGTCTCTTATACACATCTGACGCTGCCGACGA -O 5 -m 30 -q 15. Bowtie 2 (Langmead and Salzberg, 2012; Langmead et al., 2009) version 2.3.1 was used to align trimmed reads to hg19 with flags -q --phred33 -X 2000 --fr -p 8 -x hg19, followed by samtools (Li et al., 2009) sort command. Duplicates were marked with Picard-tools (Picard-tools) version 1.92, then samtools view with flags –b –f 1 -F12 –L were used to filter mitochondrial mapping reads with a bed file containing all chromosomes except chrM. SPP/phantom (Kharchenko et al., 2008; Landt et al., 2012) was run to obtain the fragment length with maximum strand cross-correlation. MACS2 (Zhang et al., 2008) callpeak function was then performed with flags -q 0.05 --nomodel –extsize=1/2 fragment length obtained from SPP. The program align2rawsignal (Consortium, 2012) (Kundaje) was used to create genome-wide signal coverage tracks with normalization to account for depth of sequencing and read mappability with flags kernel (k)=epanechnikov, fragment length (l)=150 (ATAC-seq) or (l)=1/2 fragment length from SPP for ChIP-seq, smoothing window (w)=150, normFlag(n)=5, mapFilter (f)=0. Further peak calling was performed with DiffBind (Ross-Innes et al., 2012; Stark, 2011) with summits=250 parameter contrasting ChIP-antibody to input control to obtain final enriched peak counts.
TCGA gene expression analysis:
Exploratory analysis and generation of survival plots and statistics was performed with the GEPIA resource (Tang et al., 2017).
Gene set enrichment analysis and pathway analysis:
Gene set enrichment analysis was performed with GSEA (Mootha et al., 2003; Subramanian et al., 2005). Pathway analysis was performed with DAVID (Huang da et al., 2009a, b) with FDR correction for multiple hypothesis testing.
Statistics:
Details of statistical tests can be found in the figure legends and manuscript text, including definitions of the tests used, definition and quantification of n, and quantification of measurement precision (error). Multiple-hypothesis correction was used for high-throughput sequencing studies and pathway analysis with and FDR cutoff of at least < 0.1 required to declare statistical significance. Otherwise, statistical significance was declared for p values < 0.05.
DATA AND SOFTWARE AVAILABILITY
ChIP-seq raw data and differential peak call files have been uploaded to GEO with accession number GSE117472. RNA-seq and ATAC-seq data are deposited in GEO with accession numbers GSE107280 and GSE107119, respectively. Raw data for immunoblots are deposited at Mendeley (http://dx.doi.org/10.17632/7wzp35y5mc.1).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HAT1 | Abcam | Cat# ab194296 |
| Rbap46 | Cell Signaling | Cat# 6882 |
| Rbap48 | Cell Signaling | Cat# 9067 |
| Histone H4 acetyl K12 | Abcam | Cat# ab46983 |
| Histone H4 acetyl K5 | Millipore | Cat# 07–327 |
| GCN5 | Cell Signaling | Cat# 3305 |
| PCAF | Cell Signaling | Cat# 3378 |
| Actin | Thermo Fisher | Cat# MA5–15452 |
| NBN/NBS1 | Invitrogen | Cat# PA5–78071 |
| Histone H4 | Abcam | Cat# ab7311 |
| Histone H3 | Abcam | Cat# ab18521 |
| Histone H2A | Abcam | Cat# ab18255 |
| Histone H2B | Abcam | Cat# ab1790 |
| Histone H3 acetyl K9 | Abcam | Cat# ab4441 |
| Cyclin E1 | Cell Signaling | Cat# 4129 |
| Cyclin A2 | Cell Signaling | Cat# 4656 |
| Phospho-Histone H3 (Ser 10) | Cell Signaling | Cat# 53348 |
| Bacterial and Virus Strains | ||
| n/a | ||
| Biological Samples | ||
| n/a | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2× SYBR Master Mix | KAPA | KK4618 |
| Nextera DNA Flex Library Prep Kit | Illumina | 20018704 |
| Lipofectamine RNAiMAX | Thermo-Fisher | 13778075 |
| Xtremegene 9 | Sigma-Aldrich | XTG9-RO |
| Critical Commercial Assays | ||
| Dual Luciferase Reporter Assay System | Promega | Cat# E1910 |
| QuikChange II XL Site-Directed Mutagenesis kit | Agilent | Cat# 200523 |
| NEBuilder® HiFi DNA Assembly Master Mix | NEB | Cat# E2621S |
| Deposited Data | ||
| ChIP-seq data available at GEO | This paper | GSE117472 |
| RNA-seq data available at GEO | Gruber et al. 2018 | GSE107280 |
| ATAC-seq data available at GEO | Gruber et al. 2018 | GSE107119 |
| Immunoblot primary data at Mendeley | This paper | http://dx.doi.org/10.17632/7wzp35y5mc.1 |
| Experimental Models: Cell Lines | ||
| hTert-HME1 | ATCC | Cat# CRL-4010 |
| MCF10A | ATCC | Cat# CRL-10317 |
| A549 | ENCODE | gift |
| HepG2 | ENCODE | gift |
| 4T1 | ATCC | Cat# CRL-2539 |
| Experimental Models: Organisms/Strains | ||
| Balb/c female mice (6–8-week-old) | Taconic | Cat# BALB-F |
| Oligonucleotides | ||
| HAT1 mutagenesis primers for D276Q mutation: CAGTTCTTGATATTACAGCGCAAGATCCATCCAAAAGCTAT, ATAGCTTTTGGATGGATCTTGCGCTGTAATATCAAGAACTG | IDT | Custom |
| HAT1 mutagenesis primers for shRNA-C binding site: GGTCGCAAAGAGCGTAGCTCCGTCTTTGTTGTATTTCTCAAATACTAGAAAGTAGTGCCATCTTTCATC, GATGAAAGATGGCACTACTTTCTAGTATTTGAGAAATACAACAAAGACGGAGCTACGCTCTTTGCGACC | IDT | Custom |
| KAT2B siRNA: GGUACUACGUGUCUAAGAAtt | Invitrogen | s16895 |
| KAT2B siRNA: GGUGGUAUCUGUUUCCGUAtt | Invitrogen | s16894 |
| KAT2A siRNA: CCAUUUGAGAAACCUAAUAtt | Invitrogen | s5658 |
| KAT2A siRNA: AAUGGAACCUGUAAGUGUAtt | Invitrogen | s5657 |
| HAT1 siRNA: GCAACACGCUAGAAGGGUUtt | Invitrogen | s16196 |
| Recombinant DNA | ||
| Human HAT1 cDNA in lentiviral vector (pGFP-C-shLenti) | Origene | Cat# RC209571L1 |
| Human HAT1 lentiviral shRNAs | Origene | Cat# TL312517 |
| Mouse Hat1 lentivrial shRNAs (pLKO-puro-IPTG-3xLacO): TRCN0000231310, TRCN0000039275, TRCN0000231307 | Sigma-Aldrich | custom |
| Lentiviral control shRNA (mouse) | Sigma-Aldrich | SHC332V |
| Lentiviral control shRNA (human) in pGFP-C-shLenti | Origene | TR30021 |
| pGL4.23 | Promega | E841A |
| pRL-TK | Promega | E2241 |
| Software and Algorithms | ||
| Cutadapt | Martin, 2011. | https://cutadapt.readthedocs.io/en/stable/ |
| Bowtie2 | Langmead et al., 2009 | |
| Picard-tools | n/a | http://broadinstitute.github.io/picard |
| samtools | Li et al. 2009 | http://www.htslib.org |
| SPP/phantom | Kharchenko et al., 2008; Landt et al., 2012 | https://github.com/kundajelab/phantompeakqualtools |
| MACS2 | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| align2rawsignal | Consortium, 2012 | https://github.com/akundaje/align2rawsignal |
| DiffBind | Ross-Innes et al., 2012; Stark, 2011 | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| DAVID | Huang da et al., 2009a, b | https://david.ncifcrf.gov |
| GSEA | Mootha et al., 2003; Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp |
| Other | ||
Highlights.
HAT1 is an EGF-stimulated acetyltransferase required for EGF-dependent growth
HAT1 holoenzyme specifically binds to histone H4 promoters
Histone H4 promoters contain an acetate sensitive genomic element
HAT1 expression is associated with poor cancer outcomes in humans and mice
Acknowledgements:
We wish to thank Brandon Aubrey at Dana-Farber Cancer Institute and members of the Snyder laboratory for helpful discussions and constructive criticism of this work.
Funding: This work used the Genome Sequencing Service Center by Stanford Center for Genomics and Personalized Medicine Sequencing Center, supported by the NIH grant award S10OD020141. J.J.G. was supported by fellowships from the Jane Coffin Childs Memorial Fund for Medical Research, Stanford Cancer Institute and Susan G. Komen Foundation, as well as funding from ASCO, the Conquer Cancer Foundation and the Breast Cancer Research Foundation. M.P.S. is supported by grants from the NIH including a Centers of Excellence in Genomic Science award (5P50HG00773502).
Declaration of Interests: M.P.S. is a founder and member of the science advisory board of Personalis, SensOmics, January and Qbio and a science advisory board member of Genapsys. J.J.G and M.P.S. are supported by a grant from Curis, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Agudelo Garcia PA, Hoover ME, Zhang P, Nagarajan P, Freitas MA, and Parthun MR (2017). Identification of multiple roles for histone acetyltransferase 1 in replication-coupled chromatin assembly. Nucleic acids research 45, 9319–9335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts B (2002). Molecular biology of the cell, 4th edn (New York: Garland Science; ). [Google Scholar]
- Annunziato AT (2013). Assembling chromatin: the long and winding road. Biochimica et biophysica acta 1819, 196–210. [DOI] [PubMed] [Google Scholar]
- Annunziato AT, and Seale RL (1983). Histone deacetylation is required for the maturation of newly replicated chromatin. The Journal of biological chemistry 258, 12675–12684. [PubMed] [Google Scholar]
- Basta J, and Rauchman M (2015). The nucleosome remodeling and deacetylase complex in development and disease. Translational research : the journal of laboratory and clinical medicine 165, 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulusu V, Tumanov S, Michalopoulou E, van den Broek NJ, MacKay G, Nixon C, Dhayade S, Schug ZT, Vande Voorde J, Blyth K, et al. (2017). Acetate Recapturing by Nuclear Acetyl-CoA Synthetase 2 Prevents Loss of Histone Acetylation during Oxygen and Serum Limitation. Cell reports 18, 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Sutter BM, Li B, and Tu BP (2011). Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Molecular cell 42, 426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos EI, Fillingham J, Li G, Zheng H, Voigt P, Kuo WH, Seepany H, Gao Z, Day LA, Greenblatt JF, et al. (2010). The program for processing newly synthesized histones H3.1 and H4. Nature structural & molecular biology 17, 1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, et al. (2014). Acetate dependence of tumors. Cell 159, 1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook AJ, Gurard-Levin ZA, Vassias I, and Almouzni G (2011). A specific function for the histone chaperone NASP to fine-tune a reservoir of soluble H3-H4 in the histone supply chain. Molecular cell 44, 918–927. [DOI] [PubMed] [Google Scholar]
- Eriksson PR, Mendiratta G, McLaughlin NB, Wolfsberg TG, Marino-Ramirez L, Pompa TA, Jainerin M, Landsman D, Shen CH, and Clark DJ (2005). Global regulation by the yeast Spt10 protein is mediated through chromatin structure and the histone upstream activating sequence elements. Molecular and cellular biology 25, 9127–9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evertts AG, Zee BM, Dimaggio PA, Gonzales-Cope M, Coller HA, and Garcia BA (2013). Quantitative dynamics of the link between cellular metabolism and histone acetylation. The Journal of biological chemistry 288, 12142–12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Lin SH, Ren F, Li JT, Chen JJ, Yao CB, Yang HB, Jiang SX, Yan GQ, Wang D, et al. (2016). Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nature communications 7, 11960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber JJ, Chen J, Geller B, Jager N, Lipchik AM, Ford JM, Snyder MP (2018). Epigenetic Remodeling in Response to BRCA2-Crisis. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond CM, Stromme CB, Huang H, Patel DJ, and Groth A (2017). Histone chaperone networks shaping chromatin function. Nature reviews Molecular cell biology 18, 141–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris ME, Bohni R, Schneiderman MH, Ramamurthy L, Schumperli D, and Marzluff WF (1991). Regulation of histone mRNA in the unperturbed cell cycle: evidence suggesting control at two posttranscriptional steps. Molecular and cellular biology 11, 2416–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz N (1991). The regulation of histone gene expression during the cell cycle. Biochimica et biophysica acta 1088, 327–339. [DOI] [PubMed] [Google Scholar]
- Holden P, and Horton WA (2009). Crude subcellular fractionation of cultured mammalian cell lines. BMC research notes 2, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Jackson V, Granner D, and Chalkley R (1976). Deposition of histone onto the replicating chromosome: newly synthesized histone is not found near the replication fork. Proceedings of the National Academy of Sciences of the United States of America 73, 2266–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Chung MK, Fan J, and Rabinowitz JD (2014). Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer & metabolism 2, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck KM, and Pemberton LF (2012). Histone chaperones link histone nuclear import and chromatin assembly. Biochimica et biophysica acta 1819, 277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, and Park PJ (2008). Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nature biotechnology 26, 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurat CF, Lambert JP, Petschnigg J, Friesen H, Pawson T, Rosebrock A, Gingras AC, Fillingham J, and Andrews B (2014a). Cell cycle-regulated oscillator coordinates core histone gene transcription through histone acetylation. Proceedings of the National Academy of Sciences of the United States of America 111, 14124–14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurat CF, Recht J, Radovani E, Durbic T, Andrews B, and Fillingham J (2014b). Regulation of histone gene transcription in yeast. Cellular and molecular life sciences : CMLS 71, 599–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. (2012). ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome research 22, 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Yu W, Qian X, Xia Y, Zheng Y, Lee JH, Li W, Lyu J, Rao G, Zhang X, et al. (2017). Nucleus-Translocated ACSS2 Promotes Gene Transcription for Lysosomal Biogenesis and Autophagy. Molecular cell 66, 684–697 e689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, and Marinov GK (2015). The bioenergetic costs of a gene. Proceedings of the National Academy of Sciences of the United States of America 112, 15690–15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XJ, Wu J, Altheim BA, Schultz MC, and Grunstein M (1998). Deposition-related sites K5/K12 in histone H4 are not required for nucleosome deposition in yeast. Proceedings of the National Academy of Sciences of the United States of America 95, 6693–6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnetjournal 17, 10–12. [Google Scholar]
- Marzluff WF, Gongidi P, Woods KR, Jin J, and Maltais LJ (2002). The human and mouse replication-dependent histone genes. Genomics 80, 487–498. [PubMed] [Google Scholar]
- Marzluff WF, Wagner EJ, and Duronio RJ (2008). Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nature reviews Genetics 9, 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina R, Buck T, Zaidi SK, Miele-Chamberland A, Lian JB, Stein JL, van Wijnen AJ, and Stein GS (2008). The histone gene cell cycle regulator HiNF-P is a unique zinc finger transcription factor with a novel conserved auxiliary DNA-binding motif. Biochemistry 47, 11415–11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megee PC, Morgan BA, Mittman BA, and Smith MM (1990). Genetic analysis of histone H4: essential role of lysines subject to reversible acetylation. Science 247, 841–845. [DOI] [PubMed] [Google Scholar]
- Mews P, Donahue G, Drake AM, Luczak V, Abel T, and Berger SL (2017). Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 546, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra P, Xie RL, Medina R, Hovhannisyan H, Zaidi SK, Wei Y, Harper JW, Stein JL, van Wijnen AJ, and Stein GS (2003). Identification of HiNF-P, a key activator of cell cycle-controlled histone H4 genes at the onset of S phase. Molecular and cellular biology 23, 8110–8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature genetics 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Murzina NV, Pei XY, Zhang W, Sparkes M, Vicente-Garcia J, Pratap JV, McLaughlin SH, Ben-Shahar TR, Verreault A, Luisi BF, et al. (2008). Structural basis for the recognition of histone H4 by the histone-chaperone RbAp46. Structure 16, 1077–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan P, Ge Z, Sirbu B, Doughty C, Agudelo Garcia PA, Schlederer M, Annunziato AT, Cortez D, Kenner L, and Parthun MR (2013). Histone acetyl transferase 1 is essential for mammalian development, genome stability, and the processing of newly synthesized histones H3 and H4. PLoS genetics 9, e1003518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osley MA (1991). The regulation of histone synthesis in the cell cycle. Annual review of biochemistry 60, 827–861. [DOI] [PubMed] [Google Scholar]
- Pardee AB (1974). A restriction point for control of normal animal cell proliferation. Proceedings of the National Academy of Sciences of the United States of America 71, 1286–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthun MR (2013). Histone acetyltransferase 1: more than just an enzyme? Biochimica et biophysica acta 1819, 256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthun MR, Widom J, and Gottschling DE (1996). The major cytoplasmic histone acetyltransferase in yeast: links to chromatin replication and histone metabolism. Cell 87, 85–94. [DOI] [PubMed] [Google Scholar]
- Picard-tools. http://broadinstitutegithubio/picard.
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Carrillo A, Wangh LJ, and Allfrey VG (1975). Processing of newly synthesized histone molecules. Science 190, 117–128. [DOI] [PubMed] [Google Scholar]
- Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, et al. (2015). Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer cell 27, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibahara K, Verreault A, and Stillman B (2000). The N-terminal domains of histones H3 and H4 are not necessary for chromatin assembly factor-1- mediated nucleosome assembly onto replicated DNA in vitro. Proceedings of the National Academy of Sciences of the United States of America 97, 7766–7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark R B. G (2011). DiffBind: differential binding analysis of ChIP-Seq peak data. Bioconductor. [Google Scholar]
- Stauber C, and Schumperli D (1988). 3’ processing of pre-mRNA plays a major role in proliferation-dependent regulation of histone gene expression. Nucleic acids research 16, 9399–9414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Li C, Kang B, Gao G, Li C, and Zhang Z (2017). GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic acids research 45, W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CB (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Moshkina N, Min J, Zeng H, Joshua J, Zhou MM, and Plotnikov AN (2012). Structural basis for substrate specificity and catalysis of human histone acetyltransferase 1. Proceedings of the National Academy of Sciences of the United States of America 109, 8925–8930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Zhang K, and Grunstein M (2005). Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell 121, 375–385. [DOI] [PubMed] [Google Scholar]
- Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, and DeBerardinis RJ (2009). Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer research 69, 7986–7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi N, Swinnen JV, and Smans K (2012). ATP-citrate lyase: a key player in cancer metabolism. Cancer research 72, 3709–3714. [DOI] [PubMed] [Google Scholar]
- Zhang W, Bone JR, Edmondson DG, Turner BM, and Roth SY (1998). Essential and redundant functions of histone acetylation revealed by mutation of target lysines and loss of the Gcn5p acetyltransferase. The EMBO journal 17, 3155–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome biology 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Kennedy BK, Lawrence BD, Barbie DA, Matera AG, Fletcher JA, and Harlow E (2000). NPAT links cyclin E-Cdk2 to the regulation of replication-dependent histone gene transcription. Genes & development 14, 2283–2297. [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, Carrer A, Sengupta A, Campbell SL, Kuo YM, et al. (2016). ATP-Citrate Lyase Controls a Glucose-to-Acetate Metabolic Switch. Cell reports 17, 1037–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Roeder RG, and Luo Y (2003). S phase activation of the histone H2B promoter by OCA-S, a coactivator complex that contains GAPDH as a key component. Cell 114, 255–266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-seq raw data and differential peak call files have been uploaded to GEO with accession number GSE117472. RNA-seq and ATAC-seq data are deposited in GEO with accession numbers GSE107280 and GSE107119, respectively. Raw data for immunoblots are deposited at Mendeley (http://dx.doi.org/10.17632/7wzp35y5mc.1).