

Abstract
Background & Aims:
The role of aryl hydrocarbon receptor (AHR) in liver fibrosis is controversial, because loss and gain of AHR activity each lead to liver fibrosis. The goal of this study is to investigate how the expression of AHR by different liver cell types, hepatic stellate cells (HSCs) in particular, affects liver fibrosis in mice.
Methods:
We studied the effects of AHR on primary mouse and human HSCs, measuring their activation and stimulation of fibrogenesis using RNA-seq analysis. C57BL/6J mice were given the AHR agonists TCDD or ITE, or carbon tetrachloride (CCl4), or underwent bile duct ligation. We also performed studies in mice with disruption of Ahr specifically in HSCs, hepatocytes, or Kupffer cells. Liver tissues were collected from mice and analyzed by histology, immunohistochemistry, and immunoblotting.
Results:
AHR was expressed at high levels in quiescent HSCs, but the expression decreased with HSC activation. Activation of HSCs from AHR-knockout mice was accelerated, compared to HSCs from wild-type mice. In contrast, TCDD or ITE inhibited spontaneous and transforming growth factor beta (TGFB)-induced activation of HSCs. Mice with disruption of Ahr in HSCs, but not hepatocytes or Kupffer cells, developed more severe fibrosis following administration of CCl4 or bile duct ligation. C57BL/6J mice given ITE did not develop CCl4-induced liver fibrosis, whereas mice without HSC AHR given ITE did develop CCl4-induced liver fibrosis. In studies of mouse and human HSCs, we found that AHR prevents TGFB-induced fibrogenesis by disrupting the interaction of SMAD3 with beta-catenin, which prevents the expression of genes that mediate fibrogenesis.
Conclusions:
In studies of human and mouse HSCs, we found that AHR prevents HSC activation and expression of genes required for liver fibrogenesis. Development of non-toxic AHR agonists or strategies to activate AHR signaling in HSCs might be developed to prevent or treat liver fibrosis.
Keywords: gene regulation, signal transduction, cell-specific effect, xenobiotic receptor
Graphical Abstract
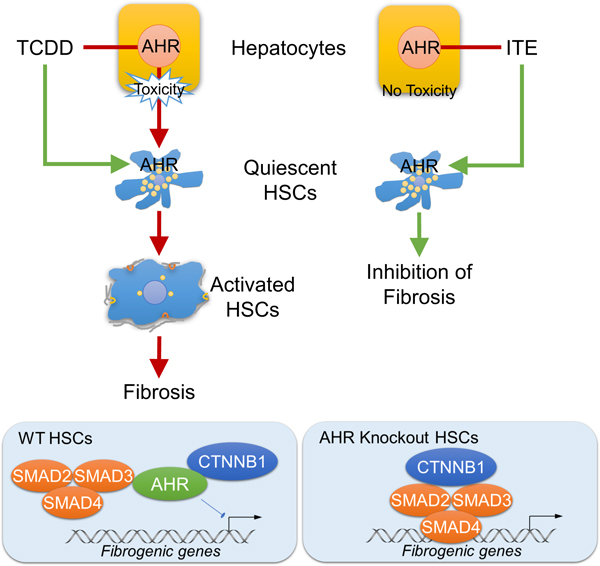
Lay Summary
Activation of hepatic stellate cells (HSCs) is key to liver fibrosis formation. This study showed that drug activation of the aryl hydrocarbon receptor can prevent HSC activation and liver fibrosis.
Introduction
Liver fibrosis, defined as the excessive intercellular accumulation of extracellular matrix (ECM) proteins in the liver, is strongly associated with chronic viral or non-viral liver injuries.1 Advanced liver fibrosis causes liver cirrhosis, leading to portal hypertension and liver failure. Liver fibrosis is also a major risk factor for the development of hepatocellular carcinoma. Activated hepatic stellate cells (HSCs) are the major cell population responsible for the production of ECM proteins and pro-fibrogenic cytokines.2 Residing in the space of Disse between the sinusoidal endothelial cells and hepatocytes, HSCs are characterized by their expression of desmin and glial fibrillary acidic protein in the quiescent state and α-smooth muscle actin (α-SMA) in the activated state. The activation of HSCs, which is characterized by increased fibrogenic gene expression and proliferation, is central to the pathogenesis of liver fibrosis. As such, understanding the molecular basis of HSC activation will help to develop strategies to prevent and treat liver fibrosis. The TGFβ pathway that integrates a myriad of injury signals is pivotal for HSC activation.3 Emerging evidence also suggested the role of Wnt-β-catenin signaling in promoting HSC activation and its crosstalk with the TGFβ pathway in the fibrotic diseases.4 However, how Wnt signaling, particularly the β-catenin-mediated canonical pathway, promotes the fibrogenic progression is not fully understood.
The aryl hydrocarbon receptor (AhR), highly expressed in the liver, is a well-established xenobiotic receptor that senses environmental toxicants and regulates xenobiotic metabolism.5 Many industrial pollutants, such as the polycyclic aromatic hydrocarbons, are AhR ligands.6 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), a prototypical xenobiotic activator of AhR, is a tool compound widely used to study the toxicological effect of AhR. As a ligand-dependent transcriptional factor, AhR signals through its DNA-binding motif and diverse protein partners.6 Upon binding by TCDD, AhR translocates into the nucleus where it heterodimerizes with AhR nuclear translocator (ARNT) on the xenobiotic response elements (XREs) to regulate the transcription of a battery of TCDD-responsive genes involved in xenobiotic catabolism, inflammatory response, and metabolic reprogramming. ChIP-sequencing analysis has revealed non-XRE binding of AhR in the genome, suggesting AhR responsive genes are not limited to those harboring XREs.7 Subsequent studies, mainly through the characterization of AhR−/− mice, have implicated AhR and its endogenous ligands in tissue development and pathophysiology, including liver fibrosis.8, 9 The AhR signaling has also been implicated in the homeostasis of energy metabolism, gut microbiota, stem cell differentiation, circadian rhythm, and adaptive immunity.10 In addition to its function as a transcriptional factor, AhR also participates in the proteasome-dependent proteolysis by functioning as a substrate-specific adaptor that targets selected proteins for degradation in the Cullin4B (CUL4B) ubiquitin E3 ligase complex CUL4BAhR.11 The reported substrate proteins for CUL4BAhR include estrogen receptor α, androgen receptor, and β-catenin.11, 12
The role of AhR in liver fibrosis has been intriguing and controversial. On one hand, AhR−/− mice exhibited spontaneous liver fibrosis.8 On the other hand, treatment of mice with TCDD or constitutive activation of AhR in the hepatocytes sensitized mice to methionine and choline deficient- or high-fat diet induced liver fibrosis.13–15 The liver is an organ of multiple cell types, including the hepatocytes, HSCs and Kupffer cells. It is unclear whether AhR has a cell-type specific role in liver fibrosis. More specifically, it has not been reported whether and how AhR plays a role in HSC activation and liver fibrosis.
In this study, we uncovered an unexpected role of AhR in preventing HSC activation and attenuating liver fibrosis. Knockout of AhR in HSCs was sufficient to cause spontaneous liver fibrosis and sensitize mice to experimental liver fibrosis. In contrast, the non-toxic AhR agonist ITE exhibited anti-fibrotic activity in vivo.
Materials and Methods
Detailed information of chemical reagents, antibodies, and real-time PCR primers is provided in Supplementary Tables 1–3.
Experimental Animals and Histology
The wildtype C57BL/6J (000664), AhRfl/fl (Ahrtm3.1Bra/J, 006203), Albumin-Cre (B6.Cg-Tg(Alb-cre)21Mgn/J, 003574), and LysM-Cre (B6.129P2-Lyz2tm1(cre)Ifo/J, 004781) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Lrat-Cre mice were previously described.2 AhRfl/fl mice were crossbred with Albumin-Cre, LysM-Cre, or Lrat-Cre mice to generate cell type-specific AhR knockout mice. For the toxicological comparison of TCDD and ITE, 6 week old male C57BL/6J mice were treated with vehicle (DMSO), TCDD (25 µg/kg), or ITE (10 mg/kg) once a week for two weeks by intraperitoneal injection. The animals were terminated 6 h after the third injection. For the carbon tetrachloride (CCl4) model of liver fibrosis, 9–10 week old male mice were intraperitoneally injected with CCl4 (1 µl/g body weight, 1:3 diluted in corn oil, twice a week) for 4 weeks. The animals were terminated 72 h after the final CCl4 injection. When ITE was used to treat CCl4-induced liver fibrosis, ITE (10 mg/kg) was administrated every other day along with CCl4 (0.5 µl/g body weight) in corn oil by intraperiton eal injection for 4 weeks. The animals were terminated 6 h after the final dose of ITE. For the bile duct ligation (BDL) model of liver fibrosis, 8–9 week old mice were subject to common bile duct ligation, and the animals were terminated 14 days after the surgery. See Supplementary Information for details of the histology. The use of mice was in accordance with the University of Pittsburgh Institutional Animal Care and Use Committee.
Primary HSC Isolation and Sorting, Adenovirus, Lentivirus, Plasmids and Cell Transfection
Primary mouse HSCs were isolated as previously reported.16 For the RNA-seq analysis, HSCs were further purified by vitamin A-based FACS sorting as described.16 See Supplementary Information for details of primary HSCs isolation and sorting, adenovirus, lentivirus, plasmids and cell transfection.
RNA Sequencing (RNA-seq) Analysis
RNA-seq was performed at the Health Sciences Sequencing Core at Children’s Hospital of Pittsburgh. Gene expression was analyzed by gene set enrichment analysis (GSEA).17 See Supplementary Information for details of bioinformatic analysis of the RNA-seq results and the Gene Expression Omnibus (GEO) datasets.
Immunofluorescence, Immunoprecipitation (IP) and Western Blot, Chromatin Immunoprecipitation (ChIP), and Quantitative Real-Time PCR
These were performed as described,18 and see Supplementary Information for details.
Statistics
Statistical analysis was performed using Prism GraphPad 7.0 (La Jolla, CA). All results were presented as means ± SEM of at least three replicates. Statistical differences between groups were determined using unpaired two-tailed Student t test or one-way ANOVA with post-hoc Tukey test. P values less than 0.05 were considered statistically significant.
Results
Treatment of mice with the non-toxic AhR ligand ITE ameliorates CCl4-induced liver fibrosis
The classical AhR ligand TCDD induces liver fibrosis in mice.14 TCDD is known to be toxic to the liver, so we speculated that the liver fibrosis might have been secondary to the TCDD-induced hepatotoxicity and inflammation. We decided to test the effect of 2-(1’H-indole-3’-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE), a non-toxic tryptophan metabolite and endogenous AhR agonist,19 on liver fibrosis. Treatment of mice with TCDD for 2 weeks caused typical hepatotoxicity as indicated by neutrophil infiltration and collagen deposition (Figure 1A), consistent with a previous report.20 In contrast, the liver of ITE-treated mice showed no signs of hepatotoxicity and fibrosis (Figure 1A). The lack of fibrogenic activity of ITE was confirmed by measuring the expression of fibrogenic marker genes (Figure 1B). We then tested the effect of ITE on CCl4-induced liver fibrosis as outlined in Figure 1C. Treatment with ITE ameliorated CCl4-induced liver fibrosis, which was supported by decreased collagen deposition and fibrogenesis as revealed by Masson’s Trichrome staining (MTS), Sirius Red staining, and immunostaining of α-SMA (Figure 1D). The suppression of α-SMA in ITE- and CCl4-treated mice was further verified by Western blotting (Figure 1E) and real-time PCR (Figure 1F). Consistent with the relief of liver fibrosis, the levels of ALT and AST were decreased in ITE-treated mice compared to their vehicle-treated counterparts (Figure 1G). The hepatic expression of Cyp1a1 and Cyp1a2, two typical AhR target genes, was induced by ITE, suggesting AhR was efficiently activated (Figure 1H). We also determined whether ITE can mitigate the existing liver fibrosis by treating mice with ITE after the initiation of the CCl4 model as outlined in Supplementary Figure 1A. The post-treatment with ITE also attenuated CCl4-induced liver fibrosis as shown by histology (Supplementary Figure 1B) and measurement of α-SMA protein expression (Supplementary Figure 1C). ITE post-treatment induced the expression of Cyp1a1 and Cyp1a2 (Supplementary Figure 1D), but had little effect on the serum ALT and AST levels (Supplementary Figure 1E).
Figure 1. Treatment of mice with the non-toxic AhR ligand ITE ameliorates CCl4-induced liver fibrosis.
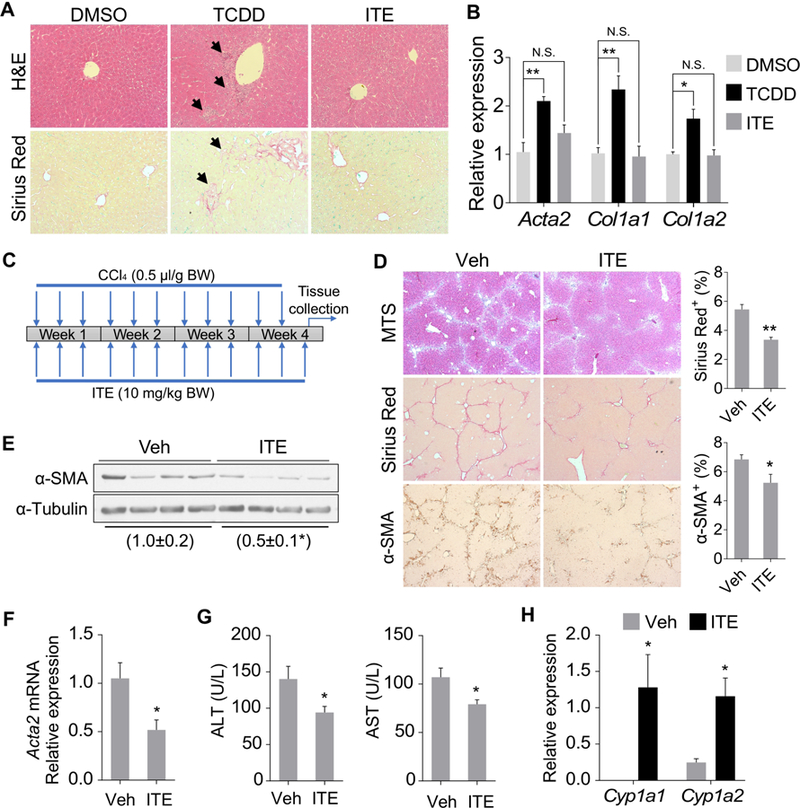
(A and B) Six week old male C57BL/6J mice were treated with vehicle (DMSO), TCDD (25 µg/kg), or ITE (10 mg/kg) once a week for two weeks (n=4 per group) before being analyzed for histology by H&E and Sirius Red staining (A, original magnification 20X) and the expression of fibrogenic genes by real-time PCR (B). Arrows indicate neutrophil infiltration in H&E and collagen deposition in the Sirius Red staining. (C-H) Eight week old male C57BL/6J mice were treated with vehicle or ITE (10 mg/kg) together with CCl4 (0.5 µl/g body weight) three times a week for four weeks (n=6 per group) as outlined in (C). Histology was analyzed by Masson’s Trichrome staining (MTS), Sirius Red staining, and α-SMA immunostaining (D, original magnification 5X) with the quantification of Sirius Red and α-SMA staining shown on the right. The hepatic protein (E) and mRNA (F) expression of α-SMA was measured by Western blotting and real-time PCR, respectively. The serum levels of ALT and AST (G) and the hepatic expression of Cyp1a1 and Cyp1a2 (H) were also measured. All data were presented as mean ± SEM. *, p<0.05; **, p<0.01; N.S., statistically not significant.
AhR is highly expressed in HSCs and the expression of AhR inversely correlates with HSC activation in vitro and in vivo
To investigate whether AhR in the HSCs mediates the anti-fibrotic effect of ITE, we compared the expression of AhR in primary hepatocytes and HSCs isolated from the same mice. There was an approximately 4-fold increase in the mRNA expression of both Ahr and its DNA-binding partner Arnt in HSCs compared to hepatocytes (Figure 2A). The basal expression of Cyp1a1 and Cyp1b1 was also markedly higher in HSCs (Figure 2A). These results were consistent with a recently reported proteomic analysis, in which HSCs were shown to express a higher level of AhR than hepatocytes.21 Treatment of primary HSCs isolated from WT mice with TCDD induced the expression of Cyp1a1, Cyp1a2 and Cyp1b1, but the induction was abolished in HSCs isolated from AhR−/− mice (Figure 2B), suggesting that AhR in HSCs is transcriptionally functional. Moreover, we found the expression of AhR was inversely correlated with the activation of HSCs. When primary HSCs were subject to spontaneous activation in culture, the expression of AhR and its target gene Cyp1a1 in primary mouse (Figure 2C) and human (Figure 2D) HSCs decreased with the onset of HSC activation in a time-dependent manner. The expression of Acta2 was measured to validate the HSC activation. Interestingly, we observed a major induction of Cyp1b1, another AhR target gene, with the onset of HSC activation (Supplementary Figure 2), suggesting that it was not a general decline in drug metabolism adipogenic differentiation in fully activated HSCs was correlated with an increased expression of AhR (Supplementary Figure 3).
Figure 2. AhR is highly expressed in HSCs and the expression of AhR inversely correlates with HSC activation.
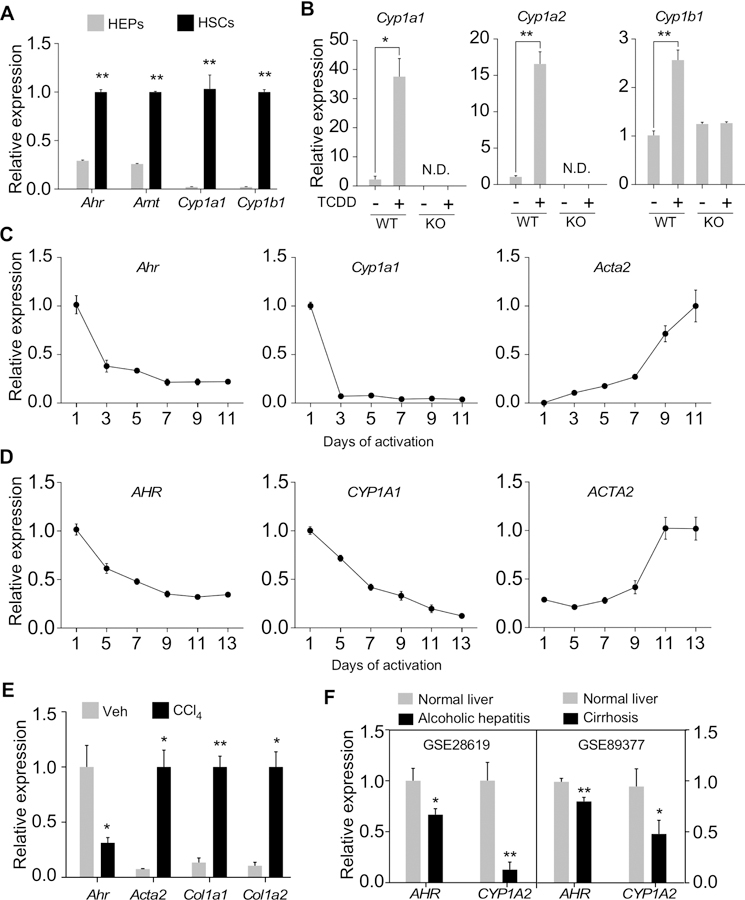
(A) Primary hepatocytes (HEPs) and HSCs were isolated from the same mouse liver. Gene expression was determined immediately after cell isolation by real-time PCR (n=4). (B) Primary HSCs were isolated from WT or AhR−/− (KO) mice and treated with TCDD (20 nM) for 6 days. Gene expression was determined by real-time PCR (n=4). (C and D) Primary mouse (C) or human (D) HSCs were culture-activated for the indicated duration. Gene expression was determined by real-time PCR (n=4). (E) Primary mouse HSCs were isolated from male mice treated with four doses of CCl4 (0.5 µl/g body weight, every 3 days, n=3) or vehicle (n=4). Gene expression was determined immediately after cell isolation by real-time PCR. (F) Two datasets (GSE28619 and GSE89377) from the GEO database were analyzed for the expression of AHR and CYP1A2. All data were presented as mean ± SEM. *, p<0.05; **, p<0.01; N.D., not detectable.
The fibrosis-responsive down-regulation of AhR in HSCs was also confirmed in vivo, as the primary HSCs isolated from CCl4-treated mice showed a decreased expression of Ahr compared to HSCs isolated from vehicle-treated mice (Figure 2E). Moreover, analysis of two Gene Expression Omnibus (GEO) datasets, alcoholic hepatitis (GSE28619) and cirrhosis (GSE89377), revealed that both fibrosis-prone liver diseases are associated with a decreased expression of AHR and its target gene CYP1A2 (Figure 2F), suggesting that the fibrosis-responsive down-regulation of AHR is conserved in humans.
Pharmacological activation or forced expression of AhR inhibits HSC activation
To determine whether the dynamic expression of AhR during HSC activation is functionally relevant, we treated primary mouse HSCs with TCDD and found it inhibited the expression of fibrogenic genes at both the protein (Figure 3A) and mRNA (Figure 3B) levels. The activation of AhR by TCDD was verified by the induction of Cyp1b1 (Supplementary Figure 4A). Treatment of primary mouse HSCs with 6-formylindolo[3,2-b]carbazole (FICZ) and ITE, two endogenous AhR agonists, also inhibited fibrogenic gene expression (Supplementary Figure 4B). TCDD or ITE had little effect on HSC proliferation and apoptosis, as shown by BrdU labeling (Supplementary Figure 4C) and TUNEL staining (Supplementary Figure 4D), respectively. The inhibitory effect of TCDD on HSC activation was AhR-dependent, because the inhibitory effect was abolished in HSCs isolated from AhR−/− mice, as evaluated by cell morphology (Figure 3C), or the expression of fibrogenic marker genes (Figure 3D). A similar pattern of inhibition was observed in primary human HSCs treated with the human AHR activator 3-methylcholanthrene (3MC) (Figure 3E). The activation of AHR by 3MC was verified by the induction of CYP1A2 (Supplementary Figure 4E). Inhibition of HSC activation was also observed in primary mouse HSCs infected with adenovirus expressing the mouse AhR (Ad-AhR), as shown by the immunofluorescence of α-SMA and Ki67 (Figure 3F) and the expression of fibrogenic genes (Figure 3G). Adenoviral overexpression of AhR achieved a similar inhibition of human HSC activation (Figures 3H and I). The adenoviral overexpression of AhR and induction of AhR target genes in the mouse (Supplementary Figure 4F) and human (Supplementary Figure 4G) HSCs was validated by real-time PCR.
Figure 3. Pharmacological activation or forced expression of AhR inhibits HSC activation.
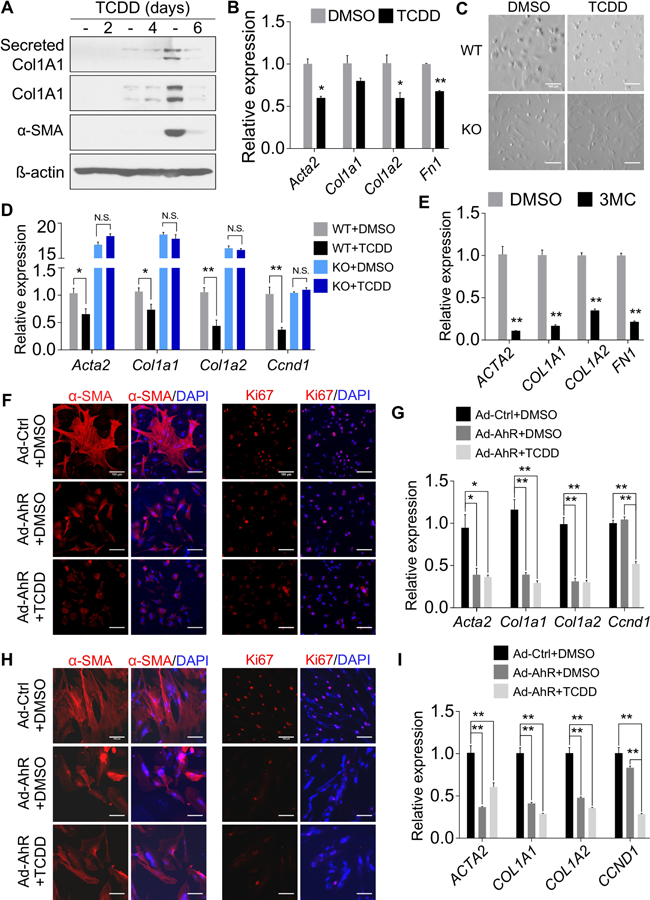
(A) Primary mouse HSCs were treated with TCDD (50 nM) for 2, 4, and 6 days, respectively. Protein levels from the culture medium or whole cell lysates were detected by Western blotting. (B) Primary mouse HSCs were treated with TCDD (20 nM) for 4 days. Gene expression was determined by real-time PCR (n=3). (C and D) Primary mouse HSCs were isolated from WT and AhR−/− mice and treated with TCDD (20 nM) for 6 days. Cell morphology was analyzed by light field microscopy (C) and gene expression was determined by real-time PCR (D, n=4). (E) Primary human HSCs were treated with 3MC (2 µM) for 6 days. Gene expression was determined by real-time PCR (n=4). (F and G) Primary mouse HSCs were infected with adenovirus overexpressing AhR (MOI=10) and treated with or without TCDD (50 nM). The expression of α-SMA and Ki67 was detected by immunofluorescence (F), and gene expression was determined by real-time PCR (G, n=3). (H and I) The experiments were the same as in (F and G) except that primary human HSCs were used. Scale bar=100 µm. All data were presented as mean ± SEM. *, p<0.05; **, p<0.01.
Knockout of AhR promotes HSC activation in vitro
Consistent with our hypothesis that the down-regulation of AhR contributes to the activation of HSCs, HSCs isolated from AhR−/− mice showed enhanced culture-induced activation as shown by cell morphology (Figure 4A) and induction of fibrogenic marker genes (Figure 4B). Increased protein expression of α-SMA and Col1A1 in AhR−/− HSCs was verified by Western blotting (Figure 4C), and induction of α-SMA and Ki67 was verified by immunofluorescence (Figure 4D).
Figure 4. Knockout of AhR promotes HSC activation in vitro.
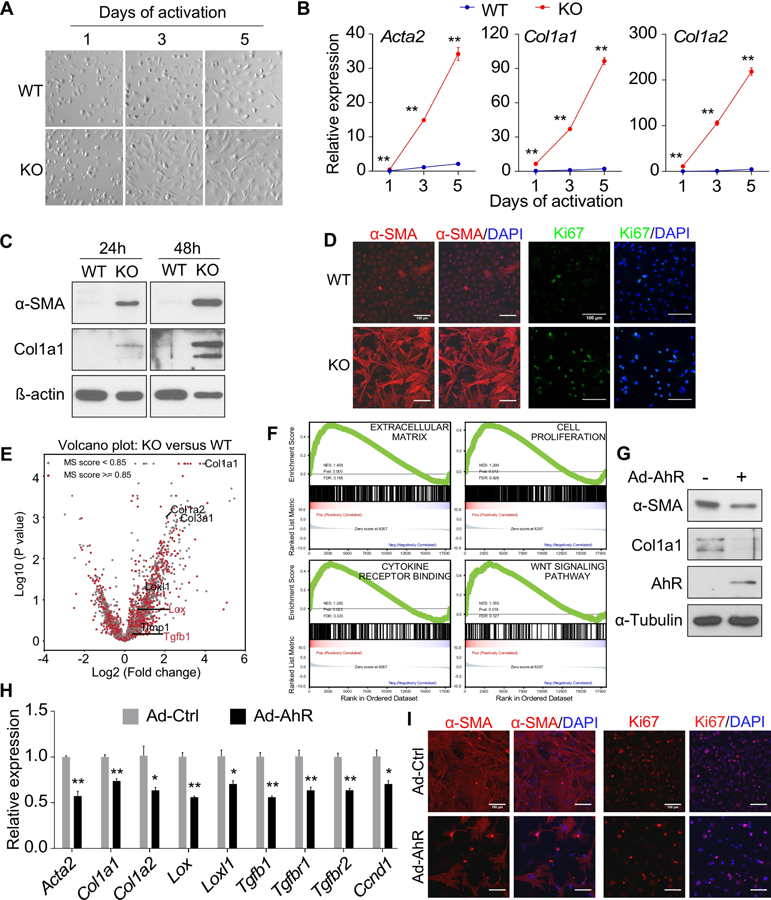
(A and B) Primary mouse HSCs were isolated from WT and AhR−/− (KO) mice and culture-activated for the indicated duration. Cell morphology was analyzed by light field microscopy (A) and gene expression was determined by real-time PCR (B, n=4). (C) Primary mouse HSCs were isolated and culture-activated for 24h or 48h. The protein expression was determined by Western blotting. (D) Primary mouse HSCs were isolated and culture-activated for 48h. The expression of α-SMA and Ki67 was detected by immunofluorescence. (E) Primary mouse HSCs were isolated and FACS purified and directly subject to RNA-seq analysis. Differentially expressed genes are shown by a volcano plot with several fibrogenic marker genes annotated. The red dots and blue dots indicate genes that are significantly up-regulated (663 genes) and down-regulated (66 genes), respectively. The cutoff was set as 0.05 for p-value and 1.5 for log2(FC) (FC: fold change, KO versus WT). (F) Gene set enrichment analysis (GSEA) of the RNA-seq results. (G-I) Primary mouse HSCs isolated from KO mice were infected with adenovirus overexpressing AhR and treated with or without TCDD (50 nM). The gene expression was determined by Western blotting (G), real-time PCR (H, n=4), or immunofluorescence (I). Scale bar=100 µm. All data were presented as mean ± SEM. *, p<0.05; **, p<0.01.
To better assess the effect of AhR knockout on HSC activation, we performed RNA-seq analysis on primary HSCs isolated from WT and AhR−/− mice following vitamin A autofluorescence-based FACS.16 The RNA-seq results are shown in Supplementary Table 4, with the major pathways affected presented in Supplementary Figure 5A. AhR−/− HSCs exhibited altered transcriptome including an elevated expression of fibrogenic markers, such as Col1a1, Col1a2, Col3a1 and Lox (Figure 4E). Several pathways related to HSC activation were selected for Gene Set Enrichment Analysis (GSEA).17 We found a significant up-regulation of “Extracell ular Matrix,” “Cell Proliferation,” “Cytokine Receptor B inding,” and “Wnt Signaling Pathway” in AhR−/− HSCs (Figure 4F). The “Cell Cycle Progression Pathways” were also si gnificantly up-regulated, whereas “TGF β Pathway” and “Epithelial Mesenchymal Transition” showed a trend of increase (Supplementary Figure 5B). Overall, the RNA-seq results supported the notion that AhR−/− HSCs were activated with an elevated Wnt signaling. When the RNA-seq results were integrated with a previously reported AHR-binding element (dioxin responsive element, or DRE) database,33 we found that 4901 out of 17128 genes have at least one potential DRE (Supplementary Table 5), but the DRE-harboring genes are not enriched in either up-regulated or down-regulated gene sets (Supplementary Table 6). It was also noted that many of the fibrogenic genes, such as Col1a2, do not contain DREs. The activation of AhR−/− HSCs was attenuated when the expression of AhR was reconstituted by adenoviral infection, as shown by the protein expression of α-SMA and Col1A1 (Figure 4G), the mRNA expression of fibrogenic genes (Figure 4H), and immunofluorescence of α-SMA and Ki67 (Figure 4I).
Knockout of AhR in HSCs sensitizes mice to liver fibrosis and abolishes the anti-fibrotic activity of ITE
To determine whether AhR suppresses HSC activation and fibrosis in vivo, we generated HSC-specific AhR knockout mice (HSC-KO) by crossbreeding the AhR floxed (AhRfl/fl, flox) mice20 with the Lrat-Cre transgenic mice expressing Cre in HSCs under the control of the lecithin-retinol acyltransferase (Lrat) gene promoter2 (Supplementary Figure 6A). The AhR expression in HSCs isolated from HSC-KO mice was decreased by 75% compared to the flox mice (Supplementary Figure 6B). Compared to the flox mice, HSC-KO mice exhibited more pronounced liver fibrosis when challenged with CCl4, as evidenced by increased collagen deposition and expression of α-SMA and Desmin (Figure 5A). The increased protein expression of α-SMA (Figure 5B) and mRNA expression of collagen genes (Figure 5C) in HSC-KO mice were verified by Western blotting and real-time PCR, respectively. The HSC-KO mice also exhibited increased sensitivity to bile duct ligation (BDL)-induced liver fibrosis, as evidenced by increased expression of α-SMA (Supplementary Figure 6C) and histological analysis (Supplementary Figure 6D), but without affecting the serum ALT and AST levels (Supplementary Figure 6E). Furthermore, the anti-fibrotic effects of ITE were abolished in HSC-KO mice as shown by histology (Figure 5D), fibrogenic gene expression (Figure 5E), and serum ALT and AST levels (Figure 5F), suggesting the HSC AhR is required for the anti-fibrotic activity of ITE.
Figure 5. Knockout of AhR in HSCs sensitizes mice to liver fibrosis and abolishes the anti-fibrotic activity of ITE.
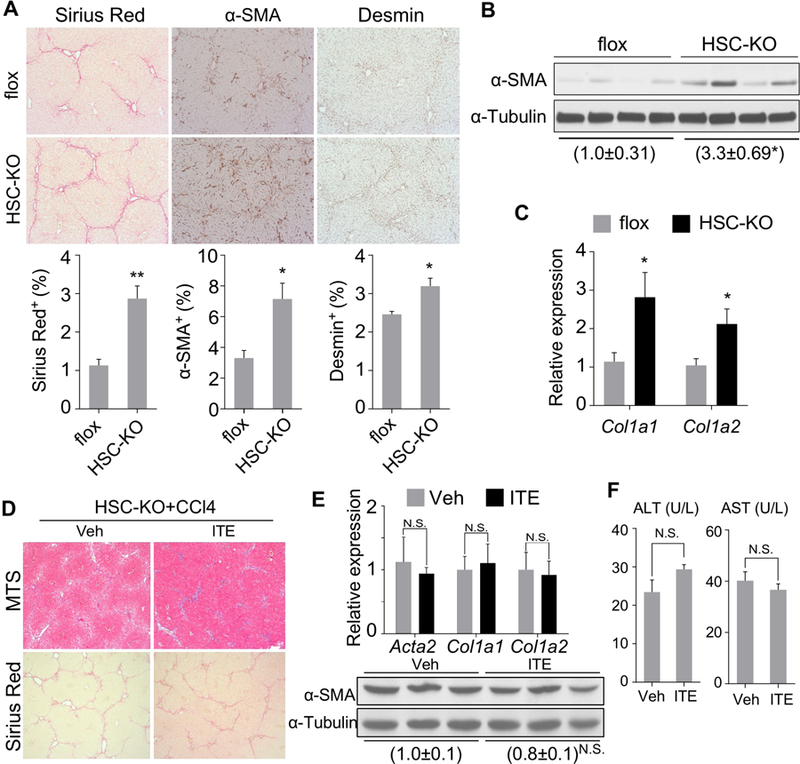
(A-C) 9–10 week old male AhRflox/flox (flox) and HSC-KO mice were treated with CCl4 (1 µl/g body weight) twice a week for 4 weeks (n=5 per group). The liver histology was analyzed by Sirius Red staining and immunostaining of α-SMA and Desmin (A, original magnification 10X with the quantifications of staining shown on the bottom). The α-SMA protein level was determined by Western blotting (B), and the mRNA expression of Col1a1 and Col1a2 was determined by real-time PCR (C). (D-F) 8–10 week old male HSC-KO mice were treated with vehicle or ITE (10 mg/kg) together with CCl4 (0.5 µl/g body weight) three times a week for four weeks (n=3 per group). Liver tissues were analyzed for histology by Masson’s Trichrome (MTS) and Sirius Red staining (D, original magnification 5X), the expression of fibrogenic gene by real-time PCR (top) or Western blotting (bottom) (E), and the serum ALT and AST levels (F). All data were presented as mean ± SEM. *, p<0.05; **, p<0.01; N.S., statistically not significant.
To determine whether the hepatocyte AhR plays a role in the development of liver fibrosis, we also generated the hepatocyte-specific AhR knockout mice (HEP-KO) by crossbreeding the AhR floxed mice with the Albumin-Cre transgenic mice (Supplementary Figure 6F). Compared to flox mice, HEP-KO mice showed a similar sensitivity to CCl4-induced liver fibrosis (Supplementary Figure 6F).
It has been reported that the whole body AhR−/− mice exhibited spontaneous periportal liver fibrosis8, 9 which was verified by our own analysis (Supplementary Figure 7). To determine the loss of AhR in which cell type is responsible, we used the HEP-KO and HSC-KO mice as well as the Kupffer cell-specific AhR knockout mice (MAC-KO). The MAC-KO mice were generated by crossbreeding the AhR floxed mice with the LysM-Cre transgenic mice. Compared to the whole body AhR−/− mice, only HSC-KO mice, but not HEP-KO or MAC-KO mice, showed spontaneous liver fibrosis as shown by Sirius Red staining and immunostaining of α-SMA (Supplementary Figure 7).
AhR attenuates TGFβ-stimulated fibrogenesis by inhibiting Smad3-mediated transcriptional activation of fibrogenic genes
The TGFβ-Smad3 signaling stimulates liver fibrogenesis and is central to HSC activation. Indeed, we showed the recruitment of pSmad3 onto the Acta2 and Col1a1 gene promoters was increased in CCl4-treated liver (Supplementary Figure 8A, left panel). Moreover, the CCl4-responsive recruitment of pSmad3 onto the Acta2 gene promoter was inhibited by the co-treatment of ITE (Supplementary Figure 8A, right panel). Having shown the inhibitory effect of AhR on the spontaneous activation of HSCs, we went on to determine whether AhR can also inhibit TGFβ-stimulated HSC activation. Treatment of primary mouse HSCs with TGFβ induced the expression of fibrogenic genes as expected, but the inductions were largely attenuated by the co-treatment of TCDD (Figure 6A). The attenuation of TGFβ-induced expression of α-SMA by TCDD was confirmed by immunofluorescence (Figure 6B). Next, we overexpressed AhR in the human stellate cell line LX2 in which the endogenous AhR had little expression and little response to AhR ligands, whereas overexpression of AhR restored the responsiveness to AhR ligands (Supplementary Figure 8B). Overexpression of AhR in LX2 cells significantly attenuated the response to the TGFβ stimulation, and ligand treatment modestly enhanced the attenuation as shown by gene expression analysis by real-time PCR (Figure 6C) or Western blotting (Figure 6D).
Figure 6. AhR attenuates TGFβ-stimulated fibrogenesis by inhibiting Smad3-mediated transcriptional activation of fibrogenic genes.
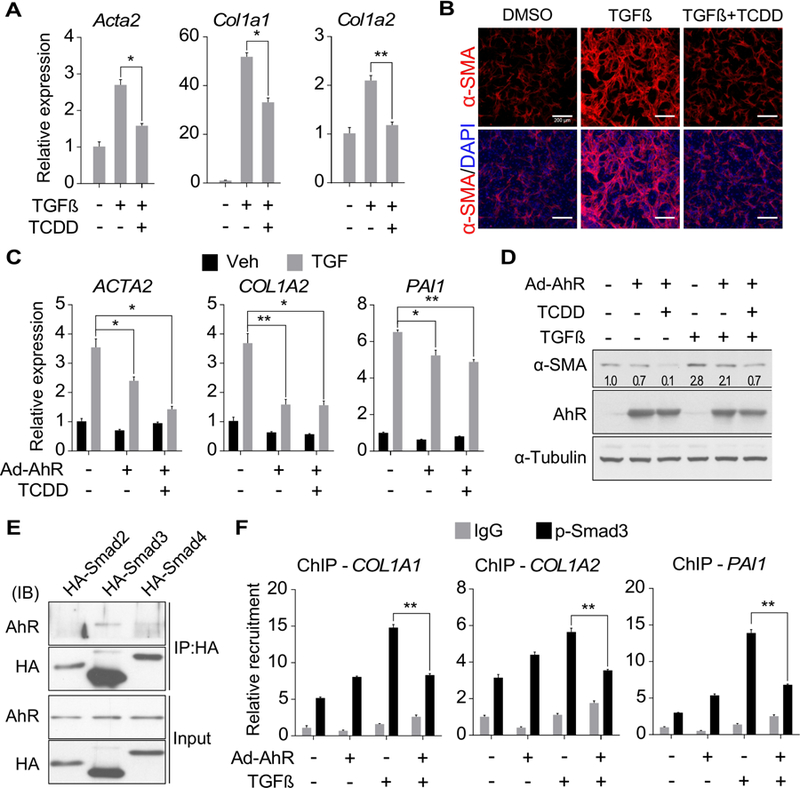
(A) Primary mouse HSCs were treated with TGFβ1 (2 ng/ml) in the absence or presence of TCDD (50 nM) for 24h. Gene expression was determined by real-time PCR (n=3). (B) Primary mouse HSCs were treated with TGFβ1 (2 ng/ml) in the absence or presence of TCDD (50 nM) for 48h. The expression of α-SMA was determined by immunofluorescence. Scale bar=200 µm. (C and D) LX2 cells were infected with adenovirus overexpressing AhR and treated with TCDD (50 nM) and/or TGFβ1 (5 ng/ml) for 24h. The gene expression at mRNA (C) and protein (D) levels were measured by real-time PCR (n=4) and Western blotting, respectively. The relative values of α-SMA protein expression in the Western blotting are labeled. (E) LX2 cells stably expressing human AhR were transfected with HA-Smad2, HA-Smad3, or HA-Smad4 expression vector. Protein interaction was determined by co-immunoprecipitation with an HA antibody followed by Western blotting. (F) LX2 cells infected with adenovirus overexpressing AhR were pre-treated with or without TCDD (50 nM) for 24h and then TGFβ1 (5 ng/ml) with or without TCDD (50 nM) for 4h. The recruitment of phosphorylated Smad3 onto the promoter regions of the COL1A1, COL1A2 and PAI1 genes were determined by ChIP coupled with real-time PCR on the recovered DNA (n=4). All data were presented as mean ± SEM. *, p<0.05; **, p<0.01.
To elucidate the molecular mechanism underlying the inhibitory effect of AhR on TGFβ-Smad stimulated fibrosis, we initially considered the transcriptional activity of AhR as a potential mechanism. However, the reported ChIP-seq studies showed that AhR does not bind to the consensus TGFβ-responsive element.7, 22 Our own ChIP analysis confirmed the lack of AhR binding to the TGFβ-responsive elements in the promoter regions of COL1A1 and COL1A2 genes (Supplementary Figure 8C). We also examined the expression of negative regulators of the TGFβ signaling pathway. There were no significant changes in the expression of genes that are known to be involved in the protein degradation (Smurf1 and Smurf2), receptor antagonism (Bambi), nuclear transport (Xpo1, Xpo4, and Ranbp3), or transcriptional repressors (Tgif, Ski, and Skil) (Supplementary Figure 8D). We then determined whether AhR has a direct interaction with the Smad proteins. Co-immunoprecipitation (co-IP) analysis showed that AhR can specifically interact with Smad3, but not Smad2 or Smad4 (Figure 6E). The interaction did not seem to inhibit the nuclear entry of Smad2/3 (Supplementary Figure 8E), but at the functional level, overexpression of AhR attenuated TGFβ responsive recruitment of phosphorylated Smad3 onto the promoter regions of fibrogenic genes (Figure 6F).
AhR disrupts the interaction between Smad3 and β-catenin
We reason the AhR-Smad3 interaction may interfere with the accessibility of Smad3 to the Smad3-interacting transcriptional factors that are involved in ECM gene expression. Among the tested proteins β-catenin, myocyte enhancer factor 2C (MEF2C), serum response factor (SRF), and specificity protein 1 (SP1), only β-catenin exhibited substantial interaction with Smad3 in the LX2 cells (Figure 7A). The interaction between β-catenin and Smad3 was decreased by the overexpression of AhR and was further reduced by the TCDD treatment, which was correlated with an increased binding of Smad3 to AhR (Figure 7B). In contrast, the Smad3-Smad4 interaction was not affected by AhR (Figure 7B). We also showed that AhR decreased β-catenin’s binding to Smad3 in a dose-dependent manner (Figure 7C), further suggesting that AhR competes with β-catenin for the binding of Smad3. Both AhR and β-catenin interact with Smad3 via the Mad homolog domain 2 (MH2) (Figure 7D). These results collectively suggested that AhR may attenuate TGFβ stimulated fibrosis by sequestering Smad3 from β-catenin.
Figure 7. AhR disrupts the interaction between Smad3 and β-catenin.
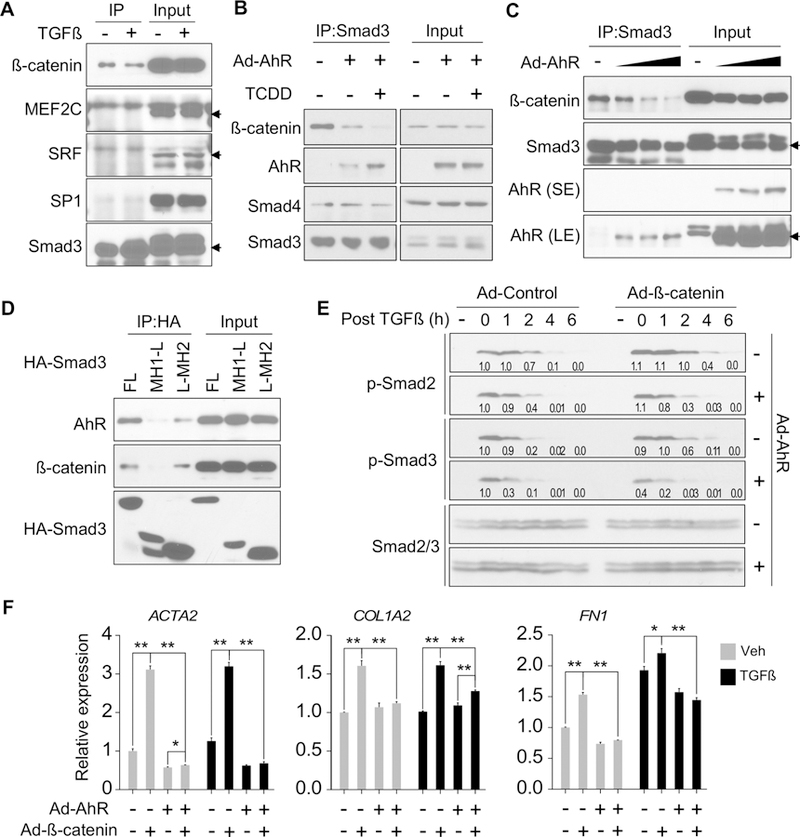
(A) LX2 cells were stimulated with TGFβ1 (5 ng/ml) for 3h. Protein interaction was determined by co-immunoprecipitation with an anti-Smad3 antibody followed by Western blotting. Arrows indicate the specific protein bands. (B) LX2 cells were infected with adenovirus overexpressing AhR and treated with or without TCDD (50 nM). Protein interaction was determined by co-immunoprecipitation with an anti-Smad3 antibody. (C) LX2 were infected with increasing doses of Ad-AhR. Protein interaction was determined by co-immunoprecipitation with an anti-Smad3 antibody. Arrows indicate the specific protein bands. SE and LE indicate short exposure and long exposure, respectively. (D) LX2 cells stably transfected with AhR were transiently transfected with HA-Smad3 plasmids (FL, full-length; MH1-L, MH1 domain plus linker; L-MH2, linker plus MH2 domain). Protein interaction was determined by co-immunoprecipitation with an anti-HA antibody. (E) LX2 were infected with Ad-AhR and/or Ad-β-catenin, and then stimulated with TGFβ1 (5 ng/ml) for 1h before harvesting at indicated time points. The levels of phosphorylated Smad2/3 were detected by Western blotting with the relative quantitative values labeled. (F) Primary human HSCs were infected with Ad-AhR with or without Ad-β-catenin and treated with TCDD (50 nM) and TGFβ1 (5 ng/ml) as indicated. Gene expression was determined by real-time PCR. All data were presented as mean ± SEM. *, p<0.05; **, p<0.01.
β-catenin is the effector of the canonical Wnt signaling pathway that is also responsive to the TGFβ stimulation.23 It has been reported that the coupling of Smad pathway and β-catenin is required for optimal induction of ECM gene expression and fibrogenesis.4 However, how β-catenin potentiates TGFβ-Smad mediated fibrogenesis and whether β-catenin is the target of the anti-fibrogenic activity of AhR are unknown. We examined the kinetics of Smad2/3 phosphorylation. In the absence of AhR, overexpression of β-catenin was efficient to prolong TGFβ-stimulated Smad2/3 phosphorylation, but this effect was largely diminished by the overexpression of AhR (Figure 7E). At the functional level, the fibrogenic gene expression induced by β-catenin was down-regulated in primary HSCs regardless of the TGFβ treatment (Figure 7F).
Mechanism by which AhR facilitates the degradation of phosphorylated Smad2/3
In our effort to determine the mechanism by which AhR facilitates the degradation of phosphorylated Smad2/3, we found that treatment of LXR2 cells with MLN4924, a pan-Cullin inhibitor, abolished the inhibitory effect of AhR on the expression of fibrogenic marker genes as shown by Western blotting (Supplementary Figure 9A) and real-time PCR (Supplementary Figure 9B). This effect of MLN4924 was also confirmed in primary human HSCs (Supplementary Figures 9C and D). These results suggested that Cullin E3 ligases may play a role in AhR-promoted degradation of phosphorylated Smad2/3. Indeed, treatment of MLN4924 or the proteasome inhibitor MG132 extended the levels of phosphorylated Smad2/3 in the presence of AhR overexpression (Supplementary Figure 9E). Because AhR has been reported as a component of the CUL4BAhR E3 ubiquitin ligase that targets the intestinal β-catenin for degradation,12 we examined whether the E3 ligase activity of AhR is required for its inhibitory effect on HSC activation. The E3 ligase-deficient AhR mutant with the deletion of the acidic domain12 remained effective in decreasing the expression of fibrogenic genes at the protein (Supplementary Figure 9F) and mRNA (Supplementary Figure 9G) levels, indicating that the E3 ligase activity of CUL4BAhR is dispensable.
Discussion
In this study, we have uncovered a novel function of AhR in HSCs and liver fibrosis. The anti-fibrogenic role of AhR in HSC activation and liver fibrosis was a surprise, because AhR has been better known for its pro-fibrogenic activity. Treatment of mice with the xenobiotic AhR activator TCDD sensitizes mice to fibrosis 13, 14, 15. TCDD and other polycyclic aromatic hydrocarbons are known to be toxic to the hepatocytes and induce neutrophil infiltration, although the mechanism for the hepatotoxicity remains to be clearly defined. For example, the Cyp1a1−/− mice showed paradoxical protection from the toxicity of BaP24 and TCDD25, whereas the expression of hepatic carboxylesterase 3 (CES3) was suggested to be important for TCDD-induced liver toxicity through the production of azelaic acid monoesters26. Nevertheless, we speculate that the fibrogenic activity of TCDD might be secondary to its hepatotoxicity and associated inflammation, a notion that has been supported by previous reports.27, 28 The hepatotoxicity and inflammation associated with TCDD exposure may have been exacerbated when animals were challenged with the MCD or HFD diet.13–15 In contrast, the non-toxic endogenous AhR ligand ITE protected mice from liver fibrosis in an AhR dependent manner. TCDD and ITE share an electron rich fused aromatic ring system with a lipophilic property, suggesting both compounds can efficiently penetrate the cell membrane. However, the metabolic stability of TCDD and ITE is different, which may have contributed to their differential effect on HSC activation and liver fibrosis. TCDD is metabolically stable, with a half-life estimated to be over seven years in the human serum,29 which renders the persistent activation of AhR. The metabolic turnover rate of endogenous AhR ligands, on the other hand, is high due to the rapid clearance by CYP1A and 1B enzymes, especially in the liver.30 Indeed, we found the AhR-activating effect of ITE, but not TCDD, was inhibited by the co-transfection of CYP1B1 in a reporter gene assay (data not shown), suggesting that ITE can be metabolized by CYP1B1 while TCDD is resistant. Future studies with the use of Cyp1a and 1b knockout mice are necessary to clarify the relevance of their regulation in the fibrogenic phenotype of AhR−/− mice. Nevertheless, it is tempting to speculate that the “sustained” activation of AhR by xenobiotic AhR ligands such as TCDD and “transient” activation of AhR by endogenous AhR ligands such as ITE may have accounted for the differential effects between the xenobiotic and endobiotic AhR ligands.
Another interesting finding is the cell type specific role of AhR in liver fibrosis, which may have explained the counterintuitive observation that both AhR knockout and xenobiotic activation of AhR sensitized mice to liver fibrosis. The liver is an organ of multiple cell types. Most of our understanding of the hepatic function of AhR has been centered on the hepatocytes. In this study, we have presented compelling evidence that AhR is highly expressed and functional in HSCs. The expression of AhR in HSCs decreased rapidly with the onset of HSC activation, and HSCs isolated from AhR−/− mice exhibited increased spontaneous activation. More importantly, we showed HSC specific knockout of AhR was sufficient to cause spontaneous liver fibrosis and sensitized mice to injury induced liver fibrosis. In contrast, mice bearing hepatocyte- or Kupffer cell-specific AhR knockout did not show spontaneous liver fibrosis.
The mechanism by which AhR inhibits HSC activation was also interesting. We showed AhR impairs β-catenin-dependent stabilization of phosphorylated Smad2/3. Treatment with the ubiquitination inhibitor MLN4924 or proteasome inhibitor MG132 attenuated the inhibitory effect of AhR on Smad2/3 phosphorylation and the expression of fibrogenic genes. AhR has been reported to have E3 ligase activity.11, 12 Interestingly, the E3-dead mutant AhR remained efficient in inhibiting HSC activation. Instead, we found AhR attenuated TGFβ-stimulated fibrogenesis by interacting with Smad3, which may have sequestered Smad3 from interacting with β-catenin. TGFβ is known to be pro-fibrogenic in HSCs and Smad3 is a key effector of the TGFβ signaling. The canonical Wnt signaling and its down-stream effector β-catenin have been suggested to play a role in fibrotic responses.4 Indeed, our RNA-seq results showed an up-regulation of both the Wnt and TGFβ pathways in AhR−/− HSCs. Our results suggested that β-catenin stabilizes the transcriptionally active phosphorylated Smad2/3. It has been reported that β-catenin interacts with the C-terminal MH2 domain of Smad3.31 This MH2 domain is also the binding site for E3 ligases.32 Therefore, it is reasonable to speculate that the stabilization of phosphorylated Smad2/3 protein by β-catenin could have been achieved through the prevention of ubiquitination. The E3 ligase responsible for AhR dependent destabilization of phosphorylated Smad2/3 remains to be defined. The AhR-Smad3 interaction and the sequestration of Smad3 from β-catenin suggest a plausible mechanism by which AhR inhibits HSC activation and liver fibrosis. Although our RNA-seq results suggested AhR may not directly inhibit HSC activation as a genomic repressor of the fibrogenic genes, we cannot exclude the transcriptional role of AhR as a mechanism to prevent HSC activation. It remains possible that certain AhR-regulated genes play a role in mediating the anti-fibrogenic effect of AhR.
Among limitations, much of our mechanistic characterization was in vitro studies. Previous reports suggested that the TGFβ-Smad pathway was activated in AhR−/− mice,34, 35 which was consistent with our results. However, future studies are necessary to determine whether fibrosis caused by AhR knockout requires the TGFβ-Smad signaling in vivo and in HSCs by using Ctnnb1 and Smad2/¾ floxed mice. In addition, it is unclear whether AhR-regulated Smad3-β-catenin interaction is also applicable in other biological events, such as embryonic stem cell differentiation (Supplementary Table 7).
In summary, we have established AhR as an anti-fibrogenic transcriptional factor in HSCs. Our results strongly suggest that development of non-toxic AHR agonists and strategies that preferentially activate AhR in HSCs may represent novel approaches to prevent and treat liver fibrosis.
Supplementary Material
What You Need to Know.
Background and Context:
The liver contains multiple cell types. We investigated how the expression of aryl hydrocarbon receptor (AHR) by different cell types affects liver fibrosis and activation of hepatic stellate cells (HSCs) in mice.
New Findings:
AHR signaling prevents activation of HSCs and expression of genes required for liver fibrogenesis.
Limitations:
Although human HSCs were used and bioinformatic analyses were performed on human liver samples, more human studies will further enhance the human relevance.
Impact:
Non-toxic AHR agonists or strategies to activate AHR signaling in HSCs might be developed to prevent or treat liver fibrosis.
Acknowledgements
We thank Dr. Jodi A. Flaws (University of Illinois Urbana-Champaign, Urbana, IL) for the Ad-AhR and control Ad-GFP adenovirus, and Dr. Tong-Chuan He (University of Chicago, Chicago, IL) for the Ad-β-catenin (S33Y).
Funding Statement: This work was supported in part by NIH grants DK083952 and ES023438 to WX.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors have declared that there is no conflict of interest.
References
- 1.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005;115:209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 2017;14:397–411. [DOI] [PubMed] [Google Scholar]
- 4.Akhmetshina A, Palumbo K, Dees C, et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun 2012;3:735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beischlag TV, Morales JL, Hollingshead BD, et al. The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr 2008;18:207–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson D, Joshi A, Elferink C. Ah receptor pathway intricacies; signaling through diverse protein partners and DNA-motifs. Toxicol Res 2015;4:1143–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lo R, Matthews J. High-resolution genome-wide mapping of AHR and ARNT binding sites by ChIP-Seq. Toxicol Sci 2012;130:349–61. [DOI] [PubMed] [Google Scholar]
- 8.Fernandez-Salguero P, Pineau T, Hilbert DM, et al. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 1995;268:722–726. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt JV, Su G, Reddy JK, et al. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci U S A 1996;93:6731–6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nebert DW. Aryl hydrocarbon receptor (AHR):“pio neer member” of the basic-helix/loop/helix per-Arnt-sim (bHLH/PAS) family of “sensors” of foreign and endogenous signals. Prog Lipid Res 2017;67:38–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohtake F, Baba A, Takada I, et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 2007;446:562–566. [DOI] [PubMed] [Google Scholar]
- 12.Kawajiri K, Kobayashi Y, Ohtake F, et al. Aryl hydrocarbon receptor suppresses intestinal carcinogenesis in ApcMin/+ mice with natural ligands. Proc Natl Acad Sci U S A 2009;106:13481–13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He J, Hu B, Shi X, et al. Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase Sirt3. Mol Cell Biol 2013;33:2047–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pierre S, Chevallier A, Teixeira-Clerc F, et al. Aryl hydrocarbon receptor–dependent induction of liver fibrosis by dioxin. Toxicol Sci 2014;137:114–124. [DOI] [PubMed] [Google Scholar]
- 15.Duval C, Teixeira-Clerc F, Leblanc AF, et al. Chronic exposure to low doses of dioxin promotes liver fibrosis development in the C57BL/6J diet-induced obesity mouse model. Environ Health Perspect 2017;125:428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mederacke I, Dapito DH, Affò S, et al. High-yie ld and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc 2015;10:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao J, Yan J, Xu M, et al. CAR suppresses hepatic gluconeogenesis by facilitating the ubiquitination and degradation of PGC1α. Mol Endocrinol 2015;29:1558–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song J, Clagett-Dame M, Peterson RE, et al. A ligand for the aryl hydrocarbon receptor isolated from lung. Proc Natl Acad Sci U S A 2002;99:14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walisser JA, Glover E, Pande K, et al. Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc Natl Acad Sci U S A 2005;102:17858–17863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Azimifar SB, Nagaraj N, Cox J, et al. Cell-type-resolved quantitative proteomics of murine liver. Cell Metab 2014;20:1076–1087. [DOI] [PubMed] [Google Scholar]
- 22.Dere E, Lo R, Celius T, et al. Integration of genome-wide computation DRE search, AhR ChIP-chip and gene expression analyses of TCDD-elicited responses in the mouse liver. BMC Genomics 2011;12:365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jian H, Shen X, Liu I, et al. Smad3-dependent nuclear translocation of beta-catenin is required for TGF-beta1-induced proliferation of bone marrow-derived adult human mesenchymal stem cells. Genes Dev 2006;20:666–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uno S, Dalton TP, Shertzer HG, et al. Benzo[a]pyrene-induced toxicity: paradoxical protection in Cyp1a1(−/−) knockout mice having increased hepatic BaP-DNA adduct levels. Biochem Biophys Res Commun 2001;289:1049–56. [DOI] [PubMed] [Google Scholar]
- 25.Uno S, Dalton TP, Sinclair PR, et al. Cyp1a1(−/−) male mice: protection against high-dose TCDD-induced lethality and wasting syndrome, and resistance to intrahepatocyte lipid accumulation and uroporphyria. Toxicol Appl Pharmacol 2004;196:410–21. [DOI] [PubMed] [Google Scholar]
- 26.Matsubara T, Tanaka N, Krausz KW, et al. Metabolomics identifies an inflammatory cascade involved in dioxin- and diet-induced steatohepatitis. Cell Metab 2012;16:634–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ozeki J, Uno S, Ogura M, et al. Aryl hydrocarbon receptor ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin enhances liver damage in bile duct-ligated mice. Toxicology 2011;280:10–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamb CL, Cholico GN, Pu X, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) increases necroinflammation and hepatic stellate cell activation but does not exacerbate experimental liver fibrosis in mice. Toxicol Appl Pharmacol 2016;311:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pirkle JL, Wolfe WH, Patterson DG, et al. Estimates of the half‐life of 2, 3, 7, 8‐ tetrachlorodibenzo‐p‐dioxin in Vietnam veterans of Operation Ranch Hand. Journal of Toxicology and Environmental Health, Part A Current Issues 1989;27:165–171. [DOI] [PubMed] [Google Scholar]
- 30.Wincent E, Bengtsson J, Bardbori AM, et al. Inhibition of cytochrome P4501-dependent clearance of the endogenous agonist FICZ as a mechanism for activation of the aryl hydrocarbon receptor. Proceedings of the National Academy of Sciences 2012;109:4479–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang M, Wang M, Tan X, et al. Smad3 prevents β-catenin degradation and facilitates β-catenin nuclear translocation in chondrocytes. J Biol Chem 2010;285:8703–8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao S, Alarcón C, Sapkota G, et al. Ubiquitin l igase Nedd4L targets activated Smad2/3 to limit TGF-β signaling. Mol Cell 2009;36:457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dere E, Forgacs AL, Zacharewski TR, et al. Genome-wide computational analysis of dioxin response element location and distribution in the human, mouse, and rat genomes. Chem Res Toxicol 2011;24:494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corchero J, Martin-Partido G, Dallas SL, et al. Liver portal fibrosis in dioxin receptor-null mice that overexpress the latent transforming growth factor-beta-binding protein-1. Int J Exp Pathol 2004;85:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andreola F, Calvisi DF, Elizondo G, et al. Reversal of liver fibrosis in aryl hydrocarbon receptor null mice by dietary vitamin A depletion. Hepatology 2004;39:157–66. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.