

Concordant with soaring obesity rates, non-alcoholic fatty liver disease (NAFLD) has become the most common chronic liver disease in the world. The obesity epidemic demands interventions to reverse obesity-associated hepatic steatosis, NAFLD, and NASH, and several new pharmacologic approaches have been developed within the last several years. Steatosis develops when energy delivery to the liver, modulated by rates of hepatic lipogenesis, exceeds its capacity to utilize or export this energy. Therefore pharmacologic approaches to reverse hepatic steatosis have focused largely – though not exclusively – on 1) reducing substrate availability to the liver, 2) reducing hepatic lipid synthesis, and 3) increasing hepatic mitochondrial fat oxidation (Figure). This perspective will discuss these three classes of emerging pharmacologic therapies against hepatic steatosis, with the ultimate intent to ameliorate NAFLD and/or NASH, and the advantages and pitfalls afforded by each strategy to treat these epidemics of obesity-associated liver disease.
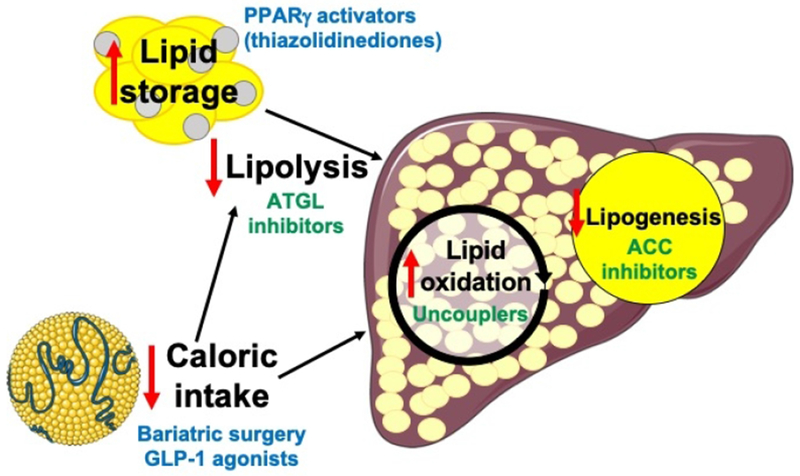
Approaches currently used in practice or under development to treat NAFLD/NASH. Agents in blue are currently approved, and those in green are under development. PPARγ, peroxisome proliferator-activated receptor-gamma; ATGL, adipose triglyceride lipase; GLP-1, glucagon-like peptide-1; ACC, acetyl-CoA carboxylase. The chylomicron image was obtained from Servier Medical Art.
Because hepatic steatosis results from a net positive balance between lipid uptake and lipid export/oxidation in the liver, approaches to reduce lipid delivery to the liver are particularly appealing, as they could in theory be utilized to prevent steatosis in addition to reversing it. This strategy is supported by evidence that increased substrate delivery to the liver is a key contributor to the increased rates of hepatic lipogenesis observed in individuals with NAFLD (1,2), Importantly, reducing steatosis by lowering substrate delivery to the liver does not depend on hepatic function, which may become compromised as NAFLD progresses to NASH, liver fibrosis, and cirrhosis. Weight loss – whether achieved by a self-imposed low-calorie diet or by bariatric surgery – reverses NAFLD and improves hepatic insulin sensitivity in humans (3, 4) and rodents (5) even without correcting body weight to the normal range. However weight loss is difficult to achieve and more difficult to maintain, with only ~20% of overweight individuals successful at long-term weight loss (6); therefore pharmacologic methods to reverse hepatic steatosis/NAFLD have been pursued with great interest. Glucagon-like peptide (GLP)-1 agonists, which are used to treat type 2 diabetes (T2D) largely due to their function as insulin secretagogues, have shown additional benefit to reduce appetite and thus food intake, leading to modest weight loss and improvement in steatosis (7). Similarly α-glucosidase inhibitors can cause modest weight loss and reduction in liver fat due to reduced absorption of dietary carbohydrates. However, both GLP-1 agonists and α-glucosidase inhibitors have significant on-target adverse effects of gastrointestinal discomfort, vomiting, and diarrhea, limiting their use. Thiazolidinediones (TZDs) activate the peroxisome proliferator-activated receptor-gamma (PPARγ), a nuclear receptor which promotes a transcriptional program resulting in increased fatty acid storage in adipose tissue. Thus TZDs increase subcutaneous adipose tissue mass, associated with both reductions in hepatic lipid content and increases in peripheral glucose disposal, although results when utilized in patients with NASH have been disappointing (8). However, potentially due to their capacity to promote adipogenesis in the setting of associated increases in fluid retention (9) and, more concerningly, appetite (10), weight gain is a class effect of TZDs. The potential cardiovascular risk of TZDs is also a concern, with rosiglitazone having been shown in some but not all studies to increase cardiovascular events, while pioglitazone, in contrast, has been associated with decreased cardiovascular risk (11). In addition, TZDs have been consistently shown to promote edema, which predisposes to an increased risk of heart failure, bone loss, and associated bone fractures (12), and which may limit the utility of TZDs in patients at higher risk of these outcomes.
With obesity common and cardiovascular risk already elevated in patients with NAFLD, there is great interest in developing alternative classes of pharmacologic agents for NAFLD. Inappropriately high rates of white adipose tissue lipolysis promote to NAFLD by increasing fatty acid delivery to the liver; thus inhibitors of lipolysis, such as atglistatin, which inhibits adipose triglyceride lipase (ATGL) and decreases hepatic steatosis and insulin resistance in mice with diet-induced obesity (13), may be attractive options. Clinical studies will be necessary to determine whether these benefits will translate to humans and whether this approach will increase cardiovascular risk as has been demonstrated with both atglistatin and whole-body ATGL knockout. In addition, further studies will be necessary to compare the impact of metabolic therapies to ameliorate NAFLD-associated NASH with inflammation/fibrosis-specific therapies currently in the pipeline, including antioxidants and immune-modulating agents (14, 15). As NASH is often considered to require two hits, hepatic steatosis and inflammation, it is possible that reversal of just one of these hits via one of the dozens of targets currently being explored may be sufficient to prevent or even reverse NASH. With both metabolic therapies (PPARα/δ/γ inhibitors, uncouplers, bile acid analogues) and immune-modulating therapies (strategies in development include farnesoid X receptor agonism, lysyl oxidase-like molecule 2 immunotherapy, and CCR-2/5 inhibition) reducing liver fibrosis in preclinical and, in some cases, clinical trials, it is likely that each class is independently capable of reversing NASH; combination metabolic and immune therapy may potentially confer even greater benefits against NASH. Further clinical studies will be required to examine this possibility.
In light of the considerable side effects of pharmacologic agents that diminish hepatic steatosis by reducing substrate supply to the liver, alternative approaches currently under development include reducing hepatic lipid synthesis. Acetyl-CoA carboxylase (ACC) inhibitors impair hepatic lipid synthesis due to inhibition of carboxylation of acetyl-CoA, the first committed step in de novo lipogenesis, while also promoting fatty acid oxidation. A small-molecule inhibitor of ACC was shown to reduce markers of NAFLD and NASH in humans (16); however these benefits have been shown by multiple investigators to occur at the cost of elevated serum triglycerides, likely due to increased hepatic very-low density lipoprotein (VLDL) export and/or impaired systemic triglyceride uptake (17). As hypertriglyceridemia would be expected to promote cardiovascular risk in patients already at risk for cardiovascular events, elevated serum triglyceride concentrations may significantly reduce the clinical utility of ACC inhibitors as an approach to reverse NAFLD/NASH.
A critical drawback of most of the pharmacologic approaches to reverse NAFLD discussed thus far, including TZDs, ACC inhibitors, and ATGL inhibitors, is that they typically do not reduce total lipid content in the body, but instead redistribute fat. In this setting, reversal of NAFLD must necessarily come at the cost of increases in lipids in other tissues. In order to reduce total lipid content, one must either reduce energy intake or promote energy utilization. Mitochondrial uncoupling has recently gained attention as a potential approach to reverse hepatic steatosis, NAFLD, and NASH: chemically modified derivatives of the mitochondrial protonophore 2,4-dinitrophenol (DNP) promote hepatic fat oxidation and thus reduce hepatic steatosis, inflammation, and fibrosis and improve hepatic insulin responsiveness associated with reductions in hepatic diacylglycerol content and protein kinase C epsilon translocation in rodents (18, 19, 20). In addition, by promoting hepatic lipid utilization, mitochondrial uncouplers also reduce hepatic VLDL export and therefore reduce plasma triglyceride and intramyocellular lipid content, thereby improving peripheral insulin sensitivity (18, 19).
Unfortunately, in part due to the potential on-target adverse effects of high doses of uncouplers (most commonly including hyperthermia, tachycardia, and arrhythmias), DNP was removed from the market by the Food and Drug Administration in 1938. In addition, numerous other adverse effects of DNP, which may be on- or off-target, have been reported, including electrolyte imbalances, agranulocytosis, shortness of breath, cataracts, hypothyroidism, and rash/pruritus, and the increase in hepatic fat oxidation that occurs secondary to uncoupler treatment would also be predicted to increase hepatic reactive oxygen species. However, animal data suggest that the toxic effects of uncouplers may be primarily attributable to peak concentrations, and that by manipulation of pharmacologic properties of these agents, it may be possible to extend drug exposure while minimizing toxicity (19). Future studies will be required to demonstrate 1) whether uncouplers are safe and effective to reverse hepatic steatosis in higher species, 2) whether improvements in hepatic steatosis following uncoupler treatment are associated with reductions in markers of NASH, and 3) how the safety and efficacy profiles of uncouplers compare to other therapies in the pipeline. Without question, due to the previously established toxicities of DNP, these agents will need to be used with care, ideally as a short course to reverse NAFLD/NASH followed by lifestyle modifications to sustain these improvements. Even so, mitochondrial uncouplers may represent an attractive class of therapy against NAFLD and NASH due to their effect to reverse hepatic steatosis while also improving peripheral metabolism, unlike many other agents used to treat NAFLD/NASH.
Despite its prevalence, affecting up to one-quarter of the world’s population, and the tremendous morbidity and mortality with which it is associated, NAFLD is not an indication for any medications at present, although several currently available pharmacologic agents may also reduce hepatic steatosis modestly. However, as discussed above, it is a critical limitation of all currently available agents that they either 1) cause lipid redistribution promoting increased subcutaneous adipose tissue mass and/or hypertriglyceridemia, or 2) are associated with unpalatable gastrointestinal side effects. However, increasing hepatic lipid oxidation by mitochondrial uncoupling represents an attractive alternative: this approach may improve NAFLD and NASH, hypertriglyceridemia, and both hepatic and peripheral insulin sensitivity by increasing energy utilization and treating the root cause of NAFLD, insulin resistance, and type 2 diabetes: excessive hepatic lipid accumulation. Ongoing research will be necessary to determine whether mitochondrial uncoupler therapy will be similarly effective to treat NAFLD/NASH in humans, or whether alternative approaches to increase energy oxidation or decrease intake may be developed to reduce hepatic lipid content without promoting lipid storage in other tissues.
Acknowledgments
Funding: The author’s research is supported by a grant from the U.S. Public Health Service (R00 CA215315), a Yale Cancer Innovators Award, Yale SPORE in Melanoma Award (P50CA121974), and Yale Diabetes Research Center Pilot (P30 DK045735), as well as an investigator-initiated award from AstraZeneca.
Footnotes
Disclosure: The author receives investigator-initiated funding from AstraZeneca for an unrelated project seeking to determine the mechanism by which SGLT2 inhibitors can predispose to ketoacidosis, and is an inventor on a patent application for liver-targeted mitochondrial uncouplers.
References
- 1.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014;146: 726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vatner DF, Majumdar SK, Kumashiro N, Petersen MC, Rahimi Y, Gattu AK, et al. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc Natl Acad Sci U S A 2015;112: 1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 2005;54: 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magkos F, Fraterrigo G, Yoshino J, Luecking C, Kirbach K, Kelly SC, et al. Effects of Moderate and Subsequent Progressive Weight Loss on Metabolic Function and Adipose Tissue Biology in Humans with Obesity. Cell Metab 2016;23: 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perry RJ, Peng L, Cline GW, Wang Y, Rabin-Court A, Song JD, et al. Mechanisms by which a Very-Low-Calorie Diet Reverses Hyperglycemia in a Rat Model of Type 2 Diabetes. Cell Metab 2018;27: 210–217 e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wing RR, Phelan S. Long-term weight loss maintenance. Am J Clin Nutr 2005;82: 222S–225S. [DOI] [PubMed] [Google Scholar]
- 7.Cuthbertson DJ, Irwin A, Gardner CJ, Daousi C, Purewal T, Furlong N, et al. Improved glycaemia correlates with liver fat reduction in obese, type 2 diabetes, patients given glucagon-like peptide-1 (GLP-1) receptor agonists. PLoS One 2012;7: e50117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 2010;362: 1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilding J Thiazolidinediones, insulin resistance and obesity: Finding a balance. Int J Clin Pract 2006;60: 1272–1280. [DOI] [PubMed] [Google Scholar]
- 10.Pickavance LC, Tadayyon M, Widdowson PS, Buckingham RE, Wilding JP. Therapeutic index for rosiglitazone in dietary obese rats: separation of efficacy and haemodilution. Br J Pharmacol 1999;128: 1570–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA 2007;298: 1180–1188. [DOI] [PubMed] [Google Scholar]
- 12.Rizos CV, Elisaf MS, Mikhailidis DP, Liberopoulos EN. How safe is the use of thiazolidinediones in clinical practice? Expert Opin Drug Saf 2009;8: 15–32. [DOI] [PubMed] [Google Scholar]
- 13.Schweiger M, Romauch M, Schreiber R, Grabner GF, Hutter S, Kotzbeck P, et al. Pharmacological inhibition of adipose triglyceride lipase corrects high-fat diet-induced insulin resistance and hepatosteatosis in mice. Nat Commun 2017;8: 14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banini BA, Sanyal AJ. Current and future pharmacologic treatment of nonalcoholic steatohepatitis. Curr Opin Gastroenterol 2017;33: 134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut 2015;64: 830–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lawitz EJ, Coste A, Poordad F, Alkhouri N, Loo N, McColgan BJ, et al. Acetyl-CoA Carboxylase Inhibitor GS-0976 for 12 Weeks Reduces Hepatic De Novo Lipogenesis and Steatosis in Patients With Nonalcoholic Steatohepatitis. Clin Gastroenterol Hepatol 2018;16: 1983–1991 e1983. [DOI] [PubMed] [Google Scholar]
- 17.Goedeke L, Bates J, Vatner DF, Perry RJ, Wang T, Ramirez R, et al. Acetyl-CoA Carboxylase Inhibition Reverses NAFLD and Hepatic Insulin Resistance but Promotes Hypertriglyceridemia in Rodents. Hepatology 2018;68: 2197–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perry RJ, Kim T, Zhang XM, Lee HY, Pesta D, Popov VB, et al. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab 2013;18: 740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 2015;347: 1253–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abulizi A, Perry RJ, Camporez JPG, Jurczak MJ, Petersen KF, Aspichueta P, et al. A controlled-release mitochondrial protonophore reverses hypertriglyceridemia, nonalcoholic steatohepatitis, and diabetes in lipodystrophic mice. FASEB J 2017;31: 2916–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]