

Abstract
Parkinson’s disease (PD) is one of the most common neurodegenerative disorders, affecting 1–1.5% of the total population. While progress has been made in understanding the neurodegenerative mechanisms that lead to cell death in late stages of PD, mechanisms for early, causal pathogenic events are still elusive. Recent developments in PD genetics increasingly point at endolysosomal (E-L) system dysfunction as the early pathomechanism and key pathway affected in PD. Clathrin-mediated synaptic endocytosis, an integral part of the neuronal E-L system, is probably the main early target as evident in auxilin, RME-8, and synaptojanin-1 mutations that cause PD. Autophagy, another important pathway in the E-L system, is crucial in maintaining proteostasis and a healthy mitochondrial pool, especially in neurons considering their inability to divide and requirement to function an entire life-time. PINK1 and Parkin mutations severely perturb autophagy of dysfunctional mitochondria (mitophagy), both in the cell body and synaptic terminals of dopaminergic neurons, leading to PD. Endolysosomal sorting and trafficking is also crucial, which is complex in multi-compartmentalised neurons. VPS35 and VPS13C mutations noted in PD target these mechanisms. Mutations in GBA comprise the most common risk factor for PD and initiate pathology by compromising lysosomal function. This is also the case for ATP13A2 mutations. Interestingly, α-synuclein and LRRK2, key proteins involved in PD, function in different steps of the endolysosomal pathway and target their components to induce disease pathogenesis. In this review, we discuss these E-L system genes that are linked to PD and how their dysfunction results in PD pathogenesis.
Keywords: Clathrin-mediated endocytosis, Autophagy, Lysosomes, Retromer complex, α-Synuclein, LRRK2
Graphical Abstract
The endolysosomal system is increasingly recognized as the key pathway affected in Parkinson’s disease (PD). Clathrin-mediated synaptic endocytosis, an integral part of the neuronal endolysosomal system, is probably the main early target. Autophagy, another important endolysosomal pathway that aids in maintaining proteostasis and a healthy mitochondrial pool is affected. Other crucial pathways such as endolysosomal sorting and trafficking, along with lysosomal degradation are also hampered. Key PD proteins such as α-synuclein and LRRK2 target the endolysosomal components to induce pathology. Here, we comprehensively discuss these mechanisms to better understand the role of endolysosomal system dysfunction in PD pathogenesis.
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, clinically identified by motor symptoms, with an increasing recognition of non-motor manifestations (Hoehn & Yahr 1967; Chaudhuri & Schapira 2009). Loss of dopaminergic neurons of the substantia nigra pars compacta (SNpc) (40–50%) and the presence of α-synuclein pathology in surviving neurons are the neuropathological hallmarks of PD. At the onset of motor symptoms, dopamine levels have decreased 60–70% in the dorsal striatum, where most SNpc dopaminergic neurons form synaptic connections (Dickson et al. 2009; Ma et al. 1997; Kordower et al. 2013; Fearnley & Lees 1991) . Under physiological conditions, α-synuclein is predominantly present in presynaptic terminals, aiding neurotransmitter release, whereas in PD, α-synuclein tends to aggregate and form Lewy bodies (the defining α-synuclein pathology in PD) (Spillantini et al. 1998; Spillantini et al. 1997). Aggregation of α-synuclein, perturbed proteostasis, mitochondrial dysfunction, oxidative stress, and impaired calcium homeostasis are the major cell-autonomous mechanisms of PD pathogenesis. Non-cell-autonomous mechanisms include cell-to-cell transmission of pathological α-synuclein and neuroinflammation (Poewe et al. 2017; Jyothi et al. 2015). So far, incomplete knowledge on causal mechanisms has limited treatment strategies to symptomatic relief. Recent studies targeting the endolysosomal (E-L) system reveal that its dysfunction can lead to the mentioned disease mechanisms, suggesting the E-L system as a key pathway in PD pathogenesis. In this review, we discuss important E-L system components, their roles in neuronal survival, and how their dysfunction causes PD.
1. Endolysosomal system in neurons: Custom-made for neuronal function
The E-L system comprises of complex, highly dynamic membrane-enclosed tubular-vesicular structures with overlapping properties and functions (Klumperman & Raposo 2014). They facilitate nutritional intake from a cell’s microenvironment through endocytosis, neutralize pathogenic materials via phagocytosis, promote cellular proteostasis through autophagy, and maintain overall cellular homeostasis through endosomal sorting and trafficking of various proteins/enzymes across organelles (Klumperman & Raposo 2014; Repnik et al. 2013). E-L system functions vary greatly depending on cell type and functional state. Neurons are unique in their highly polarized intracellular compartmentalization of cell body/soma and processes including axons, axon terminals, and dendrites. This organization enables them to form complicated networks that mediate specialized functions (Figure 1). This complexity comes at a price: burdensome demands on the E-L system, which have been increasingly implicated in neurological diseases including PD (Kett & Dauer 2016; Wang et al. 2018).
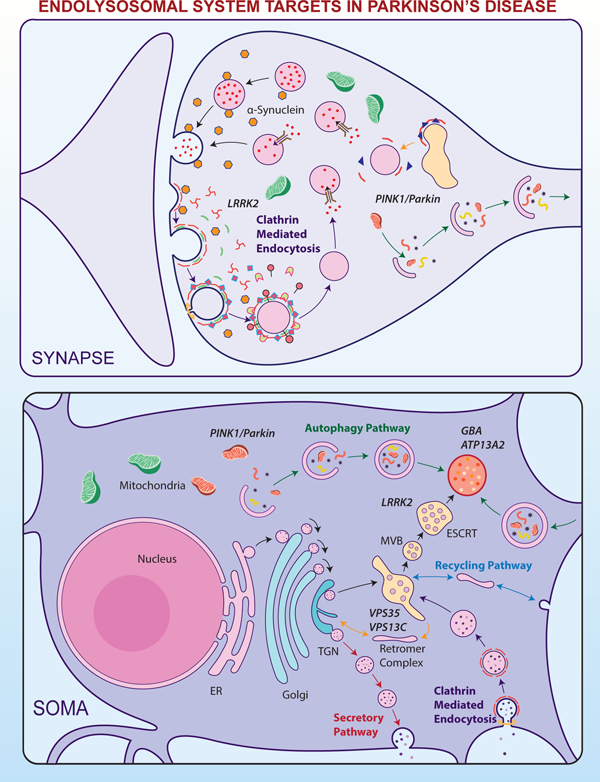
Figure 1. E-L system in the compartments of a nigrostriatal dopaminergic neuron, with the normal functional locations of E-L system proteins linked to PD.

A. Soma: E-L system proteins that form structural and functional components are synthesized in the neuronal soma. Following production in the endoplasmic reticulum (ER) and post-translational modifications in the Golgi apparatus, proteins (e.g. lysosomal hydrolases) are packed into vesicles and delivered to the trans-Golgi network (TGN). In the TGN, based on the nature of post-translational modification, the vesicles are diverted either towards lysosomes (black arrows) or to the secretory pathway (red arrows). Vesicles that are destined for lysosomes bud off from the TGN to form early endosomes. Simultaneously, early endosomes carrying nutrients and surface receptors for recycling are also derived from the plasma membrane through clathrin mediated endocytosis (CME, purple arrows). These early endosomes, with the help of the endosomal sorting complex required for transport (ESCRT), pass through highly dynamic tubulovesicular structures like multivesicular bodies (MVB) and/or late endosomes before fusing with lysosomes. Leucine-rich repeat kinase 2 (LRRK2) plays a significant role in this sorting, whereas Glucocerebrosidase 1 (GBA) and ATPase cation transporting 13A2 (ATP13A2) are crucial for normal lysosomal function. Early endosomes from the cell surface that are not sorted follow the recycling pathway back to the plasma membrane (blue arrows). Retrograde transport of receptors that are used for sorting from early endosomes to TGN occurs through the retromer complex where Vacuolar Protein Sorting 35 (VPS35) and VPS13C take part (yellow arrows). An expanding membrane sac called a phagophore sequesters malfunctioning organelles (e.g. sick mitochondria) and misfolded proteins to form a double-membraned autophagosomes. These later fuses with lysosomes and releases its contents for degradation and recycling of components; the complete process is called autophagy (green arrows). PTEN Induced Putative Kinase 1 (PINK1) and Parkin are important in identifying sick mitochondria and diverting them to a specialized form of autophagy called mitophagy.
B. Axon: Axons form a crucial bridge for retrograde transport of endosomes/autophagosomes from presynaptic terminals to soma for lysosomal degradation. A gradual development of phagophore to full-fledged autophagosome happens within the axon (green arrows) as the encapsulated cargo arrives towards proximal end.
C. Synapse: Neurotransmitter release in the presynaptic terminal is regulated principally by the synaptic vesicle cycle, an integral part of the neuronal E-L system. Initially, endosomes derived from the soma forms synaptic vesicles (SV) in the nerve terminal where RME-8 plays a role in association with clathrin (yellow arrow). Neurotransmitters are loaded into these SVs through specialized transporters. Upon arrival of an action potential in the synaptic terminal, these SVs fuse with the plasma membrane at active sites and release neurotransmitters via exocytosis (black arrows). Empty SVs are then recycled through CME (Purple arrows). α-Synuclein and endophilin A play roles here in membrane bending and SV curvature formation. Clathrin forms a layer on these invaginations with the help of adaptors, and in conjunction with dynamin leads to membrane fission forming clathrin coated vesicles (CCV). Clathrin uncoating is a prerequisite for SV recycling, which is performed by coordinated action of auxilin, Hsc70, endothelin A1, and synaptojanin 1. Phosphorylation by LRRK2 is necessary for activation of these proteins. Synaptic autophagy and mitophagy are also crucial in maintaining the health of the presynaptic terminal (green arrows), where PINK1/Parkin are crucial.
In neurons, synthesis of E-L components occurs primarily in the cell body/soma (Figure 1A). These components are trafficked through the secretory pathway to their subcellular location. A prime example of this is lysosomal hydrolase trafficking. Lysosomal hydrolases are produced in the endoplasmic reticulum, then undergo glycosylation and other post-translational modifications at the Golgi apparatus [e.g. addition of mannose-6-phosphate (M6P) groups] (Nixon & Cataldo 1995). This modification directs hydrolases to lysosomes. More specifically, hydrolases form complexes with transmembrane M6P receptors, which are sequestered into clathrin-coated vesicle (CCVs) budding from the trans-Golgi networks (TGN) to form endosomes/early endosomes (Nixon & Cataldo 1995) (Figure 1A). Inside early endosomes, M6P receptors detach from hydrolases and are recycled back to the TGN for their next round of transport (Saftig & Klumperman 2009), while hydrolases are delivered to lysosomes (discussed below).
Simultaneously, early endosomes are also derived from the cell surface through the endocytic pathway, an integral part of the E-L system. Many of the nutrients required for neuronal function and surface receptors designated for recycling are internalised through clathrin-mediated endocytosis (CME) at the plasma membrane and delivered to early endosomes (Figure 1A). Early endosomes derived from both the TGN and the plasma membrane mature into late endosomes, where Rab5 and Rab7 play a prominent role (Saftig & Klumperman 2009). This process occurs with the help of the endosomal sorting complex required for transport (ESCRT) (Figure 1A), which facilitates ubiquitin-dependent sorting of cargo/hydrolases from the outer membrane of early endosomes into interluminal vesicles (ILVs) (Wollert et al. 2009). Early endosomes derived from CME, if not sorted, follow a recycling pathway to the plasma membrane (Figure 1A) (Saftig & Klumperman 2009). ILVs are enveloped inside multivesicular bodies (MVBs), which mature into late endosomes. These late endosomes deliver their luminal contents either by forming tubular extensions or by fusing with pre-existing lysosomes to form endolysosomes (Luzio et al. 2007). These may be considered the final compartment of the endocytic pathway, where hydrolases degrade endocytic cargo and the membranes of ILVs for reuse (Klumperman & Raposo 2014; Saftig & Klumperman 2009). In addition, there is retrograde transport of receptors (such as M6P, Lamp2a, etc) from endosomes to the TGN for reuse through the retromer complex, comprised mainly of VPS35, VPS26 and VPS29 (Saftig & Klumperman 2009) (Figure 1A). Proteins that are meant for secretion follow a secretory pathway from the TGN (Saftig & Klumperman 2009; Klumperman & Raposo 2014). Selective capture and elimination of a cell’s damaged organelles and misfolded proteins happens through a process called macroautophagy (termed hereafter as autophagy) (Ohsumi 2014), an integral part of the E-L system. Double membrane vesicles called autophagosomes encapsulate unwanted/toxic cytosolic constituents including aggregated proteins and deliver them to the lysosome for degradation (Figure 1A) (Kawamata et al. 2008; Mizushima 2007). Non-selective autophagy also occurs during starvation, through which cells can recycle proteins and reuse amino acids for the synthesis of basic proteins essential for cell survival (Ohsumi 2014). Therefore, autophagy plays a pivotal role in housekeeping and maintenance of neuronal homeostasis.
Customization of the E-L system for neurons mainly happens in axons (Figure 1B) and synapses (Figure 1C). At presynaptic terminals, vesicle precursors are delivered from the cell body and undergo vesicle cycling to form functional synaptic vesicles (SVs). However, delivery of new vesicle precursors cannot match the number of SVs undergoing exocytosis to achieve neurotransmission at an active synapse (Saheki & De Camilli 2012). This deficit is solved by highly responsive CME, recapturing fused vesicular membranes from the presynaptic membrane to form CCVs, which are in turn converted back to SVs through clathrin uncoating (Figure 1C) (Heuser & Reese 1973; Morgan et al. 2000; Saheki & De Camilli 2012). Less understood clathrin-independent bulk endocytosis (Holt et al. 2003; Wu & Wu 2007) and the “kiss and run” mechanism (Fesce et al. 1994) are also parts of the synaptic endocytic repertoire. In addition, there are dense-core vesicles (DCVs) derived from the TGN that are transported down the axons. Unlike SVs, DCVs do not participate in SV recycling and are primarily used to secrete long-acting neuropeptides (Nurrish 2014). While these processes are mostly confined to the presynaptic compartment, the E-L system’s postsynaptic compartment contributes to membrane trafficking pathways primarily aimed at receptor, transporter, and ion-channel recycling (Rosendale et al. 2017).
Though this complex E-L system allows a neuron to function far away from its site of protein synthesis, the neuron is beholden to anterograde transport of proteins and membranes synthesized and modified in the soma for maintenance of axons and the synapses (Maday et al. 2014). Furthermore, constant synaptic activity, especially in continuously firing neurons like SNpc dopaminergic neurons may result in considerable build-up of damaged organelles, membranes, and misfolded proteins. These need to undergo encapsulation by an autophagosome or endosome (Figure 1C) and retrograde transport to the soma for lysosomal degradation (Figure 1B) (Yang et al. 2013). Here, long-distance axonal microtubule networks provide significant support to the neuronal E-L system (Yang et al. 2013). These spatial challenges to address a unique structural geometry, high metabolic demand, enhanced responsiveness to chemical/electrical stimulation, and the requirement for longevity due to their postmitotic nature, place a tremendous burden on the E-L system. Nigrostriatal dopaminergic neurons have an additional burden in terms of long and highly branched axons, larger surface area, selective transporters for external toxins, large fluctuations in free Ca2+, less buffering capacity, and higher oxidative stress, reducing their threshold for degeneration (Surmeier et al. 2017; Vidyadhara et al. 2019; Vidyadhara et al. 2016; Vidyadhara et al. 2017). These vulnerabilities are further exacerbated with aging. Faced with environmental and genetic risk factors, disease pathogenesis abounds.
2. E-L system dysfunction: the holy grail of PD pathophysiology?
In 1997, identification of dominant mutations in the α-synuclein (SNCA) gene (PARK1) which encodes α-synuclein as a cause of familial PD (Polymeropoulos et al. 1997), brought a conceptual shift toward the importance of genetics in disease pathogenesis. Subsequent findings revealed various other genetic mutations, with at least 10–15% of PD patients having familial PD of an absolute genetic origin. Leucine-rich repeat kinase 2 (LRRK2, PARK8) and Parkin (PRKN, PARK2) mutations are the most common genetic variants of dominant and recessively inherited PD, respectively (Deng et al. 2018). Mutations and gene multiplications of SNCA (PARK1 and PARK4) (Chartier-Harlin et al. 2004; Singleton et al. 2003) and Glucosidase Beta Acid (GBA) are also highly prevalent, whereas PD pathology was also noted with abnormal forms of Parkinsonism associated with deglycase or DJ1 (PARK7), and PTEN induced putative kinase 1 (PINK1, PARK6) (Deng et al. 2018). There is also compelling evidence for a genetic component to sporadic PD. In fact, a rigid distinction between the genetic and environmental basis of PD pathogenesis may not exist considering that cases are phenotypically indistinguishable (except for the onset) (Baba et al. 2006), implying genetic and sporadic cases share common underlying mechanisms. Even after considerable studies on these familial mutations and their interaction with environment, the core pathomechanisms of PD are still unclear, limiting treatment strategies. Over the past ten years, advancements in high-throughput sequencing, the application of genome wide association studies (GWAS), and meta-analysis, have identified new genetic mutations (refer to Deng et al. 2018 for a list of all PARK genes and risk alleles). Intriguingly, most of these identified genes have links to the E-L system (refer Table 1). Mechanistic studies also suggest that these proteins are the downstream targets of previously identified broad spectrum dysfunction in α-synuclein and LRRK2 mutations. It is now becoming clear that most PD-linked genes (Table 1) and known pathomechanisms are, in one way or another, associated with the E-L system, strongly suggesting this pathway as the key master regulator of PD pathogenesis. Recent studies suggest that neurodegeneration is initiated at the dopaminergic presynaptic terminals of striatum, years before the clinical diagnosis (Imbriani et al. 2018). Evidence that most of the E-L system genes associated with PD function at this terminal further establishes their fundamental role in PD pathogenesis. Below, we discuss our understanding of the known functions of these E-L system genes and their dysfunction in PD. We also discuss how other key PD-associated proteins target E-L pathways.
Table 1:
PD genes linked to E-L system with their PARK nomenclature, proteins they encode and their primary function
| PD Genes that are linked to E-L system | ||||
|---|---|---|---|---|
| PARK | Gene | Protein | Function | Reference |
| PARK1 | SNCA | α-Synuclein | Synaptic vesicle cycling | Polymeropoulos et al. 1997 |
| PARK2 | PRKN | Parkin | E3 Ubiquitin Ligase | Mori et al. 1998 Kitada et al., 1998 |
| PARK4 | SNCA | α-Synuclein | Synaptic vesicle cycling | Singleton et al., 2003 Chartier-Harlin et al., 2004 |
| PARK6 | PINK1 | PINK1 | Serine/Threonine Kinase | Valente et al., 2004 |
| PARK8 | LRRK2 | LRRK2 | Kinase/GTPase | Paisán-Ruíz et al., 2004 Zimprich et al., 2004 |
| PARK9 | ATP13A2 | ATPase type 13A2 | ATPase | Ramirez et al., 2006 Di Fonzo et al., 2007 |
| PARK16 | RAB29 | Ras-Related Protein Rab-7L1 | Rab GTPase | Satake et al. 2009 |
| PARK17 | VPS35 | VPS35 | Retromer | Wider et al., 2008, Vilariño-Güell et al., 2011 |
| PARK19 | DNAJC6 | Auxilin | Co-chaperone | Edvardson et al., 2012 Köroğlu et al., 2013 |
| PARK20 | SYNJ1 | Synaptojanin-1 | Polyinositol phosphatase | Krebs et al., 2013 Quadri et al., 2013 |
| PARK21 | DNAJC13 | RME-8 | Co-chaperone | Vilariño-Güell et al., 2014 |
| PARK23 | VPS13C | VPS13C | Retromer, Lipid transporter | Lesange et al., 2014, Schormair et al., 2018 |
| Risk Genes | GBA | Glucocerebrosidase 1 | Glucosidase | Sidranksy et al., 2009 |
| GAK | Cyclin-G Associated Kinase | Co-chaperone | Nalls et al., 2014 Chang et al., 2017 | |
| SH3GL2 | Endophilin A1 | Membrane bending protein | Nalls et al., 2014 Chang et al., 2017 | |
3. Clathrin-mediated synaptic vesicle endocytosis: A prime target?
SV endocytosis is predominantly mediated through CME (Figure 1C). Following exocytosis, the SV membrane is endocytosed primarily through CME, which involves formation of a clathrin coat, composed of an outer clathrin layer and an inner layer of adaptors (Saheki & De Camilli 2012). The most common adaptors such as AP-2 and AP180 binds to endocytic motifs on the SV membrane, as well as phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] present in the plasma membrane. Endophilin A1 and α-synuclein drive membrane bending and curvature formation of the developing clathrin coated pit (Westphal & Chandra 2013; Bai et al. 2010). Following this, the clathrin coated pit in conjunction with dynamin and other accessory proteins undergoes membrane fission to form a CCV (Saheki & De Camilli 2012). Regeneration of SVs from CCVs and recycling of clathrin and related coat components for further endocytosis requires un-coating of clathrin from CCVs (Figure 1C). The clathrin uncoating step is carried out by the coordinated action of the PI phosphatase synaptojanin-1 and the co-chaperone auxilin with Hsc70. Endophilin A1 and cyclin G Associated Kinase (GAK) also regulate clathrin uncoating to facilitate CME at the presynaptic terminal (Figure 1C) (Saheki & De Camilli 2012). Mutations of these E-L system genes in familial PD envisage synaptic endocytosis dysfunction as a prime trigger for PD pathogenesis.
3.1. The DNAJ family of heat shock proteins in the presynaptic terminal and beyond:
Auxilin:
DNAJC6 gene encodes auxilin, the major presynaptic endocytic protein linked to PD. Auxilin is a brain specific co-chaperone belonging to the DNAJC family (Table 1), a subclass of heat shock proteins (HSP40). Auxilin plays an important role in clathrin un-coating, recruiting ATP-activated Hsc70 to CCVs, which in turn, disassembles clathrin from vesicles facilitating their recycling (Gorenberg & Chandra 2017; Lemmon 2001) (Figure 1C). Loss-of-function autosomal recessive mutations of auxilin (PARK19) have been shown to cause juvenile/early onset PD, first observed in two teenage brothers of a Palestinian family (Edvardson et al. 2012). The study noted abnormal mRNA splicing and a decrease in auxilin mRNA levels. Other variants of auxilin mutations were later noted in a Brazilian family with similar mis-splicing of mRNA, whereas mutations affecting the J-domain (p.Q791*, p.Q846* and p.R927G) were observed in a few other consanguineous families (Koroglu et al. 2013; Olgiati et al. 2016). Along with typical PD symptoms, patients with nonsense mutations have cognitive impairment, one of the cardinal non-motor symptoms of PD. Epilepsy and mental retardation have also been noted for some PD patients with auxilin mutations (Koroglu et al. 2013). These human genetic studies suggest widespread neuronal dysfunction besides nigrostriatal dopaminergic neurodegeneration. Hence, future studies on post-mortem brains documenting PD-typical Lewy body pathology and dopaminergic neuronal loss are necessary to concretely categorize the pathology as PD or parkinsonism.
One of the few preclinical studies characterizing DNAJC6 reveals that auxilin down-regulation leads to progressive dopaminergic neuronal loss and motor disabilities in Drosophila. Furthermore, loss of auxilin exacerbates α-synuclein overexpression and paraquat (an environmental toxin)-mediated dopaminergic loss, thus aggravating previously established phenotypes in fly models of PD (Song et al. 2017). Interestingly, the brains (deep cerebellar nuclei) of adult homozygous auxilin knockout (KO) mice revealed an increased number of CCVs and empty cages at the synapse. Similar defects were noted in primary cortical and hippocampal neurons accompanied by defective SV endocytosis (Yim et al. 2010). Also, impairment was not immediately detrimental to the neurons, which may explain the post-developmental onset of PD symptoms and slow disease progression. Evaluating whether similar disruptions occur in nigrostriatal dopaminergic neurons and how it mediates pathogenesis will prove crucial in understanding the role of auxilin and the E-L system in PD. An initial step towards this suggest that LRRK2 phosphorylates auxilin, attenuating auxilin’ss normal function in human iPSC derived dopaminergic neurons. The study further revealed that phosphorylation of auxilin by overexpressing LRRK2 leads to accumulation of oxidized dopamine and α-synuclein overexpression, pathological hallmarks of PD (Nguyen & Krainc 2018). Whether loss-of-function mutations in auxilin independently exert pathology through the same mechanisms is unknown. However, this newfound link between LRRK2 and auxilin points to the crucial and widespread involvement of E-L system in PD pathogenesis.
GAK:
GAK is another DNAJC family protein (DNAJC26/Auxilin 2), homologous (~50%) to auxilin but expressed throughout the body (Lemmon 2001). In a meta-analysis of PD GWAS across 13, 708 patients and 95, 282 controls (Nalls et al. 2014), GAK was identified as a risk gene for PD which was confirmed in another recent largescale study (Chang et al. 2017) (Table 1). However, GAK’s role in PD is still contentious due to presence of mutations in TMEM175 in a nearby independent locus, which shares the same promoter as that of GAK. What adds credence is that, like auxilin, GAK functions in clathrin un-coating by recruiting Hsc70 to CCVs (Lemmon 2001) (Figure 1C). Results show that GAK overexpression can partially compensate for loss of auxilin, which is also noted to upregulate naturally in auxilin-KO mice (Yim et al. 2010). Another similarity between auxilin and GAK has been noted in their interaction with LRRK2 (Jinn et al. 2017; Nguyen & Krainc 2018). Brain-specific loss of GAK and ubiquitous KO of GAK are both lethal (postnatal and embryonic, respectively) (Lee et al. 2008a). The inability of auxilin to compensate for GAK in these mice suggests another unique role for GAK in neurons, perhaps a significant one in dopaminergic neurons. Further TMEM175 deficiency is noted to cause PD-like α-synuclein aggregation, along with impairment of lysosomal and mitochondrial function (Jinn et al. 2017). Clarity on the role of GAK will prove to be crucial not only in establishing its involvement in PD but also in consolidating CME as the prime target in PD pathogenesis.
RME-8:
Receptor mediated endocytosis 8 (RME-8) or DNAJC13 is another DNAJC family co-chaperone and principally functions in regulating retromer and clathrin un-coating dynamics on endosomes (Gorenberg & Chandra 2017) (Figure 1C). RME-8 mutations (p.N855S) in autosomal-dominant PD (PARK21) was noted first in a Dutch-German-Russian family of Mennonite origin, with multiple incidences (Table 1) (Vilarino-Guell et al. 2014; Gustavsson et al. 2015). Post-mortem brains of PD patients with RME-8 mutations revealed strong immunoreactivity for Lewy bodies in the substantia nigra, along with brain areas implicated in non-motor symptoms of PD. Nigral Lewy bodies were also found to be co-localized with RME-8 (Vilarino-Guell et al. 2014). Unlike other DNAJC proteins implicated in PD, in vitro studies have revealed that, the RME-8 mutation confers toxic gain-of-function, leading to endolysosomal cargo trafficking deficits (Norris et al. 2017). The RME-8 mutation is also predicted to affect overall endosomal trafficking, even in the soma and postsynaptic compartment (Vilarino-Guell et al. 2014), unlike the synapse-specific dysfunction predicted for auxilin. Recently, the link between RME-8 and PD has been muddied by an independent analysis of the same Mennonite family which did not find RME-8 mutations. Instead, all the affected individuals were identified to carry mutations in TMEM230 (Deng et al. 2016), which also functions in E-L system, perhaps at synapses. Hence, further studies are warranted to confirm the genetic link between RME-8 and PD. Still, evaluating the RME-8 mediated E-L system dysfunction might provide valuable insights in understanding PD.
3.2: Synaptojanin-1 and endophilin A: Partners in crime?
Two other major synaptic endocytic proteins besides the DNAJ family that are implicated in PD are synaptojanin-1 (SYNJ1) and endophilin A family. SYNJ1 is a Sac domain-containing protein with two different phosphatase domains; 5-phosphatase and Sac1 which target different phosphoinositide phosphates (PIPs) (McPherson et al. 1996). 5-phosphatase regulates SV endocytosis by dephosphorylating PI(4,5)P2 to facilitate uncoating of CCVs in co-operation with Hsc70 and auxilin (Figure 1C), whereas Sac1 domain has gained importance due to its link to PD (PARK20, Table 1) (Mani et al. 2007; Cremona et al. 1999; Saheki & De Camilli 2012). The p.R258Q missense mutations in the Sac domain were identified independently in two consanguineous families of Italian and Iranian origin. Homozygous missense mutations of SYNJ1 Sac domain are noted to cause autosomal recessive PD with extrapyramidal features identical to patients with auxilin mutations (Krebs et al. 2013; Quadri et al. 2013; Olgiati et al. 2014). Interestingly, they responded poorly to L-dopa treatment, indicating the need for preclinical studies to understand the exact pathomechanisms. Recent observations by Cao et al., suggest that the Sac domain plays a previously unappreciated role in clathrin uncoating. Mice with SynJ1 Sac mutations (knock-in) developed PD-like characteristics and featured synaptic endocytosis defects, including accumulating of CCVs. Furthermore, elevated auxilin and parkin levels were noted in these mice, suggesting the profound effect of Sac mutations on other proteins linked to PD (Cao et al. 2017). Studies on fibroblasts from patients with SYNJ1 mutations suggested defective endocytic trafficking (Quadri et al. 2013). Other studies have shed light on the involvement of SynJ1 in regulating synaptic autophagy. Mutations in the Sac1 domain of SynJ1 in both Drosophila neurons and PARK20 patient-derived iPSCs severely affected synaptic autophagosome formation but not endocytosis (Vanhauwaert et al. 2017). This discrepancy and the precise relationship between SV endocytosis and synaptic autophagy remains to be clarified. However, the Drosophila model revealed loss of dopaminergic neurons, providing evidence for the contribution of synaptic autophagy defects in pathogenesis of PD (Vanhauwaert et al. 2017). Considering its dual enzymatic activity, SynJ1 is also believed to play a role in fast endophilin-mediated, clathrin-independent endocytosis which is an alternative to CME (Watanabe et al. 2018) and several other processes. Further studies are warranted to understand if SYNJ1 exert its effects on PD pathogenesis through any of the alternative endocytic pathways.
SYNJ1 recruitment to endocytic membrane is facilitated by endophilin A (Figure 1C), a protein drawing attention in PD pathogenesis for a while, despite its locus variation being just identified as a risk factor (Table 1) (Chang et al. 2017). This is because of endophilin A’s close association with SYNJ1 (Song & Zinsmaier 2003; Cao et al. 2017) and its link to most important proteins involved in PD. One of our earlier studies indicated that overexpression of α-synuclein decreases endophilin A levels, while deletion of synucleins had the opposite effect (Westphal & Chandra 2013). As noted in SynJ1knock-ins, endophilin KO also leads to synaptic vesicle endocytosis defects, accumulation of CCVs, elevation of parkin levels, and other parkinsonian features (Cao et al. 2014; Milosevic et al. 2011). Gain-of-function mutations in LRRK2 hyper-phosphorylate endophilin A, the fly homolog, leading to synaptic endocytosis dysfunction (Matta et al. 2012). LRRK2-mediated phosphorylation of endophilin A is also noted to regulate synaptic autophagy (Soukup et al. 2016). Owing to the relatively higher frequency of LRRK2 mutations and its involvement in sporadic PD, endophilin A may prove to be another crucial protein involved in PD.
4. Autophagy in PD; cause and effect, hand in hand:
Neurons need to constantly maintain homeostasis by preserving healthy organelles and functional proteins, while selectively capturing and eliminating damaged organelles and misfolded proteins. Autophagy, an integral component of the E-L system, is an intracellular proteostasis pathway that performs this house cleaning function (Nixon 2013; Suresh et al. 2018b; Mizushima et al. 1998) (Figure 1A, C). It was initially identified in cell lines to function in maintaining energy homeostasis during critical periods of development and in response to nutrient depletion (Mizushima 2007). Intriguingly, nutrient depletion does not induce autophagy in neurons, but a complete lack of autophagy can cause lethal neurodegeneration, emphasizing its important role in survival (Hara et al. 2006; Komatsu et al. 2006). Since neurons are postmitotic and long-lived, they are particularly vulnerable to accumulation of toxic proteins (Nixon 2013). In addition, greater dependency of neurons on mitochondria for energy instead of glycolytic pathways demands constant mitochondrial turnover (Kann & Kovacs 2007), leading to many malfunctioning mitochondria. Both these factors play major roles in the pathogenesis of PD.
4.1. PINK1 and Parkin:
Role in Mitophagy and Beyond. Under physiological conditions, autophagy plays a crucial role in clearing unwanted proteins, as well as damaged mitochondria through a targeted mechanism called mitophagy (Nguyen et al. 2016). Loss-of-function mutations in PINK1 and Parkin (PARK2 and PARK6), result in recessively-inherited early-onset PD and significantly, both genes were discovered to be essential for mitophagy (Kitada et al. 1998; Valente et al. 2004; Mori et al. 1998). PINK1 is a serine/threonine protein kinase that acts as a surveillance molecule for mitochondrial health and accumulates on the outer membrane of abnormal mitochondria (Nguyen et al. 2016). PINK1 attracts recruitment of Parkin to damaged mitochondria, which through its E3 ubiquitin ligase activity (Kane et al. 2014; Narendra et al. 2010), coats the mitochondria with ubiquitin triggering autophagosomes to engulf the mitochondria (Narendra et al. 2008), culminating in lysosomal degradation (Figure 1A, C). Discoveries of PINK1 and Parkin mutations were the first genes to shed light on the direct involvement of autophagy and E-L system in PD pathogenesis (Pickrell & Youle 2015). These observations also helped in validating previous neurotoxic studies (Przedborski et al. 2004) which identified mitochondrial dysfunction as one of the primary causes of neurodegeneration. Though the symptoms are indistinguishable from sporadic cases, several studies on brains of PD patients with Parkin mutations do not show α-synuclein aggregation and Lewy body pathology (Doherty & Hardy 2013). Only one report suggests PINK1-linked parkinsonism to be associated with Lewy pathology (Samaranch et al. 2010). This prompts us to speculate if there are alternative neuropathological cascades in PD that are α-synuclein independent but centered around the E-L system. In addition to their well-defined roles in mitophagy, Parkin was identified to modulate E-L system through Rab7 ubiquitination (Song et al. 2016), whereas PINK1 phosphorylates some Rab proteins (Lai et al. 2015). It is also worth noting that several of these rab proteins (for example Rab7) regulate mitochondrial dynamics and quality control independent of Parkin/PINK1 (Wang et al., 2018). Parkin mutations are also known to directly affect retromer recruitment (Williams et al. 2018) and interact strongly with VPS35 by ubiquinating it (Martinez et al. 2017; Malik et al. 2015). Parkin binds to endophilin A and functions in its ubiquitination (Cao et al. 2014). Parkin also ubiquitinates EPS15 to regulate epidermal growth factor receptors and phosphoinositide 3-kinase (PI(3)K)-Akt signalling that plays a role in neuronal survival (Fallon et al. 2006). These developments suggest mitophagy-independent routes these proteins may take towards PD. Further investigations of Parkin and PINK1 in this direction might provide crucial insights in understanding neurodegenerative mechanisms that are Lewy-body independent.
4.2. Synaptic autophagy and axonal transport: a road less travelled in PD research
α-Synuclein aggregates initially in presynaptic termini, where the protein is enriched (Kramer & Schulz-Schaeffer 2007). α-Synuclein aggregates are a major target for autophagy-mediated degradation (Xilouri et al. 2008). In both genetic and sporadic PD, aggregated α-synuclein is likely to enhance autophagosome formation to meet the higher demand for its degradation (Wong & Holzbaur 2015). Once threshold reaches beyond compensatory limits, mutant/overexpressed α-synuclein starts to accumulate, turning detrimental to cellular homeostasis. In fact, A53T and A30P α-synuclein mutants blocked their own uptake and other substrates by lysosomes for degradation through the chaperone-mediated autophagy pathway in vitro (Cuervo et al. 2004). Dopamine-modified α-synuclein is identified to further exacerbate this process, which may explain the selective vulnerability of dopaminergic neurons in PD (Martinez-Vicente et al. 2008). A recent study on mammalian cells and transgenic mice shows that α-synuclein overexpression mediated autophagy defects are exerted through Rab1a inhibition (Winslow et al. 2010). Further understanding of the mechanisms for α-synuclein-autophagy crosstalk in PD might have crucial therapeutic applications. Backing this notion, both in vitro and in vivo studies show that enhancing autophagy and autophagosome-lysosome fusion could ameliorate PD-like features by clearing aggregated α-synuclein (Suresh et al. 2017; Suresh et al. 2018a).
Several other PD causing genes associated with the E-L system exert their pathological effects through impairing synaptic autophagy. The R258Q SynJ1 mutation directly affects autophagosome biogenesis and maturation at synaptic terminals of flies and neurites of human iPSC derived neurons. The observation of dopaminergic neuronal loss in these flies suggests autophagosome generation defects as one of the possible mechanisms through which SynJ1 mutations lead to dopaminergic neurodegeneration and PD (Vanhauwaert et al. 2017). Presynaptic endocytic membrane bending protein endophilin A also has functions in providing docking sites for autophagy proteins and facilitating autophagosome expansion. This is achieved by phosphorylation of endophilin A-BAR domain by LRRK2, as noted in a fly model (Soukup & Verstreken 2017). Congruent to these observations, mice lacking endophilin A revealed deficiency in autophagosome formation and developed a Parkinsonian phenotype (Murdoch et al. 2016). These studies, along with recent finding of clathrin involvement in autophagy regulation (Rong et al. 2012), suggest a cooperative crosstalk between SV endocytosis and autophagy.
Macromolecules and organelles in the presynaptic terminal travel a long distance from the soma, making them old and vulnerable. Rapid turnover of proteins, membranes, receptors, and vesicles in the pre-synapse assigns autophagy a major role in maintaining synaptic homeostasis. This tasks synaptic autophagy with quality control responsibilities. Though there is compelling evidence that compromised autophagy contributes to PD, these studies primarily looked at the soma from a proteinopathy or mitophagy perspective (Lynch-Day et al. 2012). Selective inactivation of autophagy by conditional knockout of autophagy protein atg7 in dopaminergic neurons leads to enhanced dopamine release and rapid presynaptic recovery with abnormal axonal profiles (Hernandez et al. 2012). Conversely, autophagy enhancement through mTOR inhibition slows dopamine release, reduces the number of synaptic vesicles, and increases autophagosome density in the axons (Hernandez et al. 2012). These results emphasize the importance of synaptic autophagy in normal dopamine transmission and further evaluation is warranted to understand if additional synaptic autophagy defects exist, which may lead to PD pathogenesis.
Another unique and stressful situation for neurons is that autophagosomes loaded with unwanted materials from an active presynaptic terminal, need to be transported a long distance to the soma for lysosomal degradation (Figure 1B). Microtubule and scaffolding proteins aid in this retrograde axonal transport of autophagosomes and are crucial for the normal functioning of E-L system (Birdsall & Waites 2019). During their transport, autophagosomes mature into autolysosomes by fusing with late-endosomes/lysosomes towards the proximal end of axon (Nikoletopoulou & Tavernarakis 2018). There are recent studies suggesting lysosomal degradation at the distal end, especially of mitophagosome mediated by Parkin-PINK1. This is predicted to provide rapid neuroprotection against oxidative stress by damaged mitochondria, without requirement for retrograde transport (Ashrafi et al. 2014; Ashrafi & Schwarz 2015). Axonal transport aids this function through anterograde transport of E-L system components required for degradation from soma towards distal end. Any defect in this to-and-fro transport machinery can completely clog the E-L system and other processes. Considering its functional significance in neuronal survival, axonal transport might hold clues for understanding pathological mechanisms. Abnormal axonal swelling with lysosomal accumulation has been noted in Alzheimer’s disease, blocking the retrograde transport of autophagosomes (Gowrishankar et al. 2015; Nixon et al. 2005). Similar understanding on the health of axonal transport machinery and its dynamics in PD is crucial towards finding commonalities amongst neurodegenerative diseases.
5. Sometimes, there are traffic issues: E-L system trafficking in PD
The multi-compartmentalised nature of neurons puts tremendous pressure on the endosomal sorting and trafficking machinery, which is also true in case of nigrostriatal neurons. The vacuolar protein sorting (VPS) group of proteins that form retromer complex comprising of VPS35, VPS26, and VPS29 are crucial in retrograde trafficking and sorting of protein cargos from endosomes to the TGN, facilitating their recycle and reuse (Figure 1A) (Haft et al. 2000; Seaman et al. 1998). The retromer complex also functions in sorting proteins/cargo to regulate endosomal and lysosomal maturation (Vagnozzi & Pratico 2018). Pedigrees with single allelic mutation in VPS35 revealed autosomal dominant, late-onset, L-dopa-responsive PD, with striking similarities to idiopathic PD (Vilarino-Guell et al. 2011; Wider et al. 2008). Broadly, it is proposed that the mutant VPS35 fails to associate with other components of the retromer complex, resulting in impaired retrograde trafficking (Williams et al. 2017). Induced loss-of-function mutations of VPS35 in vitro resulted in enlargement of late-endosomes and aggregation of α-synuclein oligomers (Follett et al. 2016). Another study on flies revealed ceramide accumulation due to VPS35 deficiency, suggesting lysosomal dysfunction (Lin et al. 2018). α-synuclein accumulation in nigral dopaminergic neurons, loss of dopaminergic neurons, reduced dopamine transporter levels in the striatum, and locomotory deficits were noted in VPS35-deficient mice (Ishizu et al. 2016; Tang et al. 2015a). Further in vitro studies of VPS35 (D620N) mutant cells revealed dysfunction in endosome-to-Golgi retrieval of lysosomal membrane proteins (LAMP2a) and accelerated lysosomal degradation of LAMP2a (Tang et al. 2015a). LAMP2a is considered important in directing misfolded α-synuclein to lysosomes, suggesting a mechanism for α-synuclein accumulation in VPS35 deficiency (Tang et al. 2015a). VPS35 mutations also affect autophagy and dendritic AMPA (α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor trafficking (Munsie et al. 2015; Zavodszky et al. 2014). VPS35 mutations bring severe abnormality in mitochondrial dynamics, as noted in cultured dopaminergic neurons, mice SNpc neurons, and in PD patient derived fibroblasts. (Tang et al. 2015b). Oxidative stress implicated in sporadic PD might mediate pathogenesis through VPS35-driven mitochondrial defects (Wang et al. 2016). VPS35 also has a role in biogenesis of mitochondria-derived vesicles, further suggesting that its dysfunction causees mitochondrial pathology and neurodegeneration (Sugiura et al. 2014). VPS35 is functionally linked to LRRK2, boosting its kinase activity (Mir et al. 2018). In fact, decreased VPS35 levels were noted in the SNpc of PD patients with LRRK2 mutations (Zhao et al. 2018). Interestingly, overexpression of VPS35 ameliorated locomotor deficits and enhanced lifespan in LRRK2 mutant models (Linhart et al. 2014; MacLeod et al. 2013), which was also seen in Parkin mutant models (Malik et al. 2015). Neuroprotection was also noted against MPP+ induced toxicity in dopaminergic neurons (Bi et al. 2013). Along with its E-L sorting functions, these links between VPS35 and α-synuclein, LRRK2, Parkin, lysosomal dysfunction, autophagy, mitochondrial perturbations, and oxidative stress strongly suggest VPS35 as a good therapeutic target.
Another VPS family member associated with PD is VPS13C. Five allelic variations in VPS13C gene can lead to autosomal recessive, early-onset PD, primarily due to premature termination of VPS13C (Lesage et al. 2016; Schormair et al. 2018). Patients were noted with cardinal PD symptoms and extrapyramidal signs. Post-mortem studies revealed α-synuclein/ubiquitin positive-Lewy bodies in the brain stem, limbic system, hippocampus, and cortical associative areas (Lesage et al. 2016). A few studies reveal that VPS13 functions in the TGN-endosomal cycle (De et al. 2017), whereas recent discoveries have also located it on the outer mitochondrial membrane (Lesage et al. 2016). In fact, siRNA-mediated silencing of VPS13C resulted in lower mitochondrial membrane potential, mitochondrial fragmentation, increased respiratory rates, exacerbated Parkin-dependent mitophagy, and upregulation of Parkin in response to mitochondrial damage (Lesage et al. 2016). A new study also revealed VPS13C’s role in forming membrane contact sites and facilitating lipid transfer between the ER and mitochondria (Kumar et al. 2018). This is noteworthy, considering lipid transfer’s increasing recognition in disease pathogenesis.
6. Lysosomes: PD may also begin at the end of the tunnel
Since lysosomes are the destination for most E-L system vesicles carrying a variety of cargos for degradation and recycling (Figure 1A), dysfunction in any of its components results in lysosomal storage disorders, severely perturbing cellular homeostasis. A growing body of evidence suggests Lysosomal Storage Disorders increase risk for PD pathogenesis (Robak et al. 2017). Among them, Gaucher disease (GD) confers the greatest risk, where 5–15% of GD patients or carriers develop PD later in life, to an extent where causal GBA mutations are the most common genetic risk factor for PD (Table 1) (Aharon-Peretz et al. 2004; Bultron et al. 2010; Simon-Sanchez et al. 2009). GBA encodes glucocerebrosidase 1 (Gcase1), which facilitates breakdown of glucosylceramide (GlcCer) into glucose and ceramide. In its absence, GlcCer accumulates and undergoes partial enzymatic deacylation to glucosylsphingosine and other more complex glycosphingolipids, which mediate disease pathogenesis. GBA is a relatively well studied E-L system gene with regards to PD, both in terms of post-mortem brains, clinical investigations, and basic research. Reduced Gcase1 activity in substantia nigra, putamen, and other brain areas of PD patients with GBA mutations have been noted along with GlcCer and glycosphingolipids accumulation (Alcalay et al. 2015; Gegg et al. 2012). Interestingly, similar observations were made even in sporadic PD patients (Alcalay et al. 2015; Rocha et al. 2015), suggesting an even broader role for GBA than just mutations and potentially involve epigenetic and environmental modifiers of GBA. Gcase1 levels decrease with aging, the major risk factor for PD (Hallett et al. 2018; Rocha et al. 2015), indicating GBA’s possible role in lowering the nigral neurons’ threshold to develop PD. Cognitive areas such as hippocampus, frontal cortex, and amygdala also showed relative decrease in GCase1 activity, along with a significant elevation of hippocampal glucosylsphingosine in sporadic cases (Gegg et al. 2012). This correlates with a higher Lewy body burden identified in the hippocampus and medial temporal regions, as well as a diffused cortical Lewy body pathology in GBA-linked PD (Hallett et al. 2018; Watson & Leverenz 2010). These pathological observations strengthen clinical findings that cognitive deterioration is severe in PD patients with GBA mutations compared to that of sporadic cases (Liu et al. 2016; Mata et al. 2016). These features indicate mouse models of PD with GBA mutations are important in studying lipid dyshomeostasis and understand mechanisms of cognitive dysfunction in PD.
A recent study by Taguchi et al., provides evidence for an association between loss of Gcase1 activity, glycosphingolipids accumulation, and α-synuclein aggregation, both in vitro and in vivo models of Gba mutation (Taguchi et al. 2017). Glucosylsphingosine was shown to promote pathological α-synuclein aggregates, capable for further self-templating, was shown to accumulate in the brains of young homozygous Gba mutant and KO mice, implicating this glycosphingolipid in PD pathogenesis. Even in heterozygous Gba mutations, the Gcase1 activity decreased with age to an extent where mice reached threshold for haploinsufficiency, mimicking the human condition. Furthermore, the Gba mutant mice, when crossed with well characterised α-synuclein transgenic PD mice, revealed accelerated PD progression including α-synuclein aggregation and motor deficits (Taguchi et al. 2017). α-Synuclein aggregation has also been seenin human iPSCs derived neurons from GD and PD patients that are exposed to Gcase1 inhibitors, and in those carrying GBA mutations (Zunke et al. 2018). Mutations in GBA also cause misprocessing of Gcase1 in the ER, leading to ER retention of Gcase1. This produces ER stress and auto-lysosomal perturbations causing an abnormal lipid profile and increased α-synuclein accumulation and secretion (Fernandes et al. 2016; Ron & Horowitz 2005). This study along with a few other observations suggest dopaminergic dysfunction due to gain-of-function mutations in Gba (Cullen et al. 2011; Fernandes et al. 2016; Sidransky & Lopez 2012). However, therapeutic strategies based on loss-of-function observations have been successful, arguing against a gain of function model. Activation of residual Gcase1 by small molecules and chaperones and ameliorating glycosphingolipid accumulation by inhibiting GlcCer synthase lowered α-synuclein pathology and reinstated lysosomal health (Richter et al. 2014; Charvin et al. 2018; Sardi et al. 2017). These strategies are already being tested in clinics (Richter et al. 2014).
ATPase cation transporting 13A2 (ATP13A2), another lysosomal protein which localises on its membrane (Figure 1A) with unknown function, has been implicated in PD (PARK9, Table 1). Autosomal recessive, early-onset parkinsonism due to ATP13A2 mutations were first noted in Kufor-Rakeb Syndrome, followed by recent studies in other forms of parkinsonism (Di Fonzo et al. 2007; Eiberg et al. 2012; Park et al. 2011; Ramirez et al. 2006). Fibroblasts from these patients showed impaired lysosomal acidification and proteostasis, leading to inefficiency in substrate degradation and autophagosome clearance. These observations were replicated in ATP13A2 knockdown dopaminergic cell lines, where its restoration re-established lysosomal homeostasis and ameliorated cell death (Dehay et al. 2012). Downregulation of ATP13A2 was also seen in nigral neurons of sporadic PD patients, along with traces in Lewy bodies (Dehay et al. 2012). A loss-of-function ATP13A2 mutant mouse model revealed age related lysosomal dysfunction, protein aggregation, and gliosis leading to motor abnormalities. Interestingly, the levels of total and oligomeric α-synuclein was normal, despite ubiquitinated protein aggregation, cathepsin-D dysfunction, and autophagy defects (Kett et al. 2015). These observations suggest that ATP13A2 mutations may lead to PD through an α-synuclein independent mechanism.
7. E-L system; a key target of key PD-associated proteins?
7.1. α-synuclein:
Misfolded α-synuclein can trigger PD pathogenesis in the presynaptic terminals of nigrostriatal neurons (Imbriani et al. 2018; Kramer & Schulz-Schaeffer 2007; Spinelli et al. 2014). This may as well be one of the earliest pathogenic events in PD, where α-synuclein aggregates target SV endocytosis, SV trafficking, and autophagy components of E-L system to initiate or exacerbate dopaminergic neuronal dysfunction.
α-Synuclein and other family members function in presynaptic terminals, where they modulate SV distribution and neurotransmission (Burre 2015; Greten-Harrison et al. 2010; Vargas et al. 2017) (Figure 1C). The biophysical properties of synucleins reveal their ability to sense and modify membrane curvature (Shen et al. 2012; Westphal & Chandra 2013; Middleton & Rhoades 2010). These functions suggest that synucleins regulate SV exocytosis and endocytosis (Vargas et al. 2014; Greten-Harrison et al. 2010). Studies in KO mice in fact show that synucleins normally function post SV exocytosis, in fusion pore expansion and/or control the kinetics of SV endocytosis by facilitating membrane curvature (Lautenschlager et al. 2017; Logan et al. 2017; Vargas et al. 2014). α-Synuclein also appears to control SV availability for release by regulating SV clustering (Diao et al. 2013; Vargas et al. 2017).
On a pathological level, several studies show that overexpression of α-synuclein inhibits neurotransmission. The locus of this consistent effect is not clear. Studies have shown that elevated α-synuclein levels and oligomeric α-synuclein inhibit SV exocytosis in a SNARE-dependent manner or affecting SV clustering (Huang et al. 2019; Nemani et al. 2010). On the other hand, considerable evidence suggests that aggregated α-synuclein primarily impacts SV endocytosis, which is of relevance to E-L system genes linked to PD. In lamprey giant synapses, injection of aggregated α-synuclein is identified to inhibit clathrin un-coating as evidenced by the accumulation of CCVs when SV recycling was evoked by high-frequency stimulation (Busch et al. 2014). A recent study also suggests that extra synuclein might sequester Hsc70 thus effect clathrin uncoating, subsequently causing SV recycling defects (Banks et al. 2019). Similar observation was made in mammalian Calyx of Held synapse, where acute elevation of monomeric and mutant α-synuclein levels decreased SV endocytosis with frequency dependent effect on SV exocytosis. This was also seen upon transgenic expression of α-synuclein (Eguchi et al. 2017; Xu et al. 2016). . Overexpression of mutant α-synuclein also affects dopamine transporter trafficking on the presynaptic membrane, leading to dopamine dyshomeostasis (Kisos et al. 2014). The observations that overexpression and deletion of synucleins have similar effects on SV exo- and endocytosis (Lautenschlager et al. 2017) suggest that SV cycling is exquisitely sensitive to α-synuclein levels. Further understanding of the precise mechanisms for interactions of α-synuclein and synaptic E-L system proteins involved in PD will provide crucial insights on early causal pathogenic events in PD.
At the soma, aggregated α-synuclein mainly targets E-L system trafficking, especially the retromer pathway. Recent studies propose that aggregated α-synuclein disrupts the retromer complex by interacting through VPS26 or VPS29, and thus preventing the recruitment of adaptors like Snx3 and SNX-BAR dimers (Chung et al. 2017; Patel & Witt 2018).. Deficiency of VPS35 in dopaminergic neurons impairs endosome-to-Golgi retrieval of receptors like Lamp2a, accelerating its lysosomal degradation. Lamp2a is a critical receptor for chaperone-mediated autophagy of misfolded α-synuclein, so its deficiency leads to a vicious cycle of α-synuclein accumulation (Tang et al. 2015a), which might be the case in patients with VPS35 mutations. Overexpression of α-synuclein also directly compromises macroautophagy through inhibition of Rab1a (Winslow et al. 2010). It is also pertinent to note that aggregated α-synuclein in the soma is a key target of treatment strategies in development through induction of macroautophagy (Suresh et al. 2017; Suresh et al. 2018a).
Recent developments suggest that α-synuclein pathology spreads in a prion-like manner through synaptically connected regions in a manner consistent with Braak staging (Braak et al. 2003; Luk et al. 2012). The E-L system plays a prominent role in cell-to-cell transmission of preformed α-synuclein filaments, leading to the spread of phospho-α-synuclein pathology. Internalization of fibrillar α-synuclein occurs via endocytosis (as well as tunnelling nanotubules) and follows the neuronal endosomal pathway to lysosomal degradation (Rodriguez et al. 2018). Heparan sulfate proteoglycans, lymophocyte-activation gene 3, α3 isoform of Na+/K+ ATPase, and a few endocytic vesicle receptors are predicted to act as receptors for α-synuclein preformed fibrils (Holmes et al. 2013; Ihse et al. 2017; Mao et al. 2016; Shrivastava et al. 2015). In vitro studies suggest that internalized α-synuclein fibrils are localized in both early endosomes and late endolysosomes (Karpowicz et al. 2017; Lee et al. 2008b). The lipid bilayer of endosomes restricts cytoplasmic spillage, so most endocytosed α-synuclein is degraded by lysosomes. How a fraction of α-synuclein preformed fibrils escapes this barrier and goes on to initiate seeding is still unclear (Bieri et al. 2018). Studies of α-synuclein axonal transport predict that retrograde transport of α-synuclein preformed fibrils is mediated by endocytic vesicles and is twice as fast and more efficient than anterograde transport (Brahic et al. 2016). This strengthens the notion that PD pathology spreads retrogradely from the synaptic terminal to soma. The E-L system is also involved in exocytosis of fibrilized α-synuclein, facilitating its prion-like spread. An ER-bound enzyme with chaperone activity USP19 (Ubiquitin carboxyl-terminal hydrolase 19) has been shown to deliver misfolded α-synuclein to late endosomes, which then fuses with the plasma membrane through a unique process called misfolding-associated protein secretion (Lee et al. 2016). Another recent study suggests that the DNAJ/Hsc70 complex could facilitate α-synuclein exocytosis (Fontaine et al. 2016). Overexpression of ATP13A2 and GBA is also noted to increase exocytosis of α-synuclein (Fernandes et al. 2016; Tsunemi et al. 2014). Together, these observations identify the E-L system as the primary conduit for the spread of α-synuclein-mediated pathology in PD.
7.2. LRRK2:
Leucine-Rich Repeat Kinase 2 (LRRK2) is a multidomain large protein with both GTPase and kinase activity along with other domains for protein-protein interaction (Kett & Dauer 2012). Epidemiological, histopathological, and clinical investigations have all established LRRK2 gene mutations as the most common genetic cause of PD (PARK8, Table 1) (Paisan-Ruiz et al. 2004; Zimprich et al. 2004), with a considerable contribution to sporadic cases (Kett & Dauer 2012). Over 8identified LRRK2 mutations cause PD. The mutation at G2019S is the most common and is known to increase LRRK2’s activity (Kett & Dauer 2012; Smith et al. 2006; West et al. 2005). LRRK2 functions have been explored through PD-related studies, in which increased G2019S kinase activity reveals its close association with the E-L system. LRRK2’s role in vesicular trafficking, lysosomal homeostasis, and autophagy are well accepted. Phosphorylation by LRRK2 is essential for a subset of RAB GTPases’ function of endolysosomal vesicular sorting and trafficking (Steger et al. 2017; Steger et al. 2016). In an overexpression system, RAB7L1 was noted to interact strongly with LRRK2 at the switch-II site (Steger et al. 2017), unlike as seen in in vitro kinase assays (Steger et al. 2016). Several LRRK2 mutations significantly affect RAB pathways, where variations in Rab7L1’s interactions with LRRK2 have been identified to increase the risk of sporadic PD. In fact, recent GWAS identify RAB29 encoding RAB7L1 (PARK16, Table 1) as an independent risk gene for PD (Satake et al. 2009). Rab7L1 upregulation could attenuate LRRK2-induced toxicity in dopaminergic neurons (MacLeod et al. 2013). This is predicted to be functioning through VPS35 and VPS26 (Linhart et al. 2014). VPS35 also cooperates with LRRK2 to regulate SV recycling and dopaminergic synaptic release (Inoshita et al. 2017), thus suggesting a prominent role for the retromer complex in LRRK2-mediated PD pathogenesis.
LRRK2 regulates lysosomal protein trafficking and morphology (Eguchi et al. 2018; Kuwahara et al. 2016). In LRRK2 mutants, perinuclear lysosomal clustering was noted, mediated by increased phosphorylation of RAB7 (Hockey et al. 2015). Two-pore channels are the calcium channels present on acidic vesicles of the E-L system and help in regulating their trafficking and fusion of lysosomes with autophagosomes/endosomes (Calcraft et al. 2009). Abnormal lysosomal morphology and density seen in fibroblasts from patients with LRRK2 mutations could be reversed by inhibiting these two-pore channels (Hockey et al. 2015). Thus, these channels might be one potential mediator of E-L system dysfunction in LRRK2-associated PD. Lysosome-related proteins involved in PD such as Gcase1, Lamp2A and ATP13A2 are also downregulated in PD patients with LRRK2 mutations (Zhao et al. 2018), suggesting lysosomes are one of LRRK2’s prime targets.
Mechanisms for LRRK2-mediated autophagy defects are still elusive. The ability of LRRK2 kinase inhibitors to reverse autophagy dysfunction suggest that kinase activity may be significant (Manzoni et al. 2013; Saez-Atienzar et al. 2014). Mutant LRRK2 is also a substrate for chaperone-mediated autophagy, impairment of which could exacerbate pathological implications (Orenstein et al. 2013). Accumulation of MVBs and malformed autophagosomes were noted in LRRK2 R1441C mutations (Alegre-Abarrategui et al. 2009). Intra-axonal autophagic vacuole defects in nigral dopaminergic projections were seen in mice expressing human LRRK2 (Li et al. 2009; Tagliaferro et al. 2015). It also regulates synaptic autophagy through phosphorylation of endophilin A (Soukup et al. 2016). G2019S mutations are known to induce autophagy through MAPK/ERK kinases (Bravo-San Pedro et al. 2012; Bravo-San Pedro et al. 2013), and some studies suggest LRRK2-mediated autophagy regulation is largely independent of mTOR (Manzoni et al. 2013). Since RAB-mediated trafficking is crucial for autophagy, which is also affected in LRRK2 mutations, it is important to evaluate if autophagy defects are the consequences of RAB dysregulation or a direct effect of LRRK2 mutations.
LRRK2 regulates the SV cycle by interacting with E-L system proteins that are also implicated in PD: auxilin, SYNJ1, and endophilin A. LRRK2 overactivity phosphorylates auxilin in its clathrin binding domain, making their interaction weaker. This leads to disruption of SV endocytosis (Nguyen & Krainc 2018). This was noted in patient iPSC-derived dopaminergic neurons, along with accumulation of oxidized dopamine, increased α-synuclein levels, and decreased Gcase1 activity (Nguyen & Krainc 2018). Hyperphosphorylation of endophilin A and synJ1 by LRRK2 significantly altered synaptic endocytosis (Islam et al. 2016; Matta et al. 2012; Pan et al. 2017). A recent study showed that LRRK2 can directly bind to clathrin-light chains through its GTPase domain, affecting Rac1-mediated endosomal dynamics (Schreij et al. 2015). LRRK2 also perturbs synaptic exocytosis through phosphorylation of N-ethylmaleimide-Sensitive Factor which upregulates SNARE disassembly (Belluzzi et al. 2016). Furthermore, LRRK2 is highly expressed in dorsal striatal medium spiny neurons (MSNs), the postsynaptic compartment of nigrostriatal pathway. G2019S mutation of LRRK2 lead to abnormally high activity of MSNs and morphological perturbations during development (Matikainen-Ankney et al. 2016). Abnormal synaptogenesis and dopamine receptor activation in the developing MSNs mediated by dysfunctional protein kinase A was seen in LRRK2 R1441C mutation (Parisiadou et al. 2014). These defects can permanently modify the circuit structure and function, predisposing the individuals to both motor and non-motor dysfunctions of PD. LRRK2 mutation-mediated endocytosis defects were noted predominantly in these MSNs, along with nigral dopaminergic neurons and not in cortical or hippocampal neurons (Pan et al. 2017; Maas et al. 2017). This suggests a heightened vulnerability in the E-L system of nigrostriatal circuit for LRRK2 mutations.
Increased LRRK2 kinase activity mediated elevation of exocytosis may also be relevant for cell-to-cell transfer of misfolded α-synuclein (Bae et al. 2018; Schapansky et al. 2018; Lin et al. 2009). LRRK2 can selectively recruit to overloaded lysosomes and divert undegraded materials out of the kidney cells, along with secretion of α-synuclein from endosomes (Eguchi et al. 2018). Studies also demonstrate heightened uptake of α-synuclein fibrils through endocytosis in the presence of pathogenic LRRK2 (Bae et al. 2018). However, LRRK2’s role in prion-like spreading of α-synuclein remains speculative, until confirmation that similar mechanisms exist in neurons.
8. Future Directions:
Clinical description and diagnosis of PD has changed little since James Parkinson’s characterization of the disease 200 years ago (Parkinson 2002). In the 1960s, identification of dopamine deficiency as the cause of motor abnormalities led to symptomatic relief for patients (Carlsson et al. 1957). Though some success has been noted with deep brain stimulation (Limousin et al. 1998), we are still relying on symptomatic relief. The last three decades of basic research have enriched our understanding of PD’s cellular and molecular mechanisms, bringing tangible hope for disease modifying treatment strategies. Identification of risk genes and conceptual shifts in our understanding through genetic models point to the E-L system dysfunction as the key mediator of PD pathogenesis. Further strengthening this concept, PD pathogenesis appears to begin at synaptic terminals where endo and exocytotic machinery is most active and where α-synuclein and LRRK2 exert their effects through the E-L system and vice-versa. Studies so far suggest that altered flux through the E-L system in nigral dopaminergic neurons that have lesser threshold for degeneration is the key trigger for neurodegeneration. Moving forward, critical elucidation of the mechanisms behind sorting, trafficking, and recycling defects demands detailed studies of auxilin and other DNAJ family proteins, along with SYNJ1, endophilin A, and the retromer complex. Studying these genes will also provide crucial insights into less explored roles of synaptic autophagy, axonal transport, and clathrin-independent endocytosis in PD. Recent surveys of PD patients note preservation of cognitive functioning as a major unmet need. Further investigation of GBA mutations implicated in PD is a promising avenue considering GBA’s known involvement in cognitive disorders like Dementia with Lewy Bodies. Along with GBA, studying ATP13A2 and other lysosomal genes will enrich our understanding of lysosomal dysfunction, potentially bringing us closer to repurposing drugs used in Lysosomal Storage Disorders for PD. Affirming ongoing clinical trials targeting autophagy, α-synuclein, and LRRK2, an enhanced understanding of E-L system-mediated pathogenesis will prove crucial in developing novel treatment strategies and diagnostic tools for PD.
Acknowledgments:
We thank lab members for fruitful discussions on this review topic and important inputs to the figure.
Funding: This work was supported by NIH (R01NS110354, R01NS083846), Nina Compagnon Hirshfield Parkinson’s Disease Research Fund and DOD (W81XWH-17–1-0564). VDJ is supported by DOD CDMRP Early Investigator Research Award (W81XWH-19–1-0264).
Abbreviations:
- PD
Parkinson’s disease
- SNpc
Substantia Nigra pars compacta
- E-L
Endolysosomal
- M6P
Mannose-6-phosphate
- TGN
Trans-Golgi Network
- CME
Clathrin-Mediated Endocytosis
- ESCRT
Endosomal Sorting Complex Required for Transport
- ILVs
Interluminal Vesicles
- MVBs
Multivesicular Bodies
- CCVs
Clathrin Coated Vesicles
- GAK
Cyclin G Associated Kinase
- GBA
Glucosidase Beta Acid
- DCVs
Dense Core Vesicles
- LRRK2
Leucine-rich repeat kinase 2
- PARK
Parkinson’s disease gene
- PRKN
Parkin
- SNCA
Alpha-synuclein gene
- GWAS
Genome Wide Association Study
- SVs
Synaptic Vesicles
- PI(4,5)P2
Phosphatidylinositol 4,5-bisphosphate
- L-dopa
Levodopa
- KO
Knockout
- RME-8
Receptor Mediated Endocytosis 8
- SYNJ1/SynJ1
Synaptojanin-1
- PIPs
Phosphoinositide Phosphates
- ATP13A2
ATPase cation transporting 13A2
- PINK1
PTEN Induced Putative Kinase 1
- VPS
Vacuolar Protein Sorting
- LAMP2a
Lysosome-Associated Membrane Glycoprotein 2
- AMPA
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- GD
Gaucher Disease
- Gcase1
Glucocerebrosidase 1
- GlcCer
Glucosylceramide
- SNARE
Soluble N-ethylmaleimide-sensitive factor Attachment protein Receptors
- HSPGs
Heparan Sulfate Proteoglycans
- USP19
Ubiquitin Carboxyl-Terminal Hydrolase 19
- MAPK/ERK
Mitogen-Activated Protein Kinase/ Extracellular Signal-Regulated Kinase
- GTPase
Guanosine Triphosphatase
- PI(4,5)P2
Phosphatidylinositol 4,5-bisphosphate
- Hsc70
Heat Shock Cognate 70
- siRNA
small interfering Ribonucleic acid
- iPSCs
induced Pluripotent Stem Cells
- HSP40
Heat Shock Protein 40
- MSNs
Medium Spiny Neurons
Footnotes
Conflict of interests: None
References:
- Aharon-Peretz J, Rosenbaum H and Gershoni-Baruch R (2004) Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med 351, 1972–1977. [DOI] [PubMed] [Google Scholar]
- Alcalay RN, Levy OA, Waters CC et al. (2015) Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 138, 2648–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alegre-Abarrategui J, Christian H, Lufino MM, Mutihac R, Venda LL, Ansorge O and Wade-Martins R (2009) LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet 18, 4022–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Schlehe JS, LaVoie MJ and Schwarz TL (2014) Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol 206, 655–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G and Schwarz TL (2015) PINK1- and PARK2-mediated local mitophagy in distal neuronal axons. Autophagy 11, 187–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba Y, Markopoulou K, Putzke JD, Whaley NR, Farrer MJ, Wszolek ZK and Uitti RJ (2006) Phenotypic commonalities in familial and sporadic Parkinson disease. Arch Neurol 63, 579–583. [DOI] [PubMed] [Google Scholar]
- Bae EJ, Kim DK, Kim C et al. (2018) LRRK2 kinase regulates alpha-synuclein propagation via RAB35 phosphorylation. Nat Commun 9, 3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Hu Z, Dittman JS, Pym EC and Kaplan JM (2010) Endophilin functions as a membrane-bending molecule and is delivered to endocytic zones by exocytosis. Cell 143, 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks SML, Medeiros AT, McQuillan M, Busch DJ, Ibarraran-Viniegra AS, Roy S, Sousa R, Lafer EM and Morgan JR (2019) Hsc70 Ameliorates the Vesicle Recycling Defects Caused by Excess α-Synuclein at Synapses. bioRxiv, 517524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belluzzi E, Gonnelli A, Cirnaru MD et al. (2016) LRRK2 phosphorylates pre-synaptic N-ethylmaleimide sensitive fusion (NSF) protein enhancing its ATPase activity and SNARE complex disassembling rate. Mol Neurodegener 11, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi F, Li F, Huang C and Zhou H (2013) Pathogenic mutation in VPS35 impairs its protection against MPP(+) cytotoxicity. Int J Biol Sci 9, 149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieri G, Gitler AD and Brahic M (2018) Internalization, axonal transport and release of fibrillar forms of alpha-synuclein. Neurobiol Dis 109, 219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdsall V and Waites CL (2019) Autophagy at the synapse. Neurosci Lett 697, 24–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN and Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24, 197–211. [DOI] [PubMed] [Google Scholar]
- Brahic M, Bousset L, Bieri G, Melki R and Gitler AD (2016) Axonal transport and secretion of fibrillar forms of alpha-synuclein, Abeta42 peptide and HTTExon 1. Acta Neuropathol 131, 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-San Pedro JM, Gomez-Sanchez R, Niso-Santano M et al. (2012) The MAPK1/3 pathway is essential for the deregulation of autophagy observed in G2019S LRRK2 mutant fibroblasts. Autophagy 8, 1537–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-San Pedro JM, Niso-Santano M, Gomez-Sanchez R et al. (2013) The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell Mol Life Sci 70, 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultron G, Kacena K, Pearson D, Boxer M, Yang R, Sathe S, Pastores G and Mistry PK (2010) The risk of Parkinson’s disease in type 1 Gaucher disease. J Inherit Metab Dis 33, 167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre J (2015) The Synaptic Function of alpha-Synuclein. J Parkinsons Dis 5, 699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch DJ, Oliphint PA, Walsh RB, Banks SM, Woods WS, George JM and Morgan JR (2014) Acute increase of alpha-synuclein inhibits synaptic vesicle recycling evoked during intense stimulation. Mol Biol Cell 25, 3926–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcraft PJ, Ruas M, Pan Z et al. (2009) NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459, 596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, Milosevic I, Giovedi S and De Camilli P (2014) Upregulation of Parkin in endophilin mutant mice. J Neurosci 34, 16544–16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, Wu Y, Ashrafi G et al. (2017) Parkinson Sac Domain Mutation in Synaptojanin 1 Impairs Clathrin Uncoating at Synapses and Triggers Dystrophic Changes in Dopaminergic Axons. Neuron 93, 882–896 e885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A, Lindqvist M and Magnusson T (1957) 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature 180, 1200. [DOI] [PubMed] [Google Scholar]
- Chang D, Nalls MA, Hallgrimsdottir IB et al. (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49, 1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C et al. (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169. [DOI] [PubMed] [Google Scholar]
- Charvin D, Medori R, Hauser RA and Rascol O (2018) Therapeutic strategies for Parkinson disease: beyond dopaminergic drugs. Nat Rev Drug Discov 17, 804–822. [DOI] [PubMed] [Google Scholar]
- Chaudhuri KR and Schapira AH (2009) Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol 8, 464–474. [DOI] [PubMed] [Google Scholar]
- Chung CY, Khurana V, Yi S et al. (2017) In Situ Peroxidase Labeling and Mass-Spectrometry Connects Alpha-Synuclein Directly to Endocytic Trafficking and mRNA Metabolism in Neurons. Cell Syst 4, 242–250 e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona O, Di Paolo G, Wenk MR et al. (1999) Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99, 179–188. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT and Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. [DOI] [PubMed] [Google Scholar]
- Cullen V, Sardi SP, Ng J et al. (2011) Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann Neurol 69, 940–953. [DOI] [PubMed] [Google Scholar]
- De M, Oleskie AN, Ayyash M, Dutta S, Mancour L, Abazeed ME, Brace EJ, Skiniotis G and Fuller RS (2017) The Vps13p-Cdc31p complex is directly required for TGN late endosome transport and TGN homotypic fusion. J Cell Biol 216, 425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Ramirez A, Martinez-Vicente M et al. (2012) Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A 109, 9611–9616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Wang P and Jankovic J (2018) The genetics of Parkinson disease. Ageing Res Rev 42, 72–85. [DOI] [PubMed] [Google Scholar]
- Deng HX, Shi Y, Yang Y et al. (2016) Identification of TMEM230 mutations in familial Parkinson’s disease. Nat Genet 48, 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fonzo A, Chien HF, Socal M et al. (2007) ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 68, 1557–1562. [DOI] [PubMed] [Google Scholar]
- Diao J, Burre J, Vivona S, Cipriano DJ, Sharma M, Kyoung M, Sudhof TC and Brunger AT (2013) Native alpha-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. Elife 2, e00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Braak H, Duda JE et al. (2009) Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol 8, 1150–1157. [DOI] [PubMed] [Google Scholar]
- Doherty KM and Hardy J (2013) Parkin disease and the Lewy body conundrum. Mov Disord 28, 702–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Cinnamon Y, Ta-Shma A et al. (2012) A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One 7, e36458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi K, Taoufiq Z, Thorn-Seshold O, Trauner D, Hasegawa M and Takahashi T (2017) Wild-Type Monomeric alpha-Synuclein Can Impair Vesicle Endocytosis and Synaptic Fidelity via Tubulin Polymerization at the Calyx of Held. J Neurosci 37, 6043–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi T, Kuwahara T, Sakurai M et al. (2018) LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc Natl Acad Sci U S A 115, E9115–E9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiberg H, Hansen L, Korbo L et al. (2012) Novel mutation in ATP13A2 widens the spectrum of Kufor-Rakeb syndrome (PARK9). Clin Genet 82, 256–263. [DOI] [PubMed] [Google Scholar]
- Fallon L, Belanger CM, Corera AT et al. (2006) A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat Cell Biol 8, 834–842. [DOI] [PubMed] [Google Scholar]
- Fearnley JM and Lees AJ (1991) Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114 (Pt 5), 2283–2301. [DOI] [PubMed] [Google Scholar]
- Fernandes HJ, Hartfield EM, Christian HC et al. (2016) ER Stress and Autophagic Perturbations Lead to Elevated Extracellular alpha-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Reports 6, 342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fesce R, Grohovaz F, Valtorta F and Meldolesi J (1994) Neurotransmitter release: fusion or ‘kiss-and-run’? Trends Cell Biol 4, 1–4. [DOI] [PubMed] [Google Scholar]
- Follett J, Bugarcic A, Yang Z, Ariotti N, Norwood SJ, Collins BM, Parton RG and Teasdale RD (2016) Parkinson Disease-linked Vps35 R524W Mutation Impairs the Endosomal Association of Retromer and Induces alpha-Synuclein Aggregation. J Biol Chem 291, 18283–18298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine SN, Zheng D, Sabbagh JJ et al. (2016) DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J 35, 1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW and Schapira AH (2012) Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol 72, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorenberg EL and Chandra SS (2017) The Role of Co-chaperones in Synaptic Proteostasis and Neurodegenerative Disease. Front Neurosci 11, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar S, Yuan P, Wu Y, Schrag M, Paradise S, Grutzendler J, De Camilli P and Ferguson SM (2015) Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc Natl Acad Sci U S A 112, E3699–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten-Harrison B, Polydoro M, Morimoto-Tomita M et al. (2010) alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A 107, 19573–19578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson EK, Trinh J, Guella I et al. (2015) DNAJC13 genetic variants in parkinsonism. Mov Disord 30, 273–278. [DOI] [PubMed] [Google Scholar]
- Haft CR, de la Luz Sierra M, Bafford R, Lesniak MA, Barr VA and Taylor SI (2000) Human orthologs of yeast vacuolar protein sorting proteins Vps26, 29, and 35: assembly into multimeric complexes. Mol Biol Cell 11, 4105–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett PJ, Huebecker M, Brekk OR, Moloney EB, Rocha EM, Priestman DA, Platt FM and Isacson O (2018) Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol Aging 67, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M et al. (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. [DOI] [PubMed] [Google Scholar]
- Hernandez D, Torres CA, Setlik W et al. (2012) Regulation of presynaptic neurotransmission by macroautophagy. Neuron 74, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE and Reese TS (1973) Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol 57, 315–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockey LN, Kilpatrick BS, Eden ER et al. (2015) Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J Cell Sci 128, 232–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehn MM and Yahr MD (1967) Parkinsonism: onset, progression and mortality. Neurology 17, 427–442. [DOI] [PubMed] [Google Scholar]
- Holmes BB, DeVos SL, Kfoury N et al. (2013) Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A 110, E3138–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt M, Cooke A, Wu MM and Lagnado L (2003) Bulk membrane retrieval in the synaptic terminal of retinal bipolar cells. J Neurosci 23, 1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Wang B, Li X, Fu C, Wang C and Kang X (2019) alpha-Synuclein: A Multifunctional Player in Exocytosis, Endocytosis, and Vesicle Recycling. Front Neurosci 13, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihse E, Yamakado H, van Wijk XM, Lawrence R, Esko JD and Masliah E (2017) Cellular internalization of alpha-synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci Rep 7, 9008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbriani P, Schirinzi T, Meringolo M, Mercuri NB and Pisani A (2018) Centrality of Early Synaptopathy in Parkinson’s Disease. Front Neurol 9, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoshita T, Arano T, Hosaka Y et al. (2017) Vps35 in cooperation with LRRK2 regulates synaptic vesicle endocytosis through the endosomal pathway in Drosophila. Hum Mol Genet 26, 2933–2948. [DOI] [PubMed] [Google Scholar]
- Ishizu N, Yui D, Hebisawa A et al. (2016) Impaired striatal dopamine release in homozygous Vps35 D620N knock-in mice. Hum Mol Genet 25, 4507–4517. [DOI] [PubMed] [Google Scholar]
- Islam MS, Nolte H, Jacob W et al. (2016) Human R1441C LRRK2 regulates the synaptic vesicle proteome and phosphoproteome in a Drosophila model of Parkinson’s disease. Hum Mol Genet 25, 5365–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinn S, Drolet RE, Cramer PE et al. (2017) TMEM175 deficiency impairs lysosomal and mitochondrial function and increases alpha-synuclein aggregation. Proc Natl Acad Sci U S A 114, 2389–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jyothi HJ, Vidyadhara DJ, Mahadevan A, Philip M, Parmar SK, Manohari SG, Shankar SK, Raju TR and Alladi PA (2015) Aging causes morphological alterations in astrocytes and microglia in human substantia nigra pars compacta. Neurobiol Aging 36, 3321–3333. [DOI] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S and Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205, 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann O and Kovacs R (2007) Mitochondria and neuronal activity. Am J Physiol Cell Physiol 292, C641–657. [DOI] [PubMed] [Google Scholar]
- Karpowicz RJ Jr., Haney CM, Mihaila TS, Sandler RM, Petersson EJ and Lee VM (2017) Selective imaging of internalized proteopathic alpha-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J Biol Chem 292, 13482–13497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata T, Kamada Y, Kabeya Y, Sekito T and Ohsumi Y (2008) Organization of the pre-autophagosomal structure responsible for autophagosome formation. Mol Biol Cell 19, 2039–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kett LR and Dauer WT (2012) Leucine-rich repeat kinase 2 for beginners: six key questions. Cold Spring Harb Perspect Med 2, a009407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kett LR and Dauer WT (2016) Endolysosomal dysfunction in Parkinson’s disease: Recent developments and future challenges. Mov Disord 31, 1433–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kett LR, Stiller B, Bernath MM et al. (2015) alpha-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2. J Neurosci 35, 5724–5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisos H, Ben-Gedalya T and Sharon R (2014) The clathrin-dependent localization of dopamine transporter to surface membranes is affected by alpha-synuclein. J Mol Neurosci 52, 167–176. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y and Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. [DOI] [PubMed] [Google Scholar]
- Klumperman J and Raposo G (2014) The complex ultrastructure of the endolysosomal system. Cold Spring Harb Perspect Biol 6, a016857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T et al. (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM and Bartus RT (2013) Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136, 2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koroglu C, Baysal L, Cetinkaya M, Karasoy H and Tolun A (2013) DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat Disord 19, 320–324. [DOI] [PubMed] [Google Scholar]
- Kramer ML and Schulz-Schaeffer WJ (2007) Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci 27, 1405–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs CE, Karkheiran S, Powell JC et al. (2013) The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat 34, 1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N, Leonzino M, Hancock-Cerutti W, Horenkamp FA, Li P, Lees JA, Wheeler H, Reinisch KM and De Camilli P (2018) VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J Cell Biol 217, 3625–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara T, Inoue K, D’Agati VD, Fujimoto T, Eguchi T, Saha S, Wolozin B, Iwatsubo T and Abeliovich A (2016) LRRK2 and RAB7L1 coordinately regulate axonal morphology and lysosome integrity in diverse cellular contexts. Sci Rep 6, 29945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YC, Kondapalli C, Lehneck R et al. (2015) Phosphoproteomic screening identifies Rab GTPases as novel downstream targets of PINK1. EMBO J 34, 2840–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautenschlager J, Kaminski CF and Kaminski Schierle GS (2017) alpha-Synuclein - Regulator of Exocytosis, Endocytosis, or Both? Trends Cell Biol 27, 468–479. [DOI] [PubMed] [Google Scholar]
- Lee DW, Zhao X, Yim YI, Eisenberg E and Greene LE (2008a) Essential role of cyclin-G-associated kinase (Auxilin-2) in developing and mature mice. Mol Biol Cell 19, 2766–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR and Lee SJ (2008b) Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol 40, 1835–1849. [DOI] [PubMed] [Google Scholar]
- Lee JG, Takahama S, Zhang G, Tomarev SI and Ye Y (2016) Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat Cell Biol 18, 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon SK (2001) Clathrin uncoating: Auxilin comes to life. Curr Biol 11, R49–52. [DOI] [PubMed] [Google Scholar]
- Lesage S, Drouet V, Majounie E et al. (2016) Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am J Hum Genet 98, 500–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu W, Oo TF et al. (2009) Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat Neurosci 12, 826–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limousin P, Krack P, Pollak P, Benazzouz A, Ardouin C, Hoffmann D and Benabid AL (1998) Electrical stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med 339, 1105–1111. [DOI] [PubMed] [Google Scholar]
- Lin G, Lee PT, Chen K, Mao D, Tan KL, Zuo Z, Lin WW, Wang L and Bellen HJ (2018) Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to alpha-Synuclein Gain. Cell Metab 28, 605–618 e606. [DOI] [PubMed] [Google Scholar]
- Lin X, Parisiadou L, Gu XL et al. (2009) Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron 64, 807–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhart R, Wong SA, Cao J et al. (2014) Vacuolar protein sorting 35 (Vps35) rescues locomotor deficits and shortened lifespan in Drosophila expressing a Parkinson’s disease mutant of Leucine-Rich Repeat Kinase 2 (LRRK2). Mol Neurodegener 9, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Boot B, Locascio JJ et al. (2016) Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann Neurol 80, 674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan T, Bendor J, Toupin C, Thorn K and Edwards RH (2017) alpha-Synuclein promotes dilation of the exocytotic fusion pore. Nat Neurosci 20, 681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ and Lee VM (2012) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzio JP, Pryor PR and Bright NA (2007) Lysosomes: fusion and function. Nat Rev Mol Cell Biol 8, 622–632. [DOI] [PubMed] [Google Scholar]
- Lynch-Day MA, Mao K, Wang K, Zhao M and Klionsky DJ (2012) The role of autophagy in Parkinson’s disease. Cold Spring Harb Perspect Med 2, a009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma SY, Roytta M, Rinne JO, Collan Y and Rinne UK (1997) Correlation between neuromorphometry in the substantia nigra and clinical features in Parkinson’s disease using disector counts. J Neurol Sci 151, 83–87. [DOI] [PubMed] [Google Scholar]
- Maas JW, Yang J and Edwards RH (2017) Endogenous Leucine-Rich Repeat Kinase 2 Slows Synaptic Vesicle Recycling in Striatal Neurons. Front Synaptic Neurosci 9, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T et al. (2013) RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 77, 425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Twelvetrees AE, Moughamian AJ and Holzbaur EL (2014) Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 84, 292–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik BR, Godena VK and Whitworth AJ (2015) VPS35 pathogenic mutations confer no dominant toxicity but partial loss of function in Drosophila and genetically interact with parkin. Hum Mol Genet 24, 6106–6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani M, Lee SY, Lucast L, Cremona O, Di Paolo G, De Camilli P and Ryan TA (2007) The dual phosphatase activity of synaptojanin1 is required for both efficient synaptic vesicle endocytosis and reavailability at nerve terminals. Neuron 56, 1004–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni C, Mamais A, Dihanich S et al. (2013) Inhibition of LRRK2 kinase activity stimulates macroautophagy. Biochim Biophys Acta 1833, 2900–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Ou MT, Karuppagounder SS et al. (2016) Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Kaushik S et al. (2008) Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest 118, 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A, Lectez B, Ramirez J, Popp O, Sutherland JD, Urbe S, Dittmar G, Clague MJ and Mayor U (2017) Quantitative proteomic analysis of Parkin substrates in Drosophila neurons. Mol Neurodegener 12, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata IF, Leverenz JB, Weintraub D et al. (2016) GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov Disord 31, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matikainen-Ankney BA, Kezunovic N, Mesias RE, Tian Y, Williams FM, Huntley GW and Benson DL (2016) Altered Development of Synapse Structure and Function in Striatum Caused by Parkinson’s Disease-Linked LRRK2-G2019S Mutation. J Neurosci 36, 7128–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta S, Van Kolen K, da Cunha R et al. (2012) LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron 75, 1008–1021. [DOI] [PubMed] [Google Scholar]
- McPherson PS, Garcia EP, Slepnev VI et al. (1996) A presynaptic inositol-5-phosphatase. Nature 379, 353–357. [DOI] [PubMed] [Google Scholar]
- Middleton ER and Rhoades E (2010) Effects of curvature and composition on alpha-synuclein binding to lipid vesicles. Biophys J 99, 2279–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milosevic I, Giovedi S, Lou X et al. (2011) Recruitment of endophilin to clathrin-coated pit necks is required for efficient vesicle uncoating after fission. Neuron 72, 587–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir R, Tonelli F, Lis P et al. (2018) The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem J 475, 1861–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N (2007) Autophagy: process and function. Genes Dev 21, 2861–2873. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M and Ohsumi Y (1998) A protein conjugation system essential for autophagy. Nature 395, 395–398. [DOI] [PubMed] [Google Scholar]
- Morgan JR, Prasad K, Hao W, Augustine GJ and Lafer EM (2000) A conserved clathrin assembly motif essential for synaptic vesicle endocytosis. J Neurosci 20, 8667–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H, Kondo T, Yokochi M, Matsumine H, Nakagawa-Hattori Y, Miyake T, Suda K and Mizuno Y (1998) Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology 51, 890–892. [DOI] [PubMed] [Google Scholar]
- Munsie LN, Milnerwood AJ, Seibler P et al. (2015) Retromer-dependent neurotransmitter receptor trafficking to synapses is altered by the Parkinson’s disease VPS35 mutation p.D620N. Hum Mol Genet 24, 1691–1703. [DOI] [PubMed] [Google Scholar]
- Murdoch JD, Rostosky CM, Gowrisankaran S et al. (2016) Endophilin-A Deficiency Induces the Foxo3a-Fbxo32 Network in the Brain and Causes Dysregulation of Autophagy and the Ubiquitin-Proteasome System. Cell Rep 17, 1071–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Pankratz N, Lill CM et al. (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46, 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF and Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR and Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA and Edwards RH (2010) Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65, 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M and Krainc D (2018) LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease. Proc Natl Acad Sci U S A 115, 5576–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TN, Padman BS and Lazarou M (2016) Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol 26, 733–744. [DOI] [PubMed] [Google Scholar]
- Nikoletopoulou V and Tavernarakis N (2018) Regulation and Roles of Autophagy at Synapses. Trends Cell Biol 28, 646–661. [DOI] [PubMed] [Google Scholar]
- Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19, 983–997. [DOI] [PubMed] [Google Scholar]
- Nixon RA and Cataldo AM (1995) The endosomal-lysosomal system of neurons: new roles. Trends Neurosci 18, 489–496. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A and Cuervo AM (2005) Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 64, 113–122. [DOI] [PubMed] [Google Scholar]
- Norris A, Tammineni P, Wang S, Gerdes J, Murr A, Kwan KY, Cai Q and Grant BD (2017) SNX-1 and RME-8 oppose the assembly of HGRS-1/ESCRT-0 degradative microdomains on endosomes. Proc Natl Acad Sci U S A 114, E307–E316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurrish S (2014) Dense core vesicle release: controlling the where as well as the when. Genetics 196, 601–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsumi Y (2014) Historical landmarks of autophagy research. Cell Res 24, 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olgiati S, De Rosa A, Quadri M et al. (2014) PARK20 caused by SYNJ1 homozygous Arg258Gln mutation in a new Italian family. Neurogenetics 15, 183–188. [DOI] [PubMed] [Google Scholar]
- Olgiati S, Quadri M, Fang M et al. (2016) DNAJC6 Mutations Associated With Early-Onset Parkinson’s Disease. Ann Neurol 79, 244–256. [DOI] [PubMed] [Google Scholar]
- Orenstein SJ, Kuo SH, Tasset I et al. (2013) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16, 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW et al. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600. [DOI] [PubMed] [Google Scholar]
- Pan PY, Li X, Wang J et al. (2017) Parkinson’s Disease-Associated LRRK2 Hyperactive Kinase Mutant Disrupts Synaptic Vesicle Trafficking in Ventral Midbrain Neurons. J Neurosci 37, 11366–11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou L, Yu J, Sgobio C et al. (2014) LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat Neurosci 17, 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Mehta P, Cooper AA et al. (2011) Pathogenic effects of novel mutations in the P-type ATPase ATP13A2 (PARK9) causing Kufor-Rakeb syndrome, a form of early-onset parkinsonism. Hum Mutat 32, 956–964. [DOI] [PubMed] [Google Scholar]
- Parkinson J (2002) An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci 14, 223–236; discussion 222. [DOI] [PubMed] [Google Scholar]
- Patel D and Witt SN (2018) Sorting Out the Role of alpha-Synuclein in Retromer-Mediated Endosomal Protein Sorting. J Exp Neurosci 12, 1179069518796215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM and Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag AE and Lang AE (2017) Parkinson disease. Nat Rev Dis Primers 3, 17013. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E et al. (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Tieu K, Perier C and Vila M (2004) MPTP as a mitochondrial neurotoxic model of Parkinson’s disease. J Bioenerg Biomembr 36, 375–379. [DOI] [PubMed] [Google Scholar]
- Quadri M, Fang M, Picillo M et al. (2013) Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat 34, 1208–1215. [DOI] [PubMed] [Google Scholar]
- Ramirez A, Heimbach A, Grundemann J et al. (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 38, 1184–1191. [DOI] [PubMed] [Google Scholar]
- Repnik U, Cesen MH and Turk B (2013) The endolysosomal system in cell death and survival. Cold Spring Harb Perspect Biol 5, a008755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter F, Fleming SM, Watson M et al. (2014) A GCase chaperone improves motor function in a mouse model of synucleinopathy. Neurotherapeutics 11, 840–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, Heutink P and Shulman JM (2017) Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140, 3191–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EM, Smith GA, Park E, Cao H, Brown E, Hallett P and Isacson O (2015) Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann Clin Transl Neurol 2, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez L, Marano MM and Tandon A (2018) Import and Export of Misfolded alpha-Synuclein. Front Neurosci 12, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron I and Horowitz M (2005) ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet 14, 2387–2398. [DOI] [PubMed] [Google Scholar]
- Rong Y, Liu M, Ma L et al. (2012) Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat Cell Biol 14, 924–934. [DOI] [PubMed] [Google Scholar]
- Rosendale M, Jullie D, Choquet D and Perrais D (2017) Spatial and Temporal Regulation of Receptor Endocytosis in Neuronal Dendrites Revealed by Imaging of Single Vesicle Formation. Cell Rep 18, 1840–1847. [DOI] [PubMed] [Google Scholar]
- Saez-Atienzar S, Bonet-Ponce L, Blesa JR, Romero FJ, Murphy MP, Jordan J and Galindo MF (2014) The LRRK2 inhibitor GSK2578215A induces protective autophagy in SH-SY5Y cells: involvement of Drp-1-mediated mitochondrial fission and mitochondrial-derived ROS signaling. Cell Death Dis 5, e1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saftig P and Klumperman J (2009) Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 10, 623–635. [DOI] [PubMed] [Google Scholar]
- Saheki Y and De Camilli P (2012) Synaptic vesicle endocytosis. Cold Spring Harb Perspect Biol 4, a005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardi SP, Viel C, Clarke J et al. (2017) Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc Natl Acad Sci U S A 114, 2699–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake W, Nakabayashi Y, Mizuta I et al. (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 41, 1303–1307. [DOI] [PubMed] [Google Scholar]
- Schapansky J, Khasnavis S, DeAndrade MP et al. (2018) Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble alpha-synuclein in neurons. Neurobiol Dis 111, 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schormair B, Kemlink D, Mollenhauer B et al. (2018) Diagnostic exome sequencing in early-onset Parkinson’s disease confirms VPS13C as a rare cause of autosomal-recessive Parkinson’s disease. Clin Genet 93, 603–612. [DOI] [PubMed] [Google Scholar]
- Schreij AM, Chaineau M, Ruan W, Lin S, Barker PA, Fon EA and McPherson PS (2015) LRRK2 localizes to endosomes and interacts with clathrin-light chains to limit Rac1 activation. EMBO Rep 16, 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN, McCaffery JM and Emr SD (1998) A membrane coat complex essential for endosome-to-Golgi retrograde transport in yeast. J Cell Biol 142, 665–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Pirruccello M and De Camilli P (2012) SnapShot: membrane curvature sensors and generators. Cell 150, 1300, 1300 e1301–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava AN, Redeker V, Fritz N et al. (2015) alpha-synuclein assemblies sequester neuronal alpha3-Na+/K+-ATPase and impair Na+ gradient. EMBO J 34, 2408–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky E and Lopez G (2012) The link between the GBA gene and parkinsonism. Lancet Neurol 11, 986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Sanchez J, Schulte C, Bras JM et al. (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41, 1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J et al. (2003) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302, 841. [DOI] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM and Ross CA (2006) Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 9, 1231–1233. [DOI] [PubMed] [Google Scholar]
- Song L, He Y, Ou J, Zhao Y, Li R, Cheng J, Lin CH and Ho MS (2017) Auxilin Underlies Progressive Locomotor Deficits and Dopaminergic Neuron Loss in a Drosophila Model of Parkinson’s Disease. Cell Rep 18, 1132–1143. [DOI] [PubMed] [Google Scholar]
- Song P, Trajkovic K, Tsunemi T and Krainc D (2016) Parkin Modulates Endosomal Organization and Function of the Endo-Lysosomal Pathway. J Neurosci 36, 2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W and Zinsmaier KE (2003) Endophilin and synaptojanin hook up to promote synaptic vesicle endocytosis. Neuron 40, 665–667. [DOI] [PubMed] [Google Scholar]
- Soukup SF, Kuenen S, Vanhauwaert R et al. (2016) A LRRK2-Dependent EndophilinA Phosphoswitch Is Critical for Macroautophagy at Presynaptic Terminals. Neuron 92, 829–844. [DOI] [PubMed] [Google Scholar]
- Soukup SF and Verstreken P (2017) EndoA/Endophilin-A creates docking stations for autophagic proteins at synapses. Autophagy 13, 971–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M and Goedert M (1998) alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A 95, 6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R and Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Spinelli KJ, Taylor JK, Osterberg VR, Churchill MJ, Pollock E, Moore C, Meshul CK and Unni VK (2014) Presynaptic alpha-synuclein aggregation in a mouse model of Parkinson’s disease. J Neurosci 34, 2037–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger M, Diez F, Dhekne HS et al. (2017) Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger M, Tonelli F, Ito G et al. (2016) Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura A, McLelland GL, Fon EA and McBride HM (2014) A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J 33, 2142–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh SN, Chavalmane AK, Dj V, Yarreiphang H, Rai S, Paul A, Clement JP, Alladi PA and Manjithaya R (2017) A novel autophagy modulator 6-Bio ameliorates SNCA/alpha-synuclein toxicity. Autophagy 13, 1221–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh SN, Chavalmane AK, Pillai M et al. (2018a) Modulation of Autophagy by a Small Molecule Inverse Agonist of ERRalpha Is Neuroprotective. Front Mol Neurosci 11, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh SN, Verma V, Sateesh S, Clement JP and Manjithaya R (2018b) Neurodegenerative diseases: model organisms, pathology and autophagy. J Genet 97, 679–701. [PubMed] [Google Scholar]
- Surmeier DJ, Obeso JA and Halliday GM (2017) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliaferro P, Kareva T, Oo TF, Yarygina O, Kholodilov N and Burke RE (2015) An early axonopathy in a hLRRK2(R1441G) transgenic model of Parkinson disease. Neurobiol Dis 82, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi YV, Liu J, Ruan J, Pacheco J, Zhang X, Abbasi J, Keutzer J, Mistry PK and Chandra SS (2017) Glucosylsphingosine Promotes alpha-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. J Neurosci 37, 9617–9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang FL, Erion JR, Tian Y, Liu W, Yin DM, Ye J, Tang B, Mei L and Xiong WC (2015a) VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for alpha-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J Neurosci 35, 10613–10628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang FL, Liu W, Hu JX, Erion JR, Ye J, Mei L and Xiong WC (2015b) VPS35 Deficiency or Mutation Causes Dopaminergic Neuronal Loss by Impairing Mitochondrial Fusion and Function. Cell Rep 12, 1631–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Hamada K and Krainc D (2014) ATP13A2/PARK9 regulates secretion of exosomes and alpha-synuclein. J Neurosci 34, 15281–15287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnozzi AN and Pratico D (2018) Endosomal sorting and trafficking, the retromer complex and neurodegeneration. Mol Psychiatry [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V et al. (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. [DOI] [PubMed] [Google Scholar]
- Vanhauwaert R, Kuenen S, Masius R et al. (2017) The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J 36, 1392–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE and Chandra SS (2014) Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci 34, 9364–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas KJ, Schrod N, Davis T, Fernandez-Busnadiego R, Taguchi YV, Laugks U, Lucic V and Chandra SS (2017) Synucleins Have Multiple Effects on Presynaptic Architecture. Cell Rep 18, 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidyadhara DJ, Sasidharan A, Kutty BM, Raju TR and Alladi PA (2019) Admixing MPTP-resistant and MPTP-vulnerable mice enhances striatal field potentials and calbindin-D28K expression to avert motor behaviour deficits. Behav Brain Res 360, 216–227. [DOI] [PubMed] [Google Scholar]
- Vidyadhara DJ, Yarreiphang H, Abhilash PL, Raju TR and Alladi PA (2016) Differential expression of calbindin in nigral dopaminergic neurons in two mice strains with differential susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Chem Neuroanat 76, 82–89. [DOI] [PubMed] [Google Scholar]
- Vidyadhara DJ, Yarreiphang H, Raju TR and Alladi PA (2017) Admixing of MPTP-Resistant and Susceptible Mice Strains Augments Nigrostriatal Neuronal Correlates to Resist MPTP-Induced Neurodegeneration. Mol Neurobiol 54, 6148–6162. [DOI] [PubMed] [Google Scholar]
- Vilarino-Guell C, Rajput A, Milnerwood AJ et al. (2014) DNAJC13 mutations in Parkinson disease. Hum Mol Genet 23, 1794–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilarino-Guell C, Wider C, Ross OA et al. (2011) VPS35 mutations in Parkinson disease. Am J Hum Genet 89, 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Telpoukhovskaia MA, Bahr BA, Chen X and Gan L (2018) Endo-lysosomal dysfunction: a converging mechanism in neurodegenerative diseases. Curr Opin Neurobiol 48, 52–58. [DOI] [PubMed] [Google Scholar]
- Wang W, Wang X, Fujioka H, Hoppel C, Whone AL, Caldwell MA, Cullen PJ, Liu J and Zhu X (2016) Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat Med 22, 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Mamer LE, Raychaudhuri S et al. (2018) Synaptojanin and Endophilin Mediate Neck Formation during Ultrafast Endocytosis. Neuron 98, 1184–1197 e1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson GS and Leverenz JB (2010) Profile of cognitive impairment in Parkinson’s disease. Brain Pathol 20, 640–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL and Dawson TM (2005) Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A 102, 16842–16847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal CH and Chandra SS (2013) Monomeric synucleins generate membrane curvature. J Biol Chem 288, 1829–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wider C, Skipper L, Solida A, Brown L, Farrer M, Dickson D, Wszolek ZK and Vingerhoets FJ (2008) Autosomal dominant dopa-responsive parkinsonism in a multigenerational Swiss family. Parkinsonism Relat Disord 14, 465–470. [DOI] [PubMed] [Google Scholar]
- Williams ET, Chen X and Moore DJ (2017) VPS35, the Retromer Complex and Parkinson’s Disease. J Parkinsons Dis 7, 219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams ET, Glauser L, Tsika E, Jiang H, Islam S and Moore DJ (2018) Parkin mediates the ubiquitination of VPS35 and modulates retromer-dependent endosomal sorting. Hum Mol Genet 27, 3189–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow AR, Chen CW, Corrochano S et al. (2010) alpha-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol 190, 1023–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollert T, Yang D, Ren X, Lee HH, Im YJ and Hurley JH (2009) The ESCRT machinery at a glance. J Cell Sci 122, 2163–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC and Holzbaur EL (2015) Autophagosome dynamics in neurodegeneration at a glance. J Cell Sci 128, 1259–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W and Wu LG (2007) Rapid bulk endocytosis and its kinetics of fission pore closure at a central synapse. Proc Natl Acad Sci U S A 104, 10234–10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xilouri M, Vogiatzi T, Vekrellis K and Stefanis L (2008) alpha-synuclein degradation by autophagic pathways: a potential key to Parkinson’s disease pathogenesis. Autophagy 4, 917–919. [DOI] [PubMed] [Google Scholar]
- Xu J, Wu XS, Sheng J et al. (2016) alpha-Synuclein Mutation Inhibits Endocytosis at Mammalian Central Nerve Terminals. J Neurosci 36, 4408–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Coleman M, Zhang L, Zheng X and Yue Z (2013) Autophagy in axonal and dendritic degeneration. Trends Neurosci 36, 418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim YI, Sun T, Wu LG, Raimondi A, De Camilli P, Eisenberg E and Greene LE (2010) Endocytosis and clathrin-uncoating defects at synapses of auxilin knockout mice. Proc Natl Acad Sci U S A 107, 4412–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavodszky E, Seaman MN, Moreau K, Jimenez-Sanchez M, Breusegem SY, Harbour ME and Rubinsztein DC (2014) Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat Commun 5, 3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Perera G, Takahashi-Fujigasaki J et al. (2018) Reduced LRRK2 in association with retromer dysfunction in post-mortem brain tissue from LRRK2 mutation carriers. Brain 141, 486–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P et al. (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. [DOI] [PubMed] [Google Scholar]
- Zunke F, Moise AC, Belur NR et al. (2018) Reversible Conformational Conversion of alpha-Synuclein into Toxic Assemblies by Glucosylceramide. Neuron 97, 92–107 e110. [DOI] [PMC free article] [PubMed] [Google Scholar]