

Abstract
Induction of the transcription factor Irf8 in the common dendritic cell progenitor (CDP) is required for classical type 1 dendritic cell (cDC1) fate specification, but the mechanisms controlling this induction are unclear. Here we identified Irf8 enhancers via chromatin profiling of DCs and used CRISPR/Cas9 genome editing to assess their roles in Irf8 regulation. An enhancer 32 kilobases downstream of the Irf8 transcriptional start site (+32 kb Irf8) that was active in mature cDC1s was required for the development of this lineage, but not for its specification. Instead, a +41 kb Irf8 enhancer previously thought to be active only in plasmacytoid DCs was found to also be transiently accessible in cDC1 progenitors, and deleting this enhancer prevented the induction of Irf8 in CDPs and abolished cDC1 specification. Thus, cryptic activation of the +41 kb Irf8 enhancer in DC progenitors is responsible for cDC1 fate specification.
Introduction
The diversification of immune cells relies upon lineage-determining transcription factors that commit multipotent progenitors to a single fate1,2. While early studies proposed that stochastic variations in the levels of these factors determined the eventual fate of progenitors3, more recent work has instead suggested that the expression of individual factors is actively induced in order to specify a particular lineage4. However, the precise mechanisms responsible for such induction remain unclear.
Gene expression is primarily controlled by cis-acting enhancers bound by transcription factors5. Certain genes are regulated entirely by a single enhancer6,7, while others, including many genes important for development, contain multiple potentially redundant enhancers as a safeguard for continued expression8,9. Further, enhancer usage in individual genes dynamically changes as progenitors mature, possibly indicating the actions of distinct transcriptional networks throughout the developmental progression of a cell type10. Analyzing the enhancers that regulate expression of lineage-determining transcription factors at developmental branch points could therefore identify the transcriptional mechanisms controlling fate choice.
Dendritic cells (DCs) are a group of immune cells critical for innate and adaptive immune responses that include ‘classical’ DCs (cDCs)11 and plasmacytoid DCs (pDCs)12. cDCs are comprised of two functionally distinct lineages called cDC1 and cDC213. cDC1s are critical for priming CD8 T cells during antiviral and antitumor immune responses14, as well as for effective responses to checkpoint blockade therapy15,16. cDC1s are also the most promising substrates for cell-based cancer vaccines17, so understanding their development is paramount.
DCs are derived from hematopoietic precursors in the bone marrow (BM), the earliest of which is the monocyte/DC progenitor (MDP)18. The MDP gives rise to a common DC progenitor (CDP)19,20, which produces distinct clonogenic progenitors, the pre-cDC1 and pre-cDC221,22. Several transcription factors regulate development of the cDC1 lineage, including Irf8, Batf3, Nfil3, and Id214,23–25. Although cDC1s can be generated in mice deficient in Nfil3, Batf3, or Id2 under inflammatory conditions26,27, Irf8−/− mice have an absolute defect in both pre-cDC1 specification and cDC1 development that cannot be rescued by such conditions. Further, the Irf8 gene contains a super-enhancer in cDC1s, and Irf8 overexpression biases bone marrow progenitors towards cDC1 output21. These properties together suggest that Irf8 is the lineage-determining transcription factor for cDC1 fate. Understanding the enhancers that regulate Irf8 could therefore provide insight into how cDC1 fate specification from its multipotent progenitor, the CDP, is achieved.
Our previous work identified two distinct enhancers within the Irf8 super-enhancer located at +32 kb and +41 kb relative to the Irf8 transcriptional start site (TSS)21. Using an integrating retroviral reporter, we demonstrated that the +32 kb Irf8 enhancer was selectively active in cDC1s, and that the +41 kb Irf8 enhancer was selectively active in pDCs. The +32 kb Irf8 enhancer contained several AP1-IRF composite elements (AICEs) that bound IRF8 and BATF3 in cDC1s by ChIP-seq, suggesting this enhancer might support Irf8 expression through autoactivation. The +41 kb Irf8 enhancer contained several E box motifs, suggesting that E proteins such as E2-2, the lineage-determining transcription factor of pDCs28, might utilize this enhancer to drive Irf8 expression in pDCs. Finally, an Irf8 enhancer located at −50 kb was identified and analyzed using bacterial artificial chromosome (BAC) reporter transgenic mice, and was predicted to be required for Irf8 expression in MDPs29. However, until now the functional requirement of these Irf8 enhancers for in vivo DC development has remained untested.
In this study, we used chromatin profiling of DCs and CRISPR/Cas9 genome editing to identify and delete enhancers regulating Irf8 in mice. We found that the +32 kb Irf8 enhancer was required for normal and compensatory cDC1 development but not for pre-cDC1 specification. We also found that the −50 kb Irf8 enhancer was not required for Irf8 expression in MDPs, as was previously predicted, but rather regulated Irf8 levels selectively in monocyte/macrophage lineages. To find other enhancers regulating the cDC1 lineage, we performed ATAC-seq on DC progenitors. We surprisingly found that the +41 kb Irf8 enhancer became transiently accessible during the transition from the MDP to the pre-cDC1 before again closing in the mature cDC1. Deletion of the +41 kb Irf8 enhancer led to decreased Irf8 expression in pDCs, as was predicted, but also surprisingly prevented Irf8 induction in CDPs and resulted in the loss of both pre-cDC1 and mature cDC1 development. Consistently, Tcf3−/− DC progenitors, which lack E2A, had reduced cDC1 potential. Thus cryptic activation of the Irf8 +41 kb enhancer within CDPs is required for the induction of Irf8 and the subsequent specification of cDC1 fate.
Results
The +32 kb Irf8 enhancer is required for cDC1 development in vivo
The +32 kb Irf8 enhancer was identified by ChIP-seq as a 547 bp region that bound the transcription factors BATF3, IRF8, and p300 in cDC1s and that contained four AICEs in the 5’ portion of the region21 (Fig. 1a). Reporter analysis indicated that the first three AICEs within this region were sufficient to confer cDC1 specific reporter activity, and that this activity could be abrogated by mutating these three AICEs (Supplementary Fig. 1a,b). The four AICEs contained in the 5’ portion of the +32 kb Irf8 enhancer also accounted for the entire activity of the full length enhancer (Supplementary Fig. 1c). Further, using Flt3L-treated BM cultures of R26Cas9/+ transgenic mice, in which Cas9 is constitutively expressed in all cells under the control of the Rosa26 promoter30, we found that expression of an sgRNA directed to the central AICE in the 5’ half of the +32 kb Irf8 enhancer caused a reduction in cDC1 development that was as large as the reduction caused by an sgRNA directed at the Irf8 coding sequence itself (Supplementary Fig. 1d). We therefore generated mice with deletions in this region by injecting zygotes with Cas9 mRNA and three sgRNAs, two flanking the first three AICEs in the 5’ half of the enhancer and a third downstream of the fourth AICE (Fig. 1a and Supplementary Fig. 1e). This generated two lines of mice with deletions within the +32 kb Irf8 enhancer, one deleting 149 bp and eliminating 3 AICEs (Irf8 +32 5’−/− mice), and a second deleting 421 bp and eliminating all 4 AICEs (Irf8 +32−/− mice) (Supplementary Fig. 1f). Both lines showed severe reductions in cDC1 development in vivo (Supplementary Fig. 1g,h). Deletion of 3 AICEs reduced cDC1 development by 10-fold compared with wildtype mice, and deletion of all 4 AICEs eliminated all residual cDC1s (Supplementary Fig. 1h). All subsequent analyses used the more complete Irf8 +32−/− strain.
Figure 1. The +32 kb Irf8 enhancer is required for cDC1 development.
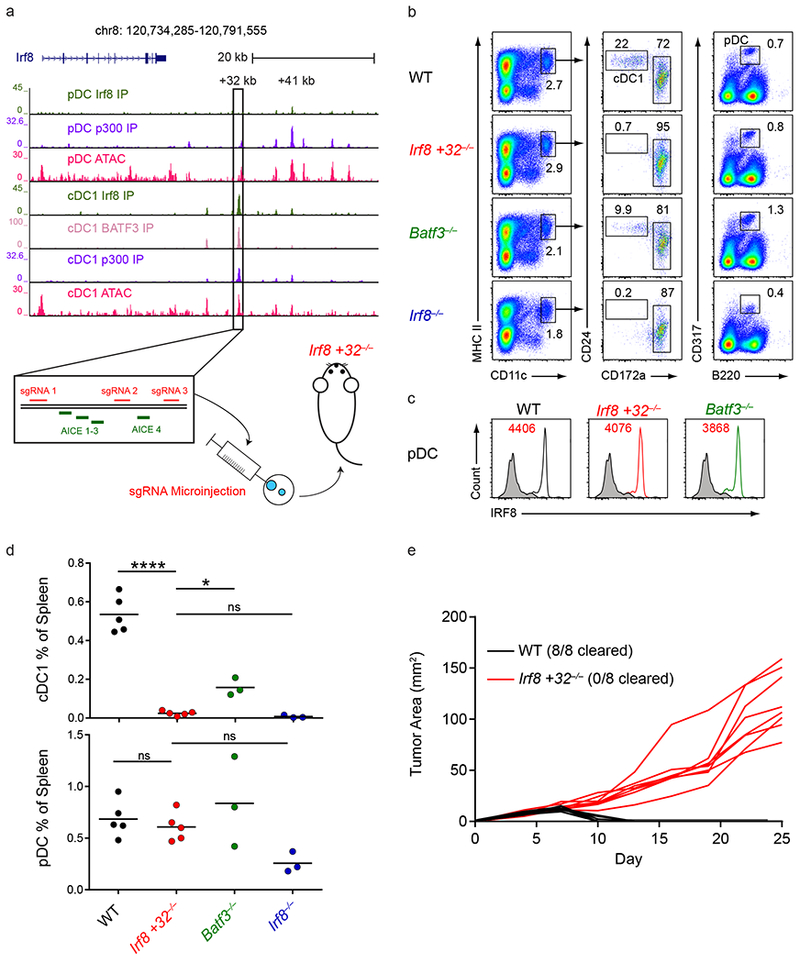
a, Normalized sequencing tracks of ChIP-seq with anti-p300, anti-BATF3, or anti-IRF8 antibodies or of ATAC-seq in the indicated populations. Boxed is the +32 kb Irf8 enhancer. Shown below is the +32 kb Irf8 enhancer with the AICE motifs and sgRNA target sequences depicted. Data are pooled from two independent experiments and the Immunological Genome Project Open Chromatin Regions (n = 1 biological replicate per population). b, Flow cytometry of live splenocytes from mice of the indicated genotypes was used to identify dendritic cell (DC) subsets. Numbers indicate the percent of cells in the indicated gates. Data are representative of four independent experiments with similar results (n = 5 mice for WT, n = 5 mice for Irf8 +32−/−, n = 3 mice for Batf3−/−, and n = 3 mice for Irf8−/−). c, Intracellular staining for IRF8 in splenic pDCs of mice of the indicated genotypes. Numbers indicate the mean fluorescence intensity (MFI) of IRF8 protein levels in pDCs. Data are representative of four independent experiments with similar results (n = 5 mice for WT, n = 5 mice for Irf8 +32−/−, n = 3 mice for Batf3−/−, and n = 3 mice for Irf8−/−). d, Statistical analysis of the frequency of splenic cDC1s and pDCs in mice of the indicated genotypes. Small horizontal lines indicate the mean. Data are pooled from four independent experiments (n = 5 mice for WT, n = 5 mice for Irf8 +32−/−, n = 3 mice for Batf3−/−, and n = 3 mice for Irf8−/−). e, Growth of 1969 regressor fibrosarcomas in WT and Irf8 +32 kb−/− mice. Data are pooled from two independent experiments (n = 8 mice for WT, n = 8 mice for Irf8 +32−/−). ns, not significant (P>0.05); *P<0.05; ****P<0.0001, ordinary one-way ANOVA (d).
The defect in splenic cDC1 development in Irf8 +32−/− mice was more complete than in Batf3−/− mice and as severe as in Irf8−/− mice (Fig. 1b,d). Irf8 +32−/− mice showed no defects in other lineages, confirming the selective activity of this enhancer in cDC1s as was predicted by our reporter assays. pDCs were normal in frequency and in intracellular IRF8 protein levels (Fig. 1c,d), as were monocytes, neutrophils, red pulp macrophages, B cells, and T cells (Supplementary Fig. 2a–c). cDC1s in Irf8 +32−/− mice were absent in all tissues, such as in the lung (Supplementary Fig. 3a,b). To test for a functional defect in Irf8 +32−/− mice, we analyzed a tumor rejection system that requires cDC1s (Fig. 1e). The regressor fibrosarcoma 1969, whose rejection relies on cross-presentation by cDC1s31, was rejected by all wildtype mice, but not by Irf8 +32−/− mice (Fig. 1e). These data indicated that the +32 kb Irf8 enhancer was absolutely required for cDC1 development and that its deletion generated a specific and functional defect restricted to this lineage.
Compensatory cDC1 development requires the +32 kb Irf8 enhancer
cDC1 development can occur in Batf3−/− mice due to compensation from BATF and BATF2, which can replace BATF3 for interactions with IRF826. Compensatory cDC1 development in Batf3−/− mice occurs in skin draining lymph nodes (SLNs) under inflammatory settings such as after bone marrow transplantation or after administration of IL-1226,27. We examined these settings to test if compensatory cDC1 development can occur in Irf8 +32−/− mice (Fig. 2). We found that cDC1s were present in SLNs from wildtype and Batf3−/− mice, but were completely absent in SLNs from Irf8 +32−/− mice (Fig. 2a,b). We further found that administration of IL-12 led to a complete restoration of splenic cDC1s in Batf3−/− mice, but not in Irf8 +32−/− mice (Fig. 2c,d). Finally, we tested for cDC1 compensation after BM transplantation27. We generated BM chimeras using wildtype, Irf8 +32−/−, or Batf3−/− donor BM transplanted into irradiated wildtype mice (Supplementary Fig. 3c,d). cDC1s developed in chimeras produced from wildtype BM, as expected, and from Batf3−/− BM, as reported27. However, cDC1s failed to develop in chimeras produced from Irf8 +32−/− BM. In summary, all compensatory cDC1 development failed to occur in mice lacking the +32 kb Irf8 enhancer.
Figure 2. The +32 kb Irf8 enhancer is required for compensatory cDC1 development but not for cDC1 specification.
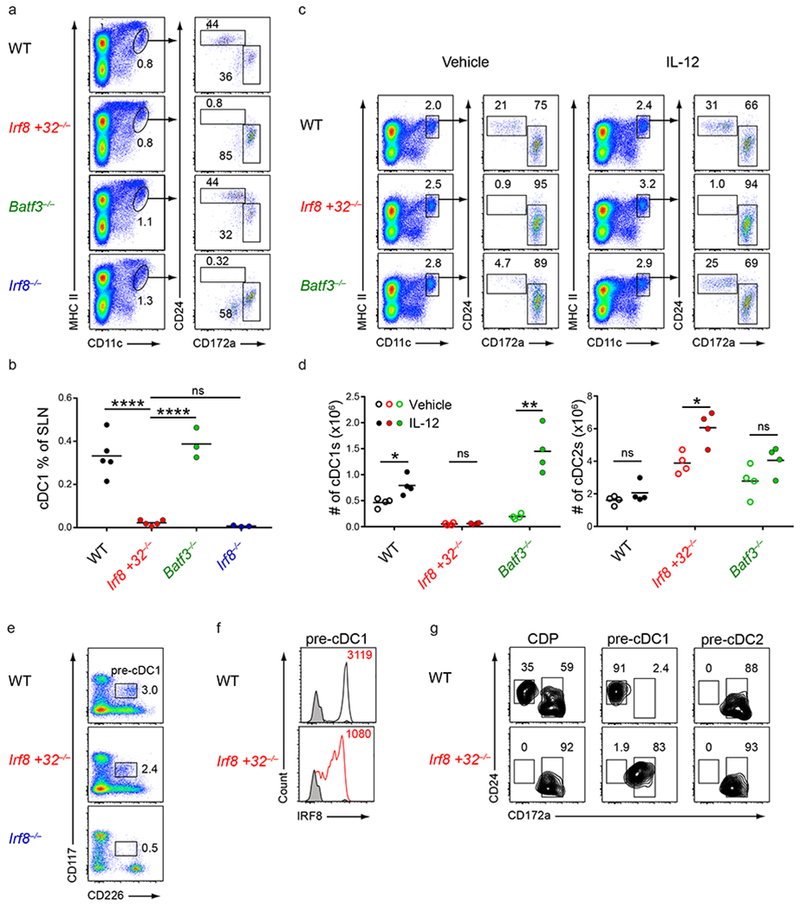
a,b, Flow cytometry of SLNs from mice of the indicated genotypes was used to identify DC subsets. Numbers indicate the percent of cells in the indicated gates (a). Statistical analysis of the frequency of cDC1s in SLNs (b). Small horizontal lines indicate the mean. Data are pooled from four independent experiments (n = 5 mice for WT, n = 5 mice for Irf8 +32−/−, n = 3 mice for Batf3−/−, and n = 3 mice for Irf8−/−). c,d, Flow cytometry of live splenocytes from mice of the indicated genotypes after administration of vehicle or IL-12 was used to identify DC subsets. Numbers indicate the percent of cells in the indicated gates (c). Statistical analysis of the absolute number of cDC1s and cDC2s in mice of the indicated genotypes treated with either vehicle or IL-12 (d). Small horizontal lines indicate the mean. Data are pooled from two independent experiments (n = 4 mice per group for WT, n = 4 mice per group for Irf8 +32−/−, n = 4 mice per group for Batf3−/−). e, Flow cytometry of Lin−CD135+ bone marrow cells from mice of the indicated genotypes was used to identify pre-cDC1s. Numbers indicate the percent of cells in the indicated gates. Data are representative of three independent experiments with similar results (n = 3 mice for WT, n = 3 mice for Irf8 +32−/−, and n = 3 mice for Irf8−/−). f, Intracellular staining for IRF8 within pre-cDC1s from mice of the indicated genotypes. Numbers indicate the MFI of IRF8 protein levels in the pre-cDC1. Black shaded histograms depict IRF8 protein levels from Irf8−/− mice. Data are representative of three independent experiments with similar results (n = 3 mice for WT, n = 3 mice for Irf8 +32−/−, and n = 3 mice for Irf8−/−). g, CDPs (Lin−CD117intCD135+CD115+), pre-cDC1s (Lin−CD117intCD135+CD226+), or pre-cDC2s (Lin−CD117loCD135+CD115+) from mice of the indicated genotypes were sorted and cultured for 5 days with Flt3L. Cells were then stained to identify DC subsets. Cells are pregated as CD11c+MHCII+ cells to identify DCs. Data are representative of three independent experiments with similar results (n = 3 mice for WT and n = 3 mice for Irf8 +32−/−). ns, not significant (P>0.05); *P<0.05, **P<0.01; and ****P<0.0001, ordinary one-way ANOVA (b) or unpaired two-tailed Student’s t-test (d).
While IL-12 injection did not restore cDC1s in Irf8 +32−/− mice, it did increase the numbers of cDC2s (Fig. 2c,d). This potentially resulted from specified pre-cDC1s failing to maintain high Irf8 levels in response to IL-12 and their subsequent diversion to the cDC2 fate, as occurs with pre-cDC1s from Batf3−/− mice21. We therefore sought to determine whether pre-cDC1 specification occurs in Irf8 +32−/− mice. Previous identification of the pre-cDC1 in BM had relied upon Zbtb46-GFP expression. However, we developed a method to identify the pre-cDC1 using CD226 expression in place of Zbtb46-GFP (Supplementary Fig. 4). CD226 was not expressed in CDPs, but became expressed in pre-cDC1s (Supplementary Fig. 4a,b). Like Zbtb46-GFP expression, CD226 expression indicated pre-cDC1 specification, as Flt3L culture of Lin−CD135+CD117intCD226+ BM cells, like Lin−CD135+CD117int Zbtb46-GFP+ BM cells, generated exclusively cDC1s (Supplementary Fig. 4c,d). Also, Lin−CD135+CD117intCD226+ cells developed in wildtype mice, but not in Irf8−/− mice known to lack pre-cDC1s (Fig. 2e)21. Using CD226 expression, we found that pre-cDC1s develop in Irf8 +32−/− mice in normal numbers but had reduced intracellular IRF8 protein levels (Fig. 2e,f, Supplementary Fig. 3e), similar to the findings in Batf3−/− mice. And in contrast to wildtype pre-cDC1s, pre-cDC1s from Irf8 +32−/− mice cultured in Flt3L become cDC2s (Fig. 2g). These cDC2s derived from Irf8 +32−/− pre-cDC1s were transcriptionally similar to cDC2s arising from wildtype pre-cDC2s and transcriptionally distinct from cDC1s arising from wildtype pre-cDC1s, including in the expression of key cDC2 transcription factors such as Irf4 (Supplementary Fig. 4e–g). Though the cDC2s derived from Irf8 +32−/− pre-cDC1s retained CD24 expression, we found that there were no transcriptional differences between CD24+ cDC2s and CD24− cDC2s (Supplementary Fig. 4f), indicating that the diverted cDC2s that developed from Irf8 +32−/− pre-cDC1s were bona fide cDC2s. This result also recapitulated the diversion of Batf3−/− pre-cDC1s to the cDC2 fate (Supplementary Fig. 4h). In summary, Irf8 +32−/− mice showed continued development of a specified pre-cDC1 progenitor in BM that fails to sustain Irf8 expression and diverts to the cDC2 lineage.
The −50 kb Irf8 enhancer regulates Irf8 expression in monocytes and macrophages
To identify enhancers regulating pre-cDC1 specification, we performed ChIP-seq of active histone marks in MDPs, CDPs, and pre-cDC1s. This analysis identified a region of active H3K27 acetylation at −50 kb relative to the Irf8 TSS that overlapped with a previously identified Irf8 enhancer (Fig. 3a)29. Analysis of BAC transgenic Irf8 reporter mice had suggested that this −50 kb region contains two PU.1 binding sites and was required for Irf8 expression in the MDP29. To test whether this enhancer regulated Irf8 induction and pre-cDC1 specification, we used CRISPR/Cas9 genome editing to generate mice with a 364 bp deletion that eliminated both of the PU.1 sites in this region (Irf8 −50−/− mice) (Supplementary Fig. 5a,b). Irf8 −50−/− mice had normal frequencies of cDCs, pDCs, neutrophils, B cells, T cells, and red pulp macrophages (Fig. 3b and Supplementary Fig. 5c), all of which had normal levels of intracellular IRF8 protein (Fig. 3c and Supplementary Fig. 5d). Frequencies and intracellular IRF8 protein levels of MDPs, CDPs, and pre-cDC1s were also normal in Irf8 −50−/− mice (Fig. 3d,e), in contrast to the predictions based on the BAC transgenic Irf8 reporter mice29.
Figure 3. The −50 kb Irf8 enhancer is not required for dendritic cell development.
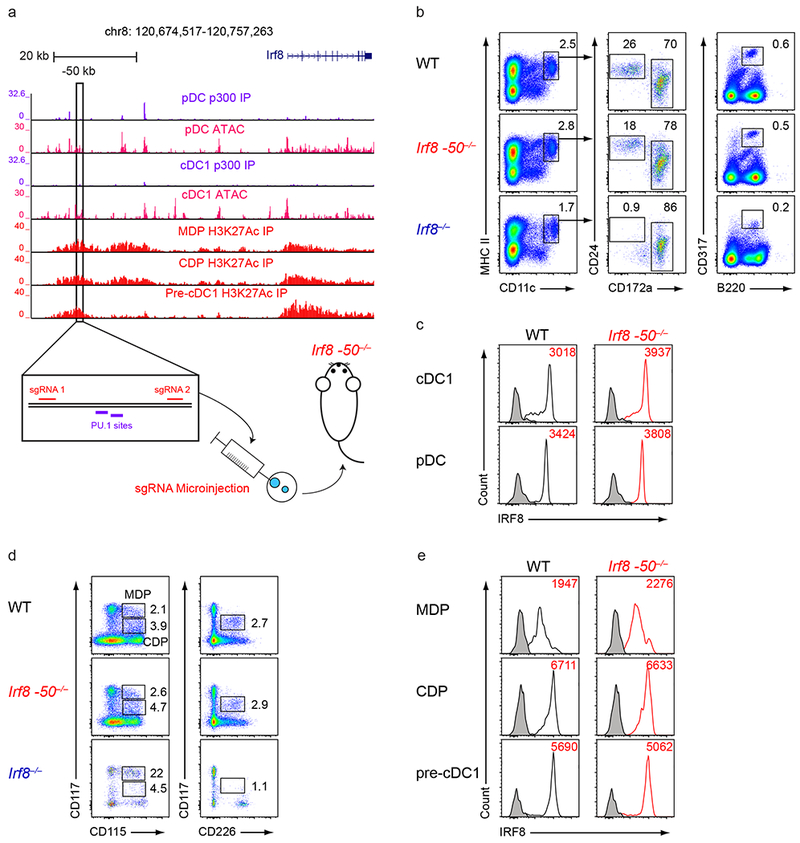
a, Normalized sequencing tracks of ChIP-seq with anti-p300 or anti-H3K27Ac antibodies or of ATAC-seq in the indicated populations. Boxed is the −50 kb Irf8 enhancer. Shown below is the −50 kb Irf8 enhancer with the PU.1 motifs and sgRNA target sequences indicated. Data are pooled from three independent experiments and the Immunological Genome Project Open Chromatin Regions (n = 1 biological replicate per population). b, Flow cytometry of live splenocytes from mice of the indicated genotypes was used to identify DC subsets. Numbers indicate the percent of cells in the indicated gates. Data are representative of four independent experiments with similar results (n = 5 mice for WT, n = 5 mice for Irf8 −50−/−, and n = 4 mice for Irf8−/−). c, Intracellular staining for IRF8 in splenic cDC1s and pDCs from mice of the indicated genotypes. Numbers indicate the MFI of IRF8 protein levels in cDC1s or pDCs as indicated. Data are representative of four independent experiments with similar results (n = 5 mice for WT, n = 5 mice for Irf8 −50−/−, and n = 4 mice for Irf8−/−). d, Flow cytometry of Lin−CD135+ bone marrow cells from mice of the indicated genotypes was used to identify DC progenitors. Numbers indicate the percent of cells in the indicated gates. Data are representative of three independent experiments with similar results (n = 3 mice for WT, n = 3 mice for Irf8 −50−/−, and n = 3 mice for Irf8−/−). e, Intracellular staining for IRF8 within MDPs, CDPs, and pre-cDC1s from mice of the indicated genotypes. Numbers indicate the MFI of IRF8 protein levels in the indicated populations. Black shaded histograms depict IRF8 levels from Irf8−/− mice. Data are representative of three independent experiments with similar results (n = 3 mice for WT, n = 3 mice for Irf8 −50−/−, and n = 3 mice for Irf8−/−).
However, ATAC-seq analysis suggested that the −50 kb Irf8 enhancer was active in F4/80+ peritoneal macrophages and Ly6C+ monocytes (Fig. 4a). In Irf8 −50−/− mice, monocytes were present at normal numbers, but showed reduced intracellular IRF8 protein levels compared to wildtype mice (Fig. 4b–d). Likewise, F4/80+ peritoneal macrophages in Irf8 −50−/− mice had greatly reduced intracellular IRF8 protein levels (Fig. 4e,f). Irf8−/− mice have been reported to have decreased survival in Salmonella enterica Typhimurium infection, possibly due to IRF8 regulation of inflammasome activity in macrophages32. However, it was not determined whether the in vivo defect in germline Irf8−/− mice was cell-intrinsic to macrophages. We found that Irf8 −50−/− mice were more susceptible to Salmonella infection compared to wildtype mice (Fig. 4g), consistent with a role for the −50 kb Irf8 enhancer in controlling IRF8 protein levels in monocytes/macrophages during infections. In conclusion, the −50 kb Irf8 enhancer was not required for DC development but did regulate Irf8 specifically in monocytes and macrophages.
Figure 4. The −50 kb Irf8 enhancer controls Irf8 expression in monocytes and macrophages.
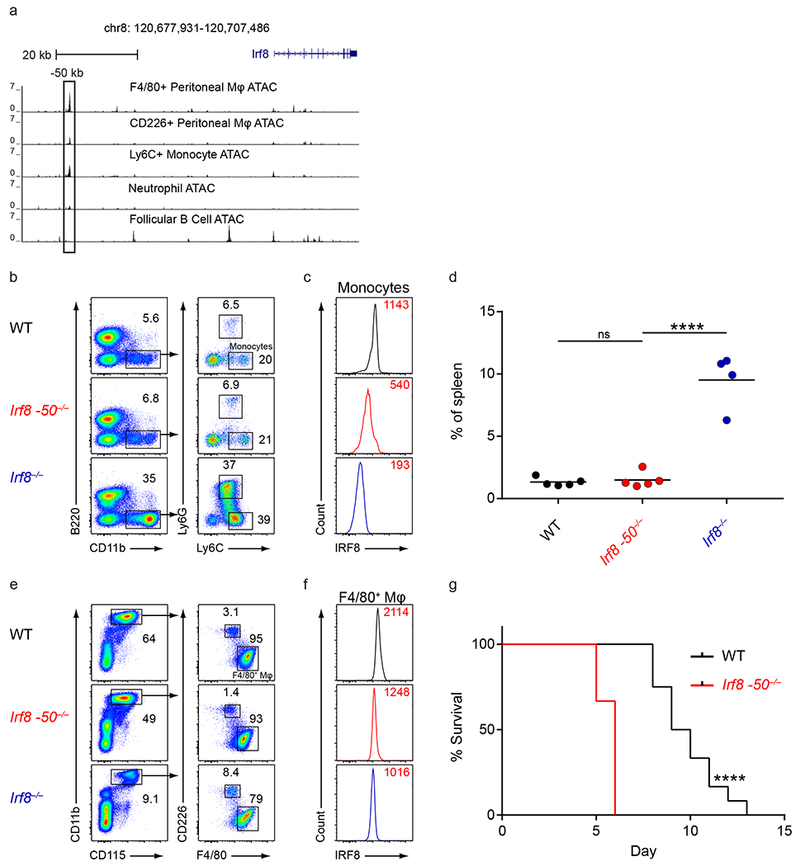
a, Normalized sequencing tracks of ATAC-seq in the indicated populations. Boxed is the −50 kb Irf8 enhancer. Data are from the Immunological Genome Project Open Chromatin Regions (n = 1 biological replicate per population). b, Flow cytometry of live splenocytes from mice of the indicated genotypes was used to identify monocytes. Numbers indicate the percent of cells in the indicated gates. Data are representative of four independent experiments with similar results (n = 5 mice for WT, n = 5 mice for Irf8 −50−/−, and n = 4 mice for Irf8−/−). c, Intracellular staining for IRF8 in splenic monocytes of mice of the indicated genotypes. Numbers indicate the MFI of IRF8 protein levels in monocytes. Data are representative of four independent experiments with similar results (n = 5 mice for WT, n = 5 mice for Irf8 −50−/−, and n = 4 mice for Irf8−/−). d, Statistical analysis of the percent of splenic monocytes in mice of the indicated genotypes. Data are pooled from four independent experiments (n = 5 mice for WT, n = 5 mice for Irf8 −50−/−, and n = 4 mice for Irf8−/−). e, Flow cytometry of peritoneal lavage cells from mice of the indicated genotypes was used to identify peritoneal macrophages. Numbers indicate the percent of cells in the indicated gates. Data are representative of four independent experiments with similar results (n = 5 mice for WT, n = 5 mice for Irf8 −50−/−, and n = 3 mice for Irf8−/−). f, Intracellular staining for IRF8 in F4/80+ peritoneal macrophages of mice of the indicated genotypes. Numbers indicate the MFI of IRF8 protein levels in peritoneal macrophages. Data are representative of four independent experiments with similar results (n = 5 mice for WT, n = 5 mice for Irf8 −50−/−, and n = 3 mice for Irf8−/−). g, WT and Irf8 −50−/− mice were infected with Salmonella typhi and survival was monitored. Data are pooled from two independent experiments (n = 10 mice for WT and n = 10 mice for Irf8 −50−/−). ns, not significant (P>0.05); ****P<0.0001, ordinary one-way ANOVA (d) or Log-rank (Mantel-Cox) test (g).
ATAC-seq reveals transient use of the +41 kb Irf8 enhancer during cDC1 development
To obtain base pair level resolution of accessible chromatin in progenitors, we next performed ATAC-seq on MDP, CDP, and pre-cDC1 populations (Fig. 5)33,34. Pearson correlation analysis of all distal ATAC-seq peaks indicated that MDPs were most closely related to CDPs and least related to pre-cDC1s (Fig. 5a). During transition from MDPs to CDPs, 687 unique peaks were gained, and 1,522 lost (Fig. 5b). By contrast, during transition from CDPs to pre-cDC1s, 14,630 peaks were gained, and 13,664 lost. Thus a large shift in chromatin accessibility occurs as CDPs transition to pre-cDC1s, which could potentially be related to the loss of alternative fate potentials in the pre-cDC1, to the differential proliferative capacities of these two cell types, or to a natural consequence of the differentiation process.
Figure 5. ATAC-seq identifies the +41 kb Irf8 enhancer as transiently active in cDC1 progenitors.
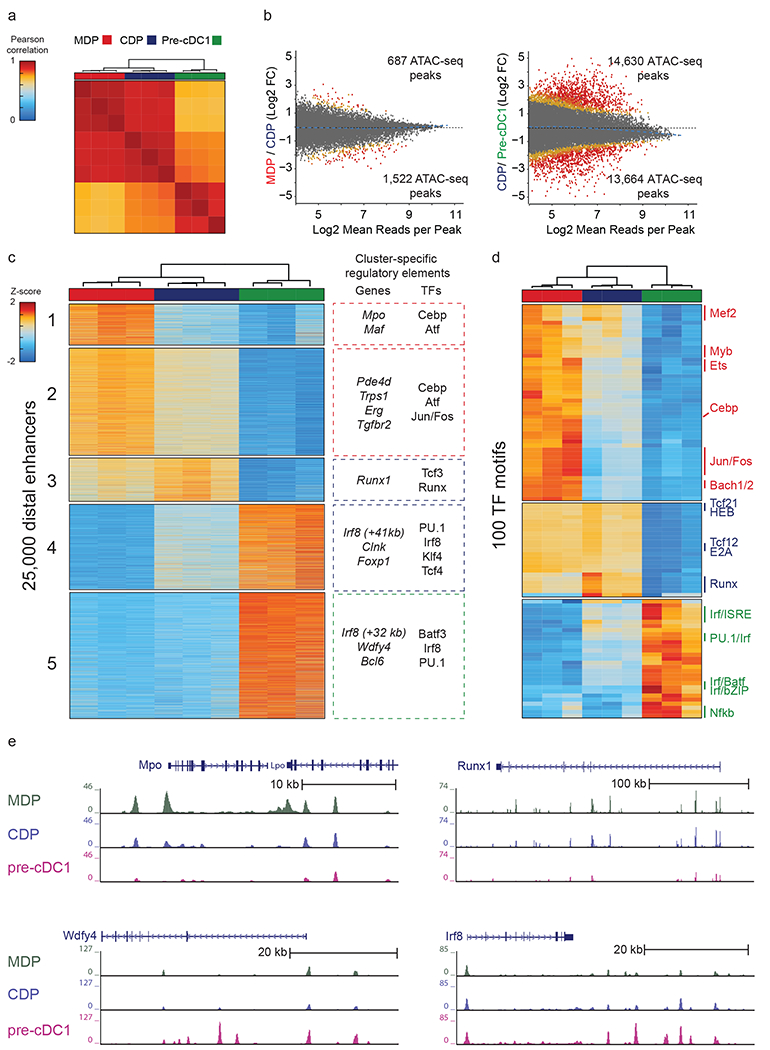
a, Pearson correlation of all distal ATAC-seq peaks for the indicated populations. Data are pooled from three independent experiments (n = 3 biological replicates per population). b, Differential ATAC-seq peaks in the indicated populations. Colors indicate orders of significance derived from DESeq2 analysis, with grey (fdr>0.1), yellow (0.1>fdr>0.01) , orange (0.01>fdr>0.001), and red (fdr<0.001) . Data are pooled from three independent experiments (n = 3 biological replicates per population). c, Heat map of k-means clusters for the top 25,000 varying distal ATAC-seq peaks. Colors indicate z-scores of reads in each peak compared with mean reads across all populations. Genes nearest to the clustered peaks and transcription factor motifs (TFs) enriched within peaks are indicated. Data are pooled from three independent experiments (n = 3 biological replicates per population). d, Heat map of k-means clusters of TF deviation z-scores for ATAC-seq profiles of the indicated populations. Data are pooled from three independent experiments (n = 3 biological replicates per population). e, Normalized sequencing tracks of ATAC-seq in the indicated populations are shown for genes in each k-means cluster. Mpo was present in k-cluster 1, Runx1 was present in k-cluster 3, Wdfy4 was present in k-cluster 4, and Irf8 was present in k-clusters 4 and 5. Data are representative of three independent experiments with similar results (n = 3 biological replicates per population).
Five main k-means clusters of peaks were identified (Fig. 5c). Cluster 1 included peaks found only in MDPs. Clusters 2 and 3 included peaks shared between MDPs and CDPs. Cluster 4 contained peaks found in both CDPs and pre-cDC1s, and cluster 5 included peaks present only in pre-cDC1s. We identified the genes closest in linear distance to the peaks within each cluster, and inferred the transcription factor motifs enriched within these peaks (Fig. 5c,d). Of note, the enrichment of transcription factors reflects the activity of any factor with a similar DNA binding motif rather than the activity of the specific factor that was enriched. For example, enrichment of Tcf3 could reflect the activity of other Tcf factors such as Tcf4 or Tcf12 within the clustered peaks. Peaks unique to MDPs (cluster 1) were in proximity to genes related to monocyte ontogeny, such as Mpo and Maf. These peaks were enriched in motifs for transcription factors such as Cebpb and Atf that regulate the macrophage/monocyte lineages35,36 (Fig. 5c,d).
The peaks identified in MDPs and CDPs (cluster 2 and 3) were enriched for motifs binding E proteins and Runx factors, both of which are required for DC development28,37,38 (Fig. 5c,d). Runx1 was also among the genes most proximal to peaks within these clusters, and the Runx1 locus contained open chromatin peaks in the MDP and CDP that were lost in the pre-cDC1 (Fig. 5e).
The peaks found in CDPs and pre-cDC1s (cluster 4) were also enriched in motifs binding E proteins (Fig. 5c). Further, cluster 4 included the +41 kb Irf8 enhancer (Fig. 5c), which contains six predicted E box motifs and was previously found to be active in pDCs but not cDC1s21 (Fig. 1a). This unexpected ATAC-seq accessibility of the +41 kb Irf8 enhancer in the pre-cDC1 (Fig. 5e) suggested that the activity of this enhancer could be important for cDC1 specification.
Finally, in the cluster of peaks found only in the pre-cDC1 (cluster 5) we found that Irf8 and Batf3 were among the genes at closest proximity to open regions and that AP1-IRF motifs and PU.1 motifs were highly enriched within these regions (Fig. 5c,d). Accessible chromatin peaks were also proximally located to genes known to be critically involved in cDC1 function, such as Wdfy439 (Fig. 5e). Finally, we found that the +32 kb Irf8 enhancer only became accessible at the pre-cDC1 stage (Fig. 5c,e). Together, our data suggests that the +41 kb Irf8 enhancer was transiently active in cDC1 progenitors before being replaced by activity at the +32 kb Irf8 enhancer in mature cDC1s.
The +41 kb Irf8 enhancer is required for cDC1 specification
By ATAC-seq analysis, the +41 kb Irf8 enhancer was inactive in MDPs, became active in CDPs and pre-cDC1s, and was inactive again in mature cDC1s (Fig. 6a). This enhancer also bound p300 in pDCs, suggesting it regulated Irf8 expression in these cells21. We targeted this enhancer using CRISPR/Cas9 genome editing with two flanking sgRNAs (Fig. 6a), producing mice with a 361 bp deletion (Irf8 +41−/−) that eliminated all six predicted E boxes (Supplementary Fig. 6a,b). As expected, Irf8 +41−/− mice had a pDC phenotype similar to the phenotype reported for Irf8−/− mice40. pDCs were present in Irf8 +41−/− mice, but had low levels of IRF8 and CD317 and increased levels of IRF4 (Fig. 6b–d). By contrast, monocytes, neutrophils, red pulp macrophages, B cells, and T cells from these mice were normal in frequency and intracellular IRF8 protein levels (Supplementary Fig. 6c,d).
Figure 6. The +41 kb Irf8 enhancer is required for cDC1 specification and development.
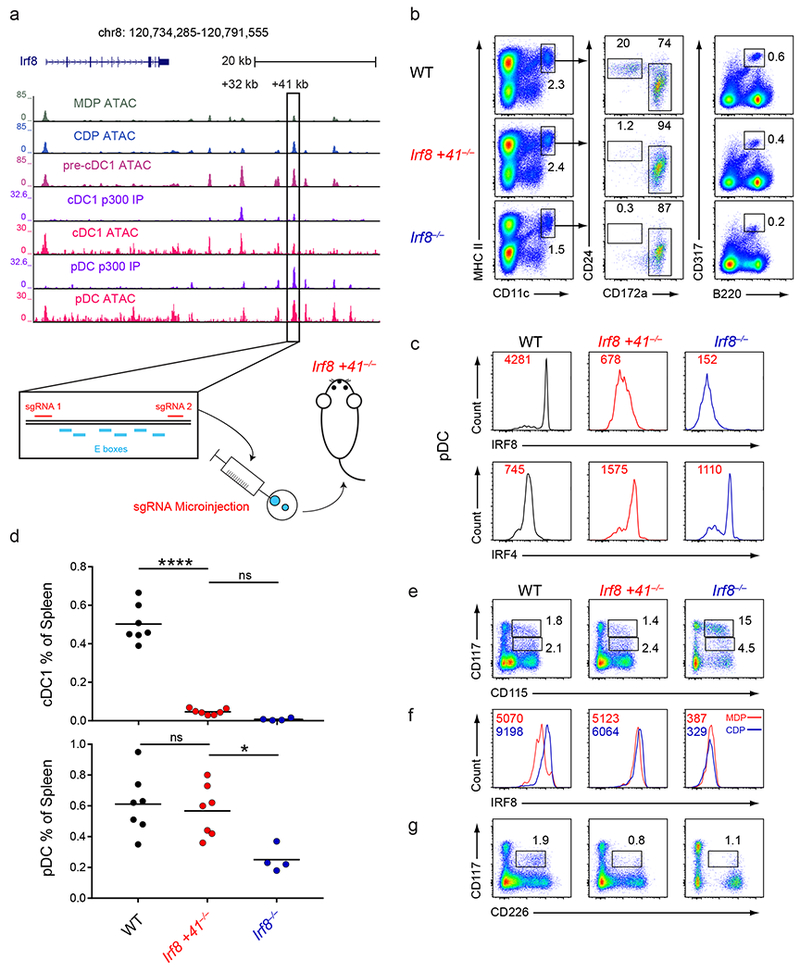
a, Normalized sequencing tracks of ChIP-seq with anti-p300 antibody or of ATAC-seq in the indicated populations. Boxed is the +41 kb Irf8 enhancer. Shown below is the +41 kb Irf8 enhancer with the E box motifs and sgRNA target sequences indicated. ATAC-seq data are representative of three independent experiments with similar results (n = 3 biological replicates for MDP ATAC, CDP ATAC, and pre-cDC1 ATAC) and ChIP-seq data are pooled from two independent experiments and the Immunological Genome Project Open Chromatin Regions (n = 1 biological replicate for cDC1 p300 IP, pDC p300 IP, cDC1 ATAC, and pDC ATAC). b, Flow cytometry of live splenocytes from mice of the indicated genotypes was used to identify DC subsets. Numbers indicate the percent of cells in the indicated gates. Data are representative of five independent experiments with similar results (n = 6 mice for WT, n = 7 mice for Irf8 +41−/−, and n = 4 mice for Irf8−/−). c, Intracellular staining for IRF8 and IRF4 in splenic pDCs of mice of the indicated genotypes. Numbers indicate the MFI of IRF8 or IRF4 protein levels in pDCs. Data are representative of four independent experiments with similar results (n = 5 mice for WT, n = 5 mice for Irf8 +41−/−, and n = 3 mice for Irf8−/−). d, Statistical analysis of the frequency of cDC1s and pDCs in spleens of mice of the indicated genotypes. Small horizontal lines indicate the mean. Data are pooled from five independent experiments (n = 6 mice for WT, n = 7 mice for Irf8 +41−/−, and n = 4 mice for Irf8−/−). e, Flow cytometry of Lin−CD135+ BM from mice of the indicated genotypes was used to identify MDPs and CDPs. Numbers indicate the percent of cells in the indicated gates. Data are representative of three independent experiments with similar results (n = 3 mice for WT, n = 3 mice for Irf8 +41−/−, and n = 3 mice for Irf8−/−). f, Intracellular staining for IRF8 within MDPs and CDPs from mice of the indicated genotypes. Red lines indicate the IRF8 protein level in MDPs and blue lines indicate the IRF8 protein level in CDPs. Numbers in red indicate the MFI of IRF8 protein levels in the MDP and numbers in blue indicate the MFI of IRF8 protein levels in the CDP. Data are representative of three independent experiments with similar results (n = 3 mice for WT, n = 3 mice for Irf8 +41−/−, and n = 3 mice for Irf8−/−). g, Flow cytometry of Lin−CD135+ BM from mice of the indicated genotypes was used to identify pre-cDC1s. Numbers indicate the percent of cells in the indicated gates. Data are representative of three independent experiments with similar results (n = 3 mice for WT, n = 3 mice for Irf8 +41−/−, and n = 3 mice for Irf8−/−). ns, not significant (P>0.05); *P<0.05; and ****P<0.0001, ordinary one-way ANOVA (d).
However, Irf8 +41−/− mice completely lacked mature cDC1s (Fig. 6b,d). We found normal frequencies of MDPs and CDPs in the bone marrow of Irf8 +41−/− mice, indicating that the +41 kb Irf8 enhancer was not required for the development of these progenitors (Fig. 6e). However, while the transition from the MDP to CDP is usually accompanied by a ~2-fold increase in intracellular IRF8 protein levels in wildtype CDPs, there was no increase in IRF8 levels in Irf8 +41−/− CDPs (Fig. 6f). Also, pre-cDC1s failed to develop in Irf8 +41−/− mice, similarly to Irf8−/− mice (Fig. 6g). In summary, the +41 kb Irf8 enhancer was only transiently active during cDC1 progenitor development, but was absolutely required for Irf8 induction in the CDP and for cDC1 specification.
E proteins are involved in cDC1 and pDC development
Since deletion of the +41 kb Irf8 enhancer abolished cDC1 specification, the factors that bound to this enhancer could be responsible for cDC1 fate commitment. The 454 bp region defining this enhancer contains predicted six E box motifs (Fig. 6a)21. ChIP-seq of LPS-activated B cells, pre-plasmablasts, and plasmablasts, in which E proteins and IRF8 are active, showed E2A binding to the +41 kb Irf8 enhancer. ATAC-Seq of these populations also found that this site was accessible in wildtype B cells, but was not in B cells doubly deficient for the E proteins E2A (Tcf3) and E2-2 (Tcf4) (Fig. 7a).
Figure 7. E proteins are involved in the development of cDC1s and pDCs.
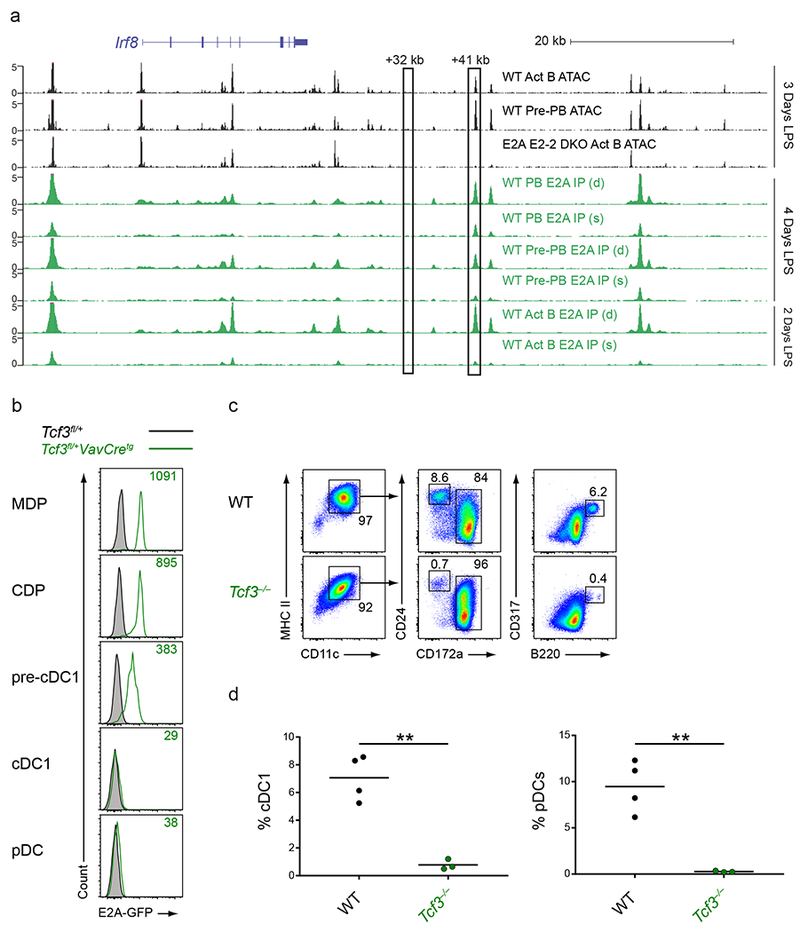
a, Normalized sequencing tracks of ChIP-seq with anti-E2A antibody or of ATAC-seq in lymph node B cells activated by LPS for the indicated number of days. Boxed are the +32 kb and +41 kb Irf8 enhancers. Populations analyzed were activated B cells (CD22+CD138−; Act B), pre-plasmablasts (CD22−CD138−; pre-PB), and plasmablasts (CD22−CD138+; PB) from either WT or Tcf3−/−Tcf4−/− (E2A E2-2 DKO) mice. B cells were either single (s)- or double (d)- crosslinked followed by ChIP with an E2A antibody as indicated. Data are pooled from two independent experiments (n = 1 biological replicate per population and per condition). b, Flow cytometry of Lin−CD135+ BM cells or live splenocytes from mice of the indicated genotypes was used to determine intracellular E2A-GFP levels in the indicated populations. Numbers indicate the MFI of E2A-GFP levels. Data are representative of four independent experiments with similar results (n = 3 mice for Tcf3fl/+ and n = 6 mice for Tcf3fl/+VavCretg). c,d, BM cells from WT or Tcf3−/− mice were cultured for 9 days in Flt3L. Cells were then analyzed by flow cytometry to identify DC subsets. Numbers indicate the percent of cells in the indicated gates (c). Statistical analysis of the frequency of cDC1s and pDCs from cultures is shown (d). Small horizontal lines indicate the mean. Data are pooled from three independent experiments (n = 3 mice for WT and n = 3 mice for Tcf3−/−). **P<0.01, unpaired two-tailed Student’s t-test (d).
The E proteins E2A and E2-2 are known to regulate B cell development and pDC development respectively28,41, but they have not been implicated in cDC development or function. Using the Tcf3fl/+VavCretg mice, which express an E2A-GFP fusion protein after Cre activity41, we found that E2A was expressed in MDPs and CDPs, was reduced in pre-cDC1s, and was completely absent in mature cDC1s and pDCs (Fig. 7b). This indicated that there was a transient period of E2A expression in DC progenitors. E2A was also expressed in pre-B cells, as has been previously described41, and also in CMPs and CLPs (Supplementary Fig. 7a). To test the relevance of this expression, we analyzed DC development from wildtype and Tcf3−/− BM using Flt3L-treated cultures. While wildtype BM showed normal cDC1 and pDC development in vitro, Tcf3−/− BM showed severely impaired cDC1 and pDC development but normal cDC2 development (Fig. 7c,d). However, cDC1s and pDCs continued to develop in Tcf3−/− mice in vivo, suggesting possible compensation for the actions of Tcf3 by another member of the Tcf family as has been previously described in B cells42 (Supplementary Fig. 7b,c). To further characterize the role of Tcf3 in vivo, we generated bone marrow chimeras with mixed wildtype and Tcf3−/− bone marrow and assessed DC development after reconstitution (Supplementary Fig. 7d,e). We found that wildtype progenitors preferentially gave rise to cDC1s and exclusively gave rise to pDCs when in competition with Tcf3−/− progenitors. These results suggest a potential role for E proteins in cDC1 and pDC development.
Discussion
Our study analyzed the in vivo function of three different Irf8 enhancers, discovering unexpected and distinct roles in DC development and monocyte/macrophage function for each. Our initial aim was to understand the development of the cDC1 subset critical for antiviral and antitumor immunity. Irf8 is the lineage-determining transcription factor responsible for cDC1 development23, but the molecular regulation of its transcription has not been well characterized. Previous studies have proposed that PU.1 regulated the induction of Irf8 in the MDP through a −50 kb Irf8 enhancer29, that BATF3 supports Irf8 autoactivation at the +32 kb Irf8 enhancer21, and that there was a pDC-specific +41 kb Irf8 enhancer as well21. However, none had tested the roles of any Irf8 enhancer elements in vivo.
In this study, we generated several targeted deletions of Irf8 enhancers using CRISPR/Cas9 genome editing in mice. Our results have confirmed the +32 kb Irf8 enhancer’s role in cDC1 development21 and excluded a role for the −50 kb Irf8 enhancer in early Irf8 expression29. However, most importantly, our analysis has established a wholly unexpected requirement for the +41 kb Irf8 enhancer in Irf8 induction within the CDP and subsequent cDC1 specification. This further suggested a previously unrecognized role for E proteins in this process as well.
We have confirmed that the +32 kb Irf8 enhancer is required for normal and compensatory cDC1 development. We had originally predicted this result because BATF3, which is required for cDC1 development, bound along with IRF8 to the +32 kb Irf8 enhancer. However, it was possible that BATF3 could also interact with other unidentified regions that serve as redundant enhancers to support cDC1 development. Our study found that not only was the +32 kb Irf8 enhancer required for normal cDC1 development, it was also required for the compensatory cDC1 development that can occur in Batf3−/− mice under various inflammatory settings. Because of this compensatory cDC1 development in Batf3−/− mice, this strain has not been useful for examining the function of cDC1s in some settings. For example, during infection by Mycobacterium tuberculosis, cDC1s reappear in Batf3−/− mice, so that the role of these cells in protection cannot be evaluated26. Also, Batf3 has functions in other lineages, such as T cells and perhaps macrophages43, so Batf3−/− mice might manifest phenotypes in other cell types that could make interpretations from this strain difficult. These limitations have been resolved by the generation of the Irf8 +32−/− mice. In these mice, Batf3 remains expressed normally in all immune lineages, but cDC1 development, both natural and compensatory, is ablated. These mice should now allow for the analysis of cDC1 function in settings where it could not previously be studied.
Our results also demonstrate how the cryptic activation of enhancers by transiently active transcriptional networks can control lineage diversification. Early models of hematopoiesis proposed that the fate choices of progenitors are resolved by stochastic fluctuations in the expression of cross-antagonistic lineage-determining transcription factors. These fluctuations cause one factor to become dominant and impose fate commitment, which is reinforced by autoactivation of this factor and cross-inhibition of opposing factors. One example is the PU.1/GATA1 circuit that regulates the choice between myeloid and megakaryocyte/erythrocyte fates. PU.1 and GATA1 are able to inhibit one another44,45 and also to activate their own transcription46,47. Further, these factors were thought to be co-expressed in progenitor cells that remained uncommitted until fluctuations in the levels of these two factors specified them to a single fate3. But this model was challenged by a recent study that used high-resolution analysis of single progenitor cells4. Using reporters for PU.1 and GATA1 expression, that study demonstrated that no progenitor cells actually co-express these factors. Rather, PU.1 levels decay before GATA1 is even expressed in progenitors destined for megakaryocyte/erythrocyte fate. This suggested that some unknown mechanism actually drives this particular cell fate by inducing GATA1, rather than stochastic fluctuations randomly generating this outcome. Our data also supports such a deterministic rather than stochastic model of fate divergence, in which transiently active transcriptional networks initiate the higher expression of lineage-determining transcription factors. Specifically, we found that activation of the +41 kb Irf8 enhancer in CDPs, potentially by E proteins, induced Irf8 expression and subsequent cDC1 fate specification. Similarly, other transiently acting networks may act to induce factors such as PU.1 or GATA1 in progenitors of other lineages. These networks may not be apparent in mature progeny, but might be revealed by the analysis of chromatin states of progenitors during development.
Our study also highlights how different enhancers of the same gene can be required at different stages of development of a single lineage. cDC1 development requires both the +41 kb and the +32 kb Irf8 enhancers, but the requirements are manifested at different developmental stages. The +41 kb Irf8 enhancer is required for the specification of pre-cDC1s from CDPs, but its activity is subsequently extinguished and is not apparent in the fully developed cDC1. On the other hand, the +32 kb Irf8 enhancer is active only after cDC1 specification, and remains active in the fully developed cDC1. The switch between these two enhancers reveals that different transcriptional networks are responsible for Irf8 expression during distinct periods of cDC1 development. Such switches in enhancer activity could occur throughout hematopoiesis, and analyzing them could help elucidate the mechanisms controlling other fate divergences that are still not fully understood.
Finally, our finding that E proteins were potentially responsible for cDC1 specification was wholly unexpected. However, since pDC development also relies on E proteins, it is unclear how cDC1 and pDC fates are separately specified. Other factors important in cDC1 development, such as Id2, could act to exclude pDC potential by blocking E protein activity, but why this would not also block cDC1 specification is unclear. Future work will need to determine the precise sequence of transcriptional events that govern cDC1 development, including our newfound role for E proteins.
Methods
Mice.
Batf3−/−, Irf8−/−, and Zbtb46GFP/+ mice have been described previously. Irf8 +32−/−, Irf8 −50−/−, and Irf8 +41−/− mice were newly generated as described below. The following mice were purchased from Jackson Laboratories: R26Cas9/+ mice (B6N.129(Cg)-Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J), CD45.1+ mice (B6.SJL-Ptprca Pepcb/BoyJ), Tcf3gfp/+ mice (B6.129-Tcf3tm1Mbu/J), and VavCretg mice (B6.Cg-Commd10Tg(Vav1-iCre)A2Kio/J). Tcf3−/− mice were a gift from B. Kee. All mice were on the C57BL6/J background except for Tcf3−/− mice, which were on the FVB/NJ background. All mice were generated, bred, and maintained in the Washington University in St. Louis School of Medicine specific pathogen-free animal facility. Animals were housed in individually ventilated cages covered with autoclaved bedding and provided with nesting material for environmental enrichment. Up to five mice were housed per cage. Cages were changed once a week, and irradiated food and water in autoclaved bottles were provided ad libitum. Animal manipulation was performed using standard protective procedures, including filtered air exchange systems, chlorine-based disinfection, and personnel protective equipment including gloves, gowns, shoe covers, face masks, and head caps. All animal studies followed institutional guidelines with protocols approved by the Animal Studies Committee at Washington University in St. Louis.
Unless otherwise specified, experiments were performed with mice between 6 and 10 weeks of age. No differences were observed between male and female mice in any assays performed and so mice of both genders were used interchangeably throughout this study. Within individual experiments, mice used were age- and sex-matched whenever possible. When mice were tracked for tumor growth or survival after Salmonella enterica Typhimurium infection, the monitoring scientist was blinded as to the genotypes of the mice in the experiment.
Generation of Irf8 enhancer deletion mice.
sgRNAs that flanked the enhancers of interest were identified using GT-Scan (http://gt-scan.braembl.org.au/gt-scan/). For deletion of the Irf8 +32 kb enhancer the following sgRNA sequences were used: Irf8 +32 5’: gttgtgatctttgaggtaga, Irf8 +32 Mid: gtctccttctgaaatttcagtt, and Irf8 +32 3’: gaactggcctggggcaggtc. For the Irf8 −50 kb enhancer the following sgRNA sequences were used: Irf8 −50 5’: ggtgacatctgtctacggag and Irf8 −50 3’: atgcacccaaggcctggctc. For the Irf8 +41 kb enhancer the following sgRNA sequences were used: Irf8 +41 5’: ggcccttgtagtttagctta and Irf8 +41 3’: aaagaagatctggggtatgt. Oligonucleotides that included these desired sgRNA sequences preceded by a T7 polymerase initiation site (ttaatacgactcactataggg) and followed by a portion of the tracrRNA sequence that annealed to the pX330 vector (gttttagagctagaaatagcaag) were then purchased (Sigma Aldrich). For example, the full Irf8 +32 5’ oligonucleotide purchased was TTAATACGACTCACTATAGGGgttgtgatctttgaggtagaGTTTTAGAGCTAGAAATAGCAAG. Each oligonucleotide was used in a PCR reaction with the PX330 Common Reverse Primer, aaaagcaccgactcggtgcc, and with the PX330 vector as a template to generate a complete DNA product containing the T7 polymerase initiation site, the sgRNA sequence, and the full tracrRNA sequence in order. RNA was then synthesized from these products using the MEGAscript T7 Transcription Kit (Thermo Fisher Scientific). RNA was purified using the MEGAclear Transcription Clean-Up Kit (Thermo Fisher Scientific) and eluted into nuclease-free injection buffer. RNA was diluted and stored at −80°C until it was used for microinjection. Purified Cas9 mRNA was a gift from W. Yokoyama.
Day 0.5 single cell embryos from C57Bl/6 mice were isolated and underwent pronuclear micro-injection at the Department of Pathology Micro-Injection Core. Each embryo was injected with 50 ng of each sgRNA and 100 ng of Cas9 mRNA. Injected embryos were then transferred into the oviducts of pseudopregnant recipient mice.
The resulting pups were screened by PCR to identify those that had successful deletion of the enhancers of interest. Mice with the desired deletion were then outcrossed to wildtype C57Bl/6 mice, and the resulting heterozygous mice were intercrossed to generate homozygous enhancer deletion mice.
Dendritic cell preparation.
Lymphoid and nonlymphoid organ DCs were harvested and prepared as described previously48. Briefly, spleens and inguinal skin-draining LNs were minced and digested in 5 mL of Iscove’s modified Dulbecco’s media (IMDM) + 10% FCS (cIMDM) with 250 µg/mL collagenase B (Roche) and 30 U/mL DNaseI (Sigma-Aldrich) for 45 min at 37⁰C with stirring. Lungs were minced and digested in 5 mL of cIMDM with 4 mg/mL collagenase D (Roche) and 30 U/mL DNaseI (Sigma-Aldrich) for 1.5 hours at 37⁰C with stirring. After digestion was complete, single cell suspensions from all organs were passed through 70-µm strainers and red blood cells were lysed with ammonium chloride-potassium bicarbonate (ACK) lysis buffer. Cells were subsequently counted with a Vi-CELL analyzer (Beckman Coulter) and 3-5×106 cells were used per antibody staining reaction.
For peritoneal cell analysis, 5 mL of MACS buffer (DPBS + 0.5% BSA + 2mM EDTA) was injected into the peritoneum of mice using a 27 g needle. After injection the mice were shaken gently to dislodge peritoneal cells. A 25 g needle was then used to collect the peritoneal fluid. Cells were ACK lysed and counted as described above.
Antibodies and flow cytometry.
Cells were stained at 4ºC in MACS buffer in the presence of Fc block (2.4G2; BD Biosciences).
The following antibodies were from BD Biosciences: CD11b PE (M1/70), CD19 Biotin (1D3), CD45R AlexaFluor488 (RA3-6B2), CD45.2 PerCP-Cy5.5 (104), CD64 AlexaFluor647 (X54-5/7.1), CD103 BV421 (M290), CD117 BUV395 (2B8), CD127 BV421 (SB/199), CD135 PE-CF594 (A2F10.1), I-A/I-E V500 (M5/114.15.2), Ly6C AlexaFluor 700 (AL-21). The following antibodies were from eBioscience: CD11b eFluor 450 (M1/70), CD11c PE (N418), CD24 PE-Cy7 (M1/69), CD172a PerCP–eFluor 710 (P84), CD317 APC (eBio927), F4/80 APC-eFluor 780 (BM8), IRF4 PE (3E4), IRF8 PerCP-eFluor 710 (V3GYWCH), Siglec H PerCP-Cy5.5 (eBio-440c). The following antibodies were from BioLegend: CD45R Biotin (RA3-6B2), CD45.2 PE (104), CD105 Biotin (MJ/718), CD115 BV711 (AFS98), CD127 biotin (A7R34), CD226 PE (10E5), F4/80 APC-Cy7 (BM8), F4/80 AlexaFluor700 (BM8), Ly6G Biotin (1A8), Ter119 Biotin (TER-119), XCR1 BV421 (ZET). The following antibodies were from Tonbo Bioscience: CD45.1 FITC (A20), CD3e Biotin (145-2c11). The following antibodies were from Invitrogen: CD11c APC-eFluor780 (N418). Cells were analyzed on a FACSCanto II or FACSAria Fusion flow cytometer (BD), and data were analyzed with FlowJo v10 software (TreeStar).
For intracellular IRF8/IRF4 staining, cells were stained for surface markers and then fixed/permeabilized with the intracellular fixation and permeabilization buffer kit (eBioscience) for 1 hour-overnight at 4⁰C. Cells were then stained for 1 hr at room temperature with intracellular antibodies. The cells were then washed and analyzed by flow cytometry.
Bone marrow isolation.
Bone marrow (BM) was harvested from the femur, tibia, and pelvis of mice. Bones were collected and fragmented by mortar and pestle in MACS buffer, and debris was removed by passing cells through a 70-µm strainer. Red blood cells were lysed with ACK lysis buffer and cells were subsequently counted on a Vi-CELL analyzer (Beckman Coulter). 3-5×106 were used per antibody staining reaction. For BM culture experiments, bulk BM cells were cultured at 37⁰C in 4 mL total volume of cIMDM supplemented with 100 ng/mL Flt3L (Peprotech) for nine days before further analysis.
Progenitor sorting and culture.
For sorting experiments, BM was isolated as described above and depleted of CD3-, CD19-, CD105-, Ter119-, and Ly6G-expressing cells by staining with the corresponding biotinylated antibodies followed by depletion with MagniSort Streptavidin Negative Selection Beads (Thermo Fisher). All remaining BM cells were then stained with fluorescent antibodies prior to sorting. Gates used to define MDPs, CDPs and pre-cDC1s were a combination of previously established markers21 and those identified in this study. MDPs were identified as Lin−CD117hiCD135+CD115+CD11c−MHCII−. CDPs were identified as Lin−CD117intCD135+CD115+CD11c−MHCII−. Pre-cDC1s were identified as either Lin−CD117intCD135+Zbtb46-GFP+ or as Lin−CD117intCD135+CD226+. Pre-cDC2s were identified as Lin−CD117loCD135+CD115+. Lineage markers included CD3, CD19, CD105, CD127, NK1.1, Ter119, and Ly6G. For retroviral reporter assays and in vitro CRISPR/Cas9 deletion CD117hi cells were sorted. A FACSAria Fusion was used for sorting and cells were sorted into cIMDM. Sort purity of >95% was confirmed by post-sort analysis before cells were used for further experiments. For culture experiments, DC progenitors were cultured at 37⁰C in 200 µL total volume of cIMDM supplemented with 100 ng/mL Flt3L (Peprotech) for five days before further analysis.
Retroviral infection and culture.
Retroviruses were produced by transfecting retroviral vectors into Plat-E cells essentially as described26 and collecting viral supernatants 2 days later. For reporter assays, Lin−CD117hi BM cells were sorted as described above and transduced with viral supernatants by ‘spin infection’ at 1800 RPM for 1 hour in the presence of 2 ug/mL polybrene. Infected cells were then cultured in Flt3L for 8 days before DCs were analyzed by flow cytometry. For in vitro CRISPR/Cas9 deletion, Lin− CD117hi BM cells from R26Cas9/+ mice were sorted and the same transduction protocol was used.
The retroviral reporter vector (Thy1.1 pA GFP CMVp_min PmeI MCS RV) was generated by replacing the XhoI-EcoRI fragment containing the IRES-GFP from the MSCV IRES GFP vector49 with a XhoI-EcoRI fragment containing IRES Thy1.1 from the MSCV-IRES-Thy1.1 vector50 to produce the Thy1.1 only RV. The SalI-BamHI fragment from hCD4 pA GFP RV51 containing GFP-Kb pA was then blunted using Pfu polymerase and inserted into the blunted EcoRI site of Thy1.1 only RV to produce a Thy1.1 pA GFP RV vector. Annealed oligos containing PmeI sites and NcoI and HindIII overhangs (CATGGTGGCATCCACTAGTTCTAGGATCCGTTTAAACA and AGCTTGTTTAAACGGATCCTAGAACTAGTGGATGCCAC) were then ligated into the NcoI-HindIII digested Thy1.1 pA GFP RV vector to produce the Thy1.1 pA GFP PmeI-MCS RV vector. A PCR product containing the minimal CMV promoter and BamHI/BglII sites was then amplified from the GFP min CMVp vector43 and ligated into the BamHI site of the Thy1.1 pA GFP PmeI-MCS RV vector to produce the final Thy1.1 pA GFP CMVp_min PmeI MCS RV vector. Enhancer regions were cloned into this vector using HindIII and BamHI digests.
The +32 kb enhancer regions were amplified from genomic DNA using Pfu polymerase. Purified PCR products were digested with HindIII and BglII and cloned into the HindIII and BamH1 digested retroviral reporter vector (Thy1.1 pA GFP CMVp RV). The following primers were used for amplification. For the +32 kb WT region: GCAAGCTTTGAGGTAGAGGGCCCA and ACGAGATCTGAGGAACACCAGGTCCCA. For the +32 kb 5’ region: GCAAGCTTTGAGGTAGAGGGCCCA and GAGCTAAGATCTCCTCAATGTCCAAGTTCACC. For the +32 kb 3’ region: TATCGATAAGCTTACGCCAGCAACTTCCTGAATC and ACGAGATCTGAGGAACACCAGGTCCCA.
A fragment of the Irf8 +32 kb enhancer containing the first three AICEs, either WT or mutated versions, were cloned into this same vector using annealed oligos with HindIII and BglII overhangs. The following primers were used. For the 3X AICE1 WT: AGCTTtctcttcttgtttctatttcaggttctccttctgaatttcagtttggctcaagttcctgcA and GATCTgcaggaacttgagccaaactgaaattcagaaggagaacctgaaatagaaacaagaagagaA. For 3X AICE1 Mut: AGCTTtctcttcttgtttctatcaaaggttcaacttctgaatcaaagtttggctcaagttcctgcA and GATCTgcaggaacttgagccaaactttgattcagaagttgaacctttgatagaaacaagaagagaA.
For in vitro CRISR/Cas9 deletion a Thy1.1-hU6-gRNA-BbsI stuffer RV vector was used (Thiesen et al, 2018). The following primers containing the sgRNA sequence and BbsI overhangs were annealed and cloned into the BbsI digested vector. For the αIrf8 sgRNA: CACCGagtttaccgaattgtccccg and AAACcggggacaattcggtaaactC. For the αIrf8 +32 kb sgRNA: CACCGgagccaaactgaaattcaga and AAACtctgaatttcagtttggctcC. For the scramble sgRNA: CACCggcactaccagagctaactca and AAACtgagttagctctggtagtgcC.
Tumor implantation.
The 1969 regressor fibrosarcoma has been previously described31. Tumor cells were thawed and propagated in R10 medium (RPMI + 10% FBS + 0.1% 2-ME). On the day of injection, cells were harvested by incubation in 0.05% trypsin-EDTA, washed three times with endotoxin-free PBS, and then 1×106 cells were injected subcutaneously in a total volume of 0.15 mL of PBS into the shaved flanks of mice. Tumor size was measured every three days beginning on day 4 and is presented as the surface area of the tumor (length X width).
IL-12 administration.
Mice were injected intraperitoneally with either saline (vehicle) or 500 µg of recombinant IL-12 (Pfizer) on two consecutive days and then analyzed three days after the second injection.
Bone marrow chimeras.
Bone marrow cells from donor mice were collected as described above. Recipient CD45.1+ mice received a single dose of 950 rads of whole-body irradiation and then received a transplant of 10×106 total BM cells the next day. For mixed BM chimeras, donor marrow from mice of each genotype was mixed at a ratio of 1:1 before transplantation. Mice were analyzed three to four weeks after transplantation for dendritic cell reconstitution.
Salmonella infection.
Salmonella enterica Typhimurium wildtype strain SB300A1 52 was used for infection. Bacteria were grown with shaking overnight at 37°C in Luria-Bertain (LB) broth, subcultured for 4 hours, and washed with with cold PBS prior to use. Intra-peritoneal infection was performed with an inoculum of 5×102 CFU bacteria.
Expression microarray analysis.
Progenitor cells or their progeny from in vitro culture were sorted as described above. RNA from sorted populations was extracted with a NucleoSpin RNA XS Kit (Machery-Nagel), then was amplified with WT Pico System (Affymetrix) and hybridized to GeneChip Mouse Gene 1.0 ST microarrays (Affymetrix) for 18 h at 45 °C in a GeneChip Hybridization Oven 640. The data was analyzed with the Affymetrix GeneChip Command Console. Microarray expression data was processed using Command Console (Affymetrix, Inc) and the raw (.CEL) files generated were analyzed using Expression Console software with Affymetrix default RMA Gene analysis settings.(Affymetrix, Inc). Probe summarization (Robust Multichip Analysis, RMA), quality control analysis, and probe annotation were performed according to recommended guidelines (Expression Console Software, Affymetrix, Inc.). Data were normalized by robust multiarray average summarization and underwent quartile normalization with ArrayStar software (DNASTAR).
ChIP.
ChIP-seq of mature DCs was described previously21. ChIP-seq of DC progenitors was performed similarly, with minor modifications. MDPs, CDPs, and pre-cDC1s were isolated from the BM of WT mice as described above and crosslinked prior to sorting. For crosslinking, cells were incubated for 8 min at room temperature with 1% formaldehyde. Reactions were then quenched with 1.25 M glycine, cells were washed twice with PBS, and pellets were “flash frozen” for storage at −80°C. Chromatin was sonicated at 4°C in sonication buffer (10 mM Tris-HCl, pH 8.0; 100 nM NaCl; 1 mM EDTA, 0.5 mM EGTA; 0.1% sodium deoxycholate; and 0.5% N-lauroylsarcosine) for 24 cycles of 20 s on and 50 s off per cycle with a Vivra-Cell VCX130PB and CV188 (Sonics & Material) to obtain DNA fragments from 140 bp to 500 bp. Chromatin was then immunoprecipitated overnight at 4°C with Dynabeads Protein A that had been pre-incubated with 2.5 µg of the appropriate antibody: anti-H3K27ac (Ab 4729; Abcam) or H3K4me1 (Ab 8895; Abcam). Beads containing protein-DNA complexes were washed seven times with RIPA buffer (50 mM HEPES, pH 7.5; 500 mM LiCl; 1 mM EDTA; 1% NP-40; and 0.7% sodium deoxycholage). DNA fragments were eluted, and crosslinking was reversed by incubation in a solution containing 50 mM Tris-HCl, pH 8.0; 10 mM EDTA; 1% SDS; and 1 mg/mL of proteinase K (New England Biolabs) for 5 hours at 65°C. DNA was purified by phenol-chloroform extraction followed by ethanol precipitation. Libraries for ChIP-Seq were prepared with a ThruPLEX-FD kit (Rubicon Genomics) and were sequenced with an Illumina HiSeq 2500.
ChIP-seq of B cells was described previously42. Briefly, in vitro LPS stimulated B cells were subjected to crosslinking at room temperature for either 10 min with 1% formaldehyde (single crosslinking) or for 45 min with 2 mM disuccinimidyl glutarate (Sigma) followed by 10 min with 1% formaldehyde (double crosslinking). The chromatin was prepared as previously described53. The pelleted genomic DNA, crosslinked with proteins, were sheared with a Bioruptor® sonicator (Diagenode) followed by immunoprecipitation using an anti-E2A antibody. The precipitated DNA (1-2 ng) was used for library preparation and sequencing.
ATAC-seq.
ATAC-seq of DC progenitors was performed using the Omni-ATAC protocol as previously described with minor modifications34. 10,000 MDPs, CDPs, and pre-cDC1s were sorted from bone marrow as described above and lysed in ice-cold ATAC-RSB buffer containing 0.1% NP40, 0.1% Tween-20, and 0.01% digitonin. Cells were incubated at 4° C for 3 min, then washed with ATAC-RSB buffer containing only 0.1% Tween-20. Nuclei were spun down by centrifugation and then incubated in 50 µL of transposition buffer (25 µL 2X TD buffer, 22.5 µL dH2O, 2.5 µL Tn5 transposase (Nextera DNA Library Prep Kit, Illumina)) and incubated at 37° C for 30 min. If 10,000 cells could not be obtained for a certain population then the quantity of Tn5 transposase was titrated down proportionately to the number of cells obtained but cells were still incubated in 50 µL total. Transposed DNA was purified with a DNA Clean & Concentrator kit (Zymo Research), eluted in 21 µL of elution buffer, and stored at −20° C until amplification. Three biological replicates for each cell population were obtained and sequenced. ATAC-seq libraries were prepared as previously described, barcoded and sequenced on an Illumina Nextseq 500.
ATAC-seq of cDC1s, pDCs, monocytes, peritoneal macrophages, neutrophils, and follicular B cells was obtained from the Immunological Genome Project Open Chromatin Regions54.
ATAC-seq of LPS activated B cells was described previously42.
Computational analysis.
For computational analysis of ATAC-seq of DC progenitors, adapter sequences were trimmed using SeqPurge and aligned to mm10 genome using bowtie2. These reads were then filtered for mitochondrial reads, low mapping quality (samtools flag “-F 1804 -f 2 -q 20”), and PCR duplicates using Picard tools MarkDuplicates. The bam was then converted to a bed and the Tn5 corrected insertion sites were obtained (“+” stranded + 4 bp, “-” stranded −5 bp)33. To identify peaks, we called peaks for each sample using MACS2 “--shift −75 --extsize 150 --nomodel --call-summits --nolambda --keep-dup all -q 0.01” using the insertion beds. To get a union peak set, the peak summits were then extended by 250 bp on either side to a final width of 501 bp, filtered by the ENCODE mm10 blacklist (https://www.encodeproject.org/annotations/ENCSR636HFF/), and filtered to remove peaks that extend beyond the ends of chromosomes. Overlapping peaks were handled using an iterative removal procedure as previously described55. First, the most significant peak (defined by MACS2 score) is kept and any peak that directly overlaps with that significant peak is removed. Then, this process iterates to the next most significant peak and so on until all peaks have either been kept or removed due to direct overlap with a more significant peak. This resulted in a union peak set of 188,509 equal width peaks. These peaks were then annotated using ChIPseeker and computed the occurrence of a TF motif using motifmatchr in R with chromVARMotifs mouse_pwms_v1 set56. All insertions that fell within each peak were then counted using “countOverlaps” in R to get a counts matrix (peak x samples). To determined differential peaks, the raw counts matrix was used as input into DESeq2 using the modelMatrixType = “expanded” and were tested for whether or not a peak was greater than a Log2FoldChange of 0.5 (lfcThreshold = 0.5, altHypothesis=“greaterAbs”)57. A cutoff of an FDR < 0.1 was used to denote a differential peak. For clustering analyses, the counts matrix was then normalized by using edgeR’s “cpm(matrix , log = TRUE, prior.count = 5)” followed by a quantile normalization using preprocessCore’s “normalize.quantiles” in R. TF motif enrichment were calculated using a hypergeometric test in R testing the representation of a motif (from motifmatchr above) in a subset of peaks vs all peaks. To compute TF motif deviations, chromVAR was used in R with raw counts in distal peaks (defined as greater than 1kb from a TSS in TxDb.Mmusculus.UCSC.mm10.knownGene) and then the top 100 variable TF motifs were determined using variability scores 56. To create sequencing tracks, the Tn5 corrected insertion sites were read into R and a coverage pileup was created that was binned every 100bp using rtracklayer and normalized by reads in peak such that they were all scaled to 30M total reads in peaks (from counts matrix).
Computational analysis of ATAC-seq of LPS activated B cells was described previously42.
ChIP-seq data sets were aligned to the mouse genome (GRCm38/mm10 assembly) by Bowtie software (version 1.1.1) with the following parameters: --sam -p 4 -t --verbose --trim5 3 -m 1 mm10 --chunkmbs 1000. Data were visualized with the ‘makeUCSCfile’ program of the Homer software package with default parameters. Peaks were identified with MACS software, version 1.4.2 (‘model based analysis for ChIP-Seq’) with a P value of 1×10−9.
Motifs in the Irf8 +32 kb and +41 kb peaks were identified using the FIMO motif-identification program at a P-value threshold of 1×10−3.
Statistical analysis.
Statistical analyses were performed with Prism (GraphPad Software). Results from independent experiments were pooled as indicated in figure legends. When comparing only two groups, unpaired two-tailed Student’s t-test was used. When comparing more than two groups, Ordinary one-way ANOVA was used. When comparing survival, Log-rank (Mantel-Cox) test was used.
Data availability.
The sequencing and microarray data generated during the course of this study have been deposited and are available on the GEO database. The ChIP-seq data of DC progenitors utilized in Figure 3 can be accessed with the following accession number: GSE132239. The ATAC-seq data of DC progenitors utilized in Figures 5 and 6 can be accessed with the following accession number: GSE132240. The microarrays utilized in Supplementary Figure 4 can be accessed with the following accession number: GSE123747.
All other primary data and materials that support the findings of this study are available from the corresponding author upon request.
Supplementary Material
Acknowledgements
This work was supported by the Howard Hughes Medical Institute (K. Murphy and H. Chang), the US National Institutes of Health (F30 DK108498 to V. Durai; K08 CA23188-01 to A. Satpathy; P50 HG007735 to H. Chang; R01 AI106352 to B. Kee; R01 DK097317 to R. Newberry), the National Science Foundation (DGE-1745038 to P. Bagadia), the Parker Institute for Cancer Immunotherapy (A. Satpathy and H. Chang), and Boehringer Ingelheim (M. Wöhner, H. Tagoh and M. Busslinger). A. Satpathy was supported by a Career Award for Medical Scientists from the Burroughs Wellcome Fund. This work benefitted from data assembled by the ImmGen consortium54. We thank the Genome Technology Access Center in the Department of Genetics at Washington University in St. Louis School of Medicine for help with genomic analysis. The Center is partially supported by NCI Cancer Center Support Grant #P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant# UL1TR000448 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH.
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Graf T and Enver T Forcing cells to change lineages. Nature 462, 587–594 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Orkin SH and Zon LI Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–644 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miyamoto T et al. , Myeloid or lymphoid promiscuity as a critical step in hematopoietic lineage commitment. Dev.Cell 3, 137–147 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Hoppe PS et al. , Early myeloid lineage choice is not initiated by random PU.1 to GATA1 protein ratios. Nature 535, 299–302 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Long HK, Prescott SL, and Wysocka J Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 167, 1170–1187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sagai T et al. , Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development 132, 797–803 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Shim S et al. , Cis-regulatory control of corticospinal system development and evolution. Nature 486, 74–79 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frankel N et al. , Phenotypic robustness conferred by apparently redundant transcriptional enhancers. Nature 466, 490–493 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osterwalder M et al. , Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554, 239–243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lara-Astiaso D et al. , Immunogenetics. Chromatin state dynamics during blood formation. Science 345, 943–949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinman RM and Cohn ZA Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp.Med . 137, 1142–1162 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cella M et al. , Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon [see comments]. Nature Medicine 5, 919–923 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Durai V and Murphy KM Functions of Murine Dendritic Cells. Immunity 45, 719–736 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hildner K et al. , Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gubin MM et al. , Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salmon H et al. , Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 44, 924–938 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saxena M and Bhardwaj N Re-Emergence of Dendritic Cell Vaccines for Cancer Treatment. Trends Cancer 4, 119–137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fogg DK et al. , A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 311, 83–87 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Naik SH et al. , Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol 8, 1217–1226 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Onai N et al. , Identification of clonogenic common Flt3(+) M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nature Immunology 8, 1207–1216 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Grajales-Reyes GE et al. , Batf3 maintains autoactivation of Irf8 for commitment of a CD8alpha(+) conventional DC clonogenic progenitor. Nat Immunol 16, 708–717 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlitzer A et al. , Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat Immunol 16, 718–728 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Schiavoni G et al. , ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp.Med . 196, 1415–1425 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kashiwada M et al. , NFIL3/E4BP4 is a key transcription factor for CD8{alpha}+ dendritic cell development. Blood 117, 6193–6197 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hacker C et al. , Transcriptional profiling identifies Id2 function in dendritic cell development. Nat Immunol 4, 380–386 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Tussiwand R et al. , Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature 490, 502–507 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seillet C et al. , CD8alpha+ DCs can be induced in the absence of transcription factors Id2, Nfil3, and Batf3. Blood 121, 1574–1583 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Cisse B et al. , Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell 135, 37–48 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schonheit J et al. , PU.1 level-directed chromatin structure remodeling at the Irf8 gene drives dendritic cell commitment. Cell Rep . 3, 1617–1628 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Platt RJ et al. , CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diamond MS et al. , Type I interferon is selectively required by dendritic cells for immune rejection of tumors. Journal of Experimental Medicine 208, 1989–2003 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karki R et al. , IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation. Cell 173, 920–933 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buenrostro JD et al. , Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corces MR et al. , An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heath V et al. , C/EBPalpha deficiency results in hyperproliferation of hematopoietic progenitor cells and disrupts macrophage development in vitro and in vivo. Blood 104, 1639–1647 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Labzin LI et al. , ATF3 Is a Key Regulator of Macrophage IFN Responses. The Journal of Immunology 195, 4446–4455 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Sawai CM et al. , Transcription factor Runx2 controls the development and migration of plasmacytoid dendritic cells. J Exp.Med 210, 2151–2159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Satpathy AT et al. , Runx1 and Cbfbeta regulate the development of Flt3+ dendritic cell progenitors and restrict myeloproliferative disorder. Blood 123, 2968–2977 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Theisen DJ et al. , WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science 362, 694–699 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sichien D et al. , IRF8 Transcription Factor Controls Survival and Function of Terminally Differentiated Conventional and Plasmacytoid Dendritic Cells, Respectively. Immunity 45, 626–640 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Kwon K et al. , Instructive role of the transcription factor E2A in early B lymphopoiesis and germinal center B cell development. Immunity 28, 751–762 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Wohner M et al. , Molecular functions of the transcription factors E2A and E2-2 in controlling germinal center B cell and plasma cell development. J Exp.Med 213, 1201–1221 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwata A et al. , Quality of TCR signaling determined by differential affinities of enhancers for the composite BATF-IRF4 transcription factor complex. Nat Immunol 18, 563–572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang P et al. , PU.1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood 96, 2641–2648 (2000). [PubMed] [Google Scholar]
- 45.Nerlov C et al. , GATA-1 interacts with the myeloid PU.1 transcription factor and represses PU.1-dependent transcription. Blood 95, 2543–2551 (2000). [PubMed] [Google Scholar]
- 46.Tsai SF, Strauss E, and Orkin SH Functional analysis and in vivo footprinting implicate the erythroid transcription factor GATA-1 as a positive regulator of its own promoter. Genes & Development 5, 919–931 (1991). [DOI] [PubMed] [Google Scholar]
- 47.Chen H et al. , PU.1 (Spi-1) autoregulates its expression in myeloid cells. Oncogene 11, 1549–1560 (1995). [PubMed] [Google Scholar]
Methods-only References
- 48.Durai V et al. , Altered compensatory cytokine signaling underlies the discrepancy between Flt3(−/−) and Flt3l(−/−) mice. J Exp.Med 215, 1417–1435 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ranganath S et al. , GATA-3-dependent enhancer activity in IL-4 gene regulation. Journal of Immunology 161, 3822–3826 (1998). [PubMed] [Google Scholar]
- 50.Sedy JR et al. , B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat.Immunol . 6, 90–98 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Zhu H et al. , Unexpected characteristics of the IFN-gamma reporters in nontransformed T cells. Journal of Immunology 167, 855–865 (2001). [DOI] [PubMed] [Google Scholar]
- 52.McKinney J et al. , Tightly regulated gene expression system in Salmonella enterica serovar Typhimurium. J Bacteriol . 184, 6056–6059 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kohwi-Shigematsu T et al. , SATB1-mediated functional packaging of chromatin into loops. Methods 58, 243–254 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heng TS, Painter MW, and Immunological Genome Project Consortium The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9, 1091–1094 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Corces MR et al. , The chromatin accessibility landscape of primary human cancers. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schep AN et al. , chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods 14, 975–978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Love MI, Huber W, and Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550–(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing and microarray data generated during the course of this study have been deposited and are available on the GEO database. The ChIP-seq data of DC progenitors utilized in Figure 3 can be accessed with the following accession number: GSE132239. The ATAC-seq data of DC progenitors utilized in Figures 5 and 6 can be accessed with the following accession number: GSE132240. The microarrays utilized in Supplementary Figure 4 can be accessed with the following accession number: GSE123747.
All other primary data and materials that support the findings of this study are available from the corresponding author upon request.