

Abstract
BACKGROUND & AIMS:
Paneth cells secrete antimicrobial proteins including lysozyme via secretory autophagy as part of the mucosal protective response. The ELAV like RNA-binding protein 1 (ELAVL1, also called HuR) regulates stability and translation of mRNAs and many aspects of mucosal physiology. We studied the posttranscriptional mechanisms by which HuR regulates Paneth cell function.
METHODS:
Intestinal mucosal tissues were collected from mice with intestinal epithelium-specific disruption of HuR (IE-HuR−/−), HuRfl/fl-Cre− mice (controls), and patients with inflammatory bowel diseases (IBD) and analyzed by histology and immunohistochemistry. Paneth cell functions were determined by lysozyme-immunostaining assays. We isolated primary enterocytes from IE-HuR−/− and control mice and derived intestinal organoids. HuR and the chaperone CNPY3 were overexpressed from transgenes in intestinal epithelial cells (IECs) or knocked down with small interfering RNAs. We performed RNA pulldown assays to investigate interactions between HuR and its target mRNAs.
RESULTS:
Intestinal tissues from IE-HuR−/− mice had reduced numbers of Paneth cells, and Paneth cells had fewer lysozyme granules per cell, compared with tissues from control mice, but there were no effects on goblet cells or enterocytes. Intestinal mucosa from patients with IBD had reduced levels of HuR and fewer Paneth cells. IE-HuR−/− mice did not have the apical distribution of TLR2 in the intestinal mucosa as observed in control mice. IECs from IE-HuR−/− mice expressed lower levels of CNPY3. Intestinal organoids from IE-HuR−/− mice were smaller and contained fewer buds compared with those generated from controls, and had fewer lysozyme-positive cells. In IECs, knockdown of HuR decreased levels of the autophagy proteins LC3-I and LC3-II, compared with control cells, and prevented rapamycin-induced autophagy. We found HuR to interact directly with the Cnpy3 mRNA coding region and increase levels of CNPY3 by increasing the stability and translation of Cnpy3 mRNA. CNPY3 bound TLR2, and cells with knockdown of CNPY3 or HuR lost membrane localization of TLR2, but increased cytoplasmic levels of TLR2.
CONCLUSIONS:
In studies of mice, IECs, and human tissues, we found HuR to increase expression of CNPY3 at the posttranscriptional level. CNPY3 is required for membrane localization of TLR2 and Paneth cell function.
Keywords: Mucosal defense, autophagy, IBD, epithelial homeostasis
Graphical Abstract
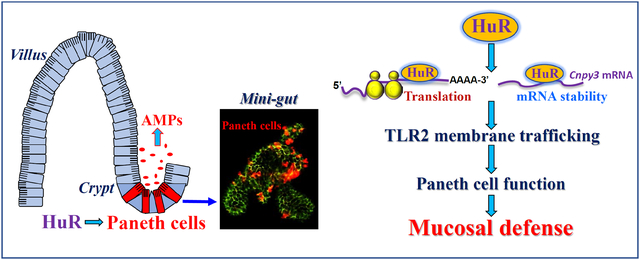
Introduction
The mammalian intestinal epithelium is a rapidly self-renewing tissue in the body and separates a wide array of luminal noxious substances and microorganisms from sterile tissue.1–3 The intestinal space is colonized by trillions of commensal bacteria, which includes pathogens that can disrupt host cellular functions. The intestinal epithelium defends against invasion of all bacteria through multiple mechanisms, including secretion of antimicrobial proteins and destruction of invading pathogens via autophagy.4,5 Paneth cells are specialized intestinal epithelial cells (IECs) that reside at the bottom of the crypts and are crucial for maintaining homeostasis of the epithelium by engendering host protection from enteric pathogens.6,7 Paneth cells produce abundant antibacterial proteins or peptides, including lysozyme, Reg3 lectins, α-defensin, and phospholipase A2. Autophagy is a conserved intracellular pathway that sequesters the cytoplasmic structures and pathogens targeted for degradation.8–10 Paneth cells secrete lysozyme through secretory autophagy to limit bacterial infection of the intestine.11 Defects in Paneth-cell secretory autophagy compromise the mucosal defense leading to a range of diseases.6,12–14
Posttranscriptional events, particularly altered mRNA turnover and translation, are major mechanisms by which mammalian cells control gene expression in response to stress.15 Changes in mRNA stability and translation are controlled primarily through processes initiated by the interaction of specific mRNA sequences (cis elements) with two major types of trans-acting factors: RNA-binding proteins (RBPs) and microRNAs (miRNAs).16,17 RBPs and miRNAs bind to cis elements on the mRNA, frequently within the 3’-untranslated region (3’-UTR), and regulate the stability and translation rates of target transcripts positively or negatively. HuR (encoded by the Elavl1 gene) is among the most prominent translation and turnover regulatory RBPs, and it associates with U- or AU-rich elements located in the 3’-UTRs and/or coding regions (CRs) of target mRNAs.18,19 Recently, HuR has emerged as a master posttranscriptional regulator of homeostasis in the intestinal epithelium.20–25 Conditional deletion of HuR in IECs inhibits the renewal of the small intestinal mucosa by inactivating the Wnt signaling pathway and decreases early rapid epithelial restitution by repressing Cdc42 translation;18,21 In addition, HuR deletion reduces intestinal tumorigenesis by controlling the levels of different proteins22 and governs gut epithelial homeostasis by interplaying with miRNAs such as miR-195 or long noncoding RNAs (lncRNAs) including H19 and SPRY4-IT1.23–25
Little is known about the in vivo function of HuR in the regulation of Paneth cells in the intestinal epithelium. Using mice bearing IEC-specific ablation of HuR we found that loss of HuR led to lysozyme granule abnormalities in Paneth cells in vivo and ex vivo, and further discovered that HuR silencing also inhibited autophagy activation in cultured IECs. Human intestinal mucosa with inflammation and injury/erosions from patients with IBD likewise exhibited both reduced HuR levels and defects in Paneth cell function. Our results indicate that HuR regulates Paneth cell function by enhancing the stability and translation of a target mRNA encoding canopy3 (CNPY3), a protein necessary for the proper subcellular localization of Toll-like receptor 2 (TLR2) on the IEC plasma membrane to carry out secretory autophagy. These findings point to HuR as a promising therapeutic target for interventions to protect the epithelium integrity in patients with massive mucosal inflammation or requiring surgical intensive care supported with total parenteral nutrition.
Materials and Methods
Animal Studies
Age- and gender-matched 8–12-week-old mice on the C57BL/6 background were used. Intestinal epithelial tissue-specific HuR deletion (IE-HuR−/−) mice were generated by crossing the HuRflox/flox (HuRfl/fl) and villin-Cre mice purchased from the Jackson Laboratory, as described in our previous studies.18,21 HuRfl/fl-Cre− mice developed and served as control littermates. Both IE-HuR−/− mice and control littermates were housed and handled in a specific pathogen-free breeding barrier and cared for by trained technicians and veterinarians. Animals were deprived of food but allowed free access to tap water for 24 h before experiments. Two portions of the small intestine taken 0.5 cm distal to the ligament of Trietz or the middle colon were removed, one for histological examination and the other for extraction of protein and RNA. The mucosa was scraped with a glass slide for various measurements as described previously.25,26 All animal experiments were performed in accordance with NIH guidelines and were approved by the Institutional Animal Care and Use Committee of University Maryland School of Medicine and Baltimore VA hospital.
Histology and Immunohistochemistry
Human tissue samples were obtained from surplus discarded tissue from Department of Surgery, University of Maryland Health Science Center and commercial tissue banks. The study was approved by the University Maryland Institutional Review Board. Dissected and opened intestines were mounted onto a solid surface and fixed in formalin and paraffin. Sections of 5 m thickness were stained with H&E for general histology. The immunofluorescence staining procedure was carried out according to the method described in our previous publications.27,28 Procedures of several methods, including plasmid construction,31,32 intestinal organoid culture,5,29 isolation of membrane and cytoplasmic protein,55 assays of newly translated protein and polysome analysis,27,32 biotin pull-down assays and RIP analysis,19,32,33 Q-PCR and immunoblotting analyses,25 and chemicals and cell culture,24 were described in Supplementary Material for more detail.
Statistical Analysis
All values were expressed as the means ± standard error of the mean (SEM). Unpaired, 2-tailed Student’s t test was used when indicated with P < .05 considered significant. When assessing multiple groups, 1-way analysis of variance (ANOVA) was utilized with Tukey’s post hoc test.34 The statistical software used was SPSS 17.1 (IBM Corp, Armonk, NY).
Results
HuR deletion causes defects in Paneth cells in the intestinal epithelium
To define the in vivo function of HuR in the regulation of Paneth cells in the mammalian intestinal epithelium, we used IE-HuR−/− mice that were generated by crossing HuRfl/fl mice with villin-Cre-expressing mice as described.18,21 HuR levels in the small intestinal and colonic mucosa were undetectable in IE-HuR−/− mice but were at wild-type levels in gastric mucosa, liver, lung, and pancreas. Targeted deletion of HuR did not alter the expression levels of other RBPs such as CUGBP1 and AUF1 in the intestinal mucosa (data not shown), as reported in our previous study.21 Conditional HuR deletion in IECs had no effect on the overall morphology and structure of the small and large intestine, although it caused mucosal atrophy in the small intestine. Interestingly, intestinal epithelium-specific HuR deletion resulted in Paneth cell defects in the small intestinal mucosa, as examined by lysozyme-immunostaining assays (Figure 1A). Staining of whole mounts of the intestine revealed that lysozyme-positive cells were normally located at the base of the crypt in littermate mice, but the numbers of these lysozyme-positive cells decreased dramatically in IE-HuR−/− mice relative to control littermate mice. The number of lysozyme granules per Paneth cell also decreased notably in the HuR-deficient intestinal epithelium (Figure 1B). On the other hand, HuR deletion failed to alter the function of Goblet cells, since there were no differences in the number of Goblet (Alcian blue-positive) cells and the levels of mucin-2 immunostaining in the small intestinal mucosa between control littermates and IE-HuR−/− mice (Supplementary Figure 1A). Ablated HuR in IECs also did not affect enterocyte differentiation in the mucosa as measured by villin immunostaining analysis (Supplementary Figure 1B).
Figure 1.

Targeted deletion of HuR in mice reduces Paneth cells and autophagy activation in the intestinal epithelium. (A) Immunostaining of Paneth cells (lysozyme-positive cells) in the small intestinal mucosa of IE-HuR−/− and control littermate mice. Red, lysozyme; and green, E-cadherin (E-cad). Scale bars, 25 μm. (B) Lysozyme fluorescence intensity per crypt in IE-HuR−/− mice compared with control littermates. Values are the means ± SEM from 5 mice. *P < 0.05 compared with littermates. (C) Growth of small intestinal organoids isolated from control littermate and IE-HuR−/− mice. (D) Immunostaining of lysozyme-positive cells in the intestinal organoids described in C. Scale bars, 100 μm. (E) Immunoblots of LC3 protiens in cultured IECs transfected with siRNA targetting HuR (siHuR) or control siRNA (C-siRNA). The levels of HuR and LC3 were examined 48 h after the transfection. (F) Effect of HuR silencing on autophagy activation induced by rapamycin in cultured IEC-6 cells. Left, immunoblots of LC3-I and LC3-II 8 h after exposure to rapamycin (50 ng/ml). Right, changes in LC3 activity at different times after treatment with rapamycin, as quantified by examining the ratio of LC3-II and LC3-I. Values are the means ± SEM (n = 3). * P < 0.05 compared with C-siRNA.
In an ex vivo model, we found that there also were obvious abnormalities in Paneth cells in intestinal organoids isolated from IE-HuR−/− mice. As shown in Figure 1C, an intestinal organoid was initiated from a single proliferating cell, but by five days after culture, the structures of organoids consisted of multiple cells and buds in the organoids isolated from control littermate mice. HuR deletion inhibited growth of the intestinal organoids markedly, since the organoids from IE-HuR−/− mice were smaller and contained fewer buds compared with those generated from control littermate mice. The surface areas of HuR-deficient organoids decreased by ~85% on day 10 after primary culture, compared to those of the organoids from control littermate mice (n = 6; P < 0.05). Moreover, DNA synthesis in HuR-deficient organoids was inhibited, as determined by the decreased BrdU incorporation (data not shown). Consistent with the observations in vivo, lysozyme-positive cells in the organoids isolated from control littermates were highly enriched, but they decreased dramatically and were almost undetectable in the organoids generated from IE-HuR−/− mice (Figure 1D). In an in vitro model, we further found that HuR silencing prevented the activation of autophagy by the pharmacologic inducer rapamycin. Decreasing HuR levels in cultured IECs by transfection with a specific siRNA targeting HuR (siHuR) not only decreased the basal levels of autophagy proteins LC3-I and LC3-II (Figure 1E), but it also inhibited rapamycin-induced autophagy (Figure 1F). As shown, treatment with rapamycin activated autophagy in control cells as indicated by increased levels of LC3-II, but this activation was abolished in HuR-silenced cells. In addition, Paneth cell defects induced by HuR deletion were associated with microbiota dysbiosis in mice (Supplementary Figure 2). The HuR-deficient intestinal epithelium exhibited a decrease in richness and biodiversity of mucosal microbiota in IE-HuR−/− mice compared with control littermate mice, although it did not display any signs of mucosal inflammation or injury/erosions (data not shown). Together, these results indicate that HuR plays an essential role in the regulation of Paneth cell function in the intestinal epithelium.
Association of decreased HuR with abnormalities in Paneth cells in patients with IBD
To investigate the impact of HuR on Paneth cell function in patients with IBD, we examined changes in the levels of HuR and Paneth cells in human intestinal mucosa. Ileal and colonic mucosal tissues in patients with IBD were collected for various measurements, whereas the tissue samples from patients without mucosal inflammation and injury/erosions served as controls. As shown in Figure 2A, HuR was found in both the cytoplasm and nucleus in normal small intestinal mucosa obtained from control individuals. However, the intestinal mucosa with inflammation and injury/erosions (Supplementary Figure 3) from patients with IBD displayed a significant decrease in the levels of HuR, particularly in the cytoplasm, when compared with those observed in control patients. In fact, there was virtually no cytoplasmic HuR fluorescence in the small intestinal mucosa of patients with IBD. Similarly, HuR levels in the colonic epithelium were also decreased in patients with IBD compared with control individuals (Figure 2B). Importantly, the decreased abundance of intestinal mucosal HuR was associated with disrupted Paneth cells in patients with IBD (Figure 2C). Lysozyme-positive cells in the ileal mucosa from patients with IBD decreased remarkably compared with those in control patients. In fact, lysozyme-positive cells were almost completely undetectable in the ileal mucosal samples obtained from IBD patients. Moreover, the decreased HuR levels and defects in Paneth cells observed in IBD patients were also accompanied by an inhibition of mucosal renewal and a reduction in tight junction expression, as reported in our previous studies.3,25 These results suggest that decreased levels of HuR and subsequent Paneth cell defects are implicated in the pathogenesis of human IBD.
Figure 2.

Association of decreased HuR with a reduction in Paneth cells in the intestinal mucosa of patients with inflammatory bowel disease (IBD). (A) Immunohistochemical staining of HuR in the ileum mucosa from control individuals (without mucosal erosions/inflammation) and patients with IBD. Green, HuR; blue, nucleus stained by DAPI. Scale bars, 25 μm. (B) Immunostaining of HuR in the colon mucosa in control individuals and IBD patients. Red, HuR; purple, nucleus stained by DAPI. (C) Immunostaining of ileum paraffin section with anti-lysozyme (red), anti-E-cadherin (green), and DAPI (blue). All these experiments were repeated in human tissue samples obtained from four control individuals or patients with IBD and showed similar results.
HuR deletion decreases the apical distribution of TLR2 in the intestinal epithelium
Given fact that TLRs are essential for normal function of Paneth cells and play an important role in activating autophagy,35,36 we tested the possibility that HuR deletion might cause Paneth cell defects by altering TLR activity. As shown in Figure 3A (top), TLR2 was found to localize primarily at the surface of villi in the small intestinal mucosa of WT mice, but was strongly concentrated along the apical area of the mucosa in control littermate mice. The fluorescence intensity of TLR2 in the cytoplasm and crypt area of the mucosa was relatively low in both mice. Targeted deletion of HuR in mice disrupted the apical distribution of TLR2 in the small intestinal mucosa (Figure 3, bottom). In mice with ablated HuR, the apical staining of TLR2 in the small intestinal mucosa disappeared completely, but it showed diffuse staining in the cytoplasm with strong intensity in the basal area of the epithelium. The most striking difference between control littermates and IE-HuR−/− mice was the appearance of many areas of small punctate foci of TLR2 in the latter. These punctate regions were located throughout the cytoplasm in the HuR-deficient intestinal epithelium and were regularly identified in every IE-HuR−/− mouse. Similarly, TLR2 distribution in the colonic mucosa was also compromised by HuR deletion, as indicated by a decrease in the apical TLR2 straining in IE-HuR−/− mice compared with control littermates (Supplementary Figure 4).
Figure 3.

HuR knockout disrupts the subcellular distribution of TLR2 in the small intestinal epithelium in vivo and ex vivo. (A) Immunostaining of the small intestinal mucosa with anti-TLR2 (red), anti-E-cadherin (E-cad, green), and DAPI (blue) in mice. Scale bars, 25 μm. The experiments were repeated in the mucosal tissues obtained from three control littermates or IE-HuR−/− mice and showed similar results. (B) Immunostaining of TLR2 and E-cadherin in the intestinal organoids isolated from control littermate and IE-HuR−/− mice. In images shown in right two panels, TLR2 fluorescence was merged with E-cadherin and DIC images, respectively. Scale bars, 100 μm.
Consistent the in vivo findings, TLR2 was normally localized at the luminal regions in primary cultures of intestinal organoids isolated from control littermate mice (Figure 3B, top), but this specific distribution of TLR2 was abolished by HuR deletion, as evidence by the fact that TLR2 staining was diffuse in HuR-deficient organoids isolated from IE-HuR−/− mice (Figure 3B, bottom). On the other hand, HuR deletion failed to alter TLR4 subcellular organization in the intestinal mucosa (Supplementary Figure 5). The subcellular distribution of TLR4 in the HuR-deficient intestinal epithelium was indistinguishable from that observed in the mucosa of control littermate mice.
To gain a deeper understanding of the abnormalities of TLR signals in IE-HuR−/− mice, we examined changes in TLR expression and demonstrated that HuR deletion did not alter the levels of total TLR proteins in the intestinal mucosa (Figure 4). As shown, TLR2 and TLR4 were highly expressed in the intestinal mucosal tissue and cultured IECs, although TLR1 and TLR6 proteins were low and almost undetectable in the intestinal epithelium in vivo as well as in culture. There were no significant differences in the levels of TLR2 and TLR4 proteins in the intestinal mucosa between control littermates and IE-HuR−/− mice (Figure 4A, B). Importantly, targeted deletion of HuR in mice decreased the levels of the endoplasmic reticulum (ER) chaperone CNPY3 in the intestinal mucosa. The HuR-deficient epithelium also exhibited decreased levels of IRGM and beclin-1 but increased NLRX1, without the effect on NOD1 abundance. In cultured IECs, decreasing HuR levels by transient transfection of siHuR also lowered cellular CNPY3 protein (Figure 4C&D), but it did not alter the protein levels of TLR2, TLR4, NOD1, NOD2, RIP2, PGRP-1α, MyD88, or gp96. HuR silencing also increased the abundance of NLRX1 in cultured cells. These findings indicate that decreasing the HuR levels in the intestinal epithelium disrupts the subcellular localization of TLR2 but does not affect its abundance.
Figure 4.

HuR deletion decreases the levels of CNPY3 in the intestinal epithelium in vivo and in vitro. (A) Immunoblots of HuR, TLRs, and their regulatory proteins such as CNPY3 and NLRX1 in the small intestinal mucosa of littermate and IE-HuR−/− mice. (B) Quantitative analysis derived from densitometric scans of immunoblots of TLR2 and CNPY3 as described in A. Values are the means ± SEM (n = 3). *P < 0.05 compared with control littermates. (C) Immunoblots of TLRs and TLR-regulatory proteins in cultured IEC-6 cells. Cells were transfected with siHuR or C-siRNA, and cell lysates were harvested 48 h thereafter. (D) Quantitative analysis of TLR2 and CNPY3 in cells described in C (n = 3). *P <0.05 compared with C-siRNA.
HuR regulates TLR2 cell surface trafficking by controlling the expression of CNPY3
In cultured IECs, CNPY3 physically interacted with TLR2 and formed CNPY3/TLR2 complexes, as assessed by IP assays using an anti-TLR2 antibody in normal cells (Figure 5A) or an anti-DDK antibody in cells transfected with the expression vector encoding DDK-tagged CNPY3 (Figure 5B). In control IP reactions, IgG did not immunoprecipitate either TLR2 or CNPY3. CNPY3 silencing by transfection with siRNA targeting the Cnpy3 mRNA (siCNPY3) failed to alter whole-cell levels of HuR or TLR2, but it impaired TLR2 subcellular distribution (Figure 5C,D). In normal cells, TLR2 was predominantly localized at the cell membrane, but this specific localization of TLR2 was impaired in CNPY3 knockdown cells. When compared with cells transfected with C-siRNA, the levels of TLR2 in membrane fractions isolated from CNPY3-silenced cells decreased remarkably, while cytoplasmic TLR2 abundance increased. Similar to what was seen after CNPY3 knockdown, HuR silencing also reduced membrane TLR2 abundance, but it increased cytoplasmic TLR2 levels (Figure 5E,F). Conversely, ectopic overexpression of CNPY3 in HuR-silenced cells rescued cell surface presence of TLR2, since the levels of membrane TLR2 in cells co-transfected with siHuR and CNPY3 expression vector were identical to those in cells transfected with C-siRNA (Figure 5G). On the other hand, overexpression of CNPY3 in control cells (without siHuR transfection) did not lead to an additional increase in the level of membrane TLR2 (Supplementary Figure 6).
Figure 5.

HuR upregulates cell surface distribution of TLR2 via CNPY3. (A) Interaction of CNPY3 with TLR2 in normal IECs as measured by immunoprecipitation (IP) assays using anti-TLR2 antibody. (B) Immunoblots of TLR2 and CNPY3 in the IP materials using anti-DDK antibody in cells transfected with a expression vector encoding DDK-taged CNPY3. (C) Immunoblots of membrane (Mem) and cytoplasimc (Cyto) TLR2 in CNPY3-silencent cells: i) total proteins; and ii) subcellular distribution of TLR2. Whole-cell lysates were harvested 48 h after transfection with siRNA targetting CNPY3 (siCNPY3) or C-siRNA. Membrane and cytoplasmic proteins were isolated, and equal loading was monitored by coomassie staining. (D) Quantitative analysis derived from densitometric scans of immunoblots of TLR2 in cells described in C (n = 3). (E) Western blot analysis of the abundance of HuR, CNPY3, and TLR2 in cells transfected with siHuR or cotransfected with siHuR and a CNPY3 expression vector for 48 h. (F) Immunoblots of membrane and cytoplasmic TLR2 in cells described in (E). (G) Quantitative analysis derived from densitometric scans of immunoblots of TLR2 in cells described in F (n = 3). (H) Activity of luciferase (Luc) NF-κB reporter gene in HuR-silenced cells (siHuR) transfected with the CNPY3 expression vector (n = 3). Twnty-four hours after transfection with the Luc-NF-κB reporter, cells were exposed to the TLR2 ligand, Pam3csk4 (10 ng/ml). The Luc activity was examined 6 h after treatment owth Pam3csk4. *P < 0.05 compared with solvent; +P < 0.05 compared with cells transfected with C-siRNA or cotransfected with siHuR and CNPY3 and then treated with Pam3csk4.
To test if the aberrant TLR2 distribution altered its function in response to the specific TLR2 agonist Pam3csk4,37,38 we examined changes in TLR2 activity in HuR-knockdown cells with or without CNPY3 overexpression by using a luciferase (Luc) NF-κB reporter assays, as described.39 Exposure of control cells to Pam3csk4 induced TLR2 activation as indicated by an increase in the Luc-NF-κB reporter activity, but this induction was abolished in HuR-silenced cells (Figure 5H). Ectopic overexpression of CNPY3 restored the response of HuR-silenced cells to Pam3csk4, and the activity of the Luc-NF-κB reporter in cells co-transfected with siHuR and CNPY3 expression vector was restored to the levels observed in control cells exposed to Pam3csk4. Collectively, these findings indicate that HuR enhances cell surface trafficking of TLR2 through a process requiring CNPY3.
HuR increases CNPY3 expression by stabilizing Cnpy3 mRNA and stimulating its translation
To investigate if HuR regulates CNPY3 expression posttranscriptionally, we first examined if HuR directly interacted with the Cnpy3 mRNA in cultured IECs. Ribonucleoprotein immunoprecipitation (RIP)40 analysis using an anti-HuR antibody followed by measurement of Cnpy3 mRNA levels by RT-Q-PCR analysis in the IP material revealed that Cnpy3 mRNA associated with HuR, as Cnpy3 mRNA was highly enriched in HuR IP relative to IgG IP samples (Figure 6A). HuR binding to the c-myc mRNA served as a positive control in this study.30 To determine if HuR binds to specific regions of the Cnpy3 5’-UTR, CR, and 3’-UTR, HuR/Cnpy3 we tested the interaction of HuR with biotinylated transcripts spanning Cnpy3 mRNA regions (Figure 6B, schematic). Following incubation with cytoplasmic lysates, the interaction between biotinylated Cnpy3 RNAs and HuR or other RBPs was examined by biotin pulldown followed by Western blot analysis.33 As shown, HuR only associated with the Cnpy3 CR, but not with Cnpy3 5’-UTR and 3’-UTR. In addition, the Cnpy3 mRNA fragments did not interact with other RBPs such as CUG-binding protein 1 (CUGBP1) and AU-binding factor 1 (AUF-1).
Figure 6.

HuR interacts with the Cnpy3 mRNA and increases its stability and translation in cultured IECs. (A) Association of endogenous HuR with endogenous Cnpy3 mRNA as measured by RIP and RT-Q-PCR analysis using either anti-HuR antibody (Ab) or control IgG. Values are the means ± SEM from triplicate samples. *P < 0.05 compared with IgG IP. (B) HuR immunoblots using the materials pulled down by biotinylated transcripts of the Cnpy3 5’-UTR, CR, and 3’-UTR. Left, schematic representation of various biotinylated Cnpy3 transcripts. (C) Levels of the Cnpy3 mRNA at steady state (i) and in decay assays (ii) 48 h after transfection with C-siRNA and siHuR. Cnpy3 mRNA levels were examined at different times after administration of actinomycin D. *P < 0.05 compared with C-siRNA. (D) Newly synthesized CNPY3 protein as measured by L-azidohomoalaine (AHA) incorporation assays in cells described in C. (E) Distribution of Cnpy3 (top) and Gapdh (bottom) mRNAs in each gradient fraction of polysomal profiles prepared from cells described in C. (F) Levels of the Luc reporter activity 48 h after transfection with siHuR. Top, schematic of different chimeric firefly luciferase reporters bearing the Cnpy3 5’-UTR, CR, or 3’-UTR. *P < 0.05 compared with C-siRNA.
Further analysis of the impact of HuR on CNPY3 expression was undertaken. We found that HuR silencing decreased Cnpy3 mRNA levels by enhancing its degradation, since silencing HuR lowered moderately the Cnpy3 mRNA half-life (Figure 6C). This effect was measured by incubating cells with actinomycin D and measuring the time required for Cnpy3 mRNA to decline to one-half of its initial abundance. As shown, silencing HuR caused a slightly faster decline in Cnpy3 mRNA levels, as evidence that Cnpy3 mRNA was less stable when HuR levels were diminished. Analysis of Cnpy3 mRNA translation revealed that de novo synthesis of CNPY3 was slower in HuR-silenced cells compared with cells transfected with C-siRNA (Figure 6D), as determined by using Click-iT chemistry (Materials and Methods) to measure nascent translation. The reduction of CNPY3 protein synthesis by HuR silencing was specific, as there was no change in nascent GAPDH synthesis in the same cell populations. To further define the role of HuR in promoting CNPY3 translation, we examined the relative sizes of Cnpy3 mRNA polysomes. After fractionating cytoplasmic lysates through sucrose gradients, we observed that decreasing HuR levels did not affect global polysomal profiles19,30; however, the abundance of Cnpy3 mRNA associated with the most actively translating fraction (fraction 9) decreased in HuR-silenced cells shifting to a smaller, lesser-translating fraction (fraction 8; Figure 6E, top). In contrast, Gapdh mRNA, encoding the housekeeping protein GAPDH, was distributed similarly in both groups (Figure 6E, bottom). Finally, HuR promoted the stability and translation of Cnpy3 mRNA by interacting with Cnpy3 CR, since HuR silencing decreased the levels of Cnpy3-CR luciferase reporter activity (Figure 6F), but it did not affect the activities of Luc-5’-UTR and Luc-3’-UTR reporter genes. Together, these results indicate that HuR increases CNPY3 expression by stabilizing the Cnpy3 mRNA and enhancing its translation.
Discussion
Activation of Paneth cells and their secretory autophagy plays an important role in the intestinal epithelium homeostasis by enhancing host defense against invading bacteria and other pathogens,8,13,41 but the exact mechanism underlying this process is poorly understood. The present study provides powerful genetic evidence demonstrating the importance of HuR in the regulation of Paneth cell function through the control of TLR2 signaling pathway. Conditional deletion of HuR in mice not only specifically decreased the levels of apical TLR2 in the intestinal mucosa but it also resulted in defects in Paneth cells. In an ex vivo model, HuR-deficient enteroids from IE-HuR−/− mice also exhibited the disrupted localization of TLR2 and dysfunction of Paneth cells. Interestingly, HuR regulated cell surface trafficking of TLR2 in IECs by posttranscriptionally controlling CNPY3 expression levels. HuR interacted directly with the Cnpy3 mRNA via the CR and increased its stability and translation. These findings represent a conceptual advance linking the RBP HuR with secretory autophagy in Paneth cells and reveal the key role of the HuR/CNPY3/TLR2 axis in the homeostasis of the intestinal epithelium, which in turn impacts upon mucosal inflammation-associated diseases.
The results reported here indicate that HuR is essential for maintaining the subcellular localization of TLR2 in the intestinal epithelium. Targeted HuR deletion in mice disrupted the apical distribution of TLR2 in the intestinal mucosa and ex vivo-prepared organoids, and HuR silencing also reduced cell surface trafficking of TLR2 in cultured cells, although neither intervention changed whole-cell TLR2 levels. On the other hand, for unknown reasons, HuR deletion in mice did not alter the localization of TLR4 in the gut epithelium. The subcellular distribution of each TLR is crucial for its ligand recognition and biological functions.42,43 TLR1, 2, 6, 5, and 10 are all present on the plasma membrane, where they are best available to contact bacterial and fungal cell wall constituents. Conversely, TLR3, 7, 8, and 9 reside intracellularly and recognize bacterial and viral nucleic acids generated during viral replication or released following microbial degradation.44,45 TLR subcellular distribution is dynamic and tightly controlled by numerous factors at multiple levels.46,47 Aberrant trafficking and localization of TLRs cause the loss of responses to specific TLR agonists and lead to the inactivation of innate immunity during bacterial infection.42,43 Paneth cells function as IECs responsible for TLR-mediated immune response in the intestinal mucosa and secrete lysozyme via secretory autophagy to limit bacterial invasion.8,10 Since TLR dysfunction inhibits autophagy,35,48 defects in Paneth cells observed in the HuR-deficient intestinal epithelium result primarily from the disruption of TLR2 localization. Consistent with our observations, targeted deletion of the autophagy genes Atg16l1 or Atg5 in mice also compromised the exocytosis pathway in Paneth cells.8,10,14 Although HuR silencing prevented the activation of autophagy in IECs, the exact mechanisms and consequences of the altered autophagic response in Paneth cell function in vivo remain to be fully investigated. In addition, Wnt signaling is necessary for inducing Paneth cell formation,56 whereas HuR deletion in IECs represses Wnt activity by lowering the expression levels of Wnt co-receptor LRP6 at the posttranscription level.18 It is likely that the inactivation of Wnt pathway may also plays a role in Paneth cell defects in the HuR-deficient intestinal epithelium.
The result presented here also show that HuR regulates TLR2 cell surface trafficking by targeting CNPY3. Although it was initially thought to be important for TLR4 expression only, CNPY3 modulates the subcellular localization of multiple TLRs by directly interacting with the general chaperone gp96.50–52 For example, TLR1 or TLR9 forms a multimolecular complex with CNPY3 and gp96, and the binding of TLR1 or TLR9 to either molecule requires the presence of the other.51,53 Genetic disruption of CNPY3/gp96 interaction abolishes their chaperone function and inhibits the activation of all TLRs except TLR3. In Cnpy3-knockout mice, cell surface expression of multiple TLRs, including TLR1, TLR2, and TLR4, and endosomal localization of TLR9, are totally disrupted, and the multiple TLR-mediated immune responses are also impaired after exposure to various challenges.44,49 Our results show that CNPY3 bound to TLR2 and this interaction was necessary for TLR2 cell surface trafficking in cultured IECs. Decreased levels of cellular CNPY3 by transfection with siCNPY3 or HuR silencing reduced the membrane TLR2 abundance. Ectopically expressed CNPY3 in HuR-silenced cells rescued the cell surface expression of TLR2, but it did not additionally increase the levels of membrane TLR2 in control cells (without siHuR transfection). These findings suggest that CNPY3 is required for TLR2 membrane distribution and that CNPY3 overexpression alone does not sufficiently induce trafficking of TLR2 to the plasma membrane in normal conditions. On the other hand, HuR knockout did not affect gp96 levels in the intestinal epithelium in vivo as well as in vitro.
Another significant finding from this study is that the Cnpy3 mRNA is a novel target of HuR and that HuR/Cnpy3 mRNA association increased Cnpy3 mRNA stability and translation. These observations are consistent with our previous results19,21,32 and work from others22,54 who have demonstrated that HuR associates with target mRNAs and increases their expression by stabilizing mRNAs and/or increasing their translation. Reporters bearing partial transcripts spanning the Cnpy3 5’-UTR, CR and 3’-UTR further showed that HuR influenced CNPY3 expression predominantly via the Cnpy3 CR but not with the 5’-UTR or the 3’-UTR. The interaction of HuR with the Cnpy3 CR was found to be functional, as HuR silencing repressed only Cnpy3-CR luciferase reporter constructs, but not with the Cnpy3-5’-UTR or Cnpy3 3’-UTR reporter constructs. Although HuR generally interacts with the 3’-UTRs of target transcripts, it also associates, in some instances, with the CRs of target mRNAs and regulates their fate. In this regard, HuR stabilized Xiap mRNA by directly interacting with its CR.55 In support of these findings, multiple HuR-binding motifs (such as AU-rich elements) were found to be only located at the CRs of human and mouse Cnpy3 mRNAs but not at their 5’- or 3’-UTRs. We did not further characterize the specific nucleotides with which HuR interacts and increases Cnpy3 mRNA stability and translation, since those experiments would require more specialized biochemical, crystallographic, and molecular methods than those used here. In addition, our results show that HuR knockout also decreased the levels of other TLR regulatory factors such as IRGM and beclin-1 but it increased NLRX1 in the intestinal epithelium. The exact mechanisms through which these factors disrupt TLR2 localization and subsequent Paneth cell defects in the HuR-deficient epithelium remain to be fully elucidated in future studies.
The findings reported here are of particular importance from a clinical point of view, since human intestinal mucosa with inflammation and injury/erosions from patients with IBD displayed both decreased levels of HuR and defects in Paneth cells. As reported, Paneth cell defects and deregulated expression of autophagy genes such as Atg16l1, Atg5, and Atg7 are commonly observed in patients with mucosal inflammation-associated diseases.8,14,41 In this study, we establish for the first time a cause-effect relationship between HuR and posttranscriptional control of Paneth cell functions in in vivo, ex vivo and cell culture models. Abnormalities in Paneth cells in the intestinal epithelium of IE-HuR−/− mouse are similar to those observed in patients with IBD. In addition, Paneth cell defects induced by HuR knockout also shaped intestinal microbiota in mice, and this microbiota dysbiosis may also in turn affect epithelial homeostasis. Taken together, the results of our current study strongly support the notion that HuR regulates Paneth cell function by modulating TLR2 localization via posttranscriptional control of CNPY3 expression. These findings help understand how Paneth cells preserve their function in response to pathogen-induced stress, and represent a novel therapeutic target to protect the intestinal epithelium integrity in patients with massive mucosal inflammation and/or critical surgical disorders.
Supplementary Material
Acknowledgements
J.Y.W. is a Senior Research Career Scientist, Biomedical Laboratory Research & Development Service, U.S Department of Veterans Affairs.
Funding
This work was supported by Merit Review Awards (to J.Y.W.; J.N.R.) from US Department of Veterans Affairs; grants from National Institutes of Health (DK57819, DK61972, DK68491 to J.Y.W.); and funding from the National Institute on Aging-Intramural Research Program, NIH (to M.G.).
Abbreviations used:
- RBP
RNA-binding protein
- CNPY3
canopy3
- TLRs
Toll-like receptors
- RNP
ribonucleoprotein
- RIP
RNP immunoprecipitation
- IE-HuR−/−
intestinal epithelium-specific HuR knockout
- E-cad
E-cadherin
- IECs
intestinal epithelial cells
- LncRNA
long noncoding RNA
- miRNAs
microRNAs
- UTR
untranslated region
- CR
coding region
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplemental Data
Included: Supplementary Methods, six Supplementary Figures, and one Supplemental Table.
Conflicts of interest
The authors disclose no conflicts.
References
- 1.Bankaitis ED, Ha A, Kuo CJ, et al. Reserve Stem cells in intestinal homeostasis and injury. Gastroenterology 2018; 155:1348–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clevers H The intestinal crypt, a prototype stem cell compartment. Cell 2013; 154:274–84. [DOI] [PubMed] [Google Scholar]
- 3.Xiao L, Wu J, Wang JY, et al. Long Noncoding RNA uc.173 promotes renewal of the intestinal mucosa by inducing degradation of microRNA 195. Gastroenterology 2018; 154:599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Backhed F, Ley RE, Sonnenburg JL, et al. Host-bacterial mutualism in the human intestine. Science 2005; 307:1915–20. [DOI] [PubMed] [Google Scholar]
- 5.Torow N, Marsland BJ, Hornef MW, et al. Neonatal mucosal immunology. Mucosal Immunol 2017; 10:5–17. [DOI] [PubMed] [Google Scholar]
- 6.Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008;456:259–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riba A, Olier M, Lacroix-Lamande S, et al. Paneth cell defects induce microbiota dysbiosis in mice and promote visceral hypersensitivity. Gastroenterology 2017; 153:1594–1606.e2. [DOI] [PubMed] [Google Scholar]
- 8.Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol 2013; 15:713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bento CF, Renna M, Ghislat G, et al. Mammalian autophagy: how does it work? Annu Rev Biochem 2016; 85:685–713. [DOI] [PubMed] [Google Scholar]
- 10.Gukovskaya AS, Gukovsky I, Algul H, et al. Autophagy, inflammation, and immune dysfunction in the pathogenesis of pancreatitis. Gastroenterology 2017; 153:1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bel S, Pendse M, Wang Y, et al. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 2017; 357:1047–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freeman JJ, Feng Y, Demehri FR, et al. TPN-associated intestinal epithelial cell atrophy is modulated by TLR4/EGF signaling pathways. FASEB J 2015; 29:2943–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mittal R, Coopersmith CM. Redefining the gut as the motor of critical illness. Trends Mol Med 2014; 20:214–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011; 474:307–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol 2007; 8:113–26. [DOI] [PubMed] [Google Scholar]
- 16.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136:215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turner M, Diaz-Munoz MD. RNA-binding proteins control gene expression and cell fate in the immune system. Nat Immunol 2018; 19:120–129. [DOI] [PubMed] [Google Scholar]
- 18.Liu L, Christodoulou-Vafeiadou E, Rao JN, et al. RNA-binding protein HuR promotes growth of small intestinal mucosa by activating the Wnt signaling pathway. Mol Biol Cell 2014; 25:3308–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu TX, Wang PY, Rao JN, et al. Chk2-dependent HuR phosphorylation regulates occludin mRNA translation and epithelial barrier function. Nucleic Acids Res 2011; 39:8472–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao L, Wang JY. RNA-binding proteins and microRNAs in gastrointestinal epithelial homeostasis and diseases. Curr Opin Pharmacol 2014; 19:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu L, Zhuang R, Xiao L, et al. HuR enhances early restitution of the intestinal epithelium by increasing Cdc42 translation. Mol Cell Biol 2017; 37:e00574–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giammanco A, Blanc V, Montenegro G, et al. Intestinal epithelial HuR modulates distinct pathways of proliferation and apoptosis and attenuates small intestinal and colonic tumor development. Cancer Res 2014; 74:5322–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhuang R, Rao JN, Zou T, et al. miR-195 competes with HuR to modulate stim1 mRNA stability and regulate cell migration. Nucleic Acids Res 2013; 41:7905–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zou T, Jaladanki SK, Liu L, et al. H19 long noncoding RNA regulates intestinal epithelial barrier function via microRNA 675 by interacting with RNA-binding protein HuR. Mol Cell Biol 2016; 36:1332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao L, Rao JN, Cao S, et al. Long noncoding RNA SPRY4-IT1 regulates intestinal epithelial barrier function by modulating the expression levels of tight junction proteins. Mol Biol Cell 2016; 27:617–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang JY, Johnson LR. Luminal polyamines stimulate repair of gastric mucosal stress ulcers. Am J Physiol 1990; 259:G584–92. [DOI] [PubMed] [Google Scholar]
- 27.Xiao L, Rao JN, Zou T, et al. miR-29b represses intestinal mucosal growth by inhibiting translation of cyclin-dependent kinase 2. Mol Biol Cell 2013; 24:3038–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung HK, Wang SR, Xiao L, et al. α4 coordinates small intestinal epithelium homeostasis by regulating stability of HuR. Mol Cell Biol 2018; 38:e00631–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindemans CA, Calafiore M, Mertelsmann AM, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 2015; 528:560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu L, Rao JN, Zou T, et al. Polyamines regulate c-Myc translation through Chk2-dependent HuR phosphorylation. Mol Biol Cell 2009; 20:4885–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Zhang Y, Xiao L, et al. Cooperative repression of insulin-like growth factor yype 2 receptor translation by microRNA 195 and RNA-binding protein CUGBP1. Mol Cell Biol 2017; 37:e00225–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu L, Ouyang M, Rao JN, et al. Competition between RNA-binding proteins CELF1 and HuR modulates MYC translation and intestinal epithelium renewal. Mol Biol Cell 2015; 26:1797–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zou T, Rao JN, Liu L, et al. Polyamines regulate the stability of JunD mRNA by modulating the competitive binding of its 3’ untranslated region to HuR and AUF1. Mol Cell Biol 2010; 30:5021–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harter HL. Critical values for Duncan’s new multiple range test. Biometrics 1960:671–685. [Google Scholar]
- 35.Delgado MA, Elmaoued RA, Davis AS, et al. Toll-like receptors control autophagy. EMBO J 2008; 27:1110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chabot S, Wagner JS, Farrant S, et al. TLRs regulate the gatekeeping functions of the intestinal follicle-associated epithelium. J Immunol 2006; 176:4275–83. [DOI] [PubMed] [Google Scholar]
- 37.Godefroy E, Gallois A, Idoyaga J, et al. Activation of toll-like receptor-2 by endogenous matrix metalloproteinase-2 modulates dendritic-cell-mediated inflammatory responses. Cell Rep 2014; 9:1856–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Round JL, Lee SM, Li J, et al. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 2011; 332:974–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson AA, Kwok LW, Porter EL, et al. Lentiviral delivery of RNAi for in vivo lineage-specific modulation of gene expression in mouse lung macrophages. Mol Ther 2013; 21:825–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lal A, Mazan-Mamczarz K, Kawai T, et al. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J 2004; 23:3092–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iida T, Yokoyama Y, Wagatsuma K, et al. Impact of autophagy of innate immune cells on inflammatory bowel disease. Cells 2019; 8(1),7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGettrick AF, O’Neill LA. Localisation and trafficking of Toll-like receptors: an important mode of regulation. Curr Opin Immunol 2010; 22:20–7. [DOI] [PubMed] [Google Scholar]
- 43.Chaturvedi A, Pierce SK. How location governs toll-like receptor signaling. Traffic 2009; 10:621–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takahashi K, Shibata T, Akashi-Takamura S, et al. A protein associated with Toll-like receptor (TLR) 4 (PRAT4A) is required for TLR-dependent immune responses. J Exp Med 2007; 204:2963–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tapping RI. Innate immune sensing and activation of cell surface Toll-like receptors. Semin Immunol 2009;21:175–84. [DOI] [PubMed] [Google Scholar]
- 46.Ey B, Eyking A, Gerken G, et al. TLR2 mediates gap junctional intercellular communication through connexin-43 in intestinal epithelial barrier injury. J Biol Chem 2009; 284:22332–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Podolsky DK, Gerken G, Eyking A, et al. Colitis-associated variant of TLR2 causes impaired mucosal repair because of TFF3 deficiency. Gastroenterology 2009;137:209–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mukherjee S, Hooper LV. Antimicrobial defense of the intestine. Immunity 2015; 42:28–39. [DOI] [PubMed] [Google Scholar]
- 49.Wakabayashi Y, Kobayashi M, Akashi-Takamura S, et al. A protein associated with toll-like receptor 4 (PRAT4A) regulates cell surface expression of TLR4. J Immunol 2006; 177:1772–9. [DOI] [PubMed] [Google Scholar]
- 50.Mutoh H, Kato M, Akita T, et al. Biallelic variants in CNPY3, encoding an endoplasmic reticulum chaperone, cause early-onset epileptic encephalopathy. Am J Hum Genet 2018; 102:321–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu B, Yang Y, Qiu Z, et al. Folding of Toll-like receptors by the HSP90 paralogue gp96 requires a substrate-specific cochaperone. Nat Commun 2010; 1:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morales C, Li Z. Drosophila canopy b is a cochaperone of glycoprotein 93. J Biol Chem 2017; 292:6657–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hart BE, Tapping RI. Cell surface trafficking of TLR1 is differentially regulated by the chaperones PRAT4A and PRAT4B. J Biol Chem 2012; 287:16550–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peng W, Furuuchi N, Aslanukova L, et al. Elevated HuR in pancreas promotes a pancreatitis-like inflammatory microenvironment that facilitates tumor development. Mol Cell Biol 2018;38:e00427–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang X, Zou T, Rao JN, et al. Stabilization of XIAP mRNA through the RNA binding protein HuR regulated by cellular polyamines. Nucleic Acids Res 2009; 37:7623–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farin HF, Van Es JH, CleversH. Redundant sources of Wnt regulate intestinal stem cells and promote formation of Paneth cells. Gastroenterology 2012; 143:1518–29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.