

SUMMARY
Rhodopsin (Rho), a prototypical G protein-coupled receptor (GPCR) in vertebrate vision, activates the G protein transducin (GT) by catalyzing GDP-GTP exchange on its α subunit (GαT). To elucidate the determinants of GT coupling and activation, we obtained cryo-EM structures of a fully functional, light-activated Rho-GT complex in the presence and absence of a G protein-stabilizing nanobody. The structures illustrate how GT overcomes its low basal activity by engaging activated Rho in a conformation distinct from other GPCR-G protein complexes. Moreover, the nanobody-free structures reveal native conformations of G protein components and capture three distinct conformers showing the GαT helical domain (⎱HD) contacting the Gβγ subunits. These findings uncover the molecular underpinnings of G protein activation by visual rhodopsin and shed new light on the role played by Gβγ during receptor-catalyzed nucleotide exchange.
Graphical Abstract
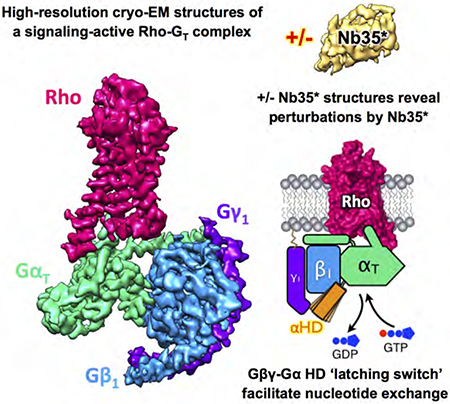
In Brief
High-resolution cryo-EM structures of a light-activated, signaling-active rhodopsin-transducin complex (Rho-GT) reveal a previously unknown mechanism involving G protein βγ subunits and the α subunit helical domain (HD) in receptor-catalyzed activation, as well as provide a structural basis for vertebrate visual phototransduction, and illustrate the structural perturbations caused by a nanobody in a GPCR-G protein complex.
INTRODUCTION
Rho, the photoreceptor evolved for dim light vision in vertebrates, is a founding member of the G protein-coupled receptor (GPCR) superfamily that includes over 800 members in humans (Fredriksson et al., 2003). GPCRs signal across the plasma membrane through the activation of heterotrimeric G proteins (Gαβγ), promoting the exchange of GDP for GTP and the dissociation of the heterotrimer into GTP-bound Gα and Gβγ subunits. The vertebrate visual phototransduction pathway (Stryer, 1991) is a prototypical G protein-coupled receptor (GPCR) signaling system, in which the signal from light is amplified through the GPCR Rho, the G protein GT (a Gi/o family member (Simon et al., 1991) with three subunits designated as GαT, Gβl, Gγ1), and phosphodiesterase 6 (PDE6), the effector enzyme that reduces the cytosolic concentration of the second messenger cGMP. Rho is composed of the apoprotein opsin and a covalently bound ligand, 11-cis retinal, which upon photon absorption, isomerizes to all-trans retinal thus activating Rho. The relatively high stability and abundance of Rho in vertebrate retinae (Nickell et al., 2003) make it an attractive model for GPCR structural studies.
Crystallization efforts on Rho have yielded the first high-resolution structures of a GPCR in its inactive, apo and agonist-bound states (Palczewski et al., 2003; Park et al., 2008; Choe et al., 2011), and in complex with arrestin (Kang et al., 2015). However, the lack of a high-resolution complex containing Rho and its physiological signaling partner GT has hindered our understanding of the molecular basis for the remarkable signal amplification attained by this system. Furthermore, while recent advances in structural biology have started to yield high-resolution structures of GPCR-G protein complexes, including a 4.5 Å cryo-EM structure of an engineered Gi bound to Rho (Kang et al., 2018), the use of dominant-negative G protein mutants (Liang et al., 2018; Draper-Joyce et al., 2018; García-Nafría et al., 2018; Kang et al., 2018) and binding partners such as nanobodies (Rasmussen et al., 2011; Zhang et al., 2017; Liang et al., 2017; Liang et al., 2018) or antibody fragments (Koehl et al., 2018; Kang et al., 2018; Kumar et al., 2019), prevents these complexes from undergoing G protein activation in the presence of GTP. Here we describe cryo-EM structures of a fully functional and signaling-active Rho-GT complex, in the presence and absence of a newly engineered nanobody that does not interfere with G protein activation. The structures reveal how Rho specifically couples to and elicits a striking stimulation of its cognate signaling partner GT, and provide new insights into the mechanism of receptor-mediated G protein activation.
RESULTS AND DISCUSSION
Isolation and structural determination of the Rho-GT complex
To form a functional complex, purified bovine rod outer segment (ROS) membranes containing native dark-state Rho were mixed in the dark with native bovine Gβ1γ1 and a recombinant GαT, in which 18 residues were replaced with the corresponding residues from Gαi1 (Skiba et al., 1996; Ramachandran et al., 2011; Milano et al., 2018) (Figure S1A), from here on referred to as rGαT. The addition of Gαi1 residues allowed rGαT to be expressed and purified from E coli and a His6-tag was introduced to the N terminus of rGαT, which is necessary for complex purification despite precluding N-terminal myristoylation. Importantly, rGαT can undergo Rho-catalyzed GDP-GTP exchange and activate the downstream enzyme PDE6 as effectively as native bovine retinal GαT (Ramachandran et al., 2011; Milano et al., 2018) (Figures S1B and S1C). Exposure to light induced Rho activation and the ensuing formation of the Rho-GT complex on ROS membranes, as depicted in Figure 1A. The complex was extracted from membranes using the detergent lauryl maltose neopentyl glycol (LMNG) (Chae et al., 2010) and subsequently purified with Ni-immobilized-metal affinity and size exclusion chromatography. Of note, the purified Rho-GT complex is fully capable of undergoing G protein activation, as it readily dissociates upon the addition of GTPγS, a non-hydrolyzable GTP analogue (Figure S1D).
Figure 1. Purification and structural determination of the rhodopsin (Rho)-transducin (GT) complex.
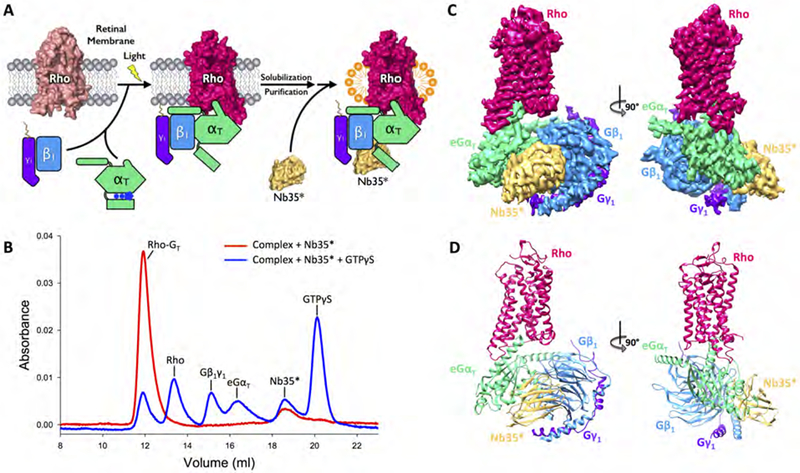
(A) Schematic illustration of the purification of the Rho-GT complex. The complex is formed on retinal membranes through light activation of Rho, solubilized with detergent, and purified through chromatography steps. The engineered nanobody (Nb35*) is then added to the Rho-GT complex in excess. (B) Size exclusion chromatography (SEC) profiles of the purified Rho-GT-Nb35* complex (red) and its dissociation upon the addition of GTPγS (blue). (C) Orthogonal views of the cryo-EM density map colored by subunit (Rho in strawberry, eGαT in lime, Gβ1 in blue, Gγ1 in purple, Nb35* in gold). (D) Structure of the Rho-GT-Nb35* complex in the same views and color scheme.
See also Figures S1–S3 and Table S1.
In the process of optimizing complex formation for structural studies we tested the use of Nb35, the nanobody that was originally used to obtain the structure of the β2 adrenergic receptor-GS protein complex (β2AR-GS) and shown to bind at the interface between the α and β subunits of the G protein GS (Rasmussen et al., 2011). The native Nb35 was unable to bind to the Rho-GT complex formed with rGαT, prompting us to perform protein engineering on both rGαT and the nanobody (the residues mutated in rGαT and Nb35 are illustrated in Figures S2A and S2B respectively). The engineered Nb35, termed Nb35*, binds to the Rho-GT complex formed with the engineered rGαT subunit, termed eGαT, in a 1:1 ratio (Figure S2C). This eGαT subunit is fully capable of undergoing Rho-catalyzed GDP-GTP exchange, exhibiting the same fold-stimulation as obtained with wild-type rGαT (Figure S2D). More importantly, unlike GPCR-G protein complexes formed with GS and Nb35, the Rho-GT-Nb35* complex readily dissociates upon the addition of GTPγS and is thus still capable of undergoing nucleotide exchange and G protein activation (Figure 1B).
Using single-particle cryo-EM, we determined three-dimensional maps of the Nb35*-bound and the Nb35*-free Rho-GT complexes with indicated global resolutions of 3.3 Å and 3.9 Å, respectively, facilitating the refinement of atomic models (Figures 1C and 1D, Figures S3 to S5, Table S1). Apart from distinct differences related to nanobody binding described further below, the two structures are overall similar, with the lower global resolution of the Nb35*-free complex reflecting the increased dynamics between the rGαT and Gβγ modules in the absence of the stabilizing nanobody.
Structural basis of Rho-GT coupling
The structure of the Rho-GT-Nb35* complex reveals that light-activated Rho forms extensive contacts with GT resulting in an interface area of 1042 Å2. This interface is contributed by transmembrane helix 3 (TM3), intracellular loop 2 (ICL2), TM5, TM6 and helix 8 (H8) of Rho, along with the α5 helix, α4-β6 loop, β2-β3 loop and αN-β1 loop of eGαT (Figure S6A). Although Rho is capable of activating G proteins from the inhibitory Gi/o family, GT is the only cognate G protein present within the physiological setting of rod photoreceptor cell outer segments. Moreover, the basal nucleotide exchange rate for GT is markedly lower than that for Gi, and as a result, the activation by Rho elicits a 107-fold increase in the nucleotide exchange rate of GT (Vuong et al., 1984). This mechanism gives rise to the striking signal-to-noise ratio that is essential to the signal amplification required for vertebrate vision (Figure 2A). In addition, only activated GT, but not Gi, is capable of activating PDE6, the downstream phosphodiesterase that is essential for phototransduction (Figure 2B).
Figure 2. Comparison between the Rho-GT and Rho-Gi complexes.
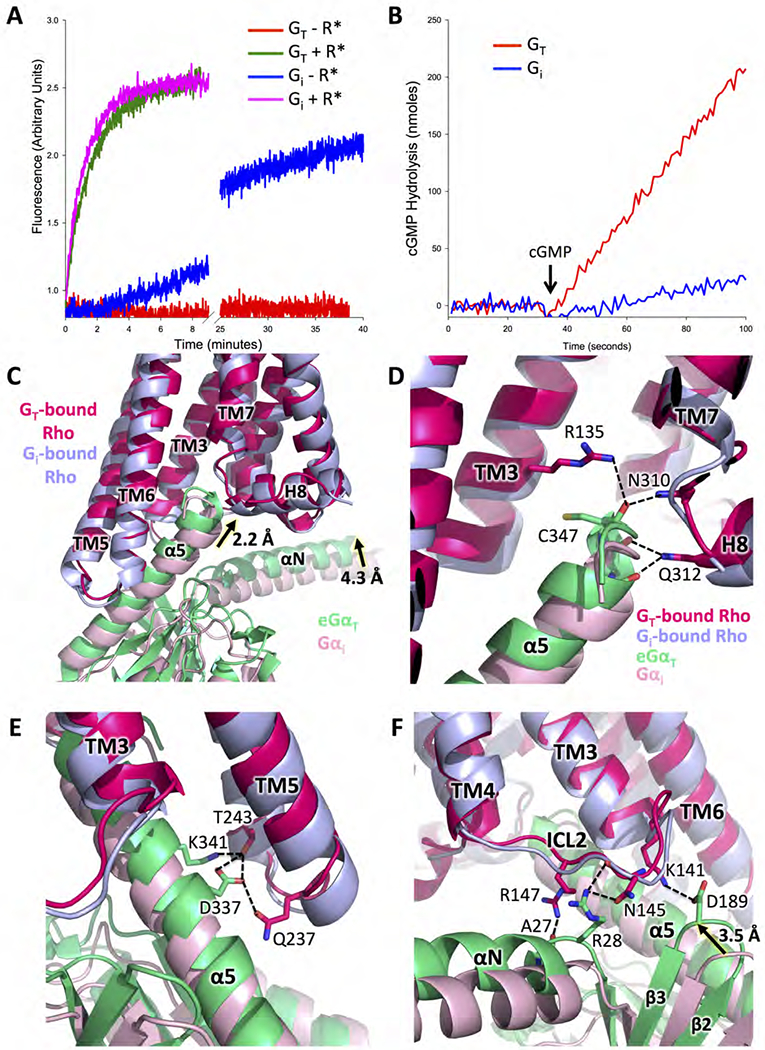
(A) Basal and Rho-catalyzed nucleotide exchange activities for GT and Gi, as monitored by the changes in Trp fluorescence that accompany G protein activation (GT: basal exchange in red, Rho-catalyzed exchange in green; Gi: basal exchange in blue, Rho-catalyzed exchange in magenta). (B) Comparisons of cyclic GMP hydrolysis by the cGMP phosphodiesterase 6 (PDE6), the physiological effector of GT, as stimulated by GTPγS-bound GT, versus GTPγS-bound Gi (GT in red, Gi in blue). (C) Comparison of Gα subunit conformation based on receptor alignment in the structures of the Rho-GT and the Rho-Gi complexes (PDB: 6CMO). eGαT inserts deeper into Rho than Gαi, allowing for more extensive interactions (GT-bound Rho in strawberry, Gi-bound Rho in light blue, eGαT in lime, Gαi in pink). (D) Interactions between the C terminus of eGαT and Rho compared to the same region in the Rho-Gi complex. (E) Interactions between the α5 helix of eGαT and TM5, TM6 of Rho compared to the same region in the Rho-Gi complex. (F) Interactions between ICL2 of Rho and eGαT compared to the same region in the Rho-Gi complex.
See also Figures S1, S2 and S6.
The 3.3 Å structure of the Rho-GT-Nb35* complex, compared to the recent 4.5 Å structure of the Rho-Gi-Fab_G50 complex (Kang et al., 2018), provides a 3D platform for understanding the highly specific interactions between Rho and its cognate G protein partner GT. In the receptor region, the conformation Rho in complex with GT is mostly similar to previous structures of Rho in complex with either the 11-residue GαT C-terminal peptide (GαTCT) (RMSD = 0.61 Å) (Choe et al., 2011) (Figure S6B) or Gi (RMSD = 1.65 Å) (Figure 2C). The outward tilt of TM6 in Rho when engaged with GT is significantly smaller than that observed in GS-coupling GPCR-G protein complexes, such as the β2AR-GS complex (Figure S6C) (Rasmussen et al., 2011), but agrees well with the conformation in Gi/o-coupling GPCR-G protein complexes like the μ-opioid receptor-Gi protein complex μOR-Gi (Figure S6D) (Koehl et al., 2018), supporting the notion that the different degrees of opening at the cytoplasmic side of the receptor contribute to selective coupling to either GS or Gi/o families of G proteins. The most significant difference between the Rho-GT and Rho-Gi complex structures lies within the G proteins. The C-terminal α5 helix in GT inserts deeper into Rho by 2.2 Å compared to Gαi (Figure 2C), in good agreement with the conformation of the GαT C-terminal peptide observed in its crystal structure with Rho (Figure S6B). Consequently, the backbone carbonyl groups of residues at the C-terminal end of the eGαT α5 helix are within hydrogen-bonding distance with side chains from N3108.47 and Q3128.49 (superscript indicating Ballesteros-Weinstein numbering for GPCRs (Ballesteros and Weinstein, 1996) at the N terminus of H8 (Figure 2D). In addition, the carbonyl group of C347G.H5.22 of eGαT (superscript indicating common Gα numbering (CGN) system (Flock et al., 2015)) interacts with R1353.50 from the conserved E(D)RY motif of Rho, thus stabilizing the active-state Rho conformation (Figure 2D). The upward shift in the position of the α5 helix leads to a rigid body movement of the entire a subunit, enabling it to make more extensive contacts with ICL2 and ICL3 of Rho. Within the ICL3 region (Figure 2E), T2436.26 and Q2375.72 from the cytoplasmic ends of TM5 and TM6 form hydrogen bonds with D337G.H5.13 and K341G.H5.17, stabilizing the conformation of the α5 helix as it exits from Rho. In the case of ICL2 (Figure 2F), the upward movement of eGαT leads to interactions between A27G.hns1.02 and R28G.hns1.03 in the αN-β1 loop, D189G.s2s3.02 in the β2-β3 loop of eGαT, and residues R14734.55, N14534.53 and K1413.56 of Rho. These interactions are not present in the Rho-Gi complex structure (Kang et al., 2018). Notably, in a recent crystal structure of the complex between Rho and a truncated version of the Go α subunit (miniGαo) (Tsai et al., 2018), although the C-terminal position of the miniGαo α5 helix C terminus agrees well with that in both the Rho-GαTCT and our Rho-GT complex, the miniGαo α5 helix tilts downwards when exiting the receptor compared to that in Rho-GT (Figure S6E). Consequently none of the interactions between the ICL2 and ICL3 of Rho and the G protein observed in Rho-GT is present in the Rho-miniGαo structure. This difference in Rho-coupling could be attributed to either the differences in sequence identities between GαT and Gαo, or the by the significant modifications introduced to miniGαo in order to allow for receptor-coupling in the absence of the Gβγ subunits, i.e. both the helical domain and N-terminal αN helix were truncated and 7 thermo-stabilizing mutations, including one dominant-negative mutation, were introduced.
It has been established that the β2-β3 loop (residue K188G.s2s3.01) is involved in maintaining low basal nucleotide exchange activity through ionic interactions with the α5 helix residues D333G.H5.09 and D337G.H5.13 in the GDP-bound state of GαT (Lambright et al., 1996; Marin et al., 2001) (Figure 3A). Our structure shows that contacts provided by ICL2 and ICL3 in the Rho-GT complex drive the disruption of these interactions (Figure 3B). Thus, these contacts are likely necessary for Rho to overcome the high affinity of GT for GDP, enabling the photoreceptor to give rise to a striking stimulation of nucleotide exchange. In addition, in the presence of Rho, the C-terminal end of eGαT, which is unstructured in the GDP-bound GT heterotrimer (Lambright et al., 1996), extends the α5 helix by one helical turn, pulling it upward into the receptor (Figures 3A and 3B). The movement of the α5 helix not only displaces the guanine-ring-binding β6-α5 loop, but also disrupts the hydrophobic pocket formed by residues from the β2 and β3 strands, and the α1 and α5 helices in the GDP-bound state. As a result, the α1 helix of eGαT tilts toward the β6-α5 loop and becomes unstructured in the nucleotide-free state. Since the α1 helix is connected to the P-loop and to the helical domain (αHD) at its N- and C-terminal ends, respectively, structural changes in this region directly perturb nucleotide binding and promote the separation of the αHD from the Ras domain.
Figure 3. Conformational changes in the α5 helix and its surrounding regions upon Rho engagement.
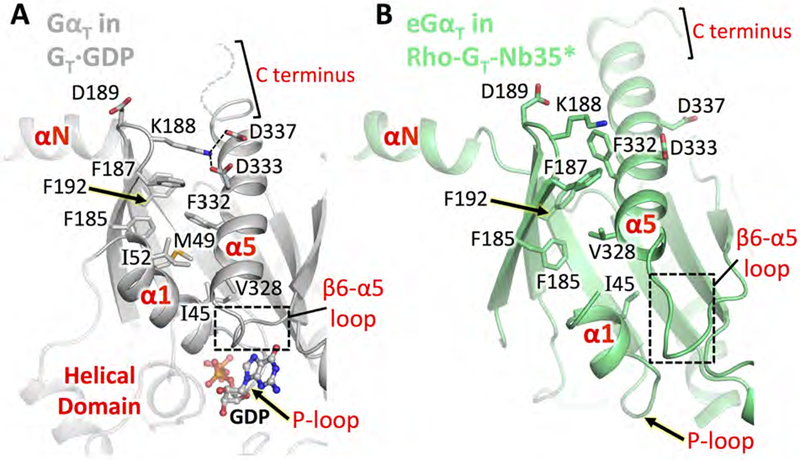
Comparison of Ras domain structural elements in GT·GDP and Rho-GT-Nb35*. (A) GαT in GT·GDP (PDB: 1GOT, grey). Residues 344-LKDCGLF-350 at the C terminus are shown as a dashed curve as they are not resolved in the GT·GDP crystal structure. The helical domain is shown as a transparent cartoon for reference. (B) eGαT in the Rho-GT-Nb35* complex (lime) aligned to GT·GDP based on Gβ1γ1.
See also Figures S3 and S5.
The ICL2 of Rho adopts an extended-loop conformation in the Rho-GT complex presenting polar residues to form electrostatic interactions with eGαT (Figure 4A). This configuration, in agreement with the structure of the Rho-Gi complex (Kang et al., 2018), is significantly different from what has been observed for other Class A GPCRs in complex with G proteins, where the ICL2 adopts an α-helical conformation (Rasmussen et al., 2011; Koehl et al., 2018) and exposes nonpolar residues to interact with a hydrophobic groove in Gα (Figures 4B and 4C). Interestingly, ICL2 of Rho is also capable of folding into an α helix as observed in the Rho-arrestin complex structure (Kang et al., 2015) (Figure 4D). In that complex, the ICL2 helix exposes nonpolar residues at identical positions (M14334.51, F14634.54) as in other GPCR-G protein complexes to interact with a hydrophobic pocket opened up by the rotation of the arrestin N- and C-terminal domains. Therefore, the conformational plasticity of the receptor intracellular loops, especially ICL2, likely not only contributes to the coupling to G proteins, but may also play a role in the selectivity of different GPCR signaling partners.
Figure 4. Comparisons of ICL2 in the Rho-GT-Nb35* complex and other GPCR complexes.
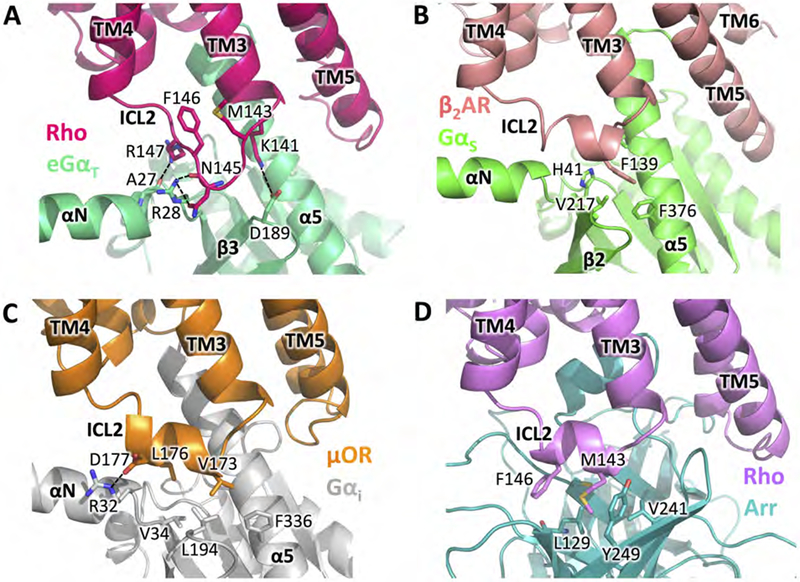
(A) Conformation of ICL2 in GT-bound Rho and its interactions with eGαT in the Rho-GT-Nb35* complex (GT-bound Rho in strawberry, GαT in lime). (B) Conformation of ICL2 in β2AR and its interactions with GαS in the β2AR-GS complex (PDB: 3SN6) (β2AR in salmon, GαS in green). (C) Conformation of ICL2 in μOR and its interactions with Gαi in the μOR-Gi complex (PDB: 6DDF) (μOR in orange, Gαi in grey). (D) Conformation of ICL2 in arrestin-bound Rho and its interactions with arrestin in the Rho-arrestin complex (PDB: 4ZWJ) (arrestin-bound Rho in purple, arrestin in teal).
See also Figure S6.
Structural effects induced by the nanobody
The addition of Nb35, which binds at the interface between the GS protein α and β subunits, was essential for obtaining the crystal structure of the β2AR-GS complex (Rasmussen et al., 2011). Nb35 has since been used in determining all subsequent GPCR-GS cryo-EM structures (Zhang et al., 2017; Liang et al., 2017; Liang et al., 2018), as it helps to rigidify the conformation of the G protein and thus assists in structure determination. However, binding of Nb35 to nucleotide-free GS prevents the GTP-induced dissociation of the complex (Rasmussen et al., 2011), raising the question of whether the Nb35-bound structures represent a bona fide state poised for G protein activation. As our engineered Nb35* binds at the same position in GT as in GS, comparisons between the Nb35*-bound and Nb35*-free Rho-GT complex structures allow for the direct visualization of the structural effects caused by nanobody binding. The most notable differences between the two structures involve the GαT subunit. In the absence of Nb35*, although the interaction between switch II (SWII) of rGαT and θβ1γ1 is maintained, the remainder of the rGαT Ras domain moves further away from Gβ1γ1 (Figure 5A). As a result, the GDP-binding modules, such as the β6-α5 loop, the αG helix and the P-loop, also move further away from their original positions in the GDP-bound GT heterotrimer (Figure 5B). We postulate that this inter-subunit separation in GT promotes the dissociation of Gβ1γ1 from GαT upon GTP binding. On the other hand, addition of the nanobody interferes with this separation and therefore potentially slows the rate of receptor-mediated nucleotide exchange. This is indeed the case with GS, where the presence of Nb35 appears to totally impede this reaction. By contrast, Nb35* does not appear to interfere with nucleotide exchange in the Rho-GT complex, likely due to the lower affinity of this engineered nanobody for the G protein compared to the original Nb35 raised in Llamas. We also note that in addition to perturbing the overall configuration of eGαT, the nanobody causes local conformational changes in the switch III (SWIII) region, which is not resolved in the Nb35*-bound complex due to clashes with the nanobody (Figure 5A).
Figure 5. Nanobody-induced structural changes in the Rho-GT complex.
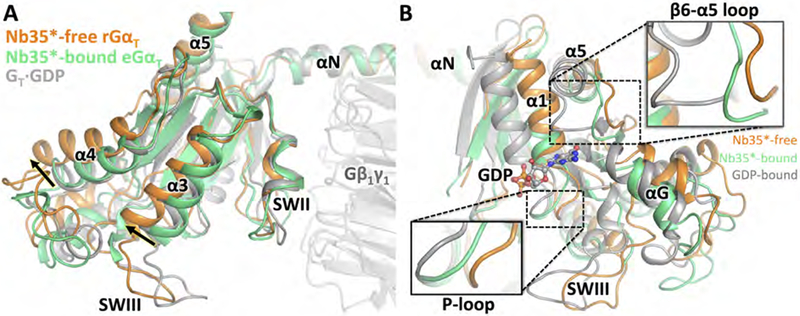
(A) Structures of eGαT in the nanobody-bound Rho-GT complex (lime), rGαT in the nanobody-free Rho-GT complex (orange), and GDP-bound GT (PDB: 1GOT, grey), overlaid based on the alignment of the Gβ1γ1 subunit. (B) Conformational changes in the nucleotide-binding pocket.
See also Figures S3, S4 and S5.
Stabilization of the GαT helical domain by Gβ1γ1 and its implications for GT activation
The lack of high resolution density for the αHD in both the Nb35*-bound and Nb35*-free Rho-GT maps suggests that it assumes variable positions, in agreement with earlier negative-stain EM (Westfield et al., 2011; Gao et al., 2017) and electron paramagnetic resonance (EPR) (Van Eps et al., 2011) studies, as well as the recently determined cryo-EM structures of other GPCR-G protein complexes. Strikingly, however, our single-particle classification reveals that a substantial population (~35%) of both the Nb35*-bound and Nb35*-free Rho-GT complex shows relatively stable density for the open αHD (Figures S3C and S4C). Further 3D classification of these particle projections from the Nb35*-free Rho-GT complex enabled us to obtain three independent 4.5 Å resolution maps showing αHD in contact with Gβ1γ1 in three distinct conformations (Figures 6A to 6D and Figures S7A to S7C). The orientation of Gβ1γ1 in these three structures is identical to that observed in the 3.9 Å structure of the Nb35*-free Rho-GT complex, except in one conformer where Gβ1γ1 rotates slightly counter-clockwise when viewed downward from Rho due to an 8° tilt of the rGαT αN helix (Figure S7D). Even though the densities for the αHD have poor resolution due to its mobility, they provide sufficient secondary structure features to enable rigid-body docking in unique conformations (Figures S7A to S7C). The αHD in the three structures is open to varying degrees compared to the crystal structure for the GDP-bound GT heterotrimer (Lambright et al., 1996) (opening by 66°, 68° and 91°, respectively, based on the orientation of the αA helix in αHD), but is not as extensively delocalized as in the crystal structure of the β2AR-GS complex (where it opens by 132°; Rasmussen et al., 2011). The low resolution models of the three conformers suggest that electrostatic interactions occur between polar residues on the outer rim of the 2nd and 3rd β propeller blades of Gβ1, and residues from three regions on the αHD, namely Q75H.HA.17, R82H.HA.24, T86H.HA.28 in the αA helix, D108H.HB.14, E111H.hbhc.03, E112H.hbhc.04 in the αB-αC loop, and E141H.HD.12, Q143H.hdhe.02 in the αD-αE loop (Figures 6B to 6D and Figures S7A to S7C). These interactions are not present in the Rho-Gi-Fab_G50 complex structure (Kang et al., 2018), in which the additional Fab fragment (Fab_G50) binds at the interface between the αHD and Gβ1, thus preventing the αHD from adopting the conformations observed in the Nb35*-free Rho-GT complex.
Figure 6. GαT Helical domain conformations in the Nb35*-free Rho-GT complex.
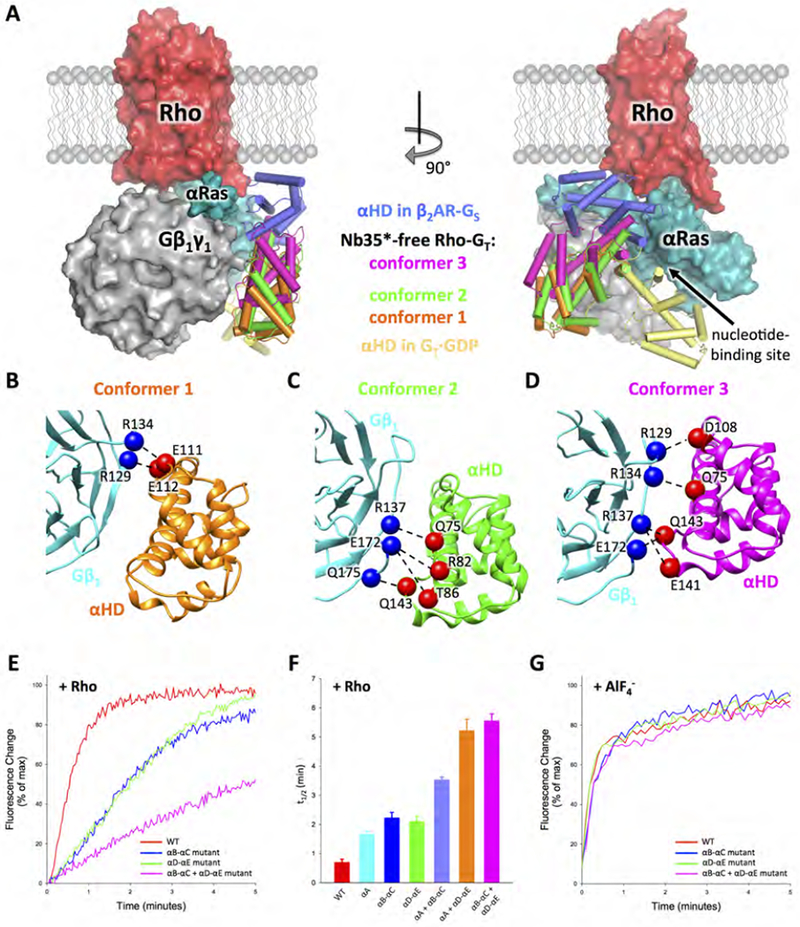
(A) Orthogonal views of the complex with the rGαT helical domain (αHD) in the three observed distinct conformations (colored in orange, green and purple respectively) compared to the conformations of the helical domain observed in the β2AR-GS complex (PDB: 3SN6, dark blue) and GDP-bound GT (PDB: 1GOT, yellow) crystal structures. The comparison is based on the Gα Ras domain alignment. Rho (red), Gβlγ1 (grey) and rGαT Ras domain (cyan) are from the 3.9 Å Nb35*-free complex structure. (B-D) Interactions between αHD and Gβ1 revealed by docking the αHD crystal structure (PDB: 1GOT) and the Gβ1 structure from the 3.9 Å Nb35*-free Rho-GT complex into the three 4.5 Å Nb35*-free complex cryo-EM maps. Gβ1 is colored in cyan. αHD is colored in orange, green and purple, respectively, in three different conformers. Residues from Gβ1 and αHD, that are positioned to form contacts, are shown as blue and red spheres respectively. (E) Trp fluorescence assay monitoring Rho-catalyzed nucleotide exchange in wild-type rGαT (red), the rGαT helical domain mutants changing αB-αC loop residues (E111A, E112A) (blue), αD-αE loop residues (E141A, Q143A) (green) or residues from both αB-αC loop and αD-αE loop (D108A, E111A, E112A, E141A, Q143A) (magenta). (F) t1/2 (time needed for 50% of the fluorescence change to occur) of Rho-catalyzed nucleotide exchange in rGαT wild-type and mutants from Figure 6E, and the rGαT helical domain mutants (fluorescence curves shown in Figure S7E) changing αA helix residues (Q75A, R82A, T86A) (cyan), residues from both αA helix and αB-αC loop (Q75A, R82A, T86A, D108A, E111A, E112A) (light blue), or residues from both αA helix and αD-αE loop (Q75A, R82A, T86A, E141A) (orange). Data represents mean ± SEM (n=3). (G) Trp fluorescence assay monitoring response to the addition of AlF4− in both wild-type and mutant GαT (color scheme the same as in Figure 6E).
See also Figures S4, S7 and Tables S2 and S3.
The αHD residues positioned to interact with Gβ1, with the exception of Q143H.hdhe.02, are all located on the solvent-exposed surface and are not involved in any interactions with either the rGαT Ras domain or Gβ1γ1 in the GT heterotrimer crystal structure (Lambright et al., 1996). Although Q143H.hdhe.02 is involved in contacting SWIII in the GDP-bound state, the Q143A point mutation in GT has been shown to have no effect on the basal or Rho-catalyzed nucleotide exchange rates (Marin et al., 2001). Three rGαT mutants, in which residues that contact Gβ1 from each of three regions of the αHD (i.e. the αA helix, αB-αC loop and αD-αE loop) were changed en bloc to Ala, each showed moderate decreases in the rate of Rho-catalyzed nucleotide exchange (Figures 6E and 6F, and Figures S7E), as monitored by changes in the intrinsic Trp fluorescence of rGαT that occur as a result of conformational changes in its SWII region (Phillips and Cerione, 1988). Moreover, these mutations have an additive effect, as mutating αHD residues from any two of the three regions in combination markedly reduced the Rho-stimulated exchange activity (Figures 6E and 6F, and Figures S7E). Furthermore, the rGαT mutants are all responsive to AlF4− in the same manner as wild-type rGαT, as monitored by the same Trp fluorescence assay (Figures 6G and S7F), demonstrating that these amino acid substitutions do not impair the ability of rGαT to undergo activating conformational changes. Taken together, these results point to a previously unappreciated role for Gβ1 in promoting Rho-catalyzed nucleotide exchange by stabilizing the αHD through a series of polar interactions. Based on these findings, we propose a ‘latching switch’ mechanism (Figure 7), where Gβ1 sequesters αHD, thus helping it to remain separated from the rGαT Ras domain upon receptor coupling, providing a clear exit route for GDP. As our assays have shown, ablating these interactions reduces the rate of receptor-induced nucleotide exchange activity of the G protein (Figures 6E and 6F, and Figures S7G), presumably because there are enhanced opportunities for the more flexible αHD to close over the Ras domain, thereby hindering its ability to rapidly release GDP and bind GTP. In addition, we postulate that these interactions with Gβ1 also maintain αHD in relatively close proximity to the Ras domain, which would ensure the rapid closure of the αHD against the Ras domain upon GTP loading.
Figure 7. Schematic illustrating the ‘latching switch’ mechanism of Rho-catalyzed nucleotide exchange in GT.
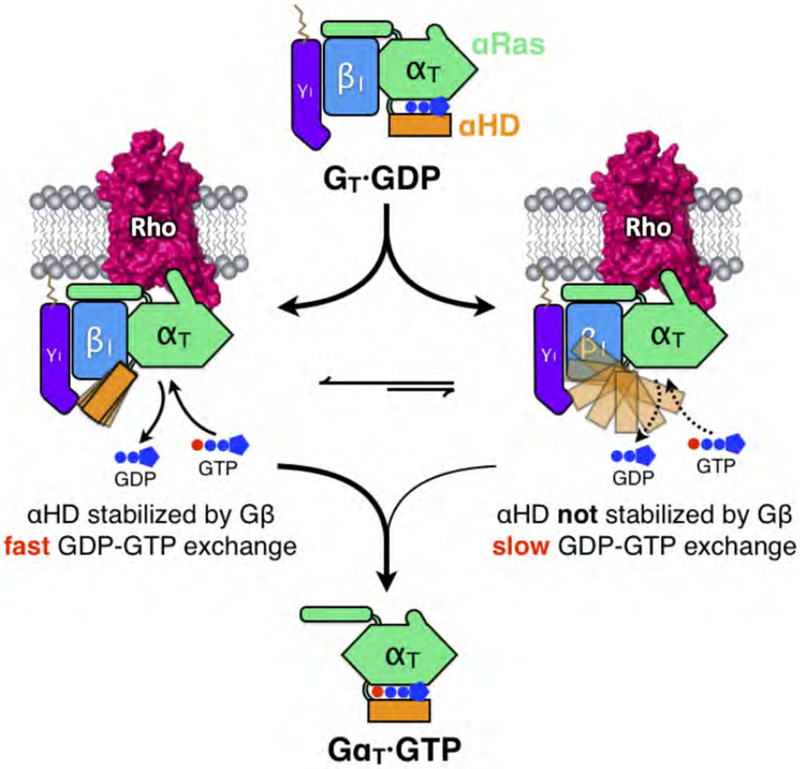
Coupling with Rho disengages αHD from the Ras domain and increases the flexibility of αHD. In a significant population of the Rho-GT complex particles, Gβ latches onto αHD and stabilizes it at an open conformation through a series of electrostatic interactions. These interactions prolong the opening of the nucleotide-binding pocket providing a clear exit route for GDP, while also positioning the αHD in close proximity to the Ras-like domain to facilitate quick closure of the clam-shell-like structure upon GTP loading. The Gβ-αHD interactions allow for fast GDP-GTP exchange and when these interactions are disrupted in the αHD mutants, Rho-catalyzed nucleotide exchange activity is markedly reduced.
The Gβ1 residues involved in the αHD interactions are highly conserved across all Gβ subtypes (Table S2), while the αHD residues involved in this interface are also highly conserved within the Gi/o family, and to varying degrees in other G protein families (Table S3). In agreement with this observation, unmasked and unsharpened cryo-EM reconstructions of our recently reported μOR-Gi (Koehl et al., 2018) and CB1-Gi (Krishna et al., 2019) complexes show the αHD occupying a similar overall position as in the Rho-GT complex, albeit with somewhat increased flexibility, which may be attributed to the Fab fragment (scFv16) making contact with the outer rim of the 2nd β propeller blade of Gβ1 (Figures S7G and S7H). Interestingly, αHD residues that contact Gβ1 are significantly less conserved in Gq/11, which may help to explain why its receptor-catalyzed nucleotide exchange rate has been reported to be slower than that of Gs or Gi (Berstein et al., 1992). Thus, specific Gα subtypes may encode the propensity of their αHD to interact with the Gβ subunit, providing an additional mechanism for modulating receptor-catalyzed nucleotide exchange rate in G proteins.
Conclusions
The cryo-EM structures of the Rho-GT complex provide important insights into the structural and mechanistic basis by which Rho promotes a striking stimulation of nucleotide exchange in GT, the critical first step in vertebrate vision. The deeper insertion of GT into Rho, when compared to Gi, gives rise to additional electrostatic interactions between the intracellular loops of Rho and the Ras domain, thereby enabling Rho to overcome the high affinity of GT for GDP. Compared to earlier structures of GPCR-G protein complexes, the Rho-GT structures highlight the conformational plasticity of ICL2 of GPCRs, which contributes to their selective coupling to different signaling partners. In addition, comparisons between the structures of the Rho-GT-Nb35* complex and the Nb35*-free complex illustrate that nanobody binding at the Gα-Gβ interface is positioned to restrict the conformational changes in Gα Ras domain that promote the inter-subunit separation, thus interfering with GTP-induced G protein subunit dissociation. These effects will need to be taken into account when interpreting existing structures of GPCR-GS protein complexes determined with the original Nb35. Finally, the cryo-EM analysis of the Nb35*-free Rho-GT complex provides direct structural evidence of the active involvement of the Gβγ subunits in nucleotide exchange. While such a role has been alluded to in various earlier biochemical (Rondard et al., 2001; Cherfils et al., 2003; Ford et al., 1998) and biophysical (Van Eps et al., 2006; Oldham et al., 2007) studies, until now, no structural evidence for such an involvement had been presented. Through sequestering the flexible αHD in a moderately open conformation while still in relatively close proximity to the Ras domain, the Gβγ subunits may help to facilitate the dissociation of GDP and then allow for the rapid closure of the nucleotide-binding pocket in the presence of GTP. Given the conservation of the Gβ and helical domain interface residues, it is very likely that similar interactions will also contribute to the receptor-catalyzed activation of other G proteins.
Star★Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reasorces and reagents should be directed to and will be fulfilled by the Lead Contact, Georgios Skiniotis (yiorgo@stanford.com).
METHOD DETAILS
Purification of retinal proteins
Urea-washed rod outer segment (UROS) membranes were isolated as described (Min et al., 2000) from dark-adapted bovine retinae (W.L. Lawson Co., Lincoln, NE), flash-frozen and stored at −80°C in HMN buffer (20 mM HEPES pH 7.5, 5 mM MgCl2, 100 mM NaCl, 1 mM DTT) at a concentration of 300 μM. UROS membranes were used as the source for Rho in Rho-GT complex formation. The Gβ1γ1 subunit complex was purified essentially as described previously (Ramachandran et al., 2011). Bovine retinae were exposed to light and subjected to sucrose gradient ultra-centrifugation to prepare purified rod outer segment (ROS) membranes. After a series of isotonic and hypotonic washes, 100 μM GTP was added to release GT subunits from the membrane. Gβ1γ1 was separated from GαT through a 5 mL HiTrap Blue HP (GE Healthcare) column and the resulting Gβ1γ1 complex was further purified by anion exchange chromatography through a 5 mL HiTrap Q HP (GE Healthcare) column, using Buffer A (20 mM HEPES pH 7.5, 5 mM MgCl2, 1 mM DTT, 10% glycerol) and Buffer B (Buffer A + 1 M NaCl) to form the gradient. The Gβ1γ1 complex typically elutes at 100 mM NaCl and was concentrated to 20 μM, flash-frozen and stored at −80°C.
Engineering and purification of rGαT and Nb35*
A homology model of rGαT was generated using SWISS-MODEL (Waterhouse et al., 2018) based on the structure of GαS in the β2AR-GS-Nb35 complex (PDB: 3SN6). Residues in GαS that are involved in interactions with Nb35 were introduced into the corresponding positions of rGαT (Figure S2A), and residues in Nb35 that may cause steric clashing with rGαT were mutated to either smaller residues or Ala (Figure S2B). Both the wild-type rGαT and engineered eGαT were expressed in E. coli BL21(DE3) competent cells and purified as described previously (Milano et al., 2018). The proteins were concentrated to 20 mM in HMN buffer with the addition of 10% glycerol, flash-frozen and stored at −80°C. The engineered nanobody, Nb35*, was expressed in the periplasm of E. coli strain WK6, then purified as described previously (Rasmussen et al., 2011), and stored at 4 °C in HN buffer (20 mM HEPES pH 7.5, 100 mM NaCl).
Rho-GT complex formation and purification
GT was formed by mixing 22 nmol of rGαT (or eGαT) with 20 nmol Gβ1γ1, and incubated on ice for 5 minutes. The Rho-GT complex was formed on ROS membranes by mixing GT with UROS containing 140 nmol Rho and illuminating the mixture under a halogen lamp covered with a UV-absorbing glass and a 495 nm long-pass filter at 4°C for 30 min. The suspension was centrifuged at 14,000g for 30 min. From here on, all subsequent steps were carried out in the dark under dim red light. The supernatant was discarded and the pellets containing the Rho-GT complex and excess Rho were re-suspended in 3 mL HMNT buffer (20 mM HEPES pH 7.5, 2 mM MgCl2, 100 mM NaCl, 100 μM TCEP) + 1% (w/v) LMNG. The mixture was incubated at 4°C with rocking for 30 min to allow for complete solubilization. HMNT buffer (12 mL) was then added to lower the LMNG concentration to 0.2% and the sample was further incubated at 4°C with rocking for 1 hour. Solubilized complex with 10 mM imidazole was loaded onto a 1 mL HisTrap HP column (GE Healthcare) pre-equilibrated with HMNT buffer + 0.02% LMNG. The Rho-GT complex was eluted from the column with an imidazole gradient in HMNT buffer + 0.02% LMNG, as a single peak at ~100 mM imidazole. Peak fractions were pooled and concentrated with an Amicon Ultra-0.5 100 kD MWCO concentrator (EMD Millipore) to 500 μL and injected onto a Superdex 200 10/300 GL column (GE Healthcare), pre-equilibrated with HMNT buffer + 0.005% LMNG. Peak fractions were pooled and concentrated with an Amicon Ultra-0.5 100 kD MWCO concentrator (EMD Millipore) to 50 μL, resulting in a complex concentration of ~40 mg/mL. For the Rho-GT-Nb35* complex, Nb35* was added to the Rho-GT complex in a 1.1:1 molar ratio and incubated at 4 °C overnight.
rGαT helical domain mutations and Trp fluorescence assays
The helical domain mutations were introduced into the rGαT construct using the R-F cloning protocol (Bond and Naus, 2012). The mutants were expressed and purified in the same way as the wild-type protein. Fluorescence measurements were carried out with a Varian eclipse spectrofluorimeter. UROS was light activated by incubation on ice under ambient light for 2 min. Rho-catalyzed nucleotide exchange on the rGαT subunit was observed by monitoring fluorescence with the excitation wavelength set at 300 nm (Slit width = 5 nm) and the emission wavelength set at 345 nm (slit width = 10 nm). Rho (10 nM), 250 nM β1γ1 and 250 nM rGαT were premixed in 1 ml of HMND buffer (20 mM HEPES pH 7.5, 5 mM MgCl2, 100 mM NaCl, 0.02% DDM) and the fluorescence emission was monitored in real-time. Subsequently, 50 μM GTPγS was added. All kinetic traces were corrected for the background fluorescence from Rho and Gβ1γ1. The initial portion of the kinetic traces monitoring the fluorescence changes were fitted to a straight line. The fitted values of the slope and intercept were used to calculate the t1/2 values, the time needed for 50% of the fluorescence change to occur. The binding of aluminum fluoride (AlF4−) was measured by monitoring the intrinsic tryptophan fluorescence of rGαT (excitation: 280 nm; emission: 340 nm). rGαT (500 nM) was mixed with 1 ml of HMND buffer at room temperature and the fluorescence emission was monitored in real-time. Subsequently, AlF4− (5 mM NaF and 50 μM AlCl3) was added.
Measurement of PDE activity
cGMP hydrolysis by PDE was measured and analyzed as described previously (Liebman and Evanczuk, 1982). Typically, in 200 μL of reaction buffer containing 10 mM Tris pH 8.0, 2 mM MgCl2, and 100 mM NaCl, 1 μM GTPγS-loaded Gα subunit was incubated with 50 nM PDE purified from bovine retinae. The pH (in mV) was monitored in real time, and then once a stable baseline was achieved, 5 mM cGMP was added and the decrease in pH was recorded for 150 sec. The buffering capacity of the mixture was obtained by adding NaOH (400 nmol). The hydrolysis rate of cGMP (nmol/sec) was determined from the ratio of the initial slope of the pH record (mV/sec) and the buffering capacity of the assay buffer (mV/nmol).
Cryo-electron microscopy data collection and processing
Cryo-EM samples of both Nb35*-bound and Nb35*-free Rho-GT complexes were prepared in the same manner. Three μL samples of each complex were applied to glow-discharged 200 mesh grids (Quantifoil R1.2/1.3) and subsequently vitrified using a Vitrobot Mark IV (Thermo Fischer Scientific). Light was avoided as much as possible during sample freezing. Cryo-EM images of the Rho-GT-Nb35* complex were collected on a Titan Krios operated at 300 kV at a nominal magnification of 130,000x with a Gatan GIF Quantum ES Imaging energy filter, using a Gatan K2 Summit direct electron camera in counting mode, corresponding to a pixel size of 1.06 Å. A total of 3837 image stacks were obtained with a defocus range of −1.2 to −2.2 μm. Each stack movie was recorded for a total of 8 seconds with 0.2s per frame. The dose rate was 7 e/Å2/s, resulting in an accumulated dose of 50 electrons per Å2. The images of Nb35*-free complex were collected on a Titan Krios operated at 300 kV at a nominal magnification of 29,000x, using a Gatan K2 Summit direct electron camera without energy filter in counting mode. The data for the Nb35*-free sample included 12,616 movie stacks at a pixel size of 0.86 Å and with an accumulated dose of 70 electrons per Å2. Dose fractionated image stacks were subjected to beam-induced motion correction and filtered according to the exposure dose using MotionCor2 (Zheng et al., 2017). The sum of each movie was applied to CTF parameters determination by Gctf v1.06 (Zhang, 2016). Particle selection, two-dimensional and three-dimensional classification, and 3D reconstruction were performed using RELION 2.1 (Scheres, 2012). In the next step, Bayesian polishing was performed in RELION 3.0 using the final particle set used for the RELION 2.1 reconstructions. In a final step, the polished particles were used to obtain the final 3D reconstructions using cisTEM (Grant et al., 2018).
For the Rho-GT-Nb35* complex, a total of 2.3 million projections were initially extracted and subjected to 2D and subsequent 3D classification. Two 3D classes accounting for 309,116 projections were used to obtain an initial map with indicated resolution of 3.7 Å resolution. The data was further processed by Bayesian polishing and were imported into cisTEM for a final round of local refinement and reconstruction using automasking in cisTEM. The indicated resolution of the final reconstruction is 3.3 Å.
For the Nb35*-free complex, a subset of 1.6 million projections obtained after 2D classification was subjected to a first round of 3D classification into 8 classes. Two of the classes showed better density features and were combined for another round of 3D classification into four classes. Projections from three classes accounting for 250,451 particles were merged to obtain an initial map using RELION 2.1. The same particle subset was subjected to Bayesian polishing in RELION3.0 and subsequently refined in cisTEM to produce a final map at an overall resolution of 3.9 Å. Five separate classes from the initial 3D classification were combined and subjected to a new round of 3D classification into eight classes. Three resulting classes were distinguished by the different positioning of the alpha helical domain, and were independently subjected to 3D refinement and reconstruction, producing three maps with an indicated resolution of 4.5 Å. The indicated global resolutions for the final reconstruction were obtained using “gold standard” FSC at 0.143 cut-off. Local resolution was determined using the Bsoft package (Heymann and Belnap, 2012) with unfiltered half-maps as input.
Model building and refinement
The initial model for the Rho-GT-Nb35* complex was constructed as poly-Ala chains based on crystal structures of Rho (PDB: 3PQR) (Choe et al., 2011), GDP-bound GT (PDB: 1GOT) (Lambright et al., 1996) and Nb35 in the β2AR-GS complex (PBD: 3SN6) (Rasmussen et al., 2011), and manually docked into cryo-EM density using Chimera (Pettersen et al., 2004). It was then subjected to iterative rounds of automated refinement using Phenix real space refine (Adams et al., 2004), and manual building in Coot (Emsley and Cowtan, 2004). Sequence assignment was guided by bulky amino acid residues, such as Phe, Tyr, Trp and Arg. The final model was subjected to global refinement and minimization in real space in Phenix. Validation was performed in MolProbity (Chen et al., 2010) and EMRinger (Barad et al., 2015). The Nb35*-free Rho-GT model was constructed and refined in the same way as the Nb35*-bound complex. For the Nb35*-free Rho-GT complex conformers based on the three 4.5 Å cryo-EM maps, the structure of the GαT helical domain from the GDP-bound GT crystal structure (PDB: 1GOT) (Lambright et al., 1996), together with components of the high-resolution Nb35*-free Rho-GT complex structure, were docked as rigid bodies into the corresponding EM densities. The final refinement statistics for both Nb35*-bound and Nb35*-free complexes are provided in Table S1.
DATA AND SOFTWARE AVAILABILITY
The cryo-EM density maps and atomic model have been deposited in EM Data Bank (EMD-20222 and EMD-20223) and Protein Data Bank (6OY9 and 6OYA), respectively.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lauryl maltose neopentyl glycol (L-MNG) | Anatrace | Cat# NG310 |
| Bovine retinae | W.L. Lawson Co | N/A |
| TCEP | Thermo Fisher Scientific | Cat# 77720 |
| Deposited Data | ||
| Rho-GT-Nb35* coordinates | This paper | PDB: 60YA |
| Rho-GT-Nb35* EM map | This paper | EMD-20223 |
| Rho-GT coordinates | This paper | PDB: 60Y9 |
| Rho-GT EM map | This paper | EMD-20222 |
Highlights.
Structures of a Rho-GT complex +/− nanobody at 3.3 Å and 3.9 Å resolution respectively
Extensive Rho-GT contacts enable striking signal-to-noise ratio in phototransduction
Rho-GT +/− nanobody structures illustrate perturbations resulting from nanobody-binding
The Gβγ-GαHD ‘latching switch’ is essential for fast GPCR-catalyzed GDP-GTP exchange
ACKNOWLEDGMENTS
We thank Brian Kobilka for valuable advice. This work is supported by the NIH (R35 GM122575 and R01 CA201402 to R.A.C., and R01 NS092695 to G.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams PD et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros JA & Weinstein H (1995). [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors in Methods in Neurosciences (ed. Sealfon SC) 25, 366–428 (Academic Press; ). [Google Scholar]
- Barad BA et al. (2015). EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berstein G et al. (1992). Reconstitution of agonist-stimulated phosphatidylinositol 4,5-bisphosphate hydrolysis using purified m1 muscarinic receptor, Gq/11, and phospholipase C-beta 1. J. Biol. Chem 267, 8081–8088. [PubMed] [Google Scholar]
- Bond SR & Naus CC (2012). RF-Cloning.org: an online tool for the design of restriction-free cloning projects. Nucleic Acids Res 40, W209–W213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae PS et al. (2010). Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat. Methods 7, 1003–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB et al. (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherfils J & Chabre M (2003). Activation of G-protein Gα subunits by receptors through Gα-Gβ and Gα-Gγ interactions. Trends Biochem. Sci 28, 13–17. [DOI] [PubMed] [Google Scholar]
- Choe H-W et al. (2011). Crystal structure of metarhodopsin II. Nature 471, 651–655. [DOI] [PubMed] [Google Scholar]
- Draper-Joyce CJ et al. (2018). Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature 558, 559–563. [DOI] [PubMed] [Google Scholar]
- Emsley P & Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Flock T et al. (2015). Universal allosteric mechanism for Gα activation by GPCRs. Nature 524, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CE et al. (1998). Molecular basis for interactions of G protein βγ subunits with effectors. Science 280, 1271–1274. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin L-G & Schioth HB (2003). The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol. Pharmacol 63, 1256–1272. [DOI] [PubMed] [Google Scholar]
- Gao Y et al. (2017). Isolation and structure-function characterization of a signaling-active rhodopsin-G protein complex. J. Biol. Chem 292, 14280–14289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Nafría J, Nehme R, Edwards PC & Tate CG (2018). Cryo-EM structure of the serotonin 5-HT 1B receptor coupled to heterotrimeric G o. Nature 558, 620–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Rohou A & Grigorieff N (2018). cisTEM, user-friendly software for single-particle image processing. eLife 7, e35383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymann JB & Belnap DM (2007). Bsoft: Image processing and molecular modeling for electron microscopy. J. Struct. Biol 157, 3–18. [DOI] [PubMed] [Google Scholar]
- Kang Y et al. (2015). Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y et al. (2018). Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 558, 553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehl A et al. (2018). Structure of the μ-opioid receptor-Gi protein complex. Nature 558, 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Krishna et al. (2019). Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell, 176, 448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambright DG et al. (1996). The 2.0 Å crystal structure of a heterotrimeric G protein. Nature 379, 311–319. [DOI] [PubMed] [Google Scholar]
- Liang Y-L et al. (2017). Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546, 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y-L et al. (2018). Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature 555, 121–125. [DOI] [PubMed] [Google Scholar]
- Liebman PA & Evanczuk AT (1982). [72] Real time assay of rod disk membrane cGMP phosphodiesterase and its controller enzymes in Methods in Enzymology 81, 532–542 (Academic Press; ). [DOI] [PubMed] [Google Scholar]
- Marin EP, Krishna AG & Sakmar TP (2001). Rapid Activation of Transducin by Mutations Distant from the Nucleotide-binding Site Evidence For A Mechanistic Model Of Receptor-Catalyzed Nucleotide Exchange By G Proteins. J. Biol. Chem 276, 27400–27405. [DOI] [PubMed] [Google Scholar]
- Marin EP, Krishna AG, Archambault V, Simuni E, Fu W-Y & Sakmar TP (2001). The Function of Interdomain Interactions in Controlling Nucleotide Exchange Rates in Transducin. J. Biol. Chem 276, 23873–23880. [DOI] [PubMed] [Google Scholar]
- Milano SK, Wang C, Erickson J, Cerione RA & Ramachandran S (2018). Gain-of-function screen of alpha transducin identifies an essential phenylalanine residue necessary for full effector activation. J. Biol. Chem 293, 17941–17952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min KC, Gravina SA & Sakmar TP (2000). Reconstitution of the vertebrate visual cascade using recombinant heterotrimeric transducin purified from Sf9 cells. Protein Expr. Purif 20, 514–526. [DOI] [PubMed] [Google Scholar]
- Nickell S, Park PS-H, Baumeister W & Palczewski K (2007). Three-dimensional architecture of murine rod outer segments determined by cryoelectron tomography. J. Cell Biol 177, 917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham WM, Van Eps N, Preininger AM, Hubbell WL & Hamm HE (2007). Mapping allosteric connections from the receptor to the nucleotide-binding pocket of heterotrimeric G proteins. Proc. Natl Acad. Sci. USA 104, 7927–7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K et al. (2000). Crystal Structure of Rhodopsin: A G Protein-Coupled Receptor. Science 289, 739–745. [DOI] [PubMed] [Google Scholar]
- Park JH, Scheerer P, Hofmann KP, Choe H-W & Ernst OP (2008). Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 454, 183–187. [DOI] [PubMed] [Google Scholar]
- Pettersen EF et al. (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Phillips WJ & Cerione RA (1988). The intrinsic fluorescence of the alpha subunit of transducin. Measurement of receptor-dependent guanine nucleotide exchange. J. Biol. Chem 263, 15498–15505. [PubMed] [Google Scholar]
- Ramachandran S & Cerione RA (2011). A Dominant-negative Gα Mutant That Traps a Stable Rhodopsin-Gα-GTP-βγ Complex. J. Biol. Chem 286, 12702–12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SGF et al. (2011). Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 469, 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SGF et al. (2011). Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondard P et al. (2001). Mutant G protein α subunit activated by Gβγ: a model for receptor activation? Proc. Natl Acad. Sci. USA 98, 6150–6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW (2012). RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MI, Strathmann MP & Gautam N (1991). Diversity of G proteins in signal transduction. Science 252, 802–808. [DOI] [PubMed] [Google Scholar]
- Skiba NP, Bae H & Hamm HE (1996). Mapping of Effector Binding Sites of Transducin α-Subunit Using Gat/Gail Chimeras. J. Biol. Chem 271, 413–424. [DOI] [PubMed] [Google Scholar]
- Stryer L (1991). Visual excitation and recovery. J. Biol. Chem 266, 10711–10714. [PubMed] [Google Scholar]
- Tsai C-J, Pamula F, Nehme R, Muhle J, Weinert T, Flock T, Nogly P, Edwards PC, Carpenter B, Gruhl T, et al. (2018). Crystal structure of rhodopsin in complex with a mini-Go sheds light on the principles of G protein selectivity. Sci. Adv 4, eaat7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eps N, Oldham WM, Hamm HE &Hubbell WL (2006). Structural and dynamical changes in an α-subunit of a heterotrimeric G protein along the activation pathway. Proc. Natl Acad. Sci. USA 103, 16194–16199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eps N et al. (2011). Interaction of a G protein with an activated receptor opens the interdomain interface in the alpha subunit. Proc. Natl. Acad. Sci. U. S. A 108, 9420–9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong TM, Chabre M & Stryer L (1984). Millisecond activation of transducin in the cyclic nucleotide cascade of vision. Nature 311, 659–661. [DOI] [PubMed] [Google Scholar]
- Waterhouse A et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfield GH et al. (2011). Structural flexibility of the Gas α-helical domain in the β2-adrenoceptor Gs complex. Proc. Natl. Acad. Sci 108, 16086–16091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y et al. (2017). Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ et al. (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM density maps and atomic model have been deposited in EM Data Bank (EMD-20222 and EMD-20223) and Protein Data Bank (6OY9 and 6OYA), respectively.