

Summary
Cellular therapy is enabling new approaches to tackle significant unmet needs in areas such as regenerative medicine and immunotherapy. The pharmacology of cell therapeutics becomes of critical importance to assure that these new drugs work reproducibly and effectively. Cell pharmacology can benefit from adapting principles of classical molecular drug pharmacokinetics (PK) and pharmacodynamics (PD) to quantitatively understand rate‐limiting constraints of cell fate after administration. Future innovations focused on improvements in drug delivery using a PK/PD perspective can aid in designing a cell therapeutic product to overcome any pharmacological barriers for a given disease application. Herein, we present a perspective on the development of an ex vivo mesenchymal stromal therapeutic using a PK/PD framework and also present examples of general cell engineering techniques that implicitly influence the PK/PD curve by genetically modifying cells to regulate their in vivo duration, biodistribution, and activity. stem cells translational medicine 2019;8:874&879
Significance Statement.
Attention to cell dosing, duration of therapeutic activity, and bioavailability are critical for the success of an emerging new class of cell therapeutics. The conventional absorption, distribution, metabolism, and excretion criteria for chemical drugs are inadequate for pharmacological evaluation of cell therapies and must be redefined. This study will illuminate pharmacokinetic/pharmacodynamic models that are cast on mesenchymal stromal cells and broadly engineering techniques that are relevant for future manipulation of cell pharmacology. Well‐characterized cell pharmacology will enable achieving controlled delivery, predict outcomes, and the risk–benefit ratio, while increasing the efficacy of early clinical studies.
Introduction
Cell therapeutics have unique mechanisms of action (MoA) that, similar to other medicinal agents, need to reproducibly engage biological targets in patients for a predictable treatment response. This perspective will focus on the pharmacology of cell therapeutics and how conventional pharmacology criteria for molecular drugs can be redefined for cell therapies. Successful development of standardized treatment protocols for cell therapies will depend on a deep understanding of pharmacokinetics (PK) and pharmacodynamics (PD). PK is classically categorized as quantifying the absorption, distribution, metabolism, excretion, and toxicity. For cell therapy, this translates into understanding the initial dose, administration route, duration of cell bioavailability, and cell clearance. Quantitative cell tracking methods in well‐designed biodistribution studies are critical in cell PK analysis. PD effects for cell therapy ideally translate to measuring biomarkers and subject endpoints that are directly affected by the MoA. An understanding of PD, and implicitly MoA, can help optimize dosing frequency, stratify responders and nonresponders, and manage side effects of the therapy on the body. For example, increasing cell dose might not proportionally increase therapeutic benefit due to natural cell trafficking after administration to organs such as lungs 1, 2, 3, 4. A PK/PD framework can, thus, help design dosing justification, clinical trial strategy, and begin to even define pharmaco‐economics for cell therapy developers. To this end, we provide a unique perspective on the development of an ex vivo mesenchymal stromal cell (MSC) therapeutic designed around an immunomodulation PK/PD paradigm. Then, we broaden the discussion into molecular targeted approaches using genetically engineered cell therapeutics.
Case Study of Pharmacological Modeling of MSC Therapeutics
The development of MSC therapeutics is an example where the development of PK/PD relationships can potentially help explain product ineffectiveness 5, risk–benefit ratio, and define potential strategies to improve MSC therapy using drug delivery techniques. It is firstly critical to specify the mechanism of action (MoA) of MSCs for pharmacological modeling. Although several therapeutic hypotheses for MSCs are reported, one MoA, in particular, has been highly reproduced and catalyzed the use of MSCs for immune‐mediated diseases: MSCs can modulate the in vitro effector function of peripheral blood mononuclear cells (PBMCs) 6, 7, 8, 9, 10, 11, 12. This reaction occurs when MSCs and PBMCs are separated by a transwell membrane, supporting the notion that MSCs can act in an indirect manner (i.e., without cell–cell contact). This immunomodulation MoA does not seem to be specific to bone marrow MSCs, although they may be more potent in this assay than other fibroblastic cell types 13. This MoA is well aligned with clinical unmet needs such as graft‐versus‐host disease (GvHD) 14, where a single modulator may not manage the disease well and a broader spectrum immunomodulator is required. Indeed, children with GvHD were responsive to MSC therapy in a recent successful trial 15.
Unlike a conventional molecular therapeutic, cells can sense and respond to their environment in a way that can be directly related to their MoA. Experiments that have blocked MSC sensing, also referred to as licensing or activating MSCs, during in vitro applications of immunomodulation have showed that the suppression of PBMCs is lost under these conditions 16, 17. These results lend credence to the importance of tracking this sensing process precisely in mechanistic studies. Whether these results hold up in clinical applications of MSCs remains of interest, although our contention is that MSC sensing and response cannot be overlooked because it objectively can cause a theoretical difference in drug action as the therapeutic has literally “changed” during use. By understanding and controlling MSC sensing, we may be able to test for patients prior to treatment that can activate this sensing mechanism to enrich for responders, if it indeed is important for effective MSC therapy. The microenvironment that has these signals in which MSCs act would, consequently, also be considered in defining the immunomodulation response at the molecular level. Activation of MSCs and extracellular molecules by local environmental cues, for example, could be critical to measure and optimize this PK/PD relationship. MSCs can become activated by inflammatory molecules and synthesize factors such as TSG‐6, PGE2, and kyneurine when in coculture 1, 16, 18, 19. Preactivation of MSCs may even boost their immunoregulatory effects 20, 21, 22, 23, particularly if this could be aligned to each patient when considering that MSC licensing is not a binary phenotype. A more recent mechanism of action being investigated for MSC cell therapy is through MSC‐derived extracellular vesicles (EVs), which include exosomes and microvesicles (MVs). EVs and MVs can influence tissue responses to injury, infection, and disease 24. MSC‐derived exosomes carry molecular content that includes cytokines and growth factors, signaling lipids, mRNAs, and regulatory miRNAs. Micro RNA‐containing exosomes have been shown to contribute toward immunomodulation by inhibiting macrophage activation through suppression of Toll‐like receptor signaling 25. EVs derived from MSCs have been shown to activate a proliferative program in surviving epithelial cells after injury via a horizontal transfer of mRNA 26. Moreover, MSCs were found to ameliorate ischemia/reperfusion‐induced damage in tissue‐specific case of epithelial cells via microRNA‐223 27. Recent studies have shown that the content of MSC exosomes is not static, but rather a product of the MSCs’ surrounding environment 28.
The description of MSCs interacting with leukocytes is a simplified framework for a microsystems’ biology problem. In biophysical terms, the interaction between MSCs and immune cells at its unit PK/PD volume can be viewed as a simplified chemical reaction mediated by MSC secreted factors (PK) resulting in an activated or differentiated cell immune population with altered cell surface markers, effector function, and/or cytokine responsiveness. At therapeutic concentrations and time of exposure, a durable immune response change can result that is measurable by a switch in host‐derived, systemic cytokine profiles, and phenotypic alteration of immune cell activation/differentiation markers (referred to as PD). IV infusion of cell therapy can be rate‐limited by low viability and long‐term engraftment of a cell transplant. This results in a brief and temporary systemic biological response. Longer‐term exposure to control the exposure of the subject to a cellular medicine is an example of a design goal for a drug delivery technology. Critical variables of this immune reaction that we recast in chemical reaction kinetic terms have been studied by many investigators, for example: (a) stoichiometry—an effective MSC‐immune cell ratio, (b) time—an effective duration of coculture, and (c) catalysis—MSC activation and response of soluble factor communication between MSCs and immune cells 29. These variables need to be controlled for an effective immunomodulation response; for example, in order to have a T‐cell suppressive response in vitro, MSCs require coculture at ratios of 1:10 or greater of MSCs:PBMCs for a period of 3 days 29, 30. These in vitro models have shown that leukocytes alter their cell surface profile after exposure to MSCs thereby defining a potential pharmacodynamic measurement that can be assessed by flow cytometry. Considering this dynamic reaction framework (albeit with simple assumptions) when translating MSCs from in vitro to in vivo applications is one approach for allometric scaling up to a human therapy.
PD, and more specifically allometric scaling, is the next topic to cover more deeply and yet another lesson from molecular therapeutics that can be adapted to guide MSC therapy. Foreknowing a MoA, we anticipate the ability for MSC therapeutics to ideally prove this therapeutic concept in vitro, in animal studies, and in larger subjects in veterinary and human trials. Yet, a paradoxical issue with the systemic use of MSC therapeutics for immunomodulation is the overall lack of predictive and reproducible measures of the body's response (PD) when scaling from successful in vitro and animal studies to human trials. Allometric scaling of drug dosing from animal studies to human studies is a paradigm that exists for molecular therapeutics 31, 32, 33, 34, 35, 36, but is not well established for MSCs. The criteria by which to allometrically scale is therefore worth further evaluation to see what drug criteria is not being satisfied when going from animal to human studies.
This perspective, thus far, summarizes a particular view of MSC therapeutics that may explain where product ineffectiveness can be explained in the case of systemic uses of this cell therapy product. Our contention is that PK does not meet PD criteria for systemic uses of MSCs, as modeled by in vitro coculture experiments. IV infusion of MSCs for systemic treatments cannot control the immunomodulation reaction kinetics that is found to be effective in smaller scale model systems. Typical clinical doses of as many as 100–200 million cells in a 70 kg patient diminish to only 10% within the first 48 hours 37. Preclinical PK/PD studies have historically revealed two important observations that have transformed the view of MSCs as a “cell replacement” therapy to a “transient” cell therapy: (a) infusion of MSCs lead to quick therapeutic effects in different injury models largely without direct MSC contact to a diseased tissue and (b) quantitative studies of MSC biodistribution after IV injection showed a short‐lived fate of MSCs 3, 19. Furthermore, IV‐MSCs are shown to get trapped in the lungs with a half‐life of 24 hours 1, 2, 38, resulting in pulmonary thromboembolism due to pro‐coagulation in transplanted MSCs 39, 40, 41, 42 and are thereby limited by a maximum tolerable dose that can be administered safely without pulmonary toxicity. Instant blood‐mediated inflammatory reaction has been noted for culture‐expanded MSCs that is also a limit to dosing and thus PK as well 43. Studies have shown that MSC differentiation can lead to pulmonary complications and, in the context of renal disease, result in significant loss of kidney function 44, 45. In addition, because transplantation depends on engraftment for therapeutic effectiveness, treatment kinetics is inherently delayed. Current literature supports that active immunologic processes may be responsible for the short in vivo persistence of MSCs. Allogeneic IV‐MSCs elicited a memory T‐cell response and rapid clearance of subsequent intravenous doses by the immune system 46. Activated natural killer cells, known to eliminate foreign cells, have also been shown to facilitate MSC lysis 47, 48. Furthermore, immunocompromised mice demonstrated higher sensitivity to MSC‐derived secreted factors after a cell transplant compared with wild‐type mice 4. Collectively, these studies suggest that recognition and immune clearance of MSCs could severely limit bioavailability, thereby greatly reducing therapeutic efficacy and duration of therapeutic activity. An alternative hypothesis, that justifies why in animal studies immunosuppression is observed despite a rapid clearance of MSCs, is that efferocytosis is an essential part of the MoA 49, 50. Studies have shown that inactive MSCs can be just as effective at inducing tolerance as live MSCs 51, but recent reports demonstrate that the potency of inactive or apoptotic MSCs is limited 52. Furthermore, direct transplantation of both live or apoptotic cells may not be the safest or most effective means of administering MSCs and their therapeutic factors. The limits on half‐life and maximal dose of IV‐MSCs may explain the failure to scale promising results from mouse models to human trials. Patient responses to IV‐MSCs in clinical testing, although not consistent in all patients, can be insightful cases to understand what aspects of the PK/PD paradigm have been tipped in favor of a therapeutic effect.
There are several alternate approaches that investigators are evaluating to better control for MSC bioavailability including local injections 53, the use of surface‐modified MSCs to improve homing to target organs 54, 55, 56, or even engineered MSCs with increased survival signaling 57. We have explored the systemic use of MSC bioreactors to control the interaction with immune cells ex vivo 58. This technology solution is designed to control and scale these critical reaction conditions, modeled by in vitro experiments, in an ex vivo bioreactor compartment. This ex vivo approach can potentially overcome dose limits, risks 59, and side‐effects of IV cell transplantation. The device allows for intimate exchange between MSCs and a patient's immune cells in a manner akin to transplantation by enabling the same sensing and response to a patients' blood activating signals. The bioreactor system was designed to provide engineering controls for critical features that were found in vitro to be associated with effective MSC immunomodulation. By immobilizing MSCs in a reactor, we confine and concentrate the interaction of MSCs to target immune cell within the volume of the bioreactor thereby decreasing the reaction volume. Using a clinical reactor with a surface area of ∼2.0 m2, we can theoretically immobilize >10 billion cells within this device without concern of escape or lung toxicity issues. In this way, we can theoretically restore therapeutic MSC: leukocyte ratios back to a <1:10 ratio very easily within the bioreactor (Table 1). Immune cells are continuously exposed ex vivo to ensure sufficient reaction condition of MSC ligands binding to immune target cell receptors during the transit time of immune cells in a single pass in the reactor. By controlling for the duration of reactor use, essentially the interaction frequency of MSCs and immune cells. We have shown that the ex vivo reactor can be used independent of cell transplantation to provide immunological and tissue‐sparing support in several models of inflammatory organ failure 60, 61. Prior to treatment, the cells are seeded and stabilized in the device. Following application, the device can then be disconnected and discarded once the MSCs have assisted in the recovery of the patient. Ultimately, this makes for a safe and favorable chemical reaction environment where we can control immune cell change as a function of bioreactor dosage (i.e., number of MSCs and duration of exposure).
Table 1.
Theoretical comparison between IV and ex vivo (EV) MSCs
| Parameter | IV MSCs | EV MSCs | Δ (EV/IV) |
|---|---|---|---|
| Biodistribution (reaction volume) | 5,000 ml (human blood volume) | 100 ml (reactor volume) | 1/50 |
| Half‐life (duration) | 1–3 hours | >>72 hours | >288× |
| Initial dose (cell number) | 200 M | >>200 M | >1–5× |
| ∼MSC: leukocyte compartment | <1:100 | >5:1 | >5,000× |
Abbreviations: MSC, mesenchymal stromal cell; EV, ex vivo; ∼, The ratio between MSCs to leukocytes is approximated in the table (rather than an absolute precision calculation).
Outgoing Perspective on Cell Engineering and Biomanufacturing
Emerging cell therapies are being combined with other molecular agents or genetically modified to “actively” control PK/PD properties with a target protein expression system in mind (Fig. 1). These cell engineering techniques, in essence, shift the pharmacological properties of a cell therapy with the intent of matching PK with PD for a reproducible treatment effect. PD will be specific for each cell therapy application so we limit the breadth of scope in discussion save for asserting that reverse engineering this process is ideal where a known therapeutic target specification is in place, for example the production of factor VIII for gene therapy applications based on a minimal level of protein expression for physiological blood clotting 62, 63. A set of “STED” parameters have been proposed 64 that highlight key PK processes for genetically modified cells defined as: spreading in the target tissue, transduction efficiency, expression strength in the transduced cells, and duration of gene expression. These PK parameters are important characterization tests in vitro and in vivo that can act as metrics by which a batch of cells with desired PK properties is released for clinical use. Chimeric Antigen Receptor (CAR)‐T‐cell therapy, for instance, can be simplified to a potency release assay that evaluates the interaction between CAR‐T cells and their binding targets. In vivo, although, CAR‐T cells need to migrate to the cancer target 65, which remains an important issue for solid tumor penetration compared with bloodborne targets. Milone and Bhoj present a thorough review on T‐cell pharmacokinetic data, factors affecting engraftment, and pharmacodynamic relationships 66. Concurrent delivery of molecular drugs may continue to be required to augment donor cell efficacy and in vivo persistence 67. Cell surface engineering has been exploited for improved targeting, cell viability, and pseudo‐autocrine stimulation of the donor cells by chemically conjugating drug loaded particles onto the plasma membrane of the cells 68. Efforts to maximize local biodistribution and decrease off‐target toxicity have also been accomplished by genetically engineering tumor tropic cells to produce a therapeutic enzyme that can convert a pro‐drug into an active tumor‐toxic effector drug 69. Alternatively, cells have been modified to deliver an oncolytic virus that selectively replicates in tumor cells amplifying the antitumor effect with subsequent rounds of infection and lysis 70. Time‐dependent elimination of therapeutically delivered cells can be achieved by engineering suicide switches into cells in an attempt to model the effect of cell elimination strategies to improve outcomes 71. Designing potency and efficacy studies while validating an underlying pharmacological model can enable predictable efficacy and optimization strategies in preclinical and clinical studies.
Figure 1.
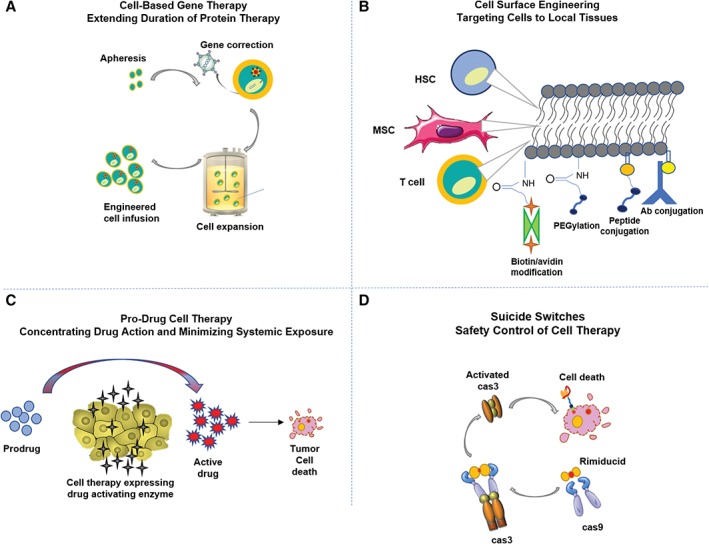
Examples of engineering techniques to control in vivo cell pharmacokinetics. Examples of various engineering techniques to improve (A) cell‐based gene therapy to extend duration of protein therapy, (B) targeting cells to local tissue via cell surface engineering 50, (C) pro‐drug cell therapy concentrates drug action within a microenvironment to minimize systemic exposure and decrease off‐target toxicities 45, and (D) engineered suicide switches as a safety control of cell therapy.
Conclusion
Finally, it is crucial to understand that drug delivery is intimately linked to the quality of drug substance as well as any drug delivery component and its reproducible manufacture in cGMP formats. Scalable and sustainable manufacturing is lacking in the field of cell therapy and is an important consideration when deploying a cell therapy solution. Combining a cell therapeutic and a drug delivery technique is best done at the earliest stages of drug development so that the quality of both components in a final product is harmonized in lock‐step. The manufacture of cell therapies becomes important to interpreting studies and including cross‐compared results when different laboratories prepare cells with significant variation. The use of reproducible in vivo data to model and find standardized cell dosage, frequency, and applications is a community effort that is enabled when the high‐quality components are robustly manufactured. A PK/PD profile can, thus, set quality targets that have a dual‐face to the manufacturer as a framework for bioprocess engineering design and optimization of an input drug substance and delivery component too.
Disclosure of Potential Conflicts of Interest
N.V. and A.A. declared employment and stock ownership with Sentien Biotechnologies, Inc. R.N.B. declared employment with ECBIO at the time the research was performed. B.P. declared patent ownership, consultant fee and stock ownership with Sentien Biotechnologies, Inc. The other author indicated no potential conflicts of interest.
Acknowledgments
This work was supported by the National Institutes of Health (R01EB012521, R01GM127353, and R21AI134116) as well as the Shriners Foundation and Rutgers TechAdvance.
References
- 1. Lee RH, Pulin AA, Seo MJ et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti‐inflammatory protein TSG‐6. Cell Stem Cell 2009;5:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schrepfer S, Deuse T, Reichenspurner H et al. Stem cell transplantation: The lung barrier. Transplant Proc 2007;39:573–576. [DOI] [PubMed] [Google Scholar]
- 3. Gao J, Dennis JE, Muzic RF et al. The dynamic in vivo distribution of bone marrow‐derived mesenchymal stem cells after infusion. Cells Tissues Organs 2001;169:12–20. [DOI] [PubMed] [Google Scholar]
- 4. Elman JS, Murray RM, Wang F et al. Pharmacokinetics of natural and engineered secreted factors delivered by mesenchymal stromal cells. PLoS One 2014;9:e89882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Squillaro T, Peluso G, Galderisi U. Clinical trials with mesenchymal stem cells: An update. Cell Transplant 2016;25:829–848. [DOI] [PubMed] [Google Scholar]
- 6. Djouad F, Plence P, Bony C et al. Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood 2003;102:3837–3844. [DOI] [PubMed] [Google Scholar]
- 7. Liu J, Lu XF, Wan L et al. Suppression of human peripheral blood lymphocyte proliferation by immortalized mesenchymal stem cells derived from bone marrow of Banna Minipig inbred‐line. Transplant Proc 2004;36:3272–3275. [DOI] [PubMed] [Google Scholar]
- 8. Corcione A, Benvenuto F, Ferretti E et al. Human mesenchymal stem cells modulate B‐cell functions. Blood 2006;107:367–372. [DOI] [PubMed] [Google Scholar]
- 9. Le Blanc K. Immunomodulatory effects of fetal and adult mesenchymal stem cells. Cytotherapy 2003;5:485–489. [DOI] [PubMed] [Google Scholar]
- 10. Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005;105:1815–1822. [DOI] [PubMed] [Google Scholar]
- 11. Jiang XX, Zhang Y, Liu B et al. Human mesenchymal stem cells inhibit differentiation and function of monocyte‐derived dendritic cells. Blood 2005;105:4120–4126. [DOI] [PubMed] [Google Scholar]
- 12. Krampera M, Glennie S, Dyson J et al. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen‐specific T cells to their cognate peptide. Blood 2003;101:3722–3729. [DOI] [PubMed] [Google Scholar]
- 13. Boregowda SV, Phinney DG. MSCs: Paracrine effects In: Hematti P, Keating A, eds. Mesenchymal Stromal Cells: Biology and Clinical Applications. New York, NY: Springer New York, 2013:145–167. [Google Scholar]
- 14. Le Blanc K, Frassoni F, Ball L et al. Mesenchymal stem cells for treatment of steroid‐resistant, severe, acute graft‐versus‐host disease: A phase II study. Lancet 2008;371:1579–1586. [DOI] [PubMed] [Google Scholar]
- 15. Kurtzberg J, Prasad V, Grimley MS et al. Allogeneic human mesenchymal stem cell therapy (Prochymal®) as a rescue agent for severe treatment resistant GVHD in pediatric patients. Biol Blood Marrow Transplant 2010;16:S169. [DOI] [PubMed] [Google Scholar]
- 16. Ren G, Zhang L, Zhao X et al. Mesenchymal stem cell‐mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem cell 2008;2:141–150. [DOI] [PubMed] [Google Scholar]
- 17. Krampera M, Cosmi L, Angeli R et al. Role for interferon‐γ in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells 2006;24:386–398. [DOI] [PubMed] [Google Scholar]
- 18. Meisel R, Zibert A, Laryea M et al. Human bone marrow stromal cells inhibit allogeneic T‐cell responses by indoleamine 2,3‐dioxygenase‐mediated tryptophan degradation. Blood 2004;103:4619–4621. [DOI] [PubMed] [Google Scholar]
- 19. Nemeth K, Leelahavanichkul A, Yuen PS et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)‐dependent reprogramming of host macrophages to increase their interleukin‐10 production. Nat Med 2009;15:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Valencic E, Piscianz E, Andolina M et al. The immunosuppressive effect of Wharton's jelly stromal cells depends on the timing of their licensing and on lymphocyte activation. Cytotherapy 2010;12:154–160. [DOI] [PubMed] [Google Scholar]
- 21. DelaRosa O, Lombardo E. Modulation of adult mesenchymal stem cells activity by toll‐like receptors: Implications on therapeutic potential. Mediators Inflamm 2010;2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Opitz CA, Litzenburger UM, Lutz C et al. Toll‐like receptor engagement enhances the immunosuppressive properties of human bone marrow‐derived mesenchymal stem cells by inducing indoleamine‐2,3‐dioxygenase‐1 via interferon‐β and protein kinase R. Stem Cells 2009;27:909–919. [DOI] [PubMed] [Google Scholar]
- 23. Polchert D, Sobinsky J, Douglas G et al. IFN‐γ activation of mesenchymal stem cells for treatment and prevention of graft versus host disease. Eur J Immunol 2008;38:1745–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karasu E, Eisenhardt SU, Harant J et al. Extracellular vesicles: Packages sent with complement. Front Immunol 2018;9:721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Phinney DG, di Giuseppe M, Njah J et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat Commun 2015;6:8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bruno S, Grange C, Deregibus MC et al. Mesenchymal stem cell‐derived microvesicles protect against acute tubular injury. J Am Soc Nephrol 2009;20:1053–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yuan X, Wang X, Chen C et al. Bone mesenchymal stem cells ameliorate ischemia/reperfusion‐induced damage in renal epithelial cells via microRNA‐223. Stem Cell Res Ther 2017;8:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Phinney DG, Pittenger MF. Concise review: MSC‐derived exosomes for cell‐free therapy. Stem Cells 2017;35:851–858. [DOI] [PubMed] [Google Scholar]
- 29. Li M, Khong D, Chin LY et al. Therapeutic delivery specifications identified through compartmental analysis of a mesenchymal stromal cell‐immune reaction. Sci Rep 2018;8:6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim J, Hematti P. Mesenchymal stem cell‐educated macrophages: A novel type of alternatively activated macrophages. Exp Hematol 2009;37:1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mager DE, Woo S, Jusko WJ. Scaling pharmacodynamics from in vitro and preclinical animal studies to humans. Drug Metab Pharmacokinet 2009;24:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Muller PY, Milton MN. The determination and interpretation of the therapeutic index in drug development. Nat Rev Drug Discov 2012;11:751. [DOI] [PubMed] [Google Scholar]
- 33. Kamath AV. Translational pharmacokinetics and pharmacodynamics of monoclonal antibodies. Drug Discov Today Technol 2016;21–22:75–83. [DOI] [PubMed] [Google Scholar]
- 34. Thiel C, Hofmann U, Ghallab A et al. Towards knowledge‐driven cross‐species extrapolation. Drug Discov Today Dis Model 2016;22:21–26. [Google Scholar]
- 35. Huang Q, Riviere JE. The application of allometric scaling principles to predict pharmacokinetic parameters across species. Expert Opin Drug Metab Toxicol 2014;10:1241–1253. [DOI] [PubMed] [Google Scholar]
- 36. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharmacy 2016;7:27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pittenger MF, Le Blanc K, Phinney DG et al. MSCs: Scientific support for multiple therapies. Stem Cells Int 2015;2015:280572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Németh K, Leelahavanichkul A, Yuen PS et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E 2‐dependent reprogramming of host macrophages to increase their interleukin‐10 production. Nat Med 2009;15:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tatsumi K, Ohashi K, Matsubara Y et al. Tissue factor triggers procoagulation in transplanted mesenchymal stem cells leading to thromboembolism. Biochem Biophys Res Commun 2013;431:203–209. [DOI] [PubMed] [Google Scholar]
- 40. Gleeson BM, Martin K, Ali MT et al. Bone marrow‐derived mesenchymal stem cells have innate procoagulant activity and cause microvascular obstruction following intracoronary delivery: Amelioration by antithrombin therapy. Stem Cells 2015;33:2726–2737. [DOI] [PubMed] [Google Scholar]
- 41. Wu Z, Zhang S, Zhou L et al. Thromboembolism induced by umbilical cord mesenchymal stem cell infusion: A report of two cases and literature review. Transplant Proc 2017;49:1656–1658. [DOI] [PubMed] [Google Scholar]
- 42. Moll G, Ankrum JA, Kamhieh‐Milz J et al. Intravascular mesenchymal stromal/stem cell therapy product diversification: Time for new clinical guidelines. Trends Mol Med 2019;25:149–163. [DOI] [PubMed] [Google Scholar]
- 43. Moll G, Ignatowicz L, Catar R et al. Different procoagulant activity of therapeutic mesenchymal stromal cells derived from bone marrow and placental decidua. Stem Cells Dev 2015;24:2269–2279. [DOI] [PubMed] [Google Scholar]
- 44. Tolar J, Nauta AJ, Osborn MJ et al. Sarcoma derived from cultured mesenchymal stem cells. Stem Cells 2007;25:371–379. [DOI] [PubMed] [Google Scholar]
- 45. Kunter U, Rong S, Boor P et al. Mesenchymal stem cells prevent progressive experimental renal failure but maldifferentiate into glomerular adipocytes. J Am Soc Nephrol 2007;18:1754–1764. [DOI] [PubMed] [Google Scholar]
- 46. Zangi L, Margalit R, Reich‐Zeliger S et al. Direct imaging of immune rejection and memory induction by allogeneic mesenchymal stromal cells (MSC). Stem Cells 2009;27:2865–2874. [DOI] [PubMed] [Google Scholar]
- 47. Sotiropoulou PA, Perez SA, Gritzapis AD et al. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells 2006;24:74–85. [DOI] [PubMed] [Google Scholar]
- 48. Spaggiari GM, Capobianco A, Becchetti S et al. Mesenchymal stem cell‐natural killer cell interactions: Evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL‐2‐induced NK‐cell proliferation. Blood 2006;107:1484–1490. [DOI] [PubMed] [Google Scholar]
- 49. Galipeau J, Sensébé L. Mesenchymal stromal cells: Clinical challenges and therapeutic opportunities. Cell Stem Cell 2018;22:824–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gonçalves FdC et al. Membrane particles generated from mesenchymal stromal cells modulate immune responses by selective targeting of pro‐inflammatory monocytes. Sci Rep 2017;7:12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Luk F, de Witte SF, Korevaar SS et al. Inactivated mesenchymal stem cells maintain immunomodulatory capacity. Stem Cells Dev 2016;25:1342–1354. [DOI] [PubMed] [Google Scholar]
- 52. Galleu A, Riffo‐Vasquez Y, Trento C et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient‐mediated immunomodulation. Sci Transl Med 2017;9:p.eaam7828. [DOI] [PubMed] [Google Scholar]
- 53. Panés J, García‐Olmo D, Van Assche G et al. Expanded allogeneic adipose‐derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn's disease: A phase 3 randomised, double‐blind controlled trial. Lancet 2016;388:1281–1290. [DOI] [PubMed] [Google Scholar]
- 54. Sackstein R, Merzaban JS, Cain DW et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med 2008;14:181–187. [DOI] [PubMed] [Google Scholar]
- 55. Sarkar D et al. Cell surface engineering of mesenchymal stem cells. Methods Mol Biol 2011;698:505–523. [DOI] [PubMed] [Google Scholar]
- 56. Huang J, Zhang Z, Guo J et al. Genetic modification of mesenchymal stem cells overexpressing CCR1 increases cell viability, migration, engraftment, and capillary density in the injured myocardium. Circ Res 2010;106:1753–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Noiseux N, Gnecchi M, Lopez‐Ilasaca M et al. Mesenchymal stem cells overexpressing Akt dramatically repair infarcted myocardium and improve cardiac function despite infrequent cellular fusion or differentiation. Mol Ther 2006;14:840–850. [DOI] [PubMed] [Google Scholar]
- 58. Li M, Tilles AW, Milwid JM et al. Phenotypic and functional characterization of human bone marrow stromal cells in hollow‐fibre bioreactors. J Tissue Eng Regen Med 2012;6:369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vulliet PR, Greeley M, Halloran SM et al. Intra‐coronary arterial injection of mesenchymal stromal cells and microinfarction in dogs. Lancet 2004;363:783–784. [DOI] [PubMed] [Google Scholar]
- 60. Parekkadan B, van Poll D, Suganuma K et al. Mesenchymal stem cell‐derived molecules reverse fulminant hepatic failure. PLoS One 2007;2:e941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. van Poll D, Parekkadan B, Cho CH et al. Mesenchymal stem cell‐derived molecules directly modulate hepatocellular death and regeneration in vitro and in vivo. Hepatology 2008;47:1634–1643. [DOI] [PubMed] [Google Scholar]
- 62. Miesbach W, Meijer K, Coppens M et al. Gene therapy with adeno‐associated virus vector 5‐human factor IX in adults with hemophilia B. Blood 2018;131:1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Arruda VR, Doshi BS, Samelson‐Jones BJ. Novel approaches to hemophilia therapy: Successes and challenges. Blood 2017;130:2251–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ylä‐Herttuala S. The pharmacology of gene therapy. Mol Ther 2017;25:1731–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang Z, Liu Q. Clinical pharmacological considerations on CAR‐T cell therapy for cancer. J Pharmacol Clin 3:001–004. [Google Scholar]
- 66. Milone MC, Bhoj VG. The pharmacology of T cell therapies. Mol Ther Methods Clin Dev 2018;8:210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Redeker A, Arens R. Improving adoptive T cell therapy: The particular role of T cell costimulation, cytokines, and post‐transfer vaccination. Front Immunol 2016;345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stephan MT, Moon JJ, Um SH et al. Therapeutic cell engineering with surface‐conjugated synthetic nanoparticles. Nat Med 2010;16:1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mooney R, Abdul Majid A, Batalla J et al. Cell‐mediated enzyme prodrug cancer therapies. Adv Drug Deliv Rev 2017;118:35–51. [DOI] [PubMed] [Google Scholar]
- 70. Howells A, Marelli G, Lemoine NR et al. Oncolytic viruses‐interaction of virus and tumor cells in the battle to eliminate cancer. Frontiers in oncology 2017;7:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shen K, Luk S, Elman J et al. Suicide gene‐engineered stromal cells reveal a dynamic regulation of cancer metastasis. Sci Rep 2016;6:21239. [DOI] [PMC free article] [PubMed] [Google Scholar]