

Abstract
Background
HER3/ErbB3 receptor deletion or blockade leads to tumor cell apoptosis, whereas its overexpression confers anti-cancer drug resistance through upregulation of protective mechanisms against apoptosis. We produced the anti-HER3 antibody 9F7-F11 that promotes HER3 ubiquitination and degradation via JNK1/2-dependent activation of the E3 ubiquitin ligase ITCH, and that induces apoptosis of cancer cells. Cellular FLICE-like inhibitory protein (c-FLIP) is a key regulator of apoptotic pathways. Here, we wanted to determine the mechanisms underlying the pro-apoptotic effect of 9F7-F11.
Methods
Anti-HER3 antibody-induced apoptosis was assessed by western blot, and by flow cytometry measurement of Annexin V/7-AAD-labelled tumor cells (BxPC3, MDA-MB-468 and DU145 cell lines). c-FLIP/ITCH interaction and subsequent degradation/ubiquitination were investigated by co-immunoprecipitation of ITCH-silenced vs scramble control cells. The relationship between ITCH-mediated c-FLIP degradation and antibody-induced apoptosis was examined by western blot and flow cytometry of tumor cells, after ITCH RNA interference or by pre-treatment with ITCH chemical inhibitor chlorimipramine (CI).
Results
Following incubation with 9F7-F11, cancer cell apoptosis occurs through activation of caspase-8, − 9 and − 3 and the subsequent cleavage of poly (ADP-ribose) polymerase (PARP). Moreover we showed that ubiquitination and proteasomal degradation of the anti-apoptotic protein c-FLIP was mediated by USP8-regulated ITCH recruitment. This effect was abrogated by ITCH- and USP8-specific RNA interference (siRNA), or by the ITCH chemical inhibitor CI. Specifically, ITCH silencing or CI blocked 9F7-F11-induced caspase-8-mediated apoptosis of tumor cells, and restored c-FLIP expression. ITCH-silencing or CI concomitantly abrogated HER3-specific antibody-induced apoptosis of Annexin V/7-AAD-labelled BxPC3 cells. 9F7-F11 favored the extrinsic apoptosis pathway by inducing TRAIL-R2/DR5 upregulation and TRAIL expression that promoted the formation of death-inducing signaling complex (DISC), leading to caspase-8-mediated apoptosis. Incubation with 9F7-F11 also induced BID cleavage, BAX upregulation and BIM expression, which initiated the caspase-9/3-mediated mitochondrial death pathway. The anti-HER3 antibody pro-apoptotic effect occurred concomitantly with downregulation of the pro-survival proteins c-IAP2 and XIAP.
Conclusions
The allosteric non-neuregulin competing modulator 9F7-F11, sensitizes tumor cells to DR5/caspase-8-mediated apoptosis through ITCH-dependent downregulation of c-FLIP.
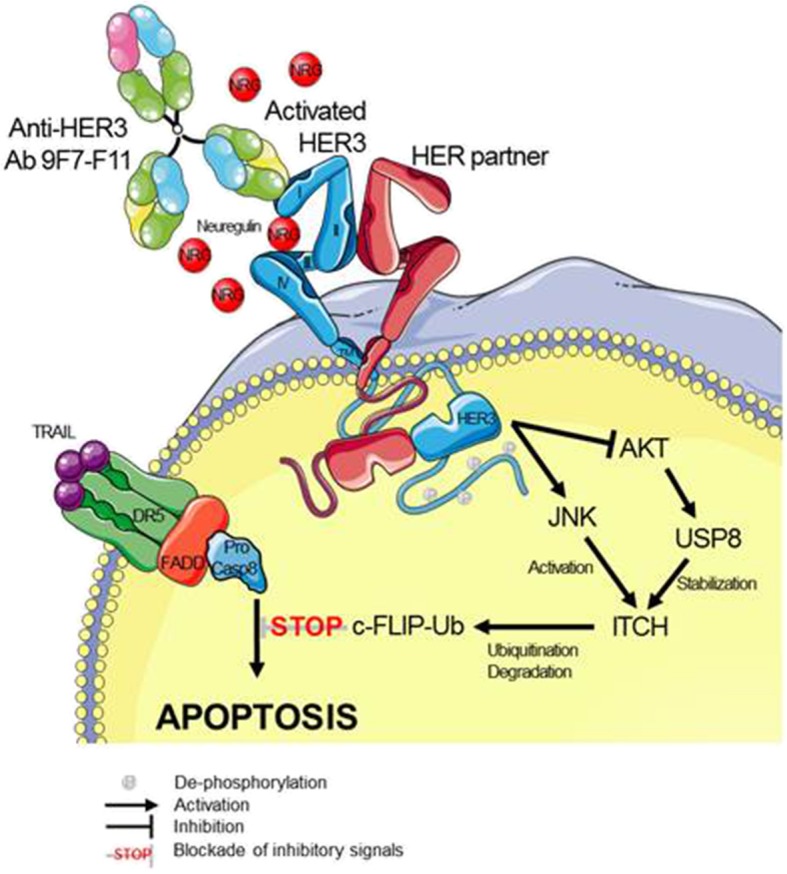
Graphical abstract
Electronic supplementary material
The online version of this article (10.1186/s12964-019-0413-8) contains supplementary material, which is available to authorized users.
Keywords: Cancer, HER3, Antibody, Apoptosis, C-FLIP, ITCH
Background
Apoptosis is a cell death type that is essential during embryonic development and for homeostasis maintenance. It is also one of the processes deregulated during cell transformation. In many cancers, activation of the HER3 (ErbB3) receptor frequently results in strong and aberrant activation of the AKT pathway, a critical oncogenic stimulus that leads to apoptosis resistance. HER3 knockdown [1] or pharmacological blockade [2] directly restores tumor cell-specific apoptosis. Conversely, HER3 overexpression confers drug resistance to paclitaxel in breast cancer via upregulation of survivin, a pro-survival protein that inhibits apoptosis [3]. In this case, targeting HER3 with antibodies indirectly counteracts drug resistance by favoring tumor cell apoptosis [4].
Apoptosis is induced through two main pathways. The extrinsic pathway is activated by the binding of specific ligands (e.g., FASL, TRAIL, and TNF) to their death receptors (DR) at the cell surface (FAS/CD95, DR4/TRAIL-R1 and DR5/TRAIL-R2, TNFR1 and TNFR2) [5–7]. These ligand/receptor complexes recruit adaptor molecules, such as Fas-associated protein with death domain (FADD), on their death effector domains (DED), leading to the formation of Death-Inducing Signaling Complexes (DISC) that in turn recruit pro-caspase 8. Following autocatalytic cleavage [8, 9], activated caspase-8 induces cleavage and activation of caspase-3, − 6 and − 7 directly in type I cells [10], or indirectly in type II cells, by BID cleavage into the truncated form tBID [11] that activates the intrinsic apoptotic machinery by formation of tBID/BAX complexes in mitochondria [12]. These complexes form mitochondrial pores for cytochrome C release that promotes apoptosome formation and then caspase-9 cleavage/activation to induce apoptosis [13]. DISC-induced apoptosis is negatively regulated by a short-lived protein called FLICE-like inhibitory protein (c-FLIP) [14], a competitive mimetic of pro-caspase 8 [15–17] with two DEDs, like the initiator caspase. However, due to the absence of the catalytic cysteine residue present in pro-caspase 8, c-FLIP is inactive, and thus is considered to be an anti-apoptotic protein. c-FLIP also blocks the intrinsic apoptotic pathway by competing with caspase-8 to prevent BID activation. c-FLIP is upregulated in many cancers (e.g., pancreatic [18], breast [19], prostatic [20], and colorectal [21] cancer, glioblastoma [22], Burkitt and non-Hodgkin lymphoma [23, 24]). c-FLIP upregulation promotes defects of DR-mediated apoptosis and resistance to several anti-cancer drugs [25]. However, c-FLIP is an unstable protein with rapid turnover via ubiquitination and proteasomal degradation by specific E3 ubiquitin ligases, such as ITCH [26–28]. Cisplatin treatment sensitizes tumor cells to apoptosis by favoring ITCH-mediated c-FLIP downregulation [29, 30]. Conversely, cystatin B inhibits TRAIL-induced apoptosis in melanoma cells by protecting c-FLIP from degradation by ITCH [31].
We previously showed that several anti-HER3 antibodies, including the allosteric non-neuregulin competing modulator 9F7-F11 [32], induce ITCH-mediated HER3 degradation [33] and promote apoptosis in tumor cells [2]. Among numerous anti-HER3 antibodies described [34, 35], 9F7-F11 is the only anti-HER3 antibody whose binding and affinity to HER3 are enhanced in the presence of neuregulin 1 (NRG1) [32]. This property translated in vivo into an efficient anti-tumor activity in NRG1-addicted and NRG1-rearranged cancer cells [32], making it a first-in-class antibody. Here, we asked whether its pro-apoptotic effect was regulated by ITCH-dependent c-FLIP expression regulation. We found that 9F7-F11 sensitizes tumor cells to DR5/caspase-8-mediated apoptosis through ITCH-dependent downregulation of c-FLIP.
Methods
Cell lines
The BxPC3 (pancreas), MDA-MB-468 (breast), and DU145 (prostate) human cancer cell lines were obtained from the American Type Culture collection (ATCC) (Rockville, MD), and grown as recommended by ATCC. All cell lines were free of mycoplasma contamination (determined by the MycoAlert™ Detection Kit (Lonza, Switzerland) and were authenticated by short tandem repeat profiling using the Promega PowerPlex 21 System. These three cell lines both expressed EGFR, HER2 and HER3 receptors, and the HER3 ligand neuregulin, and are all equipped with the ITCH/USP8/USP9X machinery (Additional file 1: Figure S1). We previously demonstrated that the anti-HER3 antibody 9F7-F11 induced HER3 ubiquitination and degradation in these three cell lines through a JNK-dependent ITCH activation [33]. Furthermore, 9F7-F11 reduced HER2/HER3 heterodimerization, increased HER3 homodimers, without affecting HER2 homodimers in HER2/HER3-transfected 3T3 cells [36].
Antibodies and other reagents
The anti-HER3 antibody 9F7-F11 has been described elsewhere [2, 32, 33, 36]. Rabbit monoclonal antibodies against HER3 and phosphorylated (p) HER3 (Tyr1289), PARP (clone 46D11), cleaved caspase-8 (Asp391; clone 18C8), caspase-9 and cleaved caspase-9, caspase-3 (clone 8G10) and cleaved caspase-3, XIAP, DR5 (clone D4E7), BIM, c-FLIP (clone D16A8), USP9X, β-actin and β-tubulin were from Cell Signaling Technology (Danvers, MA). The mouse monoclonal antibody against ITCH was from BD Biosciences (San José, CA; ref.611199). The rabbit polyclonal antibodies against HER3 (clone C17), USP8, USP9X, c-FLIPL/S (clone H-202), BAX, FAS, TRAIL, DcR2, cIAP2, and BID were from Santa Cruz Biotechnology (Santa Cruz, CA). For detection of activated ITCH, a rabbit anti-pITCH (Thr222) antibody from Millipore (Billerica, MA) was used. The human recombinant NRG1-β1 extracellular domain (ECD) was from RD Systems (Minneapolis, MN) and was used at 100 ng/ml. The proteasome inhibitor MG132 and chlorimipramine (CI) were from Sigma-Aldrich (St Louis, MO). The control IgG antibody used for co-immunoprecipitation experiments was from Santa Cruz Biotechnology.
siRNA transfections
2 × 106 BxPC3 cells were plated in 10 cm-dishes with RPMI medium without antibiotics until 50% confluence. Cells were then transfected with 50 nM of pools of four different siRNAs against human ITCH or USP8, or scramble control (ON-TARGETplus SMART pool, Dharmacon, Germany) in OptiMEM medium using Interferin™ (Life Technologies, Carlsbad, CA). After 4 h of transfection, medium was replaced by RPMI for another 48 h before using the cells for apoptosis measurement by flow cytometry, or for another 72 h before using the cells for western blot analysis.
Annexin V/7-AAD apoptosis measurement by flow cytometry
Cell apoptosis was assessed using Annexin V/7-Amino actinomycin D (7-AAD) apoptosis kit (Beckman Coulter-Immunotech, Marseille, France) according to the manufacturer’s instructions. Briefly, 105 serum-starved BxPC3 cells were transfected with 50 nM ITCH or scramble control siRNAs, as described above. Alternatively, BxPC3 cells were pre-treated with 15 μM of ITCH chemical inhibitor CI. After 48 h, cells were washed and treated with 50 μg/ml of anti-HER3 antibody 9F7-F11, with or without 100 ng/ml of NRG1 for 96 h. As positive control, 300 nM staurosporine (Sigma, Saint-Louis, MO) was incubated with BxPC3 cells for 6-20 h. After Annexin V/7-ADD labeling of treated cells, data were acquired on a Gallios flow cytometer and analyzed with the Kaluza software (Beckman Coulter). All experiments were performed in triplicates.
Cell lysis and immunoprecipitation
10 × 106 BxPC3 cells were lysed in CHAPS buffer (Sigma-Aldrich) containing the protease inhibitor cocktail V (Calbiochem, Billerica, MA) and the phosphatase inhibitor cocktail II (Sigma-Aldrich). For c-FLIPL/S immunoprecipitation (Fig. 4), 2 mg of each total cell lysate was pre-cleared by overnight addition of 50 μl of magnetic beads (Dynabeads™; Life Technologies), to capture and remove the anti-HER3 antibody 9F7-F11. Supernatants (2 mg) were then incubated with 2 μg of the anti-c-FLIPL/S antibody H-202, which recognizes both c-FLIPL and c-FLIPS, at 4 °C for 6 h before overnight incubation with 20 μl of Dynabeads magnetic beads at 4 °C under agitation. Samples were washed five times with 400 μl CHAPS buffer, re-suspended in 100 μl of 2X SDS Laemmli buffer and heated at 90 °C for 10 min before electrophoresis. No c-FLIP protein was immunoprecipitated after incubation with beads alone or with the control IgG antibody.
Fig. 4.

ITCH or USP8 silencing by siRNA inhibits 9F7-F11-induced c-FLIP ubiquitination and proteasomal degradation. a BxPC3 cells were transfected with 50 nM ITCH-specific siRNA (siITCH) or with control scramble siRNA (siSC) for 72 h, before pre-treatment with 10 μM MG132 for 4 h. Cells were then incubated with 9F7-F11, with or without NRG1, or medium as control for 4 h. After immunoprecipitation of total protein extracts (2 mg) with the anti-c-FLIP antibody H-202, c-FLIP, ITCH and USP8 expression and c-FLIP ubiquitination were analyzed by western blotting. BxPC3 cells were transfected with siSC, siITCH (b) or siUSP8 (c) for 72 h, and then incubated with 9F7-F11 for 4 h. Expression of ITCH, c-FLIP and USP8 was assessed in total protein extracts by western blotting. Protein level was measured with the ImageJ software and indicated as signal intensity (SI), relative to untreated control (SI = 1.0 ± .0). Significant increase or decrease of the densitometry, compared to control, is indicated in bold. β-tubulin was evaluated as loading control
HER3/c-FLIPL/S double immunoprecipitation was performed after NRG1 stimulation and/or 9F7-F11 incubation of BxPC3 cells (Fig. 3). First, total cell lysates (2 mg) were incubated with 2 μg of the anti-HER3 antibody 2F12, which recognizes the HER3 intracellular C-terminal tail and does not compete with 9F7-F11. The incubation was performed at 4 °C for 6 h before overnight incubation with 20 μl of magnetic Dynabeads at 4 °C under agitation. Total supernatants were recovered and then incubated with 2 μg of the anti-c-FLIP antibody H-202 at 4 °C for 6 h, before overnight incubation with 20 μl of Dynabeads magnetic beads at 4 °C under agitation. Samples were then processed as described above before electrophoresis.
Fig. 3.
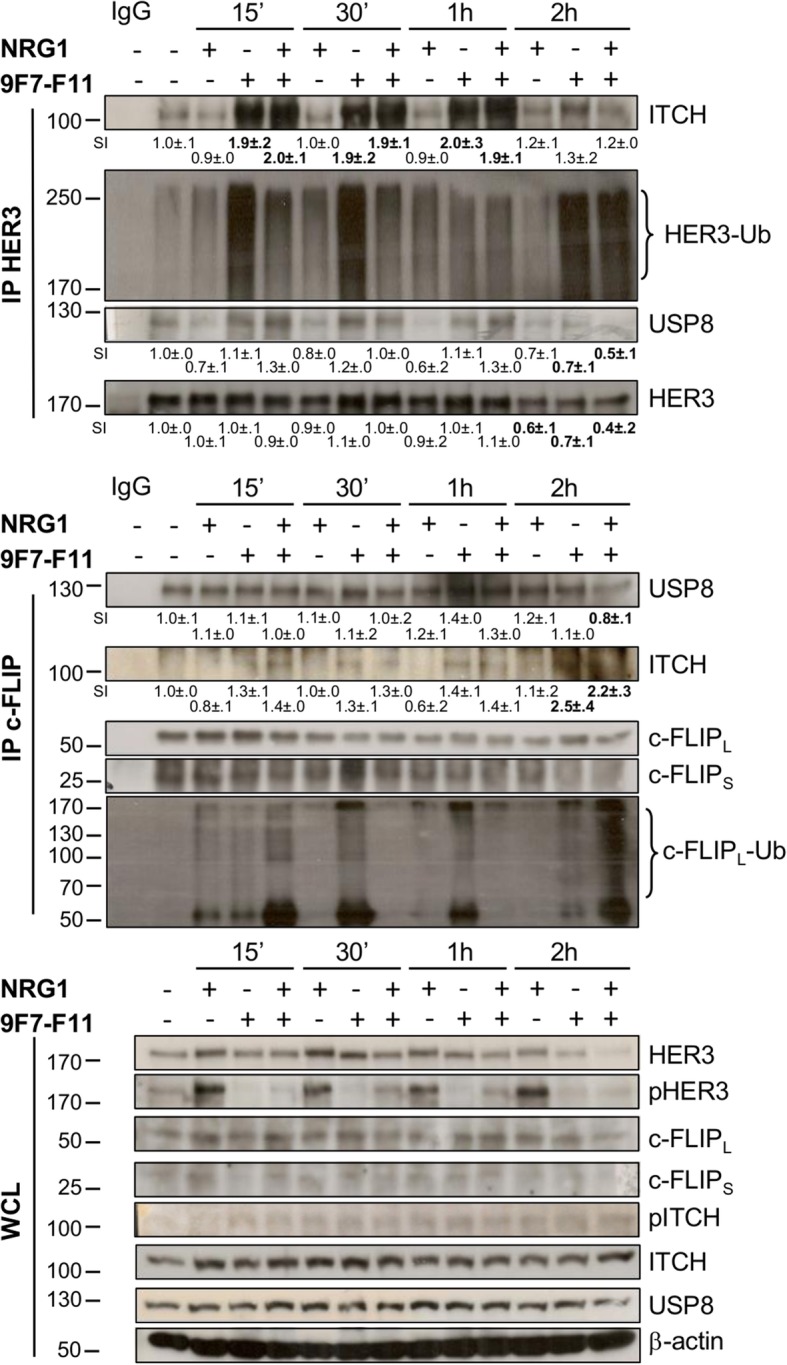
USP8-regulated ITCH interaction with c-FLIP mediates 9F7-F11-induced c-FLIP ubiquitination. BxPC3 cells were incubated with NRG1 or/and 9F7-F11 for various times. After cell lysis in CHAPS buffer, 2 mg of total protein extracts were co-immunoprecipitated with the anti-HER3 antibody 2F12 (Millipore) against HER3 C-terminal tail. Then, the first soluble supernatant was co-immunoprecipitated with the rabbit anti-c-FLIP polyclonal antibody H-202 (Santa Cruz Biotechnology) that targets both c-FLIPL and c-FLIPS. The presence of ITCH and USP8 in the two immunoprecipitates was assessed by western blotting. HER3 and c-FLIP ubiquitination status were assessed using the anti-K48 ubiquitin antibody. Whole cell lysates (WCL) were analyzed using the appropriate antibodies. Quantification of signal intensity (SI) with ImageJ software is indicated below the images, in comparison to SI = 1.0 ± .0 for untreated control. Significant increase or decrease of the densitometry, compared to control, is indicated in bold. β-actin was evaluated as loading control
DR5-DISC was isolated after immunoprecipitation (Fig. 6) using the Dynabeads system, as described by the manufacturer. Briefly, 2 mg of total cell lysates from 9F7-F11-treated BxPC3 cells were incubated with magnetic beads previously cross-linked with the anti-DR5 antibody D4E7. The Dynabeads bead-coupled DISC complexes were immunoprecipitated overnight at 4 °C in a rotator, and then washed ten times with CHAPS buffer, before electrophoresis and western blotting.
Fig. 6.

9F7-F11 activates the extrinsic apoptotic pathway through FAS and DR5 upregulation, TRAIL expression induction and DcR2 downregulation. a BxPC3, MDA-MB-468, and DU145 cells were treated with 9F7-F11 for various times. FAS, DR5, mTRAIL and DcR2 expression were detected by western blotting. b BxPC3 cells were incubated with 9F7-F11. After cells lysis at different time points, total protein extracts (2 mg) were co-immunoprecipitated with the human anti-DR5 polyclonal antibody D4E7 (Cell Signaling Technology) or with human anti-IgG as control. The presence of DISC components (caspase-8, FADD and DR5) was assessed in immunoprecipitates and whole cell lysates (WCL) by western blotting. Protein level was measured with the ImageJ software and indicated as signal intensity (SI), relative to untreated control (SI = 1.0 ± .0). Significant increase or decrease of the densitometry, compared to control, is indicated in bold. β-tubulin was evaluated as loading control
Western blotting
2 × 106 cells/dish were cultured at 37 °C for 24 h. After serum starvation in RPMI/1%serum with antibiotics for 24 h, cells were incubated with various compounds. For short kinetics, cells were stimulated with 100 ng/ml of NRG1 and/or incubated with 50 μg/ml of 9F7-F11 for 15 min to 3 h. For long kinetics, cells were incubated with 50 μg/ml of anti-HER3 antibody 9F7-F11 for 24 h to 120 h. After incubation, cells were washed, scraped and lysed with CHAPS buffer (Sigma-Aldrich), as indicated above. After washing in 1X PBS, the insoluble fraction was removed by centrifugation, and protein concentration in cell lysates was determined using the BCA assay. Two hundred micrograms of protein lysates were directly mixed with Laemmli buffer and heated at 95 °C for 5 min. After electrophoresis in reducing conditions, proteins were transferred to polyvinylidenedifluoride membranes (Millipore) and then incubated with the relevant primary and peroxidase-conjugated secondary antibodies, as previously described [33]. β-tubulin or β-actin were used as loading control.
Statistical analysis
Apoptosis values represent the means ± SD for at least three independent experiments performed in triplicates. The significance of differences between experimental variables was determined using the two-tailed Student’s t test. The significance of P values are *P < 0.05, **P < 0.01 or ***P < 0.001.
Results
The anti-HER3 antibody 9F7-F11 induces tumor cell apoptosis through caspase-8/9/3 activation and PARP cleavage
We previously demonstrated by flow cytometry analysis after Annexin V/7-AAD staining that the anti-HER3 antibodies 9F7-F11, 16D3-C1 and H4B-121 induce apoptosis of pancreatic cancer cells [2]. To precisely determine the underlying molecular mechanisms, we assessed apoptosis induction in three cancer cell lines at different time points during incubation with the allosteric NRG1 non-competing antibody 9F7-F11 [2, 32, 33]. 9F7-F11 induced caspase-8 cleavage into the p41/43 and p18 C8c fragments in BxPC3 (pancreas), DU145 (prostate) and MDA-MB-468 (breast) at 48 h (Fig. 1). The p37 C9c cleavage product, which indicates activation of caspase-9, appeared at 24 h for BxPC3 and DU145 cell lines, and 48 h for MDA-MB-468. p17/19 C3c cleavage products, which indicate activation of caspase-3, mainly occured 48 h-post 9F7-F11 treatment for MDA-MB-498 and DU145, and 72 h-post treatment for BxPC3 (Fig. 1). Caspase activation was followed by PARP cleavage from 24 h–48 h to the end of the experiment (120 h) (Fig. 1). Incubation with the HER3 ligand NRG1 alone did not induce apoptosis BxPC3 cells (Additional file 1: Figure S2). Conversely, 9F7-F11 could induce apoptosis of BxPC3 cells (caspase-8/9/3 cleavage/activation and PARP cleavage) also in the presence of NRG1 (Additional file 1: Figure S2). This demonstrated that 9F7-F11-induced apoptosis via caspase activation is a general mechanism of cancer cell death, independent of ligand stimulation.
Fig. 1.

9F7-F11-induced tumor cell apoptosis occurs through caspase-8/9/3 activation and PARP cleavage. BxPC3, MDA-MB-468 or DU145 cancer cells were incubated with the anti-HER3 antibody 9F7-F11. Caspase-8, − 9 and − 3 and PARP cleavage were analyzed by western blotting and cell lysates prepared at different time points during antibody incubation. Quantification of signal intensity (SI) with ImageJ software is indicated below the images (relative to untreated control measured as 1.0 ± .0). Significant increase or decrease of the densitometry, compared to control, is indicated in bold. M: medium alone; p41/43 and p18 C8c: caspase-8 cleavage; p37 C9c: caspase-9 cleavage; p17/19 C3c: caspase-3 cleavage; PARPc: PARP cleavage. β-tubulin was evaluated as loading control
9F7-F11 induces c-FLIP proteasomal degradation
We previously showed that 9F7-F11 promotes HER3 ubiquitination and proteasomal degradation through JNK1/2-dependent ITCH activation [33]. As c-FLIP also is an ITCH target [26, 27], we asked whether this anti-apoptotic factor was ubiquitinated and degraded concomitantly with HER3. Expression of the long isoform c-FLIPL was notably reduced in BxPC3, MDA-MB-468, and DU145 cells after 2 h-incubation with 9F7-F11, but not with NRG1 (Fig. 2a). ITCH phosphorylation was increased at 10-30 min until the end of the experiment (2 h) (Fig. 2a). Expression of USP8 and USP9X, which stabilize ITCH by preventing its auto-ubiquitination [27, 37], was globally stable during the entire experiment, except for BxPC3 cells at 2 h post-9F7-F11 treatment. Both long and short c-FLIP isoforms (c-FLIPL and c-FLIPS) and HER3 were downregulated after 3 h of incubation with 9F7-F11 (Fig. 2b). c-FLIPL expression, as well as HER3 expression, was reduced in BxPC3 and MDA-MB-468 cells after longer incubation with 9F7-F11, but not with medium or NRG1 (Fig. 2c). Then, to determine whether 9F7-F11 induced c-FLIP degradation via the proteasome, we pre-incubated, or not, cells with the proteasome inhibitor MG132 for 4 h, before addition of 9F7-F11 with or without NRG1 (Fig. 2d). Pre-incubation with MG132 inhibited the 9F7-F11-induced reduction in c-FLIPL level, as previously reported for HER3 [33]. These findings demonstrated that 9F7-F11 reduces substantially c-FLIP and HER3 expression via proteasomal degradation, and independently of ligand stimulation.
Fig. 2.

9F7-F11 induces c-FLIP downregulation by proteasomal degradation. a Cancer cells were incubated with NRG1 or 9F7-F11. After cell lysis at different time points, c-FLIPL, USP8 and USP9X expression, as well as ITCH expression and phosphorylation (p) were analyzed by western blotting. b BxPC3 cells were incubated with 9F7-F11 for 3 h. HER3, c-FLIPL and c-FLIPS expression were analyzed by western blotting. c BxPC3 and MDA-MB-468 cells were incubated with NRG1 or 9F7-F11 for 48 h or 96 h, before detection of HER3 and c-FLIPL expression by western blotting. d After pre-incubation or not with 10 μM MG132 for 4 h, BxPC3 cells were incubated with 9F7-F11 with or without NRG1. After cell lysis, expression of HER3 and c-FLIPL was assessed in whole cell lysates by western blotting. The rabbit anti-HER3 polyclonal antibody C-17 (Santa Cruz Biotechnology) was used for detection. Protein level was measured with the ImageJ software and indicated as signal intensity (SI), relative to untreated control (SI = 1.0 ± .0). Significant increase or decrease of the densitometry, compared to control, is indicated in bold. β-tubulin was evaluated as loading control
USP8-regulated ITCH binding to c-FLIP mediates c-FLIP ubiquitination in cancer cells incubated with 9F7-F11
To determine whether c-FLIP binds to ITCH following antibody treatment, we collected BxPC3 cells at different time points during incubation with NRG1 or/and 9F7-F11, and immunoprecipitated them with anti-HER3 and anti-c-FLIP antibodies. We observed ITCH co-immunoprecipitation with HER3, and HER3 ubiquitination after 15 min of incubation with 9F7-F11 (with or without NRG1), and HER3 degradation at 2 h (Fig. 3, upper panel). In c-FLIP co-immunoprecipitation, strong c-FLIPL ubiquitination at 2 h was correlated with stronger ITCH interaction (Fig. 3, middle panel). ITCH was not co-immunoprecipitated with c-FLIPL in BxPC3 cells incubated with medium or NRG1 alone (Fig. 3, middle panel), suggesting that ITCH needs to be activated for substrate binding, as shown in HEK293 cells [38] and in our previous study [33].
USP8 interacts and stabilizes c-FLIP by deubiquitination, leading to DR-mediated apoptosis suppression [39]. USP8 also interacts with and stabilizes ITCH to induce c-FLIP ubiquitination, upon AKT inhibition, leading to TRAIL-induced apoptosis [27]. We already demonstrated that USP8 controls ITCH stability during 9F7-F11-induced HER3 ubiquitination and degradation [33]. Here, we found that USP8 was present in the c-FLIPL-ITCH complex in untreated cells, but was slightly reduced after 2 h of 9F7-F11 incubation (Fig. 3, IP c-FLIP), as previously reported [39]. USP8 was constitutively present also in the HER3-ITCH complex, and its expression slightly increased upon incubation with 9F7-F11 up to 1 h (Fig. 3, IP HER3), as we previously demonstrated [33]. The strong reduction of USP8 level in the two complexes at 2 h suggests that after contributing to the formation of these complexes, USP8 leaves them to favor c-FLIPL and HER3 downregulation. Altogether, these results demonstrated that 9F7-F11 induced USP8 recruitment to stabilize ITCH, and then, the USP8-ITCH complex binds to the ITCH targets c-FLIPL and HER3, allowing their ubiquitination and proteasomal degradation.
ITCH and USP8 silencing by siRNA inhibits 9F7-F11-induced c-FLIP ubiquitination and proteasomal degradation
To confirm ITCH involvement in antibody-induced c-FLIP ubiquitination, we analyzed c-FLIP ubiquitination after immunoprecipitation using an anti-c-FLIP antibody of protein extracts from BxPC3 cells transfected with anti-ITCH (siITCH) or scramble control (siSC) siRNAs and pre-treated with MG132 before incubation with 9F7-F11 and/or NRG1 for 4 h (Fig. 4a). We observed c-FLIPL poly-ubiquitination and ITCH co-immunoprecipitation in siSC cells incubated with 9F7-F11 alone or with NRG1, but not in siITCH cells (Fig. 4a). Moreover, in siSC cells, 9F7-F11 reduced USP8 interaction with c-FLIPL and promoted ITCH recruitment, allowing c-FLIPL ubiquitination (Fig. 4a). Western blot analysis confirmed c-FLIPL and c-FLIPS degradation in siSC cells upon incubation with 9F7-F11 alone (Fig. 4b). This effect was abrogated by siRNA-mediated silencing of ITCH (Fig. 4b) and USP8 (Fig. 4c). These results underlined ITCH and USP8 main role in 9F7-F11-induced c-FLIP degradation through ubiquitination. Finally, ITCH downregulation in 9F7-F11-treated siUSP8, but not in siSC cells (Fig. 4c) suggests that USP8 deubiquitinates and stabilizes ITCH, which then promotes rapid c-FLIP ubiquitination and proteosomal degradation [33]. In control cells (medium alone), USP8 knock-down induced c-FLIP downregulation, as compared with siSC treated cells (Fig. 4c), confirming that USP8 directly deubiquitinates c-FLIP [39].
ITCH silencing or chemical inhibition blocks 9F7-F11-induced apoptosis and c-FLIP degradation
To determine whether ITCH-induced c-FLIPL degradation had a major role in anti-HER3 antibody-induced cancer cell apoptosis, we assessed PARP and caspase-8/3 cleavage by western blotting in siITCH and siSC BxPC3 cells incubated with 9F7-F11 alone (Fig. 5a) or with NRG1 (Fig. 5b) for 48 h. In siITCH cells, ITCH expression was strongly downregulated, and 9F7-F11-induced c-FLIPL degradation inhibited, compared with siSC cells (Fig. 5a). This effect was stronger in siITCH cells co-incubated with NRG1 and 9F7-F11 (Fig. 5b). Inhibition of c-FLIPL degradation repressed 9F7-F11-induced apoptosis, as indicated by the lower level of cleaved PARP, p41/43 and p18 fragments (caspase-8 cleavage), and p17/19 fragments (caspase-3 cleavage) in siITCH cells compared with siSC cells (Fig. 5a). We confirmed the requirement of ITCH-mediated c-FLIPL proteasomal degradation for 9F7-F11-induced apoptosis using chlorimipramine (Cl), a specific ITCH chemical inhibitor that is known to induce apoptosis at high dose, and irreversibly blocks ITCH by binding to its substrate pocket [40]. In the absence of 9F7-F11 antibody, incubation with high dose of CI (30 μM) induced apoptosis of MDA-MB-468 cells, as indicated by PARP and caspase-8/9/3 cleavage (Fig. 5c), as previously described [40, 41]. However, pre-incubation with 15 μM CI for 48 h completely repressed 9F7-F11-induced caspase activation and apoptosis (Fig. 5c), probably by inhibiting ITCH binding to its substrate. When we combined higher dose (30 μM) of CI with 9F7-F11, we restored PARP cleavage induced by CI, albeit antibody-induced apoptosis is still repressed. ITCH blockade by 15 μM CI in 9F7-F11-treated cells was associated with the disappearance of ITCH ubiquitination, and inhibition of antibody-induced c-FLIPL and HER3 degradation, compared with cells incubated with medium or 9F7-F11 alone (Fig. 5d). In the agreement, ITCH chemical inhibitor CI reduced the percentage of apoptotic cells from 47.5 to 21.5% in NRG1/9F7-F11-treated BxPC3 cells (Fig. 5e), with a stronger effect on late apoptosis; this reduction being also observed in CI-pre-incubated BxPC3 cells and further treated with 9F7-F11 alone, in comparison with medium-pre-incubated antibody-treated cells (Fig. 5e). Similarly, ITCH RNA silencing of NRG1/9F7-F11-treated BxPC3 cells (siITCH) decreased apoptosis to 29.4%, with regard to 47.8%-apoptosis observed in siSC cells (Fig. 5f). This reduction of apoptosis was also observed in siITCH BxPC3 cells treated with 9F7-F11 alone, compared with siSC cells (Fig. 5f). As positive control, staurosporine induced 68.4%-cell apoptosis at 20 h-post-treatment (Additional file 1: Figure S3). Taken together, these results emphasized that the anti-HER3 antibody 9F7-F11 induces ITCH activation, leading to HER3 and c-FLIP degradation to allow caspase-mediated apoptosis.
Fig. 5.

ITCH silencing or chemical inhibition blocks 9F7-F11-induced apoptosis and c-FLIP degradation. siSC- and siITCH-transfected BxPC3 cells were incubated with 9F7-F11 alone (a), or with NRG1 (b) for 48 h before detection by western blotting of ITCH and c-FLIPL expression, and PARP/caspase cleavage. MDA-MB-468 cells were incubated with increasing doses of chlorimipramine (CI) for 24 h (c), or with 15 μM CI (d) before incubation with 9F7-F11 for 24 h. Total protein extracts were analyzed by western blotting to evaluate PARP and caspase cleavage (c), ITCH ubiquitination and expression and c-FLIP and HER3 expression (d). Quantification of signal intensity (SI) with ImageJ software is indicated below the images. No protein expression was measured as 0.0 ± .0. Significant decrease or increase of the densitometry, compared to control, is indicated in bold. BxPC3 cells were left untreated or pre-incubated with ITCH chemical inhibitor CI for 48 h before treatment with 9F7-F11 with or without NRG1. Apoptosis was measured at 96 h by flow cytometry after cell labelling with Annexin V/7-AAD (e). siSC and siITCH-transfected BxPC3 cells were treated with 9F7-F11 alone or with NRG1. Western blot was performed to check ITCH reduction in siITCH-transfected BxPC3 cells. Apoptosis was measured at 96 h by flow cytometry after cell labelling with Annexin V/7-AAD (f). **P < 0.01, ***P < 0.001, ns not significant
9F7-F11 activates the extrinsic apoptotic pathway through FAS and DR5 upregulation, TRAIL expression and DcR2 downregulation
To test whether DRs were involved in 9F7-F11-induced apoptosis, we analyzed the expression of factors involved in the extrinsic pathway by western blotting after incubation of various cancer cell lines with 9F7-F11. Compared with untreated cells, the FAS and DR5 receptors were upregulated at 72 h of incubation until the end of the experiment (120 h) (Fig. 6a). TRAIL precursor (mTRAIL), which induces DR5 activation via a paracrine or autocrine loop, was also overexpressed at 72 h, except for DU145. The decoy receptor DcR2, which inhibits DR5 activation by trapping TRAIL, was already downregulated at 24-48 h (Fig. 6a). This demonstrated that 9F7-F11 induces the caspase-8-mediated extrinsic apoptotic pathway by downregulating DcR2, leading to DR5 upregulation and TRAIL expression.
To test whether activation of the extrinsic apoptotic pathway induces DISC formation, we immunoprecipitated BxPC3 cells with an anti-DR5 antibody at different time points during incubation with 9F7-F11 (Fig. 6b). In untreated cells (i.e., without caspase-8 activation to form a pre-DISC), FADD and pro-caspase 8 were weakly immunoprecipitated with DR5 compared to IgG baseline. Conversely, 9F7-F11 promoted p41/43 and p18 (caspase-8 cleavage) and FADD recruitment, indicating DR5-DISC formation. Interestingly, FADD activation, as well as p41/43 caspase 8 cleavage, seem to fluctuate with lower levels observed at 48 h and 96 h, and overexpression at 24 h, 72 h and 120 h-post 9F7-F11 treatment. Strong global activation was observed at 120 h-end of the experiment (Fig. 6b), thus correlating DR5/FADD overexpression with increased caspase-8 activation to induce the extrinsic apoptotic pathway.
9F7-F11 activates also the intrinsic mitochondrial apoptotic pathway
9F7-F11 induced caspase-8 activation followed by caspase-9/3 cleavage (Fig. 1). Analysis by western blotting of caspase-mediated apoptosis in BxPC3 cells incubated with 9F7-F11 showed that the anti-HER3 antibody also induced BID cleavage and formation of the truncated form tBID from 72 h–96 h until the end of the experiment (120 h) (Fig. 7a), suggesting that caspase-8 activation induces BID cleavage to favor the mitochondrial death pathway. 9F7-F11 also upregulated BAX and promoted dimer formation at 72 h (Fig. 7a), strengthening the involvement of mitochondria to promote complete activation of caspase-3 via caspase-9. The induction of the p53 transcriptional target BIM (at 48 h; Fig. 7a) allows mitochondria amplification of caspase-3 activation. These effects were confirmed also in MDA-MB-468 cells incubated with 9F7-F11 in a slight different time-frame (Fig. 7b). We only observed 9F7-F11-induced BIM induction in BAX-deficient DU145 cells [42] (Fig. 7b). These results demonstrated the involvement of the intrinsic apoptotic pathway (BID truncation, BAX upregulation and BIM expression to induce full caspase-9 activation) in cancer cells incubated with 9F7-F11.
Fig. 7.

9F7-F11 activates the intrinsic mitochondrial apoptosis pathway by inducing caspase-8-mediated BID cleavage, BAX activation and BIM expression. BxPC3 (a), MDA-MB-468 and DU145 (b) cells were incubated with 9F7-F11 for various times. BID cleavage (tBID), BAX activation and BIM expression were analyzed by western blotting. Quantification of signal intensity (SI) with ImageJ software is indicated below the images. No protein expression was measured as 0.0 ± .0. Significant decrease or increase of the densitometry, compared to untreated control, is indicated in bold. β-tubulin was evaluated as loading control
9F7-F11 induces downregulation of the pro-survival proteins c-IAP2 and XIAP
9F7-F11 sensitized tumor cells to caspase-mediated apoptosis through ITCH-dependent c-FLIP downregulation. Pro-survival proteins from the IAP family inhibit apoptosis [43]. For efficient apoptosis inhibition, XIAP is stabilized by interaction with the deubiquitinase USP9X and then promotes caspase-3 degradation through ubiquitination [44]. We previously demonstrated that anti-HER3 antibodies inhibit XIAP phosphorylation to favor apoptosis [2]. Here, western blotting analysis (Fig. 8) showed that in 9F7-F11-treated BxPC3 cells, cIAP2 and XIAP expression was reduced starting at 48 h–72 h and completely downregulated at 96 h. The progressive USP9X downregulation, which occurred concomitantly with that of cIAP2 and XIAP, could be responsible for XIAP degradation. We obtained similar results with other cancer cell lines (MDA-MB-468 and DU145 cells), but with slower kinetics (Fig. 8). This indicated that 9F7-F11 induces USP9X downregulation leading to XIAP degradation, which allows DR5-mediated caspase-dependent apoptosis.
Fig. 8.
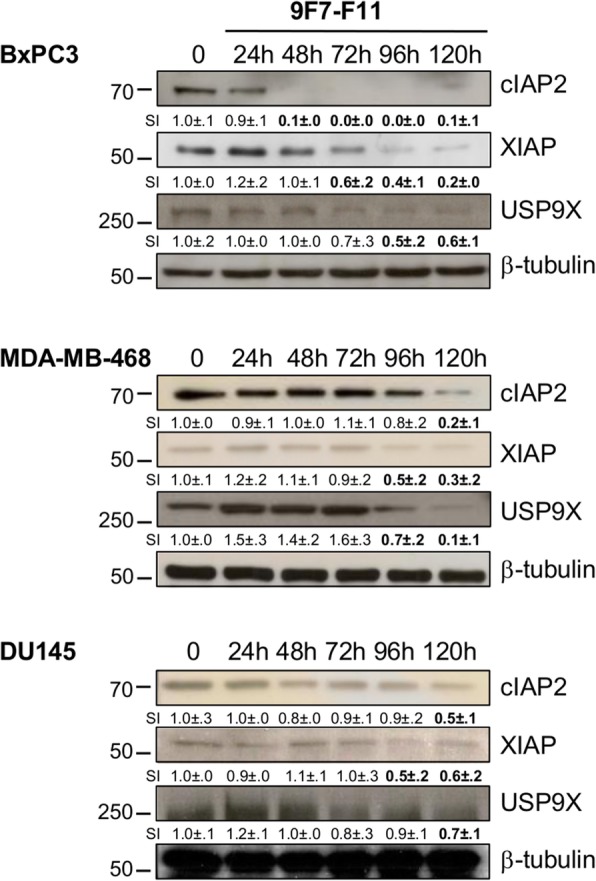
9F7-F11 induces downregulation of the pro-survival proteins c-IAP2 and XIAP. Cells were incubated with 9F7-F11, before western blot analysis of cIAP2 and XIAP (survival factors) and of UP9X (deubiquitinase) expression in protein lysates prepared at different time points. Quantification of signal intensity (SI) with ImageJ software is indicated below the images (relative to untreated control measured as 1.0 ± .0). Significant increase or decrease of the densitometry, compared to control, is indicated in bold. β-tubulin was evaluated as loading control
Discussion
Here, we report that ITCH-dependent proteasomal degradation of c-FLIP induced by the anti-HER3 antibody 9F7-F11 favors DR5/caspase 8-mediated apoptosis of tumor cells. This mechanism (as illustrated in Fig. 9) can explain how anti-HER3 antibodies directly induce cancer cell apoptosis [2, 45, 46] and counteract the HER3-mediated apoptosis inhibition observed in cancer cells resistant to chemotherapy [3, 4]. HER3 silencing [1, 47] or pharmacological inhibition [2] directly restores tumor-specific apoptosis, underlying its critical role in cell death inhibition. HER3 degradation induced by specific antibodies [2, 33, 48, 49] is a marker of pre-clinical drug efficacy [2, 32, 45, 46], and is frequently associated with cancer cell apoptosis induction [2, 45, 46]. We previously demonstrated that in cancer cells, 9F7-F11 blocks the PI3K/AKT pathway [2, 32, 33], induces HER3 downregulation and promotes cell apoptosis [2, 33], leading to in vivo tumor regression [2, 32]. The binding to HER3 and biological effects on tumor cells of 9F7-F11 are paradoxically facilitated by the natural ligand NRG1 [32]. By hijacking NRG1 addiction of cancer cells to promote its inhibitory effects on NRG1-mediated tumor growth and resistance, the allosteric non NRG1-competing 9F7-F11 displays a unique potential for targeted treatment of NRG1-positive cancers [32].
Fig. 9.

The representation illustrates a proposed model for ITCH-dependent proteasomal degradation of c-FLIP induced by 9F7-F11 to promote DR5/caspase-8 apoptosis. See text for details
HER3 ubiquitination and degradation induced by 9F7-F11 mainly occur through JNK1/2-dependent ITCH activation, and are regulated by the deubiquitinases USP8 and USP9X [33]. Here, we showed that upon incubation of cancer cells with 9F7-F11, the E3 ubiquitin ligase ITCH interacts with c-FLIP to trigger c-FLIP ubiquitination and degradation, concomitantly with early ITCH recruitment to HER3 [33]. In this setting, 9F7-F11 induces JNK1/2 activation to phosphorylate ITCH on Thr222. Other studies identified JNK1/2 as the main regulator of ITCH-induced c-FLIP degradation after TNFα stimulation [26] or AKT inhibition in glioblastoma [27]. c-FLIP degradation via JNK/ITCH activation has been recently described to sensitize tamoxifen-resistant breast cancer to TRAIL-induced cell death [28]. 9F7-F11 could be useful to bypass resistance to chemotherapy in breast cancer by favoring c-FLIP degradation via JNK/ITCH activation. In basal conditions (medium alone), the deubiquitinase USP8 contributes to stabilization of c-FLIP, as shown here (Figs. 3 and 4) and by others [39], and of ITCH [27, 33]. Conversely, during incubation with 9F7-F11, USP8 leaves the ITCH-c-FLIP complex, allowing JNK1/2-mediated ITCH activation for c-FLIP ubiquitination. ITCH and USP8 silencing experiments highlighted the role of this ubiquitination/deubiquitination process in modulating 9F7-F11-induced c-FLIP and HER3 degradation, and also in the inhibition of caspase-8-mediated apoptosis of tumor cells. This links c-FLIP downregulation by 9F7-F11 with antibody-induced caspase-8 activation. In addition, antibody treatment disrupted the basal USP8-HER3 interaction to favor ITCH-mediated HER3 ubiquitination and proteasomal degradation. In conclusion, the deubiquitinase USP8 acts by co-regulating c-FLIP (Fig. 4), ITCH and HER3 [33] stability, and this triple regulation is affected by 9F7-F11. Interestingly, it has been reported that a synthetic USP8 inhibitor also induces downregulation of receptor tyrosine kinases, including HER3 and c-MET, leading to inhibition of cell survival/proliferation and tumor regression in mice xenografted with gefitinib-resistant non-small cell lung cancer cells [50]. Similarly to 9F7-F11 activity, vitamin E analogues (α-tocopherol derivatives) inhibit the HER3-mediated AKT pro-survival pathway and the anti-apoptosis factors c-FLIP and survivin to favor caspase-mediated apoptosis in cisplatin-resistant ovarian cancer [51]. Targeting c-FLIP has been also proposed in various cancers, mainly indirectly by using chemotherapies such as cisplatin, 5-fluorouracil, gemcitabine, etoposide and paclitaxel [14].
We demonstrated that 9F7-F11 induces DR5 upregulation and TRAIL expression, leading to DR5-mediated caspase-8 activation through the formation of the DR5-DISC complex. Chemical compounds, such as dibenzylideneacetone, also upregulate DR5, via activation of ROS-mediated C/EBP homologous transcription factor (CHOP) that induces transcription of pro-apoptotic proteins [52]. CHOP-dependent DR5 upregulation and ROS production promote TRAIL-induced apoptosis through downregulation of XIAP, survivin, and c-FLIPL and c-FLIPS [52]. Similarly, the natural molecule zerumbone, a sesquiterpene from tropical ginger, induces DR5 upregulation via ROS-mediated activation of the MAP kinases ERK1/2 and p38, leading to DR5/TRAIL-mediated apoptosis [53]. Oxidative stress, through ROS production, is often associated with apoptosis via JNK1/2 activation. Some therapeutic antibodies promote apoptosis together with ROS accumulation, leading to JNK activation [54, 55]. Anti-HER3 antibodies, such as MM-121 and hMP-RM-1 that downregulate survivin [4], or 9F7-F11 that inhibits XIAP phosphorylation [2], favor apoptosis and reduce cell survival and proliferation. Here, we showed that 9F7-F11 represses the expression of the survival proteins XIAP and c-IAP2 and also of c-FLIP, concomitantly with DR5 upregulation and TRAIL expression induction. This suggests that antibody-induced caspase-8-mediated apoptosis involves DR5 activation by TRAIL autocrine loop or by ROS-dependent DR5 aggregation for DISC formation. It is worth noting that in our setting, DR5-DISC formation started early with the recruitment of FADD and pro-caspase 8, before DR5 upregulation. Therefore, anti-HER3 antibody-triggered DR5 aggregation and DISC formation might occur independently of TRAIL induction to activate caspase-8-mediated apoptosis. Our findings showed that 9F7-F11-induced apoptosis involves also the mitochondrial pathway with the induction of BID cleavage by caspase-8, BAX upregulation and BIM expression induction, leading to mitochondria-dependent caspase-9 activation and full caspase-3 activation. This is in agreement with previous work showing that siRNA-mediated HER3 downregulation induces BAX-BAK-mediated apoptosis [47]. Upon incubation with 9F7-F11, we observed BID cleavage and BIM expression also in DU145 prostate cancer cells, which are BAX-deficient [42] but express BAK that could replace BAX to induce caspase-9 activation. It has been demonstrated that BAK is preferentially activated by tBID and BAX by BIM, to modulate the response to chemotherapy [56]. Indeed, anti-HER3 antibody-induced apoptosis in DU145 cells was lower than in the other tested cancer cell lines, probably because BAX deficiency prevented BIM-mediated apoptosis.
Conclusions
We provide evidence that the allosteric non-NRG1 competing modulator 9F7-F11, sensitizes tumor cells to caspase-mediated apoptosis through ITCH-dependent degradation of c-FLIP, and independently of ligand addiction. The description of the multiple modes of action of the anti-HER3 antibody 9F7-F11 not only adds to our basic understanding of dysregulated signaling in cancer, but might help the selection of drug combinations and clinical indications for 9F7-F11 in NRG1-addicted or NRG1-rearranged cancer.
Additional file
Figure S1. (a) Flow cytometry analysis of EGFR, HER2 and HER3 expression at the membrane of BxPC3, DU-145 and MDA-MB-468 cells. (b) Western blot analysis of receptor and NRG1 expression, and USP8, USP9 and ITCH expression in whole lysates of BxPC3, DU-145 and MDA-MB-468 cell lines. (PDF 230 kb)
Acknowledgements
We thank G. Heintz and S. Bousquié for excellent technical assistance.
Abbreviations
- 7-AAD
A-Aminoactinomycin D
- BAD
Bcl-2-associated death promoter
- BID
BH3 interacting-domain death agonist
- BIM
Bcl-2-like protein 11
- c-FLIP
FLICE-like inhibitory protein
- CI
Chlorimipramine
- cIAP2
Cellular inhibitor of apoptosis protein 2
- DED
Death effector domain
- DISC
Death-inducing signaling complex
- DR
Death receptor
- FADD
Fas-associated protein with death domain
- ITCH
E3 ubiquitin-protein ligase Itchy homolog
- JNK
c-Jun N-terminal kinase
- NRG1
Neuregulin 1
- PARP
Poly (ADP-ribose) polymerase
- SD
Standard deviation
- USP8
Ubiquitin-specific peptidase 8
- XIAP
X-linked inhibitor of apoptosis protein
Authors’ contributions
CLC and TC contributed to the conception and design of the study. CLC, YL, OD, RL, and TC contributed to the development of the methodology. CLC and TC acquired, analyzed and interpreted the data. CLC, CS, CL, MAP, AP and TC wrote the manuscript. JMB, JFP, AP and TC edited and supervised the manuscript, and provided critical comments. All authors read and approved the final manuscript.
Funding
This work was supported by the program “Investissement d’Avenir” (grant agreement: LabexMabImprove, ANR-10-LABX-53-01) and by the grant AAP13 “Fonds Unique Interministeriel” FUI UmAbHER3 F120402 M. CLC was supported by INSERM through the FUI UmAbHER3. CS was supported by the Conseil Régional de Bourgogne, the European Union and the University of Burgundy.
Availability of data and materials
All data generated or analysed during this study are included in this published article (and its supplementary information files).
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
OD, JMB, and JFP are employed by GamaMabs Pharma. AP, CL and TC are inventors in the WO2012/156532 patent “Anti-human HER3 and uses thereof”, the WO2015/067986 patent “Neuregulin allosteric anti-HER3 antibody”, and the WO2016/177664 patent “Low-fucose anti-human HER3 antibodies and uses thereof”. OD, JMB and JFP are inventors in the WO2016/177664 patent “Low-fucose anti-human HER3 antibodies and uses thereof”. The other authors declare no conflict of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Christophe Le Clorennec, Email: christophe.leclorennec@yahoo.fr.
Yassamine Lazrek, Email: yassaminelazrek@gmail.com.
Olivier Dubreuil, Email: odubreuil@gamambs.com.
Carla Sampaio, Email: sampaiocarla@yahoo.fr.
Christel Larbouret, Email: christel.larbouret@inserm.fr.
Romain Lanotte, Email: romain.lanotte@inserm.fr.
Marie-Alix Poul, Email: marie-alix.poul@inserm.fr.
Jean-Marc Barret, Email: jmbarret@gamamabs.com.
Jean-François Prost, Email: jfprost@gamamabs.com.
André Pèlegrin, Email: andre.pelegrin@inserm.fr.
Thierry Chardès, Phone: (33) 467 612 404, Email: thierry.chardes@inserm.fr.
References
- 1.Lee D, Yu M, Lee E, Kim H, Yang Y, Kim K, et al. Tumor-specific apoptosis caused by deletion of the ERBB3 pseudo-kinase in mouse intestinal epithelium. J Clin Invest. 2009;119:2702–2713. doi: 10.1172/JCI36435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lazrek Y, Dubreuil O, Garambois V, Gaborit N, Larbouret C, Le Clorennec C, et al. Anti-HER3 domain 1 and 3 antibodies reduce tumor growth by hindering HER2/HER3 dimerization and AKT-induced MDM2, XIAP, and FoxO1 phosphorylation. Neoplasia N Y N. 2013;15:335–347. doi: 10.1593/neo.121960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang S, Huang X, Lee C-K, Liu B. Elevated expression of erbB3 confers paclitaxel resistance in erbB2-overexpressing breast cancer cells via upregulation of Survivin. Oncogene. 2010;29:4225–4236. doi: 10.1038/onc.2010.180. [DOI] [PubMed] [Google Scholar]
- 4.Wang S, Huang J, Lyu H, Cai B, Yang X, Li F, et al. Therapeutic targeting of erbB3 with MM-121/SAR256212 enhances antitumor activity of paclitaxel against erbB2-overexpressing breast cancer. Breast Cancer Res BCR. 2013;15:R101. doi: 10.1186/bcr3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.French LE, Tschopp J. Protein-based therapeutic approaches targeting death receptors. Cell Death Differ. 2003;10:117–123. doi: 10.1038/sj.cdd.4401185. [DOI] [PubMed] [Google Scholar]
- 6.Mahmood Z, Shukla Y. Death receptors: targets for cancer therapy. Exp Cell Res. 2010;316:887–899. doi: 10.1016/j.yexcr.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 7.Wajant H. Death receptors. Essays Biochem. 2003;39:53–71. doi: 10.1042/bse0390053. [DOI] [PubMed] [Google Scholar]
- 8.Dickens LS, Boyd RS, Jukes-Jones R, Hughes MA, Robinson GL, Fairall L, et al. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol Cell. 2012;47:291–305. doi: 10.1016/j.molcel.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schleich K, Warnken U, Fricker N, Oztürk S, Richter P, Kammerer K, et al. Stoichiometry of the CD95 death-inducing signaling complex: experimental and modeling evidence for a death effector domain chain model. Mol Cell. 2012;47:306–319. doi: 10.1016/j.molcel.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 10.Lemke J, von Karstedt S, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014;21:1350–1364. doi: 10.1038/cdd.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schug ZT, Gonzalvez F, Houtkooper RH, Vaz FM, Gottlieb E. BID is cleaved by caspase-8 within a native complex on the mitochondrial membrane. Cell Death Differ. 2011;18:538–548. doi: 10.1038/cdd.2010.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Westphal D, Kluck RM, Dewson G. Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 2014;21:196–205. doi: 10.1038/cdd.2013.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walczak H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb Perspect Biol. 2013;5:a008698. doi: 10.1101/cshperspect.a008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Humphreys L, Espona-Fiedler M, Longley DB. FLIP as a therapeutic target in cancer. FEBS J. 2018;285:4104–4123. doi: 10.1111/febs.14523. [DOI] [PubMed] [Google Scholar]
- 15.Horn S, Hughes MA, Schilling R, Sticht C, Tenev T, Ploesser M, et al. Caspase-10 negatively regulates Caspase-8-mediated cell death, switching the response to CD95L in favor of NF-κB activation and cell survival. Cell Rep. 2017;19:785–797. doi: 10.1016/j.celrep.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hughes MA, Powley IR, Jukes-Jones R, Horn S, Feoktistova M, Fairall L, et al. Co-operative and hierarchical binding of c-FLIP and Caspase-8: a unified model defines how c-FLIP isoforms differentially control cell fate. Mol Cell. 2016;61:834–849. doi: 10.1016/j.molcel.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schleich K, Buchbinder JH, Pietkiewicz S, Kähne T, Warnken U, Öztürk S, et al. Molecular architecture of the DED chains at the DISC: regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain. Cell Death Differ. 2016;23:681–694. doi: 10.1038/cdd.2015.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haag C, Stadel D, Zhou S, Bachem MG, Möller P, Debatin K-M, et al. Identification of c-FLIP(L) and c-FLIP(S) as critical regulators of death receptor-induced apoptosis in pancreatic cancer cells. Gut. 2011;60:225–237. doi: 10.1136/gut.2009.202325. [DOI] [PubMed] [Google Scholar]
- 19.Day TW, Huang S, Safa AR. C-FLIP knockdown induces ligand-independent DR5-, FADD-, caspase-8-, and caspase-9-dependent apoptosis in breast cancer cells. Biochem Pharmacol. 2008;76:1694–1704. doi: 10.1016/j.bcp.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, Zhang L, Yang H, Huang X, Otu H, Libermann TA, et al. c-Fos as a proapoptotic agent in TRAIL-induced apoptosis in prostate cancer cells. Cancer Res. 2007;67:9425–9434. doi: 10.1158/0008-5472.CAN-07-1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson TR, McLaughlin KM, McEwan M, Sakai H, Rogers KMA, Redmond KM, et al. C-FLIP: a key regulator of colorectal cancer cell death. Cancer Res. 2007;67:5754–5762. doi: 10.1158/0008-5472.CAN-06-3585. [DOI] [PubMed] [Google Scholar]
- 22.Panner A, James CD, Berger MS, Pieper RO. mTOR controls FLIPS translation and TRAIL sensitivity in glioblastoma multiforme cells. Mol Cell Biol. 2005;25:8809–8823. doi: 10.1128/MCB.25.20.8809-8823.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valente G, Manfroi F, Peracchio C, Nicotra G, Castino R, Nicosia G, et al. cFLIP expression correlates with tumour progression and patient outcome in non-Hodgkin lymphomas of low grade of malignancy. Br J Haematol. 2006;132:560–570. doi: 10.1111/j.1365-2141.2005.05898.x. [DOI] [PubMed] [Google Scholar]
- 24.Valnet-Rabier M-B, Challier B, Thiebault S, Angonin R, Margueritte G, Mougin C, et al. c-Flip protein expression in Burkitt’s lymphomas is associated with a poor clinical outcome. Br J Haematol. 2005;128:767–773. doi: 10.1111/j.1365-2141.2005.05378.x. [DOI] [PubMed] [Google Scholar]
- 25.Safa AR, Pollok KE. Targeting the anti-apoptotic protein c-FLIP for Cancer therapy. Cancers. 2011;3:1639–1671. doi: 10.3390/cancers3021639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang L, Kamata H, Solinas G, Luo J-L, Maeda S, Venuprasad K, et al. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124:601–613. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 27.Panner A, Crane CA, Weng C, Feletti A, Fang S, Parsa AT, et al. Ubiquitin-specific protease 8 links the PTEN-Akt-AIP4 pathway to the control of FLIPS stability and TRAIL sensitivity in glioblastoma multiforme. Cancer Res. 2010;70:5046–5053. doi: 10.1158/0008-5472.CAN-09-3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piggott L, Silva A, Robinson T, Santiago-Gómez A, Simões BM, Becker M, et al. Acquired resistance of ER-positive breast Cancer to endocrine treatment confers an adaptive sensitivity to TRAIL through posttranslational downregulation of c-FLIP. Clin Cancer Res Off J Am Assoc Cancer Res. 2018;24:2452–2463. doi: 10.1158/1078-0432.CCR-17-1381. [DOI] [PubMed] [Google Scholar]
- 29.Abedini MR, Muller EJ, Bergeron R, Gray DA, Tsang BK. Akt promotes chemoresistance in human ovarian cancer cells by modulating cisplatin-induced, p53-dependent ubiquitination of FLICE-like inhibitory protein. Oncogene. 2010;29:11–25. doi: 10.1038/onc.2009.300. [DOI] [PubMed] [Google Scholar]
- 30.Abedini MR, Muller EJ, Brun J, Bergeron R, Gray DA, Tsang BK. Cisplatin induces p53-dependent FLICE-like inhibitory protein ubiquitination in ovarian cancer cells. Cancer Res. 2008;68:4511–4517. doi: 10.1158/0008-5472.CAN-08-0673. [DOI] [PubMed] [Google Scholar]
- 31.Yang F, Tay KH, Dong L, Thorne RF, Jiang CC, Yang E, et al. Cystatin B inhibition of TRAIL-induced apoptosis is associated with the protection of FLIP(L) from degradation by the E3 ligase itch in human melanoma cells. Cell Death Differ. 2010;17:1354–1367. doi: 10.1038/cdd.2010.29. [DOI] [PubMed] [Google Scholar]
- 32.Le Clorennec C, Bazin H, Dubreuil O, Larbouret C, Ogier C, Lazrek Y, et al. Neuregulin 1 allosterically enhances the antitumor effects of the noncompeting anti-HER3 antibody 9F7-F11 by increasing its binding to HER3. Mol Cancer Ther. 2017;16:1312–1323. doi: 10.1158/1535-7163.MCT-16-0886. [DOI] [PubMed] [Google Scholar]
- 33.Le Clorennec C, Lazrek Y, Dubreuil O, Larbouret C, Poul M-A, Mondon P, et al. The anti-HER3 (ErbB3) therapeutic antibody 9F7-F11 induces HER3 ubiquitination and degradation in tumors through JNK1/2- dependent ITCH/AIP4 activation. Oncotarget. 2016;7(24):37013–37029. doi: 10.18632/oncotarget.9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malm M, Frejd FY, Ståhl S, Löfblom J. Targeting HER3 using mono- and bispecific antibodies or alternative scaffolds. mAbs. 2016;8:1195–1209. doi: 10.1080/19420862.2016.1212147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacob W, James I, Hasmann M, Weisser M. Clinical development of HER3-targeting monoclonal antibodies: perils and progress. Cancer Treat Rev. 2018;68:111–123. doi: 10.1016/j.ctrv.2018.06.011. [DOI] [PubMed] [Google Scholar]
- 36.Thomas G, Chardès T, Gaborit N, Mollevi C, Leconet W, Robert B, et al. HER3 as biomarker and therapeutic target in pancreatic cancer: new insights in pertuzumab therapy in preclinical models. Oncotarget. 2014;5:7138–7148. doi: 10.18632/oncotarget.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mouchantaf R, Azakir BA, McPherson PS, Millard SM, Wood SA, Angers A. The ubiquitin ligase itch is auto-ubiquitylated in vivo and in vitro but is protected from degradation by interacting with the deubiquitylating enzyme FAM/USP9X. J Biol Chem. 2006;281:38738–38747. doi: 10.1074/jbc.M605959200. [DOI] [PubMed] [Google Scholar]
- 38.Gallagher E, Gao M, Liu Y-C, Karin M. Activation of the E3 ubiquitin ligase itch through a phosphorylation-induced conformational change. Proc Natl Acad Sci U S A. 2006;103:1717–1722. doi: 10.1073/pnas.0510664103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeong M, Lee E-W, Seong D, Seo J, Kim J-H, Grootjans S, et al. USP8 suppresses death receptor-mediated apoptosis by enhancing FLIPL stability. Oncogene. 2017;36:458–470. doi: 10.1038/onc.2016.215. [DOI] [PubMed] [Google Scholar]
- 40.Rossi M, Rotblat B, Ansell K, Amelio I, Caraglia M, Misso G, et al. High throughput screening for inhibitors of the HECT ubiquitin E3 ligase ITCH identifies antidepressant drugs as regulators of autophagy. Cell Death Dis. 2014;5:e1203. doi: 10.1038/cddis.2014.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rossi M, Munarriz ER, Bartesaghi S, Milanese M, Dinsdale D, Guerra-Martin MA, et al. Desmethylclomipramine induces the accumulation of autophagy markers by blocking autophagic flux. J Cell Sci. 2009;122:3330–3339. doi: 10.1242/jcs.048181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gillissen B, Essmann F, Graupner V, Stärck L, Radetzki S, Dörken B, et al. Induction of cell death by the BH3-only Bcl-2 homolog Nbk/Bik is mediated by an entirely Bax-dependent mitochondrial pathway. EMBO J. 2003;22:3580–3590. doi: 10.1093/emboj/cdg343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silke J, Vince J. IAPs and cell death. Curr Top Microbiol Immunol. 2017;403:95–117. doi: 10.1007/82_2016_507. [DOI] [PubMed] [Google Scholar]
- 44.Engel K, Rudelius M, Slawska J, Jacobs L, Ahangarian Abhari B, Altmann B, et al. USP9X stabilizes XIAP to regulate mitotic cell death and chemoresistance in aggressive B-cell lymphoma. EMBO Mol Med. 2016;8:851–862. doi: 10.15252/emmm.201506047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fattore L, Malpicci D, Marra E, Belleudi F, Noto A, De Vitis C, et al. Combination of antibodies directed against different ErbB3 surface epitopes prevents the establishment of resistance to BRAF/MEK inhibitors in melanoma. Oncotarget. 2015;6:24823–24841. doi: 10.18632/oncotarget.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noto A, De Vitis C, Roscilli G, Fattore L, Malpicci D, Marra E, et al. Combination therapy with anti-ErbB3 monoclonal antibodies and EGFR TKIs potently inhibits non-small cell lung cancer. Oncotarget. 2013;4:1253–1265. doi: 10.18632/oncotarget.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee H, Lee H, Chin H, Kim K, Lee D. ERBB3 knockdown induces cell cycle arrest and activation of Bak and Bax-dependent apoptosis in colon cancer cells. Oncotarget. 2014;5:5138–5152. doi: 10.18632/oncotarget.2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Belleudi F, Marra E, Mazzetta F, Fattore L, Giovagnoli MR, Mancini R, et al. Monoclonal antibody-induced ErbB3 receptor internalization and degradation inhibits growth and migration of human melanoma cells. Cell Cycle Georget Tex. 2012;11:1455–1467. doi: 10.4161/cc.19861. [DOI] [PubMed] [Google Scholar]
- 49.Gaborit N, Abdul-Hai A, Mancini M, Lindzen M, Lavi S, Leitner O, et al. Examination of HER3 targeting in cancer using monoclonal antibodies. Proc Natl Acad Sci U S A. 2015;112:839–844. doi: 10.1073/pnas.1423645112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Byun S, Lee S-Y, Lee J, Jeong C-H, Farrand L, Lim S, et al. USP8 is a novel target for overcoming gefitinib resistance in lung cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2013;19:3894–3904. doi: 10.1158/1078-0432.CCR-12-3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shun M-C, Yu W, Park S-K, Sanders BG, Kline K. Downregulation of epidermal growth factor receptor expression contributes to alpha-TEA’s Proapoptotic effects in human ovarian Cancer cell lines. J Oncol. 2010;2010:824571. doi: 10.1155/2010/824571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prasad S, Yadav VR, Ravindran J, Aggarwal BB. ROS and CHOP are critical for dibenzylideneacetone to sensitize tumor cells to TRAIL through induction of death receptors and downregulation of cell survival proteins. Cancer Res. 2011;71:538–549. doi: 10.1158/0008-5472.CAN-10-3121. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Yodkeeree S, Sung B, Limtrakul P, Aggarwal BB. Zerumbone enhances TRAIL-induced apoptosis through the induction of death receptors in human colon cancer cells: evidence for an essential role of reactive oxygen species. Cancer Res. 2009;69:6581–6589. doi: 10.1158/0008-5472.CAN-09-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen C, Liu Y, Zheng D. An agonistic monoclonal antibody against DR5 induces ROS production, sustained JNK activation and Endo G release in Jurkat leukemia cells. Cell Res. 2009;19:984–995. doi: 10.1038/cr.2009.60. [DOI] [PubMed] [Google Scholar]
- 55.Stein R, Gupta P, Chen X, Cardillo TM, Furman RR, Chen S, et al. Therapy of B-cell malignancies by anti-HLA-DR humanized monoclonal antibody, IMMU-114, is mediated through hyperactivation of ERK and JNK MAP kinase signaling pathways. Blood. 2010;115:5180–5190. doi: 10.1182/blood-2009-06-228288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L, et al. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell. 2013;51:751–765. doi: 10.1016/j.molcel.2013.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (a) Flow cytometry analysis of EGFR, HER2 and HER3 expression at the membrane of BxPC3, DU-145 and MDA-MB-468 cells. (b) Western blot analysis of receptor and NRG1 expression, and USP8, USP9 and ITCH expression in whole lysates of BxPC3, DU-145 and MDA-MB-468 cell lines. (PDF 230 kb)
Data Availability Statement
All data generated or analysed during this study are included in this published article (and its supplementary information files).