

Abstract
Traumatic brain injury (TBI) is the leading cause of acquired neurologic disability in children, yet specific therapies to treat TBI are lacking. Therapies that decrease the inflammatory response and enhance a reparative immune action may decrease oxidative damage and improve outcomes after TBI. Docosahexaenoic acid (DHA) modulates the immune response to injury in many organs. DHA given in the diet before injury decreased rat pup cognitive impairment, oxidative stress and white matter injury in our developmental TBI model using controlled cortical impact (CCI). Little is known about DHA effects on neuroinflammation in the developing brain. Further, it is not known if DHA given after developmental TBI exerts neuroprotective effects. We hypothesized that acute DHA treatment would decrease oxidative stress and improve cognitive outcome, associated with decreased pro-inflammatory activation of microglia, the brain’s resident macrophages.
Methods:
17-day-old rat pups received intraperitoneal DHA or vehicle after CCI or SHAM surgery followed by DHA diet or continuation of REG diet to create DHACCI, REGCCI, SHAMDHA and SHAMREG groups. We measured brain nitrates/nitrites (NOx) at post injury day (PID) 1 to assess oxidative stress. We tested memory using Novel Object Recognition (NOR) at PID14. At PID 3 and 7, we measured reactivity of microglial activation markers Iba1, CD68 and CD206 and astrocyte marker GFAP in the injured cortex. At PID3, 7 and 30 we measured mRNA levels of inflammation-related genes and transcription factors in flow-sorted brain cells.
Results:
DHA decreased oxidative stress at PIDI and pro-inflammatory microglial activation at PID3. CCI increased mRNA levels of two interferon regulatory family transcription factors, blunted by DHA, particularly in microglia-enriched cell populations at PID7. CCI increased mRNA levels of genes associated with “pro-“ and “anti-“ inflammatory activity at PID3, 7 and 30. Most notably within the microglia-enriched population, DHA blunted increased mRNA levels of pro-inflammatory genes at PID 3 and 7 and of anti-inflammatory genes at PID 30. Particularly in microglia, we observed parallel activation of pro-inflammatory and antiinflammatory genes. DHA improved performance on NOR at PID14 after CCI.
Conclusions:
DHA decreased oxidative stress and histologic and mRNA markers of microglial pro-inflammatory activation in rat pup brain acutely after CCI associated with improved short term cognitive function. DHA administration after CCI has neuroprotective effects, which may result in part from modulation of microglial activation towards a less inflammatory profile in the first week after CCI. Future and ongoing studies will focus on phagocytic function and reactive oxygen species production in microglia and macrophages to test functional effects of DHA on neuroinflammation in our model. Given its favorable safety profile in children, DHA is a promising candidate therapy for pediatric TBI.
Keywords: Docosahexaenoic acid, developing brain, controlled cortical impact, neuroinflammation, rats
Introduction
Traumatic brain injury (TBI) is the leading cause of acquired cognitive disability in children. (Anderson et al., 2012; Langlois et al., 2005; Yeates et al., 2005) Children are particularly vulnerable to inflammation and oxidative stress, both of which contribute significantly to neurologic impairment after TBI. (Bayir et al., 2006)
Mechanical forces account for much of the primary cell death and dysfunction in the brain immediately after TBI. The extent of final injury depends upon a variety of factors, including secondary insults such as hypoxia and hypotension. A growing body of evidence suggests that the nature, intensity and duration of the immune response to TBI plays a very important role in determining the definitive effects of the initial injury on long term brain function. (Loane and Kumar, 2016)
Astrocytes and microglia (the brain’s resident macrophages), along with infiltrating macrophages (macrophages entering the brain during blood-brain barrier breakdown) are the predominant actors in the cellular immune response to TBI. The immune response is complex and dynamic. Useful, albeit simplistic, terminology categorizes the immune response into M1 and M2 types. While any benefit or detriment derived from an “M1” or “M2” response after TBI is likely to be highly contextual and time-dependent, M1 (“pro-inflammatory) responses are potentially detrimental to brain recovery after TBI, particularly if long-lasting or excessive. M1 responses include the production of reactive oxygen species, cytokines, and chemokines that create a cytotoxic environment. Immune responses that should foster brain recovery after TBI, particularly if present at the appropriate time, are termed M2 (“reparative” and “inflammation-resolving”). These actions include phagocytosis of detritus and production of cytokines, chemokines, and growth factors that promote the proliferation and survival of neurons, oligodendrocytes, axons and synapses. Microglial activation alone, indicated by increased expression of Iba-1 (ionized calcium-binding adapter protein 1) and/or change in morphology towards a larger and less ramified form), does not discriminate between microglia that display primarily phagocytic activity or reactive oxygen species production. (Imai and Kohsaka, 2002)
Persistent pro-inflammatory “M1” microglial activation is associated with lasting neurologic disability after TBI in adult rodents. (Loane et al., 2013) Microglia can resolve inflammation by reverting to a reparatory “M2” phenotype.(Dringen, 2005; Kumar and Loane, 2012) Therapies that modulate microglia away from an inflammatory towards a reparatory phenotype may decrease neurologic disability after TBI.(Hellewell et al., 2016) Data about the inflammatory response of the developing brain to TBI are scarce. In a mouse model of neonatal TBI, minocycline (an agent that decreases pro-inflammatory microglial activation) improved neuropathology at day1, but not day 5, after injury. (Chhor et al., 2017) In a rat model of pediatric TBI, minocycline improved histologic and functional outcomes in the first two weeks after injury. (Simon et al., 2018) An improved understanding of the developing brain’s inflammatory response after TBI, with or without exposure to immune modulation, could result in new and effective approaches to improve outcomes.
Docosahexaenoic acid (DHA), an essential fatty acid found in fish oil and a major component of brain cell membranes, exerts anti-inflammatory activity in the cellular and humoral immune system in vivo. (Calder, 2017; Innis, 2008) In vitro, DHA decreases M1 microglia activation and promotes M2 macrophage polarization. (Antonietta Ajmone-Cat et al., 2012; Chang et al., 2015) DHA decreased histologic injury and neurologic impairment in the adult rat after TBI. (Abdullah et al., 2014; Bailes and Mills, 2010; Begum et al., 2013; Li et al., 2014; Mills et al., 2011; Wu et al., 2004, 2011) In our rat model of pediatric TBI using controlled cortical impact (CCI), dietary DHA begun one day before injury improved cognitive function and decreased oxidative stress and white matter damage. (Schober et al., 2016) Additional information about DHA effects on the inflammatory response of the developing brain to TBI are lacking. Further, it is unknown if the therapeutic effects of DHA are retained when treatment begins acutely after developmental TBI.
The objective of the present study was to test the hypothesis that acute post-injury DHA administration decreases oxidative stress and cognitive dysfunction in rat pups after CCI, associated with decreased pro-inflammatory activation of microglia and/or infiltrating macrophages. We hypothesized that DHA would decrease expression of markers associated with pro-inflammatory microglia and infiltrating macrophages after developmental TBI. We further hypothesized that these inflammatory changes would be associated with decreased oxidative stress and improved outcomes. To test these hypotheses, we sought to assess DHA effects on oxidative stress, astrocytic and microglial activation, mRNA levels of markers of pro-inflammatory and reparative activation in microglia and infiltrating macrophages and on cognitive outcome using a translationally relevant model of DHA administration after experimental TBI in the immature rat. To meet this objective, we used our established model of pediatric TBI using CCI in 17-day old (P17) rats. We selected this age because rat brain maturation at P17 is comparable to that of the human infant/young toddler, the pediatric age group at highest risk for cognitive deficits after TBI.(Anderson et al., 2005a; Anderson et al., 2005b; Bittigau et al., 2004; Dobbing and Sands, 1979; Rice and Barone, 2000). We measured oxidative stress at post injury day (PID) 1 because oxidative injury peaks in the first 24-48 hours after TBI. (Ansari et al., 2008). We used histology and flow cytometry-sorted cells for mRNA expression of microglia/macrophage markers to assess cell populations and M1/M2 states. We assessed glial activation at PID 3, 7 and 30 because in the adult rodent brain M1 peaked in a bimodal fashion at day 3 and 28 after TBI, while M2 peaked at day 7 and macrophage infiltration resolved by day 30 . (Jin et al., 2012; Loane and Kumar, 2016) We tested memory using Novel Object Recognition (NOR) at PID14 and tissue sparing using histology at PID30 based on our previous work showing cognitive deficits at PID14 and volume loss at PID60 in our model.(Schober et al., 2013; Schober et al., 2016 )
Methods and Materials
Animals
The Animal Care and Use Committees at the University of Utah approved all experimental protocols in accordance with US NIH guidelines. Male Sprague-Dawley rats arrived at the University of Utah from Charles Rivers Laboratories (Raleigh, NC) on post-natal day (P) 7-10 and housed in litters of 10 with the lactating dam until weaning on P 24. All rats had free access to food and water and remained in a temperature- and light-controlled (12 h on/12 h off) environment. On the day of surgery, P17, rat pups randomized to Docosahexaenoic Acid (DHA) received DHA intraperitoneally (IP) 30 minutes after either controlled cortical impact (CCI) or control (SHAM) procedure ended. DHA pups returned to the dam after recovery from anesthesia. Rat pups randomized to Regular Diet (REG) received vehicle IP 30 minutes after either CCI or SHAM procedure ended. Dams fed either 0.1% DHA chow or continued on standard chow at the end of surgery. After weaning, DHACCI and SHAMDHA rats consumed the DHA diet exclusively while REGCCI and SHAMREG continued on standard rat chow.
Docosahexaenoic Acid Administration
The DHA rodent diet (Envigo Teklad, WI) substitutes 0.1% of the soybean oil in standard chow with purified DHA (U-84-A, Nu-Chek Prep, MN). This substitution results in the same macronutrient content and caloric density (3.2 kcal/g and 15% kcal derived from fat) as standard rat chow. This 0.1% DHA diet provides DHA as 3.3% of total fat. All dams and pups received REG diet (Envigo Teklad 2920), with the exception of rats in the DHA groups. P17 rats depend exclusively on dam milk for their intake. Dam milk should not reflect changes in dietary DHA intake until at least 10 hours after ingestion, based on human data, (Lauritzen et al., 2002) and gavage feeding rat pups soon after surgery carries a significant risk of pulmonary aspiration. We instead injected rats IP at 30 minutes after CCI or SHAM surgery with DHA (7.5mg/100mcL to approximate 100-150 mg/kg/day dietary intake expected from 0.1% DHA diet) or vehicle, VEH (100mcL soybean oil emulsion and ethanol equivalent to DHA injection).
To minimize litter effects in the experimental groups, Charles River culled rats from different litters prior to shipment to our laboratory to generate groups often male pups per dam. We further minimized litter effects by combining CCI and SHAM rats within the same dietary group in cages of 3-5 rats per cage after weaning.
CCI procedure
Rats underwent CCI or SHAM surgery as previously described. (Schober et al., 2016) We accomplished induction with 3% Isoflurane and maintenance with 2-2.5% Isoflurane for the duration of surgical preparation using a VetEquip Bench Top Isoflurane Anesthesia System (Pleasanton, CA). A servo-controlled heating pad maintained rats’ rectal temperature at 37±0.5 °C. We placed anesthetized rats into a stereotaxic frame (David Kopf, Tujunga, CA). After shaving, prepping with povidone-iodine and incising the scalp, we performed a craniotomy (6-mm×6-mm) over the left parietal cortex (centered at the point 4-mm posterior and 4-mm lateral to bregma) taking care not to perforate the dura. Once the craniotomy was complete, we reduced anesthesia to 1% Isoflurane for a 5-min equilibration period. CCI was then delivered using the ImpactOne™ (Leica Biosystems Inc., IL)) to the left parietal cortex using a 5-mm rounded tip to deliver a 2.0 mm deformation at 5 m/s velocity and 100 ms duration. Immediately after CCI, we increased Isoflurane to 2-2.5% and the bone flap replaced and secured with dental cement (Patterson Dental, Salt Lake City, UT). We sutured the scalp incision and applied triple antibiotic ointment and bupivacaine 0.5% topically. Rats recovered in a temperature-controlled chamber. We delivered IP DHA or VEH, as appropriate for group assignment, 30 minutes after surgery. Once fully awake, rats returned to their dams and littermates. SHAM rats underwent the same anesthetic protocol and duration, scalp incision, stereotaxic positioning and suturing procedures. At post injury days (PID) 1, 3, 7 and 30, rats received IP xylazine (8mg/kg) and ketamine (40mg/kg) followed by intracardial perfusion with ice-cold PBS and decapitation for oxidative stress, histologic and/or molecular outcomes as detailed below.
Oxidative Stress Outcomes (Nitrates/Nitrites (NOx))
At PID1, we extracted brains and dissected ipsilateral (left) hippocampi and cortices on ice, snap frozen in liquid nitrogen and stored at −80°C. Total protein samples (n=8-10 per group) were extracted by homogenizing tissues in ice-cold lysis buffer (150mM NaCl, 50mM Tris pH7.4, 1mM EDTA, 0. 5% Na-deoxycholate, 1% Igepal CA-630) incubated at 4 °C with protease inhibitors (Roche Applied Science, Indianapolis, IN). We centrifuged samples at 10,000 rpm at 4 °C for 5 min and supernatants were stored at - 80 °C until use. We used the determination of protein concentration by bicinchoninic acid or BCA, method (Pierce Protein Research Products, Rockford, IL) to calculate volume for equal protein loading. Nitrate concentrations were measured using chemiluminescence. Nitric oxide combined with oxidative species such as peroxide and superoxide produces nitrates and nitrogen radicals. The nitric oxide and nitrogen radicals react with tissue and serum proteins creating nitrates and nitrites in fluid (NOx). Concentrations of NOx provide a measure of acute/subacute nitrosative oxidative stress. We used a chemiluminescence analyzer (Sievers NOA 280i, GE Analytical Instruments, Boulder, CO) according to manufacturer’s protocol to measure NOx in picomoles normalized by protein concentration for each sample.
Histologic Outcomes
Histologic outcomes included gliosis and activation markers at PID 3 and 7 and lesion volume at PID 30. After perfusion/fixation with ice cold PBS and 4% PFA, brains were cryoprotected via placement in graded sucrose solutions (5, 15 and 30%), frozen whole using dry ice and stored at −80 for later use. We processed all slides on the same day, exposed to freshly made reagents at the same times to minimize variation in conditions between groups. Frozen brains (n=6-8 per group) were sliced into coronal sections from −2.56 to −4.8 bregma. For each animal, three serial 25micron-thick sections from dorsal hippocampus taken 125 microns apart were placed on a single slide. Each of the three sections was taken from a comparable anatomical level as determined by The Rat Brain Atlas in Stereotaxic Coordinates. (Paxinos, 2005)
For immunohistochemistry, we warmed brain sections for 1 hour at 37°C and rinsed with 0.1M PBS. Then, slides were incubated in blocking buffer: 0.1M PBS, pH 7.4 containing 5% Normal Donkey or Goat Serum (Sigma 9663/Sigma 9023); 0.1% Fish Gelatin (Sigma, G7765); 1% Bovine Serum Albumin (Fisher, BP1605); 0.1% Triton X-100 (Sigma, X-100) for 1hr at room temperature then incubated in blocking buffer containing primary antibodies overnight at 4°C. Primary antibodies included rabbit anti-GFAP (1:1000, R&D Systems, Cat# NB300-141), mouse anti-CD68 (1:1000, Bio Rad, Cat#MCA341R), rabbit anti-IBA1 (1:1000, Wako Chemicals, Cat# 019-19741), mouse anti-MHC class 2 (1:1000, BD Bioscience, Cat# 556999), goat anti-CD206 (1:1000, R&D Systems, Cat#AF2535). On the second day, we rinsed slides in 0.1M PBS and incubated with secondary antibodies at room temperature for 1 hour. All secondaries were applied at 1:1000, were non-biotinylated and raised in either donkey or goat: catalog #’s: A31573 (donkey anti rabbit IgG), A11010 (goat anti-rabbit alexa fluor 546), A11017 (goat anti-mouse alexa fluor 488), A11056 (donkey anti-goat 546), (purchased from Life Technologies). After the last rinse all slides were incubated in DAPI (Molecular Probes, Cat # D1306) for 30 minutes (to counterstain nuclei) then rinsed and cover-slipped with Immuno-Mount (Thermo Fisher Scientific). Full dilution curves were performed for each antibody and specificity confirmed using non-primary controls.
Proportional areas of gliosis and macrophage markers were quantified using techniques developed previously. (Donnelly et al., 2009; Orr et al., 2017) Briefly, 0.4mm2 boxes were used to sample the M1 area of cortex and CA1 region of the hippocampus ipsilateral (ipsi, or left sides) and contralateral (contra, or right sided) to the site of CCI or craniotomy (SHAM) on tissue sections approximately −3.00 mm to bregma as depicted in Fig. 1A. Independent analyses using multiple tissue sections per animal confirmed that a single tissue section per animal was representative of the overall pathology. When anatomical landmarks of the cortex were deformed due to injury, the penumbral region of the injured cortex directly above the CA1 region was sampled. Maximum projection confocal image for each area (ipsi cortex, ipsi hippocampus, contra cortex, contra hippocampus) was generated per subject from 3μm thick z-planes, taken with a 20x objective, with a C2 laser scanning confocal microscope (Nikon Instruments Inc, Melville, NY, USA). Threshold based proportional area measures of positive signal within each sample area were generated using Metamorph quantification software (Molecular Devices). Obvious areas of tissue folding or tears were excluded. CD206 positive individual cells were counted based upon discrete cell surface labeling. Data are reported as proportional area of immunoreactivity, or cell density (CD206), within the standard 0.4mm2 sample area. Histological outcomes were confirmed through two independent blinded analyses.
Figure 1.
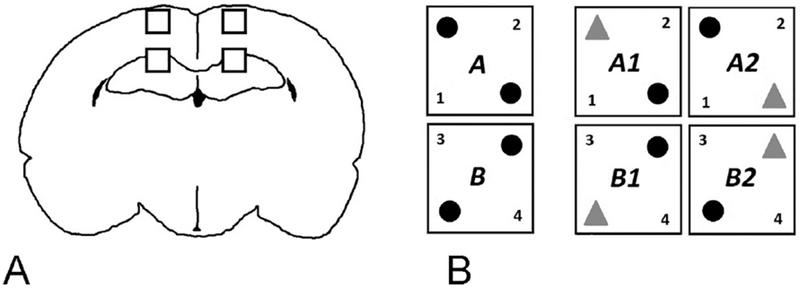
A: Histology Methods
Illustration of areas sampled for gliosis and microglia/macrophage phenotype in the M1 and CA1 regions of the cortex and hippocampus.
B: Novel Object Recognition Methods
Schematic representation of Novel Object Recognition testing paradigm: on the left, the two variations (A and B) on familiar object positioning (black spheres) during Familiarization. On the right, the two variations on novel object positioning (gray triangles in A1 and A2) or (B1 and B2) during Novel Object testing, as detailed in the text.
A modified Eriochrome Cyanine (EC-103164 Sigma Aldrich) and nuclear counter stain (Nuclear Fast Red, H-3403, Vector Laboratories) staining protocol was used to quantify spared tissue area on PID 30. The total area of intact tissue from both hemispheres was quantified for a single tissue section from each animal representing −3mm bregma. Ventricle areas were excluded from analyses. All histological analyses were conducted blindly with respect to treatment condition using a Nikon microscope (Nikon Corporation, Tokyo, Japan) and Scion imaging analysis software (Scion Corporation, Frederick, MD, USA).
Molecular Outcomes: Cellular Gene Expression
We used 8-10 rats from each of the four groups per assay and time point to assess cellular inflammation at PID3, 7 and 30. We dissected ipsilateral (left) hippocampi and cortices (300-350mg per brain) from PBS perfused brains on ice and processed them immediately using the Neural Tissue Dissociation Kit (Miltenyi Biotec Inc) in cold Hanks’ Balanced Salt Solution (HBSS W/O, Gibco ™) on ice. Following myelin and debris removal with a 70-30% Percoll gradient (Percoll ® GE Healthcare, Hibernate-A™, Thermo-Fisher and Dulbecco’s Phosphate-Buffered Saline (DPBS) Gibco™) cell counts were performed using a hemocytometer. Samples were blocked using 6mcL 3% mouse serum and 2mcL FC blocker (CD32 Rat BD Fc Block™ Cat # 550271 BD Biosciences) in DMEM Buffer (Thermo-Fisher) for 20 minutes at 4°C. We added fluorescent antibodies against CD11b (CD11b-PE, Abeam Cat# ab333816) and CD45 (CD45-FITC, BD Bioscience, Cat# 11-0461-82) 0.6 mcL and 2mcL/million cells, respectively. We then incubated samples for 20 minutes at 4°C, protected from light then washed and re-suspended in HBSS. We performed flow cytometry with a BD FACSAria™ II Cell Sorter analyzed using FlowJo software (version 9.7.5 TreeStar, Inc., Ashland, OR). We had previously determined optimal signal to noise ratio in preceding antibody titration experiments for each antibody. We showed very high specificity (99.6%) and good sensitivity (91%) for sorting CD11b+/− populations using this approach, as shown by separate flow experiments using autoMACS® Cell Separator and CD11b-PEb/c magnetic beads (Cat# 130-105-316, Miltenyi Biotech). Similarly, we confirmed accuracy of FSS/SSC for gating live versus dead cells using Propidium Iodide (BioLegend® Cat #42130) for live/dead discrimination. Cell survival ranged between 92 and 97%. We used monocytes in rat pup blood to gate CD11b+ cells into CD11b+/CD45high and CD11b+/CD45low in the initial pilot studies, and subsequently used SHAM brains instead because they had very few if any CD11b+/CD45high cells. The specificity of the signals of antibodies against specific antigens was determined using isotype-matched immunoglobulins (ab 91357 and Bioscience 114714) and unstained controls for all experiments. Rat brain samples were each sorted into three populations: LO cells (CD11b+ CD45low, enriched in resident microglia) HI cells (CD11b+ CD45 high, enriched in infiltrating macrophages) and NEG cells (CD11b- cells, enriched in astrocytes, neurons, oligodendrocytes and endothelial cells).
We extracted RNA from samples collected into HBSS-containing tubes on ice using TRIzol® Reagent (Invitrogen Cat# 15596026) and stored at - 80 °C until ready for use. RNA concentration (ng/mcL) and purity (RIN) was determined using Agilent Tape Station (Agilent 2200). We generated a custom panel for assessing gene expression (nCounter ® Gene Expression Assays, Nanostring Technologies, WA). We selected 44 genes related to inflammation, cell type and transcription factor expression and 4 housekeeping genes (see Table 1 for Accession Numbers). The NanoString nCounter ® gene expression assay uses amplification-free digital color-coded barcode technology that directly measures levels of individual target RNA molecules with high precision and sensitivity designed to measure up to 800 targets from a single sample. We used 100 nanograms of total RNA per sample in all analyses according to the manufacturer’s suggested protocol.
Table 1: Accession Numbers and Gene Names.
Each row shows one of the 48 genes assayed using the NanoString nCounter ® gene expression assay, classified in one of four groups as defined in the text and under the column heading “Gene Group”: cell type marker, housekeeping, inflammation-related, and transcription factor genes. For each gene, the name used in the text is defined under Gene, followed by the full Name and Alternative name(s) if applicable. The corresponding Accession Number gives the NCBI Reference Sequence, and the Probe name identifies the probe as made by NanoString. Each probe was made to encompass the most common mRNA variants for each gene.
| Gene Group | Gene | Name and Alternate Name(s) | Accession Number | Probe Name |
|---|---|---|---|---|
| Cell Type | Aif1 | Allograft inflammatory factor 1 also known as Iba1 (Ionized calcium- binding adapter Protein1 ) | NM_017196.2 | NM_017196.2:290 |
| Inflammation | Arg1 | Arginase 1 | NM_017134.2 | NM_017134.2:995 |
| Inflammation | Arg2 | Arginase 2 | NM_019168.1 | NM_019168.1:1095 |
| Inflammation | Ccl2 | C-C motif Chemokine Ligand2 or Monocyte Chemoattractant Protein 1 (MCP-1) | NM_031530.1 | NM_031530.1:190 |
| Inflammation | Ccl22 | C-C motif Chemokine Ligand 22 | NM_057203.1 | NM_057203.1:140 |
| Inflammation | Ccr2 | C-C motif Chemokine Receptor 2 | NM_021866.1 | NM_021866.1:55 |
| Inflammation | Ccr7 | C-C motif Chemokine receptor 7 | NM_199489.4 | NM_199489.4:390 |
| Inflammation | Cd68 | Cluster of Differentiation 68 or Macrosialin | NM_001031638.1 | NM_001031638.1:615 |
| Inflammation | Cd80 | Cluster of Differentiation 80 | NM_012926.1 | NM_012926.1:486 |
| Inflammation | Cd86 | Cluster of Differentiation 86 | NM_020081.1 | NM_020081.1:140 |
| Cell Type | Cx3cr1 | C-x3-c motif Chemokine Receptor 1 | NM_133534.1 | NM_133534.1:370 |
| Inflammation | Cxcl1 | C-x3-c motif Chemokine Ligand 1 or Fractalkine | NM_030845.1 | NM_030845.1:605 |
| Inflammation | Cybb | cytochrome b-245 beta chain or Nox2/Gp91-phox (NADPH Oxidase catalytic subunit) | NM_023965.1 | NM_023965.1:772 |
| Inflammation | Fabp4 | Fatty Acid Binding Protein 4 | NM_053365.1 | NM_053365.1:24 |
| Inflammation | Fcgr2b | Fc Fragment Of IgG Receptor IIb or CD32 | NM_175756.1 | NM_175756.1:1000 |
| Inflammation | Fcgr3a | Fc Fragment Of IgG Receptor IIIa or CD16 | NM_207603.1 | NM_207603.1:660 |
| Inflammation | Hmox1 | Heme Oxygenase 1 | NM_012580.2 | NM_012580.2:897 |
| Inflammation | Ifng | Interferon gamma | NM_138880.2 | NM_138880.2:186 |
| Inflammation | Il10 | Interleukin 10 | NM_012854.2 | NM_012854.2:185 |
| Inflammation | Il13 | Interleukin 13 | NM_053828.1 | NM_053828.1:200 |
| Inflammation | Il18 | Interleukin 18 | NM_019165.1 | NM_019165.1:110 |
| Inflammation | Il1b | Interleukin 1-Beta | NM_031512.1 | NM_031512.1:440 |
| Inflammation | Il4 | Interleukin 4 | NM_201270.1 | NM_201270.1:50 |
| Inflammation | Il4r | Interleukin 4 Receptor | NM_133380.2 | NM_133380.2:875 |
| Inflammation | Il6 | Interleukin 6 | NM_012589.1 | NM_012589.1:55 |
| Transcription Factor | Irf5 | interferon regulatory factor 5 | NM_001106586.1 | NM_001106586.1:1970 |
| Transcription Factor | Irf8 | interferon regulatory factor 8 | NM_001008722.1 | NM_001008722.1:552 |
| Inflammation | Marco | Macrophage receptor with collagenous structure | NM_001109011.1 | NM_001109011.1:1594 |
| Inflammation | Mmp9 | Matrix Metalloproteinase 9 | NM_031055.1 | NM_031055.1:2565 |
| Cell Type | Mpo | Myeloperoxidase | NM_001107036.1 | NM_001107036.1:1276 |
| Inflammation | Mrc1 | mannose receptor C-type 1 or CD206 | NM_001106123.1 | NM_001106123.1:425 |
| Inflammation | Nfe2l2 | nuclear factor, erythroid derived 2, like 2 | NM_031789.1 | NM_031789.1:1315 |
| Inflammation | Nos2 | nitric oxide synthase 2; or nitric oxide synthase, inducible | NM_012611.2 | NM_012611.2:20 |
| Transcription Factor | Pparg_v1 | Peroxisome Proliferator-activated Receptor Gamma transcript variant 1 | NM_013124.3 | NM_013124.3:69 |
| Transcription Factor | Pparg_v2 | Peroxisome Proliferator-activated Receptor Gamma transcript variant 2 | NM_001145366.1 | NM_001145366.1:18 |
| Cell Type | Mbp | Myelin Basic Protein | NM_001025291.1 | NM_001025291.1:121 |
| Inflammation | Ptgs2 | prostaglandin-endoperoxide synthase 2 or COX2 | NM_017232.3 | NM_017232.3:1605 |
| Cell Type | Rbfox3 | RNA binding fox-1 homolog 3 or NeuN | NM_001134498.2 | NM_001134498.2:374 |
| Cell Type | Gfap | Glial Fibrillary Acid Protein | NM_017009.2 | NM_017009.2:728 |
| Inflammation | Retnla | Resistin-like molecule alpha or FIZZ1 | NM_053333.1 | NM_053333.1:135 |
| Inflammation | Socs3 | Suppressor Of Cytokine Signaling 3 | NM_053565.1 | NM_053565.1:597 |
| Inflammation | Tgfb1 | Transforming Growth Factor Beta | NM_021578.2 | NM_021578.2:610 |
| Inflammation | Tnf | Tumor Necrosis Factor Alpha | NM_012675.2 | NM_012675.2:305 |
| Inflammation | Trem2 | Triggering Receptor Expressed on Myeloid Cells 2 | NM_001106884.1 | NM_001106884.1:148 |
| Housekeeping | Actb | Beta Actin | NM_031144.2 | NM_031144.2:19 |
| Housekeeping | Gapdh | Glyceraldehyde 3 Phosphate Dehydrogenase | NM_017008.2 | NM_017008.2:850 |
| Housekeeping | Hprt1 | Hypoxanthine Phosphoribosyltransferase 1 | NM_012583.2 | NM_012583.2:20 |
| Housekeeping | Rpl32 | Ribosomal Protein L32 | NM_013226.2 | NM_013226.2:384 |
The NanoString nCounter ® gene expression assay generates RNA results for twelve samples at a time, with positive and negative controls for each sample across the 48 genes assayed. We used nSOLVER™ Analysis Software (NanoString Technologies, Inc.), an R-language-based program, to generate separate data tables for each set of comparisons as recommended by the manufacturer. We normalized individual gene mRNA results using the positive controls for each sample for all samples included in the analysis of interest. We used Nano String Norm package (version1.1.21, 11/17/2015) and R Statistical Software to optimize normalization further, and found that using one or any number of the four housekeeping genes did not additionally minimize batch effects.
Novel Object Recognition Testing
We used a black Plexiglas® NOR chamber (52X52X30cms) as described for juvenile rats (P29-40) (Reger et al., 2009) housed in a small room with soft lighting. Objects used for testing were small, plastic and easily cleaned objects secured to the box using Velcro ®. Interaction of the rat with an object was measured using automated video tracking and data analysis equipment from EthoVision® vXT 13 (Noldus Information Technology, Wageningen NL) and confirmed by dual observer analysis of the videotaped session when needed. Exploration was timed for the duration that the rat’s nose was within 2 cms of the object, as determined by a digitally drawn arena. We did not consider time spent rearing, if present, as exploration. We tested rat pups (n=5 per group) at PID 14 (age P31). We cleaned the box and objects between rat pups with 70% alcohol to remove odor traces. Rat pups were acclimated to handling prior to commencing NOR procedures. During the Habituation phase, each rat pup individually explored the empty box for five minutes on day one and fifteen minutes on day two to minimize stress and ensure acclimation to the testing environment. During the Familiarization phase, we placed a single rat in the arena containing two identical objects for five minutes. We exposed half of the rats to two identical objects in one configuration (see black spheres in Fig. 1B) and the other half to the mirror image configuration (see black spheres in Fig 1B). Each rat returned to the arena 24 hours later for Novel testing over five minutes. Each rat was exposed to one object from the familiarization trial (black sphere) and to one novel object (red triangle) in the configurations shown in Fig 1B, alternating between A1 and A2 for every rat exposed to Familiarization A1 and between B1 and B2 for every rat exposed to Familiarization B. Performance on the testing phase was measured by the percent novel exploration time (100* (N/ (N+F)) where N is the time spent on the Novel object and F on the familiar. A higher exploration time is considered to reflect greater memory ability, while an exploration time of 50% is not better than chance.
Statistical Analyses
Investigators blinded to experimental groups performed all data acquisition and analyses. All analyses except gene expression were analyzed using two-way analysis of variance (ANOVA) followed by the Holm-Sidak test for multiple comparisons using GraphPad® Prism 6.0 (GraphPad® Software, CA). For immunohistochemical analyses of gliosis and macrophage markers, we used a priori planned comparisons to determine the effect of DHA treatment (SHAMREG vs SHAMDHA), the effect of injury (SHAMREG vs REGCCI) and the effect of DHA treatment on injury (REGCCI vs. DHACCI). Results were considered statistically significant at p<0.05 and presented as mean ± standard error. We analyzed gene expression results using R Statistical Software (2016). (Team, 2016) We used Kruskal-Wallis tests to compare groups at each of the three time points. For NEG cells, we compared the four groups. For CD11b + cells, we compared only two groups (REGCCI and DHACCI) because cell numbers and RNA yield from HI cells in both SHAM groups was very low. For four groups we used a cutoff of p<0.001 for significance and for two group comparisons we used p<0.025. A p-value cutoff of <0.001 was chosen to protect against false positives (type I error) when performing multiple comparisons between four groups. Such a cutoff corresponds to a limit on False Discovery Rate (FDR) across these analyses of approximately 0.5%, meaning the significant findings very likely represent real differences. We used a less stringent p-value of 0.025 for the comparison of CCI groups at genes that successfully passed the first, more stringent threshold. We assessed effects of time after injury using repeated measures ANOVA with Tukey contrasts for multiple comparison of means.
Results
DHA Decreased Nitrate/Nitrite (NOx) Concentration in the Injured Brain at PID1
In adult patients after TBI, elevated NOx in cerebrospinal fluid and microdialysate samples correlates with death and poor outcome. (Clark et al., 1996; Tisdall et al., 2013) Therefore, this indicator of reactive oxygen species (ROS) is potentially a sensitive measure of TBI-induced secondary injury. We examined NOx in response to DHA treatment after CCI. Hippocampal/cortical NOx levels decreased 5-fold in DHA treated CCI rats relative to non-treated CCI (REGCCI) rats at PID1 (0.05 ± 0.01 vs 0.25 ± 0.09 mean picomoles NOx/micrograms protein, p=0.006) as shown in Figure 2. DHACCI NOx did not differ from SHAM controls indicating that DHA treatment decreased an acute indicator of TBI morbidity.
Figure 2: DHA Decreased Nitrate/Nitrite (NOx) Concentration in the Injured Brain at PID1.
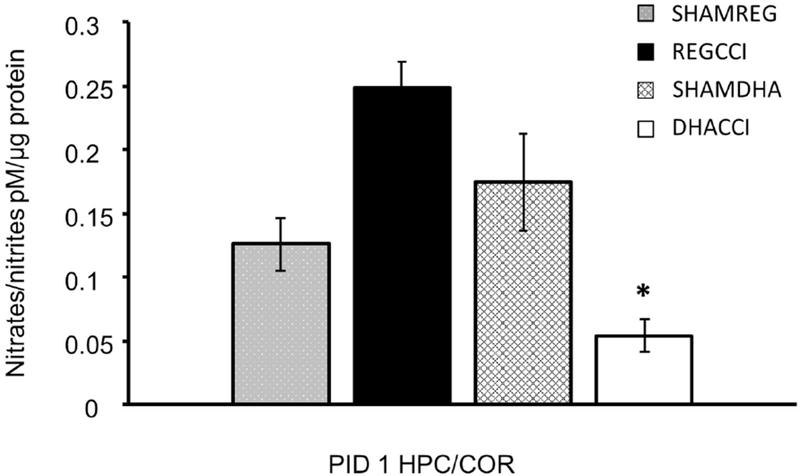
DHA blunted the CCI-induced increase in ipsilateral brain (hippocampus and cortex) NOx (nitrites/nitrates) at PID1. Cortical NOx levels increased in REGCCI rats, shown in black, relative to DHACCI , shown in white (0.25 ± 0.09 vs. 0.05 ± 0.01 mean picomoles NOx/micrograms protein, p=0.006) at PID1. DHACCI NOx did not differ from SHAM controls.
Histology
We next sought to determine if DHA led to long-term changes in inflammation and tissue preservation. To control for inter-animal variability, we initially attempted to normalize injured regions of the brain (cortex and hippocampus ipsilateral to CCI) to corresponding regions of the uninjured, contralateral hemisphere. These within-subject normalizations introduced a high degree of variability within groups likely due to the non-uniform and diffuse effects of TBI and the relatively low expression of most markers in sham controls; even slight differences in marker expression in the contralateral hemisphere introduced large variability when we expressed outcomes as ratio of hemispheres within animals. There was no significant effect of DHA treatment or TBI in the contralateral hemisphere (data not shown). Therefore, all results reported are for the side ipsilateral to injury for CCI or the left side for SHAMS. There were no significant effects of DHA in the absence of CCI (SHAMREG vs. SHAMDHA, p>0.05 for all markers; Figures 3–7) and therefore comparisons with the SHAMDHA group were not performed in the context of CCI.
Figure 3: DHA decreased CCI-induced Microgliosis.
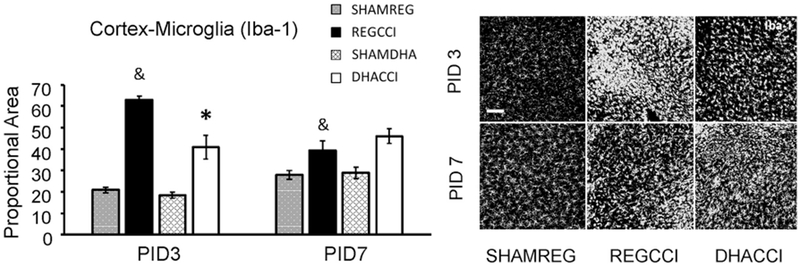
Results for cortical Iba-1 reactivity proportional area at PID 3 and PID 7 are shown graphically on the left. REGCCI and DHACCI are shown in solid black and white bars, respectively, while corresponding SHAM groups are shown as spotted gray and latticed white for SHAMREG and SHAMDHA rats. CCI significantly increased Iba-1 activation in the cortex at both PID 3 and 7 relative to SHAM controls (&p<0.0001 main effect of injury, adjusted p value p<0.0001 at PID3 and p<0.01 at PID7. DHA treatment significantly reduced microglia/macrophage activation (Iba-1 reactivity) to CCI in PID 3 cortex (REGCCI vs. DHACCI, *p=0.006). On the right, representative images of cortical Iba-1 reactivity are shown for SHAMREG, REGCCI and DHACCI. The scale bar is 100 microns.
Figure 7: DHA decreased CCI-induced CD68 activation in cortex at PID3.
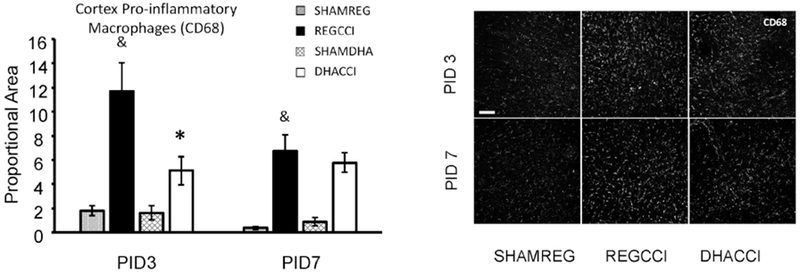
Results for cortical CD68 reactivity proportional area at PID 3 and PID 7 are shown graphically on the left. REGCCI and DHACCI are shown in solid black and white bars, respectively, while corresponding SHAM groups are shown as spotted gray and latticed white for SHAMREG and SHAMDHA rats. CCI increased CD68 immunoreactivity compared to SHAM controls (&p<0.0001, main effect of injury) with significant increases at both PID 3 (p=0.0001) and PID 7 (p=0.005). DHA significantly decreased CD68+ microglia/macrophage activation in the cortex (*p=0.03, main effect of treatment) with significant differences between REGCCI and DHACCI at PID3 (*p=0.02). In the hippocampus, CCI did not significantly increase CD68 (p=0.08), and there was no effect of DHA on CD68 expression after CCI (p=0.21, main effect of treatment). On the right, representative images of cortical CD68 reactivity are shown for SHAMREG, REGCCI and DHACCI. The scale bar is 100 microns.
DHA decreased CCI-induced Microgliosis
To determine the effect of injury and treatment on glial activation, we examined Iba-1 and GFAP (glial fibrillary acidic protein), general markers of microglia/macrophages and astrocyte activation, respectively. Interestingly, there were significant main effects of time (p<0.05) with increased reactivity at PID 7 vs. PID 3 for both markers in hippocampal and cortical regions in both SHAMDHA and SHAMREG groups. This is potentially reflective of developmental changes in microglia/macrophage and astrocyte densities (Figures 3–6).
Figure 6: CCI decreased hippocampal GFAP reactivity at PID 7.
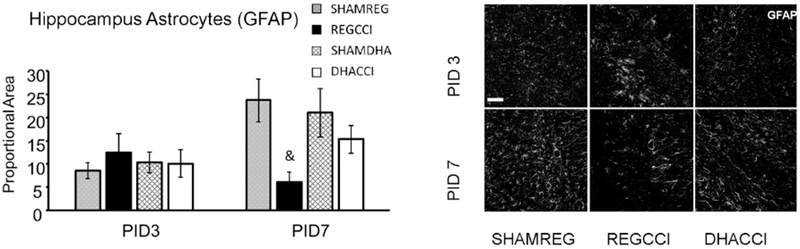
Results for hippocampal GFAP reactivity proportional area at PID 3 and PID 7 are shown graphically on the left. REGCCI and DHACCI are shown in solid black and white bars, respectively, while corresponding SHAM groups are shown as spotted gray and latticed white for SHAMREG and SHAMDHA rats. CCI significantly decreased GFAP reactivity in the hippocampus at PID 7 relative to SHAM controls (&p=0.03 main effect of CCI, adjusted p value 0.001). There was a trend toward a treatment by time interaction in the hippocampus (p=0.06) with GFAP immunoreactivity in DHACCI hippocampus returning toward SHAM levels by PID 7. On the right, representative images of hippocampal GFAP reactivity are shown for SHAMREG, REGCCI and DHACCI. The scale bar is 100 microns. DHACCI GFAP did not differ from sham at either time point.
CCI significantly increased the density of both Iba-1 and GFAP with obvious and sustained microglia/macrophage and astrocyte activation in the cortex at both PID 3 and 7 relative to SHAM controls. Specifically, CCI increased cortical Iba-1 (p<0.0001 main effect of injury, adjusted p value p<0.0001 at PID3 and p=0.005 at PID7) and cortical GFAP (p=0.001 main effect of injury, adjusted p value p=0.0073 at PID3 and p=0.047 at PID7) relative to SHAM controls (Figs. 3 and 5). In the hippocampus, CCI-induced microglia/macrophage activation was transient showing increased Iba-1 reactivity at PID 3 (p=0.03) that returned to sham levels by PID7 (p=0.40) (Fig. 4). Interestingly, CCI did not increase GFAP labeling in the hippocampus at PID3 but instead decreased it at PID 7 (p=0.03 main effect of CCI, adjusted p=0.001) (Fig. 6).
Figure 5: DHA did not affect CCI increased Astrogliosis.
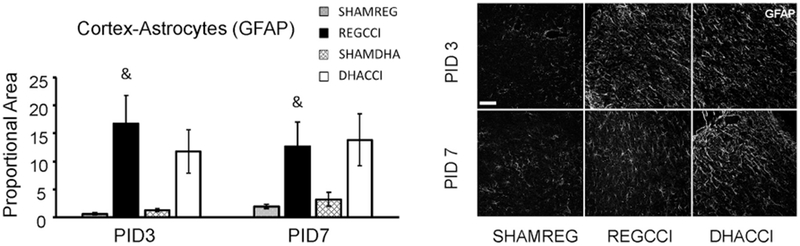
Results for cortical GFAP reactivity proportional area at PID 3 and PID 7 are shown graphically on the left. REGCCI and DHACCI are shown in solid black and white bars, respectively, while corresponding SHAM groups are shown as spotted gray and latticed white for SHAMREG and SHAMDHA rats. CCI significantly increased GFAP reactivity in the cortex at both PID 3 and 7 relative to SHAM controls (&p=0.001 main effect of injury, adjusted p value p<0.01 at PID3 and p<0.05 at PID7). DHA treatment did not affect increased GFAP reactivity at either time point (p>0.25 main effect of treatment. On the right, representative images of cortical GFAP reactivity are shown for SHAMREG, REGCCI and DHACCI. The scale bar is 100 microns.
Figure 4: CCI did not increase hippocampal microgliosis at PID 7. DHA did not decrease CCI-induced Microgliosis in the Hippocampus at PID3.
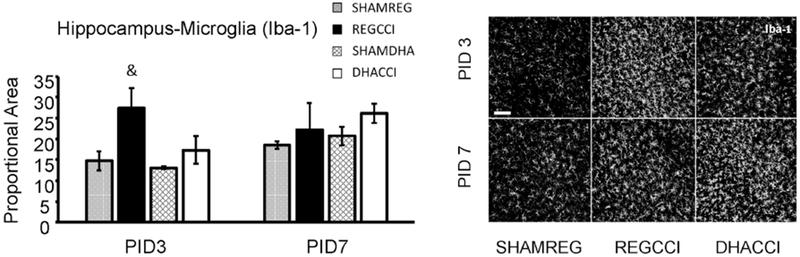
Results for hippocampal Iba-1 reactivity proportional area at PID 3 and PID 7 are shown graphically on the left. REGCCI and DHACCI are shown in solid black and white bars, respectively, while corresponding SHAM groups are shown as spotted gray and latticed white for SHAMREG and SHAMDHA rats. CCI transiently increased Iba-1 activation in the cortex at PID 3 relative to SHAM controls (& p=0.03) that returned to sham levels by PID7 (p=0.40). There was no significant effect of DHA treatment on CCI-induced microglia/macrophage activation in hippocampus. On the right, representative images of hippocampal Iba-1 reactivity are shown for SHAMREG, REGCCI and DHACCI. The scale bar is 100 microns.
DHA treatment significantly reduced microglia/macrophage activation (Iba-1 reactivity) to CCI in PID 3 cortex (REGCCI vs. DHACCI, p=0.006) (Fig. 3). There was no significant effect of DHA treatment on CCI-induced microglia/macrophage activation in hippocampus (Fig. 4).
Interestingly there was not significant effect of DHA on CCI-induced astrocyte activation (GFAP immunoreactivity) in the cortex or the hippocampus (p>0.25 main effect of treatment; Figs. 5–6), however, there was a trend toward a treatment by time interaction in the hippocampus (p=0.06) with GFAP immunoreactivity returning toward sham levels by PID 7 (Fig. 6). Collectively, CCI induced microglia/macrophage and astrocyte activation in the cortex. DHA treatment decreased cortical microglia activation at PID3, relative to REG diet.
DHA decreased CCI-induced Pro-Inflammatory Microglia/macrophage Activation
Since DHA decreased microglia/macrophage activation, we next sought to determine whether DHA was selectively affecting purportedly reparative, CD206- or Arginase-positive cells or pro-inflammatory, CD68- or Major Histocompatibility Complex II (MHC II)-positive, cells. Arginase labeling was sparse and did not appear cellular in any groups. Therefore, we used CD206 to evaluate reparative microglia/macrophage microglia/macrophage activation. CD206 (Cluster of Differentiation 206), also known as Mrc-1 (macrophage mannose receptor 1), is important for phagocytosis and for clearing inflammatory proteins in the inflammation-resolving phase. CD206 immunoreactivity increased over time in both the cortex (p=0.03, main effect of time) and hippocampus (p=0.006) in both SHAM and CCI groups; however, there was no effect of injury on CD206 expression (p>0.6) for either brain region (results not shown) at PID3 nor PID7. Although the number of CD206 positive cells increased over time in all the conditions, very few cells were positive for CD206 even at PID 7. There was no effect of DHA treatment on CD206 reactivity in the cortex or hippocampus (p>0.5) after CCI.
Next we examined two markers associated with pro-inflammatory and primed microglia activation: CD68 and MHC II. High CD68 microglial reactivity is associated with persistent inflammation and ongoing white matter degeneration for many years after TBI in humans (Johnson et al., 2013) and with a pro-inflammatory milieu after CCI in rats. (Harting et al., 2008) Decreased CD68 after injury is associated with neuroprotection afforded by candidate therapeutics in rodents. (Hellewell et al., 2013; Loane et al., 2014). MHC II is expressed by pro-inflammatory macrophages in vitro and MHCII+ microglia after TBI produce pro-inflammatory cytokines and contribute to long-term inflammatory hyperactivity. (Fenn et al., 2014; Gensel et al., 2017) CCI increased CD68 immunoreactivity in the cortex compared to SHAM controls (p<0.0001, main effect of injury) with significant increases at both PID 3 (p=0.0001) and PID 7 (p=0.005) (Fig. 7). DHA decreased CD68+ microglia/macrophage activation in the cortex (p=0.03, main effect of treatment) with significant differences between REGCCI and DHACCI at PID3 (p=0.02) (Fig. 7). In the hippocampus, CCI did not significantly increase CD68 (p=0.08), and there was no effect of DHA on CD68 expression after CCI (p=0.21, main effect of treatment). CCI increased cortical MHCII compared to SHAM controls (p=0.0004, main effect of injury) at both time points (p=0.02 at PID3, p=0.005 at PID7), however, DHA did not affect cortical MHCII expression after CCI (p=0.2 main effect of treatment) (results not shown). CCI did not increase hippocampal MHCII expression at either time point.
In a separate experiment, we examined the long-term (PID 30) histological effects of CCI and DHA. As with earlier time points, there was a significant main effect of CCI in microglia/macrophage activation (Iba-1) in the cortex (p=0.049) and the hippocampus (p=0.020) at PID 30. There were no significant treatment effects with DHA in the cortex (p=0.14, main effect of treatment) or the hippocampus (p=0.10, main effect of treatment). Interestingly, there was a trend for a treatment x injury interaction in both the cortex (p=0.08) and hippocampus (p=0.10) driven by increased immunoreactivity in the CCI-DHA vs. CCI-REG groups in both the cortex (p=0.07) and hippocampus (p=0.06) shown in Supplemental Figure 2.
For GFAP, there was no significant main effect of injury in cortex (p=0.52) or hippocampus (p=0.19). There was also no significant main effect of treatment on GFAP immunoreactivity in the cortex (p=0.62) or hippocampus (p=0.16) (results not shown). For CD68, there was also no significant main effect of injury in the cortex (p=0.19) or hippocampus (p=0.27) nor a significant main effect of treatment in either brain region (cortex, p=0.76; hippocampus, p=0.90)(results not shown). At PID 30, we did not quantify cellular labeling of MHCII nor CD206 because reactivity was very low in all groups at this chronic time point.
Previously we observed the DHA administration prior to CCI improved long-term (PID60) tissue sparing in rat pups (Schober et al., 2016). While CCI decreased cortical area ipsilateral to injury to 73% of SHAM volume at PID 30 (p=0.01), there was no effect of DHA treatment on lesion area (p=0.7) in the current study (data not shown).
Collectively, our histological analyses indicate that CCI leads to acute and long-term inflammation in the cortex and hippocampus of rat pups and that post-TBI DHA administration decreases acute indices of inflammation, specifically of CD68+ macrophages/microglia.
Flow Cytometry and Cell Population mRNA Levels
Our histological evaluation suggests that DHA may decrease pro-inflammatory microglia/macrophage activation. However, the relative activation state of microglia/macrophage after TBI is difficult to determine using a limited number of phenotypic markers. (Morganti et al., 2016) The number of antibodies for phenotypic evaluation and identification of microglia and macrophages are further limited in rats. Therefore, we sought to further determine the immunomodulatory potential of DHA treatment and the extent to which microglia (LO, or CD11b+ CD45low) vs. macrophages (HI, or CD11b+ CD45high) are affected by treatment using a combination of flow cytometry and gene-array analyses. We additionally used flow cytometry-derived cell counts to estimate changes in the size of each cell population after CCI relative to SHAM controls.
Cell Counts
As noted in the methods, we collected 300-350 mg of saline-perfused brain ipsilateral to injury in all groups at the three time points to make single-cell suspensions for each sample. We used flow cytometry to sort live single cells into populations of CD11b+ CD45 high (HI, macrophage enriched), CD11b +CD45low (LO, microglia enriched) and CD11b (−) (NEG, composed largely of oligodendrocytes, astrocytes, neurons and endothelial cells).
CCI increased the total cell yield, estimated by adding counts from the three populations, at PID 3 (REGCCI = 234 ± 36 % SHAM and DHACCI = 250 ± 31% SHAM, p<0.001) with a trend in the same direction for REGCCI, but not DHACCI, at later time points (PID7 REGCCI = 172 ± 52% SHAM, p=0.07 and PID30 REGCCI =246 ± 88%, p=0.09, respectively). Increased infiltrating macrophages, or HI cells, drove this increased total cell yield in both CCI groups at PID3 and in REGCCI rats at PID 7 and 30. At PID3, CCI increased HI cells without regard to treatment (REGCCI= 777 ± 176 % SHAM, DHACCI=964 ± 110 % SHAM, p<0.0001 for both CCI groups) and in REGCCI only at PID 7 and 30 (575 ± 318 % SHAM, p=0.04 and 362 ± 194% SHAM, p=0.03, respectively). Supplementary Figure 1 shows representative flow cytometry plots of the percentage of CD11b +CD45low and CD11b+ CD45 high cells in rat brain after CCI or SHAM injury. REGCCI rats had higher HI (infiltrating macrophages) at PID3, 7 and 30 relative to SHAM. In summary, CCI increased infiltrating macrophages relative to SHAM at PID 3, 7 and 30. DHA treatment abrogated this increase at PID 7 and 30, but not at PID 3.
mRNA Results
As shown in Table 1, we included six genes with high specificity for cell type: Cx3cr1, for microglia; Marco, for macrophages; MPO for neutrophils; MBP for oligodendrocytes, Rbfox3 for neurons and GFAP for astrocytes. As noted in the methods, NEG cells are enriched in oligodendrocytes, neurons, and astrocytes. Cx3cr1 should be high in LO cells, while MPO and Marco should be high in HI cells. These predictions were met across our samples, with one notable exception. REGCCI samples at PID3 had unexpectedly high levels of Cxrcr1 in NEG cells, even when studies were repeated on different days and samples compared between different litters. We did not find differences in CD11b mRNA expression between REGCCI and DHACCI rats using real time RT-PCR in the injured brain tissue. Based on the 91% sensitivity of the CD11b antibody (as noted in the methods) we posited that the REGCCI NEG cells included a fraction of actual CD11b+ cells in a higher proportion than did the other three groups. We therefore excluded NEG, HI and LO samples from those rat brains from which the NEG cells had Cx3cr1 mRNA counts that were statistically extreme outliers. An alternative explanation is that CCI induces co-expression of microglial markers such as CX3cr1 in astrocytes, a cell type that is CD11b negative. We are actively pursuing studies to test this possibility based on a recent study showing that injury leads to the appearance of cells that express both astrocyte and microglial markers in humans and mice, and on reports of a similar phenomenon described in a mouse model of amyotrophic lateral sclerosis. (Trias et al., 2013; Wilhelmsson et al., 2017)
DHA Decreased Transcription Factors Associated with Pro-inflammatory Activation in PID3 CD11b (−) cells and PID7 Microglia, but Increased them in PID30 Microglia. DHA did not Affect Macrophage Transcription Factor Expression
As noted in Table 1, 4 of the 48 genes are transcription factors. Figures 8, 9 and 10 show results of transcription factor mRNA analyses for PID3, 7 and 30, respectively. Neither diet nor injury affected PPARy mRNA variant 1 nor variant 2 levels at anytime point. In LO cells (microglia-enriched population) at PID7, DHA decreased irf5 and irf8 mRNA relative to REGCCI microglia (p=0.0015 for irf5 and p= p=0.0015 for irf8) as shown in Figure 9B. In HI cells (macrophage-enriched population) neither Irf5 nor irf8 mRNA differed between CCI groups at any time point (Figure 10 A–C). Injury increased Irf5 and Irf8 in NEG cells at PID3 (Figure 8A, p<0.0001) and increased Irf5 more in REGCCI than in DHACCI rats (p=0.01) albeit at levels far lower than in HI and LO cells. Analyses of these transcription factors suggest that DHA decreases interferon-related transcription factors in microglia, particularly at PID 7, but not in macrophages.
Fig 8: Transcription Factor mRNA levels in NEG cells over time.
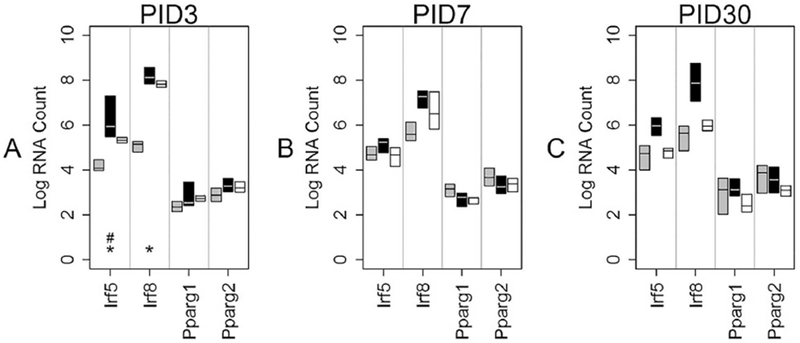
This graph shows median mRNA values, with the upper and lower quartiles for each gene of interest. REGCCI is shown in black, DHACCI in white and combined SHAM groups in gray; SHAMDHA and SHAMREG were combined because we found no differences between these groups. We performed statistical analyses using RNA values, but used log conversion for graphical representation to allow fitting a wide range of RNA values on a single y-axis. Asterisks (*) denote within group differences, driven by injury, while # denotes differences between the two CCI groups, driven by diet. A At PID3, injury increased Irf5 and Irf8 in NEG cells (* p<0.0001). REGCCI Irf5 increased in REGCCI relative to DHACCI (#p=0.01). B and C. Transcription factor mRNA levels did not differ between groups at PID7 nor at PID30.
Fig 9: Transcription Factor mRNA levels in LO cells over time.
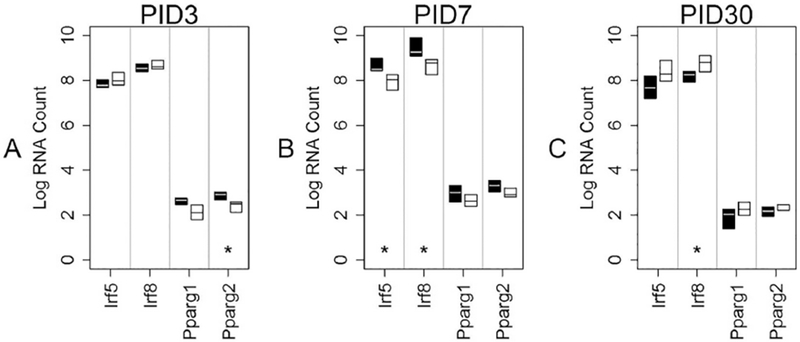
This graph shows median mRNA values, with the upper and lower quartiles for each gene of interest. REGCCI is shown in black and DHACCI in white. We performed statistical analyses using RNA values, but used log conversion for graphical representation to allow fitting a wide range of RNA values on a single y-axis. Asterisks (*) denote differences between the two CCI groups, driven by diet. A. At PID3, REGCCI pparg2 increased relative to DHACCI, p=0.01, but the very low mRNA counts in both groups make the results of unlikely significance. B. In LO cells (microglia-enriched population) at PID7, DHA decreased irf5 and irf8 mRNA relative to REGCCI microglia (p=0.0015 for irf5 and p= p=0.0015 for irf8) C. DHACCI irf 8 increased relative to REGCCI, p=0.01.
Fig 10: Transcription Factor mRNA levels in HI cells overtime.
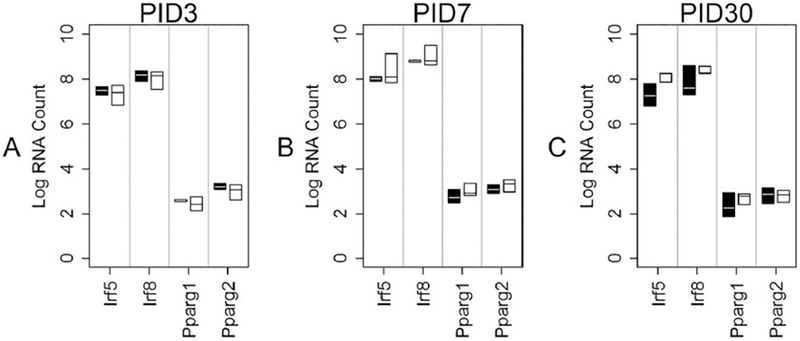
This graph shows median mRNA values, with the upper and lower quartiles for each gene of interest. REGCCI is shown in black and DHACCI in white. We performed statistical analyses using RNA values, but used log conversion for graphical representation to allow fitting a wide range of RNA values on a single y-axis. Asterisks (*) denote differences between the two CCI groups, driven by diet. A-C. Transcription factor mRNA levels did not vary between groups at any time point.
As noted in Table 1, the remaining genes are involved in inflammation. Figures 11–13 depict the mRNA results for each of these, classifying genes typically associated with inflammation as “pro-inflammatory” and those associated with repair or resolution of inflammation as “anti-inflammatory”.
Fig 11: Inflammation-related mRNA levels in NEG cells over time.
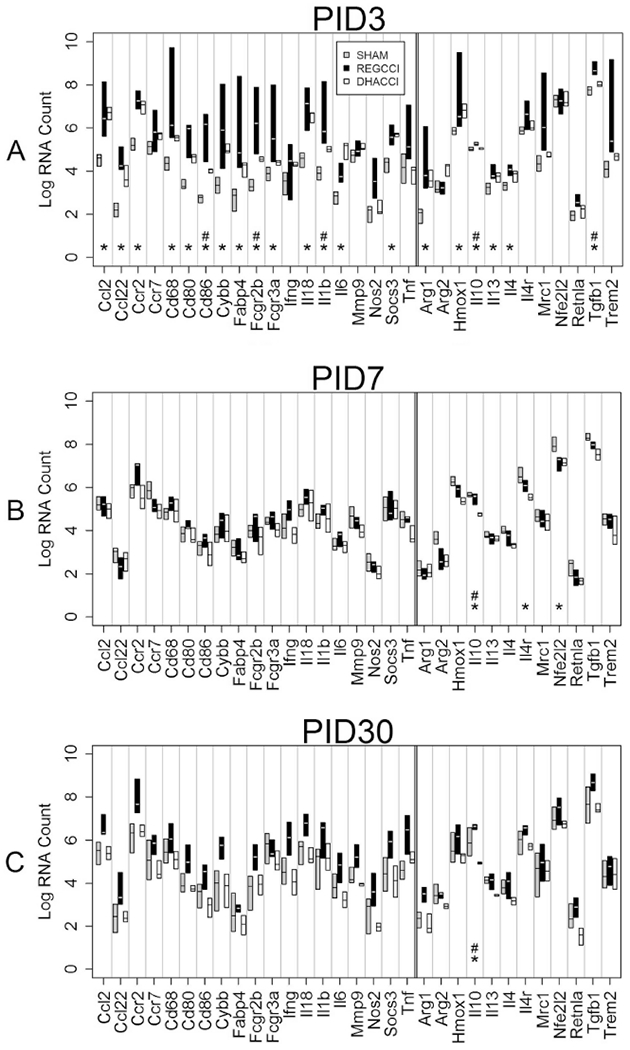
Log values of median mRNA values for each gene of interest in each of three groups (REGCCI in black, DHACCI in white and combined SHAM groups in gray). We combined SHAMDHA and SHAMREG because we found no differences between these groups. Asterisks (*) denote genes that differed significantly between CCI and SHAM, while the pound sign marks those genes that differed also between CCI groups. A. In NEG cells at PID3, pro-inflammatory Ccl2, Ccr2, CD68, CD80, CD86, Cybb, Fabp4, CD16, CD32, IL18, IL6 and IL-1 β as well as the anti-inflammatory Arg1, Hmox1, IL10, IL13, IL4, Socs3 and TGFβ genes (* p<0.0001 for all) increased in both CCI groups relative to SHAM. DHA diet blunted increased mRNA levels of several inflammatory genes after injury, namely CD86p=0.009, CD32(Fcgr2b) p=0.02, IL-1B p=0.005 and TNF α p=0.002 as well as IL10 p=0.001 and TGF β p=0.002 (# p values <0.025 relative to REGCCI) and in CD68 the decrease neared significance (p=0.027).
B. In NEG Cells at PID7, anti-inflammatory IL4r and Nfe2l2 decreased in both CCI groups (* p= 0.0008 and 0.0009) relative to SHAM. Anti-inflammatory IL-10 mRNA decreased in DHACCI but not in REGCCI rats, relative to SHAM groups (*p=0.0005) C. In NEG cells At PID 30, anti-inflammatory IL-10 mRNA decreased in DHACCI relative to SHAM and to REGCCI (*p=0.0006, #p=0.0008).
Fig 13: Inflammation-related mRNA levels in HI cells overtime.
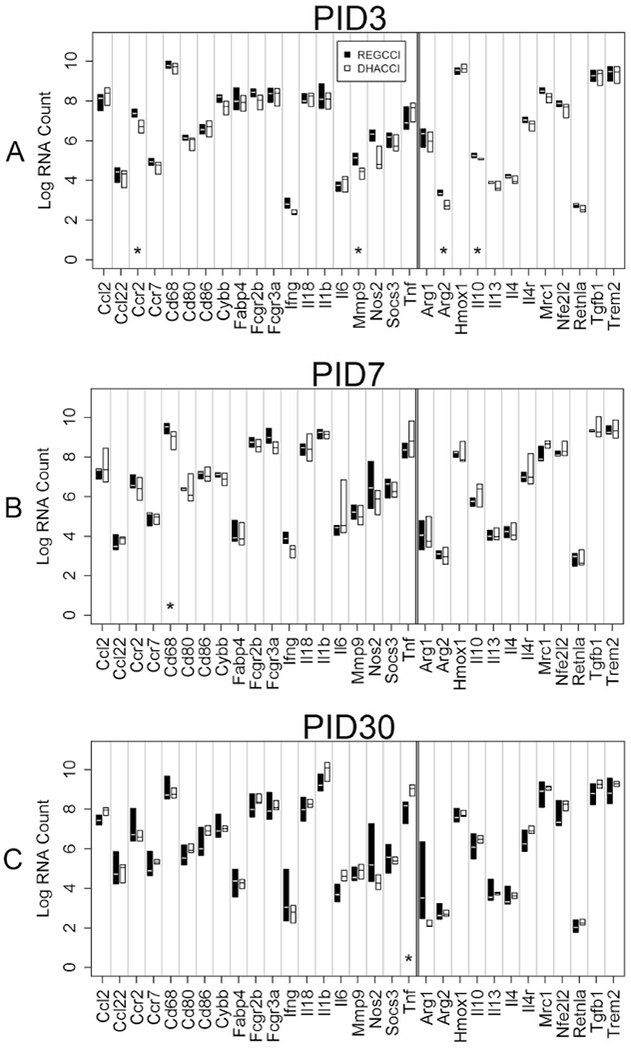
This graph shows median mRNA values, with the upper and lower quartiles for each gene of interest. REGCCI is shown in black and DHACCI in white. We performed statistical analyses using RNA values, but used log conversion for graphical representation to allow fitting a wide range of RNA values on a single y-axis. Asterisks (*) denotes differences between the two CCI groups. A. In HI cells at PID3, REGCCI had higher mRNA levels of pro-inflammatory CCr2 (p=0.02) MMP9 (p=0.02) and higher anti-inflammatory IL10 (p=0.02). B. At PID7, REGCCI had higher mRNA levels of pro-inflammatory CD68 (p=0.01). At PID30, DHACCI had higher TNFα mRNA levels (p=0.02) than REGCCI.
In CD11b (−) cells, DHA Decreased Pro-inflammatory TNFα, IL-1β, CD86 and CD32 and Anti-inflammatory TGF β at PID3. DHA decreased Anti-inflammatory IL-10mRNA at PID3 and 30, but increased it at PID7. Inflammatory Gene mRNA differed from SHAM over Time in REGCCI, but not in DHACCI Rats
In NEG cells at PID3, pro-inflammatory Ccl2, Ccr2, CD68, CD80, CD86, Cybb, Fabp4, CD16, CD32, IL18, IL6 and IL-1 β as well as the anti-inflammatory Arg1, Hmox1, IL10, IL13, IL4, Socs3 and TGFβ genes increased in both CCI groups relative to SHAM (*p<0.0001 for all) as shown in Figure 11A. Several of those genes differed between CCI groups as well. DHA treatment blunted increments in CD86 p=0.009, CD32 (Fcgr2b) p=0.02, IL-1B p=0.005 and TNF α p=0.002 as well as IL10 p=0.001 and TGF β p=0.002 (# p values <0.025 relative to REGCCI) and in CD68 the decrease neared significance (p=0.027). In NEG Cells at PID7, anti-inflammatory IL4r and Nfe2l2 decreased in both CCI groups (* p= 0.0008 and 0.0009) relative to SHAM. Anti-inflammatory IL-10 mRNA decreased in DHACCI but not in REGCCI rats, relative to SHAM groups (*p=0.0005) as shown in Figure 11B. In NEG cells At PID 30, anti-inflammatory IL-10 mRNA decreased in DHACCI relative to SHAM and to REGCCI (*p=0.0006, #p=0.0008) as shown in Figure 11C.
In repeated measures analysis for NEG cells over time, mRNA levels increased in REGCCI relative to DHACCI and to the two SHAM groups for the pro-inflammatory genes: CD68, CD86, Cybb, Fabp4, CD32, CD16 (p<0.0001 relative to SHAM and p<0.0001 to DHACCI for all) and CD80 (p<0.01 relative to all three groups). For the anti-inflammatory genes: Arg1, Hmox1, CD206 a similar pattern emerged with REGCCI mRNA greater than the other three groups over time (p<0.001 or smaller for all). DHACCI NEGS mRNA levels did not differ from SHAM over time in any of the inflammation related genes. The transcription factors irf5 and irf8 also increased relative to the other three groups over time in REGCCI, but not in DHACCI, NEGS. These data suggest that DHA decreases the pro-inflammatory environment of the injured brain in non- macrophage/microglia populations, such as astrocytes and endothelial cells.
In Microglia, both Pro- and Anti-Inflammatory Gene mRNA levels Increased at PID 3 and 7 in REGCCI relative to DHACCI, and at PID 30 in DHACCI relative to REGCCI. Pro-inflammatory Gene mRNA Increased over Time in REGCCI, but not in DHACCI Rats
To determine the extent to which DHA treatment altered microglia activation after TBI, we examined gene expression profiles in FACS sorted cells. In LO cells at PID3, pro-inflammatory MMP9 (p=0.006) and anti-inflammatory IL10 mRNA levels (p=0.004) increased in REGCCI, compared to DHACCI as shown in Figure 12A. Consistent with our findings, fish oil gavaged before injury reduced MMP9 mRNA levels in P17 rats in the first four days after CCI. (Russell et al., 2014)
Fig 12: Inflammation-related mRNA levels in LO cells overtime.
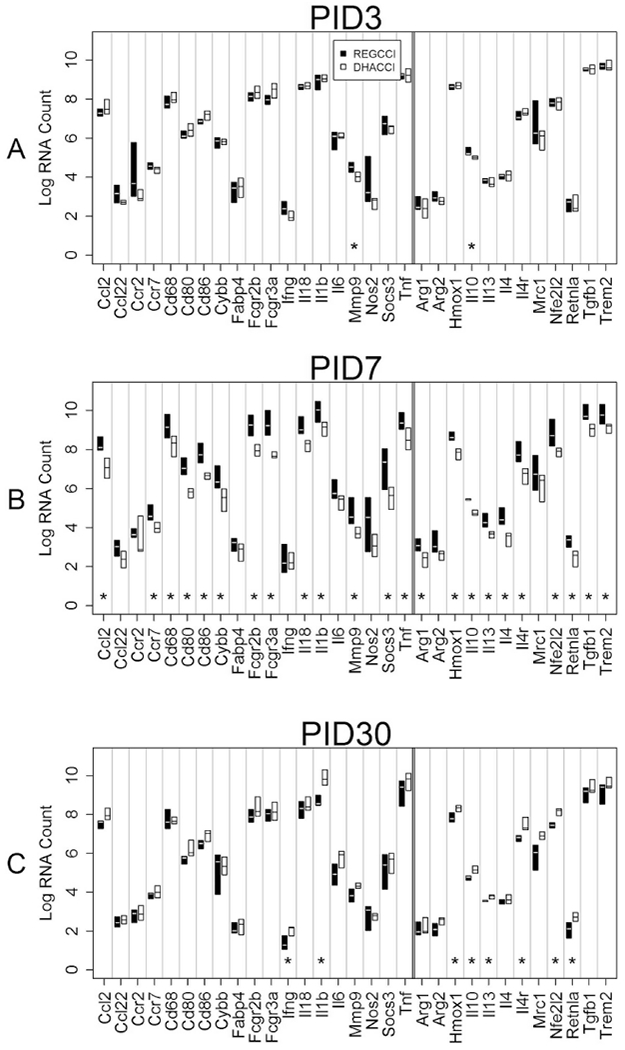
This graph shows median mRNA values, with the upper and lower quartiles for each gene of interest. REGCCI is shown in black and DHACCI in white. We performed statistical analyses using RNA values, but used log conversion for graphical representation to allow fitting a wide range of RNA values on a single y-axis. Asterisks (*) denotes differences between the two CCI groups. A. In LO cells at PID3, pro-inflammatory MMP9 (p=0.006) and anti-inflammatory IL10 mRNA levels (p=0.004) increased in REGCCI, compared to DHACCI. B. At PID7, REGCCI had higher mRNA levels of many pro-inflammatory genes: Ccr7 (p=0.02), Cybb (p=0.02), CcL2 (p=0.003), CD68 (p=0.008), CD80 (p=0.003), CD86 (p=0.001), CD16 (Fcgr2b, p=0.002), CD32 (Fcgr3a, p=0.002), MMP9 (p=0.004), IL-1β(p=0.006), IL-18(p=0.004), TNF α (p=0.005) and Socs3 r(p=0.02) relative to DHACCI. Expression of anti-inflammatory genes also increased in REGCCI: TGF β (0.001), IL10 (p=0.004), IL 13 (p=0.002), IL 4 (p=0.003) IL4r (p=0.001), Nfe2l2 (p=0.005), Retnla (p=0.005) Trem2 (p=0.02) and Hmox1 (p=0.004). C. On PID 30, fewer genes were affected but did so in the opposite direction: DHACCI had higher pro-inflammatory Inf Y (p=0.01) and IL-1 β (p=0.002), and higher anti-inflammatory Hmox1 (p=0.02), Retnla (p=0.02), IL-10 (p=0.005), IL4r (p=0.004), IL13 (p=0.005) and Nfe2l2 (p=0.0009).
In LO cells at PID7, REGCCI had increased mRNA levels of pro-inflammatory Ccr7 (p=0.02), Cybb (p=0.02), CcL2 (p=0.003), CD68 (p=0.008), CD80 (p=0.003), CD86 (p=0.001), CD16 (Fcgr2b, p=0.002), CD32 (Fcgr3a, p=0.002), MMP9 (p=0.004), IL-1β (p=0.006), IL-18(p=0.004), TNF α (p=0.005) and Socs3 r(p=0.02) relative to DHACCI. These results are consistent with our histologic findings that DHA decreased pro-inflammatory, CD68+, microglial activation after CCI. Numerous studies report that TBI is associated with expression of pro-inflammatory and proresolving markers simultaneously. (Turtzo et al., 2014) Accordingly, we found that several genes associated with inflammation resolution were also higher in REGCCI relative to DHACCI at this time point: TGF β (0.001), IL10 (p=0.004), IL 13 (p=0.002), IL 4 (p=0.003) IL4r (p=0.001), Nfe2l2 (p=0.005), Retnla (p=0.005) Trem2 (p=0.02) and Hmox1 (p=0.004) as shown in Figure 12B.
In LO cells at PID 30, DHA increased pro-inflammatory Inf Y (p=0.01) and IL-1 β (p=0.002) and also increased genes associated with inflammation resolution including Hmoxl (p=0.02), Retnla (p=0.02), IL-10 (p=0.005), IL4r (p=0.004), IL13 (p=0.005) and Nfe2l2 (p=0.0009), as shown in Figure 12C. In repeated measures analysis overtime, only two genes differed significantly. Pro-inflammatory CCR2 (p=0.003) and CCL22 mRNA levels (p=0.02) increased in REGCCI relative to DHACCI. Collectively these data indicate that DHA treatment reduced pro-inflammatory microglial activation. This could be due to changes in the pro-inflammatory environment as opposed to directly modulating microglia activation.
In Macrophages, Pro-inflammatory Gene mRNA levels Increased in REGCCI at PID 7 and in DHACCI at PID 30, while both Pro- and Anti-Inflammatory levels Increased in REGCCI at PID 3. Compared to Microglia and CD11b (−) Cells, Relatively Fewer Genes Differed between Groups
We next evaluated gene expression in recruited (CD45 high) macrophages. Differences between CCI groups were few in HI cells (Figure 13 A–C), suggesting that DHA modulated inflammation in our model directly acting on brain resident cells rather than on the circulating immune cells that entered the brain after CCI. In HI cells at PID3, REGCCI had higher mRNA levels of pro-inflammatory CCr2 (p=0.02) MMP9 (p=0.02) and higher anti-inflammatory IL10 (p=0.02). At PID7, REGCCI had higher mRNA levels of pro-inflammatory CD68 (p=0.01) but at PID30, DHACCI had higher TNFα mRNA levels (p=0.02) than REGCCI. In repeated measures analysis, three pro-inflammatory genes differed significantly: Inf Y mRNA levels increased in REGCCI (p=0.04) and CCL2 and TNF a decreased in REGCCI (p=0.03 and 0.01, respectively) relative to DHACCI. Transcription factor irf5 mRNA increased in DHACCI relative to REGCCI (p=0.04) over the three time points.
In summary, cellular mRNA levels show decreasing effects of injury on inflammation-related genes, both pro- and anti-inflammatory, over time after CCI. For macrophages and microglia, our conclusions are limited to comparisons between DHA treated and REG treated rats after CCI. DHA decreased pro-inflammatory genes in microglia, particularly at PID7, but did not affect inflammation in macrophages at PID3, 7 nor 30 after CCI.
Novel Object Recognition Testing
DHA Improved Performance on Novel Object Recognition Testing at PID14 after CCI
We previously showed improved cognitive function when we gave DHA treatment prior to CCI. (Schober et al., 2016) In the present work, we treated rats with DHA after CCI. To assess the effects of DHA on memory function, we utilized the Novel Object Recognition (NOR) task. We chose to measure NOR at PID14, based on previous data in our model showing injury effect at this time. (Schober et al., 2013) Higher novel exploration represents better memory. Scores of 50%, representing 50% of time exploring the novel object, are similarto those achieved by chance alone. Novel exploration differed between groups by injury (p=0.027) and by diet (p=0.0013). DHACCI had greater novel exploration (52.1 ± 4.7 %) than REGCCI (27.6 ± 3%), p=0.0027, as shown in Figure 14. Novel exploration did not differ between SHAMDHA and SHAMREG (63.1 ± 6.2 % and 50.2 ± 1.7%, respectively, p=0.15).
Figure 14: Novel Object (NOR) Exploration Time.
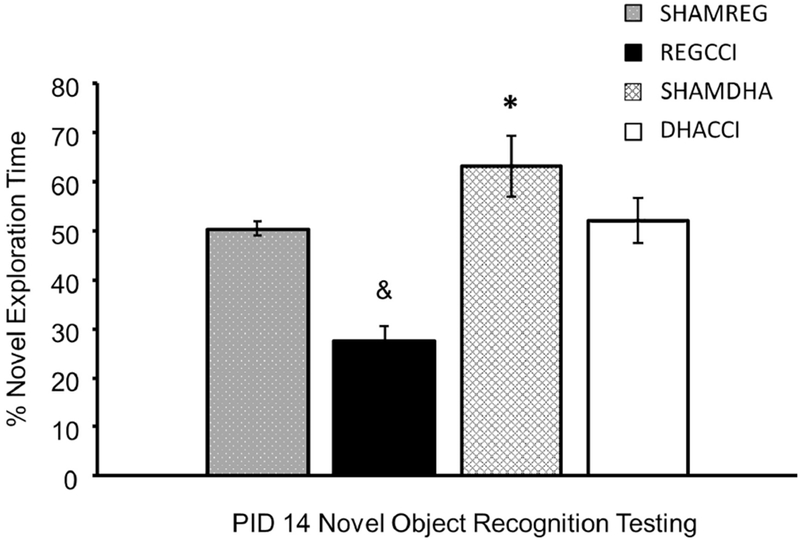
Higher percent novel exploration time represents better memory. Scores of 50%, representing 50% of time exploring the novel object and are no better than those achieved by chance alone. DHACCI rats had higher exploration time (52.1 ± 4.7 %) than REGCCI (27.6 ± 3%), p=0.02 as shown by asterisk. Exploration time did not differ between SHAMDHA and SHAMREG (63.1 ± 6.2 % and 50.2 ± 1.7%, respectively, p=0.15) and REGCCI less than SHAMREG (27.6 ± 3% vs 50.2 ± 1.7%, p=0.009 as shown by the ampersand while DHACCI did not differ from SHAM (p=0.2)
Discussion
Here, we show in a developmental model of TBI that DHA treatment after injury decreased oxidative stress, pro-inflammatory microglia activation mRNA and histologic markers, and improved recognition memory during the first two weeks after TBI. Generally, inflammatory actions of CCI and/or DHA affected pro-inflammatory and anti-inflammatory markers in parallel. Most notably within the microglia-enriched population, DHA showed a bi-phasic effect on CCI-induced changes: DHA blunted pro-inflammatory mRNA at PID 3 and 7 and anti-inflammatory genes at PID 30 after CCI.
We also show that macrophage infiltration, identified using CD11b+ CD45 high cells, persisted in rat pup brain for as long as 30 days. This persistence stands in contrast to the adult brain where CD11b+ CD45hi macrophage infiltration drops off markedly after the first week and resolves by 30 days after CCI. (Jin et al., 2012) We found that post-injury DHA treatment decreased macrophage infiltration at day 7 and 30, but not at day 3, compared to REG treated CCI rats and that DHA did not affect macrophage inflammation-related mRNA levels after CCI. In contrast, microglia (CD11b+ CD45Io) were more sensitive to DHA treatment with significant decreases in gene expression associated with pro-inflammatory and reparative microglia functions.
The observed decrease in brain NOx levels in DHA treated rats suggests that brain and/or cerebrospinal fluid NOx could be a potential biomarker of inflammation and DHA effect in future translational studies. These novel observations expand on our previous findings in which we used a pre-injury dosing paradigm and pave the way for future pre-clinical work.
We speculate that DHA improved cognitive function at PID14 by decreasing secondary injury from oxidative stress and microglial pro-inflammatory activation during the first week after CCI. DHA is a known antioxidant, shown to decrease production and/or scavenging of free radicals. In vitro, DHA decreased macrophage and microglia free radical production in response to an inflammatory stimulus. (Kielar et al., 2000) (Hjorth et al., 2013; Yang et al., 2018) In fetal rat brain, DHA decreased oxidative injury from ethanol exposure by acting as a free radical scavenger as shown by electron spin microscopy.(Green et al., 2001) Our findings suggest DHA may decrease free radical production by blunting microglial pro-inflammatory activation early after TBI. We did not study scavenging effects in our work but DHA could have decreased NOx in our model via free radical scavenging directly and/or by decreasing nitric oxide production. DHA also could have improved cognitive function via other mechanisms, such as repletion of DHA losses after TBI as shown in adult models. (Abdullah et al., 2014) Indeed, we found that DHA treatment restored brain DHA content at PID 60 (unpublished data). We did not measure DHA dynamics in the brain at earlier time points, because the presence of esterified and deesterified DHA early after CCI confounds measurement of brain DHA content using standard gas chromatography methods. In future work, we will use imaging and radiolabeled DHA to assess brain DHA content at earlier time points. Our novel observation of DHA-mediated decreases in brain NOx may provide a less-invasive means to examine DHA effects. While published data on NOx in biological samples from children after TBI are lacking, elevated NOx in cerebrospinal fluid and micro dialysate samples from adult patients after TBI correlates with death and poor outcome. (Clark et al., 1996; Tisdall et al., 2013) Therefore, NOx may be a translationally useful marker of injury and inflammation in future pre-clinical or clinical TBI studies.
The therapeutic effects of DHA could result from immunomodulatory effects on microglia. Through gene expression studies of CD11b+ CD45Io microglia and CD11b+ CD45hl macrophages, in the current study, we observed greater DHA-mediated changes in microglia than in macrophage cell populations. Specifically, transcription factors associated with pro-inflammatory activation decreased in microglia at PID 3 and 7 with DHA treatment. The IRF family is critically important in regulating microglia/macrophages maturation and polarization. In particular, irf5 is necessary for macrophage maturation and differentiation and is associated with pro-inflammatory macrophage polarization. (Chistiakov et al., 2018) Irf8 expression is necessary for microgliogenesis. Irf8 over-expression is associated with pro-inflammatory microglia activation. (Kierdorf et al., 2013; Masuda et al., 2012; Zeng et al., 2017) Both Irf5 and irf8 decreased with DHA treatment in microglia at PID 3 and 7, albeit increased at PID 30. Neither transcription factor was affected by DHA in macrophage populations. In accordance with these transcription factor expression changes, phenotypic genes associated with pro- and anti-inflammatory functions were dramatically different with DHA in microglia vs. macrophage populations.
Differences in microglia response after TBI are difficult to compare between immature and mature rodents, in part because delivering equivalent injury to animals of different sizes is challenging. Current understanding about maturational differences in how microglia respond to brain injury derives largely from a small number of studies and indirect comparisons. By some indicators, microglial response in immature and mature brain can be similar: for example, ionized calcium binding adapter protein 1 (Iba1), a microglial activation marker, did not differ between adult and P21 transgenic mice histologically at 2 days after controlled cortical impact, CCI.(Mannix et al., 2011). However, our findings are consistent with the observation that the immature brain mounts a more vigorous inflammatory response to injury. (Claus et al., 2010) While we did not use parallel experiments using adult rats, we found that CD11b, CD45hi macrophage infiltration persists at least to PID 30, in contrast to findings in adult mice after CCI that flow-sorted CD11b+ CD45hi cells resolved by day 30 after injury.(Jin et al., 2012)
In regards to the immunomodulatory effects of treatment in young and adult models, DHA decreased histologic injury and neurologic impairment in the adult rat after TBI (Abdullah et al., 2014; Bailes and Mills, 2010; Begum et al., 2013; Li et al., 2014; Mills et al., 2011; Wu et al., 2004, 2011) similar to our observations here in rat pups. Minocycline, an antibiotic that inhibits microglial activation and improves outcomes in adult stroke and TBI models, decreased microglial activation and improved outcomes in a neonatal hypoxic ischemic stroke model.(Fan et al., 2006; Garrido-Mesa et al., 2013; Kohler et al., 2013) However, disparity in response after TBI suggests that microglia at different developmental stages could respond differently to minocycline after injury. Minocycline had no effect and even aggravated damage in a neonatal (P7) TBI model, but improved outcomes in P17 rats after CCI.(Chhor et al., 2017; Hanlon et al., 2016; Hanlon et al., 2017; Simon et al., 2018) Remaining immunomodulatory agents, shown to succeed in adult models, are yet untested in developmental TBI.(Haber et al., 2018; Liu et al., 2017)
Strain measurements have revealed that CCI has greater injury effects on cortex relative to hippocampus. (Mao et al., 2011) While these studies were conducted in adult animals, in the current study we observed that CCI increased immunohistochemical labeling for microglia/macrophages, astrocytes, and CD68 in the cortex. CCI induced a significant increase in microglia/macrophage activation in the hippocampus but did not increase astrocytes nor CD68. These data indicate that CCI induced a greater inflammatory response in cortex vs. the hippocampus. Consistent with this observation, DHA treatment decreased indices of inflammation in the cortex but not the hippocampus within the first week of injury. The regionally specific therapeutic effect of DHA may be due to a greater magnitude of injury in the cortex vs. hippocampus, i.e. a greater therapeutic window for treatment effects in the cortex. Future studies evaluating the regional specificity of post DHA treatment on inflammation, using FACS and gene expression, as well as, neurogenesis and synaptic integrity will provide greater insight into DHA therapeutic potential for pediatric TBI.
Our study is not without limitations. For example, we tested males at a single developmental age (17 day old male rats) utilizing one experimental model of TBI, CCI. To address some of these limitations, we are currently conducting studies on DHA effects in female rat pups and in a fluid percussion large-animal model. The NOx assay, while useful in that it is translationally relevant, is only one measure of oxidant stress and could be confounded by oxyhemoglobin present in the sample. We used cardial saline perfusion and tissue rinsing to minimize this limitation. Similarly, cognitive outcome was evaluated using one measure. However, we have previously demonstrated DHA improved learning, as shown by performance on the Morris Water Maze. Additionally, the NOR has value as a stress-free measure of memory in experimental TBI. Antibodies for pro-inflammatory and inflammation-resolving markers are more limited in rats than in mice, and in this study options were further reduced by low reactivity to CD206 that may have been a function of time point or developmental age. Alternatively, two time points may not have been sufficient to capture peak CD206 expression, based on a study in female rats using CCI. Turtzo et al reported that CCI increased CD206 expression in the brain ipsilateral to injury relative to control only at day 5, but not at any of the other six time points evaluated up to two months after injury.(Turtzo et al., 2014) Also, mRNA levels do not imply protein expression, nor do protein markers define phenotype. We recognize these limitations and plan to conduct functional assays of DHA effects on neuroinflammation after CCI. Finally, distinguishing activated microglia from macrophages is challenging in vivo, difficult even in transgenic mice. For one, activated microglia and macrophages have overlapping biology. In addition, the immune response to injury is dynamic and markers consistent with inflammatory activation vary throughout development.
To our knowledge, this is the first study assessing effects of acute post-injury DHA administration in a developmental TBI model. Previous work by our group and others demonstrated efficacy of DHA given before injury. (Russell et al., 2014; Schober et al., 2016) While DHA likely has pleiotropic effects after TBI, our results are consistent with the possibility that DHA’s neuroprotective effects in our model are associated with modulation of microglia away from a pro-inflammatory phenotype during the first week after injury. Future areas of inquiry will include functional assays of phagocytic capacity and reactive oxygen species production in rat brain-derived macrophages/microglia to assess DHA effects on the inflammatory milieu after CCI and exploration of potential molecular mediators of DHA effect; for example, assays to evaluate potential inhibition of inducible nitric oxide synthase activation in microglia/macrophages after CCI.
Conclusions
DHA treatment after CCI decreased oxidative stress and histologic and mRNA markers of microglial pro-inflammatory activation in rat pup brain during the first week after injury, associated with improved short term cognitive function. DHA and CCI each tend to affect pro-inflammatory and anti-inflammatory markers in parallel, making it difficult to ascertain whether anti-inflammatory markers support a compensatory host response or a direct effect of the intervention. We plan to conduct phagocytosis and reactive oxygen species assays in our model to better assess the functional effects of CCI with or without DHA on the developing rat brain. Acute DHA administration after CCI has neuroprotective effects, which may result from DHA’s modulation of microglial activation towards a more reparative phenotype early after CCI. Other putative mechanisms of action include direct antioxidant effects via free radical scavenging and repletion of DHA losses after TBI during a period of rapid brain growth. DHA, or fish oil, is a common nutritional supplement for pregnant mothers and is safe for neonates. (Lien, 2009) Given its favorable safety profile in children, DHA is a promising candidate therapy for pediatric TBI.
Supplementary Material
Supplementary Figure 1: Representative Images of CD11b+ CD45 high cells at PID3, 7 and 30
Representative flow cytometry plots of the percentage of CD11b positive, CD45low and CD45hi cells in rat brain after CCI or SHAM injury. REGCCI and DHACCI had higher infiltrating macrophages (CD11b positive, CD45hi cells expressed as % of CD11b positive cells) at A) PID3 B) PID 7 and C) PID30.
Supplementary Figure 2: CCI induced Microglial Activation at PID 30
Results for cortical and hippocampal Iba-1 reactivity proportional area at PID 30 are shown graphically on the left. REGCCI and DHACCI are shown in solid black and white bars, respectively, while corresponding SHAM groups are shown as spotted gray and latticed white for SHAMREG and SHAMDHA rats. CCI significantly increased Iba-1 activation in the cortex and hippocampus relative to SHAM controls (p<0.05 main effect of injury for both regions, adjusted p value & p<0.01 for DHACCI vs. SHAM). There was no main effect of treatment for the cortex (p=0.14) or hippocampus (p=0.10). On the right, representative images of cortical Iba-1 reactivity are shown for SHAMREG, REGCCI and DHACCI. The scale bar is 100 microns.
Acknowledgements
We are grateful for the guidance of Dr. Marco Bortolato, MD, PhD, who assisted in study design and optimization of techniques related to Novel Object Recognition testing and for the NOx tissue assay and analyses performed by the laboratory of Dr. Leif D. Nielin, MD (Yi Jin).
Funding: This work was supported by the NINDS Grant number 5R21 NS090098-02, National Institutes of Health and by Pediatric Critical Care Medicine at the University of Utah.
Research reported in this publication utilized the Biorepository and Molecular Pathology Shared Resource at Huntsman Cancer Institute at the University of Utah supported by the National Cancer Institute of the National Institutes of Health under Award Number P30CA042014. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.”
Research reported in this publication utilized the University of Utah Flow Cytometry Facility through Award Number 5P30CA042014-24 supported by the National Center for Research Resources of the National Institutes of Health under Award Number 1S10RR026802-01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abdullah L, Evans JE, Ferguson S, Mouzon B, Montague H, Reed J, Crynen G, Emmerich T, Crocker M, Pelot R, Mullan M, Crawford F, 2014. Lipidomic analyses identify injury-specific phospholipid changes 3 mo after traumatic brain injury. FASEB J 28, 5311–5321. [DOI] [PubMed] [Google Scholar]
- Anderson V, Catroppa C, Morse S, Haritou F, Rosenfeld J, 2005a. Functional plasticity or vulnerability after early brain injury? Pediatrics 116, 1374–1382. [DOI] [PubMed] [Google Scholar]
- Anderson V, Godfrey C, Rosenfeld JV, Catroppa C, 2012. 10 years outcome from childhood traumatic brain injury. Int J Dev Neurosci 30, 217–224. [DOI] [PubMed] [Google Scholar]
- Anderson VA, Catroppa C, Haritou F, Morse S, Rosenfeld JV, 2005b. Identifying factors contributing to child and family outcome 30 months after traumatic brain injury in children. J Neurol Neurosurg Psychiatry 76, 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW, 2008. A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. J Neurotrauma 25, 513–526. [DOI] [PubMed] [Google Scholar]
- Antonietta Ajmone-Cat M, Lavinia Salvatori M, De Simone R, Mancini M, Biagioni S, Bernardo A, Cacci E, Minghetti L, 2012. Docosahexaenoic acid modulates inflammatory and antineurogenic functions of activated microglial cells. J Neurosci Res 90, 575–587. [DOI] [PubMed] [Google Scholar]
- Bailes JE, Mills JD, 2010. Docosahexaenoic acid reduces traumatic axonal injury in a rodent head injury model. J Neurotrauma 27, 1617–1624. [DOI] [PubMed] [Google Scholar]
- Bayir H, Kochanek PM, Kagan VE, 2006. Oxidative stress in immature brain after traumatic brain injury. Dev Neurosci 28, 420–431. [DOI] [PubMed] [Google Scholar]
- Begum G, Harvey L, Dixon CE, Sun D, 2013. ER stress and effects of DHA as an ER stress inhibitor. Transl Stroke Res 4, 635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittigau P, Sifringer M, Felderhoff-Mueser U, Ikonomidou C, 2004. Apoptotic neurodegeneration in the context of traumatic injury to the developing brain. Exp Toxicol Pathol 56, 83–89. [DOI] [PubMed] [Google Scholar]
- Calder PC, 2017. Omega-3 fatty acids and inflammatory processes: from molecules to man. Biochem Soc Trans 45, 1105–1115. [DOI] [PubMed] [Google Scholar]
- Chang HY, Lee HN, Kim W, Surh YJ, 2015. Docosahexaenoic acid induces M2 macrophage polarization through peroxisome proliferator-activated receptor gamma activation. Life Sci 120, 39–47. [DOI] [PubMed] [Google Scholar]
- Chhor V, Moretti R, Le Charpentier T, Sigaut S, Lebon S, Schwendimann L, Ore MV, Zuiani C, Milan V, Josserand J, Vontell R, Pansiot J, Degos V, Ikonomidou C, Titomanlio L, Hagberg H, Gressens P, Fleiss B, 2017. Role of microglia in a mouse model of paediatric traumatic brain injury. Brain Behav Immun 63, 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakov DA, Myasoedova VA, Revin VV, Orekhov AN, Bobryshev YV, 2018. The impact of interferon-regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology 223, 101–111. [DOI] [PubMed] [Google Scholar]
- Clark RS, Kochanek PM, Obrist WD, Wong HR, Billiar TR, Wisniewski SR, Marion DW, 1996. Cerebrospinal fluid and plasma nitrite and nitrate concentrations after head injury in humans. Crit Care Med 24, 1243–1251. [DOI] [PubMed] [Google Scholar]
- Claus CP, Tsuru-Aoyagi K, Adwanikar H, Walker B, Manvelyan H, Whetstone W, Noble-Haeusslein LJ, 2010. Age is a determinant of leukocyte infiltration and loss of cortical volume after traumatic brain injury. Dev Neurosci 32, 454–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing J, Sands J, 1979. Comparative aspects of the brain growth spurt. Early Hum Dev 3, 79–83. [DOI] [PubMed] [Google Scholar]
- Donnelly DJ, Gensel JC, Ankeny DP, van Rooijen N, Popovich PG, 2009. An efficient and reproducible method for quantifying macrophages in different experimental models of central nervous system pathology. J Neurosci Methods 181, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R, 2005. Oxidative and antioxidative potential of brain microglial cells. Antioxid Redox Signal 7, 1223–1233. [DOI] [PubMed] [Google Scholar]
- Fan LW, Lin S, Pang Y, Rhodes PG, Cai Z, 2006. Minocycline attenuates hypoxia-ischemia-induced neurological dysfunction and brain injury in the juvenile rat. Eur J Neurosci 24, 341–350. [DOI] [PubMed] [Google Scholar]
- Fenn AM, Gensel JC, Huang Y, Popovich PG, Lifshitz J, Godbout JP, 2014. Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol Psychiatry 76, 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido-Mesa N, Zarzuelo A, Galvez J, 2013. Minocycline: far beyond an antibiotic. Br J Pharmacol 169, 337–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensel JC, Kopper TJ, Zhang B, Orr MB, Bailey WM, 2017. Predictive screening of M1 and M2 macrophages reveals the immunomodulatory effectiveness of post spinal cord injury azithromycin treatment. Sci Rep 7, 40144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green P, Glozman S, Weiner L, Yavin E, 2001. Enhanced free radical scavenging and decreased lipid peroxidation in the rat fetal brain after treatment with ethyl docosahexaenoate. Biochim Biophys Acta 1532, 203–212. [DOI] [PubMed] [Google Scholar]
- Haber M, James J, Kim J, Sangobowale M, Irizarry R, Ho J, Nikulina E, Grin’kina NM, Ramadani A, Hartman I, Bergold PJ, 2018. Minocycline plus N-acteylcysteine induces remyelination, synergistically protects oligodendrocytes and modifies neuroinflammation in a rat model of mild traumatic brain injury. J Cereb Blood Flow Metab 38, 1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon LA, Huh JW, Raghupathi R, 2016. Minocycline Transiently Reduces Microglia/Macrophage Activation but Exacerbates Cognitive Deficits Following Repetitive Traumatic Brain Injury in the Neonatal Rat. J Neuropathol Exp Neurol 75, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon LA, Raghupathi R, Huh JW, 2017. Differential effects of minocycline on microglial activation and neurodegeneration following closed head injury in the neonate rat. Exp Neurol 290, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harting MT, Jimenez F, Adams SD, Mercer DW, Cox CS Jr., 2008. Acute, regional inflammatory response after traumatic brain injury: Implications for cellular therapy. Surgery 144, 803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellewell S, Semple BD, Morganti-Kossmann MC, 2016. Therapies negating neuroinflammation after brain trauma. Brain Res 1640, 36–56. [DOI] [PubMed] [Google Scholar]
- Hellewell SC, Yan EB, Alwis DS, Bye N, Morganti-Kossmann MC, 2013. Erythropoietin improves motor and cognitive deficit, axonal pathology, and neuroinflammation in a combined model of diffuse traumatic brain injury and hypoxia, in association with upregulation of the erythropoietin receptor. J Neuroinflammation 10, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjorth E, Zhu M, Toro VC, Vedin I, Palmblad J, Cederholm T, Freund-Levi Y, Faxen-Irving G, Wahlund LO, Basun H, Eriksdotter M, Schultzberg M, 2013. Omega-3 fatty acids enhance phagocytosis of Alzheimer’s disease-related amyloid-beta42 by human microglia and decrease inflammatory markers. J Alzheimers Dis 35, 697–713. [DOI] [PubMed] [Google Scholar]
- Imai Y, Kohsaka S, 2002. Intracellular signaling in M-CSF-induced microglia activation: role of Iba1. Glia 40, 164–174. [DOI] [PubMed] [Google Scholar]
- Innis SM, 2008. Dietary omega 3 fatty acids and the developing brain. Brain Res 1237, 35–43. [DOI] [PubMed] [Google Scholar]
- Jin X, Ishii H, Bai Z, Itokazu T, Yamashita T, 2012. Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PLoS One 7, e41892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W, 2013. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136, 28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielar ML, Jeyarajah DR, Penfield JG, Lu CY, 2000. Docosahexaenoic acid decreases IRF-1 mRNA and thus inhibits activation of both the IRF-E and NFkappa d response elements of the iNOS promoter. Transplantation 69, 2131–2137. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Holscher C, Muller DN, Luckow B, Brocker T, Debowski K, Fritz G, Opdenakker G, Diefenbach A, Biber K, Heikenwalder M, Geissmann F, Rosenbauer F, Prinz M, 2013. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci 16, 273–280. [DOI] [PubMed] [Google Scholar]
- Kohler E, Prentice DA, Bates TR, Hankey GJ, Claxton A, van Heerden J, Blacker D, 2013. Intravenous minocycline in acute stroke: a randomized, controlled pilot study and meta analysis. Stroke 44, 2493–2499. [DOI] [PubMed] [Google Scholar]
- Kumar A, Loane DJ, 2012. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun 26, 1191–1201. [DOI] [PubMed] [Google Scholar]
- Langlois JA, Rutland-Brown W, Thomas KE, 2005. The incidence of traumatic brain injury among children in the United States: differences by race. J Head Trauma Rehabil 20, 229–238. [DOI] [PubMed] [Google Scholar]
- Lauritzen L, Jorgensen MH, Hansen HS, Michaelsen KF, 2002. Fluctuations in human milk long-chain PUFA levels in relation to dietary fish intake. Lipids 37, 237–244. [DOI] [PubMed] [Google Scholar]
- Li L, Wu Y, Wang Y, Wu J, Song L, Xian W, Yuan S, Pei L, Shang Y, 2014. Resolvin D1 promotes the interleukin-4-induced alternative activation in BV-2 microglial cells. J Neuroinflammation 11, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien EL, 2009. Toxicology and safety of DHA. Prostaglandins Leukot Essent Fatty Acids 81, 125–132. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhao Z, Ji R, Zhu J, Sui QQ, Knight GE, Burnstock G, He C, Yuan H, Xiang Z, 2017. Inhibition of P2X7 receptors improves outcomes after traumatic brain injury in rats. Purinergic Signal 13, 529–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Kumar A, 2016. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp Neurol 275 Pt 3, 316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI, 2013. Progressive Neurodegeneration After Experimental Brain Trauma: Association With Chronic Microglial Activation. J Neuropathol Exp Neurol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Stoica BA, Tchantchou F, Kumar A, Barrett JP, Akintola T, Xue F, Conn PJ, Faden AI, 2014. Novel mGluR5 Positive Allosteric Modulator Improves Functional Recovery, Attenuates Neurodegeneration, and Alters Microglial Polarization after Experimental Traumatic Brain Injury. Neurotherapeutics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannix RC, Zhang J, Park J, Zhang X, Bilal K, Walker K, Tanzi RE, Tesco G, Whalen MJ, 2011. Age-dependent effect of apolipoprotein E4 on functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab 31, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, Guan F, Han X, Yang KH, 2011. Strain-based regional traumatic brain injury intensity in controlled cortical impact: a systematic numerical analysis. J Neurotrauma 28, 2263–2276. [DOI] [PubMed] [Google Scholar]
- Masuda T, Tsuda M, Yoshinaga R, Tozaki-Saitoh H, Ozato K, Tamura T, Inoue K, 2012. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Rep 1, 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JD, Hadley K, Bailes JE, 2011. Dietary supplementation with the omega-3 fatty acid docosahexaenoic acid in traumatic brain injury. Neurosurgery 68, 474–481; discussion 481. [DOI] [PubMed] [Google Scholar]
- Morganti JM, Riparip LK, Rosi S, 2016. Call Off the Dog(ma): M1/M2 Polarization Is Concurrent following Traumatic Brain Injury. PLoS One 11, e0148001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr MB, Simkin J, Bailey WM, Kadambi NS, McVicar AL, Veldhorst AK, Gensel JC, 2017. Compression Decreases Anatomical and Functional Recovery and Alters Inflammation after Contusive Spinal Cord Injury. J Neurotrauma 34, 2342–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G.a.W. Charles, 2005. The Rat Brain in Stereotaxic Coordinates.
- Reger ML, Hovda DA, Giza CC, 2009. Ontogeny of Rat Recognition Memory measured by the novel object recognition task. Dev Psychobiol 51, 672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice D, Barone S Jr., 2000. Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect 108 Suppl 3, 511–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell KL, Berman NE, Gregg PR, Levant B, 2014. Fish oil improves motor function, limits blood-brain barrier disruption, and reduces Mmp9 gene expression in a rat model of juvenile traumatic brain injury. Prostaglandins Leukot Essent Fatty Acids 90, 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober ME, Requena D, Block BP, Davis LJ, Rodesch C, Casper TC, Juul SE, Kesner RP, Lane RH, 2013. Erythropoietin Improved Cognitive Function and Decreased Hippocampal Caspase Activity in Rat Pups after Traumatic Brain Injury. J Neurotrauma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober ME, Requena DF, Abdullah OM, Casper TC, Beachy J, Malleske D, Pauly JR, 2016. Dietary Docosahexaenoic Acid Improves Cognitive Function, Tissue Sparing, and Magnetic Resonance Imaging Indices of Edema and White Matter Injury in the Immature Rat after Traumatic Brain Injury. J Neurotrauma 33, 390–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DW, Aneja RK, Alexander H, Bell MJ, Bayir H, Kochanek PM, Clark RSB, 2018. Minocycline Attenuates High Mobility Group Box 1 Translocation, Microglial Activation, and Thalamic Neurodegeneration after Traumatic Brain Injury in Post-Natal Day 17 Rats. J Neurotrauma 35, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team RC, 2016. in: Computing R.F.f.S. (Ed.), R: A language and environment for statistical computing, Vienna, Austria. [Google Scholar]
- Tisdall MM, Rejdak K, Kitchen ND, Smith M, Petzold A, 2013. The prognostic value of brain extracellular fluid nitric oxide metabolites after traumatic brain injury. Neurocrit Care 19, 65–68. [DOI] [PubMed] [Google Scholar]
- Trias E, Diaz-Amarilla P, Olivera-Bravo S, Isasi E, Drechsel DA, Lopez N, Bradford CS, Ireton KE, Beckman JS, Barbeito L, 2013. Phenotypic transition of microglia into astrocyte-like cells associated with disease onset in a model of inherited ALS. Front Cell Neurosci 7, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turtzo LC, Lescher J, Janes L, Dean DD, Budde MD, Frank JA, 2014. Macrophagic and microglial responses after focal traumatic brain injury in the female rat. J Neuroinflammation 11, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsson U, Andersson D, de Pablo Y, Pekny R, Stahlberg A, Mulder J, Mitsios N, Hortobagyi T, Pekny M, Pekna M, 2017. Injury Leads to the Appearance of Cells with Characteristics of Both Microglia and Astrocytes in Mouse and Human Brain. Cereb Cortex 27, 3360–3377. [DOI] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gomez-Pinilla F, 2004. Dietary omega-3 fatty acids normalize BDNF levels, reduce oxidative damage, and counteract learning disability after traumatic brain injury in rats. J Neurotrauma 21, 1457–1467. [DOI] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gomez-Pinilla F, 2011. The salutary effects of DHA dietary supplementation on cognition, neuroplasticity, and membrane homeostasis after brain trauma. J Neurotrauma 28, 2113–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Li R, Michael Greenlief C, Fritsche KL, Gu Z, Cui J, Lee JC, Beversdorf DQ, Sun GY, 2018. Unveiling anti-oxidative and anti-inflammatory effects of docosahexaenoic acid and its lipid peroxidation product on lipopolysaccharide-stimulated BV-2 microglial cells. J Neuroinflammation 15, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeates KO, Armstrong K, Janusz J, Taylor HG, Wade S, Stancin T, Drotar D, 2005. Long-term attention problems in children with traumatic brain injury. J Am Acad Child Adolesc Psychiatry 44, 574–584. [DOI] [PubMed] [Google Scholar]
- Zeng Q, Man R, Luo Y, Zeng L, Zhong Y, Lu B, Wang X, 2017. IRF-8 is Involved in Amyloid-beta1–40 (Abeta1–40)-induced Microglial Activation: a New Implication in Alzheimer’s Disease. J Mol Neurosci 63, 159–164. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Representative Images of CD11b+ CD45 high cells at PID3, 7 and 30
Representative flow cytometry plots of the percentage of CD11b positive, CD45low and CD45hi cells in rat brain after CCI or SHAM injury. REGCCI and DHACCI had higher infiltrating macrophages (CD11b positive, CD45hi cells expressed as % of CD11b positive cells) at A) PID3 B) PID 7 and C) PID30.
Supplementary Figure 2: CCI induced Microglial Activation at PID 30
Results for cortical and hippocampal Iba-1 reactivity proportional area at PID 30 are shown graphically on the left. REGCCI and DHACCI are shown in solid black and white bars, respectively, while corresponding SHAM groups are shown as spotted gray and latticed white for SHAMREG and SHAMDHA rats. CCI significantly increased Iba-1 activation in the cortex and hippocampus relative to SHAM controls (p<0.05 main effect of injury for both regions, adjusted p value & p<0.01 for DHACCI vs. SHAM). There was no main effect of treatment for the cortex (p=0.14) or hippocampus (p=0.10). On the right, representative images of cortical Iba-1 reactivity are shown for SHAMREG, REGCCI and DHACCI. The scale bar is 100 microns.