

Abstract
RIP3 activation leads to activation of necroptosis and the NLRP3 inflammasome pathways. The activation of RIP3 in lupus nephritis (LN) has not been investigated. In this study, RIP3 and necroptosis pathway activations were demonstrated in podocytes in renal biopsies from patients with class IV LN and in the diseased kidneys from lupus-prone NZM2328 and MRL/lpr mice. RIP3 activation was accompanied with the activation of MLKL, the effector molecule of the necroptosis pathway, and activation of caspase-1, the effector of the NLRP3 inflammasome pathway. Podocyte activation of RIP3 was detected readily with the development of LN in NZM2328 mice, suggesting this activation may play a significant role in the pathogenesis of LN. GSK872, a RIP3 specific inhibitor, inhibited the development of LN in MRL/lpr mice with down-regulation of RIP3 activation in podocytes, decreased the splenic sizes and weights and anti-dsDNA antibody titers. IgG from pooled sera of diseased NZM2328 mice succumbing to LN induced both the necroptosis pathway and NLRP3 inflammasome activation in a podocyte cell line and this activation was specifically blocked by GSK872. These results indicate that the necroptosis pathway and the RIP3 dependent NLRP3 inflammasome pathway are activated in podocytes during LN. Inhibition of RIP3 kinase may be a novel therapeutic approach to treat LN and systemic lupus erythematosus (SLE).
Keywords: RIP3, Necroptosis, NLRP3 inflammasome, Lupus nephritis, Podocyte
1. Introduction
Necroptosis is a regulated cell necrosis process sharing characteristics unique to necrosis and apoptosis (1). With the inhibition of caspase-8, receptor interacting protein kinase 1 (RIP1) and RIP3 are recruited and auto-phosphorylated (2). The RIP1-RIP3 complex recruits the mixed lineage kinase domain like pseudokinase (MLKL) and promotes its phosphorylation (2). The phosphorylated MLKL inserts itself into the plasma membrane, compromising its integrity of the plasma membrane (2). This mechanism may lead to cell death (3). NLRP3 inflammasome activation is often accompanied with the necroptosis pathway activation (4). RIP3 is the essential molecule for both necroptosis pathway and NLRP3 inflammasome activation (5). Although the NLRP3 inflammasome activation was initially thought to be dependent on the necroptotic pathway activation, RIP3 dependent NLRP3 inflammasome activation has been shown to be independent of necroptotic cell death (5).
Cell death has been considered a major contributing factor in the pathogenesis of SLE and lupus nephritis (LN) (6, 7). Of all the mechanisms that lead to cell death, necroptosis has been the least interrogated regarding its contribution in LN. In this study, the RIP3 dependent necroptosis pathway and NLRP3 inflammasome activation are shown to be present in the kidneys of patients with LN and in the kidneys of lupus-prone mice. In addition, inhibition of RIP3 kinase is effective in treating LN in mice, suggesting that RIP3 is a target for the treatment of lupus and LN.
2. Materials and Methods
2.1. Animals
Female 10-week-old MRL/lpr mice were purchased from SLAC Laboratory Animal Company (Shanghai, China). Both MRL/lpr and NZM2328 (8) were maintained in a specific pathogen-free condition at the Experimental Animal Center of Sun Yat-sen University. The study protocol was approved by the Ethics Committee of Sun Yat-sen University and all experiments were performed in accordance with the National Institutes of Health Guide for Care and Use of Animals. Only female mice were used in this study.
2.2. GSK872 treatment
A RIP3 kinase inhibitor GSK872 (Merck) was used in this study and diluted at 1mg/ml in vehicle (saline) solution. 20-week-old NZM2328 mice were randomized into 2 groups (N = 6 per group) and were treated with either GSK872 (1mg/kg) or an equal volume of vehicle every day intraperitoneally for 2 weeks. After treatment, kidneys were collected for Western blot and flow cytometry analysis. In addition, MRL/lpr mice were randomized into 2 groups, vehicle group and GSK872 group (N = 8 per group). Mice were treated as above from the age of 12 weeks to 18 weeks. Urine was collected every week. At the end of treatment, mice were anesthetized. Sizes of spleens were evaluated and blood and kidneys were obtained for subsequent analysis.
2.3. Single cell suspensions of glomeruli for flow cytometry (FACS)
Isolation of glomeruli and single cell suspension preparations of NZM2328 mice were performed as previously described by Takemoto et al (9) and modified by Sung et al (10). In brief, kidneys were perfused with PBS and then with magnetic beads (Invitrogen) in PBS. The kidneys were minced and digested with collagenase (Sigma). Iron bead containing glomeruli were captured with a DynaMag 15 magnet (Invitrogen). The glomeruli were then digested with Liberase TM (Roche) for 30 minutes to obtain a single cell suspension. Glomerular single cell suspensions were stained for glomerular resident cells using anti-mouse CD26-PE (Biolegend), anti-mouse Nephrin-Alexa Fluor 647 (Bioss), anti-mouse CD105-PE-Cy7 (Ebioscience), anti-mouse CD31-PerCP-Cy5.5 (Biolegend) and anti-mouse CD45-APC-efluor 780 (Ebioscience). The FAM-FLICA Caspase-1 Assay Kit (Immunochemistry Technology) was used to detect intracellular active caspase-1.
2.4. Evaluation of glomerulonephritis
Kidneys sections were stained with periodic Acid-Schiff (PAS) and histopathologic lesions were evaluated as previously described (11). In brief, glomerular lesions were graded on a scale of 0 – 3, with 0 being normal, 1 being mild (cell proliferation and /or cell infiltration), 2 being moderate (cell proliferation and/or cell infiltration with membrane proliferation), and 3 being severe (cell proliferation and/or cell infiltration, membrane proliferation and crescent formation and/or hyalinosis). Renal vascular lesions were graded on a scale of 0–3, where 0 being normal, 1 being mild with perivascular cell infiltration, 2 being moderate with cellular infiltration of the arterial wall, and 3 being severe with intimal thickening,
2.5. Cell culture and stimulation
An immortalized podocyte cell line was generated from a NZM2328 mouse with severe proteinuria as previously described (12). For maintenance, undifferentiated podocytes were grown at 33°C, 5% CO2, RPMI 1640 supplemented with 10% fetal calf serum, 100U/ml penicillin, 100mg/ml streptomycin on plates coated with rat tail type I collagen (Sigma). For differentiation, podocytes were cultured at 37°C, 5% CO2, RPMI 1640 supplemented with 10% fetal calf serum, 100U/ml penicillin, 100mg/ml streptomycin for 14 days.
For dose-response experiments involving GSK872, 20 ng/ml TNF-α (Invivogen), 5µM SMAC mimetic (Selleck) and 20µM Z-VAD-FMK (Selleck) were used to activate the necroptotic pathway (13, 14) . Well-differentiated podocytes were cultured in 6-well plates and treated with vehicle (PBS), 0.03µM, 0.3µM or 3µM GSK872 for 1 hour before the addition of TNF-α/SMAC mimetic/Z-VAD-FMK for 24 hours. The cells were treated with trypsin and processed for Western blot analysis.
For RIP3 kinase inhibition, well-differentiated podocytes were cultured and primed with 3µM RIP3 kinase inhibitor GSK872 for 1 hour. Then podocytes were stimulated with PBS, heat-inactivated pooled sera from 12-week-old NZM2328 mice without proteinuria and negative for anti-dsDNA antibody, heat-inactivated serum from 36-week-old NZM2328 mice with 3+ proteinuria and positive anti-dsDNA antibody or sera from diseased NZM2328 mice with IgG depleted by Protein A/G Agarose (Beyotime). After incubation, cells were subjected to the proximity ligation assay, flow cytometry and Western blot analysis.
For activation of NLRP3 inflammasome, podocytes were treated with 3µM GSK872 for 1 hour before the addition of 1 µg/ml LPS (Sigma) for another 4 hours. 5 mM ATP was then added for the last 0.5 hour of incubation. Cells and supernatants were collected and subjected to Western blot analysis.
2.6. Flow cytometry
Well differentiated podocytes were primed with 3µM GSK872 for 1 hour. PBS, control sera and diseased sera from NZM2328 mice with severe proteinuria were added for the next 24 hours. After stimulation, cells were collected and stained with FAM-FLICA Caspase-1 assay kit. Flow cytometry was used to assess the levels of active caspase-1 in podocytes.
2.7. Immunofluorescence
Human kidney biopsies were obtained from 8 LN (class IV) patients for diagnostic purposes. Renal tissues from a non-tumor involved area obtained from kidneys removed from patients undergoing nephrectomy for a therapeutic reason were used as “normal” kidney tissue. For the isolation of urinary podocytes, fresh morning urine samples (100 ml) were collected from 4 LN (class IV) patients and 4 healthy volunteer donors. The urine samples were subjected to centrifugation for 10 minutes at 1500 rpm. The pellets were washed with PBS 3 times and suspended in a small volume of DMEM culture medium (Life Technologies). The cells were spun onto slides in a Cytospin. Kidneys from 12-week-old, 36-week-old NZM2328 mice and vehicle or GSK872 treated MRL/lpr mice were fixed in acetone. Cryostat sections (4µm) and urinary cell smear slides were washed 3 times with PBS and blocked for 1 hour with 5% BSA in PBS. For the staining of human kidney sections and urinary podocytes, primary antibodies of mouse anti-human phospho-RIP3 antibody (p-RIP3) (Ab) (Abcam) and rabbit anti-human phospho-MLKL (p-MLKL) Ab (Abcam) were used. Secondary antibodies included Alexa Fluor 488-conjugated anti-mouse IgG Ab (Thermo Fisher) and Alexa Fluor 488-conjugated anti-rabbit IgG Ab (Thermo Fisher). For the staining of mouse kidney sections, primary antibodies of mouse anti-mouse p-RIP3 Ab (Abcam) and rabbit anti-mouse p-MLKL Ab (Abcam) were used. Secondary antibodies were the same as above. FAM-FLICA Caspase-1 assay kit was used to detect caspase-1 activation. Alexa Flour 555-conjugated anti-synaptopodin antibody (Santa Cruz) was used to label podocytes. For detection of complement and immune complex deposition in kidneys of vehicle or GSK872 treated mice, FITC-conjugated anti-mouse IgG (Santa Cruz) and anti-mouse complement C3 (Cedarlane) were used. The mean intensity of fluorescence was scored on a scale of 0~3 as previously described (15) . A Zeiss LSM800 confocal microscope was used for analysis.
2.8. Proximity ligation assay (PLA)
Stimulated podocytes were washed 3 times with PBS and blocked for 1 hour with 5% BSA in PBS. Then the cells were incubated with mouse anti-RIP1 Ab (Abcam) and rabbit anti-RIP3 Ab (Abcam) or rabbit anti-RIP3 Ab and mouse anti-NLRP3 Ab (Adipogen) at 4°C overnight. The RIP1-RIP3 interactions and RIP3-NLRP3 interactions were detected using the PLA kit (Duolink, Sigma) according to manufacturer’s protocol (16). Podocytes were labeled with anti-mouse synaptopodin Ab (Bioss) and stained with DAPI. Mounting media (Vector) was used. A Zeiss LSM 800 confocal microscope was used for analysis.
2.9. Western blot
For immunoblotting analysis, electrophoresis of kidney cell lysates or cell lysates from the NZM2328 podocyte cell line was conducted with 10% running gels and 5% stacking gels. Proteins from the cell lysates were then transferred onto a polyvinylidene fluoride membrane. After blockade with 5% non-fat dry milk in Tris buffered saline with 0.1% Tween 20 (TBST), membranes were incubated overnight at 4°C with the following primary antibodies: rabbit anti-RIP3 Ab (Abcam), anti-p-RIP3 Ab (Abcam), anti-MLKL (Abcam), anti-p-MLKL Ab (Abcam), mouse anti-NLRP3 (Adipogen), anti-caspase-1 p20 (Adipogen), rabbit anti-caspase-8 (Abcam) and anti-GAPDH Ab (Cell Signaling Technology). After washing 3 times with TBST, membranes were incubated with their corresponding secondary antibodies. The signals on the membranes were detected by a chemiluminescence analysis kit (Millipore).
2.10. Measurement of proteinuria and blood urea nitrogen (BUN)
Mouse urine samples were tested with proteinuria analysis strips (Albustix). Six different color can be distinguished: negative, trace, 30, 100, 300, and ≥2000mg/dl protein. Mouse urine samples with protein≥300 mg/dl was designated as severe proteinuria. BUN in serum samples obtained when mice were sacrificed was determined by a commercial autoanalyzer (Beckman Coulter).
2.11. Enzyme linked immunosorbent assay (ELISA)
Levels of IL-1β were measured by IL-1β ELISA kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. Mouse serum anti-double strands DNA (anti-dsDNA) antibody analysis was performed as previously described (11). The absorbance of each well was detected at optical density 450nm.
2.12. Statistical analysis
Data were recorded as the mean ± SD. Statistical analysis was performed using SPSS 20.0. The differences were assessed by t test, or one-way ANOVA or the nonparametric Wilcoxon rank-sum test. Two-tailed p < 0.05 was considered statistically significant.
2.13. Informed Consents
Written informed consents were obtained from all subjects from whom urine samples or kidney biopsies were collected. The study protocol was approved by the Ethics Committee of First Affiliated Hospital, Sun Yat-sen University.
3. Results
3.1. Activation of RIP3 and the necroptosis pathway in podocytes of class IV LN
In order to demonstrate the activation of RIP3 and the necroptosis pathway in podocytes, we took advantage of the availability of Abs to p-RIP3 and p-MLKL, which are the phosphorylated active forms of RIP3 and MLKL. As shown in Figure 1A and 1B, Abs to p-RIP3 and p-MLKL stained the glomeruli from a patient with Class IV LN. With anti-synaptopodin Ab to identify podocytes, the staining of p-RIP3 and p-MLKL were localized in the podocytes. From the staining patterns by the anti-synatopodin Ab and those by the anti-p-RIP3 and anti-p-MLKL Abs, the majority of the podocytes and most likely all the podocytes in the glomeruli of patients with class IV LN expressed both p-RIP3 and p-MLKL. Similar results were obtained in the biopsies from an additional seven patients with class IV LN. Podocytes from a non-involved kidney specimen from a patient with renal tumor did not stain with Abs to either p-RIP3 or p-MLKL (Figure 1A and 1B). Similar results were obtained from two additional “normal” kidneys.
Figure 1. Activation of RIP3 and the necroptosis pathways in podocytes of class IV LN.
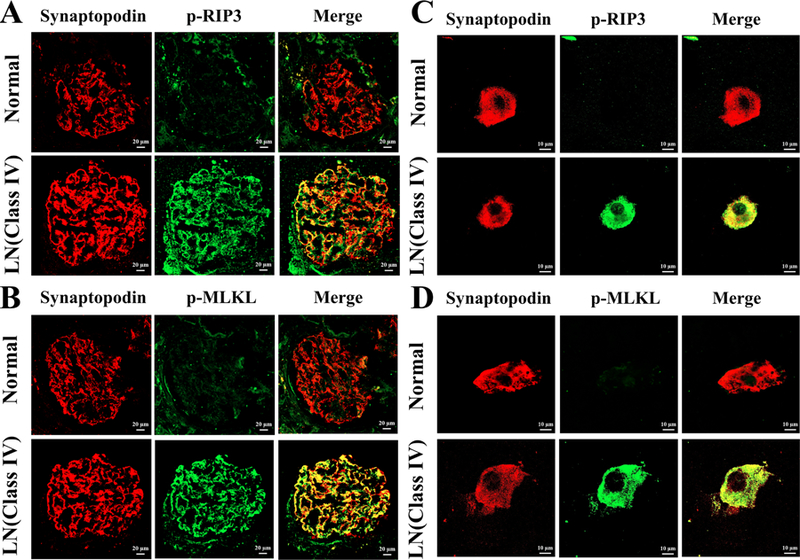
Representative p-RIP3 (A) and p-MLKL (B) stained kidneys biopsy sections from normal and class IV LN patients are shown. In A, podocytes positive for synaptopodin (red fluorescence) in a normal kidney did not stained for p-RIP3 while the podocytes in a class IV LN patient stained with p-RIP3 (green fluorescence) as shown with yellow fluorescence in the merge photo. In B, podocytes in a Class IV LN patient expressed p-MLKL while those from normal kidney tissue did not. In A and B, the original magnification was × 200. Similarly, Abs to p-RIP3 (C) and p-MLKL (D) stained urinary podocytes from class IV LN patients but not from a normal individual. In C and D, original magnification was × 630.
To collaborate the above results, urinary podocytes from patients with class IV LN were prepared and cytospun onto a glass slide. They were fixed and stained with Abs to synaptopodin, p-RIP3 and p-MLKL. As shown in Figure 1C, the urinary podocytes from a patient with class IV LN were identified by positive staining for synaptopodin (red fluorescence). Anti-p-RIP3 Ab that was labeled with green fluorescence stained the same urinary podocyte. The merged figure showed a yellow color for the colocalization of the two chromophores. This result provides undisputable evidence that podocytes in class IV LN expressed activated RIP3. Similarly, p-MLKL was shown in a podocyte (Figure 1D). Similar results were seen in the urinary podocytes of three additional patients with LN. In addition, p-RIP3 and p-MLKL were not detected in podocytes from four normal healthy volunteers.
3.2. Activation of RIP3 and the necroptosis pathway in podocytes of diseased NZM2328 mice
Kidney lysates from 12-week-old and 36-week-old NZM 2328 female mice were analyzed for evidence for RIP3 and MLKL activation. The 12-week-old mice did not have proteinuria while the 36-week-old mice had 3+ proteinuria. As shown in Figure 2A-2C, the kidney lysates of 12-week-old mice did not show significant levels of p-RIP3 and p-MLKL. In contrast there were considerable amounts of p-RIP3 and p-MLKL in the lysates from 36-week-old mice. The increases in p-RIP3 and p-MLKL were correlated with the decreases in caspase-8 expression in the lysates of 36-week-old mice (Figure 2D and 2E). The expression of p-RIP3 and p-MLKL in the podocytes of the mice with severe proteinuria were confirmed by confocal immunofluorescence (Figure 2H and I). The podocytes identified by the expression of synaptopodin were shown to stain for p-RIP3 and p-MLKL. These results support the conclusion that RIP3 and the necroptosis pathway are activated in podocytes within the diseased glomeruli as the mice developed severe proteinuria with glomerulonephritis (GN).
Figure 2. Activation of RIP3 and the necroptosis pathway in podocytes of diseased NZM2328 mice.
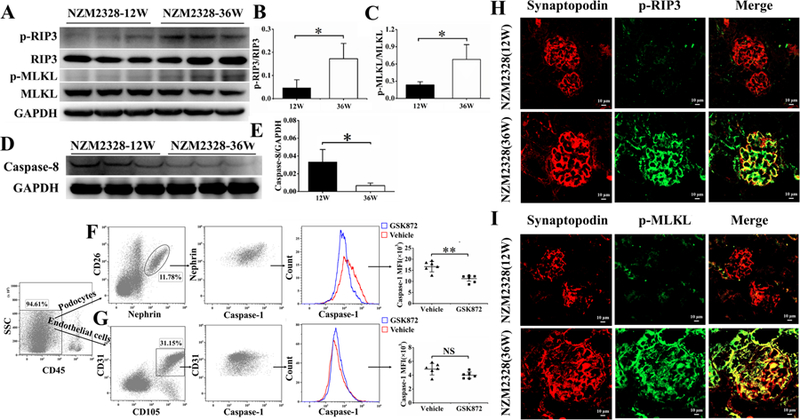
Kidneys from 12-week-old (N = 3) and 36-week-old (N = 3) NZM2328 mice were collected and prepared for Western blot analysis. (A) p-RIP3 and p-MLKL were expressed more in the kidneys of 36-week-old NZM2328 mice. The quantitative increases in p-RIP3 and p-MLKL were depicted in B and C, respectively. Decrease in caspase-8 expression was detected in 36-week-old NZM2328 mice kidneys as shown by Western blot analysis (D). These decreases were shown quantitatively in E. Podocytes (F) and endothelial cells (G) from a 20-week-old NZM2328 mice treated with GSK872 for 2 weeks were isolated and analyzed by flow cytometry after they were stained with relevant Abs. Caspase-1 activation was detected by the FAM-FLICA Caspase-1 assay. Caspase-1 activation was detected in podocytes but not in endothelial cells. In H and I, kidney sections from 12-week-old and 36-week-old NZM2328 mice were analyzed for activation of RIP3 and MLKL. Podocytes labeled by Alexa Flour 555-conjugated anti-synaptopodin Ab (red) were stained with anti-mouse p-RIP3 Ab (green) (H) or anti-mouse p-MLKL Ab (green) (I). Podocytes in a 36-week-old mouse expressed p-RIP3 and p-MLKL but not in podocytes of a 12-week-old mouse. Results in B, C, E, F and G were shown as mean ± SD. *p<0.05, **p<0.01, and NS = not significant.
In an additional experiment shown in Figure 2F, 20-week-old NZM2328 mice were treated either with GSK872, a specific RIP3 kinase inhibitor or vehicle for two weeks. The mice were sacrificed. Single cell suspension was prepared from the isolated glomeruli. The cells were stained for either nephrin or CD31. Nephrin positive podocytes and CD31 positive endothelial cells were sorted. They were interrogated for the expression of activated caspase-1 by the FAM-FLICA Caspase-1 assay. The brief treatment with GSK872 abolished the expression of activated caspase-1. Since caspase-1 activation is a biomarker of NLRP3 inflammasome activation, this result supports the conclusion that activation of RIP3 is accompanied by the activation of NLRP3 inflammasome in the diseased glomerulus.
3.3. Inhibition of RIP3 dependent necroptosis and NLRP3 inflammasome pathways activation by GSK872 in MRL/lpr mice with reduction of autoimmunity and alleviation of disease activity
MRL/lpr mice have renal disease at an earlier age within a shorter period. These mice were used to demonstrate the therapeutic effect of targeting RIP3 with its specific inhibitor GSK872. 12-week-old MRL/lpr female mice were either treated with GSK872 or with vehicle (saline). The mice were sacrificed at 18 weeks of age. As shown in Figure 3A, the kidney lysates from mice treated with vehicle had elevated p-RIP3 and p-MLKL activities. In contrast, p-RIP3 and p-MLKL were markedly diminished in the kidney of GSK872 treated mice. Activation of caspase-1, a biomarker of NLRP3 inflammasome activation was readily detected in the kidney lysates of mice treated with vehicle (Figure 3B). The activation of caspase-1 was much less in mice treated with GSK872. Kidney lysates of vehicle-treated mice expressed more NLRP3 inflammasome in comparison with those from GSK872 treated mice. These results indicate that treatment with RIP3 inhibitor is effective in inhibiting both NLRP3 inflammasome and necroptosis pathways activation.
Figure 3. Inhibition of RIP3 dependent necroptosis and NLRP3 inflammasome pathways, amelioration of nephritis and reduction of splenic sizes and auto-Ab formation in MRL/lpr mice by GSK872.
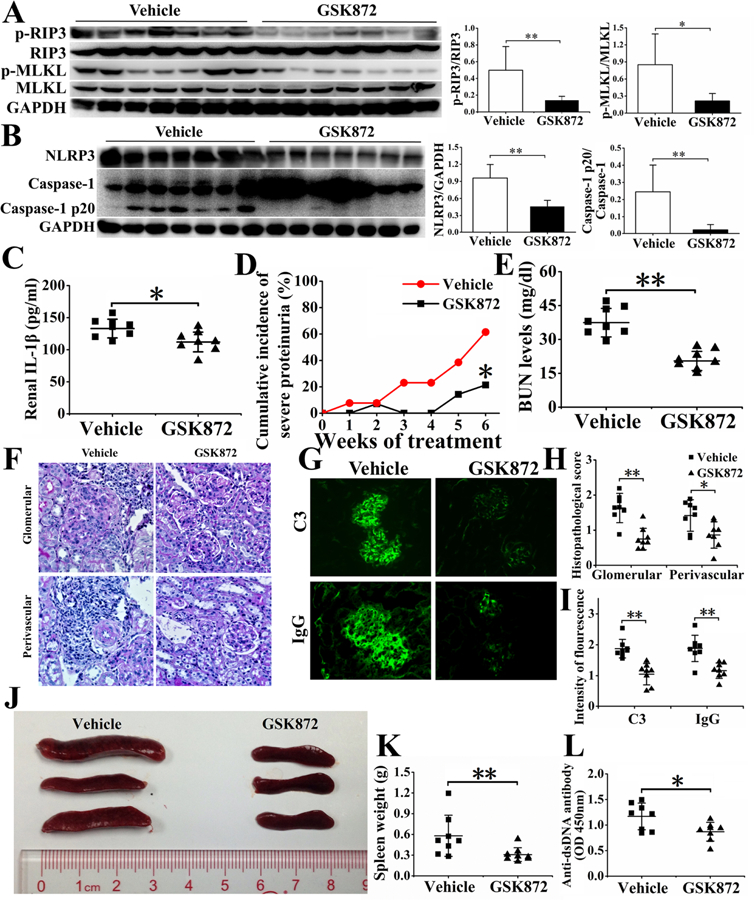
12-week-old MRL/lpr were treated with GSK872 for 6 weeks or vehicle (saline). The mice were analyzed for the effects of GSK872 treatment. In A, the expression of p-RIP3 and p-MLKL were reduced in GSK872 treated mice (n=7) but not in vehicle treated mice (n=7). In B, NLRP3 and caspase-1 p20 expression were reduced in GSK872 treated mice. In C, IL-1β in the kidneys was determined by ELISA. Severity of proteinuria was recorded weekly. Cumulative incidence of severe proteinuria was determined in D. (E). Serum levels of BUN were analyzed using a commercial auto-analyzer. Renal sections were stained with PAS (F) to assess renal pathology. C3 and IgG depositions were shown by immunofluorescence in G. Histopathological scores were shown in H. Intensities of fluorescence of IgG and C3 depositions were shown in I. Spleen sizes (J) and weights (K) were recorded. Serum levels of anti-dsDNA antibody were measured by ELISA (L). Results in relevant figures were recorded as mean ± SD. *p < 0.05 and **p < 0.01.
Renal IL-1β was significantly reduced in GSK872 treated mice (Figure 3C). This treatment delayed the onset and severity of proteinuria (Figure 3D) and reduced blood BUN (Figure 3E). The kidneys of GSK872 treated mice had less cell infiltration and smaller glomeruli with less perivascular cellular infiltration (Figure 3F and 3H). The glomeruli of mice treated with GSK872 had less IgG and C3 deposition detected by immunofluorescence (Figure 3G and 3I). There were reductions of splenic sizes and weights in the GSK872 treated mice (Figure 3J and 3K) with reduction of anti-dsDNA Abs (Figure 3L). These results show the effectiveness of targeting RIP3 in treating lupus mice with reduction of renal pathology and autoimmunity.
The effectiveness of GSK872 in treating LN was also illustrated by immunofluorescence in Figure 4. GSK872 effectively abolished the activation of RIP3, MLKL and caspase-1, a marker for NLRP3 inflammasome activation, in the podocytes within the diseased glomeruli.
Figure 4. Inhibition of RIP3 dependent necroptosis and NLRP3 inflammasome pathways activation in podocytes of MRL/lpr mice by GSK872 as shown by immunofluorescence.
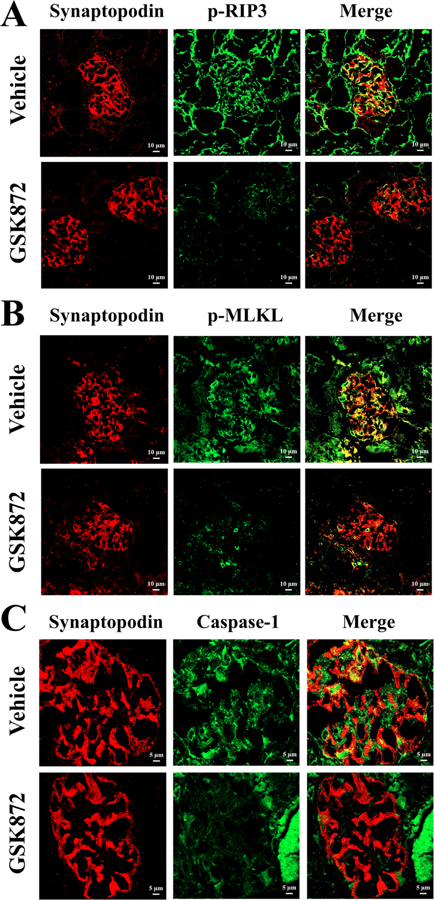
Cryostat sections of kidneys were stained with Alexa Flour 555-conjugated anti-synaptopodin Ab (red) to label podocytes, stained with anti-mouse p-RIP3 Ab (green) (A) or anti-mouse p-MLKL Ab (green) (B). FAM-FLICA Caspase-1 assay kit was used to detect NLRP3 inflammasome activation in podocytes (C). Original magnification × 200.
3.4. Activation of RIP3 dependent necroptosis and NLRP3 inflammasome pathways by IgG from sera of mice with severe immune-complex mediated nephritis
A podocyte cell line generated from NZM2328 was stimulated in vitro with sera from mice with severe nephritis (diseased sera). As shown in Figure 5A and 5B, the diseased sera induced the interaction of RIP3 with RIP1 and RIP3 with NLRP3 as shown by the proximity ligation assay (PLA). This interaction is the result of RIP3 activation. The activation of RIP3 was then accompanied by the phosphorylation of RIP3, MLKL and increased caspase-1 p20, the marker of activation of NLRP3 inflammasome (Figures 5E-I). Removal of IgG from the diseased sera abolished the ability of the diseased sera to activate RIP3 that resulted in the activation of necroptosis and NLRP3 inflammasome (Figures 5C and 5D).
Figure 5. Activation of RIP3 dependent necroptosis and NLRP3 inflammasome pathways by IgG from sera of diseased lupus-prone mice.
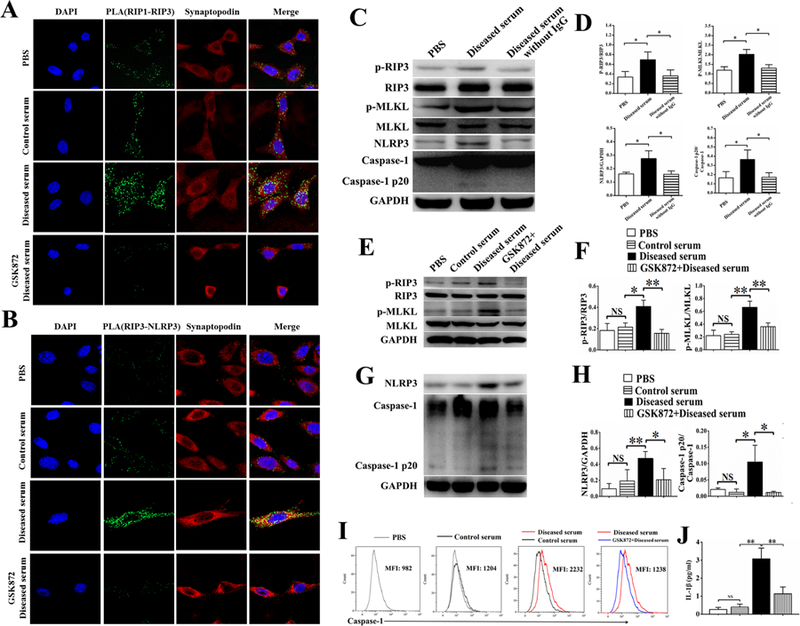
The NZM2328 podocyte cell line was stimulated with control serum or diseased serum, with or without GSK872 for 24 hours. After stimulation, interactions between RIP1-RIP3 (A) and RIP3-NLRP3 (B) in podocytes were detected by PLA. Podocytes were labelled with synaptopodin (A, B). In A, podocytes from the NZM2328 cell line incubated with control sera did not show interaction between RIP1 and RIP3 whereas podocytes incubated with diseased sera showed interaction between RIP1 and RIP1 that initiate the necroptosis pathway. This interaction was inhibited by GSK872. In B, podocytes from the NZM2328 cell line incubated with the diseased sera expressed interaction between RIP3 and NLRP3 showing interaction between RIP3 and NLRP3 that led to the activation of NLRP3 inflammasome. This interaction was inhibited by GSK872. In C, podocytes from the cell line stimulated with the diseased mouse sera for 24 hours expressed more p-RIP3, p-MLKL, NLRP3 and caspase-1 p20 as detected by Western blot analysis. Depletion of IgG from the diseased sera failed to detect these increases. The quantitative expressions of p-RIP3, p-MLKL, NLRP3 and caspase-1 p20 were shown in D. The increases in p-RIP3/RIP3 and p-MLKL/MLKL induced by diseased sera were inhibited by GSK872 as shown by Western blot analysis (E). These changes are quantified in F. The increases in NLRP3 and caspase1-p20 induced by the diseased sera were abolished by GSK872 as shown by Western Blot analysis (G). The quantitative aspect of the increases in G is depicted in H. The FAM-FLICA Caspase-1 assay kit and flow cytometry analysis were used to assess activation of caspase 1. Mean fluorescence intensity was recorded in I showing that caspase-1 activation induced by the diseased sera was inhibited by GS872. Levels of IL-1β in the supernatants following different stimulations were measured in J. IL-1β secretion induced by the diseased sera was inhibited by GSK872. Original magnifications of immunofluorescence photos were × 630. Each experiment was repeated for three times. When applicable, results were recorded as mean ± SD, with *p < 0.05, **p < 0.01 and NS = not significant.
The effects of the diseased sera on the NZM2328 podocyte cell line were blocked by GSK872. GSK872 blocked the interaction between RIP3 and RIP1 or NLRP3 (Figure 5A and 5B), the phosphorylation of RIP3 and MLKL (Figures 5E and 5F), caspase-1 activation (Figures 5G-I) and the induction of IL-1β (Figure 5J).
3.5. Inhibition of RIP3 dependent activation of NLRP3 inflammasome by GSK872
Although GSK872 is a highly specific RIP3 inhibitor, it was important to ascertain that its action was solely on RIP3. In a preliminary experiment, TNF-α, SMAC mimetic and Z-VAD-FMK, a caspase-1 inhibitor, were used to stimulate the NZM2328 podocyte line. These reagents induced robust phosphorylation of RIP3, MLKL and caspase-1 (Supplemental Figure 1). 3µM of GSK872 completely inhibited these processes.
As shown in Figures 6A-6F, LPS plus ATP stimulated the activation of RIP3 and the RIP3 dependent necroptosis pathways in the NZM2328 podocyte line (Figure 6A). LPS plus ATP delivered a strong stimulus of activation of the NLRP3 inflammasome and IL-1β secretion (Figure 6D). GSK872 at 3µM inhibited completely the phosphorylation of RIP3 and MLKL (Figures 6B and 6C) with a small but significant reduction of activated caspase-1 and NLRP3 (Figures 6E and 6F). In addition, the reduction of IL-1β in the culture supernatant was modest (Figure 6D). These results show that GSK872 is indeed specific for RIP3.
Figure 6. Specific inhibition of the RIP3 dependent necroptosis and NLRP3 inflammasome pathways activation of the podocytes from the NZM2328 cell line activated by LPS plus ATP.
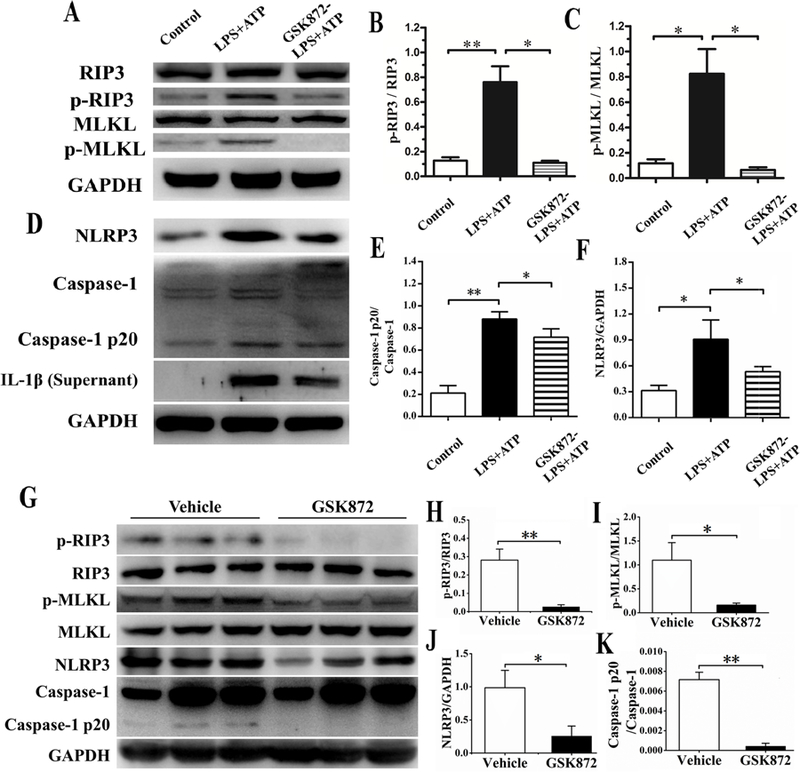
The increases in p-RIP3 and p-MLKL induced by LPS plus ATP in the podocytes from the cell line were inhibited by GSK872 by Western blot analysis (A) and confirmed by scanning the band intensities in the gel in B and C. LPS plus ATP induced increases in NLRP3, caspase-1 p20 and IL-1β secretion were partially inhibited by GSK872 as shown by Western blot analysis (D) and in the band densities in the gels for caspase-1 p20/caspase1 (E) and for NLRP3 (F). Following 2 weeks of treatment with GSK872 or vehicle, NZM2328 mouse kidneys were collected for Western blot analysis. P-RIP3, p-MLKL, NLRP3, and caspase-1 p20 were greater in vehicle treated mice (n=3) than in GSK872 treated mice (n=3). The relative expressions of pRIP3/RIP3, p-MLKL/MLKL, NLRP3 and caspase-1 p20/caspase-1 are shown in H-K respectively. Relevant results were recorded as mean ± SD with *p<0.05 and **p<0.01.
To determine whether RIP3 dependent activation of NLRP3 inflammasome is largely responsible for the NLRP3 activation in vivo in mice with LN, kidney lysates were prepared from 20-week-old NZM2328 female mice treated for two weeks with either GSK872 or vehicle (saline). As shown in figure 6G, there was a significant presence of p-RIP3, p-MLKL, NLRP3 and activated caspase-1 in the kidney lysates of vehicle treated mice. In the GSK872 treated mice, p-RIP3, p-MLKL, NLRP3 and activated caspase-1 were either undetectable or markedly reduced (Figures 6G-6K). These results support the thesis that activation of RIP3 is largely responsible for NLRP3 activation in the kidney.
4. Discussion
Since the discovery of necroptosis, its roles in various kidney diseases have been studied. Mulay et al reported that the necroptosis pathway was activated and the pharmacological inhibition of RIP3 or MLKL or specific gene knockout of RIP3 or MLKL exerted protective effects and limited tissue injury and organ failure in a crystal induced acute kidney injury (AKI) model (17). The necroptosis pathway has also been identified in tubular cells in renal ischemia/reperfusion injury (18) and in cisplatin-induced acute AKI (14). In addition, Lau et al showed that recipients receiving a RIP3−/− kidney had longer survival and improved renal function, suggesting that the necroptosis pathway might contribute to allograft rejection (19). However, no investigation has been reported on the activation of the necroptosis pathway and its role in podocyte injury and in LN. The present study represents the first report on the activation of the necroptosis pathway in LN.
As discussed in the Introduction, RIP3 activation leads to both the activation of necroptosis and NLRP3 inflammasome pathways. In the present study, we show that RIP3 is activated in podocytes of patients with Class IV LN and in the podocytes of lupus-prone mice with immune complex mediated nephritis. In addition, the diseased sera of NZM2328 were shown to activate RIP3 leading to both activation of the necroptosis pathway and the NLRP3 inflammasome activation. Although not directly documented, the activation of NLRP3 inflammasome is inferred as a consequence of RIP3 activation. The serum-induced increase in the phosphorylation of RIP3 and MLKL is abolished with the depletion of IgG from the sera. These observations support the conclusion that circulating antibodies are responsible for the activation of the two RIP3 dependent pathways. This observation is similar to that by Ichinose et al (20). In their study, IgG in sera from patients with LN up-regulates calcium/calmodulin-dependent protein kinase IV (CaMKIV) in podocytes and leads to podocyte injury (20). In both our study and the study by Ichinose et al, the targets for the reactive IgG have not been identified. Thus, further studies are needed in the elucidation of the targets on the cell surface. In our case, TNF-α receptor may be a prime target for these autoantibodies. In addition, our data with GSK872, a specific RIP3 kinase inhibitor and with diseased sera from NZM2328 mice with nephritis suggest that the major pathway of NLRP3 inflammasome activation in LN is via RIP3 activation. This information together with the effectiveness of GSK872 identifies RIP3 as a suitable therapeutic target for LN.
It is of note that following treatment with RIP3 inhibitor, GSK872, both necroptosis and NLRP3 inflammasome pathways were inhibited. Renal damage was significantly improved. It is well established that the NLRP3 inflammasome plays a central role in the activation of caspase-1 resulting in the activation of IL-1β and IL-18 (21, 22). There are many pathways to activate the NLRP3 inflammasome. In this regard, we have demonstrated that brilliant blue G that inhibits P2X7 activation suppresses NLRP3/ASC/caspase-1 assembly and the subsequent release of IL-1β (23) and that glycogen synthase kinase 3β activation causes renal damage through activation of the NLRP3 inflammasome in lupus-prone mice (11). Together with the findings reported here, many different signaling pathways for the activation of NLRP3 inflammasome are operative in the pathogenesis of LN. It is surprising that inhibition of one of these activated pathways would result in amelioration of LN and the reduction of auto-Abs production, suggesting down regulation of many other NLRP3 inflammasome activation pathways. It appears that many of the NLRP3 inflammasome activation pathways are interconected and may share a common inhibitory pathway. In this regard, COPS and POPs which play significant roles in down-regulating NLRP3activation may be relevant (24). Nevertheless, the exact molecular mechanism accounting for this observation remains to be elucidated.
In a previous report (12), we provided evidence that IgG from diseased NZM2328 mice was able to induce activation of the NLRP3 inflammasome pathway. In this study, this activation is shown to be through RIP3. Although multiple pathways have been identified to activate necroptosis (25), the major pathway is through the interaction of TNF-α and its receptor. Thus it is likely that IgG in the sera of diseased MZN2328 mice have the capability to activate RIP3 by targeting the TNF-α receptor. The identification of the specificity of these IgG molecules will be of interest and will provide further insight in our understanding of the pathogenesis of LN.
RIP1-RIP3 complex formation is a hallmark of activation of the necroptosis pathway (26). Once activated, downstream molecules are phosphorylated initiating the necroptosis pathway. By immunofluorescence, both p-RIP3 and p-MLKL were shown to be present within the glomeruli in both kidney biopsies from class IV LN patients and in lupus prone NZM2328 and MRL/lpr mice, indicating that the necroptosis pathway is activated in LN. We further show that the necroptosis pathway is activated in podocytes in the diseased kidney. In view of the importance of the necroptosis pathway in diseases and in homeostasis of the immune system (27), this pathway is undoubtedly activated in other cells during the normal immune response and autoimmunity. In the present study, the issues of the relative contribution of necroptosis to the pathogenesis of LN and whether the activation of the necroptosis pathway on cells other than the podocytes have not been addressed. This aspect of investigation is hindered by the absence of specific inhibitor of mouse MLKL and the absence of tissue a specific deletion mutants involving RIP3 and MLKL. Nevertheless, in view of the current interest in the role of cell death in the pathogenesis of SLE in general and LN in particular (6, 7), the necroptosis pathway should be interrogated further.
It is of note that GSK872 significantly decreases the splenic sizes of MRL/lpr mice and reduces anti-ds-DNA antibody titers. These observations are congruent with the observation that RIP3 plays a significant role in modulating T cell function by limiting the magnitude of T cell proliferation after antigenic stimulation (28). In view of the recent observation that complement driven NLRP3 inflammasome activation is important in Th1 immunity (29), it would be of interest to determine whether RIP3 activation is present in T cells and whether its activation has a role in determining the magnitude of an immune response to an antigen stimulus. Further understanding of RIP3 activation may help us to devise a scheme to limit ongoing autoimmune responses.
5. Conclusion
Our results demonstrate that RIP3 dependent necroptosis and NLRP3 inflammasome pathways are activated in podocytes during LN. IgG from diseased lupus-prone mice induced both necroptosis and NLRP3 inflammasome pathways activation in a podocyte cell line and this activation was blocked by GSK872, a RIP3 inhibitor. Inhibition of RIP3 kinase may be a novel therapeutic approach to treat LN and SLE.
Supplementary Material
Supplementary Figure 1. GSK872 inhibited necroptosis and NLRP3 inflammasome pathways induced by TNF-α + SMAC mimetic + Z-VAD-FMK. The NZM2328 podocyte cell line was stimulated with or without TNF-α + SMAC mimetic + Z-VAD-FMK (TSZ) in the presence of different concentrations of GSK872 for 24 hours. Cells were collected and subjected to Western blot.(A) Representative bands of RIP3, p-RIP3, MLKL, p-MLKL, caspase-1 p20 and GAPDH are shown. Relative expressions of p-RIP3 (B), p-MLKL (C) and caspase-1 p20 (D) normalized to RIP3, MLKL and GAPDH were are shown. Relevant results were recorded as mean ± SD with *p < 0.05, **p < 0.01 and NS = not significant.
Highlights.
Necroptosis and NLRP3 inflammasome pathways are activated in podocytes in LN;
RIP3 regulates both NLRP3 inflammasome and necroptosis activation;
IgG from LN activates RIP3 dependent necroptosis and NLRP3 inflammasome pathways;
Inhibition of RIP3 reduces autoimmunity and alleviates clinical manifestation of LN.
Acknowledgments
Funding
This work was supported by grants from the National Natural Science Foundation of China (81671593, 81701595, 81801614), Guangdong Natural Science Foundation (2014A030313096, 2016A030310172), Guangzhou Science and Technology Planning Program (201707010093), NIH grant (NIH R01 AR-047988) and Alliance for Lupus Research (TIL332615 and DIA481517.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G, Molecular mechanisms of necroptosis: an ordered cellular explosion, Nat Rev Mol Cell Biol 2010;11(10):700–14. 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 2.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. , RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis, Science 2009;325(5938):332–6. 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 3.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. , Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase, Cell 2012;148(1–2):213–27. 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 4.Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D, Activation of the NLRP3 inflammasome by proteins that signal for necroptosis, Methods Enzymol 2014;545:67–81. 10.1016/B978-0-12-801430-1.00003-2. [DOI] [PubMed] [Google Scholar]
- 5.Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, et al. , RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL, Nat Commun 2015;6:6282 10.1038/ncomms7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mistry P, Kaplan MJ, Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis, Clin Immunol 2017;185:59–73. 10.1016/j.clim.2016.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fenton K, The effect of cell death in the initiation of lupus nephritis, Clin Exp Immunol 2015;179(1):11–6. 10.1111/cei.12417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waters ST, Fu SM, Gaskin F, Deshmukh US, Sung SS, Kannapell CC, et al. , NZM2328: a new mouse model of systemic lupus erythematosus with unique genetic susceptibility loci, Clin Immunol 2001;100(3):372–83. 10.1006/clim.2001.5079. [DOI] [PubMed] [Google Scholar]
- 9.Takemoto M, Asker N, Gerhardt H, Lundkvist A, Johansson BR, Saito Y, et al. , A new method for large scale isolation of kidney glomeruli from mice, Am J Pathol 2002;161(3):799–805. 10.1016/S0002-9440(10)64239-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sung SJ, Ge Y, Dai C, Wang H, Fu SM, Sharma R, et al. , Dependence of Glomerulonephritis Induction on Novel Intraglomerular Alternatively Activated Bone Marrow-Derived Macrophages and Mac-1 and PD-L1 in Lupus-Prone NZM2328 Mice, J Immunol 2017;198(7):2589–601. 10.4049/jimmunol.1601565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao J, Wang H, Huang Y, Zhang H, Wang S, Gaskin F, et al. , Lupus nephritis: glycogen synthase kinase 3beta promotion of renal damage through activation of the NLRP3 inflammasome in lupus-prone mice, Arthritis Rheumatol 2015;67(4):1036–44. 10.1002/art.38993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu R, Guo C, Wang S, Huang Y, Jin O, Hu H, et al. , Podocyte Activation of NLRP3 Inflammasomes Contributes to the Development of Proteinuria in Lupus Nephritis, Arthritis Rheumatol 2017;69(8):1636–46. 10.1002/art.40155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hussain M, Zimmermann V, van Wijk SJL, Fulda S, Mouse lung fibroblasts are highly susceptible to necroptosis in a reactive oxygen species-dependent manner, Biochem Pharmacol 2018;153:242–7. 10.1016/j.bcp.2018.01.025. [DOI] [PubMed] [Google Scholar]
- 14.Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, et al. , A Role for Tubular Necroptosis in Cisplatin-Induced AKI, J Am Soc Nephrol 2015;26(11):2647–58. 10.1681/ASN.2014080741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L, Yang N, Wang S, Huang B, Li F, Tan H, et al. , Adenosine 2A receptor is protective against renal injury in MRL/lpr mice, Lupus 2011;20(7):667–77. 10.1177/0961203310393262. [DOI] [PubMed] [Google Scholar]
- 16.Qing DY, Conegliano D, Shashaty MG, Seo J, Reilly JP, Worthen GS, et al. , Red blood cells induce necroptosis of lung endothelial cells and increase susceptibility to lung inflammation, Am J Respir Crit Care Med 2014;190(11):1243–54. 10.1164/rccm.201406-1095OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mulay SR, Desai J, Kumar SV, Eberhard JN, Thomasova D, Romoli S, et al. , Cytotoxicity of crystals involves RIPK3-MLKL-mediated necroptosis, Nat Commun 2016;7:10274 10.1038/ncomms10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linkermann A, Brasen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U, et al. , Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury, Kidney Int 2012;81(8):751–61. 10.1038/ki.2011.450. [DOI] [PubMed] [Google Scholar]
- 19.Lau A, Wang S, Jiang J, Haig A, Pavlosky A, Linkermann A, et al. , RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival, Am J Transplant 2013;13(11):2805–18. 10.1111/ajt.12447. [DOI] [PubMed] [Google Scholar]
- 20.Ichinose K, Ushigusa T, Nishino A, Nakashima Y, Suzuki T, Horai Y, et al. , Lupus Nephritis IgG Induction of Calcium/Calmodulin-Dependent Protein Kinase IV Expression in Podocytes and Alteration of Their Function, Arthritis Rheumatol 2016;68(4):944–52. 10.1002/art.39499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Latz E, Xiao TS, Stutz A, Activation and regulation of the inflammasomes, Nat Rev Immunol 2013;13(6):397–411. 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tschopp J, Schroder K, NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production?, Nat Rev Immunol 2010;10(3):210–5. 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 23.Zhao J, Wang H, Dai C, Zhang H, Huang Y, Wang S, et al. , P2X7 blockade attenuates murine lupus nephritis by inhibiting activation of the NLRP3/ASC/caspase 1 pathway, Arthritis Rheum 2013;65(12):3176–85. 10.1002/art.38174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Indramohan M, Stehlik C, Dorfleutner A, COPs and POPs Patrol Inflammasome Activation, J Mol Biol 2018;430(2):153–73. 10.1016/j.jmb.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petrie EJ, Czabotar PE, Murphy JM, The Structural Basis of Necroptotic Cell Death Signaling, Trends Biochem Sci 2019;44(1):53–63. 10.1016/j.tibs.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 26.Rayamajhi M, Miao EA, The RIP1-RIP3 complex initiates mitochondrial fission to fuel NLRP3, Nat Immunol 2014;15(12):1100–2. 10.1038/ni.3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Donnell JA, Lehman J, Roderick JE, Martinez-Marin D, Zelic M, Doran C, et al. , Dendritic Cell RIPK1 Maintains Immune Homeostasis by Preventing Inflammation and Autoimmunity, J Immunol 2018;200(2):737–48. 10.4049/jimmunol.1701229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaiha GD, McKim KJ, Woods M, Pertel T, Rohrbach J, Barteneva N, et al. , Dysfunctional HIV-specific CD8+ T cell proliferation is associated with increased caspase-8 activity and mediated by necroptosis, Immunity 2014;41(6):1001–12. 10.1016/j.immuni.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. , T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells, Science 2016;352(6292):aad1210 10.1126/science.aad1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. GSK872 inhibited necroptosis and NLRP3 inflammasome pathways induced by TNF-α + SMAC mimetic + Z-VAD-FMK. The NZM2328 podocyte cell line was stimulated with or without TNF-α + SMAC mimetic + Z-VAD-FMK (TSZ) in the presence of different concentrations of GSK872 for 24 hours. Cells were collected and subjected to Western blot.(A) Representative bands of RIP3, p-RIP3, MLKL, p-MLKL, caspase-1 p20 and GAPDH are shown. Relative expressions of p-RIP3 (B), p-MLKL (C) and caspase-1 p20 (D) normalized to RIP3, MLKL and GAPDH were are shown. Relevant results were recorded as mean ± SD with *p < 0.05, **p < 0.01 and NS = not significant.