

Abstract
Here we report the structure of the widely utilized calmodulin (CaM)-dependent protein kinase II (CaMKII) inhibitor KN93 bound to the Ca2+-sensing protein CaM. KN93 is widely believed to inhibit CaMKII by binding to the kinase. The CaM-KN93 interaction is significant as it can interfere with the interaction between CaM and it’s physiological targets, thereby raising the possibility of ascribing modified protein function to CaMKII phosphorylation while concealing a CaM–protein interaction. NMR spectroscopy, stopped-flow kinetic measurements, and x-ray crystallography were used to characterize the structure and biophysical properties of the CaM-KN93 interaction. We then investigated the functional properties of the cardiac Na+ channel (NaV1.5) and ryanodine receptor (RyR2). We find that KN93 disrupts a high affinity CaM-NaV1.5 interaction and alters channel function independent of CaMKII. Moreover, KN93 increases RyR2 Ca2+ release in cardiomyocytes independent of CaMKII. Therefore, when interpreting KN93 data, targets other than CaMKII need to be considered.
Keywords: calcium calmodulin dependent kinase type II (CaMKII), CaMKII inhibition, calcium signaling, KN93, KN92, autocamtide-2- inhibitor peptide
Graphical Abstract
1. INTRODUCTION
In excitable cells such as cardiomyocytes, Ca2+ sensing proteins transduce changes in intracellular Ca2+ concentration into protein-protein interactions that modulate and regulate aspects of cellular function. Calmodulin (CaM) and Ca2+ calmodulin dependent protein kinase II, (CaMKII)) have significant roles in these processes [1,2] and as such have been the topic of immense investigation. To date, PubMed contains more than forty thousand reports on various aspects of CaM structure and function, and greater than twenty one thousand reports pertaining to CaMKII. Intriguingly, these two proteins are ascribed to directly regulating and modulating many of the same proteins [1,3,4].
Structurally, CaM consists of two globular domains connected by a flexible linker [5]. Each domain contains two EF-hand motifs which bind a total of 4 Ca2+ ions per CaM molecule [6]. In isolation there is a 6–10-fold difference in Ca2+ binding affinity between the -N and -C domains [7–9] as well as differences in kinetics of Ca2+ binding [10] that allow CaM to sense and transduce changes in Ca2+ over a wide range of concentrations (nM to mM). Ca2+ binding to CaM drives conformational changes that expose hydrophobic residues used for interacting with a wide range of cellular proteins. Many CaM interactions have been characterized, and patterns in binding sequence, and conformations used for interaction have been identified and described [2,11].
In addition to directly modifying the function of many proteins (including ion channels), CaM can also activate CaM dependent kinases, such as CaMKII. CaMKII is comprised of 12 monomeric subunits that arrange to form a dodecameric structure. In the presence of Ca2+, one CaM molecule can bind to each CaMKII monomer. This interaction can release a CaMKII domain, which can then phosphorylate a target protein. Once CaM is bound to CaMKII, a domain can auto-phosphorylate a neighboring CaMKII domain and thereby remove the CaM binding dependence for activity [12]. Effects of CaMKII phosphorylation can be controversial and the mechanistic details of CaMKII interactions and how phosphorylation modifies the target protein function are often unclear or unknown.
The small molecule KN93 and the autocamtide-2-related inhibitory peptide (AIP) are commonly used in experiments for inhibiting CaMKII activity. KN93 has been one of the most widely used inhibitors for investigating in vivo function of CaMKII [13]. This CaMKII inhibitor is also often used in cellular assays as it has the advantage of being membrane permeable and readily soluble in DMSO at high concentrations (up to 10 mM). Until recently it had been generally accepted that KN93 inhibits CaMKII function by competing with CaM for the kinase CaM binding site [14]. This assumed CaMKII-KN93 interaction is believed to block CaM from binding to CaMKII and prevent the release of the catalytic kinase domain, thereby inhibiting phosphorylation activity. KN93 has a relatively high degree of inhibition specificity amongst kinases, however several reports note additional interactions with other ion channels [13]. Given the awareness of KN93 modifying the function of other cellular targets, a control molecule (KN92) is often used to provide information about these off-target interactions. KN92 is very similar to KN93 with the main difference between the two molecules being a change in stereo chemistry at a central nitrogen and the addition of a 2-hydroxyethyl to the sulfonamide nitrogen (Figure 1). The alterations in the structure of the KN92 molecule are presumed to prevent KN92 from binding in the CaMKII-CaM binding site.
Figure 1.

Structures of a) KN93 (CAS No. : 139298–40-1) and b) KN92 (CAS No. : 1431698–47-3). AIP peptide sequence: KKALRRQEAVDAL.
AIP is a short peptide derived from the CaMKII auto-phosphorylation site. It contains a Thr to Ala mutation that blocks the ability of CaMKII to phosphorylate itself and thereby prevents release of the catalytic domain used for phosphorylation of targets. AIP is regarded as being a more specific and potent inhibitor of CaMKII compared to KN93 [15]. Complete inhibition of CaMKII activity can be achieved at 1 μM of AIP; where as for KN93,10 μM is often used. Interestingly, an IC50 value (concentration that achieves 50% inhibition) of 20 μM has been reported for KN93 [15], suggesting that under the reported conditions even 10 μM may not achieve complete CaMKII inhibition. By contrast the AIP IC50 was found to be 40 nM [15]. Despite these advantages, AIP is often a less attractive approach compared to KN93, as it is not readily membrane permeable which can limit the types of experiments that can be performed.
Structurally both KN93 and KN92 contain three hydrophobic rings that are spaced ~4 and ~8 Å apart. We noticed that these hydrophobic rings structurally resemble hydrophobic side chains of amino acids that CaM often interacts with in the presence of Ca2+. Given the controversial results regarding many CaMKII findings we posited that CaM may interact with the KN93 and KN92 molecule in the presence of Ca2+. To our surprise we found that CaM interacts with both KN molecules. Moreover our resulting structure of the CaM-KN93 complex is such that it could render CaM unavailable for interaction with other proteins. We then investigated CaM-modulation of several CaMKII modulated cardiomyocyte proteins using both KN93 and AIP. Given the large number of proteins that CaM and CaMKII interact with, we have focused our initial investigation to a small subset of CaM/CaMKII targets; the cardiac sodium channel (NaV1.5), and the ryanodine receptor (RyR2).
In the literature, the effects of CaM and CaMKII on NaV1.5 and RyR2 are starting to reach consensus. Our recent report [16] identified a direct role for CaM in modulating NaV1.5 function. Specifically we found that CaM engages part of the channel required for gating with high affinity, and impairment of this interaction increased the time required for channels to recover from inactivation [16]. Others have shown that CaMKII can also directly modulate NaV1.5 function. When activated by CaM, CaMKII can phosphorylate parts of the NaV1.5 αsubunit [17], and mice containing a phosphomimetic mutation display an enhanced late NaV1.5 current [18]. CaM and CaMKII also both directly interact with RyR2 to modulate channel function. Phosphorylation of RyR2 by CaMKII enhances spark-detected Ca2+ leak [19]. Conversely direct binding of CaM to RyR2 has the opposite effect and reduces spark-detected Ca2+ leak [20]. The detailed mechanisms for how these RyR2 processes occur are complex and still the subject of active inquiry [21]. Stabilization of the CaM-RyR2 interaction is being explored and developed as a potential therapeutic approach for treating arrhythmia [22].
Here we present the structure of the CaM-KN93 complex and demonstrate that KN93 alters NaV1.5 and RyR2 function in a manner that is consistent with impairment of CaM modification through different mechanisms. Taken together our results provide structural insight into the CaM-KN93 interaction and highlight appropriate considerations for future investigations that will enhance our understanding of Ca2+ signaling in excitable cells.
2. RESULTS
2.1. CaM engages multiple KN93 molecules using both domains
NMR resonance frequencies are sensitive to the local electronic environment. Changes in resonance frequency (known as chemical shift perturbations) can be used to identify binding sites or conformational changes. In the presence of Ca2+, isotopically enriched 15N CaM displays a well-dispersed 15N-1H HSQC NMR spectrum, where each cross peak corresponds to an amide proton from the CaM backbone or side chain. Upon the addition of the small molecule KN93 (dissolved in DMSO) the majority of the cross-peaks were affected (Figures 2a -left, and S1a). Both the CaM -N and -C domains, as well as the linker that connects them displayed numerous chemical shift perturbations (Figure S1g). No changes to the spectrum were observed during a control titration with DMSO alone (Figure S1d), indicating that the effects are due to CaM interacting with KN93. Next we examined the very similar KN92 control molecule. We found that KN92 also affects the CaM NMR spectrum as many of the cross peaks disappear. However, unlike KN93, a new set of peaks did not appear with further addition of a KN92 (Figure S1b). Both the KN93 and KN92 molecules contain three aromatic rings (Figure 1) that likely are the source of interaction with the hydrophobic side chains of CaM that are accessible in the Ca2+ saturated state. We anticipated that CaM would not engage either of these molecules in the absence of Ca2+. Indeed, in the absence of Ca2+ neither KN93 nor KN92 had any effect on the 15N-1H HSQC CaM spectrum (Figures S1e, and S1f). By contrast no changes to the spectrum were observed in the presence of AIP (1 μM), indicating that there is no interaction between CaM and AIP (Figure S1c).
Figure. 2. Biophysical characterization of CaM-KN93 interaction.

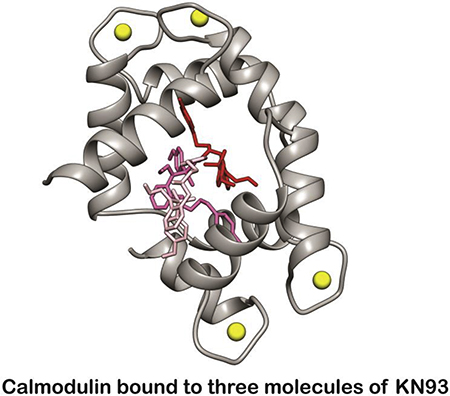
a) Overlay of isotopically enriched CaM 15N-1H HSQC spectra in the absence (black) and presence of KN93 (red) (1:2 molar ratio CaM : KN93) (left). Overlay of 1D 1H NMR spectra filtered by 15N-CaM during substoichiometric titration (right). Spectra display reductions in peak intenisty follow by the emergence of a new set of peaks as well as resonance shifting, consistent with the presence of multiple exhange processes. b) Quin 2 fluorescence data from stopped-flow measurements of CaM in the absense (black) and presence of KN93 (red). Ca2+ release rates for CaM in the presence of TFP (blue) or KN93. A fast and slow component are observed, consistent with properties of the CaM -N and -C domains. Error bars are shown as standard error measurements. Replicate values are shown in Table S2. c) Crystal structure of CaM engaging three molecules of KN93 (pink, purple, and green) (PDB ID 6M7H). Side views of the individual CaM-C domain (dark grey) and -N domain (light grey).
Having established that Ca2+ saturated CaM interacts with both KN93 and KN92, we wanted to understand if this finding was relevant for interpretation of experimental CaMKII data. Given that CaM displayed numerous chemical shift perturbations upon the addition of KN93 and KN92, we performed sub-stoichiometric titrations to gain insight into the CaM-KN93 and KN92 affinities. For high affinity interactions, NMR spectra often display a reduction in peak intensity followed by the emergence of a second set of signals (slow exchange). For modest affinity interactions cross-peaks typically broaden and disappear (intermediate exchange). Weaker binding events can produce shifts in the cross peaks during the course of the titration (fast exchange) [23]. As shown in Figure 2a -right, titrating KN93 into CaM initially resulted in a decrease in peak intensity. Further titration of KN93 gave rise to a second population of cross peaks (Figure S2) and, titrating beyond a 2:1 molar ratio resulted in sharpening and shifting of cross peaks (Figures 2a -right and S2). We interpreted these results as CaM binding two molecules of KN93 with moderate to high affinity (consistent with intermediate and slow exchange processes), and a potential third KN93 molecule interacting with weaker affinity. Titrating with KN92, we observed the peak width to broaden and disappear, consistent with an intermediate mode of exchange and a modest affinity. Addition of up to 15-fold molar excess KN92 did not yield any new cross peaks.
2.2. KN93 stabilizes Ca2+ binding to the CaM EF hand loops
The presence of several exchange modes complicates quantifying the overall CaM-KN93 affinity with our NMR data (we note that at a 1:0.5 CaM: KN93 stoichiometry we did not observe the presence of two populations of peaks that typically allow for Kd determination of high affinity interactions using a ZZ exchange NMR experiment). To gain insight into the apparent affinity of the CaM-KN93 interaction we used stopped-flow measurements to probe the Ca2+ release rate from CaM. This approach was particularly appealing as it is an orthogonal technique compared to NMR, yet similar to NMR it also does not require mutagenesis or attachment of a probe to CaM. When CaM binds a target protein the positions of the CaM helixes are reoriented and this in turn affects the EF hand amino acids that CaM uses for binding Ca2+. In this way CaM Ca2+ affinity can be altered up or down by an order of magnitude depending on the binding target [24]. For our experiments, quin-2 Ca2+ indicator was rapidly mixed with Ca2+-CaM and the fluorescence signal was monitored over time. This process was repeated in order to determine CaM Ca2+ release rates for a range of KN93 concentrations. The individual measurements each displayed a fast and slow component (Figure 2b and Table S1), consistent with our reported Ca2+ release rates for the CaM -N and -C domains respectively [25]. Sequential additions of KN93 resulted in slowing of both the fast and slow components. Plotting the KN93 concentration against the Ca2+ release rate allowed for fitting the data in a dose response manner, which provided information about the EC50 (concentration required to achieve half of an effect) as well as the number of molecules involved for each observed component (Figure 2b and Table S1). We find that KN93 has an EC50 of 5.1 ± 1.4 μM with a Hill coefficient of −1.5 ± 0.3 for the fast component (we attribute this to the CaM-N domain) and the slow component has an EC50 of 6.3 ± 0.5 with a Hill coefficient of −1.2 ± 0.1 (we attribute this to the CaM–C domain). Because Ca2+ dissociation rates from CaM are highly dependent on temperature and solution conditions [10] we also quantified the effect of another CaM binding molecule (Trifluoroperazine (TFP) to provide a reference for our calculated values. We note that TFP has been used by others for enhancing the removal of Troponin C (highly homologous to CaM) in sarcomere sample preparations [26]. Using TFP we determined a fast component EC50 of 1.9 ± 0.5 |JM with a Hill coefficient of −1.9 ± 0.4, and a slow component EC50 of 0.9 ± 0.2 with a Hill coefficient of −1.0 ± 0.1. Overall, we find that similar to TFP, KN93 engages CaM in a manner that stabilizes Ca2+ binding, but with a slightly weaker EC50.
2.3. The structural underpinnings of the CaM-KN93 complex
To understand the architecture and molecular mechanism of the CaM-KN93 interaction, we crystallized the Ca2+/CaM-KN93 complex (PDB ID 6M7H) (Figures 2c and S3). In the absence of a binding partner, Ca2+/CaM has a high degree of flexibility [5] that hampers the formation of an ordered crystalline lattice. Upon the addition of KN93 we observed the rapid formation of 2D plates for many conditions in the commercially available Hampton PEG ion screen. However, we were unable to phase the native data set using molecular replacement. To overcome this we produced selenium-enriched methionine-labeled CaM. Following optimization of the crystal conditions, the diffraction data resolved to 1.6 Å and the phase was determined using single anomalous dispersion (SAD). Consistent with our NMR data, we observe CaM engaging three molecules of KN93 with density for a Ca2+ ion in each of the CaM EF hand motifs (Figures 2c, 3, and S3). The identity of the Ca2+ ion is supported by the EF hand amino acid side orientations used for coordination. Positions 1, 3, 5, 9 and 12 face inward towards the ion, and density for a water molecule is present at position 7. This structure is also consistent with our stopped flow and NMR data; both CaM domains are involved in the interaction as CaM wraps around to encircle three molecules of KN93 (Figure 2c). Similar to other CaM interactions numerous hydrophobic residues from both the CaM-N and -C domain create hydrophobic pockets and patches for interacting with the substrate (Figure 3). The CaM-N domain hydrophobic pocket is formed by several Phe, Ile, Leu, and Met residues (F19, F68, I27, I63, L32, L39 and M36, M51, M71) and contains the Cl -ring of the KN93 molecule 201 (pink molecule, Figure 3a and b). The other end of KN93 201 (methyoxy-substituted phenyl group) lays along the hydrophobic patch of the CaM-C domain that is created by Phe, Leu, and Met residues (F92, L105, L112 M109, M124, M145) (Figure 3f and i). A second molecule of KN93 (202 - purple) sits adjacent to KN93 (201 -pink) in an antiparallel manner. The Cl - ring from KN93 202 is embedded in a CaM-C domain hydrophobic pocket (F92, F141, I100, I125, M124, and M145) (Figure 3f and g) while the KN93 202 methyoxy-substituted phenyl group lays along the CaM-N domain hydrophobic patch (F12, F19, F68, M71 M72, and M76) (Figure 3a and d). These crystal contacts are validated in solution by our NMR data, where the CaM backbone amide resonances of these Phe, Ile, Leu, and Met residues are broadened and / or significantly shifted in the presence of KN93 (Figure 3e and j). The third molecule of KN93 (203 -green) sits more towards the outside of the hydrophobic CaM cavity, sandwiched in-between the two CaM domains (Figure 3a and f). Only a single modest Leu side chain from the CaM-N domain, and a few Met amino acids from the CaM-C domain (L18, M109, M124 and M144) are utilized for this interaction (Figure 3c and h). The deeply nested rings of KN93 molecules 201 (pink) and 202 (purple) into the respective CaM -N and -C domain pockets suggest these KN93 molecules interact with higher affinity compared to the third molecule KN93 203 (green). This is consistent with our sub-stoichiometric NMR titration data where we observe line broadening (intermediate exchange) and appearance of a second set of peaks (slow exchange) during addition of approximately the first two molar equivalents of KN93, and more significant resonance shifting (fast exchange) for additions beyond a 2:1 stoichiometry (Figures 2a and S2). We note that the presence of three molecules of KN93 in our crystal structure is consistent with the Hill coefficients determined from our stopped-flow titration data. Given CaM’s limited side chain contacts to the peripheral KN93 binding site (203-green); we anticipate this third site is likely unoccupied in solution at KN93 concentrations (~10 μM) typically used for experimental investigation of CaMKII activity.
Figure 3. Numerous hydrophobic amino acids from both the CaM-N and -C domains are involved in binding three molecules of KN93.

a) & f) Surfaces of CaM-N and -C domains color coded by hydrophobicity (ranging from yellow = hydrophobic to red = hydrophilic). KN93 molecules are shown in pink, purple and green, b-d), and g -i) Side chains of amino acids that are within 5 Å of the KN93 molecules are shown as licorice and labeled. e) & j) Overlay of CaM cross peaks (2D 15N-1H HSQC NMR) corresponding to CaM amino acids at the KN93 binding interface in the absence (blue) and presence (red) of KN93. NMR spectra and corresponding chemical shift perturbation map are shown in Figure S1.
Several crystal structures of CaM bound to TFP have been reported. LigPlot analysis for our CaM-KN93 structure and the available CaM-TFP structures are shown in Figures S4 and S5. The CaM-TFP structures highlight CaM’s ability to bind small hydrophobic molecules in different orientations with different stoichiometry. In a similar manner, other CaM-KN93 binding orientations and stoichiometries are likely possible, and we anticipate these are influenced by the experimental conditions. Overall we note that the CaM-KN93 interactions involve the majority of CaM’s hydrophobic amino acids that are commonly utilized for interaction with proteins (Figure 2).
2.4. KN93 impairs direct CaM modulation of NaV1.5
Having established that CaM engages KN93 in a manner that could render CaM unavailable, or disrupt interaction with other proteins, we set out to investigate the effects of KN93 on several CaM modulatory functions. Our recent work demonstrated that in the presence of Ca2+, CaM directly modifies two properties of cardiac NaV channel (NaV1.5) function [16]. This is accomplished by a high affinity interaction between CaM and the NaV1.5 channel inactivation gate (IG) (Kd = 66 nM). In our previous report we used structure guided mutations to impair the CaM-IG interaction, which resulted in impaired recovery from inactivation and enhanced kinetics of inactivation [16]. Intriguingly we find that the addition of 10 μM KN93 (concentration most commonly reported for many biochemical assays) has similar effects on wild type NaV1.5 function (Figures 4a, b, and S6).
Figure. 4. KN93 affects NaV1.5 function similar to structure guided mutations that impair the CaM-NaV-IG interaction.

a) Recovery from fast inactivation and b) inactivation kinetics in presence of DMSO control (black) or 10 μM KN93 (red). In the presence of KN93 recovery from fast inactivation was impaired (τ = 16.5 ± 3.4 ms), compared to the control group (τ = 5.8 ± 0.9 ms; P=0.004). c) Recovery from fast inactivation and d) inactivation kinetics in the absence (filled circle) and presence of 1 μM AIP (open circle).
To understand if these effects arise form alterations to the CaM-IG interaction, we again turned to NMR to characterize the CaM-IG complex in the presence of KN93. Addition of KN93 to isotopically enriched 15N CaM that was bound to an IG peptide construct resulted in numerous chemical shift perturbations corresponding to amino acids located in both the CaM - N and -C domains (Figure 5 and S7). Close inspection of our CaM-IG structural model [16] and the CaM-IG-KN93 spectra revealed that many of these differences are for CaM amino acids used for interaction with IG -site A as well as -site B (Figure 5a, b, and c). We note that many of the CaM amino acids at the IG binding interfaces are buried in hydrophobic interaction and are not directly accessible for interaction with KN93 (Figure 5b). Therefore we conclude that the observed change to these CaM resonance frequencies (Figure 5c) arise from modification of the local environment of these amino acids; i.e. the chemical shift perturbations establish that KN93 changes how CaM interacts with the IG construct. To test if KN93 removes CaM from the NaV1.5-IG (analogous to TFP enhanced removal of CaM from skinned smooth muscle [27]), we compared the NMR spectra of CaM-IG-KN93 with CaM-KN93. We find that the NMR spectra do not match (Figure S7). Because KN93 modifies the CaM-IG complex, but does not strip CaM away from the IG, we conclude that KN93 forms a ternary complex with the CaM-IG proteins (CaM-IG-KN93). We note that our previous structural characterization revealed CaM does not rigidly bind to the IG. There is flexibility between the two CaM domains as well as in-between the IG construct binding sites, which could allow for modification of CaM interaction without complete displacement of the CaM-IG proteins [16].
Figure 5. KN93 alters CaM’s interaction with the NaV1.5 IG.

a) Our structural model of CaM bound to the cardiac sodium channel inactivation gate [16]. CaM’s surface is color coded according to hydrophobic properties of amino acids side chains, ranging from hydrophobic (yellow) to hydrophilic (red). b) Structural features of the CaM-IG binding interfaces. Important NaV1.5 IG amino acids at each binding interface are labeled (blue) and corresponding hydrophobic CaM side chains that are within 5 Å are shown as licorice. c) Overlay of (2D 15N-1H HSQC NMR) cross peaks of CaM amino acids at the CaM-IG interface in the absence (blue) and presence (red) of KN93. NMR spectra and corresponding chemical shift perturbation map are shown in Figure S7.
As a control, and to ensure that the changes in NaV1.5 recovery from inactivation or kinetics of activation were not influenced by CaMKII inhibition, we investigated NaV1.5 function in the presence of AIP. No differences to either recovery from inactivation or the kinetics of inactivation were observed with the addition of 1 μM AIP (Figure 4c and d). This is consistent with a (1) phosphomimetic Glu mutation or (2) an Ala mutation at the CaMKII site (S571) having no effect on NaV1.5 inactivation kinetics or recovery from inactivation [18]. Based on our data we conclude that KN93 disrupts direct CaM modulation of NaV1.5 by altering the CaM-IG interaction.
2.5. KN93 enhances RyR2 Ca2+ leak
Next, we tested whether KN93 altered CaM’s interaction with another ion channel, the cardiac ryanodine receptor (RyR2). RyR2 is an intracellular ion channel responsible for releasing Ca2+ from the sarcoplasmic reticulum (SR) into the cytosol. During diastole, RyR2 releases Ca2+ in small aliquots detected spectroscopically as sparks. Hence RyR2 activity can be monitored by confocal microcopy in real time by measuring the rate and size of Ca2+ sparks. Spark-detection of Ca2+ release is often used as an index of Ca2+ leak. Based on Ca2+-spark measurements, phosphorylation of RyR2 by CaMKII at S2814 enhances basal RyR2 spark leak [19]. On the other hand, binding of CaM to RyR2 reduces the frequency of sparks [20]. The structural mechanisms of how CaM binding modulates RyR2 activity are not known, but it appears that RyR2 needs to be phosphorylated either at residue 2808 (PKA / CaMKII site) or S2814 (CaMKII site) for CaM to reduce spark frequency [21].
To test if KN93 impairs CaM modulation of RyR2 channels, we utilized mouse cardiomyocytes containing a homozygous phosphomimetic S2814D mutation, which maximizes the inhibitory efficacy of CaM on RyR2. The cell membrane was permeabilized with saponin and cytosolic [Ca2+] was clamped at 100 nM. The bath solution for both groups contained 100 nM CaM (estimated free CaM concentration in cytosol), as well as 1 μM AIP to ensure that CaMKII function was inhibited and did not influence the results. KN93 increased spark frequency by 33% compared to the DMSO control (Figures 6 and S8). Neither the spark amplitude nor spark mass were altered (Figure 6b). The increase in spark frequency resulted in an elevation in spark-detected Ca2+ leak. Since spark frequency can be increased by higher [Ca2+] in the SR, we next measured SR Ca2+ content by application of caffeine. No significant differences in SR Ca2+ content were found between the two groups (Figure S8). Taken together, these results indicate that KN93 increases RyR2 channel activity in cardiomyocytes, possibly by interfering with CaM’s ability to inhibit of RyR2 channels.
Figure. 6. KN93 increases RyR2-mediated Ca2+ spark activity in cardiomyocytes.

a) Representative Ca2+ sparks recorded from mouse permeabilized cardiomyocytes containing the homozygous RyR2 S2814D phosphomometic mutation in the presence of DMSO (control) or KN93. The bath solution contained 1 μM AIP to block CaMKII effects. b) Properties of Ca2+ sparks. Complete data sets are shown in Figure S8.
2.6. KN93 does not sequester CaM from RyR2
Given that the CaM-KN93 interaction relies on a significant amount of CaM’s hydrophobic residues that are typically used for protein-protein interactions, we investigated if KN93 could enhance removal of CaM from RyR2 (similar to TFP removal of CaM in skinned smooth muscle preparations [27]. To test our hypothesis and probe the KN93 mechanism of enhanced diastolic Ca2+ leak, we investigated CaM-RyR2 co-localization in the absence and presence of KN93. For this, we used suramin, which is known to displace CaM from RyR2 [28], however the mechanistic details for this process are unknown as the suramin RyR binding site has yet to be elucidated. Fluorescently-labeled (Alexa 568) CaM (A-CaM) was continually washed over RyR2 (transfected into REx 293 cells) and the A-CaM fluorescence of the cells was monitored over time in the absence and presence of KN93 followed by application of suramin (Figure 7a). No differences in A-CaM fluorescence were observed during the application of KN93 compared to the control. For groups that were exposed to KN93, the fluorescence intensity of CaM decreased more rapidly upon the addition of suramin compared to the control, however we note that this could due, in part to, variability in suramin diffusion or mixing. With the application of suramin, a similar decrement in intensity was observed irrespective of KN93 treatment. These results indicate that, similar to the NaV1.5-CaM interaction, KN93 does not sequester CaM from RyR2, i.e. prevent CaM binding to RyR2 channels.
Figure. 7. KN93 does not alter the CaM-RyR2 interaction.

a) Alexa 568 labeled CaM (A-CaM) washed over T-REx 293K cells transfected with amino fused RyR2-GFP in the absence and presence of KN93. Application of suarmin dissociates CaM, thereby allowing for quantification and normalization. Error bars are shown as SEM. b) Overlay of 2D 15N 1H HSQC NMR spectra of CaM-RyR2 peptide complex in the absence (black) and presence of KN93 (red). Overlays of the NMR spectra are shown in both directions to highlight the lack of changes upon addition of KN93. c) A-CaM binding to intact RyR2 isolated from pig heart preparations. RyR2 was bound to FRET donor labelled FKBP12.6. For attaching Alexa 568 to CaM a single Cys was substitued at residues T26 or at T34, to ensure that the CaM mutation and the labelling position did not alter the results.
2.7. KN93 does not alter the NMR spectra of CaM bound to a RyR2 peptide
To determine if KN93 disrupts CaM RyR2 modulation by altering or perturbing the CaM-RyR2 binding interface we investigated the effects of KN93 on the CaM -RyR2 peptide (RyR2p) complex using NMR. Using a peptide corresponding to the RyR2 human sequence derived from an alignment with the CaM-RyR binding site [29] we collected 2D 15N-1H HSQC NMR spectra of the CaM complex in the absence and presence of KN93. The addition of up to 10 fold molar excess KN93 had no effect on the CaM-RyR2p spectra. To confirm this result we prepared a sample of CaM-KN93 complex and then titrated in the RyR2p. The resulting spectrum was indistinguishable from that of CaM-RyR2p in the absence of KN93 (Figure 7b).
2.8. KN93 does not alter CaM binding to full-length RyR2 channels
Given that CaM has been shown to interact with RyR2 peptides using multiple conformations [30], we wanted to determine if KN93 altered CaM binding to full-length RyR2. Fluorescently labeled CaM (Alexa 568, (A-CaM)) was titrated into full-length functional RyR2 (in native SR membranes isolated from pig hearts) that was decorated with the RyR2 accessory protein FKBP12.6 labelled with Alexa 488 donor (D-FKBP) (Figure 7c). The FRET signal between D-FKBP and A-CaM was monitored as previously described [31,32]. As previously shown, FKBP and CaM bind at RyR2 sites that are well within FRET range to each other [33]. Accordingly, we have detected strong FRET between D-FKBP and A-CaM in isolated SR samples [31,32] and cardiomyocytes [34]. Here too, we observed strong FRET between D-FKBP and A-CaM bound to RyR2. No significant differences in the FRET signal were observed during titrations of KN93 in either the absence or presence of 30 nM or 30 μM Ca2+. Based on our extensive investigation of CaM binding to RyR2, we conclude that KN93 alters RyR2 function by a mechanism that does not involve direct disruption of the CaM-RyR2 interaction.
3. DISCUSSION
CaMKII experimental results and interpretation of data often vary between laboratories. Understanding these discrepancies and controversies can be problematic as the structure and function of CaMKII are highly complex and the mechanistic details for many CaMKII interactions unknown. Moreover, the details for how the addition of a phosphate group modifies the target protein function are often lacking or unclear. A wide array of inhibitors are available for probing CaMKII modulation and regulation, with the most commonly used being the small molecule KN93 and the peptide AIP. We questioned if differences in the molecular mechanisms of these two inhibitors could explain some of the inconsistencies and controversies in the CaMKII and CaM literature.
A recent investigation challenged the widely accepted mechanism of KN93-CaMKII inhibition by demonstrating that KN93 lacks affinity for CaMKII [35]. Consistent with our previous work they confirmed that KN93 can directly interact with CaM in the presence of Ca2+ [35,36]. Moreover it is intriguing that genetically ablated CaMKII models do not directly recapitulated phenotypes that are always consistent with ascribed CaMKII activity [37–39]; suggesting that CaMKII data interpretation is more complex than previously thought. Given the overwhelming number of investigations pertaining to CaM and CaMKII regulation, these findings highlight a knowledge gap that merits our investigation of the molecular underpinning of the CaM-KN93 interaction and its physiological implications.
In this study, we utilized NMR spectroscopy, x-ray crystallography and stopped flow kinetic measurements to quantify the structure and biophysical properties of the CaM-KN93 interaction. We find that in the presence of Ca2+, CaM utilizes an extensive amount of its hydrophobic residues to wrap around and engage three molecule of KN93, while no interaction is observed between CaM and AIP.
We recently demonstrated that CaM engages a peptide corresponding to the sodium channel inactivation gate (IG) with high affinity (Kd = 66 nM) [16]. Intriguingly, this is two orders of magnitude greater than the EC50 that we determined for the CaM-KN93 interaction using stopped-flow kinetics; yet our NMR data clearly demonstrate that KN93 is able to alter CaM’s interaction with the IG construct. While these data initially appear to conflict, a deeper understanding of CaM interaction and consideration of kinetics provide a rationale. Certain CaM interactions (such as NaV1.5 IG-CaM) derive their high affinity from a cooperation of weaker CaM -N and -C domain interactions [40]. The central helix that connects the two CaM domains is unstructured in solution [41] and functions as a flexible tether that links the two CaM domains together [42]. Similar to troponin C, the binding of one CaM domain can pre-localize the other CaM domain in close proximity to a second binding site, and thereby generate a high affinity interaction (μM × μM = nM) [43,44]. Typically, protein-protein dissociation constants (Kd) are dependent on koff rates, as kon is often assumed to be diffusion limited for small spherical proteins, such as an isolated CaM domain. However, because CaM domains are tethered together, pre-localization does not allow an individual domain to diffuse far away. This enhances kon, and therefore smaller Kd values (koff/kon) (high affinity) can be obtained with faster koff rates [43,45]. We speculate that the dynamics of some CaM interactions allow the small KN93 molecule (599 Daltons) to intercalate into a CaM domain-binding interface, while others do not. We suggest that specific CaM domain lifetimes are likely a more accurate parameter for evaluating if KN93 can interject and disrupt a CaM interaction. As our data show, not every CaM-protein interaction will be disrupted by KN93. The structural features of each CaM interaction as well as the experimental conditions should be considered when evaluating if and how a particular CaM modulated process will be affected by KN93.
We chose to investigate the functional consequences of the CaM-KN93 interaction on two independent systems; NaV1.5 CaM Facilitated Recovery from Inactivation (CFRI)[46], and RyR2 Ca2+ leak. We chose NaV1.5 because the structural details and mechanisms of Ca2+ modulation by direct CaM binding and CaMKII phosphorylation have been well characterized [16,18]. We recently demonstrated that NaV1.5 channels with impaired CaM-IG binding display (1) faster kinetics of inactivation (~33% faster) and (2) require more time to recover from inactivation (~ 270% longer). Here we find that KN93 has a similar effect on recovery from inactivation for wild type NaV1.5 channels (~ 280 % longer), but does not alter the kinetics of inactivation. This is consistent with our NMR data and can be explained by the method used for disrupting the CaM-IG complex. In both cases we measure the function of NaV1.5 in the presence of fixed 1 μM free [Ca2+] and therefore CaM is bound to the IG during both channel inactivation and recovery from inactivation processes. In our previous report, structure-guided NaV1.5 mutations reduced the CaM-IG affinity and allowed the IG to more rapidly undergo conformation changes during the channel inactivation process. For wild-type NaV1.5 channels in the presence of KN93, our NMR data reveal that KN93 does not remove CaM from the IG. Instead it forms a ternary complex (Ca2+-CaM-IG-KN93) (Figure S7). This alteration impairs CaM from facilitating recovery from inactivation, but it does not remove CaM from the IG and therefore does not enhance the rate of the inactivation process.
Modification of voltage gated sodium channel function by calcium is a topic of significant inquire and controversy [46,47]. Given that both CaM and CaMKII have been reported to modify NaV1.5 function; (yet if and how these processes are related remains unknown and requires further investigation) consideration of the CaM-KN93 interaction will be important for future investigations aimed at understanding the role(s) of CaM-NaV and CaMKII-NaV interactions in cardiomyocyte physiology and pathophysiology.
Characterization of the RyR2 system was appealing given its prominent role in disease, and because the effects of CaM binding and CaMKII phosphorylation are known, while the structural and mechanistic details or these processes remain to be resolved. Functionally the addition of KN93 to permeabilized cardiomyocytes containing a CaMKII phosphomimetic mutation (2814D) enhances the frequencies of spark-detected Ca2+ release, thereby suggesting that KN93 could interfere with the CaM-RyR2 interaction(s). Our data indicate that this KN93 effect on RyR2 involves a mechanism that is independent of disrupting the CaM-RyR2 interaction.
The lack of CaM-RyR2 complex disruption by KN93 result is a particularly important result as it underscores the necessity for empirical data to determine if KN93 directly disrupts a CaM interaction. A recent study reports that the RyR2 channel must be phosphorylated by CaMKII in order for CaM to exert an inhibitory effect, however, the same study implicitly indicates that CaMKII phosphorylation does not alter the mode of CaM binding or the affinity of CaM for RyR2 [21]. These results suggest the presence of a complex network of allosteric signaling used for transmitting structural changes imparted by CaM-RyR2 binding to the RyR2 channel pore. It is well noted that KN93 can interact with many proteins, (particularly ion channels) and it will require further investigation to determine if KN93 modifies RyR2 Ca2+ leak by: 1) inhibiting allosteric communication between the CaM binding sites and the channel pore, 2) disrupting other RyR2 accessory protein interaction(s), and / or 3) interacting directly with the RyR2 channel and modifying gating independent of CaM and / or CaMKII modulation.
In summary many of the proteins that are modified by CaM and CaMKII (such as NaV1.5 and RyR2) have important roles in normal physiology, and dysfunction of these proteins, or the proteins that modulate or regulate them have been associated with disease. Ca2+ modulation by CaM and CaMKII proceeds through a series of complex interactions and affect numerous proteins and processes through the entire body. This study starts foundational work that should stimulate further investigations towards determining the full implications of the CaM-KN93 interaction and its consequences to our understanding of mechanisms of Ca2+ signaling.
4. Online Methods
4.1. Protein Production
Unlabeled and isotopically enriched CaM were produced as described previously [48]. The human CALM3 gene was sub-cloned into a pET15b vector. Proteins were expressed in BL21 (DE3) cells and purified by hydrophobic affinity chromatography followed by reverse phase HPLC. Proper protein folding was verified by 2D 15N-1H HSQC NMR spectroscopy. Purity and molecular weight were confirmed by SDS gel electrophoresis and electrospray mass spectroscopy. The NaV1.5 IG peptide (GPGS-QDIFMTEEQKKYYNAMKKLGSKKPQKPIPRPLNKYQGFIFDIVTKQA) was produced as described by [16]. A peptide corresponding to the human sequence of RyR2 (RSKKAVWHKLLSKQRKRAVVACFRMAPLYN) (derived from an alignment with CaM-RyR peptide complex [29]) was purchased from InnoPep.
4.2. NMR spectroscopy
KN93 and KN92 were purchased from calbiochem and dissolved in DMSO. CaM samples were prepared by dialyzing overnight in either: 20 mM BISTRIS, 100 mM KCl, 2 mM CaCl2, pH 6.7 or 50 mM HEPES, 100 mM KCl, 2 mM CaCl2, pH 7.4 Concentration was determined using a Cary100 UV-VIS spectrometer with molar extinction coefficients of ε = 3006 M−1cm−1 for CaM, and 4470 M−1cm−1 for the IG construct. 450 μl samples containing 40 μM isotopically enriched 15N CaM or 15N CaM in the presence of a 1:1 molar equivalence of IG construct were titrated with 0.5 mM and 5 mM stocks of KN93 or 0.5 mM and 2 mM stocks of KN92. Addition of DMSO (greater than 10 fold the amount used for a 1:1 molar ratio) had no appreciable effect on the CaM spectra in the absence or presence of the IG construct. 1D 1H, and 2D 1H-15N HSQC spectra were recorded on a Bruker 800 and 900 MHz spectrometers equipped with triple resonance cryoprobes at 298 K and 310 K. Ca2+ CaM and Ca2+ CaM-IG backbone resonance assignments were transferred from previously determined data sets Johnson et al., 2018 [16]
450 μl of 40 μM isotopically enriched 15N CaM was titrated with the unlabeled RyR2 peptide. Saturation was observed at a 1:2 molar ratio. A second sample at a 1:2 molar ratio of CaM-RyR2 peptide was prepared and titrated up to 10 × molar excess with KN93 (400 μM). The 10 x KN93 spectra contains only a few very subtle changes. Comparing this spectra to a third sample titrated with the same volume of DMSO confirmed that the very subtle alterations arise from the addition of DMSO and are not due to the addition of KN93.
4.3. Stopped-flow kinetic measurements
Ca2+ dissociation kinetics were measured using an Applied Photophysics Ltd. (Leatherhead, UK) model SX.18 MV stopped-flow instrument with a dead time of 1.4ms at 4 ° C. Direct Ca2+ dissociation rates were measured from unlabeled CaM using the fluorescent Ca2+ chelator Quin-2. Solution conditions were 3.0 μM CaM, 10 mM TRIS pH = 7.0, 150 mM KCl, 30 μM CaCl2 and data was collected at 4 °C. Quin-2 was excited at 330 nm with its emission monitored through a 510-nm broad band-pass interference filter (Oriel, Stratford, CT, USA). Each Ca2+ dissociation event rate represents an average of at least two traces. The data were fit with a single or double exponential equation after mixing was complete. The Ca2+ release rates were plotted against the log of the KN93 or TFP concentration and fit to the four parameter variable slope equation: Y=Bottom + (Top-Bottom)/(1+10^((LogEC50-X)*HillSlope)) using prism 5. Four individual titrations were collected for both KN93 and TFP and EC50 values and hill coefficients are reported as the average ± the standard deviation.
4.4. X-ray Crystallography
Selenomethionine (SeMet) substituted CaM was produced in BL21 (DE3) cells using minimal media supplemented with selenomethionine (0.5 g/L) and an amino acid mixture that provides feedback inhibition of methionine synthesis [49]. Selenomethionine CaM (10 mg/mL) was combined with a 10 fold molar excess of KN93 and crystallized by vapor diffusion from a solution of 100 mM Bis-Tris pH 6.5, 25% PEG 3350. Crystals were flash frozen in liquid nitrogen after incubating in cryoprotection solution comprised of the mother liquor supplemented with 20% glycerol. X-ray diffraction data were collected at the Advanced Photon Source at Argonne National Lab, beamline 21-ID-F (LS-CAT) with a X = 0.97872 Å (Table S1). Data were processed with XDS [50], the space group was verified with Pointless and data were scaled with Aimless from the CCP4 software suite. Crystals belonged to space group P6i22 with cell dimensions a = 40.511, b = 40.511, c = 348.785, α = 90°, β = 90°, γ = 120°. The phase was solved using single anomalous dispersion using Autosol. A total of 11 sites were found with a figure of merit = 0.70. After building the CaM model, the Fo-Fc map was inspected to identify 3 KN93 molecules. Several rounds of refinement with Phenix [51] and module building with COOT [52] were required to produce the final model (Rwork = 23.1% Rfree = 27.4%). Validation of the model was confirmed by Molprobity [53]. Coordinates and structure factors were uploaded to the Protein Data Bank under accession code 6M7H.
4.5. Plasmid construct, cell culture and transfection
Plasmid encoding human WT-NaV1.5 was sub-cloned into pRcCMV and co-transfected with a plasmid vector encoding GFP as a fluorescent marker. Cultured cells (tsA201) were transiently transfected with 1.5 μg of plasmid using Lipofectamine™ (Invitrogen) according to the manufacturer’s instructions. Cells were maintained in tissue culture dishes using DMEM with 10% FBS and 1% penicillin-streptomycin. Green fluorescent cells were selected for electrophysiological analysis 24–48 hours after transfection.
4.6. Electrophysiology
Sodium currents were recorded from transiently transfected HEK293T cells at room temperature 24–48 hours after transfection using whole-cell patch clamp. Pipettes were pulled from thin-wall borosilicate glass (OD: 1.5 mm, Warner Instrument Corp., Hamden, CT, U.S.A.) on a P-97 multistage Flaming-Brown micropipette puller (Sutter Instruments, Novato, CA, U.S.A) and fire polished to a resistance between 1.0 and 2 MΩ. The larger pipet size was advantageous for maintaining voltage clamp as well as aiding in delivery of 1 μM auto inhibitory peptide (AIP). All data was collected 5 minutes after cell rupture. Pipette solution contained (in mM): 10 NaF, 100 CsF, 20 CsCl, 20 BAPTA, 10 HEPES, adjusted to pH 7.35 with CsOH. Extracellular (bath) recording solution contained (in mM): 145 NaCl, 4 KCl, 1 MgCl2, 10 HEPES, and 1.8 CaCl2, adjusted to pH 7.35 with CsOH. Series resistance was compensated at 80%. Recovery from inactivation was measured by a standard two-pulse protocol consisting of a depolarizing (−20 mV) 20 ms pulse to engage fast inactivation, followed by a variable duration recovery step to −120 mV and a final test pulse (−20 mV, 20 ms). Channel availability after the end of the recovery interval was normalized to initial values and plotted against the recovery time. The time course of recovery from fast inactivation in the absence or presence of 10 μM KN93 exhibited a mono-exponential time course described by a single time constant (τ). The kinetics of inactivation was assessed by fitting the inactivation of the channel during a 200 ms step to −20 mV. The time course of the fast inactivation in the presence or absence of KN-93 exhibited a double-exponential time course described by two time constant: a fast (τf) and a slow (τs). Data acquisition was carried out using an Axopatch 200B patch clamp amplifier and pCLAMP 10.4 software (Molecular Devices, Sunnyvale, CA). All data were analyzed using pCLAMP 10.0 or Microsoft Excel 2013 and plotted using SigmaPlot 12.5 (Systat Software, Inc., San Jose, CA). Patch-clamp measurements are presented as the means ± SEM. Groups (KN93, control (DMSO) and AIP) were compared using Student’s t-test, with P < 0.05 considered significant.
4.7. Structure of the CaM NaV1.5 IG interaction
We recently published a model of CaM binding to the complete cardiac sodium NaV1.5 channel IG. The final structure was derived by fitting components of two crystal structures (CaM-N domain, (PDB ID 4DJC) and -C domain, (PDB ID 5DBR)) to small angle x-ray scattering data of the full length CaM-NaV1.5 IG complex collected in solution. The resulting structural model satisfied ITC, NMR, SECMALS, and other biophysical data [16].
4.8. Spark measurements in permeabilized cardiomyocytes
Animal use was approved by the Animal Care and Use Committee of Vanderbilt University, USA (animal protocol #M1600259–00) and performed in accordance with NIH guidelines. Ventricular cardiomyocytes were isolated as previously described [54] from 12 to 22 week old C57BL/6 mice containing a RyR2-S2814D homozygous mutation. Cells were permeabilized with saponin (40 μg/mL) for 45 seconds and then bathed for 30 minutes in a freshly-made internal solution (pH = 7.2) containing (mM): K-aspartate (120), KCl (15), K2HPO4 (5), MgCl2 (5.6), HEPES (10), dextran (4% w/v), MgATP (5), Phosphocreatine-Na2 (10), creatine phosphokinase (10 U/mL), reduced L-glutathione (10), EGTA (0.5), CaCl2 (0.12), calmodulin (100 nM), autocamtide-2-related inhibitory peptide (AIP, 1 μM), and Fluo-4 (0.03). The internal solution contained either dimethyl sulfoxide (DMSO) or 10 μM KN-93 in DMSO. Confocal imaging and analysis was performed as previously described [55].
4.9. CaM binding to RyR2 in T-REx 293 cells
Flp-In T-REx-293 cells were transfected with the plasmid containing amino fused GFP-RYR2 cDNA. Cells were cultured on a laminin-coated coverslip and washed with a solution containing (in mM): potassium aspartate 150; MgCl2 0.25; EGTA 0.1; HEPES 10 and pH 7.2. Cells were permeabilized with internal solution containing saponin (0.005%) followed by a wash out with saponin-free internal solution comprised of (in mM): K-aspartate 100; KCl 15; KH2PO4 5; MgATP 5; EGTA 0.35; CaCl2 0.22; MgCl2 0.75; HEPES 10; and 2% dextran (MW: 40,000), (pH 7.2). Free [Ca2+] and [Mg2+] were calculated to be 200 nM and 1 mM respectively. Permeabilized cells were incubated with CaM containing Alexa 568 (A-CaM) (300 nM) dissolved in internal solution for 10 minutes, and then equilibrated with A-CaM (60 nM). The A-CaM signal co-localized with GFP-RyR2, and T-REx 293 cells without RyR2 expression did not display any A-CaM signal, indicating that the majority of A-CaM was bound to RyR2. Chamber solution was replaced with internal solution containing KN-93 (10 μM) and A-CaM (60 nM) for a 10 min incubation. The fluorescence of A-CaM bound to RyR2 was measured with laser scanning confocal microscopy (Radiance 2000 MP, Bio-Rad, UK) equipped with a ×40 oil-immersion objective lens (N.A.=1.3). Background fluorescence was subtracted and bound A-CaM fluorescence was calculated by F (A-CaM) = (F-Fmin)/(F0-Fmin). F0 is an average fluorescence of A-CaM after the equilibration in A-CaM (60 nM). To an obtain Fmin value, cells were treated with internal solution containing 100 μM Suramin. Normalized fluorescence of bound A-CaM was plotted as a function of time. Fractions of the A-CaM dissociated from RyR2 after KN-93 treatment (n=7 cells) were compared with the corresponding values from non-treated cells (n=3 cells). All 2-D images were analyzed with ImageJ software (NIH, USA). Groups were compared using the Student’s t test for unpaired data sets and differences were considered statistically significant at p < 0.05. Statistical analysis and graphical representation of averaged data was carried out on Origin 2016 SR2 software (OriginLab, USA).
4.10. FRET measurements for CaM binding to intact RyR2 channels
SR membrane vesicles were isolated from porcine cardiac ventricle by differential centrifugation of homogenized tissue, as described [56]. All vesicles were flash-frozen and stored at − 80°C. Immediately prior to the fluorescence binding studies, the SR vesicles were stripped of residual endogenous CaM by incubation with 300 nM myosin light chain kinase-derived CaM binding peptide, followed by sedimentation [57]. Cardiac SR (0.5 mg/ml) membranes were pre-incubated with 70 nM AF488-labeled FKBP12.6 (donor, D-FKBP, for 90 min, at 37°C, in a solution containing 150 mM KCl, 5 mM GSH, 0.1 mg/mL BSA, 1μg/mL Aprotinin/Leupeptin, 1 mM DTT and 20 mM PIPES (pH 7.0)). To remove unbound D-FKBP, the SR membranes were spun at 110,000×g for 20 min. A range of [AF568-labeled CaM (acceptor, A-CaM)] was pre-incubated for 30 min at 21°C with 10uM KN93 in a solution containing 150 mM KCl, 5 mM GSH, 0.1 mg/mL BSA, 1μg/mL Aprotinin/Leupeptin, 2 mM DTT, 1mM EGTA, 20 mM PIPES (pH 7.0), and either 0.038 mM CaCl2 to give 30 rM free Ca2+ or 1.02 mM CaCl2 to give 30 μM free Ca2+ (calculated by MaxChelator). Resuspended SR was added to A-CaM assay samples for 1 hr incubation at 21 °C. Samples were loaded onto a 384-well plate, fluorescence decays were acquired, and the analyzed fluorescence lifetime values were used to calculate FRET values in accordance with our previous reports [58,59]
Supplementary Material
Highlights.
Ca2+ /Calmodulin (CaM) binds the CaM dependent kinase II inhibitor (CaMKII) KN93.
KN93 modifies NaV1.5 and RyR2 function independent of CaMKII.
KN93 disrupts a high affinity interaction between NaV1.5 and CaM.
When evaluating KN93 data, targets other than CaMKII need to be considered.
Acknowledgements
We thank Dr. Christopher George (University of Cardiff, UK) for providing cDNA of the human ryanodine receptor type 2 (RyR2) fused with amino-terminal GFP tag (GFP-RyR2). We thank Walter Chazin for provided resources for recombinant protein production and NMR time (NIH R35 GM118089). This work was also supported in part by: the National Science Foundation (NSF) grant HRD-1547757 (S.M.D.), grants for NMR instrumentation from the NSF (0922862), NIH (S10 RR025677), and Vanderbilt University matching funds. Imaging was supported by Vanderbilt University Cell Imaging Shared Resources. NIH grants CA68485, DK20593, DK58404, DK59637 and EY08126. NIH HL138579 (S.G. J.P.D.) and HL127299 (P.B.R.) American Heart Association Postdoctoral Fellowship (16POST31010019) (R.T.R.). NIH grants R01HL092097 and R01HL138539 (R.L.C.) Foundation Leducq (A.L.G., F.P.), NIH T32 NS007491 (D.J.B.), R01 HL130231 (to A.V.Z.) Argonne National Laboratory contract DE-AC02-06CH11357. Use of LS-CAT Sector 21 supported by the Michigan Economic Development Corporation (Grant 085P100817).
ABBREVIATIONS
- AIP
autocamtide-2-related inhibitor peptide
- CaM
calmodulin
- CaMKII
CaM dependent protein kinase type II
- DMSO
dimethylsulfoxide
- HSQC
heteronuclear single quantum coherence
- NaV1.5
voltage gated sodium channel 1.5
- RyR2
ryanodine receptor calcium release channel isoform 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Sorensen AB, Søndergaard MT, Overgaard MT, Calmodulin in a heartbeat., FEBS J. 280 (2013) 5511–32. doi: 10.1111/febs.12337. [DOI] [PubMed] [Google Scholar]
- [2].Kursula P, The many structural faces of calmodulin: a multitasking molecular jackknife., Amino Acids. (2014). doi: 10.1007/s00726-014-1795-y. [DOI] [PubMed] [Google Scholar]
- [3].Bers DM, ed., Excitation-Contraction Coupling and Cardiac Contractile Force, 2002.
- [4].Pitt GS, Calmodulin and CaMKII as molecular switches for cardiac ion channels, 73 (2007) 641–647. doi: 10.1016/j.cardiores.2006.10.019. [DOI] [PubMed] [Google Scholar]
- [5].Barbato G, Ikura M, Kay LE, Bax A, Pastor RW, Backbone Dynamics of Calmodulin Studied by 15N Relaxation Using Inverse Detected Two-Dimensional NMR Spectroscopy: The Central Helix Is Flexible, Biochemistry. 31 (1992) 5269–5278. doi: 10.1021/bi00138a005. [DOI] [PubMed] [Google Scholar]
- [6].Babu YS, Bugg CE, Cook WJ, Structure of calmodulin refined at 2.2 Å resolution, J. Mol. Biol 204 (1988) 191–204. doi: 10.1016/0022-2836(88)90608-0. [DOI] [PubMed] [Google Scholar]
- [7].Sorensen BR, Shea MA, Interactions between Domains of Apo Calmodulin Alter Calcium Binding and, 2960 (1998) 4244–4253. [DOI] [PubMed] [Google Scholar]
- [8].Tsalkova TN, Privalov PL, Thermodynamic study of domain organization in troponin C and calmodulin, J. Mol. Biol 181 (1985) 533–544. doi: 10.1016/0022-2836(85)90425-5. [DOI] [PubMed] [Google Scholar]
- [9].Teleman A, Drakenberg T, Forsén S, Kinetics of Ca2+binding to calmodulin and its tryptic fragments studied by43Ca-NMR, Biochim. Biophys. Acta (BBA)/Protein Struct. Mol 873 (1986) 204–213. doi: 10.1016/0167-4838(86)90047-6. [DOI] [PubMed] [Google Scholar]
- [10].Bayley P, Ahlstrom P, Martin SR, Forsen S, The kinetics of calcium binding to calmodulin: Quin 2 and ANS stopped-flow fluorescence studies, Biochem. Biophys. Res. Commun 120 (1984) 185–191. doi: 10.1016/0006-291X(84)91431-1. [DOI] [PubMed] [Google Scholar]
- [11].Kovalevskaya NV, van de Waterbeemd M, Bokhovchuk FM, Bate N, Bindels RJM, Hoenderop JGJ, Vuister GW, Structural analysis of calmodulin binding to ion channels demonstrates the role of its plasticity in regulation., Pflugers Arch 465 (2013) 1507–19. doi: 10.1007/s00424-013-1278-0. [DOI] [PubMed] [Google Scholar]
- [12].Chao LH, Stratton MM, Lee I-H, Rosenberg OS, Levitz J, Mandell DJ, Kortemme T, Groves JT, Schulman H, Kuriyan J, A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin-dependent kinase II holoenzyme., Cell. 146 (2011) 732–45. doi: 10.1016/j.cell.2011.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pellicena P, Schulman H, CaMKII inhibitors: From research tools to therapeutic agents, Front. Pharmacol 5 February (2014) 1–10. doi: 10.3389/fphar.2014.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H, The newly synthesized selective Ca2+calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells, Biochem. Biophys. Res. Commun 181 (1991) 968–975. doi: 10.1016/0006-291X(91)92031-E. [DOI] [PubMed] [Google Scholar]
- [15].Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H, A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II, Biochem. Biophys. Res. Commun 212 (1995) 806–812. doi: 10.1006/bbrc.1995.2040. [DOI] [PubMed] [Google Scholar]
- [16].Johnson CN, Potet F, Thompson MK, Kroncke BM, Glazer AM, Voehler MW, Knollmann BC, George AL, Chazin WJ, A Mechanism of Calmodulin Modulation of the Human Cardiac Sodium Channel, Structure. 26 (2018) 683–694.e3. doi: 10.1016/j.str.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Herren AW, Weber DM, Rigor RR, Margulies KB ||, Phinney BS, Bers DM, CaMKII Phosphorylation of Na V 1.5: Novel in Vitro Sites Identified by Mass Spectrometry and Reduced S516 Phosphorylation in Human Heart Failure HHS Public Access, J Proteome Res. J Proteome Res 14 (2015) 2298–2311. doi: 10.1021/acs.jproteome.5b00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Glynn P, Musa H, Wu X, Unudurthi SD, Little S, Qian L, Wright PJ, Radwanski PB, Gyorke S, Mohler PJ, Hund TJ, Voltage-Gated Sodium Channel Phosphorylation at Ser571 Regulates Late Current, Arrhythmia, and Cardiac Function In Vivo., Circulation. 132 (2015) 567–77. doi: 10.1161/CIRCULATIONAHA.114.015218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida C, Skapura DG, Sc B, Anderson ME, Donald M, Wehrens XHT, Ryanodine Receptor Phosphorylation by CaMKII Promotes Life-Threatening Ventricular Arrhythmias in Mice with Heart Failure, Circulation. 122 (2010) 2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298.Ryanodine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Walweel K, Oo YW, Laver DR, The emerging role of calmodulin regulation of RyR2 in controlling heart rhythm, the progression of heart failure and the antiarrhythmic action of dantrolene, Clin. Exp. Pharmacol. Physiol 44 (2017) 135–142. doi: 10.1111/1440-1681.12669. [DOI] [PubMed] [Google Scholar]
- [21].Walweel K, Gomez-Hurtado N, Rebbeck RT, Oo YW, Beard NA, Molenaar P, dos Remedios C, van Helden DF, Cornea RL, Knollmann BC, Laver DR, Calmodulin inhibition of human RyR2 channels requires phosphorylation of RyR2-S2808 or RyR2-S2814, J. Mol. Cell. Cardiol (2019). doi: 10.1016/j.yjmcc.2019.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu B, Walton SD, Ho HT, Belevych AE, Tikunova SB, Bonilla I, Shettigar V, Knollmann BC, Priori SG, Volpe P, Radwański PB, Davis JP, Gyorke S, Gene transfer of engineered calmodulin alleviates ventricular arrhythmias in a calsequestrin-associated mouse model of catecholaminergic polymorphic ventricular tachycardia, J. Am. Heart Assoc 7 (2018). doi: 10.1161/JAHA.117.008155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fielding L, NMR methods for the determination of protein-ligand dissociation constants, Prog. Nucl. Magn. Reson. Spectrosc 51 (2007) 219–242. doi: 10.1016/j.pnmrs.2007.04.001. [DOI] [Google Scholar]
- [24].Gaertner TR, Putkey JA, Waxham MN, RC3/neurogranin and Ca2+/calmodulin-dependent protein kinase II produce opposing effects on the affinity of calmodulin for calcium, J. Biol. Chem 279 (2004) 39374–39382. doi: 10.1074/jbc.M405352200. [DOI] [PubMed] [Google Scholar]
- [25].Davis JP, Tikunova SB, Walsh MP, Johnson JD, Characterizing the response of calcium signal transducers to generated calcium transients, Biochemistry. 38 (1999) 4235–4244. doi: 10.1021/bi982495z. [DOI] [PubMed] [Google Scholar]
- [26].Metzger JM, Variations in cross-bridge attachment rate and tension with phosphorylation of myosin in mammalian skinned skeletal muscle fibers. Implications for twitch potentiation in intact muscle, J. Gen. Physiol 93 (2004) 855–883. doi: 10.1085/jgp.93.5.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Van Lierop JE, Wilson DP, Davis JP, Tikunova S, Sutherland C, Walsh MP, David Johnson J, Activation of smooth muscle myosin light chain kinase by calmodulin. Role of LYS30 and GLY40, J. Biol. Chem. 277 (2002) 6550–6558. doi: 10.1074/jbc.M111404200. [DOI] [PubMed] [Google Scholar]
- [28].Sigalas C, Bent S, Kitmitto a, O’Neill S, Sitsapesan R, Ca(2+)-calmodulin can activate and inactivate cardiac ryanodine receptors., Br. J. Pharmacol 156 (2009) 794–806. doi: 10.1111/j.1476-5381.2008.00092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Maximciuc AA, Putkey JA, Shamoo Y, MacKenzie KR, Complex of Calmodulin with a Ryanodine Receptor Target Reveals a Novel, Flexible Binding Mode, Structure. 14 (2006) 1547–1556. doi: 10.1016/j.str.2006.08.011. [DOI] [PubMed] [Google Scholar]
- [30].Her C, Thompson AR, Karim CB, Thomas DD, Structural dynamics of calmodulin-ryanodine receptor interactions: Electron paramagnetic resonance using stereospecific spin labels, Sci. Rep 8 (2018) 1–11. doi: 10.1038/s41598-018-29064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cornea RL, Nitu F, Gruber S, Kohler K, Satzer M, Thomas DD, Fruen BR, FRET-based mapping of calmodulin bound to the RyR1 Ca 2+ release channel, 106 (2009) 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rebbeck RT, Nitu FR, Rohde D, Most P, Bers DM, Thomas DD, Cornea RL, S100A1 protein does not compete with calmodulin for ryanodine receptor binding but structurally alters the ryanodine receptoro·calmodulin complex, J. Biol. Chem 291 (2016) 15896–15907. doi: 10.1074/jbc.M115.713107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cornea RL, Nitu FR, Samsó M, Thomas DD, Fruen BR, Mapping the ryanodine receptor FK506-binding protein subunit using fluorescence resonance energy transfer, J. Biol. Chem 285 (2010) 19219–19226. doi: 10.1074/jbc.M109.066944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Guo T, Fruen BR, Nitu FR, Nguyen TD, Yang Y, Cornea RL, Bers DM, FRET detection of calmodulin binding to the cardiac RyR2 calcium release channel, Biophys. J 101 (2011) 2170–2177. doi: 10.1016/j.bpj.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wong MH, Samal AB, Lee M, Vlach J, Novikov N, Niedziela-Majka A, Feng JY, Koltun DO, Brendza KM, Kwon HJ, Schultz BE, Sakowicz R, Saad JS, Papalia GA, The KN-93 Molecule Inhibits Calcium/Calmodulin-Dependent Protein Kinase II (CaMKII) Activity by Binding to Ca 2+ /CaM, J. Mol. Biol 431 (2019) 1440–1459. doi: 10.1016/j.jmb.2019.02.001. [DOI] [PubMed] [Google Scholar]
- [36].Johnson CN, Gomez-Hurtado N, Kryshtal DO, Pattanayek R, Haynes J, Damo S, Knollmann BC, Sorting out Ca2+ regulation by calmodulin dependent kinase II (CaMKII) and calmodulin, J. Mol. Cell. Cardiol 109 (2017) 33. [Google Scholar]
- [37].Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel-Duby R, Maier LS, Olson EN, The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload., Proc. Natl. Acad. Sci. U. S. A 106 (2009) 2342–7. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Joiner MLA, Koval OM, Li J, Julie He B, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS, Anderson ME, CaMKII determines mitochondrial stress responses in heart, Nature. 491 (2012) 269–273. doi: 10.1038/nature11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Nickel AG, Kohlhaas M, Bertero E, Wilhelm D, Wagner M, Sequeira V, Kreusser MM, Dewenter M, Kappl R, Hoth M, Dudek J, Backs J, Maack C, CaMKII does not control mitochondrial Ca 2+ uptake in cardiac myocytes, J. Physiol 0 (2019) 1–16. doi: 10.1113/JP276766. [DOI] [PubMed] [Google Scholar]
- [40].Bayley PM, Findlay WA, Martin SR, Target recognition by calmodulin: Dissecting the kinetics and affinity of interaction using short peptide sequences, Protein Sci. 5 (1996) 1215–1228. doi: 10.1002/pro.5560050701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Barbato G, Ikura M, Kay LE, Bax A, Pastor RW, Backbone Dynamics of Calmodulin Studied by15N Relaxation Using Inverse Detected Two-Dimensional NMR Spectroscopy: The Central Helix Is Flexible, Biochemistry. 31 (1992) 5269–5278. doi: 10.1021/bi00138a005. [DOI] [PubMed] [Google Scholar]
- [42].James P, Vorherr T, Carafoli E, Calmodulin-binding domains: just two faced or multi-faceted?, Trends Biochem. Sci. 20 (1995) 38–42. doi: 10.1016/S0968-0004(00)88949-5. [DOI] [PubMed] [Google Scholar]
- [43].Siddiqui JK, Tikunova SB, Walton SD, Liu B, Meyer M, de Tombe PP, Neilson N, Kekenes-Huskey PM, Salhi HE, Janssen PML, Biesiadecki BJ, Davis JP, Myofilament Calcium Sensitivity: Consequences of the effective concentration of troponin I, Front. Physiol 7 (2016) 1–14. doi: 10.3389/fphys.2016.00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Van Valen D, Haataja M, Phillips R, Biochemistry on a leash: The roles of tether length and geometry in signal integration proteins, Biophys. J 96 (2009) 1275–1292. doi: 10.1016/j.bpj.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hulme EC, Trevethick MA, Ligand binding assays at equilibrium: Validation and interpretation, Br. J. Pharmacol 161 (2010) 1219–1237. doi: 10.1111/j.1476-5381.2009.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Johnson CN, Calcium modulation of cardiac sodium channels, J. Physiol 0 (2019) 1–12. doi: 10.1113/JP277553. [DOI] [PubMed] [Google Scholar]
- [47].Nof E, Vysochek L, Meisel E, Burashnikov E, Antzelevitch C, Clatot J, Beinart R, Luria D, Glikson M, Oz S, Remme CA, Mutations in Na V 1.5 Reveal Calcium-Calmodulin Regulation of Sodium Channel, 10 (2019) 1–18. doi: 10.3389/fphys.2019.00700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Crotti L, Johnson CN, Graf E, De Ferrari GM, Cuneo BF, Ovadia M, Papagiannis J, Feldkamp MD, Rathi SG, Kunic JD, Pedrazzini M, Wieland T, Lichtner P, Beckmann B-M, Clark T, Shaffer C, Benson DW, Kääb S, Meitinger T, Strom TM, Chazin WJ, Schwartz PJ, George AL, Calmodulin mutations associated with recurrent cardiac arrest in infants., Circulation. 127 (2013) 1009–17. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3834768&tool=pmcentrez&rendertype=abstract (accessed March 27, 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J, Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin, J. Mol. Biol 229 (1993) 105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- [50].Kabsch W, Xds, Acta Crystallogr. Sect. D Biol. Crystallogr 66 (2010) 125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD, Towards automated crystallographic structure refinement with phenix.refine, Acta Crystallogr. Sect. D Biol. Crystallogr 68 (2012) 352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot, Acta Crystallogr. Sect. D Biol. Crystallogr 66 (2010) 486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC, MolProbity: All-atom structure validation for macromolecular crystallography, Acta Crystallogr. Sect. D Biol. Crystallogr 66 (2010) 12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BEC, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K, Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia, J. Clin. Invest 116 (2006) 2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Batiste SM, Blackwell DJ, Kim K, Kryshtal DO, Gomez-Hurtado N, Rebbeck RT, Cornea RL, Johnston JN, Knollmann BC, Unnatural verticilide enantiomer inhibits type 2 ryanodine receptor-mediated calcium leak and is antiarrhythmic, Proc. Natl. Acad. Sci 116 (2019) 4810–4815. doi: 10.1073/pnas.1816685116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Fruen BR, Bardy JM, Byrem TM, Strasburg GM, Louis CF, Differential Ca 2+ sensitivity of skeletal and cardiac muscle ryanodine receptors in the presence of calmodulin, Am. J. Physiol. Physiol 279 (2000) C724–C733. doi: 10.1152/ajpcell.2000.279.3.c724. [DOI] [PubMed] [Google Scholar]
- [57].Fruen BR, Black DJ, Bloomquist RA, Bardy JM, Johnson JD, Louis CF, Balog EM, Regulation of the RYR1 and RYR2 Ca 2+ release channel isoforms by Ca 2+ -insensitive mutants of calmodulin, Biochemistry. 42 (2003) 2740–2747. doi: 10.1021/bi0267689. [DOI] [PubMed] [Google Scholar]
- [58].Stroik DR, Yuen SL, Janicek KA, Schaaf TM, Li J, Ceholski DK, Hajjar RJ, Cornea RL, Thomas DD, Targeting protein-protein interactions for therapeutic discovery via FRET-based high-throughput screening in living cells, Sci. Rep 8 (2018) 1–13. doi: 10.1038/s41598-018-29685-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Schaaf TM, Peterson KC, Grant BD, Bawaskar P, Yuen S, Li J, Muretta JM, Gillispie GD, Thomas DD, High-Throughput spectral and lifetime-based FRET screening in living cells to identify small-molecule effectors of SERCA, SLAS Discov. 22 (2017) 262–273. doi: 10.1177/1087057116680151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.