

Abstract
Current assumption for assessing carcinogenic risk of polycyclic aromatic hydrocarbons (PAHs) is that they function through a common mechanism of action; however, recent studies demonstrate that PAHs can act through unique mechanisms potentially contributing to cancer outcomes in a non-additive manner. Using a primary human 3D bronchial epithelial culture (HBEC) model, we assessed potential differences in mechanism of toxicity for two PAHs, benzo[a]pyrene (BAP) and dibenzo[def,p]chrysene (DBC), compared to a complex PAH mixture based on short-term biosignatures identified from transcriptional profiling. Differentiated bronchial epithelial cells were treated with BAP (100-500 μg/mL), DBC (10 μg/mL), and coal tar extract (CTE 500-1500 μg/mL, SRM1597a) for 48 hrs and gene expression was measured by RNA sequencing or quantitative PCR. Comparison of BAP and DBC gene signatures showed that the majority of genes (~60%) were uniquely regulated by treatment, including signaling pathways for inflammation and DNA damage by DBC and processes for cell cycle, hypoxia and oxidative stress by BAP. Specifically, BAP upregulated targets of AhR, NRF2, and KLF4, while DBC downregulated these same targets, suggesting a chemical-specific pattern in transcriptional regulation involved in antioxidant response, potentially contributing to differences in PAH potency. Other processes were regulated in common by all PAH treatments, BAP, DBC and CTE, including downregulation of genes involved in cell adhesion and reduced functional measurements of barrier integrity. This work supports prior in vivo studies and demonstrates the utility of profiling short-term biosignatures in an organotypic 3D model to identify mechanisms linked to carcinogenic risk of PAHs in humans.
Keywords: Benzo[a]pyrene, Polycyclic Aromatic Hydrocarbons, Toxicogenomics, Mixtures, Bronchial Epithelial Cells, Organotypic Culture
1. Introduction
Polycyclic aromatic hydrocarbons (PAHs) are a chemically diverse class of environmental pollutants found in air, water, and soil, emitted by incomplete combustion of natural and anthropogenic sources, with over 1500 species of substituted and unsubstituted PAHs in the environment. Anthropogenic sources of PAHs include fossil fuel burning, vehicle exhaust, wood burning, and coal-tar pitch and asphalt production (Abdel-Shafy & Mansour, 2016). As such, inhalation is a primary route of exposure to PAHs in ambient air, indoor air, and cigarette and tobacco smoke. Humans are primarily exposed to PAHs as complex mixtures, which are dependent on the amount and type of combustion. Higher molecular weight PAHs (containing 4 or more fused benzene rings) tend to have lower aqueous solubility and greater lipophilicity (Ravindra et al., 2008). These can be found in the particle phase in ambient air. A number of high MW PAHs are linked to carcinogenic and mutagenic effects in humans, including benzo[a]pyrene (BAP; classified by International Agency for Research on Cancer (IARC) as a Class 1 known human carcinogen.) (Benbrahim-Tallaa et al., 2012; IARC, 2010, 2014; Rengarajan et al., 2015) Additionally, dibenzo[def,p]chrysene (DBC), formerly known as dibenzo[a,l]pyrene, is classified as a class 2A probable human carcinogen (IARC, 2010). Both BAP and DBC are environmental carcinogens linked with multiple cancer types, particularly lung and skin (Boffetta et al., 1997).
The U.S. Environmental Protection Agency Integrated Risk Information System’s (IRIS) 2010 release of a relative potency factor (RPF) approach for cancer risk assessment of PAH mixtures provided recommendations for quantitative cancer risk assessment by scaling doses and potency relative to benzo[a]pyrene (BAP), the index carcinogen (EPA, 2010). Because BAP is the index carcinogen to which potency is estimated to be greater or less than, the first assumption of similar toxicological mode-of-action (MOA) must be true for this approach to accurately estimate cancer risk. While some evidence indicates that the mutagenic and tumor-initiating MOA of BAP (involving aryl hydrocarbon receptor-modulated gene expression and metabolic activation to reactive intermediates) is shared by other PAHs, the structural diversity of PAHs provokes concern for assuming similarities in MOA (Jarvis et al., 2014).
The aryl hydrocarbon receptor (AhR) is well established as a key modulator in genotoxic PAH toxicity (W. M. Baird et al., 2005; Moorthy et al., 2015). The MOA involves binding and activating the AhR, translocation from the cytosol to the nucleus, and subsequent binding of xenobiotic response elements. This activates the transcription of many genes, some of which metabolize xenobiotics. For PAHs containing a “bay” and/or a “fjord” region (as is the case for both BAP and DBC), cytochrome P450 (CYP450)-dependent metabolism into diol epoxides is necessary for the genotoxic and mutagenic effects of PAHs. As diol epoxide metabolites, the PAHs are then able to bind DNA and other cellular constituents, which can lead to DNA strand breakage, protein damage, redox cycling, and/or oxidative stress. While the method of PAH toxicity through DNA damage is shared by genotoxic PAHs, the pathways can be markedly different and complex for potentially non-genotoxic carcinogenic PAHs, or complex PAH mixtures. DBC is the most potent carcinogenic PAH studied (in vivo rodent skin tumor assay), with an RPF estimated 30-100 (30-100 times the potency of BAP, RPF of 1) (Siddens et al., 2012). While RPF correlates well with DNA adduct formation, it does not correlate well with tumor incidence for DBC, indicating non-genotoxic mechanisms may contribute to carcinogenesis (Tilton et al., 2015). There is substantial transcriptomics evidence that BAP and DBC can act through unique mechanisms leading to tumor development in mouse skin in vivo (Tilton et al., 2015). Previous work in an in vivo rodent model determined that key mechanisms and pathways able to discriminate between low, medium, and high tumorigenicity in rodent in vivo utilized genes belonging to inflammation signaling, oxidative stress, and cell adhesion and barrier integrity processes.
Toxicity testing in animal models is often resource-intensive, time-consuming, and inconsistently predictive of human responses. While humans and rodents share common cytochrome P450 enzymes for detoxification and elimination of toxicants, the rates of phase I reactions and preferred pathways of phase II reactions vary significantly (Smith, 1991). There is evidence that the CYP450 activities of model organisms used for toxicity (mouse, rat, rabbit, dog, micropig, monkey) do not resemble human CYP450 activity profiles well (Bogaards et al., 2000; Turpeinen et al., 2007). Focusing research efforts towards advancing human cell cultures allows evaluation of human-specific xenobiotic metabolism and responses, and the capacity for high throughput screening and bioassays. Increasingly, 3D cell cultures and organoid culture models are used for toxicological and pharmacological studies for their ability to better recapitulate in vivo-like cellular heterogeneity and physiological response. Their multicellularity, cell-cell interactions and cell-matrix interactions support establishment and maintenance of a cellular microenvironment homeostasis, which may be why cells cultured in 3D tend to exhibit greater resistance to cytotoxic injury (Zavala et al., 2016). Cells cultured in 3D with multiple cell types are found to better withstand oxidative stress rather than monoculture, and the cell-cell interactions and crosstalk allow further toxicological evaluation of processes such as disruptions in cellular adherence, cell migration, differentiation, and healing (Astashkina et al., 2012; Sun et al., 2006).
Organotypic culture of human bronchial epithelial cells (HBEC) offer a physiologically relevant, sensitive model to study the effects of air pollutant chemicals and mixtures in vitro. The human airway epithelium is a major target zone of toxicity for inhaled air pollutants, and the expression of CYP450 metabolizing enzymes in HBECs make them ideal for studying PAH toxicity in vitro. In the present study, we employed the EpiAirway™ bronchial epithelium model, in which cells are differentiated at the air-liquid interface (ALI), to assess toxicity profiles of BAP and DBC, two potent carcinogens commonly found in air pollutant mixtures, in comparison to a PAH mixture. The 3D culture of primary bronchial epithelial cells was shown to be more toxicologically resistant than the 3D culture of immortalized bronchial epithelial cells in tobacco smoke toxicity evaluations (Zavala et al., 2016). In response to environmental toxicants, HBEC differentiated at the ALI exhibit degradation of tight junctions, decreased cell viability, and compensatory or protective responses at sub-toxic concentrations, including increased mucus secretion, and goblet cell hyperplasia and hypertrophy similar to clinically documented changes in the bronchial epithelium of smokers (Balharry et al., 2008; Innes et al., 2006). Transcriptional signatures were profiled short-term after treatment with BAP and DBC using a systems biology approach in a differentiated primary 3D HBEC model. Functional epithelial barrier integrity and cytotoxicity were also evaluated and compared to transcriptional data. Next-generation sequencing techniques provided a rapid, effective method to uniformly detect thousands of changes in the HBEC model after short-term PAH treatment.
2. Materials and Methods
2.1. Chemicals and Reagents
Cell culture media and phosphate buffered saline (PBS) were provided by MatTek Corporation (Ashland, MA). Benzo[a]pyrene (BAP; CAS# 50-32-8) and dibenzo[def,p]chrysene (SBC; CAS# 189-64-0) were purchased from MRIGlobal (Kansas City, MO). Coal Tar Extract,SRM 1597a,(CTE) was purchased form the National Institute of Standards & Technology (Gaithersburg, MD.) DNase I, TRIzol® reagent, Superscript® III First Strand Synthesis System, qPCR primers, and Pierce™ LDH Cytotoxicity Assay Kit were from Thermo Fisher Scientific (Waltham, MA). 2X SsoAdvanced™ Universal SYBER®Green Supermix was purchased from BioRad Laboratories, Inc. (Hercules, CA.)
2.2. Tissue Culture and Treatments
Primary HBEC differentiated on transwell inserts at the ALI (EpiAirway™ 100, MatTek, Ashland, MA) were transferred to 6-well plates each well containing 1 ml of assay medium and equilibrated for 24 hours at 37°C, 5% CO2 followed by a change of fresh medium before treatment. Inserts were washed with phosphate buffered saline (PBS, pH 7.4) and then treated with PAHs in acetone vehicle (n=4 per treatment) on apical surface for up to 48 hrs, BAP (1-500 ug/ml), DBC (1-50 ug/ml) and CTE (250 -1500 ug/ml). Dosing was chosen based on relative potency in BAP equivalents (BAPeq) for DBC (~50 BAPeq) and CTE (0.4 BAPeq) as previously reported (Siddens et al., 2012; Siddens et al., 2015). At the end of each exposure regimen, the matrix and tissue from each well insert was carefully peeled away with forceps, placed in cryovials containing 0.5ml TRIzol® reagent and snap frozen in liquid nitrogen. Frozen tissues were stored at −80°C until further analysis. Basal media was transferred to clean, sterile tubes and stored at −80°C for evaluation of cytotoxicity.
2.3. Histology
HBEC cultures were washed briefly with phosphate-buffered saline (PBS, pH 7.4) and fixed in 10% neutral buffered formalin for 48 h. The membranes were excised from the culture inserts with a surgical blade, fixed in 10% formalin, processed and embedded in paraffin. For staining and immunohistochemistry, 4 to 5-μm-thick sections were cut, mounted on slides, and deparaffinized by processing through a series of xylene and ethanol solutions. Sections were stained with hematoxylin and eosin (H&E) and analyzed by light microscopy using a Nikon Eclipse E400. Deparaffinized sections of cell culture membranes also were stained for tumor protein 63 (p63), proliferation marker protein Ki-67 (Ki67), and mucus-producing goblet cells identified by periodic acid Schiff’ s (PAS) staining. For Ki67 staining, paraffin sections were high-temperature antigen retrieved with BDTM Retrieval A solution (Dako). For Ki67 and p63 staining, endogenous peroxidase activity was blocked by immersing slides in methanol containing 3% hydrogen peroxide for 10 minutes. The following primary anti-human antibodies were applied for 30 minutes at room temperature: rabbit polyclonal antiserum against human p63 (1:100; PA5-36069; ThermoFisher, Rockford, IL) and Ki-67 (1:20; PA5-16785; ThermoFisher, Rockford, IL). MaxPoly-One Polymer HRP Rabbit Detection solution (MaxVision Biosciences, Bothell, WA) was applied for 7 minutes at room temperature and Nova Red (SK-4800; Vector Labs, Burlingame, CA) as chromagen was used with Dako hematoxylin (S3302) as counterstain. Serial sections of formalin-fixed paraffin-embedded cell culture membranes incubated with Dako Universal negative serum served as negative controls. Mucus-producing goblet cells were identified by periodic acid Schiff’ s (PAS) staining. Slides were mounted and examined by light microscopy. For immunofluorescence, membranes were fixed in ice-cold MeOH + Acetone and incubated with primary antibody (Monoclonal anti-human gastric Mucin-5B (MUC5B), sigma M5293, 1:250; monoclonal anti- β- tubulin, Sigma T7941, 1:500) overnight at 4° C, following with second antibody (1:200; Anti-mouse IgG (H+L), F(ab')2 Fragment (Alexa Fluor® 647 Conjugate, Cell Signaling Technology). The inserts were removed and mounted on slides using a Prolong Gold Antifade Reagent® (Thermos Fisher) for imaging with a Zeiss LSM 780 confocal microscope.
2.4. Transepithelial Electrical Resistance (TEER)
Transepithelial electrical resistance (TEER) measurements were made using an epithelial volt-ohmmeter (EVOM2, World Precision Instruments, Sarasota, FL). The EVOM2 was calibrated using a test electrode prior to the measurements. At time zero and 48 hrs (n=4) after treatment with BAP (1-500 ug/ml), DBC (1-50 ug/ml), CTE (250-1500 ug/ml), PBS, pH 7.4, was added to both apical and basal chambers and resistance was measured (ohms) for each insert. An empty culture insert was used to correct for the background resistance. Four cultures were used for each treatment concentration and time point. Percent TEER of control were calculated by subtracting background from TEER, then calculating the difference between 48 hr TEER minus background and 0 hr TEER minus background. Treatment effects on TEER were evaluated for significance (p<0.05) by one-way ANOVA with Dunnett’ s multiple testing correction.
2.5. Cell Viability
Lactate dehydrogenase (LDH) leakage was measured in media after treatment with PAHs (n=4) for 48 hrs using Pierce LDH Cytotoxicity Assay Kit (Thermo Scientific) following manufacturer instructions. The 48 hr timepoint was the longest PAH exposure period tested in HBEC, during which media is not changed. Briefly, basal medium samples (40μl) were aliquoted into the wells of a 96-well plate. LDH reaction reagent (40μl) was added to each sample and incubated at room temperature for 30 minutes while protected from light. Finally, 40μl of stop solution was added to each well and mixed. LDH activity was determined by subtracting absorbance at 680nm (background) from absorbance at 490nm (Synergy HTX plate reader, BioTek, Winooski, VT). A negative control of fresh cell culture medium, a positive control of medium from lysed cells, and vehicle only controls were included. Cytotoxicity was evaluated by one-way ANOVA with Dunnett’s multiple testing correction (p<0.05).
2.6. RNA Isolation and Quality Control
Total RNA was isolated from HBEC (n=4) using TRizol® reagent and was quantified on a Synergy HTX plate reader equipped with a Take3 module (BioTek). RNA quality was evaluated based on 260/280 ratio (acceptable range 2.0-2.1) and by examining 18S and 28S peaks (Bioanalyzer 2100, Agilent, Santa Clara, CA). Acceptable RNA quality was based on RIN ≥ 8.5.
2.7. mRNA-Sequencing and Analysis
Total RNA from HBEC treated with BAP (500 ug/ml; 19.8 nmol), DBC (10 ug/ml, 0.35 nmol) and vehicle control (n=4 per treatment) for 48 hr were sent to the Oregon State University Center for Genome Research and Biocomputing Core facilities for library preparation and sequencing. mRNA was poly-A selected, and libraries were prepared with the PrepXTM mRNA and Illumina sequencing workflow (Wafergen Biosystems, Fremont, CA). Paired-end sequencing (150 bp) was conducted with an Illumina HiSeq 3000 sequencer, which yielded ~30 million reads per sample. Sequenced reads were first put through Cutadapt (version 1.8.1) to trim adapter sequences from the paired-end reads. The human genome assembly GRCh38.84 was indexed using bowtie2-build (version 2.2.3) while the transcriptome was indexed using TopHat (version 2.1.1.) (Trapnell et al., 2009). TopHat was used again to align the trimmed reads to indexed transcriptome and genome (Trapnell et al., 2010). Differential expression was determined in CuffDiff (version 2.2.1) compared to vehicle control (Nookaew et al., 2012). The differentially expressed gene lists (q<0.05) for oppositely regulated, commonly regulated, and uniquely regulated genes between BAP and DBC were used for pathway enrichment analysis in MetaCore (Clarivate Analytics, Philadelphia, PA). Statistical significance of over-connected interactions was calculated using a hypergeometric distribution, where the p value represents the probability of a particular mapping arising by chance for experimental data compared to the background (Nikolsky et al., 2009). Heatmaps were generated in MultiExperiment Viewer TM4 (Saeed et al., 2003). Network visualizations were generated in Cytoscape (v3.5.1) (Shannon et al., 2003).
2.8. Quantitative PCR (qPCR)
cDNA was synthesized using a Superscript® III First Strand Synthesis Supermix kit per manufacturer’s instructions. Reactions were diluted 1:5 with nuclease –free water and stored at - 80°C until used for qPCR. A BioRad Laboratories, Inc. (Hercules, CA.) CFX96 Touch™ Real-Time PCR Detection System was used for running 20μ1 qPCR reactions to survey key gene targets. Each reaction contained 2μ1 cDNA template (10ng RNA), 150 nM of each primer, 10 μl 2X SsoAdvanced™ Universal SYBER®Green Supermix, and nuclease-free water. A list of primer sequences is reported in Supplemental Table 1. The thermocycler was programmed for 1 cycle 95°C for 1 minute initial denaturing, 40 cycles 95°C for 15 sec denaturing, 60°C for 30 sec annealing/elongation, and a melt curve 65–95°C/0.5° per 5 sec for validating single product amplification. The relative expression differences among treatments were calculated using the ΔΔCt comparative method and normalized to housekeeping genes beta-actin (ACTB) and peptidylprolyl isomerase A (PPIA). Genes significantly regulated by PAH treatment (p<0.05), including BAP (100-500 ug/ml), DBC (5-10 ug/ml) and CTE (500-1500 ug/ml) for 6-48 hr (48 hr only for CTE), were identified by one-way ANOVA with Tukey’s multiple testing correction.
3. Results
3.1. Evaluation of differentiated HBEC cultured at ALI
The morphology of differentiated HBEC cultures was evaluated by histological methods prior to treatments. H&E staining of tissue sections indicated that cultures were fully differentiated into a pseudostratified mucociliary epithelium showing goblet cells and ciliated cells along the apical side (Fig. 1A). Epithelial cells were distinguished by cell-specific markers. Immunohistochemistry was used to visualize Ki67, which is a marker of actively proliferating cells identified exclusively on the basal side, (Fig 1B) and p63, which is a marker of basal epithelial cells (Fig 1C). Mucus producing goblet cells were visualized on the apical side with PAS stain (Fig. 1D). MUC5B (marker of glandular mucous cells) and B-tubulin (ciliated respiratory cells) were visualized by immunofluorescence (Fig. 1E). Cells treated with BAP, DBC and CTE at concentrations ranging 1-1500 ug/ml applied to the apical surface for 48 hours resulted in no increase in toxicity as measured by release of LDH into media (Fig. 2).
Figure 1. Morphological characterization of primary 3D human bronchial epithelial cells (HBEC) in culture.
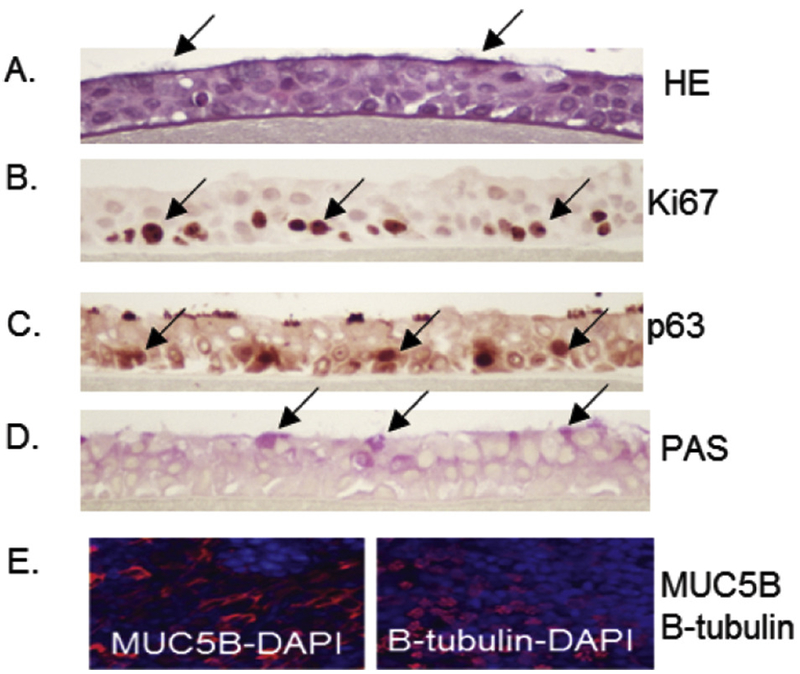
HBEC cultures were fixed in 10% formalin and tissue sections were stained with H&E (A) to observe ciliated cells, Ki67 (B) for actively proliferating cells, p63 (C) for basal cells, and PAS (D) for goblet cells. These features are noted by arrows showing positively stained cells in panels A-D. (E) Immunofluorescence of MUC5B and β-tubulin (red with DAPI counterstain in blue) as markers of glandular mucous cells and ciliated respiratory cells, respectively.
Figure 2. Cell viability measured by lactate dehydrogenase (LDH) leakage in cell media 48 hrs after exposure to PAHs.

Values are presented as LDH leakage in media for treated cells compared to acetone vehicle control (% vehicle control ± standard error). LDH leakage from HBEC treated with benzo[a]pyrene (BAP), dibenzo[def,p]chrysene (DBC) and coal tar extract (CTE) for 48 hr were not significantly different (p>0.05) from vehicle control at any concentration (one-way ANOVA).
3.2. Global gene expression analysis
We investigated the effects of BAP and DBC treatment on 3D HBEC on the molecular level by analyzing global gene expression patterns using RNAseq. Raw and normalized sequencing files are available online at NCBI Gene Expression Omnibus (GSE128471). Of the more than 60,000 transcripts mapped, there were 3486 statistically significant (q<0.05). A heatmap of 3486 differentially expressed genes (DEG) was generated using an unsupervised hierarchical clustering analysis (Fig. 3A). BAP treatment resulted in 2244 DEG and DBC treatment resulted in 2126 DEG compared to vehicle control. The dendrogram shows that biological replicates for each treatment cluster together indicating the variance between treatments is greater than the variance measured within replicates for each treatment. Among the total DEG, about three-quarters of genes were unique to each treatment, and not differentially regulated by the other treatment (1360 DEG were unique to BAP, and 1242 DEG were unique to DBC). About a quarter of the DEG (884) were common between BAP and DBC as shown by the Venn diagram in Fig. 3B. Gene expression analysis supports the hypothesis that while BAP and DBC function through a subset of commonly regulated genes and pathways, each also have chemical-specific profiles that contribute to toxicity in human bronchial epithelial cells.
Figure 3. HBEC transcriptional response to BAP and DBC.

Global gene expression was measured in HBEC 48 hrs after treatment with 500 ug/ml (19.8 nmol) benzo[a]pyrene (BAP) and 10 ug/ml (0.35 nmol) dibenzo[def,p]chrysene (DBC) by RNA sequencing. (A) Bidirectional hierarchical clustering by Euclidean distance of genes differentially expressed (q<0.05) by BAP and DBC compared to vehicle control. Values are log2 fold change for all treatments compared with control; red, green, and black represent increased, decreased and unchanged gene expression, respectively. (B) Venn diagram showing overlap of significantly regulated (q <0.05) genes by BAP and DBC in HBEC. (C) Functional enrichment of gene processes in HBEC using MetaCore network processes (GeneGo) for genes commonly regulated by BAP and DBC (q<0.05). Black bars represent functions for genes up-regulated and gray bars represent functions for genes down-regulated in common by BAP and DBC (q<0.05). The dashed line indicates the threshold for significance (p<0.05).
3.3. Barrier function toxicity as a common mechanism of toxicity in HBEC by BAP and DBC
To further analyze the treatment responses between BAP and DBC for mechanistic similarities and differences, the DEG were analyzed for significantly enriched pathways. Genes regulated in common by BAP and DBC, which means they were significantly different from control (q<0.05) for both treatments, were split into upregulated and downregulated subsets for analysis (Fig. 3C). Overall, pathways associated with cell cycle and nuclear receptor signaling were significantly enriched for genes increased by both BAP and DBC, while many pathways related to cell adhesion and barrier function were significantly enriched for decreased gene sets. In fact, pathways related to barrier integrity were some of the most significantly enriched pathways affected by BAP and DBC treatment compared to control and broadly include pathways associated with cell adhesion, epithelial-to-mesenchymal transition, TGF-beta signaling and cytoskeleton. Genes specifically associated with three cell adhesion pathways (Cell adhesion_Cell junctions, Cell adhesion_Cadherins, and Cell adhesion_Integrin-mediated cell-matrix adhesion) were compiled into a gene list related to barrier function and visualized in a heatmap (Fig. 4A). Most genes associated with these pathways were significantly decreased (q<0.05) in common by BAP and DBC treatment compared to control and support functional measurements of barrier integrity in HBEC (Fig. 4B). One pathway (nuclear receptor transcriptional regulation) was significant for both the increased and decreased gene sets (compared to control) indicating this pathway, which broadly includes a number of nuclear signaling pathways, was significantly enriched by both datasets.
Figure 4. Decrease in barrier integrity of HBEC after treatment with BAP and DBC.

(A) Bidirectional hierarchical clustering by Euclidean distance of differentially expressed genes associated with gap junction and tight junction signaling (q<0.05) after 48 hr treatment with benzo[a]pyrene (BAP) and dibenzo[def,p]chrysene (DBC) compared to vehicle control. Values are log2 fold change for all treatments compared with control; red, green, and black represent up-regulated, down-regulated and unchanged genes, respectively. (B) Transepithelial electrical resistance (TEER) measured in HBEC 48 hr after treatment with BAP, DBC and coal tar extract (CTE). Values are TEER (Ω-cm2) normalized to vehicle control (scaled to 100%). *Indicates significant reduction in TEER (p<0.05; one-way ANOVA with Tukey’s pairwise comparison). Expression of genes associated with barrier integrity were measured by quantitative PCR in HBEC after 48 hr treatment with BAP and DBC (C) and CTE (D), respectively. Values are expressed as fold change (Log2; mean ± SE) compared to vehicle control. Asterisks indicate significance compared to vehicle control (*p<0.05; **p<0.001; one-way ANOVA with Tukey’s pairwise comparison).
TEER in HBEC was measured as a functional indicator of barrier integrity 48 hours after chemical treatment and results are normalized to vehicle control (Fig. 4B). A significant decrease in TEER (p<0.05) was observed at the highest concentrations tested for BAP and DBC (500 ug/ml and 50 ug/ml, respectively) indicating a loss of cell barrier integrity. The effects of BAP and DBC on barrier integrity were compared to CTE as a mixture of PAHs that includes both BAP and DBC (Siddens et al., 2012). The dose-response range for BAP, DBC and CTE were broadly overlapping based on reported relative potency with DBC ranging 30-100 BAPeq and CTE 0.4 BAPeq (Siddens et al, 2012; 2015). HBEC were more sensitive to barrier function toxicity by CTE, which resulted in significant decreases in TEER (66-73%) at concentrations as low as 500 ug/ml. While the reduction in TEER from BAP and DBC treatment correlate with a decrease in gene expression for barrier function as measured by RNASeq, specific gene expression biomarkers of tight junction and gap junction integrity were significantly decreased at concentrations lower than those that significantly reduced TEER suggesting that gene expression may be a more sensitive endpoint or there may be a threshold associated with gene level changes that contribute to functional integrity measures. (Figs. 4C and 4D). Specifically, tight junction protein 2 (TJP2) and gap junction 1 (GJA1) were decreased following 48 hour exposure to BAP and DBC at concentrations lower than those required for functional impairment of barrier integrity as measured by TEER (Fig. 4C). Coal tar extract treatments also significantly (p<0.05) downregulated a number of barrier genes by qPCR, including TJP2, tight junction protein 1 (TJP1), GJA1, and claudin 1 (CLDN1). These genes were chosen based on relevance to tight junction and gap junction structures and BAP/DBC RNASeq results. Overall, these data show that the BAP, DBC and CTE all inhibit barrier integrity mediated by down regulation of gap junction and tight junction processes.
3.4. BAP and DBC uniquely regulate transcriptional targets of AHR and NRF2 in HBEC
Significantly enriched pathways (p<0.05) were also analyzed for DEG uniquely up and down regulated by BAP and DBC (Fig. 5). These data highlight a number of important biological processes that are regulated in a chemical-specific manner in HBEC. For example, BAP uniquely upregulates pathways related to cell cycle, hypoxia and oxidative stress while processes upregulated by DBC were related to inflammation and DNA damage signaling. DBC uniquely down-regulated genes from mitochondrial apoptosis pathways and the initiation and elongation processes of translation. BAP uniquely downregulated several pathways that DBC did not, notably the inflammation, IL-10, IL-2 signaling pathways and cell cycle G1-S regulation. BAP also downregulated several pathways related to barrier integrity that were observed to also be important for DBC (Fig. 3C).
Figure 5. Genes uniquely regulated by BAP and DBC in HBEC.

Global gene expression was measured in HBEC 48 hrs after treatment with 500 ug/ml (19.8 nmol) benzo[a]pyrene (BAP) and 10 ug/ml (0.35 nmol) dibenzo[def,p]chrysene (DBC) by RNA sequencing. Venn diagram shows genes uniquely regulated (q <0.05) by BAP and DBC in HBEC. Statistical enrichment of biological network processes (MetaCore) is shown for genes uniquely up-regulated (right-panel) and down-regulated (left-panel) by BAP (light gray bars) and DBC (dark gray bars). The dashed line indicates the threshold for significance (p<0.05).
Given the importance of oxidative stress as a mechanism of PAH toxicity, we focused on this pathway, which was only enriched in BAP, for confirmation by qPCR. Further, we noticed a subset of genes significantly regulated in opposite directions by BAP and DBC that were associated with oxidative stress and antioxidant signaling processes. In general, these genes were upregulated by BAP and down regulated by DBC and included phase I and II metabolizing enzymes and known transcriptional targets of the arylhydrocarbon receptor (AHR) and nuclear factor erythroid 2–relatedfactor 2 (NRF2). Dose-dependent CYP1A1 and CYP1B1 induction were observed in HBEC after treatment by BAP and CTE for time points ranging 6-48 hr (Fig. 6A). However, during the same time range, DBC significantly decreased CYP1A1 and CYP1B1 in a dose-dependent manner (Fig. 6A). This unique pattern of response for CYP1A1 AND CYP1B1 by BAP and DBC, which is consistent with that previously published in mouse epithelium (Siddens et al., 2012), was confirmed using overlapping concentrations for BAP and DBC in a subsequent experiment (Supplemental Figure 1). Similar results were observed for NAD(P)H quinone dehydrogenase (NOOl), aldehyde dehydrogenase 3 family member A1 (ALDH3A1), and glutathione S-transferase alpha (GSTA) in which dose-dependent increases were measured 48 hr after BAP and CTE treatment (by RNAseq and qPCR), while genes were either downregulated or not significantly changed (by qPCR or RNAseq) after treatment with DBC (Fig. 6B). One notable exception is the downregulation of GSTA by CTE, while BAP increased expression of this gene supporting that PAHs and PAH mixtures result in exposure-specific gene expression profiles in lung cells.
Figure 6. Unique regulation of AHR- and NRF2-mediated genes by BAP and DBC in HBEC.

(A) Expression of CYP1A1 and CYP1B1 was measured by quantitative PCR in HBEC 6-48 hrs after treatment with BAP, DBC and CTE (48 hr only). (B) Expression of NQO1, ALDH3A1 and GSTA was measured by quantitative PCR in HBEC 48 hrs after treatment with BAP, DBC and CTE. (C) Transcription factor signaling networks for AHR and NRF2 in HBEC after treatment with 500ug/ml (19.8 nmol) BAP (right, red) and 10 ug/ml (0.35 nmol) DBC (left, green). Target nodes are up-regulated by BAP (red) and down-regulated by DBC (green). For panesl A and B, values are expressed as fold change (Log2; mean ± SE) compared to vehicle control. Asterisks indicate significance compared to vehicle control (*p<0.05; **p<0.001; oneway ANOVA with Tukey’s pairwise comparison). For panel C, as indicated in the legends, the size of nodes is associated with significance (larger is more significant) and the color intensity represents magnitude of response (darker is larger response) for BAP and DBC compared to vehicle control.
The extent of the differences in AHR and NRF2 signaling by BAP and DBC in HBEC is represented in the transcription factor signaling network in Fig. 6C. The 3486 DEG were filtered for genes significantly regulated in opposite directions by BAP and DBC. These genes were uploaded into Metacore for network building, and we found that NRF2 and AHR are major hubs connecting genes that are oppositely regulated by BAP and DBC treatment. Using MetaCore’s Interactome tool, an Interactions by Protein function analysis returned the following results for overconnected objects in the active dataset: the top 5 overconnected genes for BAP unique DEG list (up/down) were CAMP responsive element binding protein 1 (CREB1), androgen receptor (AR), MYC proto-oncogene (MYC), RELA proto-oncogene (RELA), and estrogen receptor 1 (ESR1). The top 5 overconnected genes for the DBC unique DEG list (up/down) were MYC, CREB1, YY1 transcription factor (YY1), tumor protein 53 (TP53), ESR1. While they differ slightly in top interconnected genes, it is evident that many of the uniquely regulated genes are interconnected with common hub genes, or transcription factors (MYC, CREB1, RELA.) After filtering the DEG list to include only genes significantly regulated (q<0.05) by either BAP or DBC opposite to the other treatment, the Interactome Interactions by Protein analysis was rerun and returned the following top 5 overconnected genes: NRF2, kruppel like factor 4 (KLF4), RELA, cystatin B (CSTB), TP53. These genes, along with AHR, were added to build a network (Fig 6C) of oppositely regulated genes. Based on the top 5 overconnected gene networks, the opposite regulation suggests that NRF2 and KLF4 may play a role in modulating BAP and DBC response in a way that is unique between the two PAH treatments.
4. Discussion
This study employed a toxicogenomics approach for comparison of transcriptional differences between BAP and DBC in a 3D HBEC model to evaluate chemical biosignatures after exposure and inform known differences in toxicity to human airway epithelium. We have previously identified PAH-specific biosignatures in mouse skin epithelium in vivo linked to carcinogenic risk for PAHs and mixtures (Tilton et al., 2015). The EpiAirway™ bronchial epithelial model was chosen for its ability to recapitulate in vivo phenotypes of tissue structures, cellular responses, signaling, and functional barriers. The complexity of this model makes it an ideal tool to study perturbations in the complex and dynamic processes involved in the mechanisms of PAH toxicity. Global gene signatures for BAP and DBC were further compared to transcriptional biomarkers in HBEC cells after treatment with CTE, a complex mixture of PAHs, and biological and functional changes in cells. BAP and DBC treatment in the bronchial epithelium resulted in over three thousand genes significantly altered between these two treatments. While a quarter of the genes were regulated in common between these two PAHs, the majority of genes are uniquely regulated by one PAH and not the other. A key strength of this study design is the ability to compare the response of thousands of genes in the human bronchial epithelial model to identify chemical-specific mechanisms and pathways.
4.1. PAH-related barrier integrity alteration
While transcriptional profiling highlighted major differences in regulated pathways and processes between BAP and DBC, the results also supported a degree of similarities between the PAHs. A quarter of the genes significantly regulated were regulated in common between BAP and DBC, and these genes were present in translation, inflammation, cell adhesion, and regulation of cell proliferation processes (Fig 3C). These shared pathways have been reported in studies assessing PAH transcriptional profiles in other models including the zebrafish and Mutamouse, notably processes pertaining to cell adhesion, inflammation, and translation (Goodale et al., 2013; Labib et al., 2016). These commonly enriched pathways by different single PAHs across models and tissues supports the hypothesis that PAHs can share subsets of commonly regulated genes and pathways, yet ultimately function through chemical-specific bioactivity signatures that can contribute to their different toxicities in the target tissues.
In the present study, functional and gene expression assays were used in order to evaluate barrier function integrity after exposing HBECs to various PAH treatments. In pathway enrichment analysis, the pathways related to barrier integrity were some of the most significantly enriched pathways affected by BAP and DBC treatment compared to control. The majority of genes in the barrier integrity gene heatmap were downregulated by BAP and DBC treatment, supporting barrier integrity dysregulation as a shared mechanism of action between BAP and DBC, including epithelial to mesenchymal transition and cell adhesion. Key genes involved in cell adhesion and epithelial mesenchymal transition have also been reported in PAH-treated HepG2 cells as well, indicating that short-term PAH treatment can result in hundreds of different genes expressed, between PAHs also of varying potencies, yet can share the traits of dysregulating cell junctions and promoting cellular migration (Song et al., 2011).
PAHs have been found to decrease barrier function integrity, which in turn can lead to dysregulated inflammation and oxidative stress (Devalia et al., 1997; Schamberger et al., 2014). Regulation of tight junction and gap junction proteins is essential in maintaining homeostasis in the lungs. Tight junction structures comprised of transmembrane proteins and plaques maintain tissue permeability and intercellular adhesion (Cao et al., 2015; Schamberger et al., 2014). Some of these structural proteins, particularly the occludins, have been found to be redox-sensitive in their assembly and breakdown (Blasig et al., 2011). Elevated oxidative stress and pro-inflammatory cytokines can also disrupt tight junction function, leading to increased airway inflammation (Coyne et al., 2002). Because dysregulation of tight junctions are associated with asthma and lung cancer, barrier integrity is evaluated as a target of PAH treatment and potential predictor of toxicity (Cao et al., 2015). Tight junction downregulation tends to be correlated with TEER reductions, as well as reductions in cingulin, claudins, and other barrier proteins (Cao et al., 2015).
Trans-epithelial electrical resistance (TEER) measurements were taken to indirectly measure tight junction functional integrity and assess epithelial layer disruption. We found significantly reduced TEER measurements (p<0.05) by treatments of BAP, DBC, and CTE. The tight junction and gap junction markers TJP2 and GJA1 were also found to be downregulated by treatment to BAP and DBC, and correlated with reductions in TEER at those doses. Similarly, CTE treatment resulted in the significant downregulation of TJP1, TJP2, GJA1, and CLN1. The resulting epithelial barrier dysfunction from BAP, DBC, and CTE exposure, measured through TEER and qPCR, further support this as a mechanism of action likely to be shared by PAHs.
4.2. Uniquely regulated hub genes and pathways between BAP and DBC treatment may be the key to understanding DBC’s heightened carcinogenic potential
Through global gene expression analysis, a total of 2602 transcripts/genes were identified to be uniquely regulated between BAP and DBC. Approximately half of these genes were uniquely regulated by BAP, while the other half was uniquely regulated only by DBC. Because BAP and DBC exhibit such large differences in their tumorigenic potency in vivo (Siddens et al., 2012) and RPF estimate (IARC, 2010), we further investigated the specific roles of the uniquely regulated genes to gain insight on potential contributors to DBC’s mechanism of action.
Through the pathway enrichment analysis, we found that BAP and DBC each uniquely regulated subsets of pathways, further indicating that these chemicals may act through dissimilar mechanisms of action. There is mounting evidence that PAHs can have individual chemical signatures that can be used to discriminate chemicals by their properties, including genotoxicity and bioactivity (Goodale et al., 2013; Tilton et al., 2015; van Delft et al., 2004). To date, there are a small but valuable number of studies analyzing the transcriptional profiles of PAH exposures on a number of models. These studies have found that across the developing zebrafish, mouse lung, mouse spleen, mouse skin, and currently the human airway epithelium, PAHs continue to exhibit unique transcriptional profiles with subsets of common, but often unique, enriched pathways and transcription factors (Chepelev et al., 2016; Goodale et al., 2013; Tilton et al., 2015; White, 2002). In the present study using a human bronchial epithelial model, we found that while BAP uniquely upregulates pathways related to hypoxia and oxidative stress, and DBC uniquely upregulated pathways related to inflammation and DNA damage signaling (Fig. 3C). These enriched pathways are a result of the over 1200 genes that are uniquely regulated by one PAH treatment, but not the other.
Through hub gene network building, we found shared and unique top over-connected genes (hub genes) for the BAP and DBC unique and oppositely regulated gene lists (Fig6C, 6D). The shared hub genes were CREB1, RELA, and MYC, while BAP notably had AR as a unique hub gene and DBC had TP53 as a unique hub gene. After filtering for significance, the top overconnected genes included NRF2, KLF4, and TP53. The network visualizations built relate two key points: BAP and DBC are regulated by a number of similar transcription factors that are known regulators of cell cycle, proliferation, and oxidative stress responses. All these processes related to cancer; however they are not regulated the same way. The genes are oppositely regulated by the same transcription factors, indicating chemical-specific processes occurring at the transcriptional level that are unable to be explained by what is currently known about classic PAH mechanisms of action. These data are consistent with unique differences for BAP and DBC reported in mouse skin epithelium in vivo and include processes previously identified as predictive of PAH carcinogenic risk suggesting that PAH-specific gene regulation linked to cancer outcome are maintained in human bronchial epithelial cells in vitro (Larkin et al., 2013; Siddens et al., 2012; Tilton et al., 2015).
4.3. DBC dysregulates NRF2-mediated oxidative stress detoxification mechanisms
The mechanisms of action of BAP have been researched extensively, resulting in a wealth of knowledge on its effects in in vitro and in vivo systems (William M. Baird & Mahadevan, 2004; Phillips et al., 2008; Tung et al., 2014). BAP toxicity requires a series of CYP-dependent metabolic activation steps to become toxic metabolites, which then damage cellular constituents, including proteins and DNA through adduct formation and redox cycling (Rengarajan et al., 2015; Tung et al., 2014). The findings of this study support the hypothesis that DBC may act through dysregulation of oxidative stress detoxification mechanisms, particularly by downregulating NRF2 targets. The nuclear factor-erythroid 2-related factor-2 (NRF2) is a transcription factor plays major roles in cellular antioxidant defense by regulating Phase II detoxification genes and activating protective antioxidant responses in the cell. Targets of NRF2 include NQO1, heme oxygenase (HMOX1), superoxide dismutase (SOD1), sulfiredoxin (SRX1), thioredoxin (TXN), and glutathione peroxidases (GPXs), all of which are enzymes crucial for protective detoxification of oxidative burden. These genes were all downregulated in the DBC-treated HBEC, yet up-regulated in the BAP-treated HBEC. Particularly, the DBC-downregulated enzymes involved in redox reactions (oxidoreductases and redoxins, including multiple glutaredoxins, peroxiredoxins, thioredoxin, sulfiredoxin) typically catalyze reductions of intracellular biomolecules and reactive oxygen species to confer oxidative stress resistance. A loss in antioxidant activity is likely to increase cellular damage, since a key compensatory mechanism for chemical insult is hindered.
NRF2-deficient zebrafish morphants were found to have greater levels of cadmium-related oxidative injury, and NRF2 deficient mice are also more sensitive to chemical-induced toxicity, inflammatory stressors, and carcinogenesis, particularly in the lung (Chorley et al., 2012). Therefore, it is unsurprising that BAP and cigarette smoke exposures have been reported to cause more toxicity in NRF2 deficient mice (Kensler et al., 2007). Finally, NRF2 is thought to be protective against airway disorders as it is associated with loss of function mutations in patient cohorts with acute respiratory distress syndromes or lung cancers (Cho & Kleeberger, 2010). Overall, NRF2 is an important transcription factor in regulating a protective antioxidant response, and the observed downregulation of NRF2-regulated antioxidant genes by DBC but not by BAP suggests that suppressing this antioxidant gene network may be a potential major difference in mechanism of toxicity between BAP and DBC. It is possible that the inhibition of protective cellular mechanisms acts as a contributor to DBC’s heightened carcinogenic potential, likely by compounding DBC toxicity from reactive metabolites.
We also found and confirmed, through qPCR, that a number of genes encoding enzymes involved in AHR-mediated PAH metabolism were oppositely regulated by BAP and DBC. Phase I enzymes CYP1A1, CYP1B1, ALDH3A1, and NOOl were dissimilarly regulated by BAP and DBC treatment. While BAP and CTE treatment significantly increased CYP1A1 and ALDH3A1, DBC treatment significantly downregulated CYP1A1 and CYP1B1 expression. Because these genes are inducible by AhR activation in response to environmental contaminant exposure, the effects of these genes in response to PAH treatment may be a contributing difference in how different environmental carcinogens exert their toxicity. It is very likely that parent PAHs and their various metabolites induce unique expression patterns of Phase I and Phase II enzymes that regulate further metabolism into either reactive, toxic compounds, or less toxic metabolites. This pattern of response is consistent with that previously reported in mouse skin epithelium in a limited dosing study (Larkin et al., 2013; Siddens et al., 2012) and was confirmed in airway epithelium in vitro across dose and time. Further, studies investigating the role of inflammation on BAP pulmonary toxicity found that co-exposure of mice with BAP and lipopolysaccharide led to elevation of inflammatory pathways, inhibition of Phase I and II enzymes and decreased cell adhesion that correlated with increased genotoxicity compared to BAP treatment alone. (Arlt et al., 2015; Shi, Fijten, et al., 2017; Shi, Godschalk, et al., 2017). These studies highlight the importance of inflammation on PAH-mediated lung disease and suggest that unique processes regulated by DBC related to inflammation, metabolism and cell-cell communication may mediate the enhanced carcinogenicity of DBC compared to BAP.
4.4. Advantages of profiling short-term bioactivity signatures of chemicals in 3D human airway epithelium
The results from this study support the use of systems approaches in broadly assessing and comparing mechanisms of chemical toxicity. This approach is particularly useful in comparing mechanisms of PAH exposure in a human in vitro model, both between chemicals and across model species. The RPF approach uses mouse in vivo data to derive cancer risk values to assess human cancer risk, effectively if mouse skin in vivo studies are representative of human PAH toxicity. While there is evidence of similar, short-term transcriptional responses observed across human airway epithelium and mouse skin, more research is necessary to investigate the mechanisms of PAHs in humans, and whether these mechanisms are reflected in the mouse models used to derive RPFs.
The wealth of data generated from assessing short-term gene signatures can also be useful for comparing between specific chemical treatments and longer-term patient-derived gene signatures. From our study, the short-term gene signatures observed in BAP-treated human bronchial epithelium are consistent with specific transcriptional changes observed in the bronchial epithelium of smokers. Transcriptomic profiles in the lung epithelium of cigarette smokers share similarities with the BAP-treated human bronchial epithelium, particularly with similarly regulated Phase I and Phase II genes (Boyle et al., 2010; Chari et al., 2007; Sridhar et al., 2008; Team, 2017).
5. Conclusion
This work supported the utility of transcriptomic approaches in evaluating chemical-specific profiles of PAHs. Despite treatment for only 48 hours, the chemical signatures were markedly unique between BAP and DBC, and were consistent with differences between the chemicals across species (in vivo rodent). This work supports and demonstrates that efforts using short-term, systems biology data to inform mechanism of action profiling can be applied towards in-depth mechanistic assessments of PAH mixtures.
While prior research has focused on the liver and skin response to PAHs, we demonstrated that the human in vitro pulmonary epithelium cultured at the air-liquid interface is a toxicologically sensitive model to PAH transcriptional perturbation. The knowledge gained from studying additional mechanisms of toxicity, such as inflammation, oxidative stress response, and barrier integrity dysregulation, are valuable contributors to the growing field of predictive toxicology, as researchers can utilize knowledge of unique mechanisms to evaluate mechanistic contributors to cancer risk. In the current study, short-term transcriptional responses of PAH treatment provided evidence supporting barrier integrity dysregulation as a shared mechanism of action by PAHs, and AHR and NRF2-related gene networks as potentially responsible for differences in Phase I, Phase II, and antioxidant responses to PAHs. These findings support research scrutinizing the applicability of the RPF, where the assumption of similar mode of action is necessary for quantitative PAH cancer risk assessment.
Supplementary Material
Supplemental Table 1. Primer Sequences for Quantitative PCR
HIGHLIGHTS.
BAP and DBC regulate unique biological processes in human organotypic lung culture
DBC regulation of AhR and NRF2-mediated signaling may mediate enhanced potency
PAHs alone and in mixtures resulted in disruption of epithelial barrier integrity
in vitro studies using organotypic culture models supports prior in vivo studies
Chemical-specific biosignatures in vitro may be useful for risk assessment
Acknowledgements
This study was funded by Public Health Service grants P42ES016465 and T32ES07060.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare that there are no conflicts of interest.
References
- Abdel-Shafy HI, & Mansour MSM (2016). A review on polycyclic aromatic hydrocarbons: Source, environmental impact, effect on human health and remediation. Egyptian Journal of Petroleum, 25(1), 107–123. doi: 10.1016/j.ejpe.2015.03.011 [DOI] [Google Scholar]
- Arlt VM, Krais AM, Godschalk RW, Riffo-Vasquez Y, Mrizova I, Roufosse CA, Corbin C, Shi Q, Frei E, Stiborova M, van Schooten FJ, Phillips DH, & Spina D (2015). Pulmonary Inflammation Impacts on CYP1A1-Mediated Respiratory Tract DNA Damage Induced by the Carcinogenic Air Pollutant Benzo[a]pyrene. Toxicol Sci, 146(2), 213–225. doi: 10.1093/toxsci/kfv086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astashkina A, Mann B, & Grainger DW (2012). A critical evaluation of in vitro cell culture models for high-throughput drug screening and toxicity. Pharmacology & Therapeutics, 134(1), 82–106. doi: 10.1016/j.pharmthera.2012.01.001 [DOI] [PubMed] [Google Scholar]
- Baird WM, Hooven LA, & Mahadevan B (2005). Carcinogenic polycyclic aromatic hydrocarbon-DNA adducts and mechanism of action. Environ Mol Mutagen, 45(2–3), 106–114. doi: 10.1002/em.20095 [DOI] [PubMed] [Google Scholar]
- Baird WM, & Mahadevan B (2004). The uses of carcinogen-DNA adduct measurement in establishing mechanisms of mutagenesis and in chemoprevention. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 547(1), 1–4. doi: 10.1016/j.mrfmmm.2003.10.008 [DOI] [PubMed] [Google Scholar]
- Balharry D, Sexton K, & BeruBe KA (2008). An in vitro approach to assess the toxicity of inhaled tobacco smoke components: nicotine, cadmium, formaldehyde and urethane. Toxicology, 244(1), 66–76. doi: 10.1016/j.tox.2007.11.001 [DOI] [PubMed] [Google Scholar]
- Benbrahim-Tallaa L, Baan RA, Grosse Y, Lauby-Secretan B, El Ghissassi F, Bouvard V, Guha N, Loomis D, & Straif K (2012). Carcinogenicity of diesel-engine and gasoline-engine exhausts and some nitroarenes. The Lancet Oncology, 13(7), 663–664. doi: 10.1016/S1470-2045(12)70280-2 [DOI] [PubMed] [Google Scholar]
- Blasig IE, Bellmann C, Cording J, Vecchio G. d., Zwanziger D, Huber O, & Haseloff RF (2011). Occludin Protein Family: Oxidative Stress and Reducing Conditions. Antioxidants & Redox Signaling, 15(5), 1195–1219. doi: 10.1089/ars.2010.3542 [DOI] [PubMed] [Google Scholar]
- Boffetta P, Jourenkova N, & Gustavsson P (1997). Cancer risk from occupational and environmental exposure to polycyclic aromatic hydrocarbons. Cancer Causes & Control, 8(3), 444–472. doi: 10.1023/a:1018465507029 [DOI] [PubMed] [Google Scholar]
- Bogaards JJP, Bertrand M, Jackson P, Oudshoorn MJ, Weaver RJ, Van Bladeren PJ, & Walther B (2000). Determining the best animal model for human cytochrome P450 activities: a comparison of mouse, rat, rabbit, dog, micropig, monkey and man. Xenobiotica, 30(12), 1131–1152. doi: 10.1080/00498250010021684 [DOI] [PubMed] [Google Scholar]
- Boyle JO, Gumus ZH, Kacker A, Choksi VL, Bocker JM, Zhou XK, Yantiss RK, Hughes DB, Du B, Judson BL, Subbaramaiah K, & Dannenberg AJ (2010). Effects of cigarette smoke on the human oral mucosal transcriptome. Cancer Prev Res (Phila), 3(3), 266–278. doi: 10.1158/1940-6207.Capr-09-0192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Lin H, Muskhelishvili L, Latendresse J, Richter P, & Heflich RH (2015). Tight junction disruption by cadmium in an in vitro human airway tissue model. Respir Res, 16, 30. doi: 10.1186/s12931-015-0191-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari R, Lonergan KM, Ng RT, MacAulay C, Lam WL, & Lam S (2007). Effect of active smoking on the human bronchial epithelium transcriptome. BMC Genomics, 5(1), 297. doi: 10.1186/1471-2164-8-297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chepelev NL, Long AS, Williams A, Kuo B, Gagné R, Kennedy DA, Phillips DH, Arlt VM, White PA, & Yauk CL (2016). Transcriptional Profiling of Dibenzo[def,p]chrysene-induced Spleen Atrophy Provides Mechanistic Insights into its Immunotoxicity in MutaMouse. Toxicological Sciences, 149(1), 251–268. doi: 10.1093/toxsci/kfv232 [DOI] [PubMed] [Google Scholar]
- Cho HY, & Kleeberger SR (2010). Nrf2 protects against airway disorders. Toxicol Appl Pharmacol, 244(1), 43–56. doi: 10.1016/j.taap.2009.07.024 [DOI] [PubMed] [Google Scholar]
- Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, Xue P, Pi J, Kleeberger SR, & Bell DA (2012). Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res, 40(15), 7416–7429. doi: 10.1093/nar/gks409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne CB, Vanhook MK, Gambling TM, Carson JL, Boucher RC, & Johnson LG (2002). Regulation of Airway Tight Junctions by Proinflammatory Cytokines. Molecular Biology of the Cell, 13(9), 3218–3234. doi: 10.1091/mbc.e02-03-0134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devalia JL, Bayram H, Rusznak C, Calderon M, Sapsford RJ, Abdelaziz MA, Wang J, & Davies RJ (1997). Mechanisms of pollution-induced airway disease: in vitro studies in the upper and lower airways. Allergy, 52(38 Suppl), 45–51; discussion 57–48. [DOI] [PubMed] [Google Scholar]
- EPA, U. S. (2010). Development of a Relative Potency Factor (Rpf) Approach for Polycyclic Aromatic Hydrocarbon (PAH) Mixtures (External Review Draft). (EPA/635/R-08/012A). Washington, DC [Google Scholar]
- Goodale BC, Tilton SC, Corvi MM, Wilson GR, Janszen DB, Anderson KA, Waters KM, & Tanguay RL (2013). Structurally distinct polycyclic aromatic hydrocarbons induce differential transcriptional responses in developing zebrafish. Toxicology and applied pharmacology, 272(3), 656–670. doi: 10.1016/j.taap.2013.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC. (2010). Some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures Monographs on the evaluation of carcinogenic risks to humans. IARC MONOGRAPHS ON THE EVALUATION OF CARCINOGENIC RISKS TO HUMANS, 92. [PMC free article] [PubMed] [Google Scholar]
- IARC. (2014). Diesel and gasoline engine exhausts and some nitrarenes. IARC MONOGRAPHS ON THE EVALUATION OF CARCINOGENIC RISKS TO HUMANS, 105. [PMC free article] [PubMed] [Google Scholar]
- Innes AL, Woodruff PG, Ferrando RE, Donnelly S, Dolganov GM, Lazarus SC, & Fahy JV (2006). Epithelial Mucin Stores Are Increased in the Large Airways of Smokers With Airflow Obstruction. Chest, 130(4), 1102–1108. doi: 10.1378/chest.130.4.1102 [DOI] [PubMed] [Google Scholar]
- Jarvis IW, Dreij K, Mattsson A, Jernstrom B, & Stenius U (2014). Interactions between polycyclic aromatic hydrocarbons in complex mixtures and implications for cancer risk assessment. Toxicology, 321, 27–39. doi: 10.1016/j.tox.2014.03.012 [DOI] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, & Biswal S (2007). Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol, 47, 89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046 [DOI] [PubMed] [Google Scholar]
- Labib S, Williams A, Guo CH, Leingartner K, Arlt VM, Schmeiser HH, Yauk CL, White PA, & Halappanavar S (2016). Comparative transcriptomic analyses to scrutinize the assumption that genotoxic PAHs exert effects via a common mode of action. Arch Toxicol, 90(10), 2461–2480. doi: 10.1007/s00204-015-1595-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin A, Siddens LK, Krueger SK, Tilton SC, Waters KM, Williams DE, & Baird WM (2013). Application of a fuzzy neural network model in predicting polycyclic aromatic hydrocarbon-mediated perturbations of the Cyplbl transcriptional regulatory network in mouse skin. Toxicol Appl Pharmacol, 267(2), 192–199. doi: 10.1016/j.taap.2012.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorthy B, Chu C, & Carlin DJ (2015). Polycyclic aromatic hydrocarbons: from metabolism to lung cancer. Toxicol Sci, 145(1), 5–15. doi: 10.1093/toxsci/kfv040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolsky Y, Kirillov E, Zuev R, Rakhmatulin E, & Nikolskaya T (2009). Functional analysis of OMICs data and small molecule compounds in an integrated "knowledge-based" platform. Methods Mol Biol, 563, 177–196. doi: 10.1007/978-1-60761-175-2_10 [DOI] [PubMed] [Google Scholar]
- Nookaew I, Papini M, Pornputtapong N, Scalcinati G, Fagerberg L, Uhlen M, & Nielsen J (2012). A comprehensive comparison of RNA-Seq-based transcriptome analysis from reads to differential gene expression and cross-comparison with microarrays: a case study in Saccharomyces cerevisiae. Nucleic Acids Res, 40(20), 10084–10097. doi: 10.1093/nar/gks804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips DH, Arit VM, Aimová D, Stiborová M, Wolf CR, Henderson CJ, Thiemann M, Schmitz OJ, Frei E, Farmer PB, Singh R, & Gamboa da Costa G (2008). Metabolic activation of benzo[ a ]pyrene in vitro by hepatic cytochrome P450 contrasts with detoxification in vivo : experiments with hepatic cytochrome P450 reductase null mice. Carcinogenesis, 29(3), 656–665. doi: 10.1093/carcin/bgn002 [DOI] [PubMed] [Google Scholar]
- Ravindra K, Sokhi R, & Van Grieken R (2008). Atmospheric polycyclic aromatic hydrocarbons: Source attribution, emission factors and regulation. Atmospheric Environment, 42(13), 2895–2921. doi: 10.1016/j.atmosenv.2007.12.010 [DOI] [Google Scholar]
- Rengarajan T, Rajendran P, Nandakumar N, Lokeshkumar B, Rajendran P, & Nishigaki I (2015). Exposure to polycyclic aromatic hydrocarbons with special focus on cancer. Asian Pacific Journal of Tropical Biomedicine, 5(3), 182–189. doi: 10.1016/S2221-1691(15)30003-4 [DOI] [Google Scholar]
- Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Stum A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, & Quackenbush J (2003). TM4: a free, open-source system for microarray data management and analysis. Biotechniques, 34(2), 374–378. doi: 10.2144/03342mt01 [DOI] [PubMed] [Google Scholar]
- Schamberger AC, Mise N, Jia J, Genoyer E, Yildirim AÖ, Meiners S, & Eickelberg O (2014). Cigarette Smoke-Induced Disruption of Bronchial Epithelial Tight Junctions Is Prevented by Transforming Growth Factor-β. American Journal of Respiratory Cell and Molecular Biology, 50(6), 1040–1052. doi: 10.1165/rcmb.2013-00900C [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, & Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res, 13. doi: 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Fijten RR, Spina D, Riffo Vasquez Y, Arit VM, Godschalk RW, & Van Schooten FJ (2017). Altered gene expression profiles in the lungs of benzo[a]pyrene-exposed mice in the presence of lipopolysaccharide-induced pulmonary inflammation. Toxicol Appl Pharmacol, 336, 8–19. doi: 10.1016/j.taap.2017.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Godschalk RWL, & van Schooten FJ (2017). Inflammation and the chemical carcinogen benzo[a]pyrene: Partners in crime. Mutat Res, 774, 12–24. doi: 10.1016/j.mrrev.2017.08.003 [DOI] [PubMed] [Google Scholar]
- Siddens LK, Larkin A, Krueger SK, Bradfield CA, Waters KM, Tilton SC, Pereira CB, Löhr CV, Arlt VM, Phillips DH, Williams DE, & Baird WM (2012). Polycyclic aromatic hydrocarbons as skin carcinogens: Comparison of benzo[a]pyrene, dibenzo[def,p]chrysene and three environmental mixtures in the FVB/N mouse. Toxicology and applied pharmacology, 264(3), 377–386. doi: 10.1016/j.taap.2012.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DA (1991). Species Differences in Metabolism and Pharmacokinetics: Are We Close to an Understanding? Drug Metabolism Reviews, 23(3–4), 355–373. doi: 10.3109/03602539109029764 [DOI] [PubMed] [Google Scholar]
- Song MK, Kim YJ, Song M, Choi HS, Park YK, & Ryu JC (2011). Polycyclic aromatic hydrocarbons induce migration in human hepatocellular carcinoma cells (HepG2) through reactive oxygen species-mediated p38 MAPK signal transduction. Cancer Sci, 102(9), 1636–1644. doi: 10.1111/j.1349-7006.2011.02000.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar S, Schembri F, Zeskind J, Shah V, Gustafson AM, Steiling K, Liu G, Dumas YM, Zhang X, Brody JS, Lenburg ME, & Spira A (2008). Smoking-induced gene expression changes in the bronchial airway are reflected in nasal and buccal epithelium. BMC Genomics, 9, 259. doi: 10.1186/1471-2164-9-259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Jackson S, Haycock JW, & MacNeil S (2006). Culture of skin cells in 3D rather than 2D improves their ability to survive exposure to cytotoxic agents. Journal of Biotechnology, 122(3), 372–381. doi: 10.1016/j.jbiotec.2005.12.021 [DOI] [PubMed] [Google Scholar]
- Team, A. S. (2017). Shared Gene Expression Alterations in Nasal and Bronchial Epithelium for Lung Cancer Detection. J Natl Cancer Inst, 109(7). doi: 10.1093/jnci/djw327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilton SC, Siddens LK, Krueger SK, Larkin AJ, Lohr CV, Williams DE, Baird WM, & Waters KM (2015). Mechanism-Based Classification of PAH Mixtures to Predict Carcinogenic Potential. Toxicol Sci, 146(1), 135–145. doi: 10.1093/toxsci/kfv080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, & Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics, 25(9), 1105–1111. doi: 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, & Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol, 28(5), 511–515. doi: 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung EW, Philbrook NA, Belanger CL, Ansari S, & Winn LM (2014). Benzo[a]pyrene increases DNA double strand break repair in vitro and in vivo: a possible mechanism for benzo[a]pyrene-induced toxicity. Mutat Res Genet Toxicol Environ Mutagen, 760, 64–69. doi: 10.1016/j.mrgentox.2013.12.003 [DOI] [PubMed] [Google Scholar]
- Turpeinen M, Ghiciuc C, Opritoui M, Tursas L, Pelkonen O, & Pasanen M (2007). Predictive value of animal models for human cytochrome P450 (CYP)-mediated metabolism: A comparative study in vitro. Xenobiotica, 37(12), 1367–1377. doi: 10.1080/00498250701658312 [DOI] [PubMed] [Google Scholar]
- van Delft JHM, van Agen E, van Breda SGJ, Herwijnen MH, Staal YCM, & Kleinjans JCS (2004). Discrimination of genotoxic from non-genotoxic carcinogens by gene expression profiling. Carcinogenesis, 25(7), 1265–1276. doi: 10.1093/carcin/bgh108 [DOI] [PubMed] [Google Scholar]
- White PA (2002). The genotoxicity of priority polycyclic aromatic hydrocarbons in complex mixtures. Mutation Research Genetic Toxicology and Environmental Mutagenesis, 575(1), 85–98. doi: 10.1016/S1383-5718(02)00017-7 [DOI] [PubMed] [Google Scholar]
- Zavala J, O’Brien B, Lichtveld K, Sexton KG, Rusyn I, Jaspers I, & Vizuete W (2016). Assessment of Biological Responses of EpiAirway(™) 3-D Cell Constructs vs. A549 Cells for Determining Toxicity of Ambient Air Pollution. Inhalation toxicology, 28(6), 251–259. doi: 10.3109/08958378.2016.1157227 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Primer Sequences for Quantitative PCR