

Abstract
Integration of multi-omics in cardiovascular diseases (CVDs) presents high potentials for translational discoveries. By analyzing abundance levels of heterogeneous molecules over time, we may uncover biological interactions and networks that were previously unidentifiable. However, to effectively perform integrative analysis of temporal multi-omics, computational methods must account for the heterogeneity and complexity in the data. To this end, we performed unsupervised classification of proteins and metabolites in mice during cardiac remodeling using two innovative deep learning (DL) approaches. First, long short-term memory (LSTM)-based variational autoencoder (LSTM-VAE) was trained on time-series numeric data. The low-dimensional embeddings extracted from LSTM-VAE were then used for clustering. Second, deep convolutional embedded clustering (DCEC) was applied on images of temporal trends. Instead of a two-step procedure, DCEC performes a joint optimization for image reconstruction and cluster assignment. Additionally, we performed K-means clustering, partitioning around medoids (PAM), and hierarchical clustering. Pathway enrichment analysis using the Reactome knowledgebase demonstrated that DL methods yielded higher numbers of significant biological pathways than conventional clustering algorithms. In particular, DCEC resulted in the highest number of enriched pathways, suggesting the strength of its unified framework based on visual similarities. Overall, unsupervised DL is shown to be a promising analytical approach for integrative analysis of temporal multi-omics.
Keywords: Cardiovascular, Clustering, Multi-Omics, Time-Series, Unsupervised Deep Learning, Integrative Analysis
1. Introduction
Cardiovascular diseases (CVDs), as the leading cause of death in the U.S, are the subject of significant research investigation [1]. CVDs are complex biological phenomena with molecular mechanisms that are largely unknown. With the rise in new experimental technologies over the last three decades, a large volume of omics data describing CVDs has been accumulated [2–4]. To elucidate their complex biological mechanisms and identify key molecules associated with different disease phenotypes, we often analyze high-dimensional measurements on DNA sequences, RNA, proteins, metabolites and others. Conventional analyses tend to focus on single-omics datasets. While single-omics analyses have helped improve our understanding of CVDs [5,6], a lack of translational discoveries for multifaceted diseases like CVDs suggests the need for integrated molecular investigations [7]. A comprehensive picture of complex molecular mechanisms related to a CVD of interest can be constructed by simultaneous analysis of multiple molecular data types that are so-called multi-omics [8,9]. In particular, new computational approaches are required to overcome the challenges in integrating heterogenous and temporal data [10–13].
From predicting clinical outcomes from biomedical datasets [3,14,15] to analyzing multiple data types simultaneously [10–12], machine learning (ML) has emerged as a novel and essential component of modern biomedical research. In supervised learning with known group labels (such as controls and experimental conditions), we may use ML algorithms to classify samples based on clinical and molecular variables and build prognostic models [15,16]. With omics datasets where labels are often unknown or unmeasured, unsupervised ML algorithms such as auto-encoders (AEs) [17,18] can be employed to extract hidden sources of variation and help discover patient sub-groups [19]. Recently, deep neural networks have been developed and applied successfully in a wide range of data-intensive fields [20–23]. With the continuous availability of efficient computational resources, deep learning (DL) is being proven to outperform conventional ML approaches in areas, such as, automatic feature engineering, representation learning, sequence modelling, etc. [24,25]. In particular, we are interested in unsupervised deep learning that can help us uncover molecular signatures of CVDs. Using multiple hidden layers that may be seen as performing data-driven non-linear transformations, deep neural networks can obtain high-level abstractions that are characteristics of underlying phenotypes. However, unsupervised DL in the context of CVDs with multi-omics has been understudied.
In order to better understand CVDs, we have been using diverse genetic strains of mouse and an experimental treatment to induce cardiac hypertrophy. A landmark proteomics dataset based on this mouse model is publicly available [26] and has been analyzed for biomarker identifications [27]. This dataset comprises thousands of proteins quantified at several time points under control (CTRL) and isoproterenol-induced hypertrophy (ISO) across six genetic strains [26]. Based on the same model, we have also quantified plasma concentration levels of 610 metabolites over a period of 14 days. The temporal nature of proteomics and metabolomics data makes the integrative multi-omics analysis challenging and presents new opportunities for adapting DL approaches. We are interested in investigating relationships among proteins and metabolites during cardiac remodeling in an unsupervised manner. Therefore, we implemented two unsupervised DL approaches for the integrative analysis of our time-series multi-omics data.
First, we implemented a two-step approach where low-dimensional embeddings from a variational autoencoder (VAE) [28] are used for clustering. As a popular representation learning method, VAE extracts low-dimensional embeddings, which may shed new insights on molecular mechanisms. As proteins and metabolites were measured over time, we implemented a long short-term memory (LSTM) [29–32]-based VAE. LSTM is a type of recurrent neural network (RNN) that can capture time-dependent behavior but without the vanishing gradient problem [32]. Then, we applied clustering on the embeddings to identify important temporal trends in the data, and identify groups of molecules (proteins and metabolites) with similar temporal characteristics. Second, we utilized images of abundance levels of proteins and metabolites over 14 days under ISO condition to find clusters based on their visual similarities. To achieve this, we employed deep convolution embedded clustering (DCEC). Unlike two-step approaches, DCEC performs joint optimization for extracting low-dimensional embeddings and assigning cluster memberships [33–35].
Integrative analysis using unsupervised deep learning could contribute to characterizing multi-omics data. Specially for complex diseases such as CVDs, heterogeneous data from proteomics and metabolomics could possess hidden groups of biomarkers. We investigated whether DL may extract more biologically meaningful clusters than conventional clustering approaches. The biological pathway analysis using Reactome knowledgebase [36] revealed that the clusters obtained from DL methods harbored more significant pathways than from K-means clustering [37,38], hierarchal clustering (HC) [39] and partitioning around medoids (PAM) [40] methods. Furthermore, we investigated whether scale-free images of temporal trends as inputs could help better cluster the molecules based on visual similarities. We found that DCEC which uses image data as input performed much better than conventional clustering approaches as well as LSTM-VAE using time-series numeric data.
2. Materials and Methods
We implemented and compared three conventional clustering algorithms and two unsupervised DL approaches on temporal proteomics and metabolomics datasets. Both large-scale datasets are based on six genetically diverse mouse strains with induced cardiovascular conditions, recently generated by our lab. In proteomics dataset [26], six genetic mouse strains (FVB/NJ, CE/J, C57BL/6J, DBA/2J, BALB/cJ, and A/J; 26 mice per strain), purchased from The Jackson Laboratory, were selected for their wide relevance to CVDs, cancer, metabolic and other diseases. The mice were all juvenile males between 9-12 weeks of age. One group (72 mice of six genetic strains) received 15 mg·kg−1·d−1 isoproterenol (ISO) (subcutaneous micro-osmotic pumps; Alzet) treatment, whereas the second group (84 mice of six genetic strains) received sham treatment (CTRL). Tissue samples were collected at 0, 1, 3, 5, 7, 10, and 14 days. Cardiac protein samples were prepared and liquid chromatography tandem-mass spectrometry (LC-MS/MS) analysis for protein half-life and steady-state abundance was performed. In this study, we focused on protein abundance levels quantified as normalized spectral abundance factor (NSAF).
The metabolomics dataset is based on a similar cardiovascular mouse model for plasma metabolomics profiling. Six genetic mouse strains (FVB/NJ, CBA/J, C57BL/6J, DBA/2J, BALB/cJ, and A/J; 54 mice per strain) with divergent susceptibilities towards ISO stimulation were continuously treated with 15 mg·kg−1·d−1 ISO for 14 days using mini-osmotic pump. At baseline (day 0) and during ISO treatment (days 1, 3, 5, 7, and 14), blood samples and whole hearts were collected for plasma metabolomics profiling, cardiac phenotypic assessments, and annotation analyses of metabolic pathways. The absolute concentrations of 610 plasma metabolites were quantified in six genetic mouse strains during ISO stimulation. Over time, changes in concentrations of these metabolites represent the underlying metabolic responses associated with the progression of ISO-induced maladaptive remodeling of the heart. Note that CBA/J and CE/J strains are genetically closely related [41] and have very similar response to ISO (i.e., isoproterenol-induced lethality and cardiac fractional shortening) [42]. The other five strains are the same in both datasets.
For proteomics dataset, we normalized all abundance values under ISO with respect to corresponding CTRL. Concentration levels of metabolites in ISO were normalized with levels at day 0. Proteins and metabolites with quantified values available at 3 timepoints or more were included in the analysis. The missing values were imputed using a multiple imputation (MI) technique; fully condition specifications (FCS) with predictive mean matching (PMM) [43,44]. PMM does not make any assumption regarding the normality of the distribution and MI takes into account the uncertainty associated with missing data by generating multiple imputed values for each missing value. Smoothing splines were fitted to each molecule across time points and the degree of smoothing was automatically determined by leave-one-out cross-validation. The aggerated dataset has complete time-series data for 3,479 proteins and 513 metabolites. The abundance and concentration values were scaled so that they are in the same range between −1 and 1. As a baseline, we employed three highly popular clustering algorithms: K-means clustering, hierarchical clustering (HC) and partitioning around medoids (PAM). Then, we employed two clustering approaches based on DL; the first approach uses the time-series numeric data while the second approach uses images of temporal trends as input. Figure 1 provides an overview of our integrated clustering workflow for the multi-omics data.
Figure 1. Integrative Clustering Workflow for Temporal Multi-Omics Data.
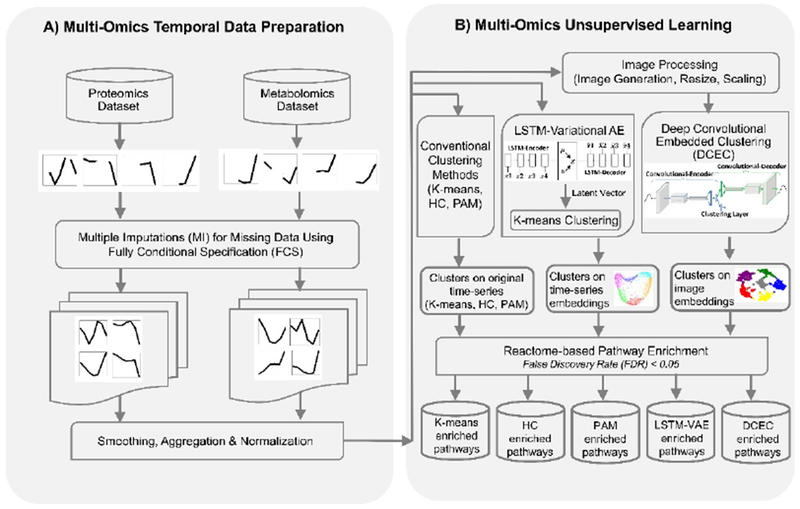
Missing values in proteomics and metabolomics datasets are completed by fully conditional specification (FCS)-based multiple imputations. Cubic splines were employed to smooth the temporal trends. Images were also generated based on the temporal trends over 14 days. As a baseline, K-means clustering, hierarchical clustering (HC), partitioning around medoids (PAM)) were employed. In contrast, two deep neural network architectures for unsupervised learning were also implemented. First, long short-term memory (LSTM)-based variational autoencoder (LSTM-VAE) was used to extract low-dimensional embeddings from the time-series numeric data and K-means clustering was employed on the embeddings. Second, deep convolutional embedded clustering (DCEC) was employed to perform clustering of molecules based on image data. The results obtained from the clustering methods were fed to Reactome knowledgebase for pathway enrichments analyses.
The first DL approach uses low-dimensional embeddings from variational autoenconder (VAE) [28] to form clusters. In autcoencoders (AEs), an encoder uses the input data to extract low-dimensional embeddings whereas a decoder reconstructs the input data from these embeddings. Improving upon standard AEs whose low-dimensional embeddings may not be structured in a meaningful manner, VAE imposes a distributional assumption on low-dimensional embeddings that is latent vectors. In the encoder of VAE, a constraint is added so that the latent vectors closely follows a unit Gaussian distribution. Consequently, beyond minimizing the reconstruction error such that the input data may be reconstructed by the decoder, low-dimensional embeddings from VAE are more robust, meaningful, and generative. The loss term in VAE comprises two errors; the reconstruction error and the latent error. The reconstruction error is the mean square error, that evaluates the network’s performance in recontructing the input data. The latent loss measures how well the latent vectors follows the assumed distribution, using Kullback-Liebler divergence (KL). Given the input data, the encoder of VAE generates vectors of means and standard deviation. This allows one to sample from the corresponding distribution and to reconstruct the input data using the decoder.
Furthermore, for temporal multi-omics data, we propose a VAE architecture based on long short-term memory (LSTM) which directly models time- or sequence-dependent behavior [29–32]. The input to an LSTM-based encoder is a time-series and the output are the low-dimensional embeddings. These embeddings are then fed to an LSTM-based decoder which tries to reconstruct the original time-series (Figure S1). Given enough iterations, the decoder accurately reconstructed the input data. Subsequently, the low-dimensional embeddings obtained from LSTM-VAE were fed to a K-means clustering algorithm. We refer to this clustering approach as LSTM-VAE throughout this paper. LSTM-VAE was implemented utilizing TensorFlow [45] and Keras [46] libraries in python.
The second DL approach utilizes images of the temporal trends of 3,992 molecules to cluster them based on their visual similarities. Specifically, deep convolutional embedded clustering (DCEC) [33] method was employed to perform joint optimization for feature learning and clustering. DCEC is a recently proposed version of improved DEC (IDEC) [34,35] with convolutional layers since these layers are better suited for images (Figure S2). To adapt DCEC to temporal multi-omics data, the numeric time-series data must be visualized. Therefore, we investigated multiple combinations of line widths and image sizes. Figure S3 shows different images generated for the same molecule. Generally, one would like to choose the image size such that it is large enough to be clearly visualized by human eye, and small enough to keep the computational complexity at a reasonable level. Similarly, the line width needs to be dense enough so the signal pattern is emphasized, and thin enough so the entire signal lies within the image frame. The kernel size is another parameter which may affect the deep convolutional learning in DCEC. Therefore, we performed a systematic analysis to select the best combination of line width, image and kernel sizes. All images were generated in portable network graphics (PNG) format.
Note that we are interested in the method that can obtain clusters which are biologically more meaningful. Therefore, once the cluster assignments were obtained from K-means, HC, PAM, LSTM-VAE and DCEC, we performed biological pathway enrichment analysis to identify the method enriching the highest number of significant pathways. In general, the clustering methods that identify a high number of significantly enriched pathways can be considered biologically important. The functional annotations on cellular pathway information were performed through Reactome knowledgebase (release v67, 2018_12) (https://reactome.org/) [36]. Reactome links proteins and metabolites to their molecular functions, describing hierarchical relationships of molecules in biological processes during cardiac hypertrophy progression [27]. All the molecules in a given cluster were used together as input for the Reactome pathway enrichment analysis. Specifically, UniProt identifiers [47] and KEGG identifiers [48] were used for proteins and metabolites, respectively. The significance of biological pathways identified by Reactome is determined by false discovery rate (FDR), calculated from corrected over-representation probability and providing statistical validation of molecular enrichment within the given pathways [49].
The clustering algorithms were implemented on a server with Intel Xeon Processor E5-2643 v4, 128 GB RAM and NVIDIA Quadro M4000 GPU. K-means and HC algorithm were implemented using scikit-learn [50] library in python programming language while PAM was implemented using ‘cluster’ package in R programming language. K-means, HC and PAM clustering assignments are provided in supplemental Tables S1 to S3, respectively.
3. Results
Unsupervised classification of multi-omics data helps us dissect the molecular basis for the complex diseases such as cardiovascular diseases (CVDs). Using the integrated proteomics and metabolomics data from mice undergoing cardiac remodeling, we investigated diverse clustering approaches, including K-means, HC, PAM, LSTM-VAE, and DCEC.
Clustering typically requires an appropriate number of clusters, K. Generally, a low value of K may extract very general patterns while a high value may be severely affected by noise. This number can be selected based on prior biological knowledge and/or computational analysis. We selected K = 6 based on prior biological knowledge, the within-cluster sum of squares, and the silhouette analysis. The within-cluster sum of squares visualized in Figure S4, suggests at least 3 or 4. However, from our biological understanding of the integrated dataset and its temporal patterns, we anticipated more distinct patterns. Evaluating the K-means clustering performance using multiple K values with the silhouette analysis [51], we decided to proceed with K = 6 (Figure S5). While a range of K values extracted clusters with above the average scores, there are wide fluctuations when K > 6. Particularly, with K > 6, the differences in cluster sizes are large and smaller clusters would contain limited numbers of molecules. Therefore, the number of clusters is set to K = 6 for all the five clustering methods.
Beyond the number of clusters K, deep neural networks require hyperparameters that must be selected for particular applications. Our LSTM-VAE network naturally has the input layer of dimension 7×1 since each molecule is associated with a vector of length 7. Input time-series data were processed through an encoder LSTM layer with an intermediate dimension n, followed by fully-connected layers for obtaining means and standard deviations, from which the latent vectors may be sampled in VAE. We set the embedding dimension to 3 as it reconstructed the input signal faithfully. It was observed when the dimension of latent vectors was set to 2, the time-series reconstructed by the decoder was too smooth and failed to capture all the variations in the original. In the decoder, we used two LSTM layers with units n (intermediate dimension) and 1 (input dimension) respectively. Therefore, the first layer generated 7×n sequences while the second layer reconstructed the time-series matching the original input dimension of 7×1. The number of epochs were set to 1000 with adadelta optimizer [52]. Finally, K-means clustering was employed on the low-dimensional embeddings extracted by LSTM-VAE. We tried a range of different values for n from 3 to 10 to be used in the LSTM layers. The average cluster losses on the original time-series obtained from different LSTM-VAE architectures demonstrated that n = 6 is a suitable number of units in intermediate LSTM layers (Figure S6). Overall, we selected a 7-6-3 (input-intermediate-embedding) architecture for the encoder layers. The LSTM-VAE model with number of hidden units in each layer (right cell) is shown in Figure S7 and the source code is available at https://github.com/bilalmirza8519/LSTM-VAE. Figure 2 shows the visualization of the 6 clusters in embedded feature space of LSTM-VAE. The clustering assignments from LSTM-VAE are reported in Supplemental Table S4.
Figure 2: 3D Visualization of 6 clusters in the Embedded Feature Space of LSTM-VAE.
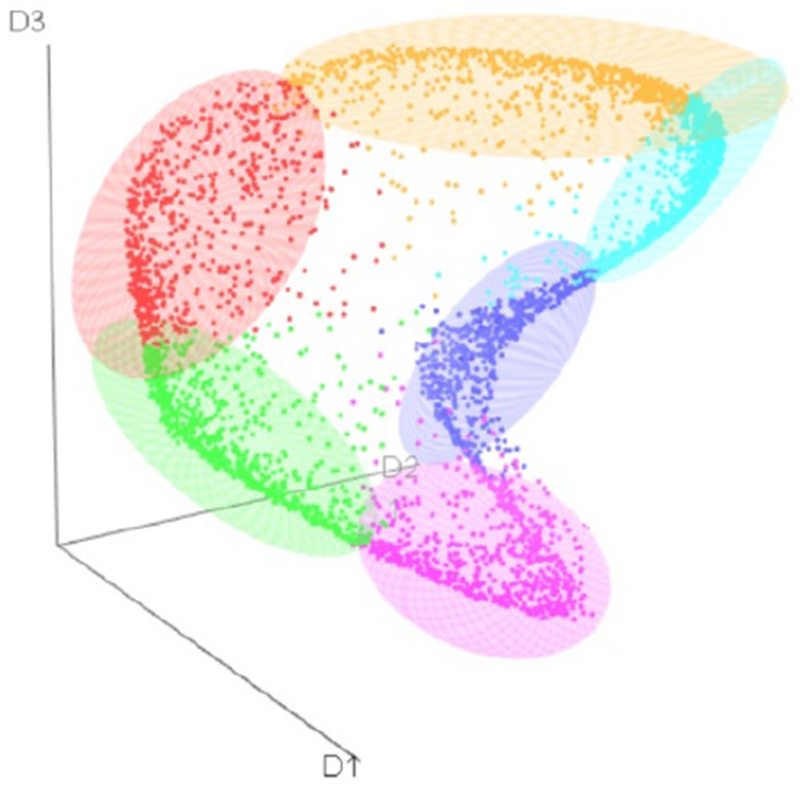
Three axes represent the three dimensions of the latent space obtained from LSTM-VAE. Different colors denote six clusters identified by K-means clustering. The ellipsoids represent 80% of the concentration for each cluster.
For the DCEC method, we used the same architecture as in the original paper [33]. The encoder has 3 convolutional layers with dimensions of 32, 64, 128, followed by a fully-connected embedding layer of dimensions 10. The stride length was set to 2. However, instead of a fixed kernel size from [33], we tried 3 different values, 3×3, 4×4, 5×5 as we wanted to find the best combination of image size, line width and kernel size. Note that the last layer has a kernel size of 3×3 and the decoder is a mirror of the encoder. The autoencoder in DCEC was pre-trained with 300 epochs with adadelta optimizer. The source code for DCEC was obtained from https://github.com/XifengGuo/DCEC. Figure S8 shows the average cluster loss calculated on the original time-series as a function of image size and line width for three different kernel sizes. Generally, a line width of > 4 with an image size greater than 40×40 produced better results than with thinner lines and smaller image sizes. The method was found to be not very sensitive to the kernel size. Note that the lowest clustering loss does not necessarily mean the best results, and therefore we also visually validated that the clusters have distinct characteristics. We selected an image size of 80×80, a kernel size of 3×3 and a line width of 7 as this combination consistently provided low clustering loss over 3 trials (Figure S9). Image sizes greater than 80×80 did not achieve any better results but increased computational time. Figure 3 shows the t-distributed stochastic neighbor embedding (t-SNE) visualization of the embeddings obtained from DCEC. Clustering assignments from DCEC are reported in Supplemental Table S5.
Figure 3: t-SNE Visualization of DCEC Embeddings.
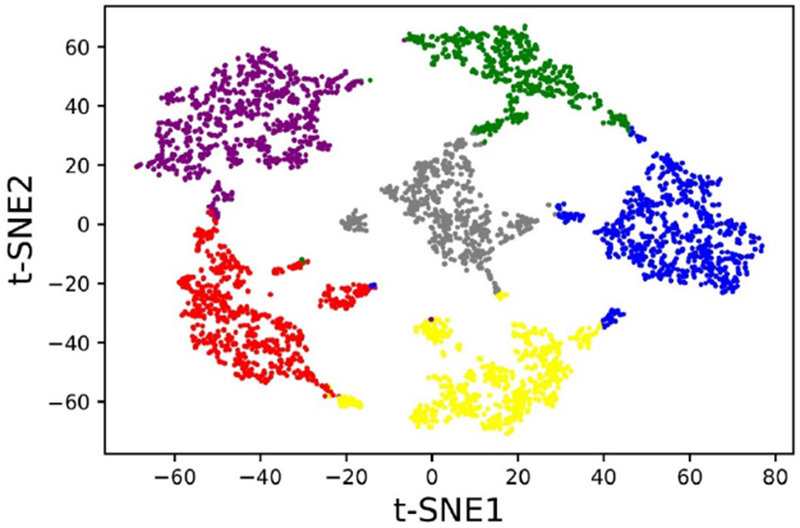
The t-SNE visualization of the embeddings, optimized for both reconstruction and cluster losses, clearly shows the separation of samples into six clusters. Each cluster obtained by DCEC is represented by a different color.
We are interested in the temporal patterns identified from different clustering methods. Visualization of six cluster centers from the five methods shows how similar (or different) their patterns are (Figure 4). Even though the clusters centers have relatable patterns across clustering methods, there are some notable differences, which may hold key biological characteristics. Nonetheless, we labeled these clusters as “increase”, “decrease”, “increase-decrease”, “decrease-increase”, “late increase” and “late decrease”, accordingly to the most dominant trends. We performed pathway enrichment analysis for the five clustering methods separately using Reactome knowledgebase [36,49]. Table 1 lists the number of significant pathways (and number of molecules in parenthesis) enriched with FDR < 0.05, by each method. The highest numbers of pathways are reported in bold while the highest numbers of molecules are reported in Italics. As can be observed, DCEC enriched more pathways in total compared to LSTM-VAE, K-means, HC and PAM. Moreover, DCEC enriched the highest number of significant pathways in most of the clusters in comparison with other four methods. K-means and PAM enriched highest number of pathways for one cluster each, but the differences in comparison with other methods are not substantial.
Figure 4. Cluster Centers for Different Clustering Methods.
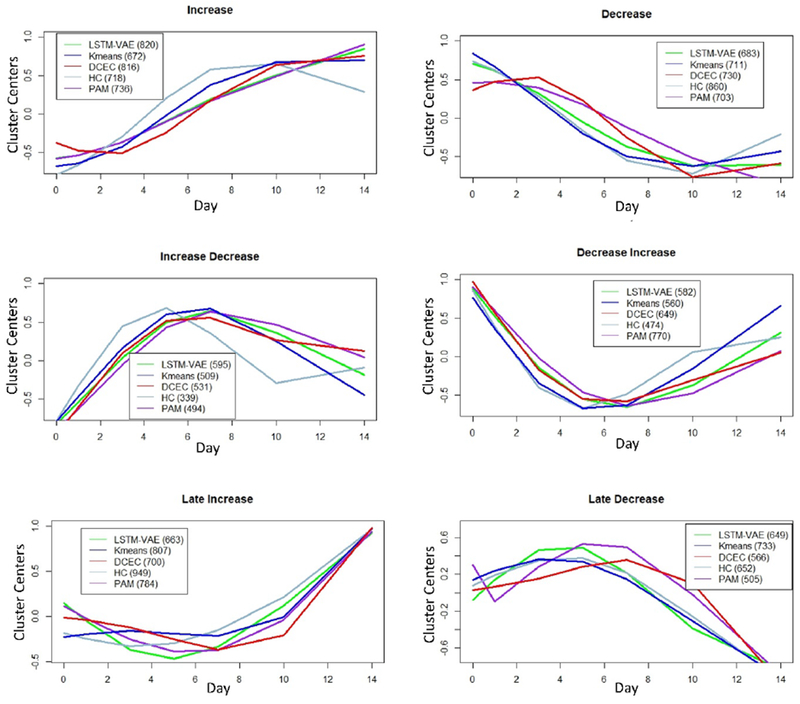
Cluster centers for the each of the 6 clusters obtained by K-means, HC, PAM, LSTM-VAE and DCECE methods are plotted. Based on the temporal similarities of cluster centers across methods, we labeled the clusters as increase, decrease, increase-decrease, decrease-increase, late increase and late decrease. However, there are some obvious differences, which can be attributed to the differences in learning algorithms and input data types (e.g., time-series numeric or image).
Table 1.
Number of Significant Pathways Enriched by Each Method with FDR < 0.05
| K-means | HC | PAM | LSTM-VAE | DCEC | |
|---|---|---|---|---|---|
| Increase | 2 (672) | 4 (718) | 28 (736) | 17 (820) | 71 (816) |
| Decrease | 17 (711) | 4 (860) | 3 (703) | 9 (683) | 6 (730) |
| Increase-Decrease | 3 (509) | 7 (339) | 5 (494) | 6 (595) | 10 (531) |
| Decrease-Increase | 32 (560) | 50 (474) | 37 (770) | 35 (582) | 56 (649) |
| Late Increase | 8 (807) | 4 (949) | 12(784) | 9 (663) | 4 (700) |
| Late Decrease | 1 (733) | 0 (652) | 2 (505) | 7 (649) | 80 (566) |
| Total | 63 (3,992) | 69 (3,992) | 87 (3,992) | 83 (3,992) | 227 (3,992) |
The temporal patterns of these molecules may be both positively or negatively correlated, as biological homeostasis is commonly maintained by regulatory feedback loops. Therefore, we also merged the complementing clusters together: increase with decrease, increase-decrease with decrease-increase, and late increase with late decrease. We refer to these merged clusters as “one-directional change”, “two-directional change” and “late change”, respectively. Table 2 shows the number of significant pathways enriched by the merged clusters at an FDR threshold of 0.05. DCEC enriched the highest number of significant pathways followed by LSTM-VAE in total. Among a total of 52 unique pathways enriched by DCEC, we found Mitochondrial Translation Termination and Mitochondrial Translation Elongation in one-directional change cluster. These are the essential cellular pathways of mitochondrial protein synthesis, significantly relevant to the cardiac remodeling and heart failure [53,54]. Furthermore, pathways of programmed cell death (e.g, Apoptotic Execution Phase) and PTEN regulation (e.g., Regulation of PTEN Stability and Activity), which were previously reported as important factors of cardiac remodeling and homeostasis [53,55–58], were detected solely by DCEC in two-directional and one-directional change clusters, respectively. Supplemental Tables S6–S8 list the unique pathways enriched by K-means, HC, PAM, LSTM-VAE, and DCEC in one-directional change, two-directional change and late change clusters, respectively. Figure 5 shows the number of shared and unique molecules across different methods for the three merged clusters. The majority of the molecules in each merged cluster are shared in the clustering results of at least three methods. Approximately 8.8% of molecules are unique to each method, that demonstrate its particular way to treat the molecules with less clear pattern.
Table 2.
Number of Significant Pathways Enriched for Merged Clusters with FDR < 0.05
| K-means | HC | PAM | LSTM-VAE | DCEC | |
|---|---|---|---|---|---|
| One-directional Change | 1 (1,383) | 1 (1,578) | 14 (1,439) | 5 (1,503) | 50 (1,546) |
| Two-directional Change | 17 (1,069) | 33 (813) | 10 (1,264) | 13 (1,177) | 29 (1,180) |
| Late Change | 8 (1,540) | 7 (1,601) | 7 (1,289) | 66 (1,312) | 71 (1,266) |
| Total | 26 (3,992) | 41 (3,992) | 31 (3,992) | 84 (3,992) | 150 (3,992) |
Figure 5. Shared and Unique Molecules Across Different Clustering Methods in Merged Clusters.
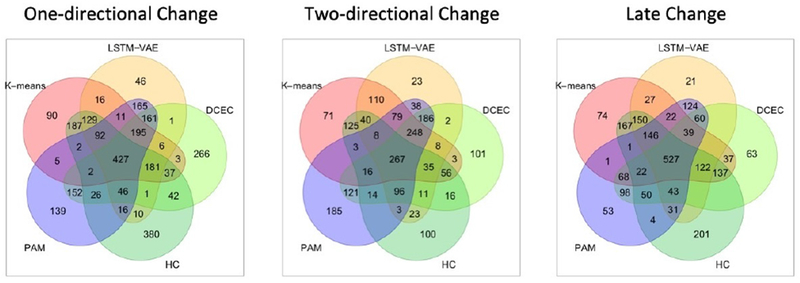
In each merged cluster, majority of the molecules are shared by at least three clustering methods. All the methods have some unique molecules in each cluster due to differences in learning algorithm and/or input data type. Hierarchal clustering generally has a high concentration of unique molecules while LSTM-VAE has the least.
Lastly, to summarize multiple pathway enrichment analyses, we ranked five clustering methods based on the number of significant pathways in both original and merged clusters. For a given cluster, the method enriching the highest number of pathways was ranked 1, the second highest 2 and so on. If the same number of pathways are enriched for more than one method, an average rank was assigned. Table 3 provides the ranking of the five methods for each individual cluster as well the average rank over nine clusters. Both deep learning methods are ranked higher than the conventional clustering methods. Specifically, DCEC is the highest ranked methods with an average rank of 1.72 and LSTM-VAE is the second highest ranked method with an average rank of 2.77. The average ranks of PAM, HC and K-means are 3.16, 3.61 and 3.72 respectively.
Table 3.
Ranking of Clustering Methods based on Number of Significant Pathways Enriched
| K-means | HC | PAM | LSTM-VAE | DCEC | |
|---|---|---|---|---|---|
| Increase | 5 | 4 | 2 | 3 | 1 |
| Decrease | 1 | 5 | 4 | 2 | 3 |
| Increase-Decrease | 5 | 2 | 4 | 3 | 1 |
| Decrease-Increase | 5 | 2 | 3 | 4 | 1 |
| Late Increase | 3 | 4.5 | 1 | 2 | 4.5 |
| Late Decrease | 4 | 5 | 3 | 2 | 1 |
| One-directional Change | 4.5 | 4.5 | 2 | 3 | 1 |
| Two-directional Change | 3 | 1 | 5 | 4 | 2 |
| Late Change | 3 | 4.5 | 4.5 | 2 | 1 |
| Average Rank | 3.72 | 3.61 | 3.16 | 2.77 | 1.72 |
4. Discussion
Temporal multi-omics helps us dissect the dynamic molecular basis of complex diseases, such as CVDs. As molecular processes tend to be regulated in a temporal fashion, we expect a network of molecules to exhibit distinct temporal patterns under a given environmental or experimental condition. However, methodologically it is unclear how to classify heterogeneous molecules in multi-omics data without using their annotations. In this temporal multi-omics study, we applied three well-known clustering algorithms (K-means, HC, and PAM) and two recent approaches based on deep learning (LSTM-VAE and DCEC) to systematically classify responses of proteins and metabolites during cardiac remodeling. Pathway enrichment analysis using Reactome knowledgebase provided an in silico biological validation and comparative evaluation. We found that deep learning approaches of LSTM-VAE and DCEC help reveal more biologically meaningful clusters. Particularly, the clusters from DCEC contain the highest numbers of significant biological pathways.
Unsupervised classification requires a number of hyperparameters, that must be set using domain expertises and computational analyses. Finding the optimal number of meaningful clusters has long been a challenging issue. We explored using a smaller number of clusters than K = 6. However, it was observed that the highly plausible delayed responses were lost in such cases since there were no clusters for late increase and late decrease. In contrast, larger values lead to a greater imbalance in the cluster sizes, where a small cluster likely comprises a pattern specific to limited molecules or even noise. We further investigated the dimensions of intermediate layers and latent vectors, as well as how to generate images based on numeric time-series. However, the possible combinations of hyperparameters are endless and may warrant investigations beyond the scope of this paper. Nonetheless, we suggested a workflow and heuristics for a systematic exploration.
For interpretability and comparison of results across five methods, the clusters were grouped and labeled based on the dominant temporal trends. However, the temporal trends from different methods have subtle differences. Such that some molecules may be classified differently, and clusters may exhibit unique biological characteristics. For example, the ‘increase’ cluster of DCEC contained much more significant pathways than that of other methods. Many of the molecules unique in this DCEC cluster start to follow increase pattern at day 3 rather than day 1. Most of the other methods classified those molecules to the ‘late increase’ cluster. On the other hand, many molecules in the ‘late increase’ and ‘late decrease’ clusters of DCEC generally have a slow or no change until day 7 or day 10, followed by a sharp change. These differences can also be observed from the cluster centers plotted for each method in Figure 4. Similarly, it can be observed from the figure that clusters corresponding to two-directional change are different for DCEC in comparison with most of other methods. These clusters from DCEC include molecules with an initial sharp change followed by a slow change in the other direction.
Pathway analysis evaluates whether a cluster of molecules may share similar functions, according to their annotations. This in silico biological validation allows us to evaluate unsupervised classification methods, as we expect good clustering would result in biologically meaningful groups. Notably, the individual clusters obtained by DCEC not only contained more enriched pathways than that by other methods, they also represented much more enriched pathways in total. Moreover, both DL approaches generated clusters with higher numbers of significant pathways than three conventional clustering methods, resulting in DCEC and LSTM-VAE ranking the highest in Table 3. Therefore, we expect that DL-based clustering approaches will be highly useful for temporal multi-omics studies. Moreover, as DCEC was originally developed for computer vision, it may be wise for molecular biologists and bioinformaticians to stay multi-disciplinary.
Multi-omics data are often imbalanced due to technological and biological differences. Our integrated dataset comprises approximately 87% proteins and 13% metabolites, such that most of the clusters and their associated pathways are dominated by proteins. Thus, it is of our interest to increase the number of metabolites in subsequent studies to extract more pathways enriched by proteins and metabolites in concert. Similarly, genomics and others on the same mouse model would expand our multi-omics study. Moreover, further biological significance related to CVDs may be understood by consulting other pathway knowledgebases such as KEGG Pathway [48] and SMPDB [61].
Conclusions
We have performed integrative analysis of temporal proteomics and metabolomics data in the context of CVDs using unsupervised deep learning. We utilized both time-series numeric data and scale-free images of the temporal trends obtained from heterogenous molecules. It was found that unsupervised methods based on deep learning generally obtained clusters that are biologically more relevant than those extracted by conventional methods such as K-means, HC and PAM. Specifically, we utilized Reactome knowledgebase to identify the number of significantly enriched biological pathways by each clustering method. The clusters obtained from DCEC, a method incorporating deep convolutional layers to effectively learn from images, contained the highest number of significant pathways. DCEC method clusters molecules based on the visual similarity of their temporal trends, demonstrating that deep convolutional learning from images is an effective approach for integrative analysis of temporal multi-omics data. We also provided a systematic approach to image generation for molecules, and found the best combination of line width, image size and kernel size for deep convolutional learning. Due to the generality of the proposed unsupervised deep learning workflow, it can be straightforwardly extended to other complex diseases.
Supplementary Material
Metabolites and proteins responded to cardiac remodeling with coherent patterns
Unsupervised deep learning (DL) helped uncover the temporal trends in multi-omics
DL methods generated more biologically meaningful clusters than conventional methods
DCEC optimizing featuring learning and clustering jointly outperformed other methods
Acknowledgements
This work was supported by the National Institutes of Health grants R35 HL 135772; T32 HL 139450; U54 GM 114833 to P.P.; the National Institutes of Health grant NIH R01 GM115833 to W.W.; the National Science Foundation grant DBI-1565137 to W.W.; and the Narodowe Centrum Nauki (National Science Center in Poland) grant 2016/23/D/ST6/03613 to N.C.C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Benjamin EJ; Virani SS; Callaway CW; Chamberlain AM; Chang AR; Cheng S; Chiuve SE; Cushman M; Delling FN; Deo R Heart disease and stroke statistics—2018 update: a report from the American Heart Association. Circulation 2018, 137, e67–e492. [DOI] [PubMed] [Google Scholar]
- 2.Fernandes M; Patel A; Husi H C/VDdb: A multi-omics expression profiling database for a knowledge-driven approach in cardiovascular disease (CVD). PloS one 2018, 13, e0207371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lau E; Wu JC Omics, Big Data, and Precision Medicine in Cardiovascular Sciences; Am Heart Assoc, 2018; ISBN 0009-7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perez-Riverol Y; Bai M; da Veiga Leprevost F; Squizzato S; Park YM; Haug K; Carroll AJ; Spalding D; Paschall J; Wang M Discovering and linking public omics data sets using the Omics Discovery Index. Nature biotechnology 2017, 35, 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lau E; Cao Q; Lam MP; Wang J; Ng DC; Bleakley BJ; Lee JM; Liem DA; Wang D; Hermjakob H Integrated omics dissection of proteome dynamics during cardiac remodeling. Nature communications 2018, 9, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGarrah RW; Crown SB; Zhang G-F; Shah SH; Newgard CB Cardiovascular metabolomics. Circulation Research 2018, 122, 1238–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Azimzadeh O; Sievert W; Sarioglu H; Merl-Pham J; Yentrapalli R; Bakshi MV; Janik D; Ueffing M; Atkinson MJ; Multhoff G Integrative proteomics and targeted transcriptomics analyses in cardiac endothelial cells unravel mechanisms of long-term radiation-induced vascular dysfunction. Journal of proteome research 2015, 14, 1203–1219. [DOI] [PubMed] [Google Scholar]
- 8.Ryan CJ; Cimermančič P; Szpiech ZA; Sali A; Hernandez RD; Krogan NJ High-resolution network biology: connecting sequence with function. Nature Reviews Genetics 2013, 14, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karczewski KJ; Snyder MP Integrative omics for health and disease. Nature Reviews Genetics 2018, 19, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Argelaguet R; Velten B; Arnol D; Dietrich S; Zenz T; Marioni JC; Buettner F; Huber W; Stegle O Multi-Omics Factor Analysis—a framework for unsupervised integration of multi-omics data sets. Molecular systems biology 2018, 14, e8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoadley KA; Yau C; Wolf DM; Cherniack AD; Tamborero D; Ng S; Leiserson MD; Niu B; McLellan MD; Uzunangelov V Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin E; Lane H-Y Machine learning and systems genomics approaches for multi-omics data. Biomarker research 2017, 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan J; Risacher SL; Shen L; Saykin AJ Network approaches to systems biology analysis of complex disease: integrative methods for multi-omics data. Briefings in bioinformatics 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Obermeyer Z; Emanuel EJ Predicting the future—big data, machine learning, and clinical medicine. The New England journal of medicine 2016, 375, 1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deo RC Machine learning in medicine. Circulation 2015, 132, 1920–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Libbrecht MW; Noble WS Machine learning applications in genetics and genomics. Nature Reviews Genetics 2015, 16, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinton GE; Salakhutdinov RR Reducing the dimensionality of data with neural networks. science 2006, 313, 504–507. [DOI] [PubMed] [Google Scholar]
- 18.Rumelhart DE; Hinton GE; Williams RJ Learning representations by back-propagating errors, nature 1986, 323, 533. [Google Scholar]
- 19.Ma T; Zhang A Multi-view Factorization AutoEncoder with Network Constraints for Multi-omic Integrative Analysis. arXiv preprint arXiv:1809.01772 2018. [Google Scholar]
- 20.Bengio Y Learning deep architectures for AI. Foundations and trends® in Machine Learning 2009, 2, 1–127. [Google Scholar]
- 21.LeCun Y; Bengio Y; Hinton G Deep learning, nature 2015, 521, 436. [DOI] [PubMed] [Google Scholar]
- 22.Min S; Lee B; Yoon S Deep learning in bioinformatics. Briefings in bioinformatics 2017, 18, 851–869. [DOI] [PubMed] [Google Scholar]
- 23.Nguyen TV; Mirza B Dual-layer kernel extreme learning machine for action recognition. Neurocomputing 2017, 260, 123–130. [Google Scholar]
- 24.Ching T; Himmelstein DS; Beaulieu-Jones BK; Kalinin AA; Do BT; Way GP; Ferrero E; Agapow P-M; Zietz M; Hoffman MM Opportunities and obstacles for deep learning in biology and medicine. Journal of The Royal Society Interface 2018, 15, 20170387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan J; Ung M; Cheng C; Greene CS Unsupervised feature construction and knowledge extraction from genome-wide assays of breast cancer with denoising autoencoders. In Proceedings of the Pacific Symposium on Biocomputing Co-Chairs; World Scientific, 2014; pp. 132–143. [PMC free article] [PubMed] [Google Scholar]
- 26.Lau E; Cao Q; Ng DC; Bleakley BJ; Dincer TU; Bot BM; Wang D; Liem DA; Lam MP; Ge J A large dataset of protein dynamics in the mammalian heart proteome. Scientific data 2016, 3, 160015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J; Choi H; Chung NC; Cao Q; Ng DC; Mirza B; Scruggs SB; Wang D; Garlid AO; Ping P Integrated Dissection of Cysteine Oxidative Post-translational Modification Proteome During Cardiac Hypertrophy. Journal of proteome research 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kingma DP; Welling M Auto-encoding variational bayes. arXiv preprint arXiv: 1312.6114 2013. [Google Scholar]
- 29.Hochreiter S; Schmidhuber J Long short-term memory. Neural computation 1997, 9, 1735–1780. [DOI] [PubMed] [Google Scholar]
- 30.Graves A; Schmidhuber J Framewise phoneme classification with bidirectional LSTM and other neural network architectures. Neural Networks 2005, 18, 602–610. [DOI] [PubMed] [Google Scholar]
- 31.Greff K; Srivastava RK; Koutník J; Steunebrink BR; Schmidhuber J LSTM: A search space odyssey. IEEE transactions on neural networks and learning systems 2017, 28, 2222–2232. [DOI] [PubMed] [Google Scholar]
- 32.Hochreiter S; Bengio Y; Frasconi P; Schmidhuber J Gradient flow in recurrent nets: the difficulty of learning long-term dependencies; A field guide to dynamical recurrent neural networks. IEEE Press, 2001; [Google Scholar]
- 33.Guo X; Liu X; Zhu E; Yin J Deep clustering with convolutional autoencoders. In Proceedings of the International Conference on Neural Information Processing; Springer, 2017; pp. 373–382. [Google Scholar]
- 34.Xie J; Girshick R; Farhadi A Unsupervised deep embedding for clustering analysis. In Proceedings of the International conference on machine learning; 2016; pp. 478–487. [Google Scholar]
- 35.Guo X; Gao L; Liu X; Yin J Improved deep embedded clustering with local structure preservation. In Proceedings of the International Joint Conference on Artificial Intelligence (IJCAI-17); 2017; pp. 1753–1759. [Google Scholar]
- 36.Fabregat A; Jupe S; Matthews L; Sidiropoulos K; Gillespie M; Garapati P; Haw R; Jassal B; Korninger F; May B The reactome pathway knowledgebase. Nucleic acids research 2017, 46, D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacQueen J Some methods for classification and analysis of multivariate observations. In Proceedings of the Proceedings of the fifth Berkeley symposium on mathematical statistics and probability; Oakland, CA, USA, 1967; Vol. 1, pp. 281–297. [Google Scholar]
- 38.Hartigan JA; Wong MA Algorithm AS 136: A k-means clustering algorithm. Journal of the Royal Statistical Society. Series C (Applied Statistics) 1979, 28, 100–108. [Google Scholar]
- 39.Johnson SC Hierarchical clustering schemes. Psychometrika 1967, 32, 241–254. [DOI] [PubMed] [Google Scholar]
- 40.KAUFMANN L Clustering by Means of medoids. In Proceedings of the Proc. Statistical Data Analysis Based on the L1 Norm Conference, Neuchatel, 1987; 1987; pp. 405–416. [Google Scholar]
- 41.Petkov PM; Ding Y; Cassell MA; Zhang W; Wagner G; Sargent EE; Asquith S; Crew V; Johnson KA; Robinson P An efficient SNP system for mouse genome scanning and elucidating strain relationships. Genome research 2004, 14, 1806–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rau CD; Wang J; Avetisyan R; Romay M; Martin L; Ren S; Wang Y; Lusis AJ Mapping genetic contributions to cardiac pathology induced by Beta-adrenergic stimulation in mice. Circulation: Genomic and Precision Medicine 2014, CIRCGENETICS. 113.000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Buuren S; Brand JP; Groothuis-Oudshoorn CG; Rubin DB Fully conditional specification in multivariate imputation. Journal of statistical computation and simulation 2006, 76, 1049–1064. [Google Scholar]
- 44.Buuren S van; Groothuis-Oudshoorn, K. mice: Multivariate imputation by chained equations in R. Journal of statistical software 2010, 1–68. [Google Scholar]
- 45.Abadi M; Barham P; Chen J; Chen Z; Davis A; Dean J; Devin M; Ghemawat S; Irving G; Isard M Tensorflow: a system for large-scale machine learning. In Proceedings of the OSDI; 2016; Vol. 16, pp. 265–283. [Google Scholar]
- 46.Chollet F Keras; 2015; https://github.com/keras-team/keras;
- 47.Consortium U UniProt: the universal protein knowledgebase. Nucleic acids research 2018, 46, 2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kanehisa M; Furumichi M; Tanabe M; Sato Y; Morishima K KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic acids research 2016, 45, D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fabregat A; Sidiropoulos K; Viteri G; Forner O; Marin-Garcia P; Arnau V; D’Eustachio P; Stein L; Hermjakob H Reactome pathway analysis: a high-performance in-memory approach. BMC bioinformatics 2017, 18, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pedregosa F; Varoquaux G; Gramfort A; Michel V; Thirion B; Grisel O; Blondel M; Prettenhofer P; Weiss R; Dubourg V Scikit-learn: Machine learning in Python. Journal of machine learning research 2011, 12, 2825–2830. [Google Scholar]
- 51.Rousseeuw PJ Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. Journal of computational and applied mathematics 1987, 20, 53–65. [Google Scholar]
- 52.Zeiler MD ADADELTA: an adaptive learning rate method. arXiv preprint arXiv:1212.5701 2012. [Google Scholar]
- 53.McClatchy DB; Ma Y; Liem DA; Ng DC; Ping P; Yates JR III Quantitative temporal analysis of protein dynamics in cardiac remodeling. Journal of molecular and cellular cardiology 2018, 121, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lam MP; Wang D; Lau E; Liem DA; Kim AK; Ng DC; Liang X; Bleakley BJ; Liu C; Tabaraki JD Protein kinetic signatures of the remodeling heart following isoproterenol stimulation. The Journal of clinical investigation 2014, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Narula J; Haider N; Virmani R; DiSalvo TG; Kolodgie FD; Hajjar RJ; Schmidt U; Semigran MJ; Dec GW; Khaw B-A Apoptosis in myocytes in end-stage heart failure. New England Journal of Medicine 1996, 335, 1182–1189. [DOI] [PubMed] [Google Scholar]
- 56.Saraste A; Voipio-pulkki L; Parvinen M; Pulkki K Apoptosis in the heart. The New England journal of medicine 1997, 336, 1025–1026. [DOI] [PubMed] [Google Scholar]
- 57.Roe ND; Xu X; Kandadi MR; Hu N; Pang J; Weiser-Evans MC; Ren J Targeted deletion of PTEN in cardiomyocytes renders cardiac contractile dysfunction through interruption of Pink1–AMPK signaling and autophagy. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2015, 1852, 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang W; Wu Z; Yang K; Han Y; Chen Y; Zhao W; Huang F; Jin Y; Jin W BMI1 promotes cardiac fibrosis in ischemia-induced heart failure via the PTEN-PI3K/Akt-mTOR signaling pathway. American Journal of Physiology-Heart and Circulatory Physiology 2018. [DOI] [PubMed] [Google Scholar]
- 59.Kapustian LL; Vigontina OA; Rozhko OT; Ryabenko DV; Michowski W; Lesniak W; Filipek A; Kroupskaya IV; Sidorik LL Hsp90 and its co-chaperone, Sgt1, as autoantigens in dilated cardiomyopathy. Heart and vessels 2013, 28, 114–119. [DOI] [PubMed] [Google Scholar]
- 60.Datta R; Bansal T; Rana S; Datta K; Chaudhuri RD; Chawla-Sarkar M; Sarkar S Myocyte-derived Hsp90 modulates collagen upregulation via biphasic activation of STAT-3 in fibroblasts during cardiac hypertrophy. Molecular and cellular biology 2017, 37, e00611–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jewison T; Su Y; Disfany FM; Liang Y; Knox C; Maciejewski A; Poelzer J; Huynh J; Zhou Y; Arndt D SMPDB 2.0: big improvements to the Small Molecule Pathway Database. Nucleic acids research 2013, 42, D478–D484. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.