

Abstract
Intervertebral disc degeneration is an important contributor to chronic low back and neck pain. Although many environmental and genetic factors are known to contribute to disc degeneration, age is still the most significant risk factor. Recent studies have shown that senescence may play a role in age-related disc degeneration and matrix catabolism in humans and mouse models. Clearance of p16Ink4a-positive senescent cells reduces the degenerative phenotype in many age-associated diseases. Whether p16Ink4a plays a functional role in intervertebral disc degeneration and senescence is unknown. We first characterized the senescence status of discs in young and old mice. Quantitative histology, gene expression and a novel p16tdTom reporter mice showed an increase in p16Ink4a, p21 and IL-6, with a decrease in Ki67 with aging. Accordingly, we studied the spinal-phenotype of 18-month-old mice with conditional deletion of p16Ink4a in the disc driven by Acan-CreERT2 (cKO). The analyses of discs of cKO and age-matched control mice showed little change in cell morphology and tissue architecture. The cKO mice exhibited changes in functional attributes of aggrecan as well as in collagen composition of the intervertebral disc. While cKO discs exhibited a small decrease in TUNEL positive cells, lineage tracing experiments using ZsGreen reporter indicated that the overall changes in cell fate or numbers were minimal. The cKO mice maintained expression of NP-cell phenotypic markers CA3, Krt19 and GLUT-1. Moreover, in cKO discs, levels of p19Arf and RB were higher without alterations in Ki67, γH2AX, CDK4 and Lipofuscin deposition. Interestingly, the cKO discs showed lower levels of SASP markers, IL-1β, IL-6, MCP1 and TGF-β1. These results show that while, p16Ink4a is dispensable for induction and maintenance of senescence, conditional loss of p16Ink4a reduces apoptosis, limits the SASP phenotype and alters matrix homeostasis of disc cells.
Keywords: p16, p19, Ink4a, senescence, mouse models, intervertebral disc degeneration, aggrecan, extracellular matrix, nucleus pulposus, aging, SASP
Introduction
The intervertebral disc provides flexibility and a range of motion to the spine and accommodates compressive and tensile biomechanical forces. The disc comprises three tissue compartments: the central nucleus pulposus (NP) - rich in aggrecan and populated with cells derived from the notochord, the circumferential, ligamentous annulus fibrous (AF) - that encompasses the NP and is primarily composed of collagens, and the cartilaginous endplates (EP) bordering the NP and AF on cranial and caudal surfaces [1]. The high concentration of negatively-charged glycosaminoglycans present on aggrecan, the predominant proteoglycan in NP and other proteoglycans such as versican is responsible for tissue hydration and osmotic properties of the disc. Furthermore, due to the disc architecture, the NP is a completely aneural and avascular tissue. Consequently, NP cells are adapted to a hypoxic microenvironment [1,2]. In fact, several studies have associated intervertebral disc degeneration with disruption of pathways that allow NP cells to adapt to hypoxia and cell stress [3,4]. The etiology of disc degeneration is multifactorial; abnormal loading, genetics, obesity, have all been shown to contribute to the disease, yet aging is considered the main risk factor [4–6]. With age, there is a decrease in levels and quality of extracellular matrix proteins, biomechanical properties, as well as an increase in inflammatory cytokine expression, catabolic processes, and cell death [5,7,8]. In both human and mouse models, age-related senescence has been correlated with degenerative phenotypes of the disc [9,10]. Despite a known relationship between senescence and disc degeneration, the contribution of cellular senescence to disease progression is not defined [11].
Several molecular networks are involved in promoting the senescent phenotype; among these, p53 and p16Ink4a are considered the master regulators of cell growth arrest, apoptotic-resistance, and senescence-associated secretory phenotype (SASP) [11,12]. Furthermore, many factors, including telomere attrition, oncogenes, and cell stress (e.g. oxidative, genotoxic, cytokine) contribute to p53 and p16Ink4a activation [11,12]. Activation of these pathways and ensuing senescence leads to the expression of cytokines, chemokines, and other SASP proteins culminating in inflammation and tissue degeneration [11]. NP cells have been shown to respond to oxidative stress and cytokines, like TNF, resulting in increased senescence and apoptosis, and decreased matrix production [13,14]. However, the molecular pathways that control senescence in the NP in response to these stressors, and their contribution to the degenerative cascade in vivo is unknown.
p16Ink4a is a potent inhibitor of proliferation that disrupts cell cycle progression by altering the association between cyclin dependent kinase 4 and 6 (CDK4/6) and cyclin D1 [15]. Due to its robust correlation with other senescence markers like excessive lysosomal activity analyzed by β-galactosidase (SA-βgal), phosphorylated histone H2A (γH2AX) and Ki67 expression, p16Ink4a is accepted as the key maker of chronic senescence [16,17]. Recent studies have shown that clearance of p16Ink4a-expressing cells mitigates some aspects of degenerative aging [18–21]. Accordingly, senolytic molecules that induce apoptosis of senescent cells have been effective in ameliorating age-related pathologies, including disc degeneration in a models of accelerated aging [22] and post-traumatic osteoarthritis [23]. However, it is still unknown whether p16Ink4a drives senescence-related dysfunction or whether other features of senescent cells mediate these effects.
To understand the importance of p16Ink4a in onset and maintenance of senescence and in the progression of age-associated disc degeneration, we characterized senescence status of mouse discs with aging and analyzed the spinal phenotype of 18-month old Acan-CreERT2-p16Ink4a conditional knockout (cKO) mice.
Results
Senescence and p16Ink4a expression and collagen fiber thickness increase with age
Increased p16Ink4a expression in aged and degenerated human intervertebral discs has been reported [9]. However, the relationship between p16Ink4a and aging is not characterized in murine disc. We performed analysis of discs from 5-and 18-month-old mice to investigate senescence and p16Ink4a expression. Histological studies showed changes in disc architecture as characterized by a noticeable decrease in NP cell vacuoles and thinning of cell band in the 18-month-old mice (Fig. 1A–C’). Picro-Sirius Red staining and polarized microscopy showed decrease in content of thin collagen fibers with concomitant increase in medium and thick fibers with aging (Fig. 1D–F). Lipofuscin, which represents deposition of oxidized molecules and correlates with senescence, showed increased accumulation in the 18-month-old mice (Fig. 1G–G’) [24]. There was an increase in p21 expression with decrease in Ki67 levels in 18-month-old mice (Fig. 1H–I’). Likewise, while the expression of IL-1β (Fig. 1J–J’) showed no change, there was a significant increase in IL-6 expression in aged animals (Fig. 1K–L). We performed gene expression analysis and found no changes in p53 and p19Arf expression, but a significant increase in p16Ink4a expression in NP of old mice (Fig. 1M–O). We also analyzed localization and levels of p16Ink4a protein in discs of 18-month-old mice that contained tdTom reporter driven by the native p16Ink4a promoter and lox-stop-lox ZsGreen reporter driven by a tamoxifen-inducible Aggrecan-Cre driver (Acantm1(cre/ERT2)Crm) [25]. Compared to young mice (Fig. 1P–P’), there was a robust p16Ink4a expression as evidence by tdTom staining in the NP compartment of 18-month-old mice (Fig. 1Q–Q’). All p16tdTom positive cells co-expressed ZsGreen driven by Acan-CreERT2. The specificity of tdTom signal was confirmed by using mice that lacked tdTom reporter but expressed ZsGreen (Suppl. Fig. 1). These results clearly showed an overall increase in senescence status and p16Ink4a expression with aging.
Figure 1. Senescence and p16Ink4a expression increase with age. (A-C’).

18-month-old mice showed a decrease in vacuoles and cell band width (arrows) and comparable features of the NP/AF junction. (D-F) Picrosirius red staining (D-D’) and quantitative polarized imaging (E, E’) showed a decrease in thin collagen fibers along with an increase of medium and thick fibers compare to 5-month-old mice (F). P < 0.05, χ2 test, N = 4–6 mice/group, 4 discs/animal. (G-G’) Sudan-Black-B staining showed an increase in Lipofuscin aggregates deposition in NP and AF of old mice (arrows). (H-I’) There was a significant decrease in Ki67 and an increase of p21 expression in all disc compartments (arrows). (J-J’) Analysis of SASP showed comparable levels of IL-1β with aging. (K-K’) IL-6 expression was significantly higher in the older mice. (L) Quantification of % positive staining for KI67, p21, IL-1β and IL-6 expression. (M-N) p53 and p19Arf mRNA levels were similar between 5-and 18-month old mice(O) p16Ink4amRNA levels were significantly higher in 18-month old mice compared to 5-month old animals.(P-Q’) Acantm1(cre/ERT2)Crm; p16tdTom;Rosa-ZsGreen 18-month-old mice showed robust expression of p16Ink4a (tdTOM/Red) in the NP with strong colocalization with Aggrecan-Cre positive cells (ZsGreen), p16Ink4a levels were low in 5-moth-old mice. For quantitative immunohistology and RNA analysis, Mann-Whitney test was used for comparing differences between the groups. NS = not significant; p ≤ 0.05 *; p ≤ 0.01 **; N=6 animals/genotype were analyzed. Scale bar A-A’, P-Q’ = 200 µm; Scale bar B-E’’, G-K’ = 50 µm.
Loss of p16Ink4a in the disc does not protect mice from age-dependent degeneration
Due to the established correlation between p16Ink4a expression, aging, and disc degeneration, we examined the effects of conditional loss of p16Ink4a on the intervertebral disc health. We analyzed spines from 18-month old mice containing alleles for the inducible knock-out of p16Ink4a (p16L)[26] driven by an inducible Aggrecan Cre driver (Acantm1(cre/ERT2)Crm)[27] (Fig. 2A). Use of a lox-stop-lox ZsGreen reporter clearly showed a robust tamoxifen induced Cre recombination driven by Acan-CreERT2 in NP, inner AF, and EP as well as in the growth-plate (Fig. 2B). The functional effects of p16Ink4a loss in the disc were investigated by analyzing 18-month-old animals with or without p16Ink4a deletion. Histological analysis showed no differences in overall disc architecture and cell morphology in the NP, AF, or EP compartments in the tamoxifen treated wild-type (WT): Acan-CreERT2; p16Ink4a+/+, conditional knock-out (cKO): Acan-CreERT2;p16Ink4af/f, or animals without tamoxifen-treatment (nT): Acan-CreERT2;p16Ink4af/f (Fig. 2C–F’’). Picro-Sirius Red staining and polarized microscopy showed comparable collagen content, architecture and fiber thickness between wild type and cKO mice (Fig. 2G–I). However, tamoxifen treated groups (WT and cKO) exhibited lower levels of thicker collagen fibers, when compared to non-treated animals. The disc histology was quantified using the modified Thompson Grading Scale [4]. There were no significant differences in the average grades of NP and AF tissue among all the groups (Fig. 3A, B). Likewise, level-by-level analysis of histopathological grades of NP and AF tissues for L¾, L4/5, L5/6 and L6/S1 showed comparable scores among all the groups except for the NP scores at L6/S1 level. At this lowest lumber level, tamoxifen treated wild type and cKO animals showed similar histological scores, but they were higher than the untreated animals. This difference in histological score at L6-S1 in WT and cKO groups compared to non-treated animals, suggested some negative effects of tamoxifen on disc health and possibly a positional contribution to this susceptibility (Fig. 3C, D).
Figure 2. Loss of p16Ink4a in the disc does not protect mice from age-dependent degeneration.

(A) Schematic showing protocol for generation of experimental and control mice. Analysis was performed using tamoxifen treated 18-month-old wild-type (Acan-CreERT2-p16Ink4a wt/wt) and p16Ink4a conditional knock-out (Acan-CreERT2-p16Ink4a f/f) and untreated (Acan-CreERT2-p16Ink4af/f) animals. (B) Specificity of Acan-CreERT2 for targeting different compartments within the intervertebral disc in skeletally mature mice is shown. Acan-Cre showed a high recombination in NP, inner AF, endplate (EP) as well as growth plate (GP), when tamoxifen was administered at 4-month. (C-F’’) There are no noticeable differences in overall intervertebral disc (IVD) architecture or cellular morphology in the tamoxifen treated wild-type (WT) (C-F), conditional knock-out (cKO) (C’-F’), and animals without tamoxifen-treatment (nT) (C’’-F’’) groups as shown by representative histological images. Higher magnification images of NP tissue (D-D”) and tissue interfaces between NP/AF (E-E’’) and NP/EP (F-F’’) show comparable cell morphology and tissue architecture between all the groups. Picrosirius red staining of discs showed similar collagen content among the groups (G-G’’). There were no differences in collagen organization (H-H’’) and distribution of collagen fibers thickness (I) between the WT and cKO mice as seen by quantitative polarized microscopy. Non-treated animals showed higher content of mature collagen than tamoxifen treated WT and cKO animals. P < 0.05, χ2 test, N = 6 mice/group, 4 discs/animal. Scale bar B-C” and G-H´´= 200 µm; Scale bar D-F” = 50 µm.
Figure 3. Loss of p16Ink4a in the disc does not protect mice from age-dependent degeneration.
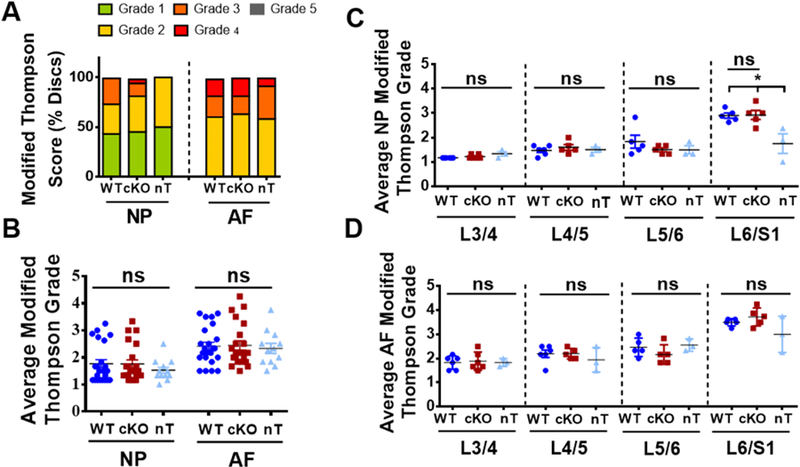
(A) Modified Thompson Grading showed no differences in the distribution of histopathological grades for both NP and AF amongst the groups. (B) The average NP and AF grades were similar for WT, cKO and nT animals. (C, D) Level-by-level analysis showed similar average grades for both NP (C) and AF (D) across the four lumbar levels L¾, L4/5, L5/6 and L6-S1 amongst all groups except for the average NP grade at L6/S1, where untreated animals showed a lower grade of degeneration than tamoxifen treated groups. 4 lumbar discs/mouse, 6 mice for WT and cKO and 3 mice for nT groups were analyzed for comparisons. Kruskal-Wallis test followed by Dunn’s multiple comparison test was used to test differences between groups showing average Thompson grading data. χ2 test was used to analyze differences between groups showing distribution of percent-degenerated-discs. NS = not significant; p ≤ 0.05 *.
p16Ink4a cKO mice have decreased NP cell death without significantly altering overall cell number and molecular phenotype of cells
We investigated if loss of p16Ink4a in the disc affects cell viability, and phenotype of NP cells. To assess cell death, we performed TUNEL staining. While the overall percentage of TUNEL positive cells was low, 18-month old cKO animals had fewer positive cells, most notably in the NP compartment than the aged matched WT controls (Fig. 4A–B). Importantly, the majority of NP and AF cells in cKO and WT animals were healthy and viable at 18-months. To further investigate the effects of p16ink4a deletion on disc compartments, we performed a fate mapping experiment where cells expressing Cre recombinase under the control of aggrecan promoter following tamoxifen treatment were marked by ZsGreen with or without deletion of p16Ink4a. It was evident that in 18-month-old mice, the ZsGreen labeled cells were retained in the NP and inner AF of both the control and cKO mice and the number of marked cells in tissue compartments were comparable between the genotypes (Fig. 4C–D’). To assess the health and integrity of the NP cells in the cKO animals, we evaluated the expression of NP phenotypic markers GLUT1, CA3, and Krt19 [28,29]. The NP cells from both the p16Ink4a cKO and WT mice robustly expressed NP phenotypic makers, and there were no discernable differences in levels of expression between the genotypes (Fig. 4E–G’). These studies clearly suggested that while the deletion of p16Ink4a provided some protection from cell death, it did not result in a substantial alteration in disc cell number or their fate.
Figure 4. p16Ink4a cKO mice have decreased NP cell death without significant altering cell number and phenotype.

(A-A’’) TUNEL assay showed a decrease in number of dying cells (arrows) in NP compartment of the cKO mouse. (B) There was a decrease in TUNEL positive cells in cKO mice. t-test for normally distributed data, Mann-Whitney test for non-normally distributed data, Shapiro-Wilk normality test was done to check the distribution. NS = not significant; p ≤ 0.05 *; p ≤ 0.01 **; N=6 animals/genotype were analyzed. (C, D’) Fate mapping of disc cells in p16Ink4a cKO and control mice. Tamoxifen treatment of mice (at 4 and 12-months) induced Cre-recombinase activity in aggrecan expressing cells resulting in simultaneous marking of cells by ZsGreen with or without deletion of p16Ink4a. Analysis of these mice at 18-months showed that almost all cells in NP, inner AF and CEP were ZsGreen positive. Cell numbers of ZsGreen positive cells in tissue compartments were comparable between cKO and control animals. (E-G’) Characterization of NP phenotypic markers in p16Ink4a cKO mice. Localization and expression levels of glucose transporter 1 (GLUT1), carbonic anhydrase 3 (CA3) and Keratin19 (Krt19) in the NP were comparable between control and cKO animals. Mann-Whitney test was used for comparing differences between the groups. NS = not significant; p ≤ 0.05 *; p ≤ 0.01 **; N=6 animals/genotype were analyzed. Scale bar = 200µm.
p16Ink4a cKO mice evidenced changes in functional attributes of aggrecan and collagen composition of the disc
Extracellular matrix molecules are integral for the biomechanical and osmotic properties of the healthy intervertebral disc [30]. The progression of disc degeneration is closely linked to alteration in matrix composition, elevated matrix catabolism, and expression of enzymes such as A Desintegrin-Like And Metalloprotease (Reprolysin Type) WithThrombospondin Type 1 (ADAMTS) and matrix metalloproteases (MMPs) which degrade proteoglycans and collagens. Therefore, we assessed whether p16Ink4a cKO altered the matrix composition of the disc by evaluating the expression and localization of both collagens and aggrecan [31]. It was evident that p16Ink4a cKO animals evidenced decreased expression of both collagen I and II in the NP compartment (Fig. 5A–B’, D). On the other hand, collagen X, a maker of hypertrophic chondrocytes often expressed during disc degeneration, was expressed at a higher level in cKO than WT controls (Fig. 5C–D) suggesting changes in the collagen composition of the NP [4]. We assessed the aggrecan content and quality in the discs by evaluating expression levels of aggrecan core protein, chondroitin sulfate (the major glycosaminoglycan of aggrecan), and the aggrecan degradation product ARGXX [31]. Interestingly, while overall levels of aggrecan in cKO were robust and comparable to the WT controls, there was a decrease in staining for both chondroitin sulfate and ARGXX (Fig. 5E–G’’’, I). Additionally, we measured the expression of cartilage oligomatrix protein (COMP) which stabilizes collagens [32]. Both cKO and WT mice showed comparable expression of COMP with prominent localization in outer AF and the NP compartments. Taken together, these results showed that while the levels aggrecan core protein were maintained in cKO mice, there were differences in the functional attributes of the resident aggrecan molecules with changes in collagen composition of the NP (Fig. D and I).
Figure 5. p16Ink4a cKO mice alter aggrecan functionality and expression of major extracellular matrix collagens of the disc.

(A-B’) cKO mice show decreased levels of collagen I and collagen II expression in the NP. (C-D) Collagen X was expressed in both groups with an increasing expression in cKO. (E-E) Analysis of aggrecan (ACAN) and its functional attributes. Expression of ACAN core protein was similar between the two genotypes (E-E’). Levels of chondroitin sulfate (CS) in NP (F-F’, I) and aggrecanases generated degradation product, ARGxx in AF were lower cKO group (G-G’’’, I). (H-I’). Mann-Whitney test was used for comparing differences between the groups. NS = not significant; p ≤ 0.05 *; p ≤ 0.01 **. N = 6 animals/genotype, 1–2 discs per animal, were analyzed. Scale bar = 200 µm (A-A’, B-C’, E-G’, H-H’ ) and 20 µm (A’’-A’’’, C’’-C’’’, G’’-G’’).
p16Ink4a cKO shows activation of p19Arf and RB without affecting the prevalence of senescent cells in the intervertebral disc
To evaluate the presence of senescent cells in the discs of cKO mice we examined the expression of several markers associated with the senescent phenotype. Sudan-Black B staining was used to specifically mark lipofuscin deposition, a hallmark of senescent cells [24,33]. There was no difference in lipofuscin deposition between the WT and cKO mice (Fig. 6A–B’’). Interestingly, neither WT nor cKO animals expressed appreciable levels of γH2AX, a known marker of senescence and DNA damage, in the NP but showed staining in the AF compartment (Fig. 6C–C’). On the other hand, CDK4 and Ki67 expression, which mark proliferating cells and are negatively correlated with senescence, was only seen in the proliferative bone marrow, and absent from NP, AF, or EP tissue compartments (Fig. 6D–F). These results suggested that deletion of p16Ink4a did not decrease cell senescence in the intervertebral disc. We then evaluated the expression of p21, a downstream effector of p16Ink4a and a marker of senescence, along with other mediators of senescence. p21 was comparably expressed in all disc compartments, again suggesting maintenance of this downstream effector even in the absence of p16Ink4a (Fig. 6G–G’, J). Interestingly, Retinoblastoma (RB) protein, a potent cell cycle inhibitor and a key downstream mediator of senescence induction, was considerably higher in cKO mice (Fig. 6H–H’). Consequently, we explored status of a potential upstream compensatory pathway that could account for the persistence of senescent cells in the p16Ink4a cKO mice. The p19Arf pathway, consisting of p19Arf and its effector protein p21, has been shown to compensate for loss of p16Ink4a. Discs of cKO animals showed an increase in p19Arf expression in the AF compartment (Fig. 6I–J). Given these results, it is plausible that loss of p16Ink4a did not affect the senescent phenotype of the intervertebral disc cells, possibly due to compensation by other drivers of cell senescence such as p19Arf and RB [34,35].
Figure 6. p16Ink4a cKO shows activation of p19Arf and RB without affecting the prevalence of senescent cells in the intervertebral disc.

(A-B’’) Sudan-Black-B staining showed comparable lipofuscin aggregates in WT and cKO mice. (C-F) There were no discernable changes in γH2AX or CDK4 and KI67 expression levels in the discs of the cKO and WT mice. (G-G’, J) p21 was expressed in all disc compartments in cKO and control animals. (H-H’) RB was higher expressed in overall disc of cKO mice. (I-J) cKO mice showed pronounced increase in p19Arf staining in the cells of inner AF and GP cells. For quantitative analysis, Mann-Whitney test was used for comparing differences between the groups. NS = not significant; p ≤ 0.05 *; N=6 animals/genotype were analyzed. Scale bar = 200 µm.
p16Ink4a cKO exhibit decreased expression of major regulators of senescence-associated secretory phenotype (SASP)
Although our data suggested that the deletion of p16Ink4a does not decrease the incidence of senescent cells in the disc, we sought to determine if the p16Ink4a null animals were protected from SASP. Activation of SASP proteins leads to overexpression of inflammatory cytokines that contribute to a progressive and degenerative cascade [11]. We observed that expression of MCP1, IL-1β, and IL-6 were all significantly decreased in the discs of cKO mice compared to age-matched controls (Fig. 7A–D). Metalloproteases have been described as an important component of SASP as well as TGFβ regulation [36]. We however found low and comparable expression of MMP-13 in both WT and p16Ink4a cKO animals (Fig. 7E–E’). In contrast, TGF-β1 expression was significantly downregulated in the cKO mice (Fig. 7F–F’). To assess the correlation between autophagic flux and SASP, we measured LC3 positive puncta in cKO mice. There were no overt differences between the groups (Fig. 7G–H,). Taken together, this data clearly suggested that while there is a compensation of p16Ink4a by other inducers of senescence, p16Ink4a deletion results in altered secretory phenotype, suggesting a novel role of p16Ink4a in modulating SASP.
Figure 7. p16Ink4a cKO exhibit decreased expression of major regulators of senescence-associated secretory phenotype (SASP).

(A-C’) cKO animals showed a pronounced decrease in MCP1 (A, A’), IL1β (B, B’) and IL-6 (C, C’) staining compared to control animals (D). (E-E’) There were comparable levels of MMP13 expression between cKO mice and the WT. (F-F’, H) TGF-β1 expression was decreased in cKO discs. Autophagy analysis showed no differences between LC3 Puncta between WT and cKO (G-H). Mann-Whitney test was used for comparing differences between the groups. NS = not significant; p ≤ 0.05 *; N=6 animals/genotype were analyzed. Scale bar = 200 µm. (L) Schematic summarizing contribution of p16Ink4a, p19Arf and RB for senescence status and SASP maintenance in the intervertebral cells.
Discussion
Although a multitude of genetic and environmental factors are known to contribute to the pathogenesis of intervertebral disc degeneration, one of the most prevalent risk factors is age. Several studies have shown that aging causes a degenerative phenotype in the disc, namely loss of water binding aggrecan and altered collagen composition, and an increase in matrix degrading proteases [37–39]. Importantly, it has been reported that both degeneration and aging in human discs is correlated with increased senescence and elevated expression of the known senescence marker, p16Ink4a [9]. Likewise, in a mouse model of progeria due to deficiency in DNA repair enzyme ERCC1, intervertebral discs showed higher p16Ink4a expression [10]. The increase in p16Ink4a was accompanied by up-regulation of SASP and changes in extracellular matrix homeostasis [40]. In this study, we clearly showed that there was an increase in senescence status and p16Ink4a expression in aged mice. Interestingly, detailed analysis of p16Ink4a cKO mice showed an overall normal tissue and cell morphology in the intervertebral discs, including sustained expression of NP phenotypic markers, and healthy levels of aggrecan at 18-months of age. Interestingly, there were changes in the functional attributes of aggrecan molecules and altered collagen composition in cKO mice. TUNEL staining also indicated that the conditional deletion of p16Ink4a offered some protection from cell death. However, while p16Ink4a was redundant for induction of the senescent phenotype in the disc through potential compensatory activation of p19Arf and RB, loss of p16Ink4a decreased SASP, a well-known initiator of inflammatory cytokine expression and downstream matrix catabolism [12]. Based on these novel findings, we concluded that while p16Ink4a alone was not sufficient for controlling the senescent fate of intervertebral disc cells it was involved in controlling the matrix homeostasis and SASP in aging spine.
As expected, discs of 18-month-mice showed increased collagen fiber thickness and altered cell morphology evident by a decrease in the number of vacuoles in comparison to 5-month-old mice. These observations were in line with previous studies investigating aging of the disc and aorta [37,38,41]. Thicker collagen fibers were recently reported in the intervertebral discs of a mouse model of spontaneous, early onset disc degeneration and were correlated with compromised mechanical function [4]. This suggested that the increase in collagen fiber thickness in discs of 18-month-old mice was likely associated with age-dependent changes in structure and biomechanical function. Furthermore, we characterized the senescent phenotype of disc cells using validated markers for: cell cycle (Ki67); cell cycle inhibitors (p21, p19Arf, p53 and p16Ink4a); SASP (IL-1β and IL-6) and oxidized molecular aggregates (Lipofuscin) [11,12,16,24,42]. Our data clearly showed that with aging there was increased evidence of cell senescence in the all the disc compartments. Interestingly, our finding that only p16Ink4a levels in the disc increased with aging without appreciable change in p53 and p19Arf levels, underscores the importance of p16Ink4a as a key marker of age-dependent senescence in murine disc. These results corroborated studies by LeMaitre et al. showing a correlation between p16Ink4a expression, intervertebral disc degeneration, and aging in humans, suggesting the utility of mouse models to assess the causative relationship between p16Ink4a and senescence during intervertebral disc aging [43].
Studies of p16Ink4a cKO suggested that loss of p16Ink4a did not result in overt alteration in disc degeneration and collagen fiber thickness. In addition, the p16Ink4a deletion did not affect expression of NP phenotypic markers. These studies were further supported by fate mapping experiments that employed a Cre-inducible ZsGreen reporter. Most of the cells in the NP and AF compartments of the 18-month-old animals were labeled, and the cellularity of both the compartments in cKO animals was not appreciably different from controls. This was not surprising given a small rescue from cell death as evidenced by fewer TUNEL positive cells was seen in cKO animals. Moreover, the lack of overt changes in cellularity as a consequence of higher cell proliferation in the wild type animals as could be ruled out from low and comparable index of proliferation seen between genotypes by Ki67 and CDK4 staining. It is important to note that there were some changes in the functional attributes of the resident aggrecan molecules. These changes manifested as a decrease in chondroitin sulfate, a critical glycosaminoglycan important for maintenance of the water content of the NP, along with lower accumulation of ARGXX neoepitope in the AF, the product of aggrecanase activity. It is plausible that loss of p16Ink4a in the disc altered the protease activity (ADAMTS, MMPs) with or without concomitant changes in biosynthesis of chondroitin sulphate but not aggrecan core protein [44]. Additionally, the change in aggrecan structure could further hasten its turnover in the cKO mice by increasing the accessibility of its core protein to proteolytic enzymes, altering the mechanical properties of the individual molecules and affecting supra-macromolecular assembly critical for tissue osmotic function [39,45–47]. It is important to note that decreased chondroitin sulfate and increased aggrecan degradation in disc aging models has been reported [40,48]. This change in aggrecan properties coupled with changes in the collagen composition in cKO, decreased collagens I and II and increased collagen X, may affect the overall biomechanics of the spinal motion segment. Although, senescence has been linked to collagen expression in disc cells, the underlying mechanisms of p16Ink4a dependent collagen regulation remains to be explored [49,50]. Noteworthy, other researchers have reported changes in mechanical properties of the disc with age and degeneration due to alteration in the structure and composition of the extracellular matrix [4,51]. Taken together, these observations highlight the novel role of p16Ink4a in matrix homeostasis in the disc [40,49,52].
The absence of overt morphological changes in the cKO mice may be attributed to slow matrix turnover in disc and compensatory activation of an alternative senescent pathway. The INK4a/ARF locus is potently activated with aging, leading to an increase of both p16Ink4a and p19Arf [53]. While there was no change in p19Arf expression in discs during aging, our results clearly showed that in the p16Ink4a cKO animals, there was an increase of both p19Arf and RB expression. This functional compensation is in agreement with studies by Sharpless et al. using the same mouse model of p16Ink4a loss [35]. Supporting this idea further, Feng and colleagues, have shown a p16Ink4a independent increase in senescence in NP cells through activation of p21 and RB when cells were exposed to mechanical stretching [54]. Likewise, recent studies showed that SIRT1 alleviates disc senescence by blocking the p53/p21 pathway in a p16Ink4a independent fashion [55]. Interestingly, p21 levels in cKO mice showed a robust and comparable expression to controls. Thus, the persistence of p21, γH2AX, along with the lack of CDK4 and Ki67 in the p16Ink4a cKO mice may reflect possible activation of the p19Arf and RB pathway that maintains the senescent cells in these discs. This is not unexpected as it has been shown that the positive benefits of p16Ink4a silencing can be masked by p19Arf retention, even causing a predisposition for tumorigenesis [34].
Recent studies have reported SASP as the key mediator of senescent induced degenerative phenotype [12,56]. IL-1β was reported as the trigger for SASP activation, leading to overexpression of both IL-6 and MCP1, which maintain this secretory signature [57]. Interestingly, and in contrast to observations by Coppe et al., our data shows that p16Ink4a plays a role in promoting the expression of IL-1β, IL-6 and MCP1 in the intervertebral disc [58]. In addition to the inflamatory component of SASP, metalloproteases and TGF-β secretion have been shown to contribute to the senescence secretory phenotype [36,59]. Accordingly, TGF-β1 expression, which increases during disc degeneration, was decreased in the p16Ink4a cKO mice, suggesting that p16Ink4a controls pro-fibrotic pathways during aging [60]. Since MMP13 expression, collagen turnover, and the autophagy index was not altered in the cKO animals, this suggests a selective role of p16Ink4a in altering SASP targets and matrix homeostasis [2]. Despite noticeable changes in levels of SASP molecules, inflamatory cytokines and TGF-β1, as well as increased secretion of proteins such as collagen X, there was no major detrimental effect on the morphology of the disc or the grade of degeneration.
Studies in other tissue types have shown that deletion of p16Ink4a expressing cells rescued aging phenotypes by altering multiple cellular senescence pathways [18,20,22,56]. In our study, conditional deletion of p16Ink4a in intervertebral disc cells, that in principle differs from clearance of p16Ink4a-positive cells, seems to affect matrix homeostasis and SASP. This is not uncommon, as in other cell types, such as macrophages, high p16Ink4a expression is not directly associated with senescence, and in articular chondrocytes p16Ink4a is dispensable for induction and maintenance of senescence during aging [16,27]. Although p16Ink4a is widely accepted as a robust marker of cellular senescence in many cell types, including chondrocytes and disc cells, our results confirm that p16Ink4a deletion can be functionally compensated by increased p19Arf and RB without changing the prevalence of senescent cells or the degenerative phenotype. Importantly, our results in the intervertebral disc echo those of articular cartilage and osteoarthritis development, where is reported that expression of p16Ink4a increases with age in both humans and mice, but the conditional loss of p16Ink4a did not prevent senescence in chondrocytes or the development of osteoarthritis [27]. Nevertheless, findings from the current study emphasize that a relationship exists among p16Ink4a, secretory pathways, and matrix functionality and composition in the disc. Therefore, further characterization of role of p16Ink4a in the intervertebral disc is warranted, as it is important to understand its relevance as a marker of senescence and a regulator of SASP. In this context, p16Ink4a could potentially serve, as an indicator of the senescence status of cells of the intervertebral disc, whose health is essential for secretion of water binding proteoglycans and collagens and thus biomechanical function of this complex tissue.
Experimental Procedures
Mice
All animal experiments were performed under protocols approved by the Institutional Animal Care and Use Committees of University of North Carolina and Thomas Jefferson University. Young (5 month) and old (18 month) C57BL/6 mice from NIA aged Rodent colony were analyzed. Wild-type (Acantm1(cre/ERT2)Crmp16Ink4awt/wt) and p16Ink4a conditional knock-out (Acantm1(cre/ERT2)Crmp16Ink4af/f) mice on C57BL/6 background were generated as shown in Figure 1A and as recently described[27]. To determine expression pattern of Cre recombinase and to perform fate mapping experiments, loxP-stop-loxP ZsGreen reporter mouse (Gt(ROSA)26Sortm6(CAG-ZsGreen1)Hze/J (Stock # 007906, Jackson Labs) was crossed with mice with or without conditional p16Ink4a loss. At 4 and 12-months of age, mice received three consecutive doses of 25 mg/kg tamoxifen (Sigma-Aldrich, St. Louis, MO, USA) in corn oil (Sigma-Aldrich) by intra-peritoneal injection to activate the Cre recombinase and analyzed at 18-months to investigate effects of p16Ink4a loss on disc health and fate of cells. For, delineating Cre targeting activity in disc, Acantm1(cre/ERT2)Crm;Rosa-ZsGreen mice were analyzed 2 weeks following tamoxifen injections at 4 month. For investigating the localization and expression of p16Ink4a, we crossed Acantm1(cre/ERT2)Crm;;Rosa-ZsGreen mice with p16tdTom reporter mice to generate Acantm1(cre/ERT2)Crm; p16tdTom;Rosa-ZsGreen mice. p16tdTom reporter mice were generated using the strategy of previous p16Ink4a-based reporter alleles wherein DNA encoding the fluorescent molecule tdTomato was targeted to the native Ink4a locus to replace exon 1alpha[61]. This approach leaves p19Arf intact while replacing one copy of native p16Ink4a with tdTomato. The details and characterization of this allele are described by Liu et al.[25].
Preparation of Mouse Spines for Histological Analysis
Spines were dissected enbloc, and immediately fixed in freshly made 4% PFA in PBS for 48 hours and decalcified in 20% EDTA for 18 days. Intact lumbar motion segments were processed and embedded in paraffin. Spines used for frozen sections were fixed for 2h, followed by 12h 30% Sucrose immersion, OCT embedding and snap freezing using liquid nitrogen. Mid coronal section, 7 µm in thickness, were cut from lumbar levels (L3–4, L4–5, L5-L6, L6-S1) of each mouse. Sections were stained with Safranin-O (proteoglycans)/Fast Green (collagen)/Hematoxylin (nuclei) for assessing gross histology or Picrosirius red, to visualize the collagen content in the intervertebral disc. Sundan-Black-B/Fast Red staining was used for detection of lipofuscin deposition. Staining was using a light microscope (Axio Imager 2, Carl Zeiss) or a polarizing microscope (Eclipse LV100 POL, Nikon). Imaging of Safranin O/Fast Green/hematoxylin and Sundan-Black-B/Fast Red stained tissues were performed using 5×/0.15 N-Achroplan (Carl Zeiss) or 20×/0.5 EC Plan-Neofluar (Carl Zeiss) objectives, Axiocam 105 color camera (Carl Zeiss), and Zen2™ software (Carl Zeiss). For Picrosirius red stained tissues, 10×/0.25 Pol/WD 7.0 objective (Nikon), Digital Sight DS-Fi2 camera (Nikon), and NIS Elements Viewer software (Nikon) were used. To evaluate degeneration of disc, mid-coronal sections from all 4 lumbar disc levels per mouse were scored using a modified Thompson grading scale by 4 blinded observers. Histopathological scores were obtained from n = 6 mice per group with 4 lumbar discs per mouse (total 24 discs per group).
Immunohistological staining
Coronal lumbar disc tissue sections of 7 μm thickness were de-paraffinized and incubated either in microwaved citrate buffer for 20 min, with proteinase K for 10 min at room temperature, or with Chondroitinase ABC for 30 min at 37 °C for antigen retrieval. Then the sections were blocked in 5% normal serum (Thermo Fisher Scientific, 10000 C) in PBS-T (0.4% Triton X-100 in PBS), and incubated with antibodies against Collagen I (1:100, Abcam ab34710), collagen II (1:400, Fitzgerald 70R-CR008), collagen X (1:500, Abcam ab58632), aggrecan (1:50, Millipore AB1031); chondroitin sulfate (1:300, Abcam ab11570 ) and COMP (1:200, Abcam ab231977); NP phenotypic markers: CA3 (1:150, SantaCruz) and Keratine19 (1:50, DSHB TROMA-III); senescent markers: tdTOM/RFP (1:100, Rockland 600–401-379); p19Arf (1:200, Novus NB200–106), p21 (1:200, Novus NB100–1941), RB (1:100, Abcam, ab85607), CDK4 (1:50, Santa Cruz DCS-35), Ki67 (1:100, Abcam ab15580) and γH2AX (1:200, Cell Signaling 9718) and SASP: IL1β (1:100, Novus NB600–633), IL-6 (1:50, Novus NB600–1131), MCP1 (1:150, Abcam ab25124), TGF-β1 (1:100, Abcam ab92486) and MMP13 (1:200, Abcam ab39012); autophagy evaluation: LC3 (1:200, Novus NB100–2220) in blocking buffer at 4 °C overnight. For GLUT-1 (1:200, Abcam, ab40084) and ARGxx (1:200, Abcam, ab3773) staining, Mouse on Mouse Kit (Vector laboratories, BMK-2202) was used for blocking and primary antibody incubation. Tissue sections were thoroughly washed and incubated with Alexa Fluor-594 conjugated secondary antibody (Jackson ImmunoResearch Lab, Inc.), at a dilution of 1:700 for 1 h at room temperature in dark. The sections were washed again with PBS-T (0.4% Triton X-100 in PBS) and mounted with ProLong® Gold Antifade Mountant with DAPI (Thermo Fisher Scientific, P36934). All mounted slides were visualized with Axio Imager 2 (Carl Zeiss) using 5×/0.15 N-Achroplan (Carl Zeiss) or 20×/0.5 EC Plan-Neofluar (Carl Zeiss) objectives, X-Cite® 120Q Excitation Light Source (Excelitas), AxioCam MRm camera (Carl Zeiss), and Zen2™ software (Carl Zeiss). LC3-positive puncta were examined with Nikon A1R confocal microscope. Positive staining for markers and LC3 puncta were measured by area (pixel2/area) using ImageJ software (http://rsb.info.nih.gov/ij/).
TUNEL assay
TUNEL assay was performed on lumbar disc sections using “In situ cell death detection” Kit (Roche Diagnostic). Briefly, sections were de-paraffinized and permeabilized using Proteinase K (20 μg/mL) for 15 min at room temperature. TUNEL assay was then carried out per manufacturer’s protocol. The sections were washed and mounted with ProLong® Gold Antifade Mountant with DAPI (Thermo Fisher Scientific) and were visualized with Axio Imager 2 microscope (Carl Zeiss).
Tissue RNA isolation and Real time RT-PCR analysis
NP tissue was dissected from lumbar and caudal discs of 5-and 18-month C57BL/6 mice, and immediately placed in RNAlater® Reagent (INVITROGEN). 7 mice per genotype were sacrificed for RNA isolation and NP tissue pooled from single animal served as an individual sample. NP tissue was collected in RNAlater® Reagent and homogenized with a Pellet Pestle Motor (Sigma Aldrich, Z359971). Total RNA was extracted from the tissue lysates using RNeasy® Mini kit (Qiagen). The purified, DNA-free RNA was converted to cDNA using EcoDry™ Premix (Clontech). Template cDNA and gene-specific primers were added to Power SYBR Green master mix (Applied Biosystems) and mRNA expression was quantified using the Step One Plus Real-time PCR System (Applied Biosystems). GAPDH was used to normalize gene expression. Melting curves were analyzed to verify the specificity of the RT-PCR and the absence of primer dimer formation. Thermal cycle was programmed for 20 s at 95 °C as initial denaturation, followed by 40 cycles of 30 s at 95 °C, and 30 s at 60 °C, with final melt curve and extension for 15 s at 95 °C, 1 min. at 60 °C, and 15 s at 95°C. Custom PCR primers specific to murine p53 (F: TAGGTAGCGACTACAGTTAGGG; R: CATGGCAGTCATCCAGTCTT), p19Arf (F: TGAGGCTAGAGAGGATCTTGAGAA; R: GTGAACGTTGCCCATCATCATC); and p16Ink4a (F: CGGTCGTACCCCGATTCAG; R: GCACCGTAGTTGAGCAGAAGAG) were reported earlier[27].
Statistical analysis
Statistical analysis was performed using Prism7 (GraphPad, La Jolla, CA, USA). Data is represented as mean ± SEM. Data distribution was checked with Shapiro-Wilk normality test and the differences between two groups were analyzed by Mann-Whitney for nonnormally distributed data. Statistical analysis of Modified Thompson Grading was performed using Kruskal-Wallis test followed by Dunn’s multiple comparison test. χ2 test was used to analyze differences between distribution of percent-degenerated-discs. P≤0.05 was considered a statistically significant difference.
Supplementary Material
Supplemental Figure 1: (A-A’) Acantm1(cre/ERT2)Crm;p16fl/+;Rosa-ZsGreen mice were used as negative control to assess specificity of the TdTom detection. Robust expression of Aggrecan-Cre positive cells (ZsGreen) is seen in disc compartments without tdTOM/Red signal. Scale bar = 200 µm
Highlights.
p16Ink4a expression increased in the mouse intervertebral disc with aging
p16Ink4a conditional deletion does not alter cell morphology and tissue architecture of 18-month mouse intervertebral disc
p16Ink4a deletion results in small decrease in TUNEL positive cells in disc
p16Ink4a deletion results in changes in aggrecan functional attributes, collagen composition and lower levels of SASP markers
p16Ink4a is dispensable for induction and maintenance of senescence in the intervertebral disc
Acknowledgments:
The authors would like to thank Drs. Andrzej Fertala and Jolanta Fertala for help with the polarized microscopy. This study is supported by the grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) AR055655 and AR064733 to MVR and the National Institute on Aging F32 AG050399 to BOD (BOD). Part of this research was conducted while Brian Diekman was an Arthritis and Aging Research Grant recipient from the Arthritis National Research Foundation and the American Federation for Aging Research. EJN receives a PhD fellowship (PD/BD/128077/2016) from MD/PhD Program of the University of Minho funded by the Fundação para a Ciência e a Tecnologia (FCT).
Footnotes
Conflicts of Interest:
Nothing to disclosure.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- [1].Huang Y-C, Urban JPG, Luk KDK, Intervertebral disc regeneration: do nutrients lead the way?, Nat. Rev. Rheumatol 10 (2014) 1–6. doi: 10.1038/nrrheum.2014.91. [DOI] [PubMed] [Google Scholar]
- [2].Choi H, Merceron C, Mangiavini L, Seifert EL, Schipani E, Shapiro IM, V Risbud M, Hypoxia promotes noncanonical autophagy in nucleus pulposus cells independent of MTOR and HIF1A signaling, Autophagy 12 (2016) 1631–1646. doi: 10.1080/15548627.2016.1192753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schoepflin ZR, Silagi ES, Shapiro IM, Risbud MV, PHD3 is a transcriptional coactivator of HIF-1α in nucleus pulposus cells independent of the PKM2-JMJD5 axis, FASEB J 31 (2017) 3831–3847. doi: 10.1096/fj.201601291R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Choi H, Tessier S, Silagi ES, Kyada R, Yousefi F, Pleshko N, Shapiro IM, Risbud MV, A novel mouse model of intervertebral disc degeneration shows altered cell fate and matrix homeostasis, Matrix Biol 70 (2018) 102–122. doi: 10.1016/j.matbio.2018.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].V Vo N, Hartman RA, Patil PR, V Risbud M, Kletsas D, Iatridis JC, Hoyland JA, Le Maitre CL, Sowa GA, James D, Molecular Mechanisms of Biological Aging in Intervertebral Discs, J Orthop Res 34 (2016) 1289–1306. doi: 10.1002/jor.23195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhang Y, Xiong C, Kudelko M, Li Y, Wang C, Wong YL, Tam V, Rai MF, Cheverud J, Lawson HA, Sandell L, Chan WCW, Cheah KSE, Sham PC, Chan D, Early onset of disc degeneration in SM/J mice is associated with changes in ion transport systems and fibrotic events, Matrix Biol 70 (2018) 123–139. doi: 10.1016/j.matbio.2018.03.024. [DOI] [PubMed] [Google Scholar]
- [7].Risbud MV, Shapiro IM, Role of cytokines in intervertebral disc degeneration: Pain and disc content, Nat. Rev. Rheumatol 10 (2014) 10(1):44–56. doi: 10.1038/nrrheum.2013.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tang X, Jing L, Chen J, Changes in the Molecular Phenotype of Nucleus Pulposus Cells with Intervertebral Disc Aging, PLoS One 7 (2012) e52020. doi: 10.1371/journal.pone.0052020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].LeMaitre AJ, Freemont C, Hoyland JA, Research article Accelerated cellular senescence in degenerate intervertebral discs : a possible role in the pathogenesis of intervertebral disc degeneration, Arthritis Res. Ther 9 (2007) 1–12. doi: 10.1186/ar2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vo N, Seo HY, Robinson A, Sowa G, Bentley D, Taylor L, Studer R, Usas A, Huard J, Alber S, Watkins SC, Lee J, Coehlo P, Wang D, Loppini M, Robbins PD, Niedernhofer LJ, and Kang JK, James Niedernhofer, Accelerated Aging of Intervertebral Discs in a Mouse Model of Progeria, J Orthop Res 100 (2012) 130–134. doi: 10.1016/j.pestbp.2011.02.012.Investigations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Muñoz-espín D, Serrano M, Cellular senescence : from physiology to pathology, Nat. Rev. Mol. Cell Biol 15 (2014) 482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- [12].He S, Sharpless NE, Senescence in Health and Disease, Cell 169 (2017) 1000–1011. doi: 10.1016/j.cell.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li P, Gan Y, Xu Y, Song L, Wang L, Ouyang B, Zhang C, Zhou Q, The inflammatory cytokine TNF-α promotes the premature senescence of rat nucleus pulposus cells via the PI3K/Akt signaling pathway, Sci. Rep 7 (2017) 1–12. doi: 10.1038/srep42938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen D, Xia D, Pan Z, Xu D, Zhou Y, Wu Y, Cai N, Tang Q, Wang C, Yan M, Zhang JJ, Zhou K, Wang Q, Feng Y, Wang X, Xu H, Metformin protects against apoptosis and senescence in nucleus pulposus cells and ameliorates disc degeneration in vivo, Cell Death Dis 7 (2016) e2441. doi: 10.1038/cddis.2016.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Boquoi A, Arora S, Chen T, Litwin S, Koh J, Enders GH, Reversible cell cycle inhibition and premature aging features imposed by conditional expression of p16Ink4a, Aging Cell 14 (2015) 139–147. doi: 10.1111/acel.12279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sharpless NE, Sherr CJ, Forging a signature of in vivo senescence, Nat. Rev. Cancer 15 (2015) 397–408. doi: 10.1038/nrc3960. [DOI] [PubMed] [Google Scholar]
- [17].Jurk D, Lawless C, Wang C, Jurk D, Merz A, Von Zglinicki T, Passos JF, Quantitative assessment of markers for cell senescence, Exp. Gerontol 45 (2017) 772– 778. doi: 10.1016/j.exger.2010.01.018. [DOI] [PubMed] [Google Scholar]
- [18].Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, Van Deursen JM, Naturally occurring p16Ink4a-positive cells shorten healthy lifespan, Nature 530 (2016) 184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Geiger H, Depleting senescent cells to combat aging Tau toxicity feeds forward in frontotemporal dementia, Nat. Med 22 (2016) 23–24. doi: 10.1038/nm.4024. [DOI] [PubMed] [Google Scholar]
- [20].Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, Zhu Y, Schafer MJ, Tchkonia T, Kirkland JL, Miller JD, Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice, Aging Cell 15 (2016) 973–977. doi: 10.1111/acel.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, LeBrasseur NK, Cellular senescence mediates fibrotic pulmonary disease, Nat. Commun 23 (2017) 14532. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Hara SPO, Larusso NF, Jordan D, Roos CM, Verzosa GC, Nathan K, Wren JD, Farr JN, Stout MB, Mcgowan SJ, Gurkar AU, Zhao J, Dorronsoro A, Ling YY, Amira S, Navarro DC, Sano T, Paul D, Niedernhofer LJ, Kirkland JL, The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs, Aging Cell 14 (2015) 644–658. doi: 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jeon OH, Kim C, Laberge R, Demaria M, Rathod S, Vasserot AP, Chung JW, Kim DH, Poon Y, David N, Baker DJ, Van Deursen JM, Campisi J, Elisseeff JH, Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment, Nat. Med 23 (2017) 775–781. doi: 10.1038/nm.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ea G, T.K.E. K, F.M. Pj, Zoumpourlis V, Specific lipofuscin staining as a novel biomarker to detect replicative and stress ‐ induced senescence . A method applicable in cryo ‐ preserved and archival tissues, 5 (2013) 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].G.A. and S.N. Liu JY, Sourourllas GP, Diekman BO, Krishnamurthy J, Hall BM, Sorrentino JA, Parker JS, Sessions GA, Cells exhibiting strong p16INK4a promoter activation in vivo display features of senescence. Under Revision., Proc Natl Acad Sci U S A (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Monahan KB SN, Rozenberg GI, Krishnamurthy J, Johnson SM, Liu W, Bradford MK, Horner J, Depinho RA, Somatic p16(INK4a) loss accelerates melanomagenesis., Oncogene 29 (2010) 5809–5817. doi: 10.1038/onc.2010.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Diekman BO, Sessions GA, Collins JA, Knecht AK, Strum SL, Mitin NK, Carlson CS, Loeser RF, Sharpless NE, Expression of p16INK4ais a biomarker of chondrocyte aging but does not cause osteoarthritis, Aging Cell 17 (2018). doi: 10.1111/acel.12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Silagi ES, Batista P, Shapiro IM, Risbud MV, Expression of Carbonic Anhydrase III, a Nucleus Pulposus Phenotypic Marker, is Hypoxia-responsive and Confers Protection from Oxidative Stress-induced Cell Death, Sci. Rep 8 (2018) 4856. doi: 10.1038/s41598-018-23196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Risbud MV, Schoepflin ZR, Mwale F, Kandel RA, Grad S, Iatridis JC, Sakai D, Hoyland JA, Defining the phenotype of young healthy nucleus pulposus cells: Recommendations of the spine research interest group at the 2014 annual ORS meeting, J. Orthop. Res 33 (2015) 283–293. doi: 10.1002/jor.22789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Johnson ZI, Shapiro IM, V Risbud M, Extracellular osmolarity regulates matrix homeostasis in the intervertebral disc and articular cartilage : Evolving role of TonEBP, Matrix Biol 40 (2014) 10–16. doi: 10.1016/j.matbio.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Silagi ES, Shapiro IM, Risbud MV, Glycosaminoglycan synthesis in the nucleus pulposus: Dysregulation and the pathogenesis of disc disease, Matrix Biol 71–72 (2018) 368–379. doi: 10.1016/j.matbio.2018.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Halász K, Kassner A, Mörgelin M, Heinegård D, COMP Acts as a Catalyst in Collagen Fibrillogenesis, J. Biol. Chem 282 (2007) 31166–31173. doi: 10.1074/jbc.M705735200. [DOI] [PubMed] [Google Scholar]
- [33].Jung T, Höhn A, Grune T, Lipofuscin: Detection and quantification by microscopic techniques, Methods Mol. Biol 594 (2010) 173–193. doi: 10.1007/978-1-60761-411-1_13. [DOI] [PubMed] [Google Scholar]
- [34].Sharpless NE, Bardeesy N, Lee K, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, Depinho RA, Loss of p16 Ink4a with retention of p19 Arf predisposes mice to tumorigenesis, Nature 413 (2001) 4–9. [DOI] [PubMed] [Google Scholar]
- [35].Sharpless NE, Ramsey MR, Balasubramanian P, Castrillon DH, DePinho RA, The differential impact of p16INK4a or p19ARF deficiency on cell growth and tumorigenesis, Oncogene 23 (2004) 379–385. doi: 10.1038/sj.onc.1207074. [DOI] [PubMed] [Google Scholar]
- [36].Coppé J-P, Desprez P-Y, Krtolica A, Campisi J, The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression, Annu. Rev. Pathol. Mech. Dis 5 (2010) 99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ohnishi T, Sudo H, Tsujimoto T, Iwasaki N, Age-related spontaneous lumbar intervertebral disc degeneration in a mouse model, J. Orthop. Res 36 (2018) 224–232. doi: 10.1002/jor.23634. [DOI] [PubMed] [Google Scholar]
- [38].Alvarez-Garcia O, Matsuzaki T, Olmer M, Masuda K, Lotz MK, Age-related reduction in the expression of FOXO transcription factors and correlations with intervertebral disc degeneration, J. Orthop. Res 35 (2017) 1–102682–2691. doi: 10.1002/jor.23583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang J, Markova D, Anderson DG, Zheng Z, Shapiro IM, Risbud MV, TNF and IL-1 promote a disintegrin-like and metalloprotease with thrombospondin type I motif-5-mediated aggrecan degradation through syndecan-4 in intervertebral disc, J. Biol. Chem 286 (2011) 39738–39749. doi: 10.1074/jbc.M111.264549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ngo K, Patil P, McGowan SJ, Niedernhofer LJ, Robbins PD, Kang J, Sowa G, Vo N, Senescent intervertebral disc cells exhibit perturbed matrix homeostasis phenotype, Mech. Ageing Dev 166 (2017) 16–23. doi: 10.1016/j.mad.2017.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wheeler JB, Mukherjee R, Stroud RE, Jones JA, Ikonomidis JS, Relation of murine thoracic aortic structural and cellular changes with aging to passive and active mechanical properties, J. Am. Heart Assoc 4 (2015) e001744. doi: 10.1161/JAHA.114.001744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Van Deursen JM, The role of senescent cells in ageing, Nature 509 (2014) 439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].LeMaitre C, A. F., Hoyland J, Accelerated cellular senescence in degenerate intervertebral discs: a possible role in the pathogenesis of intervertebral disc degeneration, Arthritis Res. Ther 9 (2007) R45. doi: 10.1186/ar2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Vamvakas SS, Mavrogonatou E, Kletsas D, Human nucleus pulposus intervertebral disc cells becoming senescent using different treatments exhibit a similar transcriptional profile of catabolic and inflammatory genes, Eur. Spine J (2017). doi: 10.1007/s00586-017-5198-0. [DOI] [PubMed] [Google Scholar]
- [45].Hayes AJ, Hughes CE, Ralphs JR, Caterson B, Chondroitin sulphate sulphation motif expression in the ontogeny of the intervertebral disc, Eur. Cells Mater 21 (2011) 1–14. doi:vol021a01 [pii]. [PubMed] [Google Scholar]
- [46].Sivan SS, Wachtel E, Roughley P, Structure, function, aging and turnover of aggrecan in the intervertebral disc, Biochim. Biophys. Acta - Gen. Subj (2014). doi: 10.1016/j.bbagen.2014.07.013. [DOI] [PubMed] [Google Scholar]
- [47].Little CB, Meeker CT, Golub SB, Lawlor KE, Farmer PJ, Smith SM, Fosang AJ, Blocking aggrecanase cleavage in the aggrecan interglobular domain abrogates cartilage erosion and promotes cartilage repair, J. Clin. Invest (2007). doi: 10.1172/JCI30765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Collin EC, Carroll O, Kilcoyne M, Peroglio M, See E, Hendig D, Alini M, Grad S, Pandit A, Ageing affects chondroitin sulfates and their synthetic enzymes in the intervertebral disc, Signal Transduct. Target. Ther (2017). doi: 10.1038/sigtrans.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kouroumalis A, Mavrogonatou E, Savvidou OD, Papagelopoulos PJ, Pratsinis H, Kletsas D, Major traits of the senescent phenotype of nucleus pulposus intervertebral disc cells persist under the specific microenvironmental conditions of the tissue, Mech. Ageing Dev (2018). doi: 10.1016/j.mad.2018.05.007. [DOI] [PubMed] [Google Scholar]
- [50].Pan H, Strickland A, Madhu V, Johnson ZI, Chand SN, Brody JR, Fertala A, Zheng Z, Shapiro IM, Risbud MV, RNA binding protein HuR regulates extracellular matrix gene expression and pH homeostasis independent of controlling HIF-1α signaling in nucleus pulposus cells, Matrix Biol (2018). doi: 10.1016/j.matbio.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Holguin N, Aguilar R, Harland RA, Bomar BA, Silva MJ, The aging mouse partially models the aging human spine: lumbar and coccygeal disc height, composition, mechanical properties, and Wnt signaling in young and old mice, J. Appl. Physiol 116 (2014) 1551–1560. doi: 10.1152/japplphysiol.01322.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Binch ALA, Shapiro IM, V Risbud M, Syndecan-4 in intervertebral disc and cartilage: Saint or synner?, Matrix Biol 52 (2016) 355–362. doi: 10.1016/j.matbio.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].K.M.L. and Burd CE, The Molecular Balancing Act of p16INK4a in Cancer and Aging, Mol Cancer Res 27 (2014) 320–331. doi: 10.1002/nbm.3066.Non-invasive. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Feng C, Yang M, Zhang Y, Lan M, Huang B, Liu H, Zhou Y, Cyclic mechanical tension reinforces DNA damage and activates the p53-p21-Rb pathway to induce premature senescence of nucleus pulposus cells, Int. J. Mol. Med (2018). doi: 10.3892/ijmm.2018.3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhou N, Lin X, Dong W, Huang W, Jiang W, Lin L, Qiu Q, Zhang X, Shen J, Song Z, Liang X, Hao J, Wang D, Hu Z, SIRT1 alleviates senescence of degenerative human intervertebral disc cartilage endo-plate cells via the p53/p21 pathway, Sci. Rep (2016). doi: 10.1038/srep22628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Capell BC, Drake AM, Zhu J, Shah PP, Dou Z, Dorsey J, Simola DF, Donahue G, Sammons M, Rai TS, Natale C, Ridky TW, Adams PD, Berger SL, MLL1 is essential for the senescence-associated secretory phenotype, Genes Dev 2 (2016) 321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].De Keizer PLJ, The Fountain of Youth by Targeting Senescent Cells?, Trends Mol. Med 23 (2017) 6–17. doi: 10.1016/j.molmed.2016.11.006. [DOI] [PubMed] [Google Scholar]
- [58].Coppé JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J, Tumor suppressor and aging biomarker p16 INK4a induces cellular senescence without the associated inflammatory secretory phenotype, J. Biol. Chem 286 (2011) 36396–36403. doi: 10.1074/jbc.M111.257071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhang K, Huang L, Sun H, Zhu Y, Xiao Y, Role of Notch expression in premature senescence of murine bone marrow stromal cells, Prog. Nat. Sci 19 (2009) 557–562. doi: 10.1016/j.pnsc.2008.09.005. [DOI] [Google Scholar]
- [60].Tran CM, Markova D, Smith HE, Susarla B, Ponnappan RK, Anderson DG, Symes A, Shapiro IM, V Risbud M, Regulation of CCN2/CTGF expression in the nucleus pulposus of the intervertebral disc: Role of smad and AP1 signaling, Arthritis Rheum (2010) NA-NA. doi: 10.1002/art.27445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM, Bardeesy N, Castrillon DH, Beach DH, Sharpless NE, Monitoring tumorigenesis and senescence in vivo with a p16 INK4a-luciferase model, Cell 152 (2013) 340–351. doi: 10.1016/j.cell.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: (A-A’) Acantm1(cre/ERT2)Crm;p16fl/+;Rosa-ZsGreen mice were used as negative control to assess specificity of the TdTom detection. Robust expression of Aggrecan-Cre positive cells (ZsGreen) is seen in disc compartments without tdTOM/Red signal. Scale bar = 200 µm